 Yi Yuan1†
Yi Yuan1† Zhihua Lin
Zhihua Lin Wenfang Dai
Wenfang Dai- 1Zhejiang Key Laboratory of Aquatic Germplasm Resource, College of Biological and Environmental Sciences, Zhejiang Wanli University, Ningbo, China
- 2Ninghai Institute of Mariculture Breeding and Seed Industry, Zhejiang Wanli University, Ningbo, China
Introduction: Razor clam (Sinonovacula constricta) is a bivalve of economic importance that widely resides in coastal and estuarine areas where salinity fluctuates rapidly. Gut microbiota, which plays a pivotal role in the disease resistance against pathogens and the growth and development of aquatic animals, is affected by salinityfluctuation in ambient environment. However, there is limited knowledge about the underlying mechanism of how clam gut microbiota respond to alternating salinity stress.
Methods: Here, we exposed S. constricta to different salinity treatments including normal salinity-acclimated (NN, 20 psu), low salinity-acclimated (LL, 5 psu), low salinity-stressed (NL, transferred from 20 to 5 psu) and normal salinity-stressed (LN, transferred from 5 to 20 psu) groups. The former two groups were used as the control, and the latter two groups aimed to investigate the effect of alternating salinity stress on clam growth and gut microbiota by 16S rRNA gene amplicon sequencing.
Results: Alternating salinity stress caused faster and higher mortality compared with the two control groups, and disrupted the gut microbiota including altered α-diversity, composition and structure. The salinity stress, hours post stress and their interaction had significant impact on the gut microbiota, which contributed 4.5%, 9.5% and 6.0%, respectively, to the variance in the gut communities. Alternating salinity stress increased the proportion of stochastic processes in governing the gut microbiota to a certain extent, and the stochasticity aggravated with the increase of stress time. Furthermore, the gut bacterial interspecies interaction networks exhibited more complex and higher connected topology in two alternating salinity stress groups compared with the control group (NN), and the interaction relationships between core OTUs in gut networks were altered after salinity stress. The functional potentials involved in immune system and metabolisms of amino acid, energy, carbohydrate and lipid remarkably increased in LN and NN groups after salinity exposure for 96 h compared to corresponding 0 h, while these pathways exhibited the opposite pattern in NL and LL groups.
Discussion: Collectively, our findings provide evidence that alternating salinity stress has potentially negative effect on the growth of S. constricta from an ecological viewpoint.
1 Introduction
Razor clam (Sinonovacula constricta), a typical filter-feeder and burrowing bivalve, is widely resided in coastal and estuarine zones where salinity fluctuates rapidly (Chen et al., 2021; Dai et al., 2023). It is an economically important bivalve worldwide, and is one of four principal aquatic bivalve species in China. In recent years, artificial breeding becomes the mainstream for clam aquaculture (Liu et al., 2009). In indoor hatchery, the physicochemical factors enable well governed, especially for salinity concentration that plays crucial role for larval metamorphosis, development and survival (Davis, 1958; Peng et al., 2019; Chen et al., 2021). However, when grown into juveniles, the larvae will be instantly sowed into natural mudflats where salinity might be dramatically distinct from that of the hatchery (Narasimham and Laxmilatha, 1996). Due to typhoon, tidal fluctuation or heavy rain, the change of salinity concentration in marine environment is inevitable. These changes could result in severe salinity challenges for marine animals living in these areas, including S. constricta. In response to the rapid changes in salinity, the clams must develop extraordinary adaptations and biological properties. Yet, little is known about the underlying response mechanisms for alternating salinity stress in clams.
The gut symbionts, as an immune and digestive organ, contribute important roles in maintaining host homeostasis and touching off immune response against invasive pathogens (Pickard et al., 2017; Dai et al., 2023). Increasing studies have evidenced that gut microbiota are closely associated to the growth, development, immunity and nutrient absorption of aquatic animals (Xiong et al., 2019; Dai et al., 2022a, 2022b; Wu et al., 2024). Notably, the change of physicochemical factors in ambient environment such as salinity can disturb balance in gut microbial community, which in turn affects the health and fitness of marine organisms (Dai et al., 2023). Salinity is proved to be an key factor in shaping gut microbial communities in host (Hou et al., 2020; Hieu et al., 2022). However, it is still unclear how and to what extent salinity variation influences gut microbiota in clams that are inhabited in coastal and estuarine zones.
To examine how alternating salinity stress influences clam survival and the composition and dynamics of gut microbiota, we exposed S. constricta to different salinity treatments including normal salinity-acclimated (20 psu), low salinity-acclimated (5 psu), low salinity-stressed (transferred from 20 to 5 psu) and normal salinity-stressed (transferred from 5 to 20 psu) groups (Figure 1A). Thus, we presented experimental data depicting the short-term gut microbial dynamics in response to abiotic factor stress in razor clams. This study aimed to investigate the following concerns: (i) how salinity variation influenced the structures, network stability, and functional potentials of clam gut microbiota; (ii) unveiling the ecological mechanisms underlying the susceptibility to abiotic factor after salinity challenge; and (iii) attempting to bridge the variation in gut microbial communities to distinct salinity stressors and host survival. Our findings would broaden our knowledge on how abiotic factor influences the gut microbiota of bivalve species, and provide scientific basis for the development of bivalve industry.
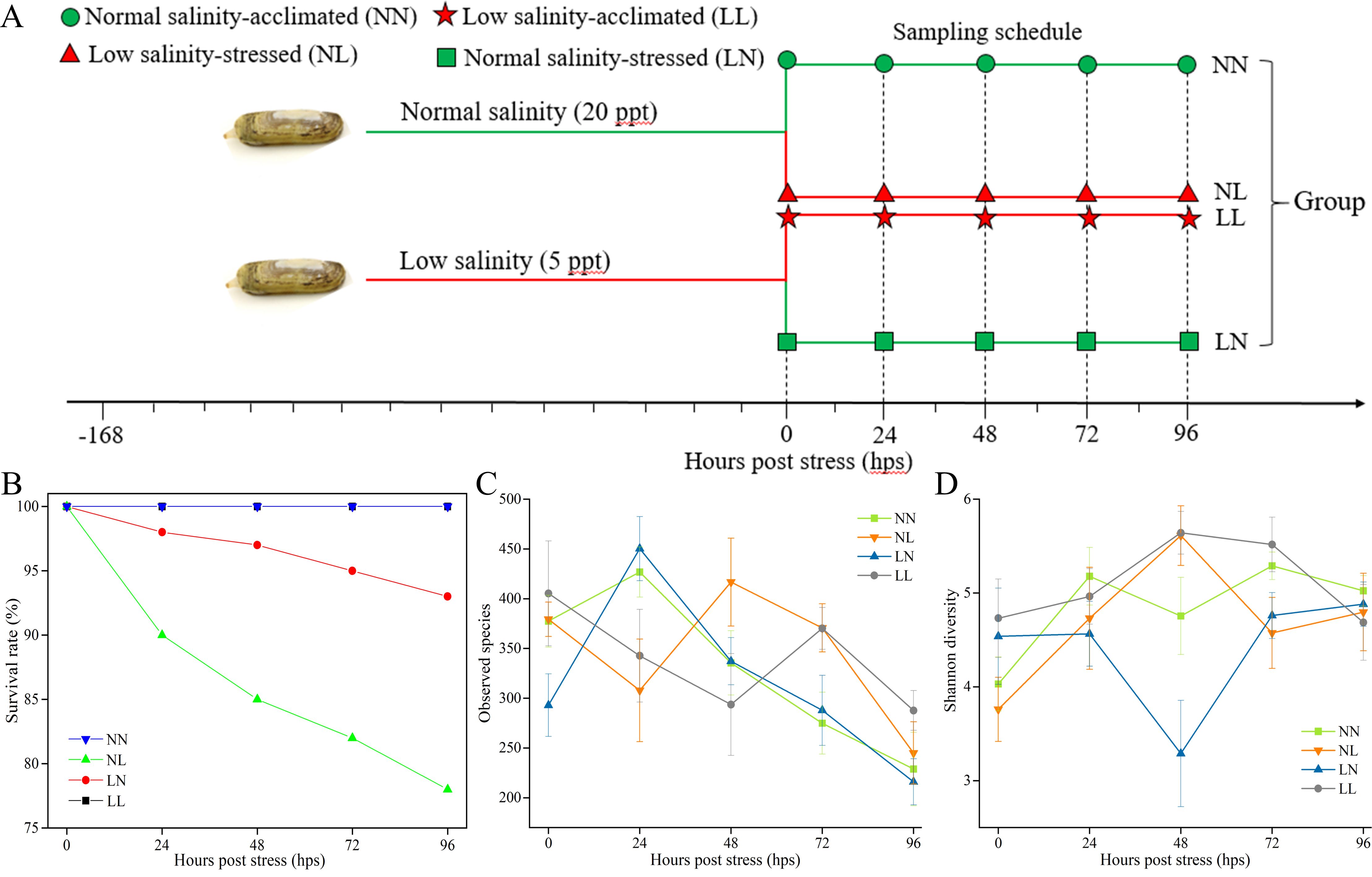
Figure 1. The experimental design (A), change of clam survival rate (B) and gut bacterial community α-diversity indices including observed species (C) and Shannon diversity (D) with stress time.
2 Materials and methods
2.1 Experimental design and gut sampling
The experiment was designed to examine the effects of normal- and low-salinity, and alternating salinity stress on the gut microbiota in razor clam (S. constricta) in a fully crossed two-factorial design for duration of 96 h. Adult razor clams with average shell length of 5.12 ± 0.31 cm and average body weight of 6.68 ± 0.56 g were sampled from a shellfish aquaculture farm located at Ningbo city, Zhejiang Province, China. After transportation to the lab, the clams were respectively acclimatized under two salinity concentrations (i.e., 20 and 5 psu) for 7 days in filtered flowing seawater (temperature at 22.35 ± 0.21°C and dissolved oxygen at 5.60 ± 0.34 mg/L by constant aeration) with 50% daily water exchange, and fed once daily with microalgae Chaeroeeros moelleri containing rich nutritional composition such as carbohydrate, fat and protein. This microalgae was gained from the Marine Biotechnology Laboratory of Ningbo University. The clams were kept singly in aerated glass jars after being randomly divided into experimental treatments. The clams were either left at their acclimation salinity or stressed to the opposite salinity, resulting in four salinity treatments: normal salinity-acclimated (20 psu, selected as control group, abbreviated as NN), low salinity-acclimated (5 psu, selected as control group, abbreviated as LL), low salinity-stressed (transferred from normal salinity to low salinity, abbreviated as NL) and normal salinity-stressed (transferred from low salinity to normal salinity, abbreviated as LN) (Figure 1A). Each group was set with three replicate tanks. In order to monitor the responses of gut microbiota to four salinity treatments, four clams from each tank from each group were sampled at 0, 24, 48, 72 and 96 hours post stress (hps), respectively (Figure 1A). Correspondingly, the whole guts from every two clams were dissected aseptically and pooled to form one biological sample due to that inadequate DNA could be obtained from a single clam in trial runs. Ultimately, 120 samples (5 samplings × 6 replicates × 4 treatments) were collected for gut microbial analysis.
2.2 Bacterial DNA extraction, PCR amplification, Illumina MiSeq sequencing and sequencing data processing
The total gDNA (genomic DNA) of clam gut microbiota was extracted using the FastDNA™ SPIN Kit (MO BIO Laboratories) following the instruction manual. The concentrations of gDNA extracts were quantified, and the purity of gDNA was examined using the NanoDrop ND-2000 spectrophotometer. We amplified the V3-V4 regions of gut bacterial 16S rRNA genes with bacterium-specific primer pair: 338F and 806R. Each sample was amplified in 50 μL reaction system in triplicate to minimize PCR-induced bias. For each sample, the PCR cycle condition was carried out based on the description in our previous study (Dai et al., 2023). Further, PCR products were visualized in a 1.5% agarose gel to validate the expected size. Then, we purified and quantified the PCR product from each sample with the commercial PCR fragment purification kit and Quant-iT PicoGreen dsDNA assay, respectively. Each sample was pooled in equimolar amount, and the final amplicons were sent to sequence on Illumina MiSeq platform.
After sequencing, the raw data was processed with an integrated Dix-seq pipeline (Wei et al., 2020). In detail, the pipeline performed the Trimmomatic to filter the barcode and low-quality sequences. After filtering, the atlas-utilis was employed to remove chimeric reads. Paired-end reads were merged with default parameters by performing USEARCH (Edgar, 2010). Finally, the bacterial phylotypes were discerned with UCLUST and binned into operational taxonomic unit (OTU) table at 97% similarity threshold (Edgar, 2010). After taxonomical assignment, the OTUs attached to archaea, eukaryota, chloroplasts, erroneous sequences and those unassigned at the bacteria domain were ruled out from the dataset. We identified the representative sequences and taxonomy classification for each OTU via RDP Classifier in the SILVA database, and constructed a phylogenetic tree of 16S rRNA genes from the filtered alignment with FastTree and IQ-TREE (Price et al., 2010; Lan et al., 2012). Finally, the α-diversity indices of clam gut microbial samples were generated, and the bacterial β-diversity distance between clam gut microbial samples was evaluated based on the OTU clustering results.
2.3 Estimating the ecological processes of gut microbiota
We constructed a Sloan’s neutral community model (NCM) to estimate the contribution of stochasticity governing the clam gut microbial community structure (Sloan et al., 2006). The nonlinear least-squares between the OTUs frequency and their abundances were assessed via the Östman’s approach, in which R2 determines the overall fit to the neutral community model (Chen et al., 2019). A R2 value is close to 1, indicating that the community assembly is fully stochastic.
2.4 Inferring the gut microbial interaction networks
To assess the effect of alternating salinity stress on the interspecies interactions within clam gut microbiota, bacterial network analyses were performed with MetaMIS (Metagenomic Microbial Interaction Simulator) software by introducing time-series dataset of gut bacterial compositions at OTU level for NN, NL, LN and LL group samples, respectively. MetaMIS is a tool based on the generalized Lotka Volterra equations, and is designed to infer underlying microbial interactions based on the metagenomic abundance profiles (Shaw et al., 2016). Positive or negative interactions between two nodes (i.e., OTUs) in each microbial interaction network were inferred. This approach systematically detects the interaction patterns of each node with other nodes, such as mutualism (+/+), competition (−/−), parasitism or predation (+/−), commensalism (+/0), amensalism (−/0), and no effect (0/0) in four salinity stress groups, respectively.
2.5 Predicting the functional potentials of gut microbiota
Function potentials of clam gut bacterial communities were inferred using PICRUSt2 v2.4.1 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) according to the KEGG (Kyoto Encyclopedia of Genes and Genomes) database for phenotype prediction (Douglas et al., 2019). Significantly differential functional pathways between NN96 and NN0, between NL96 and NL0, between LN96 and LN0 and between LL96 and LL0 were selected using response ratio analysis at a confidence interval (CI) of 95% (Dai et al., 2022a).
2.6 Statistical analysis
One-way analysis of variance (ANOVA) was used to detect the significant differences in the clam gut bacterial compositions at family level among four salinity stress groups at same stress time (Churchill, 2004). Based on Bray-Curtis distance, a principal component analysis (PCA) and analysis of similarity (ANOSIM) were performed to assess the differences in the β-diversity of clam gut bacterial communities among four salinity stress groups with the increase of stress time (Mishra et al., 2017). A permutational multivariate analysis of variance (perMANOVA) was carried out to qualify the contributions of salinity stress, hps, and their interaction on the variation in clam gut bacterial communities based on “vegan” package in R (R Development Core Team, 2013). Venn diagram was applied to count the number of shared and unique OTUs among four salinity stress groups at same time point and at each salinity stress group over stress time.
3 Results
3.1 Alternating salinity stress affecting the survival of S. constricta
The survival rate of S. constricta was unaffected by the 96-hour salinity stress in NN and LL groups (Figure 1B). In contrast, the mortality was firstly observed on 24 hps and showed a continuously decreased trend in NL and LN groups. Notably, the cumulative survival rate in NL group was 78% on 96 hps, which was significantly lower than that in LN group (93%) (Figure 1B).
3.2 Responses of gut bacterial community to alternating salinity stress
A total of 17,689,879 high-quality sequences were gained with an average of 147,416 ± 55,299 (mean ± standard deviation) sequences per sample, which generated 1784 OTUs across our enrolled 120 samples corresponding to NN, NL, LN and LL groups. Among these OTUs, there were 35, 42, 52, 34 and 56 OTUs shared among four salinity stress groups at 0, 24, 48, 72 and 96 hps, respectively, while unique OTU number varied accordingly in each salinity stress group (Supplementary Figure S1A). In addition, the NN and LL groups displayed more shared OTUs than the NL and LN groups during 96-hour salinity stress period (Supplementary Figure S1B). The trend of unique OTU numbers was ordered as NN48 (125) > NN96 (88) > NN24 (87) > NN0 (76) > NN72 (66), NL24 (178) > NL48 (155) > NL96 (102) > NL0 (85) > NL72 (77), LN72 (155) > LN96 (136) >LN24 (98) > LN0 (95) > LN48 (32), LL72 (165) > LL24 (132) > LL48 (114) > LL0 (75) > LL96 (65) within 96 hps (Supplementary Figure S1B).
Alternating salinity stress affected the α-diversity of clam gut bacterial communities. The α-diversity index such as observed species in NN and LN groups increased at 24 hps and then dropped continuously with lowest level at 96 hps (Figure 1C). However, an opposite trend was observed in NL and LL groups at 24 hps. Notably, the observed species was lower in NL and LL groups at 96 hps than that at 0 hps (Figure 1C). The Shannon diversity in NL and LL groups kept an increased trend from 0 to 48 hps and then decreased at 72 and 96 hps (Figure 1D). A completely opposite pattern was detected in LN group within 96 hps. For NN group, the Shannon diversity firstly raised at 24 hps but declined at 48 hps, then increased at 72 hps and finally decreased at 96 hps (Figure 1D).
The bacterial phyla were predominant by Tenericutes (20.4% ± 12.7%, mean ± standard deviation) and Gammaproteobacteria (16.1% ± 7.0%), followed by Alphaproteobacteria (14.1% ± 13.2%), Campilobacterota (12.5% ± 5.8%), Bacteroidetes (5.3% ± 4.9%), Actinobacteria (4.6% ± 3.3%), and Firmicutes (4.1% ± 5.4%) across the samples. Additionally, Betaproteobacteria (0.5% ± 0.3%), Fusobacteria (0.3% ± 0.7%), Planctomycetes (0.3% ± 0.4%), and Verrucomicrobia (0.1% ± 0.1%) were identified across the samples at low abundances (Supplementary Figure S2A). These large standard deviations might be due to the highly temporal succession of gut bacterial compositions (Figure 2). The relative abundance of Tenericutes decreased significantly (P < 0.05) in four salinity stress groups at 24, 72 and 96 hps compared with corresponding 0 hps. In contrast, the relative abundances of Alphaproteobacteria and Gammaproteobacteria drastically increased (P < 0.05) at 24 hps but decreased at 96 hps in the four salinity stress groups. Notably, the relative abundances of Firmicutes, Actinobacteria, Fusobacteria, Planctomycetes, Betaproteobacteria and Verrucomicrobia exhibited consistent declined trend in four salinity stress groups at 96 hps (Supplementary Figure S2A). At finer family level, the dominant families were Arcobacteraceae (12.4% ± 5.8%), Vibrionaceae (5.5% ± 4.3%), Pseudomonadaceae (5.2% ± 3.8%), Sphingomonadaceae (4.7% ± 6.8%), Methylobacteriaceae (4.5% ± 6.7%), Shewanellaceae (2.3% ± 3.4%), Flavobacteriaceae (2.3% ± 1.9%) and Rhodobacteraceae (2.2% ± 1.8%) (Supplementary Figure S2B). However, alternating salinity stress led to an apparent divergence in dominant taxa distribution in clam gut within 96 hps (Figure 3). It is worth noting that the relative abundances of potentially pathogenic Pseudomonadaceae and Shewanellaceae increased in LN and LL groups at 96 hps relative to that at 72 hps. An opposite trend for this phenomenon was observed in NN and NL groups (Figure 3).
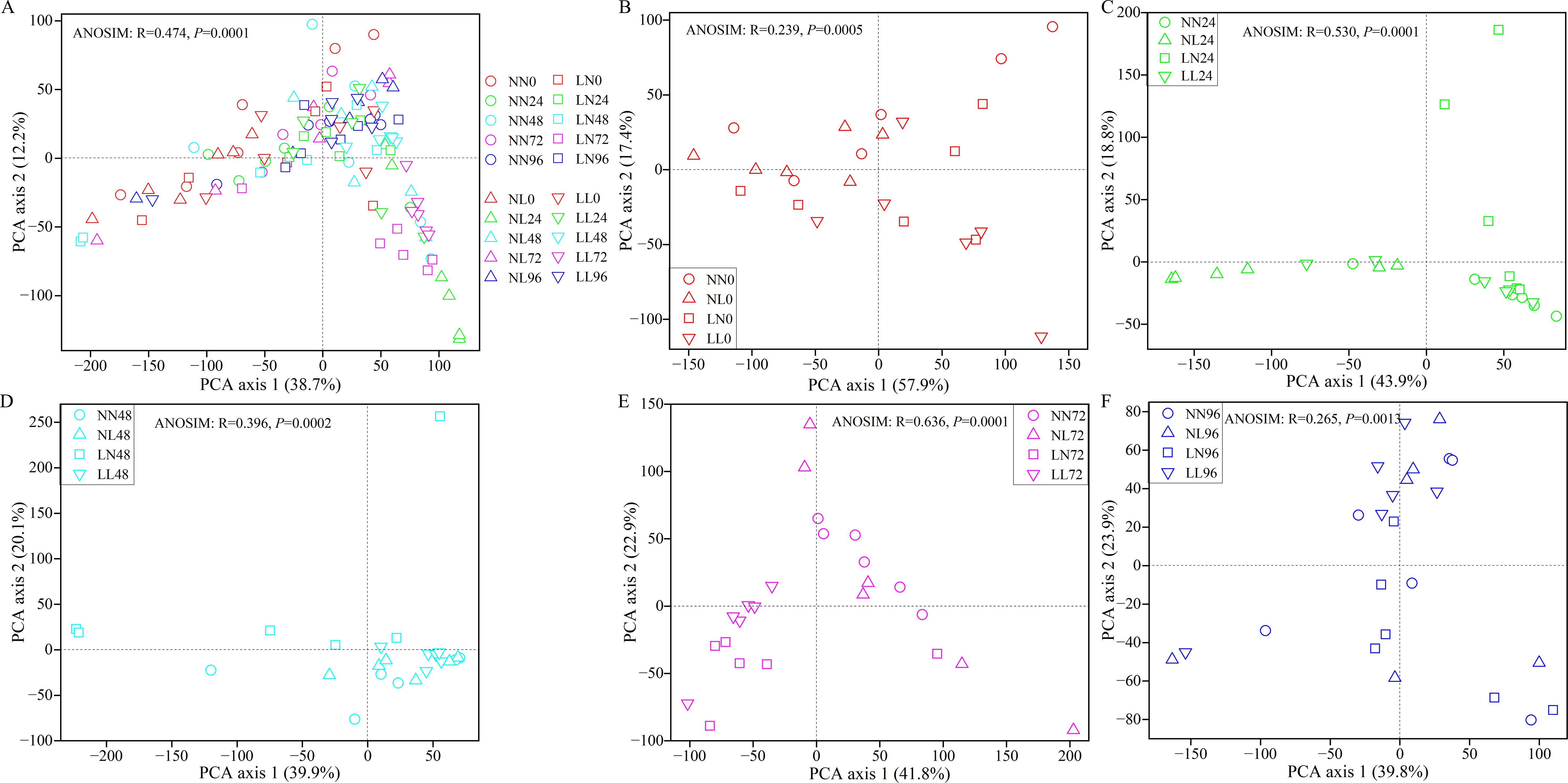
Figure 2. Principal Component Analysis (PCA) of the dissimilarity in gut bacterial community compositions among four salinity stress groups. (A) clam gut bacterial communities in NN, NL, LN and LL groups over stress time. (B–F) represent gut communities of four salinity stress groups at 0, 24, 48, 72 and 96 hps, respectively. N, normal salinity; L, low salinity. The numbers represent hours post stress. P < 0.05 indicates significant difference in gut bacterial community structures among four salinity stress groups at same hps.
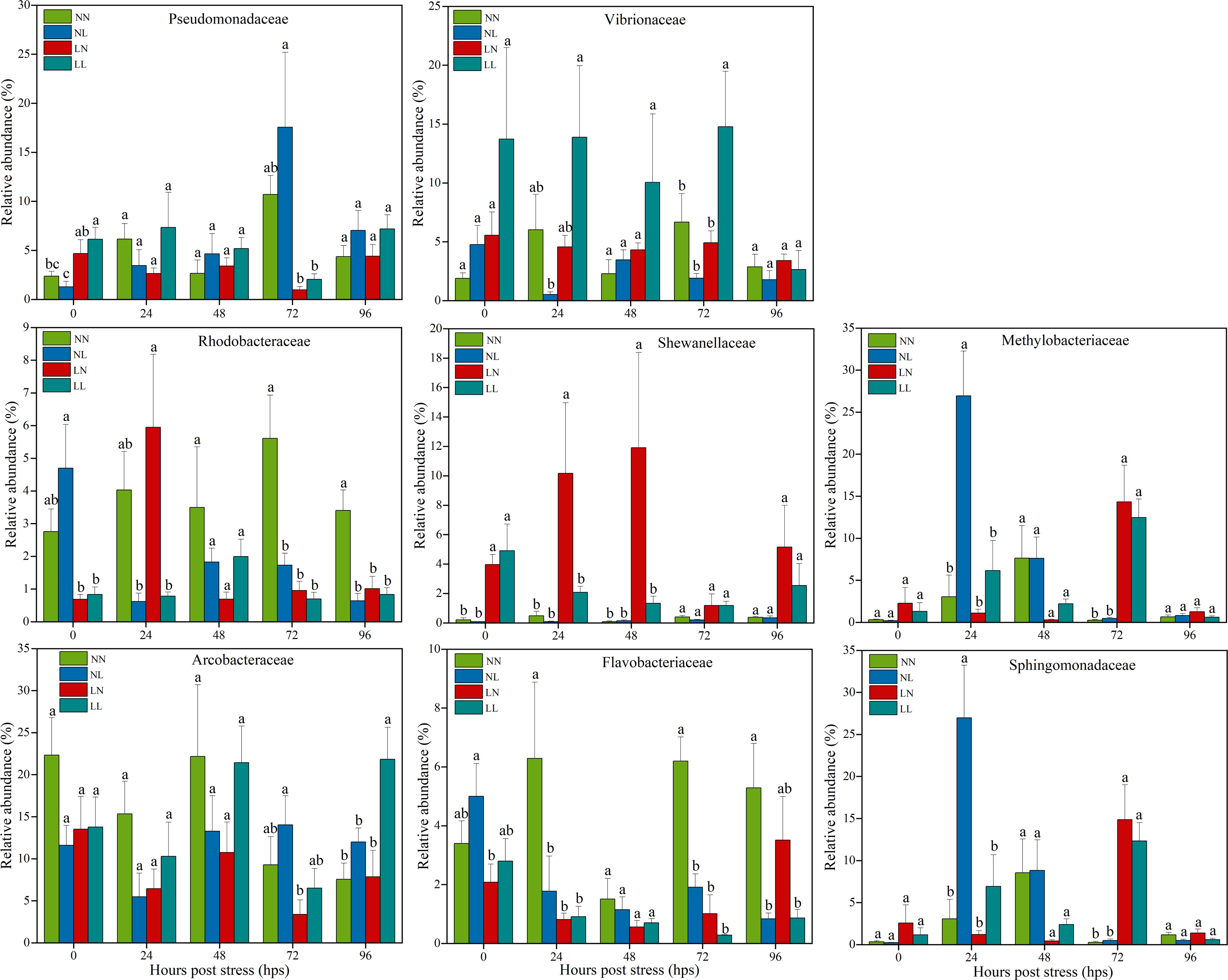
Figure 3. Bar plots illustrating the dynamics of the relative abundances of key gut bacterial families in NN, NL, LN and LL groups over stress time. Different lowercase letters above the histograms indicate significant differences among groups at same hps using one-way ANOVA with P < 0.05 considered significant.
The PCA plot illustrated dissimilarities in varying degree in the gut microbiota among four salinity treatments and among different stress time (Figure 2A). For instance, overall, the four salinity treatments markedly contributed to the divergence of the gut bacterial community at each time point (Figures 2B–F). At 0 hps, a significant difference in gut bacterial community was observed only between NL and LN groups. In the NN group, apparent separation was observed between each pair of groups among stress time, with an exception between NN24 and NN48 and between NN72 and NN96. For the NL group, the gut bacterial structures were dramatically different between each pair of groups within 96 hps, with an exception between NL72 and NL96. For LN group, the gut microbiota in LN0 was clustered away from that in LN24, LN72 and LN96, and each pair of the latter three groups exhibited apparent separation. For LL group, the gut microbiota were remarkably distinct between each pair of groups within 96 hps, with an exception between LL24 and LL0/LL48 (Figure 2). These findings were further confirmed by a dissimilarity test (Supplementary Table S1). Further, PerMANOVA revealed that salinity stress, hours post stress and their interaction dramatically contributed to 4.5%, 9.5% and 6.0% variation in the gut bacterial communities, respectively (P < 0.01 for the three variables) (Table 1).
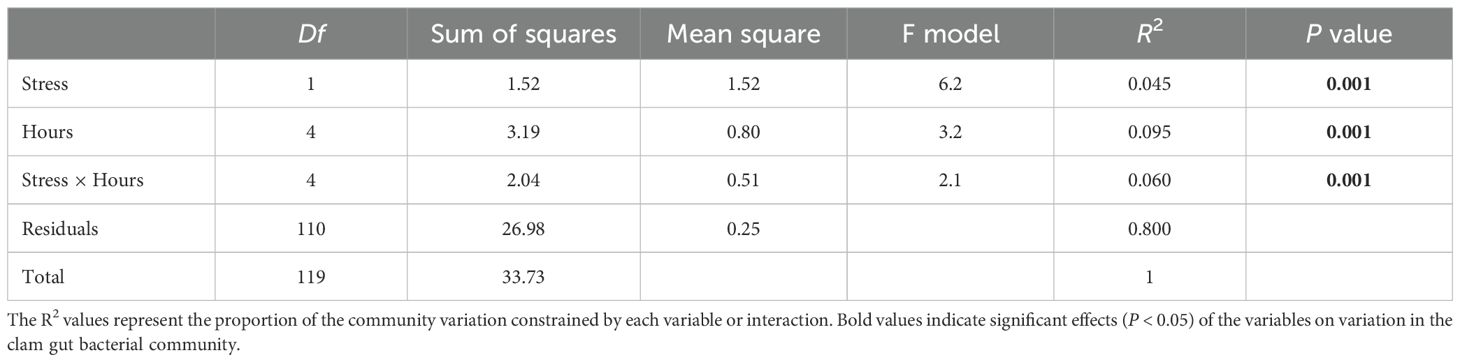
Table 1. Quantitative effects of salinity stress and hours post stress on the variation in clam gut bacterial communities based on permutational multivariate analysis of variance (perMANOVA).
3.3 Alternating salinity stress shifting the ecological processes that controlled the gut bacterial assembly
The R2 value was 0.279 in NN group, which increased to 0.491 and 0.448 in LN and LL groups, respectively, but was comparable with that NL group (0.270) on 0 hps (Figure 4). Therefore, low salinity stress enhanced stochasticity underlying the gut microbiota. The subsequent alternating salinity stress aggravated stochasticity. For example, the R2 value in NL (R2 = 0.382) group at 96 hps was higher than that at corresponding 0 hps (Figures 4B, D). However, the ecological processes did not change in NN group at 96 hps due to the R2 value (0.270) was comparable with that at 0 hps (Figure 4A).
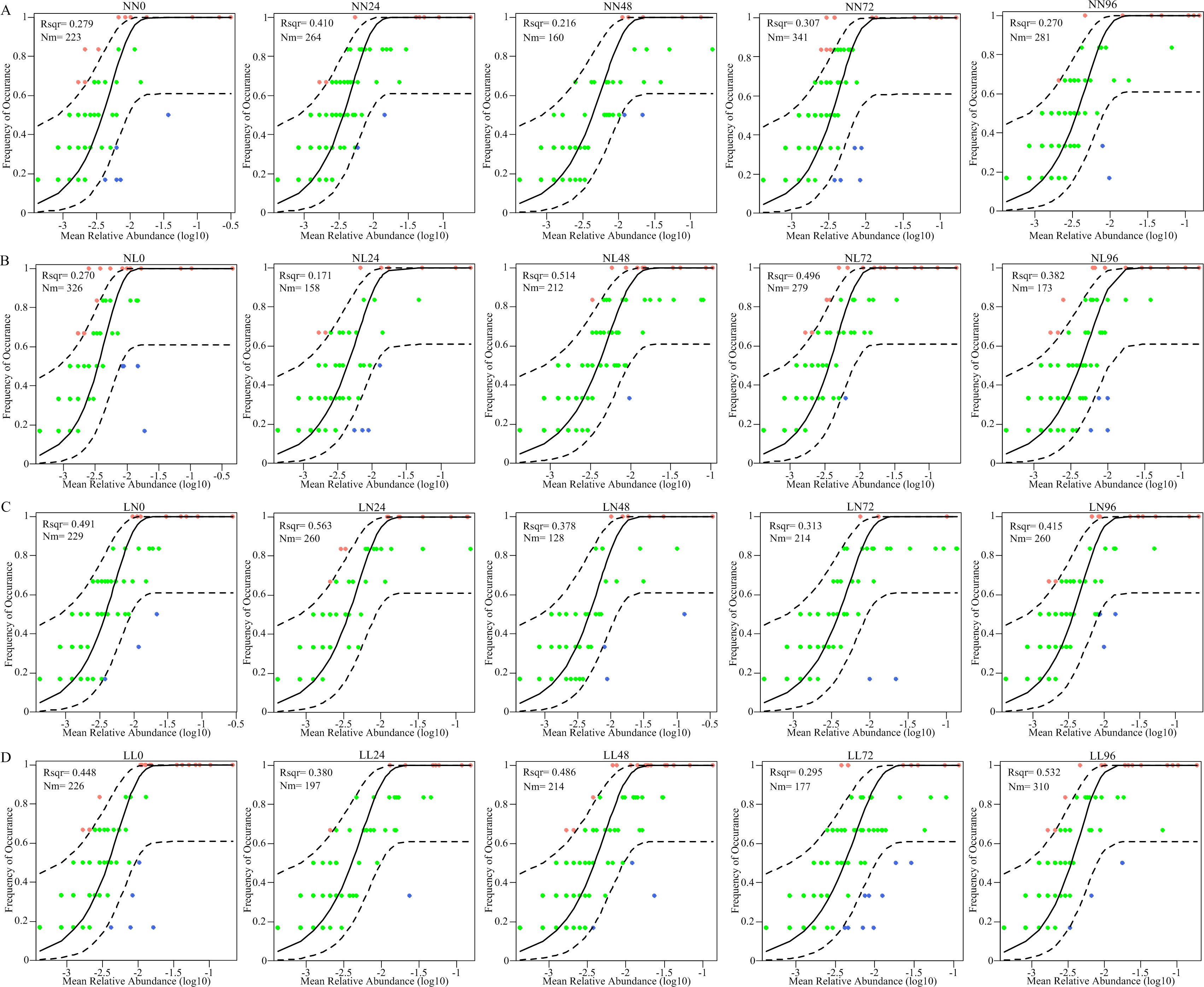
Figure 4. Salinity stress effects on the ecological processes shaping clam gut microbiota. Ecological processes governing the gut microbiota in NN (A), NL (B), LN (C) and LL (D) groups at 0, 24, 48, 72 and 96 hps using the neutral community model (NCM), respectively. The best fit to the NCM is shown by the solid black lines. The 95% confidence intervals around the model prediction are shown as dashed black lines.
3.4 Effects of alternating salinity stress on gut bacterial interaction relationships
To estimate the effects of alternating salinity stress on the species-to-species interactions within gut microbiotas, bacterial network analyses based on time-series dataset of gut microbial compositions were performed with MetaMIS for the samples in NN, NL, LN and LL groups, respectively (Figure 5). We exhibited the gut bacterial interaction relationships among core taxa in four salinity stress groups. Compared to that in the NN group (66.7%), the positive interactions decreased in NL (48.1%), LN (59.0%) and LL (51.9%) groups, suggesting a lower degree of cooperative activities in the gut microbiota of latter three groups. In contrast, interspecies interaction networks presented more complex and higher connected topology in NL, LN and LL groups compared to NN group (Figure 5). Notably, there were more number of core OTUs that were attached to Vibrio, Pseudomonas, and Shewanella taxa (core ratio = 100%, Supplementary Table S2) in the gut network of LL group in relation to NN group (Supplementary Table S3). In the four microbial networks, a variety of interaction types were observed between core OTUs. These included competition (-/-), parasitism/predation (+/-), commensalism (+/0), and amensalism (-/0) (Supplementary Figure S3). For instance, the most frequent interactive relationship for OTU3 was mutualism (+/+) in NN group, which was shifted to parasitism or predation (+/-) in NL, LN and LL groups. For OTU5, the most frequent interaction was competition (−/−) in NL and LL groups, but was mutualism (+/+) in NN group and parasitism or predation (+/-) in LN group (Supplementary Figure S3). These results indicated that alternating salinity stress affected the interspecies interaction relationships of gut microbial networks.
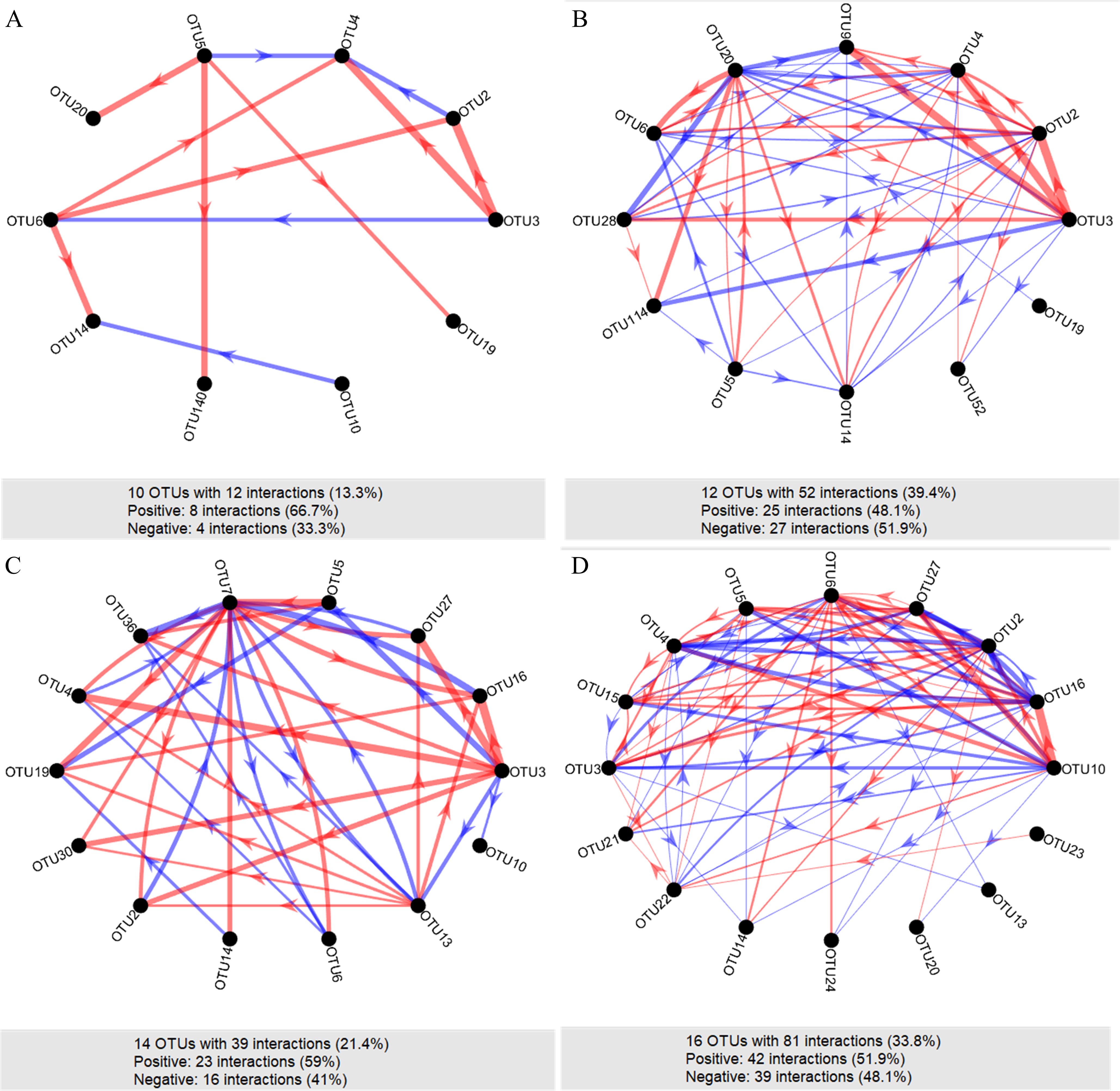
Figure 5. Interaction networks of clam gut bacterial communities in NN (A), NL (B), LN (C) and LL (D) groups. The arrow represents facilitative interaction between the two individual taxa. A red edge shows a positive interaction, whereas a blue edge shows a negative interaction between two individual taxa. The sizes of the edges were proportional to the strength of interaction.
3.5 Alternating salinity stress altering the functional potentials of gut microbiota
To evaluate how the functional potentials of gut microbiota were influenced by alternating salinity stress, the KEGG pathways were compared among four salinity stress groups (Figure 6). We showed the functional pathways that prominently varied between each two pair groups in four salinity stress groups at 0 and 96 hps. Notably, the functional potentials exhibited an opposite pattern in two alternating salinity stress groups (i.e., NL and LN). For instance, the pathways related with the metabolisms of amino acid (lysine biosynthesis), carbohydrate (amino sugar and nucleotide sugar metabolism, pentose phosphate pathway), energy (carbon fixation in photosynthetic organisms, photosynthesis), lipid (glycerolipid metabolism), and nucleotide (purine metabolism, pyrimidine metabolism), replication and repair (DNA replication), and immune system (NOD-like receptor signaling pathway) dramatically decreased (P < 0.05) in NL group at 96 hps compared to corresponding 0 hps, whereas these pathways performed opposite tendency in LN group. Similar findings for this phenomenon were also observed in the two control groups (i.e., LL and NN) (Figure 6).
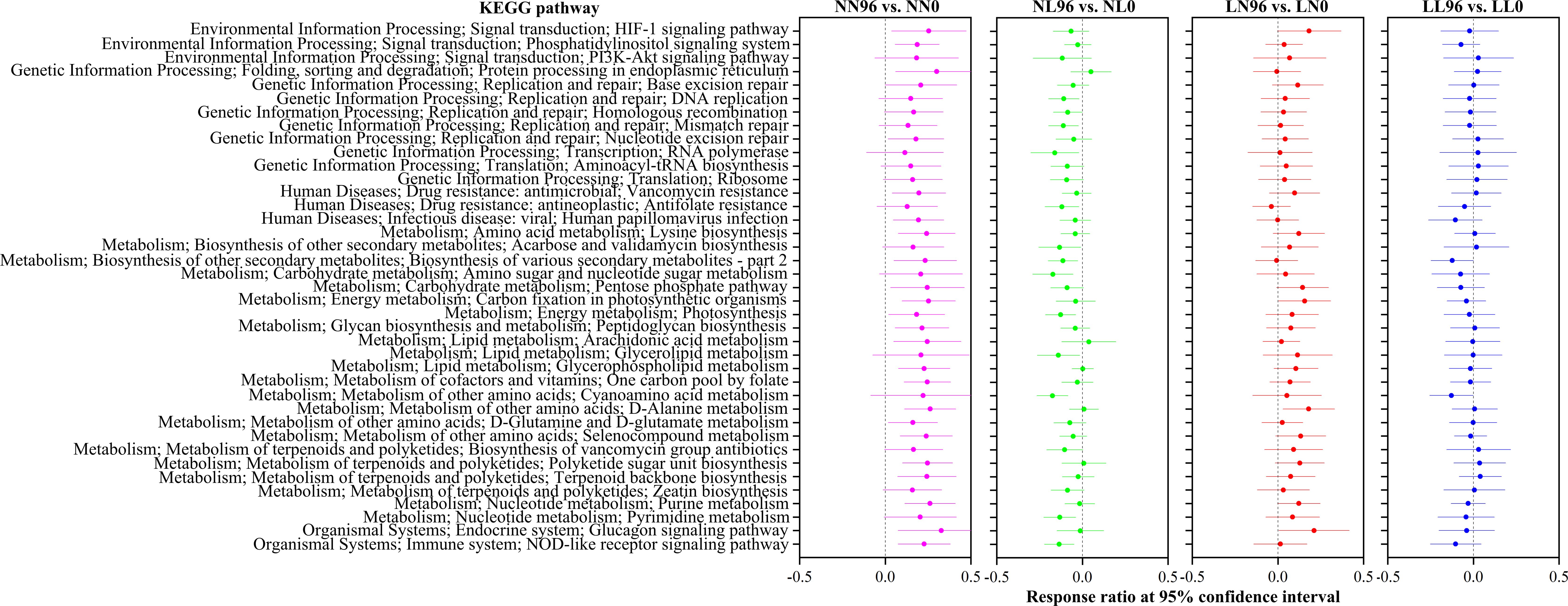
Figure 6. Comparison of the clam gut bacterial KEGG pathways between NN groups (pink circles), between NL groups (green circles), between LN groups (red circles), and between LL groups (blue circles) at 0 hps and corresponding 96 hps using response ratio method.
4 Discussion
Salinity is an important stressor that influences the survival, growth, development and gut microbial compositions of aquatic organisms (Davis, 1958; Xiong et al., 2017; Dai et al., 2022a, 2023). However, up to now, there is little data regarding the effect of alternating salinity stress on razor clams. In this study, the impact of alternating salinity challenge on razor clams was investigated via analyzing the clam survival and gut microbial dynamics under distinct salinity treatment.
In the salinity challenge test, razor clams were exposed for 96 h in different salinity treatment. We found that all clams in NN and LL groups survived for over 96 h, suggesting that clams can adapt to normal- and low-salinity conditions, which was consistent with strong tolerance ability of razor clams to low salinity stress (Chen et al., 2022). However, the razor clams began to die when culture condition was transferred from normal to low salinity or from low to normal salinity (corresponding to NL and LN groups, respectively), and the survival rate of the former was lower that of the latter during alternating salinity stress for 96 h. These findings indicated that clams were more susceptible under NL condition compared to LN condition. Additionally, the survival rate of razor clams in NL and LN groups decreased with the increase of stress time. Taken these together, alternating salinity stress resulted in varying degrees of effect on the clam growth.
Gut is the main organ responsible for nutrient absorption, which is attributed to symbiotic gut microbiota in aquatic organisms (Wu et al., 2024). Here, we found that razor clam gut microbial compositions changed correspondingly when facing salinity variation. The dominant bacterial phyla/classes in razor clams were Tenericutes, Gammaproteobacteria, Alphaproteobacteria, Campilobacterota, Bacteroidetes, Actinobacteria and Firmicutes. However, the relative abundances of these dominant gut microbes varied prominently over the increase of stress time. Specially, the proportions of potentially pathogenic taxa belonging to Shewanellaceae and Vibrionaceae increased in LN and LL groups, respectively, inferring a higher risk for developing disease in razor clams under these scenarios. Accordingly, the community structure of gut microbiota in razor clams exhibited dynamic change with stress time. These findings show that alternating salinity stress affects the number of dominant microbes in the gut, thereby disrupting the stability of gut microbiota in razor clams.
It has been proposed that an imbalanced gut microbiota further interrupts the stability of host-associated microbiota, and causes the loss of competitors against invading pathogens (Dai et al., 2019; Dong et al., 2021). Accordance with the notion, alternating salinity stress decreased the α-diversity index of clam gut microbiota such as observed species. This reduction was persistent with the increase of stress time. As reported, a microbial community with greater diversity possesses better stability and ability to withstand ambient stress and disturbance from an ecological perspective (Girvan et al., 2010; Cadotte et al., 2012). In this regard, the loss of gut bacterial diversity in razor clams under alternating salinity challenge could compromise this ability, which in turn affects its growth. Notably, the functional potentials of gut microbiota in razor clams dramatically altered after salinity stress. For instance, the functional pathways involved in the metabolisms of amino acid, carbohydrate, energy and lipid enhanced remarkably in LN group after exposure for 96 h compared to corresponding 0 h, while these pathways exhibited an opposite pattern in NL group. Therefore, we propose that alternating salinity stress disrupts functional potentials of gut microbiota, causing the allocation of energy to immune defense in resisting ambient stress. Whereas the ability for energy conversion was weakened under NL and LL scenarios. One explanation for these difference is that the increase in salinity concentration could result in gut lesions, and excessive salinity concentration could cause the gut villi to lose water and shrink (Lynch et al., 2022), which in turn impair the gut digestion and absorption ability of razor clams.
Growing evidence has showed that functional diversity depend on stable gut microbiota (Lozupone et al., 2012; Clerissi et al., 2020). In other words, gut microbial homeostasis contributes indispensable roles in host development, immunity and colonization resistance. Herein, we further investigated to what extent alternating salinity stress affected the community assembly of gut microbiota in razor clams. After salinity stress for 96 h, the stochasticity in controlling calm gut microbiota was dramatically potentiated in NL and LL groups, which was enhanced with the increase of stress time. These findings indicate that host selection on invasive pathogens could be compromised. The observed phenotype closely matched this assertion, that is, the proportion of potential pathogens enriched in NL group, and NL group was accompanied with higher mortality than NN group. Taken these together, alternating salinity stress suppresses clam resistance to environmental factor by weakening gut microbiota stability and host filtering on pathogens.
The stability of gut microbial interspecies interactions determines the resistance and resilience of a community to ambient stressor (Shade et al., 2012; Dai et al., 2023). In the present study, we found that the gut networks in razor clams were reshaped under the conditions of NL, LN and LL compared to NN, as supported by enhanced proportions of negative interspecies interaction. Notably, the core OTUs in gut networks exhibited different interaction relationships with other OTUs. For example, OTU5 belonging to Arcobacter taxa was mainly mutualism relationship with other OTUs in the gut network of NN group, which was shifted to competition relationship with other OTUs in the gut network of NL group; contrarily, parasitism or predation relationship with other OTUs was observed in the gut network of LN and LL groups. These findings suggest that salinity stress influences the interspecies interaction of clam gut networks by changing the relationships among core bacterial taxa. Vibrio and Pseudomonas taxa as potential pathogenic bacteria have been reported to be responsible for most bacterial diseases affecting marine bivalves including oysters and clams (Colwell and Sparks, 1967; Elston et al., 1981; Beaz-Hidalgo et al., 2010). In this regard, the two taxa as core OTUs in the gut network of LL group infer that opportunistic bacteria become dominant under low salinity condition, which could be a risk of developing disease for razor clams.
In summary, alternating salinity stress alters the gut microbial homeostasis and underlying ecological processes in razor clams. The susceptibility increased after alternating salinity stress, as supported by more rapid and higher mortality in NL and LN groups. Additionally, alternating salinity stress disrupted the community structure, functional potential, and network stability of clam gut microbiota. The gut microbiota of razor clams in NL and LL groups was more strongly governed by stochasticity, which may cause compromised host selection on pathogens, as observed the increased proportion of Vibrio, Pseudomonas and Shewanella taxa in the gut network of LL group. To conclude, these findings provide updated understanding of the abiotic factor impacts on bivalve growth from an ecological viewpoint.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by Institutional Review Board of Institutional Animal Care and Use Committee (IACUC) of Zhejiang Wanli University, China. The study was conducted in accordance with the local legislation and institutional requirements. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
YY: Data curation, Formal analysis, Visualization, Writing – original draft. QJ: Formal analysis, Investigation, Visualization, Writing – review & editing. QX: Conceptualization, Data curation, Funding acquisition, Methodology, Writing – review & editing. ZL: Conceptualization, Methodology, Visualization, Writing – review & editing. WD: Funding acquisition, Investigation, Project administration, Resources, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was financially supported by the Zhejiang Province Natural Science Foundation (LQ22C190005), National Natural Science Foundation of China (32202989), Zhejiang Science and Technology Project (Grant No. 2023C02011), China Agriculture Research System of MOF and MARA.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2024.1500347/full#supplementary-material
References
Beaz-Hidalgo R., Balboa S., Romalde J. L., Figueras M. J. (2010). Diversity and pathogenecity of Vibrio species in cultured bivalve molluscs. Environ. Microbiol. Rep. 2, 34–43. doi: 10.1111/j.1758-2229.2010.00135.x
Cadotte M. W., Dinnage R., Tilman D. (2012). Phylogenetic diversity promotes ecosystem stability. Ecology 93, S223. doi: 10.1890/11-0426.1
Chen Y. H., Du X. N., Dong Z. G., Li Y. F., Niu D. H. (2022). Heritability estimation of high salt tolerance in razor clam (Sinonovacula constricta). Aquaculture 559, 738423. doi: 10.1016/j.aquaculture.2022.738423
Chen W. D., Ren K. X., Isabwe A., Chen H. H., Liu M., Yang J. (2019). Stochastic processes shape microeukaryotic community assembly in a subtropical river across wet and dry seasons. Microbiome 7, 138. doi: 10.1186/s40168-019-0749-8
Chen Y. H., Ye B., Niu D. H., Li J. L. (2021). Changes in metabolism and immunity in response to acute salinity stress in Chinese razor clams from different regions. Aquacult Rep. 19, 100624. doi: 10.1016/j.aqrep.2021.100624
Churchill G. A. (2004). Using ANOVA to analyze microarray data. Biotechniques 37, 173–175. doi: 10.2144/04372TE01
Clerissi C., de Lorgeril J., Petton B., Lucasson A., Escoubas J. M., Gueguen Y., et al. (2020). Microbiota composition and evenness predict survival rate of oysters confronted to Pacific oyster mortality syndrome. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.00311
Colwell R. R., Sparks A. K. (1967). Properties of Pseudomonas enalia, a marine bacterium pathogenic for the invertebrate Crassostrea gigas (Thunberg). Appl. Microbiol. 15980-986. doi: 10.1128/am.15.5.980-986.1967
Dai W. F., Chen J., Xiong J. B. (2019). Concept of microbial gatekeepers: Positive guys? Appl. Microbiol. Biotechnol. 103, 633–641. doi: 10.1007/s00253-018-9522-3
Dai W. F., Dong Y. H., Ye J., Xue Q. G., Lin Z. H. (2022a). Gut microbiome composition likely affects the growth of razor clam Sinonovacula constricta. Aquaculture 550, 737847. doi: 10.1016/j.aquaculture.2021.737847
Dai W. F., Ye J., Liu S., Chang G. Q., Xu H. Q., Lin Z. H., et al. (2022b). Bacterial community dynamics in Kumamoto oyster Crassostrea sikamea hatchery during larval development. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.933941
Dai W. F., Zhang Z. J., Dong Y. H., He L., Xue Q. G., Lin Z. H. (2023). Acute salinity stress disrupts gut microbiota homeostasis and reduces network connectivity and cooperation in razor clam sinonovacula constricta. Mar. Biotechnol. 25, 1147–1157. doi: 10.1007/s10126-023-10267-8
Davis H. C. (1958). Survival and growth of clam and oyster larvae at different salinities. Biol. Bull. 114, 296–307. Available at: http://www.jstor.org/stable/1538986.
Dong P. S., Guo H. P., Wang Y. T., Wang R. Y., Chen H. P., Zhao Y. J., et al. (2021). Gastrointestinal microbiota imbalance is triggered by the enrichment of Vibrio in subadult Litopenaeus vannamei with acute hepatopancreatic necrosis disease. Aquaculture 533, 736199. doi: 10.1016/j.aquaculture.2020.736199
Douglas G. M., Maffei V. J., Zaneveld J., Yurgel S. N., Brown J. R., Taylor C. M., et al. (2019). PICRUSt2: An improved and extensible approach for metagenome inference. BioRxiv 2019, 672295. doi: 10.1101/672295
Edgar R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Elston R., Leibowitz L., Relyea D., Zatila J. (1981). Diagnosis of vibriosis in commercial oyster hatchery epizootics. Aquaculture 24, 53–62. doi: 10.1016/0044-8486(81)90043-0
Girvan M. S., Campbell C. D., Killham K., Prosser J. I., Glover L. A. (2010). Bacterial diversity promotes community stability and functional resilience after perturbation. Environ. Microbiol. 7, 301–313. doi: 10.1111/j.1462-2920.2005.00695.x
Hieu D. Q., Hang B. T. B., Lokesh J., Garigliany M. M., Huong D. T. T., Yen D. T., et al. (2022). Salinity significantly affects intestinal microbiota and gene expression in striped catfish juveniles. Appl. Microbiol. Biotechnol. 106, 3245–3264. doi: 10.1007/s00253-022-11895-1
Hou D. W., Zhou R. J., Zeng S. Z., Wei D. D., Deng X. S., Xing C. G., et al. (2020). Intestine bacterial community composition of shrimp varies under low-and high-salinity culture conditions. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.589164
Lan Y., Wang Q., Cole J. R., Rosen G. L. (2012). Using the RDP classifier to predict taxonomic novelty and reduce the search space for finding novel organisms. PloS One 7, e32491. doi: 10.1371/journal.pone.0032491
Liu G. F., Chen H., Li J. L., Shen H. D., Lin G. W., Wu W. J. (2009). Artificial propagation of razor clam Sinonovacula constricta by alga Chlorella sp. Fisheries Sci. 28, 192–195. Available at: https://api.semanticscholar.org/CorpusID:81155168.
Lozupone C. A., Stombaugh J. I., Gordon J. I., Jansson J. K., Knight R. (2012). Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230. doi: 10.1038/nature11550
Lynch S. A., Rowley A. F., Longshaw M., Malham S. K., Culloty S. C. (2022). “Diseases of molluscs,” in Invertebrate pathology (Oxford University Press), 171–216. doi: 10.1093/oso/9780198853756.003.0008
Mishra S., Srakar U., Taraphder S., Datta S., Swain D., Saikhom R. (2017). Multivariate statistical data analysis-principal component analysis (PCA). Int. J. Livest Res. 7, 60–78. doi: 10.5455/ijlr.20170415115235
Narasimham K. A., Laxmilatha P. (1996). “Clam culture,” in CMFRI Bulletin-Artificial reefs and Seafarming technologies. 48, 76–87. Available at: http://eprints.cmfri.org.in/id/eprint/2913.
Peng M. X., Liu X. J., Niu D. H., Ye B., Lan T. Y., Dong Z. G., et al. (20192819). Survival, growth and physiology of marine bivalve (Sinonovacula constricta) in long-term low-salt culture. Sci. Rep. 9, 2819. doi: 10.1038/s41598-019-39205-2
Pickard J. M., Zeng M. Y., Caruso R., Núñez G. (2017). Gut microbiota: role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 279, 70–89. doi: 10.1111/imr.12567
Price M. N., Dehal P. S., Arkin A. P. (2010). FastTree 2-approximately maximum-likelihood trees for large alignments. PloS One 5, e9490. doi: 10.1371/journal.pone.0009490
R Development Core Team (2013). R: a language and environment for statistical computing (Vienna: The R Foundation for Statistical Computing). Available at: http://wwwR-projectorg/.
Shade A., Peter H., Allison S. D., Baho D. L., Berga M., Bürgmann H., et al. (2012). Fundamentals of microbial community resistance and resilience. Front. Microbiol. 3. doi: 10.3389/fmicb.2012.00417
Shaw G. T. W., Pao Y. Y., Wang D. (2016). MetaMIS: a metagenomic microbial interaction simulator based on microbial community profiles. BMC Bioinf. 17, 488. doi: 10.1186/s12859-016-1359-0
Sloan W. T., Lunn M., Woodcock S., Head I. M., Nee S., Curtis T. P. (2006). Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 8, 732–740. doi: 10.1111/j.1462-2920.2005.00956.x
Wei Y., Ren T., Zhang L. (2020). Dix-seq: an integrated pipeline for fast amplicon data analysis. BioRxiv. doi: 10.1101/2020.05.11.089748
Wu S. G., Pan M. J., Zan Z. Y., Jakovlić I., Zhao W. S., Zou H., et al. (2024). Regulation of lipid metabolism by gut microbiota in aquatic animals. Rev. Aquacult 16, 34–46. doi: 10.1111/raq.12819C
Xiong J. B., Nie L., Chen J. (2019). Current understanding on the roles of gut microbiota in fish disease and immunity. Zool Res. 40, 70–76. doi: 10.24272/j.issn.2095-8137.2018.069
Keywords: Sinonovacula constricta, gut microbial community, alternating salinity stress, ecological process, interspecies interaction
Citation: Yuan Y, Jiang Q, Xue Q, Lin Z and Dai W (2024) Effect of alternating salinity stress on the gut microbiota of razor clam Sinonovacula constricta. Front. Mar. Sci. 11:1500347. doi: 10.3389/fmars.2024.1500347
Received: 23 September 2024; Accepted: 21 November 2024;
Published: 11 December 2024.
Edited by:
Wei Zhu, Chinese Academy of Sciences (CAS), ChinaCopyright © 2024 Yuan, Jiang, Xue, Lin and Dai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhihua Lin, emhpaHVhOTk4OEAxMjYuY29t; Wenfang Dai, ZGFpd2VuZmFuZzYyODNAMTYzLmNvbQ==
†These authors have contributed equally to this work