 Man Wu
Man Wu Liyang Zhan
Liyang Zhan Jian Liu
Jian Liu Jiexia Zhang
Jiexia Zhang Wangwang Ye
Wangwang Ye Bruno Delille
Bruno Delille- 1Key Laboratory of Global Change and Marine-Atmospheric Chemistry, Third Institute of Oceanography, Ministry of Natural Resources, Xiamen, China
- 2Unité d’océanographie chimique, The Freshwater and OCeanic science Unit of reSearch (FOCUS), Universite de Liege, Liege, Belgium
N2O is one of the most important greenhouse gases and ozone depletor, which was a matter of more and more concerned. The Southern Ocean was considered as one of the most important N2O source and was believed to account for ~1/4 of oceanic budget. However, there is uncertainty about this budget due to limited data availability. In this study, field and lab works were conducted for better understanding of N2O dynamics during sea ice melting and sea ice formation. In the field study, taking advantage of the Chinese Antarctic cruise, a 10 days’ time series study was carried out at a station in the Prydz Bay, Antarctica, where, surface water N2O was observed continuously, and the adjacent ice cores were taken for N2O analysis. In the lab, an ice growing simulation system was constructed to study the N2O dynamics during the sea ice formation. The result of endmember mixing models and calculation of N2O partition in three phases during sea ice formation provide important information about the dynamics of N2O during ice melting and sea ice formation processes, that is, the sea ice melting regulated N2O concentration and saturation status, which can be an explanation for reported N2O undersaturation observed in polar oceans, whereas during the sea ice formation, most of the N2O will be expelled to the deeper water while a small amount of retain the sea ice and less amount of N2O release to the atmosphere.
1 Introduction
Nitrous oxide (N2O) is one of the most important greenhouse gases, whose radiative forcing is about 300 time as higher as that of CO2 on molecular basis. As a result of anthropogenic emission, atmospheric N2O concentration increase stably in the troposphere with rate of about 0.25% yr-1, according to model results, emission of N2O, will threaten the 2°C limit of the Paris Agreement if no action is taken to reduce its emission in the future even the CO2 emission is well controlled (Tian et al., 2020). Moreover, N2O also plays an important role in ozone depletion, it has become the fastest increasing ozone depletor since the emission of halocarbons is reduced (Ravishankara et al., 2009).
The ocean is a significant N2O source, contributing about 3.4 - 4.2 Tg N a-1 to the atmosphere (Tian et al., 2020; Yang et al., 2020), which account 1/4-1/3 of global N2O budget, and aroused wide concern (Bange et al., 2019). Based on studies of the past decades, general cognition was achieved that most of the surface open ocean water are slightly oversaturated (104%-130%) with N2O, while oceanic N2O emission hot spots were identified at the Eastern Tropical Pacific and the Arabian Sea, where tremendous amounts of N2O are released into the atmosphere (Arevalo-Martínez et al., 2015; Bange et al., 1996).
Nonetheless, there are still regions, such as the Southern Ocean and Arctic Ocean, which are not well studied due to fierce in situ conditions. However, according to the model result (Freing et al., 2012; Nevison et al., 2005), the Southern Ocean was considered as a significant N2O source, which contribute about 1/4 of the oceanic N2O source strength. However, there are some studies revealed presence of at least temporary sink characteristics in the Southern Oceans. Rees et al. (1997) first reported surface N2O undersaturation in the Bellingshausen Sea and suggested that the source and sink characteristics of the Southern Ocean may need to be re-evaluated. Zhan et al. (2015) and Farías et al. (2015) observed N2O undersaturation in the surface Southern Ocean. This phenomenon also observed in the Nordic Sea (Zhan et al., 2016) and Arctic Ocean (Kitidis et al., 2010). Oxygenated denitrification was proposed as a mechanism that responsible for the undersaturation observed at the Nordic sea (Rees et al., 2021). However, there are more studies suggest that physical process including the ice melting may be an important reason for the N2O undersaturation in polar oceans (Kitidis et al., 2010; Rees et al., 1997; Zhan et al., 2016). More recently, a study revealed a low N2O concentration in Arctic sea ice (Randall et al., 2012). This low N2O concentration in sea ice may result from N2O rejection during sea ice formation. Therefore, when the sea ice melt in summer, surface water N2O concentration will be diluted and result in N2O undersaturation if the air sea exchange is not fast enough to replenish the surface water N2O concentration. However, there are also study (Fripiat et al., 2014) suggested the nitrification and denitrification may exist in the sea ice, both may be source of N2O and contribute N2O to the surface ocean. Hence, knowledge about the sea-ice interaction on N2O budget is still not well understood.
For better understanding of above question, the field work was carried out during 33th CHINARE Antarctic expedition, and the in the lab simulation work was designed and conducted. The N2O dynamic during the summer sea ice melting process and in lab ice growing simulation were studied, in order to address above knowledge gaps.
2 Material and methods
2.1 Surface ocean N2O observations and ice core sampling in the Prydz Bay
During the 33rd Chinese National Antarctic Research Expedition (CHINARE), when the R/V Xuelong remained near the sea ice shelf in the Prydz Bay, the surface water N2O concentration was monitored continuously at a station (76.14°E, 69.10° S; the location is shown by the red dot in Figure 1) from November 31 to December 10, 2016, using a fully automatic system for underway N2O measurements deployed onboard R/V Xuelong. This automatic system consisted of a cavity ring-down spectrometer (Picarro G5101i N2O isotope analyzer) and an upstream device developed in our laboratory, described in detail by Zhan et al. (2018). Briefly, the upstream device included a bubble-type equilibrator, hydrophobic membrane PTFE filter, moisture trap, air pump, mass flow controller and 4-position valve. Surface seawater from ~4.5 m below the surface was pumped continuously and sent into the equilibrator. The headspace gas in the equilibrator was sent into the Picarro analyzer for measurement of the N2O concentration. The response time of the system for N2O was 3.4 min. The N2O concentration in the surface water was calculated based on the concentration of the headspace gas using the equation of Weiss and Price (1980). The precision of this method for N2O measurements was better than 0.5% (relative standard deviation, RSD). The automatic system for N2O measurement recorded the water temperature and salinity.
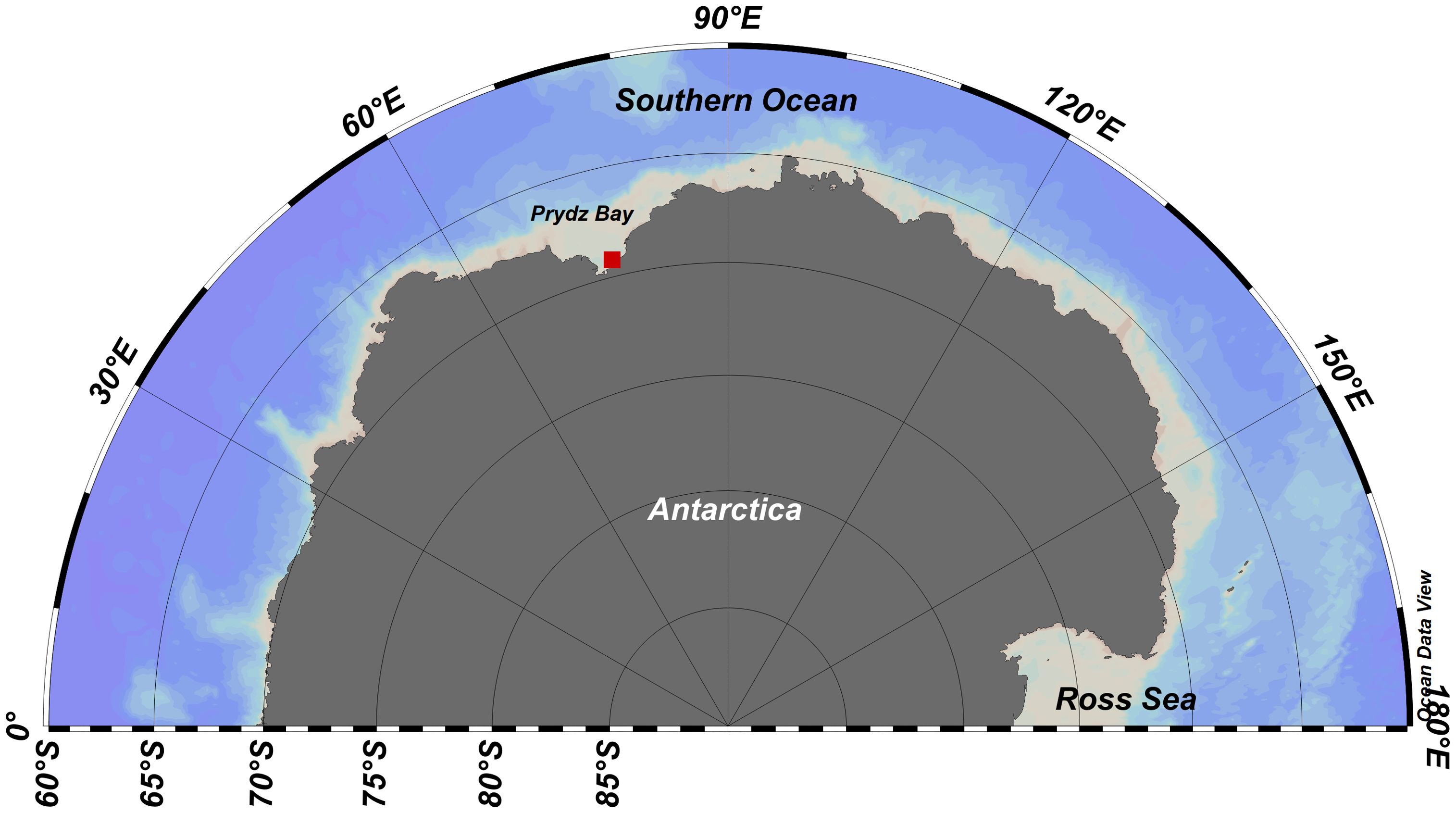
Figure 1. Station for N2O continuous observation and ice core sampling in the Prydz Bay during the 33rd CHINARE.
Two sea ice cores were collected by a manual ice core sampler on the sea ice shelf in the Prydz Bay; these cores were approximately 30~40 meters away from the N2O observation station on December 1, 2016. Ice cores were collected in first-year ice (average thickness: 130 cm). After sampling, ice cores were kept at -20°C until they were cut into sections with lengths of 10-20 cm at the home laboratory. Ice sections were then placed into a Tedlar® bag for gas sampling. Air was removed from the bag through the outlet using a vacuum pump as soon as possible to avoid contamination from the atmosphere. The ice sections in the Tedlar® bag were melted in the dark at room temperature (25°C). The salinity of the sea ice melt water was measured by a conductivity meter (WTW Cond3110).
2.2 Measurement of N2O concentration in the water and ice samples
The concentration of N2O in the water samples was determined by a system for the automated static headspace analysis, described in detail by Zhan et al. (2013). The ice samples were melted in a Tedlar® bag at room temperature in the dark, and then the N2O concentration in the melted water was measured. In brief, the water sample was transferred into 20-ml vials sealed with PTFE-coated silicone rubber septa and crimped with iron caps. High-purity nitrogen gas was introduced in the 20-ml sample vial to replace a portion of the water (approximately 12 ml) by a headspace-introducing apparatus. After sample equilibration was reached under heating and agitation using a CTC headspace autosampler, the headspace gas in the vials was injected into the GC-ECD system (Shimadzu GC-2010 gas chromatography) for determination of the N2O mixing ratio (ppb). The N2O concentration in water was then calculated based on the ratio of N2O in headspace gas. The accuracy and precision of this method were both approximately 2%.
2.3 Mixing model of N2O in the study region
To reveal the effect of sea ice melting on the N2O in the sea water, a two- endmembers mixing model was used. This mixing model base on an assumption that the sea ice will inhibit the air sea exchange, so with presence of the sea ice only the two endmembers, the sea ice melting water and surface sea water. Accordingly, the mixing process could be described using following equation:
where S10d and C10d refer to the salinity and the concentration of N2O of the surface sea water after 10 days of experiment, xi and Si are the fraction of sea ice melting water and salinity of sea ice end member; Ss and Cs is the salinity and concentration of N2O in the surface water at the beginning of the study, and Ci is the concentration of N2O in the sea ice.
2.4 Simulation experiments of the formation of artificial sea ice
The field work provided information only on the final state of the sea ice, whereas the dynamics of N2O during sea ice growth were unknown. An artificial sea ice growth experiment was set up in a custom-made polymethyl methacrylate cylindrical tank with a diameter of 30 cm and a height of 40 cm; the tank was placed in an air-cooled refrigerator. A diagram of the experimental device is shown in Figure 2.
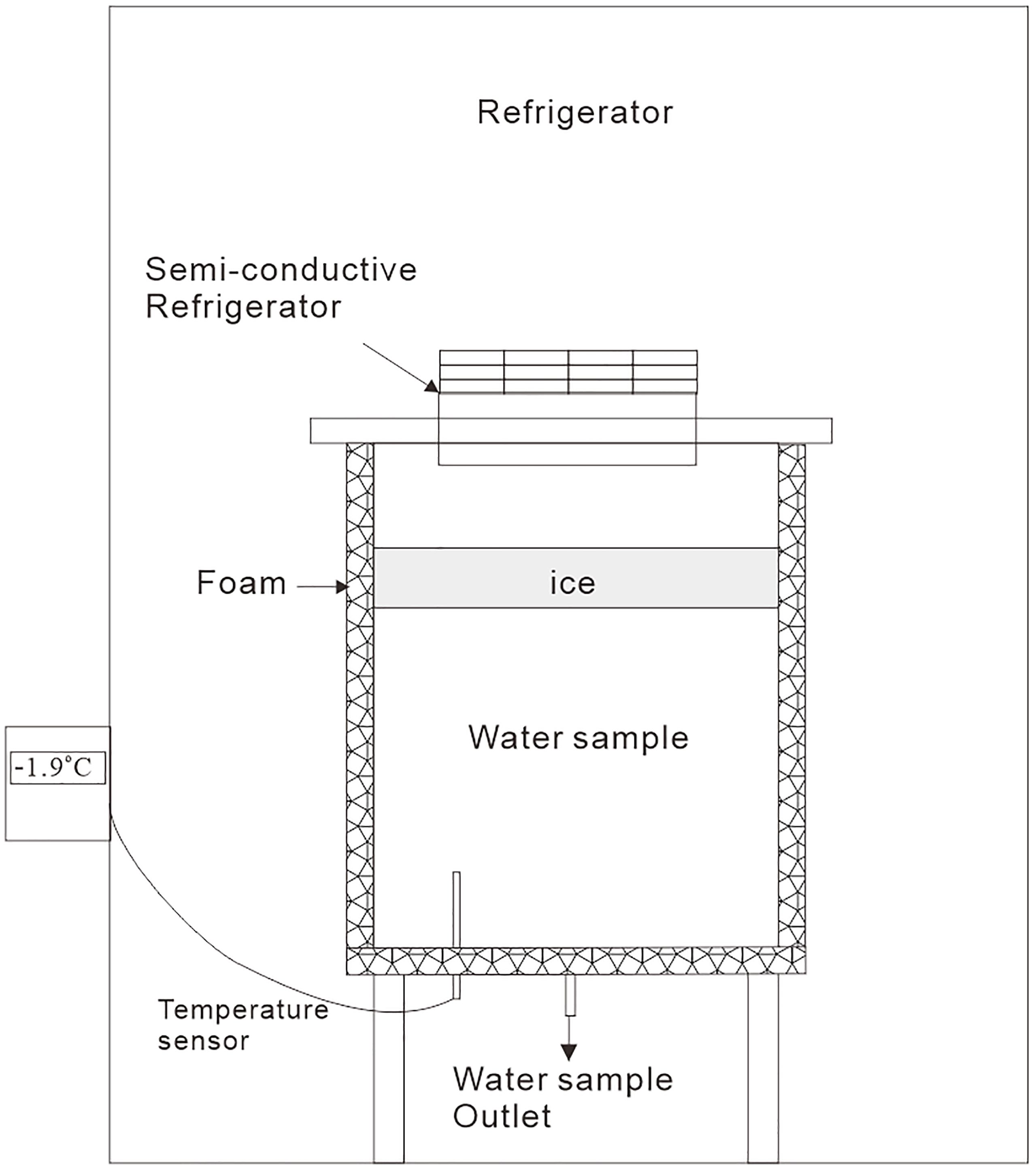
Figure 2. Diagram of the experimental device for the simulation of artificial sea ice growth.
A layer of foam insulation was wrapped outside of the wall and on bottom of the tank to avoid sea ice growth on the side and bottom, and a semiconductive refrigerator was embedded into the lid of the tank. The temperature difference between the two sides of the chilling plate of the semiconductive refrigerator was as high as 40°C. The cold side of the plate was placed inside the tank to cool the headspace air above the artificial seawater in the tank. Therefore, the surface water in the tank was initially cooled to the freezing point, and the ice could grow from the surface; this process simulated the natural process of sea ice growth. A temperature sensor was also installed in the bottom of the tank to monitor the change in temperature of the seawater in the tank.
The artificial seawater was prepared using analytically pure chemicals (Sinopharm Chemical Reagent Co., Ltd, China) according to the formula of Lyman and Fleming (1940). A batch of chemicals, including NaCl, MgCl2, Na2SO4, CaCl2, KCl, NaHCO3, KBr, H3BO3, SrCl2, and NaF, was added to pure water according to the outlined proportion and stirred until the salts dissolved completely. The salinity of the artificial seawater was between 33 and 34. This study focused only on the physical process; therefore, saturated HgCl2 solution was added to the artificial seawater with a volume ratio of 0.1% (v/v) to inhibit biological activity. The artificial seawater was stored at room temperature. Approximately 15 kg of artificial seawater at 24°C was poured into the water tank in the refrigerator and filled approximately half of the water tank volume. The refrigerator temperature was then set to -2°C. Under the cooling effect of the refrigerator, the artificial seawater in the tank became colder and colder. The temperature of the surface seawater was measured every three hours by a Cond3310 conductivity meter (WTW, Germany) until the temperature dropped to below 0°C and no ice appeared on the surface. Surface seawater was sampled into three 20-ml vials with a silicon transfer tube, and the concentration of N2O in the seawater was determined before freezing. After sampling, the refrigerator was set to -9°C, and the semiconductor cooling device began to cool the headspace air in the tank. After approximately 24 hours, a thin layer of sea ice formed on the surface water. When the sea ice grew to a thickness of approximately 10 cm for approximately 6 days, a hole was drilled into the sea ice by a handheld electro drill; the seawater samples under the sea ice were collected immediately through a silica tube inserted into the ice hole. Then, the ice layer was broken by a hammer, and the sea ice samples were collected immediately. Three sea ice pieces weighing between 500 g and 1000 g were placed into three polyvinyl fluoride (PVF, brand name Tedlar®) bags for gas sampling. The bags were sealed, and the air in the bags was pumped out through an outlet using a vacuum pump as soon as possible to avoid contamination from the atmosphere. The sea ice samples were stored at -20°C in the refrigerator until analysis. The water and ice samples along with the remaining water and ice in the tank were weighed to calculate the distribution ratio of N2O in the ice and water during sea ice growth.
The concentrations of N2O and salinity in the artificial sea ice samples were melted and determined by the same methods as the ice samples from the Prydz Bay.
To study the dynamic of N2O during the simulation sea ice formation, a set equation was developed as follow:
where, N2Oinitial, N2Oice and N2Ousw are the amount of N2O in the initial water for experiment, the sea ice formed and rejected into the under ice water when experiment finished; Wice and Wusw are the weights (kg) of the artificial ice and under ice water and C0 (nmol kg-1) of N2O in the seawater before sea ice formation; Cice and Cusw (nmol kg-1) is the concentration of N2O in the formed sea ice; Rice and Rusw are the percentage of N2O partition into the sea ice, and under ice water at the end of simulation experiment; With these parameters obtained, percentage of N2O release into the atmosphere could be derived using Equation 8.
3 Results and discussion
3.1 N2O in water and ice and their mixture in the study site
3.1.1 N2O in surface water and its variability
The variabilities of the N2O concentration, SA, salinity (SSS) and temperature in the surface water in the Prydz Bay during the 10-day observation period in early summer are shown in Figure 3. The observations were interrupted on the first and the seventh days due to the blocking of the water inlet by sea ice and temporary device failure. The concentration of N2O generally showed a downward trend with time, dropping from 17.0 nmol L-1 on November 31 to 16.5 nmol L-1 on December 10, 2016. The saturation anomaly (SA) varied between 3% and 5% and showed greater fluctuations than that of the concentration. Salinity also showed a downward trend from 34.15 to 33.48, while the temperature did not show a similar trend and varied between -1.2°C and 0.9°C.
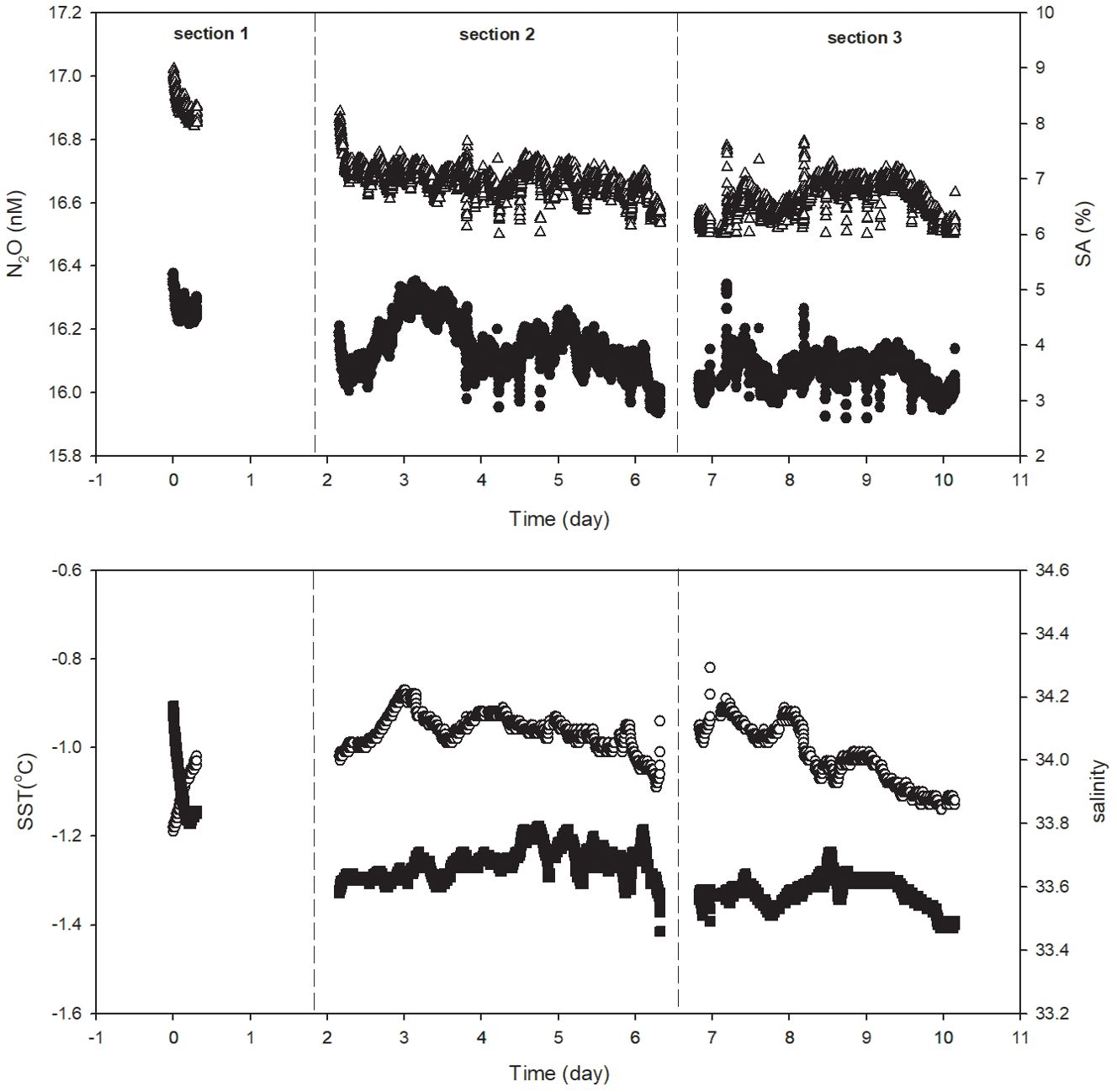
Figure 3. Variability in the N2O concentration (open triangle), saturation anomaly (SA, black dot), temperature (SST, open circule) and salinity (SSS, solid square) in the surface water of the Prydz Bay from November 31 to December 10, 2016. The continuous data were divided in three sections as shown in the figure.
To further reveal the detail of variabilities of surface water N2O, the time series stations is divided into three sections, day 0 to day 2.5, day 2.5 to day 6.5 day and day 6.5 to day 10.
For the first section, it could be seen from Figure 3 that the N2O and SA are at their highest values of 17.0 nM and 5.3%, corresponding to these values is the highest salinity of the time series station. The highest salinity at the beginning of time series station may result from the latest brine rejection during sea formation (Roden et al., 2013). However, since it is summer, and the SA correspond to this highest N2O concentration is about 5%, a saturation state fell well within the range of global open ocean value, it could be regarded as a normal surface ocean value. For the first two days, though the data is discontinuous because the inlet was clogged by the sea ice, the sharp decline of SSS from 34.17 to 33.58 could be identified from Figure 3, meanwhile, SST increase from -1.2 to -1.0°C, suggesting a fast sea ice melting process occurred, which generally occurs in late November in high latitude Southern Ocean (Lei et al., 2010; Zheng and Shi, 2011). Correspondingly, the N2O concentration decline to about 16.8 nM, and SA value decline to its first minimum of about 3.2% on day 2.5. Generally, 1°C temperature changes will lead to more than 4% of N2O solubility change at high latitude Ocean, therefore the SST change of about 0.2°C could only account for about half of the SA variability, whereas the remain should be accounted for by dilution caused by fast melting of sea ice in this season.
For the second section, the N2O concentrations shows a relative decline pattern and the SA raise to its maximum (Figure 3), and then decline to the end of this section. the SSS are relatively stable during the first half and turn to be more variable between 33.5 and 33.8 on the second half, whereas the SST reach it maximum of -0.88°C, and then remain relative stable before it declines to -1.08°C at the end of this section. When the N2O concentrations of the second section plotted against time, a rate of N2O concentration declines of about - 0.01 nM d-1 could be derived (figure not shown), indicating that the N2O concentration keeps declining at a slow rate, whereas the salinity shows a slightly patterns of rise before day 4.5, and decline in a relative fluctuating pattern till the end of section 2. However, the salinity and N2O concentrations of whole section even the data between day 2 and day 4.5 shows and significant positive correlationship (Figure 4, p<0.001, R2 = 0.47).
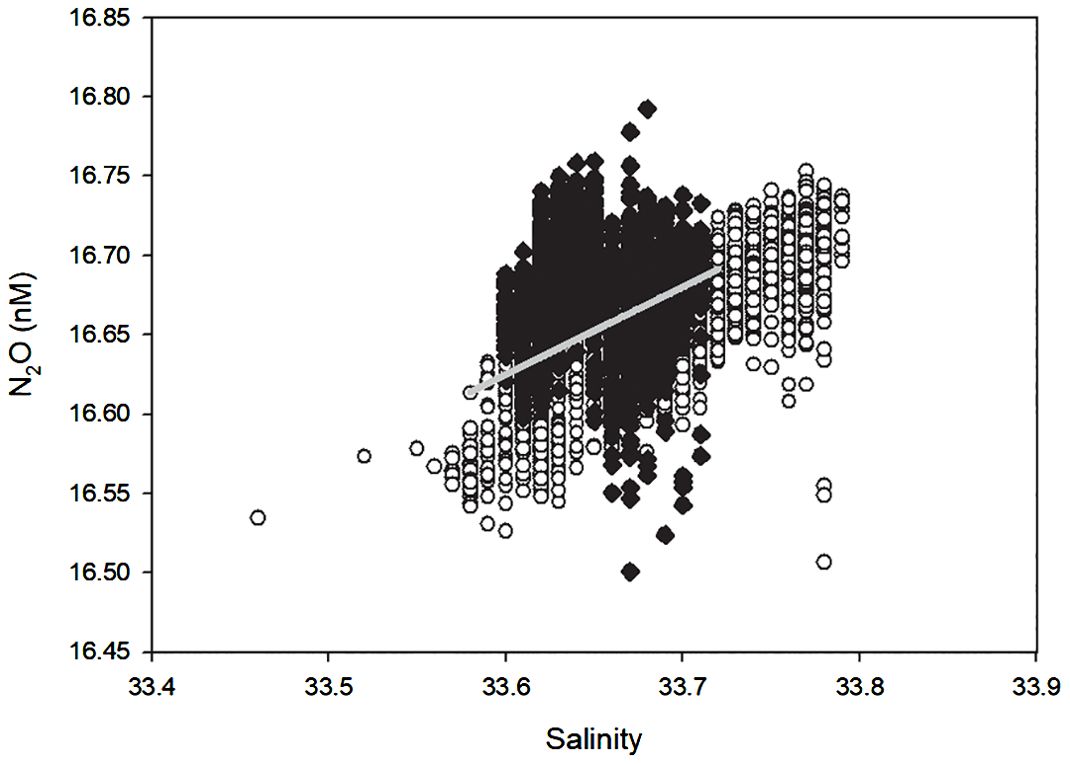
Figure 4. The N2O and Salinity plot in section 2. Black dots are data obtained from day 2 to day 4.5. Gray line is the linear fitting curved of day 2 to day 4.5. white circle is the data of the whole section 2.
The SA show two maximums in this section. The first one approaching the highest SA value (5.1%) of the time series station and corresponds to the SST maximum of this section on day 3, whereas in the rest of this section, the SA fluctuated between 3% and 4.5%, where the SA maximum does not correspond to temperature maximum.
For the last section, both the N2O concentration shows a pattern of rising at first and declining later, whereas the SA is relative stable. Again, the N2O concentration are significantly correlated with SSS (p<0.001, R2 = 0.58). Though the highest SST on day 7 could be observed, and relative high SST in the first half of this section presence, most of the SA value does not excess 4.5%. As a whole, the average SA value of the third section is the lowest of the time series station, showing a trend that surface water N2O saturation in the Prydz Bay keep declining during the time series study.
From Figure 3, it could be seen that the positive correspondence between the N2O concentration and salinity exist throughout the time series station observation. Among the possible processes that regulate the surface water N2O distribution pattern, the mixing process between different water mass endmembers, for example, sea ice melting water and shelf water, may be the most important process. To further disclose the influence of the mixing process during the whole study period, the SA value is chosen to plotted against the SSS, and the SA is chosen because it is a parameter that also take the influence of SST into account. As could be seen from Figure 5, there is significant correlation (p<0.001) between SA and SSS during the whole study period, however, the mixing pattern is non-conservative. In this Figure, temperature of each data point was marked with color, with warm color represents the higher temperature and vice versa. It shows that the most of SA in warmer water are above regression line, and that in the colder water are below the regression line. There some SA maximum deviate from the conservative regression line about 0.5-3.0% on day 1, Day 3 and D7, suggesting that introduction of N2O on these days. Especially on day 3, when SST raise to its maximum during this study. This SST maximum did not seem to be result of upwelling, since the SSS shows no increasing. Therefore, is this warming result from seasonal surface temperature rising due to the solar irradiation? Rising of SST corresponding with the highest SA, however, could not account for all of this increase, since at this temperature range, a degree of temperature rising will lead to about 4-5% of SA increment, whereas the temperature rising here is only about 0.2°C, that means only lead to a SA increment of less than 1%. Considering the precision of this underway method is about 0.5%, it could be considered that there may be other reason for the left SA rising. There studies imply that warming of the ocean may change the community of the bacterial and result in increasing of N2O production (Freing et al., 2012), however, since there are only limit information available, the possible contribution to this oversaturation is left for future studies.
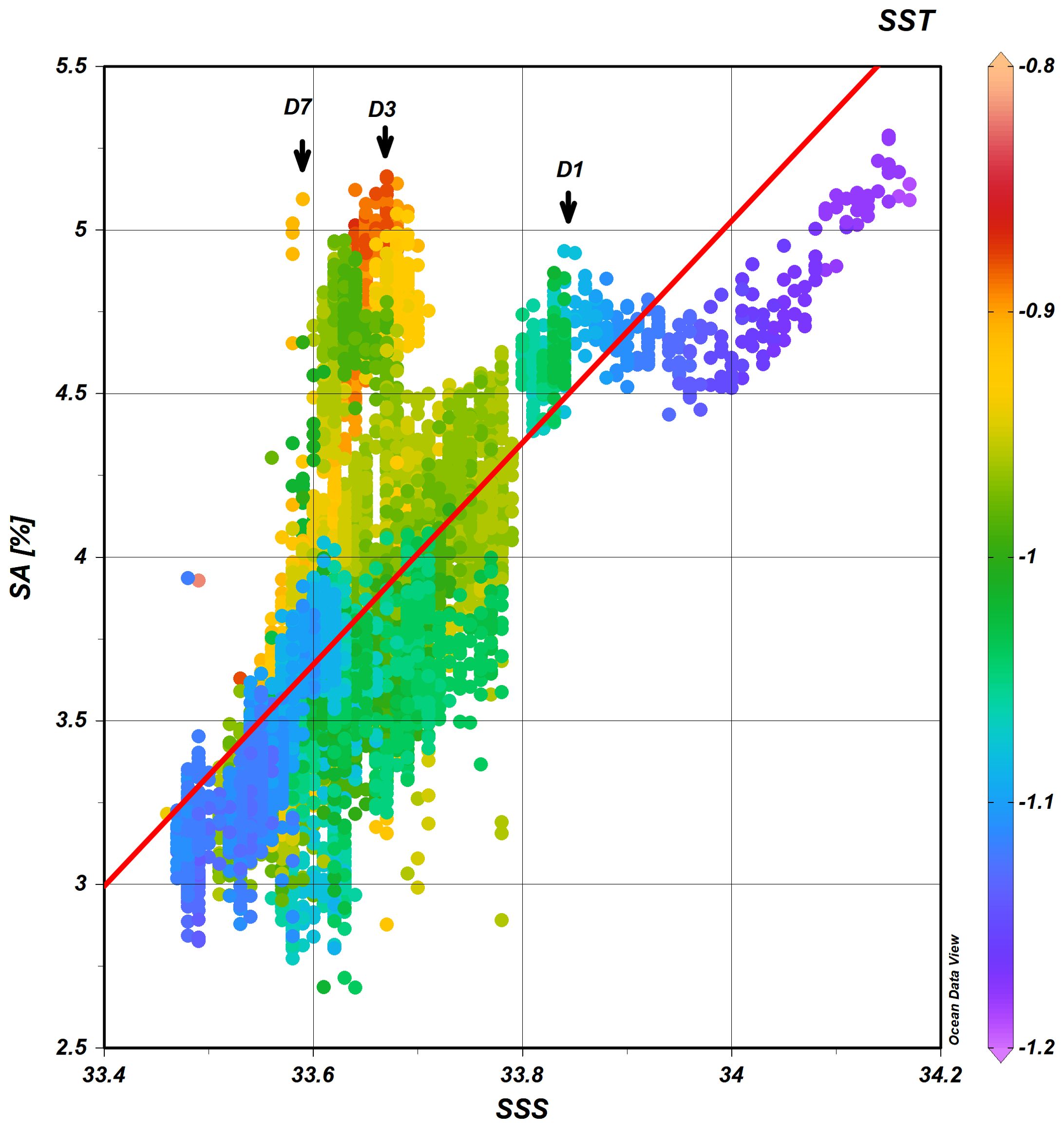
Figure 5. The SA plotted against surface water salinity (SSS). The color indicates surface water temperature.
Above all, it could be concluded that, during the seasonal transit, though N2O production may promoted somehow when sea water temperature rising, sea ice melting process should contribute to the decline of N2O saturation and SA, however, the mechanism of this production and its contribution to the balance need further study.
3.1.2 N2O concentration in the sea ice
The concentrations of N2O in two sea ice cores collected during the N2O continuous observation were far lower than those in the surface water, ranging from 1.6 nmol L-1 to 2.5 nmol L-1 (shown in Figure 6). The salinities of both ice cores were between 4.2 and 7.5 and exhibited a general C-shaped profile, with a slight increase in salinity at 50 cm. The N2O concentration patterns were similar to the salinity patterns. This presumably suggested an overall conservative behavior of N2O with salinity. The N2O concentration was normalized to a salinity of 5 (N2O@5); this was calculated from the following equation: N2O@5 = CN2O*S/5, where CN2O and S are the N2O concentration and salinity of a certain sea ice section. Accordingly, the N2O@5 ranged from 2.1 nmol L-1 to 2.7 nmol L-1 at the bottom of the ice and was relatively close to the N2O@5 values of surface water derived from continuous measurement, which were approximately 2.5 nmol L-1. This further suggested that the behavior of N2O may be conservative during ice growth, nevertheless, there are study identified that active nitrification in the sea ice (Fripiat et al., 2014), a mechanism that can produce N2O. Though the observation does not show obvious accumulation of N2O in the sea ice, this possibility and their contribution to the regional budget should be further studied. Moreover, N2O@5 at the top of the ice was approximately 1.6 nmol L-1, 35% less than the value of surface waters. This decrease at the top of the ice possibly suggested a release of N2O similar to CO2, as has been shown in many studies (Geilfus et al., 2013; Van Der Linden et al., 2020).
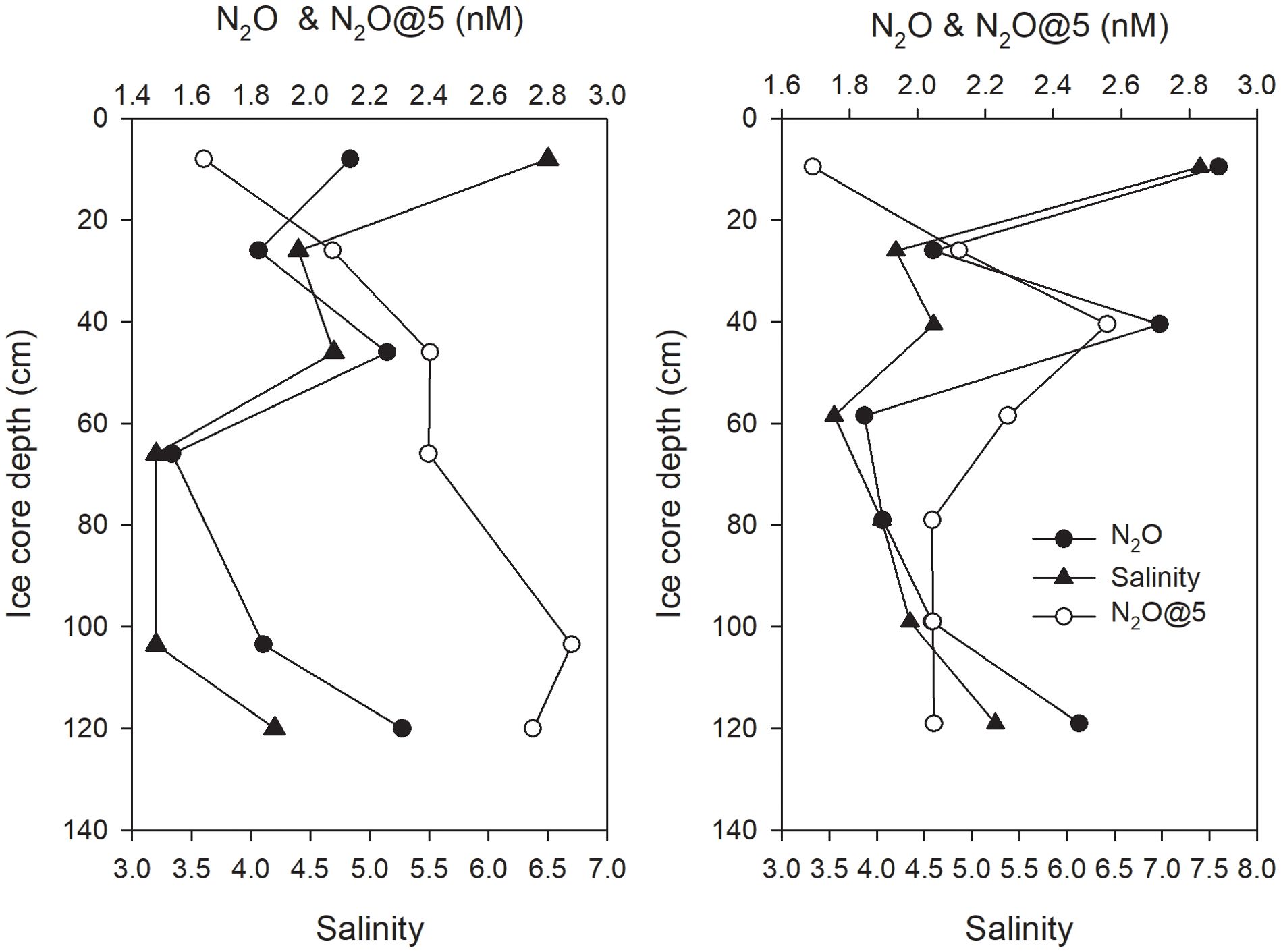
Figure 6. Concentrations of N2O and salinity in two Antarctic ice cores (N2O@5 means the concentration of N2O normalized to a salinity of 5.).
As a whole, the result of ice N2O analysis shows that the sea ice is rather depleted in N2O when compared with the sea water. This result agrees with previous studies of the low sea ice N2O concentrations (Randall et al., 2012). Sea ice melting water was then considered as one of the reason that result in surface sea water N2O undersaturation (Priscu et al., 1990; Rees et al., 1997).
3.1.3 Mixing behavior between sea ice melting water and shelf water
The depletion of N2O and salinity in sea ice cores demonstrated the dilution of N2O in the surface water by melting ice water in the warm season in the Prydz Bay. A two-end member mixing model described in method was used to calculate the contribution of melting ice water to the surface water in the Prydz Bay. One member was the melting ice water, and the other was the surface water. The average concentration of N2O (N2Oi =2.0 nmol L-1) and average salinity (si = 4.6) in the two sea ice cores were used as the end-members of the melting ice water. The initial concentration of N2O (N2O0 = 17.0 nmol L-1) and salinity (s0 = 34.09) in surface water were taken as the end-members of the surface sea water. After 10 days, at the end of this observation, the salinity (s10d = 33.48) in the surface water was the result of the mixing of the melting ice water and initial surface water. The mixing ratio of the melting ice water, xi, was 2.08%, calculated by Equation 1 based on the salinity.
When the mixing ratio of the melting ice water, 2.08%, was used in Equation 2 to calculate the theoretical N2O concentration at the end of the 10 days of observation and considering only the mixing process of the melting ice water and the surface seawater, a calculated concentration of 16.66 ± 0.08 nmol L-1 was obtained; this was very close to the observed concentration of 16.52 ± 0.08 nmol L-1.
The sea ice remained intact before the R/V Xuelong left the station; therefore, we assumed that the exchange of N2O was inhibited since our work suggested that even a thin layer of sea ice (approximately 20 cm) may be effectively inhibited (unpublished data) the N2O exchange between the sea and the air.
In other words, although the Prydz Bay is a N2O source at the beginning of the melting season, though there may be N2O production during that period, the dilution of the surface seawater by the melting ice water weakens the signal of this source. As more sea ice melts, the N2O concentration presumably decreases to an undersaturation state, as reported by Zhan et al. (2015) and Zhan et al. (2017).
3.2 Behavior of N2O during the formation of artificial sea ice
The simulation experiment of artificial sea ice formation provides additional information for sea ice N2O dynamics. The results of the simulation experiments are listed in Table 1. The initial concentration of N2O in the seawater before freezing was 11.6~12.7 nmol kg-1, and the seawater was undersaturated against the atmosphere. The initial salinity was 33.2~33.7. After sea ice growth, the concentration of N2O and salinity in the underlying water markedly increased up to 19.4~28.3 nmol kg-1 and 64.1~89.7, respectively, whereas the N2O concentration and salinity in the sea ice significantly decreased to 3.5~3.7 mol kg-1 and 6.3~10.1, respectively. Randall et al. (Randall et al., 2012) reported a similar phenomenon in the Arctic Ocean that showed low N2O in sea ice (~ 6 nmol L-1) and high N2O in the underlying seawater (11.0 nmol L-1 ~18.8 nmol L-1). This phenomenon was also observed in section 3.1.1, where a lower N2O concentration in the Antarctic Sea ice was observed, which may due to longer sea ice age, and corresponding more completely N2O rejection.
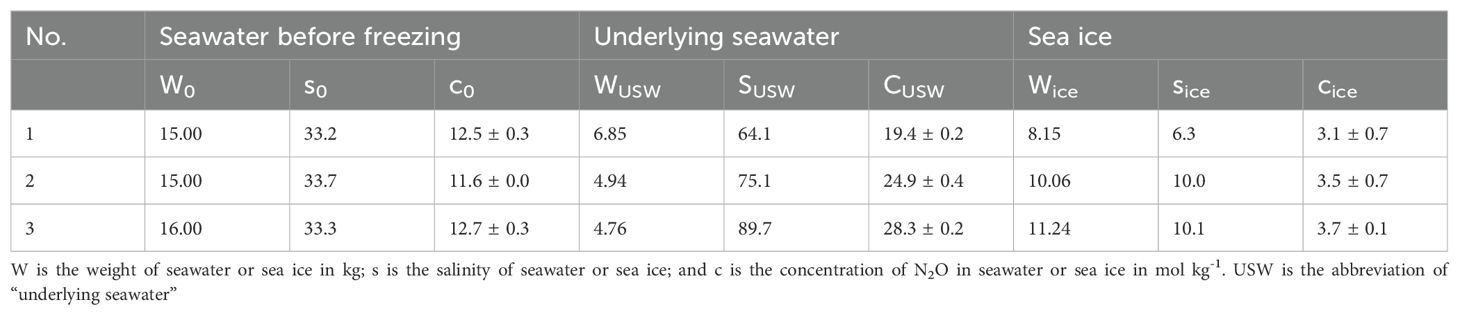
Table 1. N2O and salinity of seawater and sea ice before and after the formation of sea ice in the simulation experiments.
These results indicated that the freezing process resulted in the rejection of N2O to the underlying seawater; this is similar to the brine rejection process with a small portion of N2O remaining in the sea ice. Based on the simulation experimental data, we calculated the amounts of N2O rejected into the underlying seawater (N2Ousw) during sea ice growth and the N2O remaining in the forming ice (N2Oice); the ratios of N2Ousw and N2Oice (Rusw and Rice) to the amount of N2O originally present in the seawater that finally formed the sea ice (N2Oinitial) were also calculated using Equations 3-8.
Through the above calculations, the results from all three experiments indicated that the sum of N2Oice and N2Ousw was less than N2Oinitial; this result suggested that a fraction of N2O probably emitted to the atmosphere during ice formation. Although N2O exchange between sea ice and the atmosphere has not been reported, air-ice gas exchange of other gases, such as CO2, O2, Ar, etc., has been reported (Crabeck et al., 2016; Delille et al., 2015; Odile et al., 2014). The decrease in N2O@5 at the top of the ice cores from Prydz Bay suggested that a release of N2O from the ice to the atmosphere was plausible. The missing faction of N2O during freezing of the surface water was attributed to the release to the atmosphere (Ratmosphere). Ratmosphere is the ratio of N2O emission in the atmosphere, calculated by Equation 8.
The results of the calculations showed that for the formation of 10 cm sea ice, approximately 70% of N2O (69.3% ± 2.4%) in the surface water was rejected into the underlying seawater, approximately 20% of N2O (18.1% ± 4.0%) still remained in the ice, and a small fraction of N2O (12.6% ± 3.2%) was released into the atmosphere. However, it should be noted that when sea ice formed in the simulation system, the surface water had not yet achieved N2O equilibrium, and only approximately 10 cm of sea ice formed. This experiment provided important semi-quantitative information for N2O dynamics; that is, only a small amount of N2O in the sea water was released to the atmosphere during sea ice formation, and most of the N2O was rejected into the underlying water.
4 Conclusion
In this work, a time-series station in the Prydz Bay was studied. The N2O in the surface water was monitored for approximately 10 days, and the N2O concentrations in two ice cores were measured. The result of 10 days in situ observation shows that the N2O concentration decline with the decline of salinity. This is probably caused by contribution ice melting water, which is rather depleted with N2O. With the above sea water and sea ice N2O concentrations and salinities as two endmembers, the result two endmembers mixing model derived a mixture whose N2O concentration agrees with the field observation result, suggesting that the ice melting and mixing processes regulate the N2O dynamics in this surface water, though there may be N2O production presents, however, the contribution is probably limited.
Although the ice grown during the sea ice growth simulation experiment only had a thickness of approximately 10 cm, it cast a hint on the N2O dynamics during sea ice formation. That is, approximately 10% of the N2O in the original water body was expelled during this 10-cm sea ice formation, whereas approximately 20% was retained in the sea ice and approximately 70% was rejected with brine beneath the sea water. This result agrees with other studies that most of the N2O in the sea water will rejected to the water below, while a small fraction of N2O will release to the atmosphere, when the sea ice is not thick enough to prohibit the exchange of N2O between the ice, the sea water below and the atmosphere above.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Author contributions
MW: Writing – original draft, Conceptualization, Data curation, Funding acquisition, Methodology. LZ: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Supervision, Writing – review & editing. JL: Data curation, Investigation, Methodology, Writing – review & editing. JZ: Methodology, Validation, Writing – review & editing. WY: Formal analysis, Investigation, Methodology, Visualization, Writing – review & editing. BD: Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Scientific Research Foundation of the Third Institute of Oceanography, MNR (Grant No. 2018031) and National Natural Science Foundation of China (No. 41506225). This study was also supported by Cultivating Innovation Team Program of Third Institute of Oceanography.
Acknowledgments
We also received support from the FRS-FNRS through the bilateral grant Aquatic (Budgeting source and sinks of CH4 and N2O in polar oceans - R.M003.21), research credit Sonatina (J.0060.22) and research project Seaspray (T.0061.23). BD is a research associate of the FRS-FNRS.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Arevalo-Martínez D. L., Kock A., Löscher C., Schmitz R. A., Bange H. W. (2015). Massive nitrous oxide emissions from the tropical South Pacific Ocean. Nat. Geosci. 8, 530–533. doi: 10.1038/ngeo2469
Bange H. W., Arévalo-Martínez D. L., de la Paz M., Farias L., Kaiser J., Kock A., et al. (2019). A harmonized nitrous oxide (N2O) ocean observation network for the 21st century. Front. Mar. Sci. 6, 157. doi: 10.3389/fmars.2019.00157
Bange H. W., Rapsomanikis S., Andreae M. O. (1996). Nitrous oxide emissions from the Arabian Sea. Geophysical Res. Lett. 23, 3175–3178. doi: 10.1029/96GL03072
Crabeck O., Galley R., Delille B., Else B., Geilfus N. X., Lemes M., et al. (2016). Imaging air volume fraction in sea ice using non-destructive X-ray tomography. Cryosphere 10, 1125–1145. doi: 10.5194/tc-10-1125-2016
Delille B., Vancoppenolle M., Geilfus N., Tilbrook B., Lannuzel D., Schoemann V., et al. (2015). Southern Ocean CO2 sink: The contribution of the sea ice. J. Geophysical Res. Oceans 119, 6340–6355.
Farías L., Florez-Leiva L., Besoain V., Sarthou G., Fernández C. (2015). Dissolved greenhouse gases (nitrous oxide and methane) associated with the naturally iron-fertilized Kerguelen region (KEOPS 2 cruise) in the Southern Ocean. Biogeosciences 12, 1925–1940. doi: 10.5194/bg-12-1925-2015
Freing A., Wallace D. W., Bange H. W. (2012). Global oceanic production of nitrous oxide. Philos. Trans. R. Soc. B: Biol. Sci. 367, 1245–1255. doi: 10.1098/rstb.2011.0360
Fripiat F., Sigman D. M., Fawcett S. E., Rafter P. A., Weigand M. A., Tison J. L. (2014). New insights into sea ice nitrogen biogeochemical dynamics from the nitrogen isotopes. Global Biogeochemical Cycles 28, 115–130. doi: 10.1002/2013GB004729
Geilfus N. X., Carnat G., Dieckmann G. S., Halden N., Nehrke G., Papakyriakou T., et al. (2013). First estimates of the contribution of CaCO3precipitation to the release of CO2to the atmosphere during young sea ice growth. J. Geophysical Res.: Oceans 118, 244–255. doi: 10.1029/2012JC007980
Kitidis V., Upstill-Goddard R. C., Anderson L. G. (2010). Methane and Nitrous Oxide in surface water along the North-West Passage, Arctic Ocean. Mar. Chem. 121, 80–86. doi: 10.1016/j.marchem.2010.03.006
Lei R., Li Z., Cheng B., And Z. Z., Heil P. (2010). Annual cycle of landfast sea ice in Prydz Bay, east Antarctica. J. Geophysical Res.: Oceans. 115, CO2006. doi: 10.1029/2008jc005223
Nevison C. D., Keeling R. F., Weiss R. F., Popp B. N., Jin X., Fraser P. J., et al. (2005). Southern Ocean ventilation inferred from seasonal cycles of atmospheric N2O and O2/N2 at Cape Grim, Tasmania. Tellus B 57, 218–229. doi: 10.1111/j.1600-0889.2005.00143.x
Odile C., Delille B., Rysgaard S., Thomas D. ,. N., Geilfus N.-X. (2014). First “in situ” determination of gas transport coefficients (DO2, DAr, and DN2) from bulk gas concentration measurements (O2, N2, Ar) in natural sea ice. J. Geophysical Res. Oceans. 119, 6655–6668. doi: 10.1002/2014JC009849
Priscu J., Downes M., Priscu L., Palmisano A., Sullivan C. (1990). Dynamics of ammonium oxidizer activity and nitrous oxide(N sub (2) O) within and beneath Antarctic sea ice. Mar. Ecol. Prog. series Oldendorf 62, 37–46. doi: 10.3354/meps062037
Randall K., Scarratt M., Levasseur M., Michaud S., Xie H., Gosselin M. (2012). First measurements of nitrous oxide in Arctic sea ice. J. Geophysical Res.: Oceans 117. doi: 10.1029/2011JC007340
Ravishankara A. R., Daniel J. S., Portmann R. W. (2009). Nitrous oxide (N2O): The dominant ozone-depleting substance emitted in the 21st century. Science 326, 123. doi: 10.1126/science.1176985
Rees A. P., Brown I. J., Jayakumar A., Lessin G., Somerfield P. J., Ward B. B. (2021). Biological nitrous oxide consumption in oxygenated waters of the high latitude Atlantic Ocean. Commun. Earth Environ. 2, 1–8. doi: 10.1038/s43247-021-00104-y
Rees A. P., Owens N. J. P., Upstill-Goddard R. C. (1997). Nitrous oxide in the bellingshausen sea and drake passage. J. Geophysical Res. 102, 3383–3392. doi: 10.1029/96JC03350
Roden N. P., Shadwick E. H., Tilbrook B., Trull T. W. (2013). Annual cycle of carbonate chemistry and decadal change in coastal Prydz Bay, East Antarctica. Mar. Chem. 155, 135–147. doi: 10.1016/j.marchem.2013.06.006
Tian H., Xu R., Canadell J. G., Thompson R. L., Winiwarter W., Suntharalingam P., et al. (2020). A comprehensive quantification of global nitrous oxide sources and sinks. Nature 586, 248–256. doi: 10.1038/s41586-020-2780-0
Van Der Linden F. C., Tison J. L., Champenois W., Moreau S., Carnat G., Kotovitch M., et al. (2020). Sea ice CO 2 dynamics across seasons: impact of processes at the interfaces. J. Geophysical Res.: Oceans 125, e2019JC015807. dio: 10.1029/2019JC015807
Weiss R. F., Price B. A. (1980). Nitrous oxide solubility in water and seawater. Mar. Chem. 8, 347–359. doi: 10.1016/0304-4203(80)90024-9
Yang S., Chang B. X., Warner M. J., Weber T. S., Bourbonnais A. M., Santoro A. E., et al. (2020). Global reconstruction reduces the uncertainty of oceanic nitrous oxide emissions and reveals a vigorous seasonal cycle. Proc. Natl. Acad. Sci. 117, 11954–11960. doi: 10.1073/pnas.1921914117
Zhan L., Chen L., Zhang J., Jinpei Y., Li Y., Wu M. (2016). A permanent N2O sink in the Nordic seas and its strength and possible variability over the past four decades. J. Geophysical Res. Oceans 121, 5608–5621. doi: 10.1002/2016JC011925
Zhan L.-Y., Chen L.-Q., Zhang J.-X., Lin Q. (2013). A system for the automated static headspace analysis of dissolved N2O in seawater. Int. J. Environ. Analytical Chem. 93, 828–842. doi: 10.1080/03067319.2012.702273
Zhan L., Chen L., Zhang J., Yan J., Li Y., Wu M., et al. (2015). Austral summer N2O sink and source characteristics and their impact factors in Prydz Bay, Antarctica. J. Geophysical Res.: Oceans 120, 5836–5849. doi: 10.1002/2015JC010944
Zhan L., Wu M., Chen L., Zhang J., Li Y., Liu J. (2017). The air-sea nitrous oxide flux along cruise tracks to the arctic ocean and Southern Ocean. Atmosphere 8, 216. doi: 10.3390/atmos8110216
Zhan L., Zhang J., Li Y., Man W., Jian L., Qi L., et al. (2018). A fully automatic system for underway N2O measurements based on cavity ring-down spectroscopy. Int. J. Environ. Analytical Chem. 98, 709–724. doi: 10.1080/03067319.2018.1499902
Keywords: sea ice, nitrous oxide, Prydz Bay, Polar ocean, greenhouse gas
Citation: Wu M, Zhan L, Liu J, Zhang J, Ye W and Delille B (2024) A study on the role of sea ice in the nitrous oxide cycle in the Prydz Bay, Antarctica. Front. Mar. Sci. 11:1495913. doi: 10.3389/fmars.2024.1495913
Received: 13 September 2024; Accepted: 22 November 2024;
Published: 10 December 2024.
Edited by:
Yuanyuan Feng, Shanghai Jiao Tong University, ChinaCopyright © 2024 Wu, Zhan, Liu, Zhang, Ye and Delille. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liyang Zhan, emhhbmxpeWFuZ0B0aW8ub3JnLmNu