 Tae-Ho Yoon
Tae-Ho Yoon Hye-Eun Kang
Hye-Eun Kang Sarifah Aini
Sarifah Aini Arief Wujdi
Arief Wujdi Hyun-Woo Kim
Hyun-Woo Kim Shantanu Kundu
Shantanu Kundu- 1KNU G-LAMP Project Group, KNU Institute of Basic Sciences, Kyungpook National University, Daegu, Republic of Korea
- 2Institute of Marine Life Science, Pukyong National University, Busan, Republic of Korea
- 3Department of Marine Biology, Pukyong National University, Busan, Republic of Korea
- 4Interdisciplinary Program of Marine and Fisheries Sciences and Convergent Technology, Pukyong National University, Busan, Republic of Korea
- 5Research Center for Fishery, National Research and Innovation Agency (BRIN), Bogor, Indonesia
- 6Marine Integrated Biomedical Technology Center, National Key Research Institutes in Universities, Pukyong National University, Busan, Republic of Korea
- 7Ocean and Fisheries Development International Cooperation Institute, College of Fisheries Science, Pukyong National University, Busan, Republic of Korea
- 8International Graduate Program of Fisheries Science, Pukyong National University, Busan, Republic of Korea
Introduction: The large-scale mullet, Parachelon grandisquamis (Teleostei: Mugilidae), is a monotypic species endemic to the eastern Atlantic Ocean, playing a crucial role in tropical ecosystems. Despite its ecological significance, the systematic classification of Mugilidae remains unresolved, largely due to their diverse morphology, which necessitates the integration of molecular data.
Methods: This study aimed to achieve a comprehensive molecular characterization of the species and establish its matrilineal taxonomic placement using complete mitogenome data. Next-generation sequencing was employed to generate the de novo mitogenome of P. grandisquamis, which spans 16,859 bp and includes 13 protein-coding genes (PCGs), 22 transfer RNAs, two ribosomal RNAs, and a non-coding AT-rich control region (CR).
Results: Most PCGs use ATG as the start codon, with the exception of COI, which begins with GTG. Analysis of amino acids abundance revealed high frequencies for leucine, serine, proline, threonine, and alanine with distinctive codon usage. The proportion of nonsynonymous and synonymous substitutions suggests strong purifying selection in most PCGs, except for ND4L, ND5, and ND6. Most transfer RNAs exhibited typical cloverleaf secondary structures, with the exception of tRNA-Ser1 (GCT), which lacks base pairing in the DHU arm. Mitogenome-based phylogenetic analysis using the Bayesian approach revealed that the monotypic P. grandisquamis is closely related to the genera Chelon and Planiliza within Mugilidae. Furthermore, analysis of the CRs with polymorphic nucleotides in conserved blocks provides additional insight into the development of distinct molecular markers for species identification and population structure analysis of mullets.
Discussion: Overall, this study provides a comprehensive analysis of the mitogenomic structure and variation of P. grandisquamis and other mullets, confirming its maternal evolutionary relationships and offering valuable insights for advancing SNP-based species discrimination within the Mugilidae lineage.
1 Introduction
The grey mullets, a group of ray-finned fishes within the order Mugiliformes and family Mugilidae, are widely distributed across tropical, subtropical, and temperate regions globally (Thomson, 1966). They play a vital ecological role in aquatic ecosystems and serve as an important food resource. Mugilids currently encompasses 76 valid species across 25 genera, with two new species described within the last decade (Fricke et al., 2024). Despite their ecological significance, the taxonomy and evolutionary relationships of mugilids remain largely unresolved. This is primarily due to the morphological dimorphism observed among species, which complicates their systematic classification (Harrison et al., 2007; Schultz, 1946; Thomson, 1997). As a result, the number of recognized species may be overestimated, as taxonomic evaluations have often been based on local specimens without adequate comparison to those from other regions (Thomson, 1954).
The first comprehensive taxonomic revision of the Mugilidae family, based on mouth anatomy, validated 13 genera (Schultz, 1946; Ghasemzadeh et al., 2004). However, subsequent studies have indicated that the systematics of this group remain incomplete (Nelson, 2006). Within the extant species, three genera—Chelon, Mugil, and Planiliza—are particularly species-rich, encompassing 41 valid species and accounting for 53.94% of the family’s total diversity. The remaining genera exhibit less diversity, with 16 being monotypic (Fricke et al., 2024). The high proportion of monotypic genera may reflect the challenges in their classification due to the limited presence of diagnostic or synapomorphic characters, potentially indicating a prolonged period of static ancestral radiation (Durand et al., 2012). These morpho-anatomical phylogenetic hypotheses have consistently encountered difficulties in resolving the cladistic relationships among mugilid species (Harrison and Howes, 1991; Schultz, 1946; Ghasemzadeh, 1998; Senou, 1988; Thomson, 1997).
Over the past two decades, DNA-based research has significantly advanced the resolution of systematic challenges in fish taxonomy across various levels (Chen and Mayden, 2010). Initially, due to shared plesiomorphic characteristics, mugilids were positioned in an intermediate spot within the Acanthomorph phylogeny (Stiassny, 1993). However, more refined teleost cladistic analyses have since clarified that mugilids are evolutionarily closer to other advanced teleosts within the Percomorpha (Chen et al., 2003; Miya et al., 2003; Chen et al., 2007; Mabuchi et al., 2007). The phylogenetic placement of mugilids within the Acanthomorph lineage has been further investigated using molecular data (Setiamarga et al., 2008; Li et al., 2009). Despite this, phylogenetic analyses specifically targeting mugilids have often relied on partial molecular markers, both nuclear and mitochondrial, sourced from various geographical regions (Caldara et al., 1996; Rossi et al., 1998; Turan et al., 2005; Fraga et al., 2007; Papasotiropoulos et al., 2007; Semina et al., 2007; Blel et al., 2008; Erguden et al., 2010; Lee et al., 1995). These efforts have predominantly focused on species-rich genera such as Mugil, Chelon, and Planiliza to elucidate their evolutionary relationships and lineage diversification (Aurelle et al., 2008; Heras et al., 2009). Additionally, molecular data have been effectively employed in phylogeographic studies of mugilids, with particular emphasis on the species Mugil cephalus and Mugil curema (Crosetti et al., 1994; Jamandre et al., 2009; Ke et al., 2009; Livi et al., 2011; Shen et al., 2011; Rocha-Olivares et al., 2000).
Recently, the integration of next-generation sequencing (NGS) into biodiversity research has greatly enhanced the resolution of teleost phylogeny, including that of mugilids, through complete mitogenome-based assessments (Miya et al., 2001, 2003; Yamanoue et al., 2007). Ichthyologists worldwide have analyzed the complete mitogenomes of numerous fish species, leveraging their genetic traits to reconstruct evolutionary hypotheses and trace lineage diversification across time and space (Iwasaki et al., 2013; Satoh et al., 2016). Currently, the global GenBank database includes mitogenomes from 24 Mugilidae species across 16 genera, including eight monotypic taxa. However, this taxonomic coverage is skewed by the inclusion of just three species (Chelon labrosus, Oedalechilus labeo, and Mugil curema), which are all distributed in the eastern Atlantic Ocean. Notably, the monotypic and sympatric mugilids Neochelon falcipinnis and Parachelon grandisquamis, which are endemic to the eastern Atlantic, have yet to be assessed from this unique marine environment. As a result, mitogenome-based phylogenetic hypotheses may offer a biased perspective of mugilid evolution, highlighting the critical need to include the remaining 50% of monotypic taxa on a global scale.
The large-scaled mullet, Parachelon grandisquamis, is distributed along the eastern Atlantic coast, from Senegal south to Congo, including the islands of Bioko (Equatorial Guinea), São Tomé, and Príncipe (Fricke et al., 2024; Froese and Pauly, 2024). This species inhabits shallow coastal waters, estuaries, and brackish lagoons, including mangroves, creeks, inundated mudflats, and freshwater rivers (Harrison, 2008). The taxonomic classification of P. grandisquamis has undergone several revisions; originally described under the genus Mugil, it was later placed into the genus Liza due to key morphological features, including its large sized scale count, the presence of two pharyngobranchial valves, and the yellowish colorization of its anal and lower caudal fin lobe (Harrison, 2016; Trape et al., 2012). More recently, phylogenetic analyses based on mitochondrial DNA placed the species in a monotypic genus, Parachelon, distinguishing it from other mugilids (Durand et al., 2012). Beyond taxonomy, molecular data from several nuclear genes (RAG1, ENC1, Myh6, Ptr, Glyt, SH3PX3, Sidkey, Rhodopsin, TMO-4C4, plagl2, serb2, RNF213) and mitochondrial genes (12S rRNA, 16S rRNA, COI, and Cytb) have been generated for P. grandisquamis to elucidate the multilocus-based phylogeny of mugilids and address persistent taxonomic challenges (Durand et al., 2012; Xia et al., 2016). However, to achieve a comprehensive matrilineal phylogeny of mugilids, the generation and characterization of the complete mitogenome are crucial for clarifying the taxonomic placement of this monotypic taxon. This study aims to generate and characterize the complete mitogenome of P. grandisquamis from the eastern Atlantic Ocean to illuminate its maternal evolutionary relationships with other mugilids. In addition to provide evolutionary insights, this genetic data will be critical for understanding the population structure and evolutionary patterns of this species, contributing to conservation genetics. Such studies are essential for all extant Mugiliformes species globally, offering new perspectives on the roles of various mitochondrial genes in the evolution of these enigmatic fish across their marine habitats.
2 Materials and methods
2.1 Sampling and species identification
A single specimen of the large-scaled mullet, P. grandisquamis, was collected from the muddy seabed along the coast of Cameroon, Africa (3.6322°N, 9.8005°E) (Figure 1). The identification was confirmed based on taxonomic characteristics described in previous studies, as these often overlap with its closest congener, Chelon bandialensis. However, P. grandisquamis can be distinguished by having large size of scales, 25-30 scales in the longitudinal series and 8-10.5 scales in the transverse series (Trape et al., 2012). Additionally, the buccal teeth of P. grandisquamis are slightly smaller, and its dorsal, anal, and caudal fins are less distinctly yellowish compared to those of C. bandialensis. Muscle tissue was collected from the ventral thoracic region for molecular analysis and novel mitogenome generation. To validate the morphology-based species identification, we amplified a partial fragment of the mitochondrial COI gene using previously published primer pairs and protocols (Ward et al., 2005; Kundu et al., 2019). The amplified sequence showed a 99.79% nucleotide similarity in a BLAST search with the GenBank sequence (Accession number JQ060476) generated in the previous study where the genus and species were reassigned (Durand et al., 2012). The specimen was stored in 10% formaldehyde at the Fisheries and Animal Industries (MINEPIA) facility in Yaoundé, Cameroon. The experimental protocol was approved by the host institute Animal Care and Use Committee (PKNUIACUC-2022-72). The global distribution map of P. grandisquamis was obtained from FishBase and generated using the CMAR c-squares mapper, hosted at https://www.obis.org.au (Figure 1).
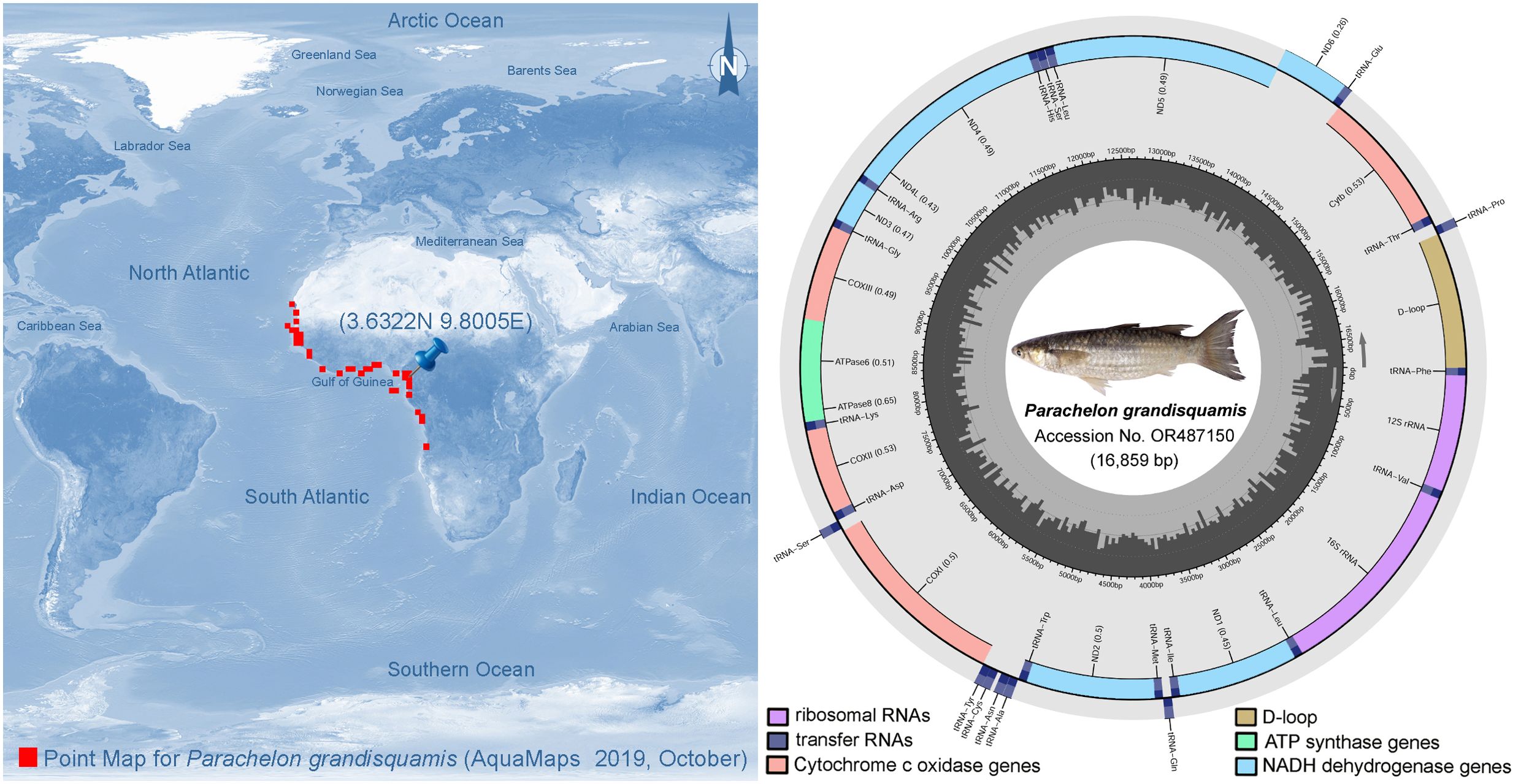
Figure 1. Global distribution pattern and complete mitogenome of P. grandisquamis. The sampling locality is denoted by a blue pin in Cameroon, eastern Atlantic Ocean. The spherical representation of gene structure and arrangement is provided and annotated by the MitoAnnotator. Various color arcs delineate the occurrence of PCGs, tRNAs, rRNAs, and CR.
2.2 DNA extraction and sequencing
The total genomic DNA was extracted by using an AccuPrep® DNA isolation kit with standardized protocol (Bioneer in Daejeon, Republic of Korea). Both the quantity and quality of the DNA were checked through a NanoDrop Microvolume spectrophotometer (Thermo Fisher Scientific D1000, Waltham, MA, USA). The complete mitogenome of P. grandisquamis was sequenced through a next-generation sequencing approach on the NovaSeq platform (Macrogen, Daejeon, Republic of Korea). The TruSeq Nano DNA high-throughput library preparation kit was used to formulate the sequencing library (Illumina, Inc., San Diego, CA, USA). Primarily, 100 ng of genomic DNA was fragmented through an adaptive-focused acoustic tool (Covaris, Woburn, MA, USA). Further, abiding by the end-repair process, DNA fragments were chosen via a bead-based process, adapted by inserting an ‘A’ base, and later ligated through TruSeq DNA UD indexing adapters. To yield the final library, the resulting products endured purification and subsequent PCR enrichment. The quantification of the library was measured using qPCR, ensuing the typical procedure (KAPA Quantification Kits for Illumina Sequencing), and quality judgment was accomplished through the 4200 TapeStation D1000 screentape (Agilent Technologies, Santa Clara, CA, USA). Lastly, the paired-end (2 × 150 bp) sequencing was directed through the NovaSeq platform (Illumina, Inc., San Diego, CA, USA).
2.3 Mitogenome assembly and annotation
More than 20 million raw reads were screened using the Cutadapt tool (http://code.google.com/p/cutadapt/), applying a Phred quality score threshold (Q score > 20). The target mitogenome was assembled using high-quality paired-end reads with the Geneious Prime version 2023.0.1 software. The complete mitogenome of M. cephalus (Accession No. AP002930) served as the reference sequence during assembly, utilizing default mapping algorithms (Miya et al., 2001). Gene boundaries and strand orientations were validated with MitoAnnotator (http://mitofish.aori.u-tokyo.ac.jp/annotation/input/) (Iwasaki et al., 2013) and MITOS Version 1.1.6, integrated with the Galaxy Version 1.1.6 online servers (https://mitos.bioinf.uni-leipzig.de/) (Bernt et al., 2013). Overlapping regions were further scrutinized using MEGA X to confirm the accuracy of the assembled mitogenome (Kumar et al., 2018). The putative amino acid sequences for each protein-coding gene (PCG) were identified using the Open Reading Frame Finder web tool (https://www.ncbi.nlm.nih.gov/orffinder/), employing the vertebrate mitochondrial genetic code.
2.4 Validation of control region
To obtain the full length of the control region (CR), a new primer pair (5′-CGTCGTAATTCTTACCTGAATTGG-3′ and 5′-GCCTGATACCAGCTCCTTGTC-3′) was designed from the segment between Cytb and 12S rRNA genes. The targeted CR was amplified using a TaKaRa Verity Thermal Cycler with a reaction mixture containing 1 µL of P. grandisquamis DNA template, 1X PCR buffer, 1 U Taq polymerase, 10 pmol of each primer, and 2.5 mM dNTPs. The amplicon was purified using the AccuPrep® PCR/Gel Purification Kit (Bioneer, Daejeon, Republic of Korea). Subsequently, the purified amplicon was amplified using the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) and sequenced bidirectionally on an ABI PRISM 3730XL DNA analyzer via the Sanger sequencing method (Macrogen, Daejeon, Republic of Korea). The consensus sequence was screened using SeqScanner version 1.0 (Applied Biosystems Inc., Foster City, CA, USA) to eliminate noisy base pairs, and CR validation was confirmed by aligning overlapping regions with MEGA X. The final mitochondrial genome sequence of P. grandisquamis was deposited in the global GenBank database, where it received an accession number.
2.5 Genomic characterization and comparative analyses
In this study, a circular representation of the assembled mitochondrial genome was generated using the MitoAnnotator online tool (http://mitofish.aori.u-tokyo.ac.jp/annotation/input/). Intergenic spacers between adjacent genes and overlapping sections were manually identified. The nucleotide composition of PCGs, ribosomal RNA (rRNA), transfer RNA (tRNA), and CR was determined using MEGA X. Base composition skew was calculated using the formulas AT-skew = [A − T]/[A + T] and GC-skew = [G − C]/[G + C] (Perna and Kocher, 1995). The mitochondrial genetic code of vertebrates was utilized in MEGA X to verify the initiation and termination codons of each PCG. Additionally, DnaSP 6.0 was employed for comparative analysis, including the calculation of relative synonymous codon usage (RSCU) and the assessment of amino acid abundance (Rozas et al., 2017). The boundaries of tRNA and rRNA genes were confirmed using ARWEN 1.2 and tRNAscan-SE Search Server 2.0 (Chan et al., 2021; Laslett and Canbäck, 2008). To investigate critical components such as PCG substitution patterns and the structure of conserved blocks in CRs, comparative analyses were performed with P. grandisquamis and other mugilids (Supplementary Table S1). The DnaSP 6.0 program was also used to estimate pairwise substitution patterns of PCGs, including non-synonymous (Ka) and synonymous (Ks) substitutions. As detailed in previous studies, CLUSTAL X alignment was used to identify structural features within the conserved blocks in CRs (Thompson et al., 1997; Kundu et al., 2024).
2.6 Dataset preparation and phylogenetic analyses
To elucidate the evolutionary relationships among species in the Mugilidae family, including P. grandisquamis, we conducted a phylogenetic analysis using the mitochondrial genomes of 21 species representing 15 genera. The Black Mouth Cameroon Tilapia, Coptodon camerunensis (Cichliformes: Cichlidae) was included as an outgroup taxon (Access number OQ696044) (Supplementary Table S1). The phylogenetic tree was constructed using a dataset comprising 13 PCGs, assembled with iTaxoTools 0.1 (Vences et al., 2021). The optimal substitution model, ‘GTR + G + I,’ was identified based on the Bayesian information criterion (BIC) using PartitionFinder 2 and JModelTest v2 (Darriba et al., 2012; Lanfear et al., 2016). Bayesian analysis (BA) was performed with MrBayes 3.1.2, employing a model with nst = 6, one cold and three hot chains of the Metropolis-coupled Markov chain Monte Carlo (MCMC). The analysis ran for 1,000,000 generations, with tree samples collected every 100 generations and 25% of the initial samples discarded as burn-in (Ronquist and Huelsenbeck, 2003). The resulting Bayesian tree was visualized using the iTOL v4 web server (https://itol.embl.de/login.cgi) (Letunic and Bork, 2007).
3 Results
3.1 Mitogenome structure and organization
In this study, we illustrated the mitogenome of P. grandisquamis, revealing a length of 16,859 base pairs (bp) with access numbers in GenBank OR487150 (Figure 1). The mitogenome for P. grandisquamis contains 37 genes, consisting of 13 PCGs, 22 tRNAs, two rRNAs, and AT-rich CR. The positive strand contains 12 PCGs, 14 tRNAs, and two rRNAs, while the negative strand contains a single PCG (ND6) as well as eight tRNAs (tRNA-Gln, tRNA-Ala, tRNA-Asn, tRNA-Cys, tRNA-Tyr, tRNA-Ser, tRNA-Glu, and tRNA-Pro) (Table 1). The overall mitogenome base composition of P. grandisquamis showed an AT bias of 55.08% (A = 28.30%, T = 26.70%, G = 15.42%, and C = 29.43%), resulting an AT-skew of 0.030 and a GC-skew of -0.312 (Table 2). Further investigation on the gene boundaries and arrangement revealed 15 intergenic spacers with a total of 122 bp and five overlapping regions, totaling of 23 bp in the mitogenome of P. grandisquamis. The longest intergenic spacer with 32 bp was identified between tRNA-Thr (T) and tRNA-Pro (P), while the most extensive overlap region of 10 bp was shared between the genes ATP8 and ATP6 (Table 1).
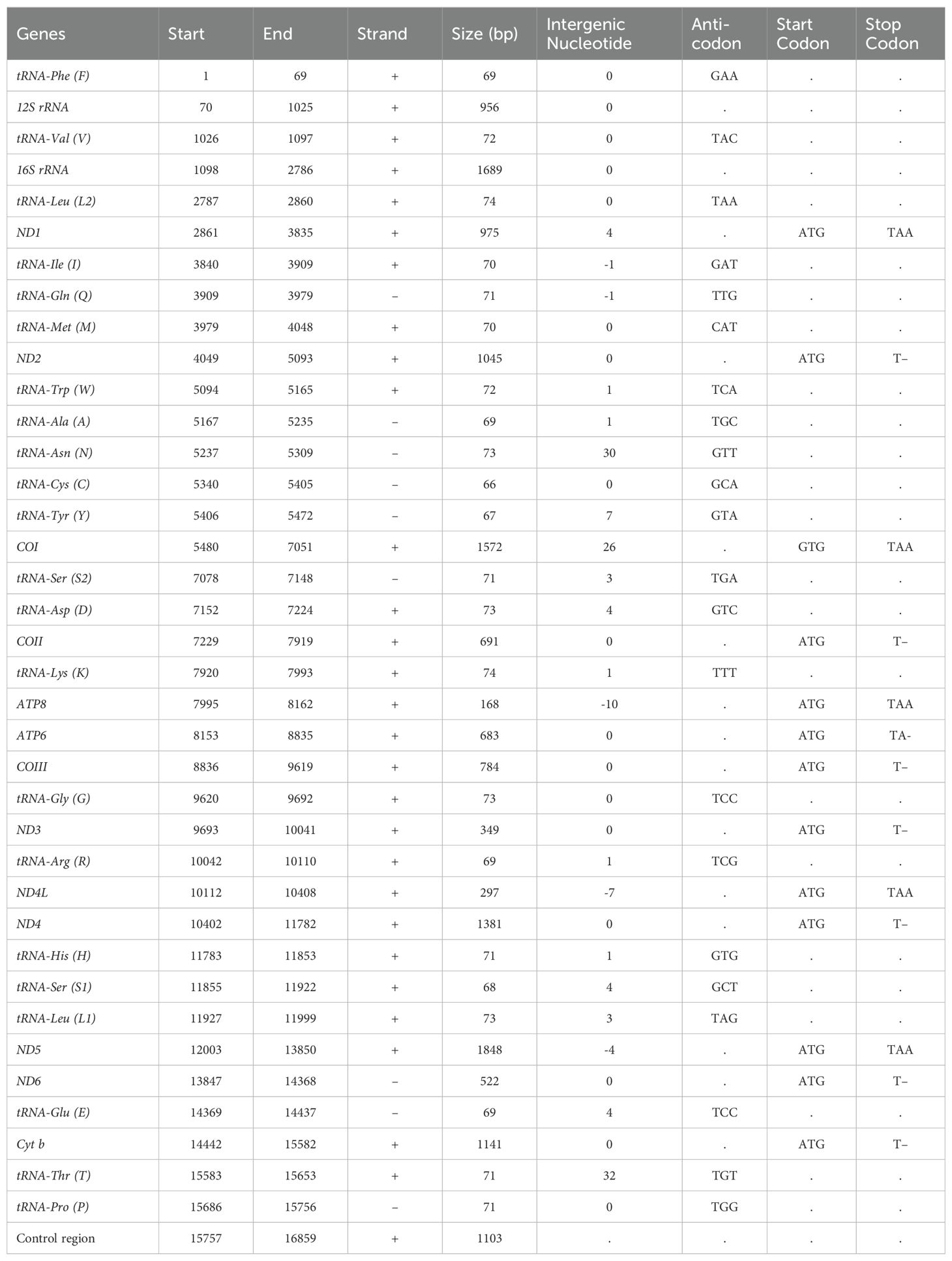
Table 1. List of annotated mitochondrial genes, including their boundaries, sizes, and intergenic nucleotides for P. grandisquamis.
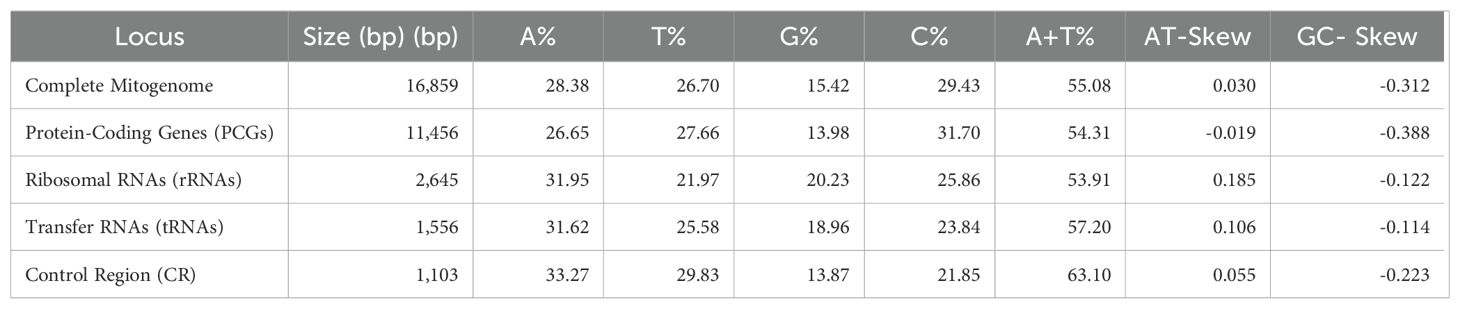
Table 2. Nucleotide composition of mitochondrial genomes in P. grandisquamis.
3.2 Features of protein-coding genes
The mitogenomes of P. grandisquamis contain 13 PCGs with a total length of 11,456 bp (68% of the complete mitogenome). Among that, the ND5 gene has the longest length with 1,824 bp, while the shortest was ATP8 with 168 bp (Table 1). Additionally, the PCG of P. grandisquamis has an AT bias of 54.31% with an AT-skew of -0.019 and a GC-skew of -0.388. In the mitogenome of this species, most PCGs start codons beginning with ATG, except in COI which starts with GTG. Notably, five PCGs (ND1, COI, ATP8, ND4L, ND5) ended with TAA, while the remaining PCGs terminated by incomplete stop codon either TA- or T– (Supplementary Table S1).
The analysis of Relative Synonymous Codon Usage (RSCU) revealed that the codons in the 13 PCGs were conserved and specifically translated to amino acids. The total number of codon transcriptions for the 20 amino acids was 3,808 for P. grandisquamis, excluding the termination codons. The amino acid composition of P. grandisquamis PCGs was predominantly characterized by two hydrophobic amino acids, leucine (14.29%) and alanine (5.86%), along with three neutral amino acids, serine (10.87%), proline (10.06%), and threonine (8.46%). In contrast, three hydrophobic amino acids—tryptophan (0.63%), cysteine (1.02%), and methionine (1.08%)—one hydrophilic amino acid, aspartic acid (1.73%), and one neutral amino acid, glycine (2.42%), were found to be less abundant (Figure 2A, Supplementary Table S2). Notably, the RSCU analysis revealed a noteworthy six variation of codon in Arginine, Leucine and Serine, enabling an increasing its frequency. Moreover, several codons showed its augmentation (> 1.5) to translate a specific amino acid: GGC in Glycine, TCC in Serine, CCC in Proline, and GCC in Alanine, indicating as the most frequent compared to the other codons (Figure 2B, Supplementary Table S3).
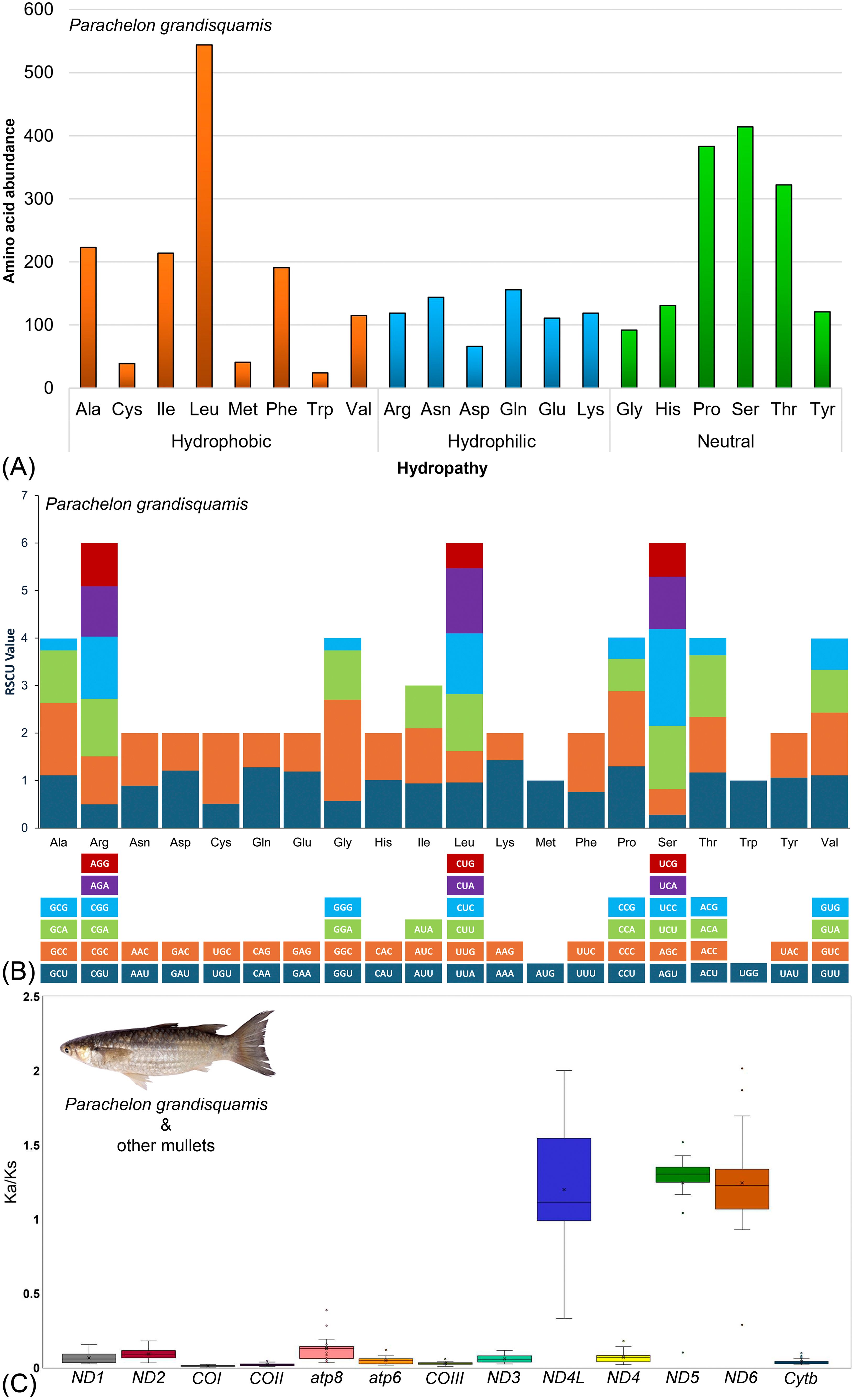
Figure 2. Structural characteristics of P. grandisquamis Protein-coding genes. (A) Amino acids abundance, (B) Relative Synonymous codon usage pattern of PCGs in mitogenomes of P. grandisquamis, and (C) Box plot displays the pairwise divergence in the Ka/Ks ratio for each PCG across all available Mugilids species mitogenomes.
The calculation of the nonsynonymous (Ka) and synonymous (Ks) substitution proportions demonstrated that PCGs of P. grandisquamis and its relative species within the family Mugilidae are subject to the different experience of selection pressure. The average pairwise Ka/Ks ratios ranged from the lowest of 0.0124 ± 0.0034 for COI to a high of 1.2976 ± 0.1017 for ND5, following the order: COI < COII < COIII < Cytb < ATP6 < ND3 < ND1 < ND4 < ND2 < ATP8 < ND4L < ND6 < ND5. Most of PCGs showed Ka/Ks ratio value less than one, except ND4L, ND5, and ND6 that have value greater than one (Figure 2C, Supplementary Table S4).
3.3 Ribosomal RNA and transfer RNA genes
The mitogenome in this study has two ribosomal RNA molecules, explicitly 12S rRNA of 956 bp and 16S rRNA of 1,689 bp which collectively contribute 16% of the complete mitogenome. The rRNA gene displayed an AT bias of 53.91% with AT-skew and GC-skew contributing 0.185 and -0.122, respectively (Table 2). The mitogenome of P. grandisquamis also contained 22 tRNA dispersedly in between rRNA and PCGs. It has a cumulative length of 1,556 bp, contributing 9% of total mitogenome. The tRNA genes of this species exhibited an AT bias of 57.20% with AT-skew and GC-skew of 0.106 and -0.114, respectively (Table 2). Furthermore, most of tRNA showed conventional form of cloverleaf secondary structure, except for tRNA-Ser1 due to lack of nucleotide bond in DHU arm. Fifteen tRNA genes (tRNA-Phe, tRNA-Gln, tRNA-Met, tRNA-Trp, tRNA-Ala, tRNA-Asn, tRNA-Cys, tRNA-Tyr, tRNA-Ser2, tRNA-Asp, tRNA-Gly, tRNA-Arg, tRNA-Ser1, tRNA-Glu, and tRNA-Pro) were constructed through a combination of conventional Watson-Crick base pairs (A=T and G≡C) and wobble base pairs (G-T), while the other seven tRNA genes exclusively use Watson-Crick base pairs. Among those 15 tRNA genes, wobble base pairing was observed prominently in tRNA-Ala and tRNA-Glu. Moreover, Wobble base pairings were identified in the DHU stem of six tRNAs, the TψC stem of eight tRNAs, the anticodon stem of five tRNAs, and the acceptor stem of nine tRNAs (Figure 3).
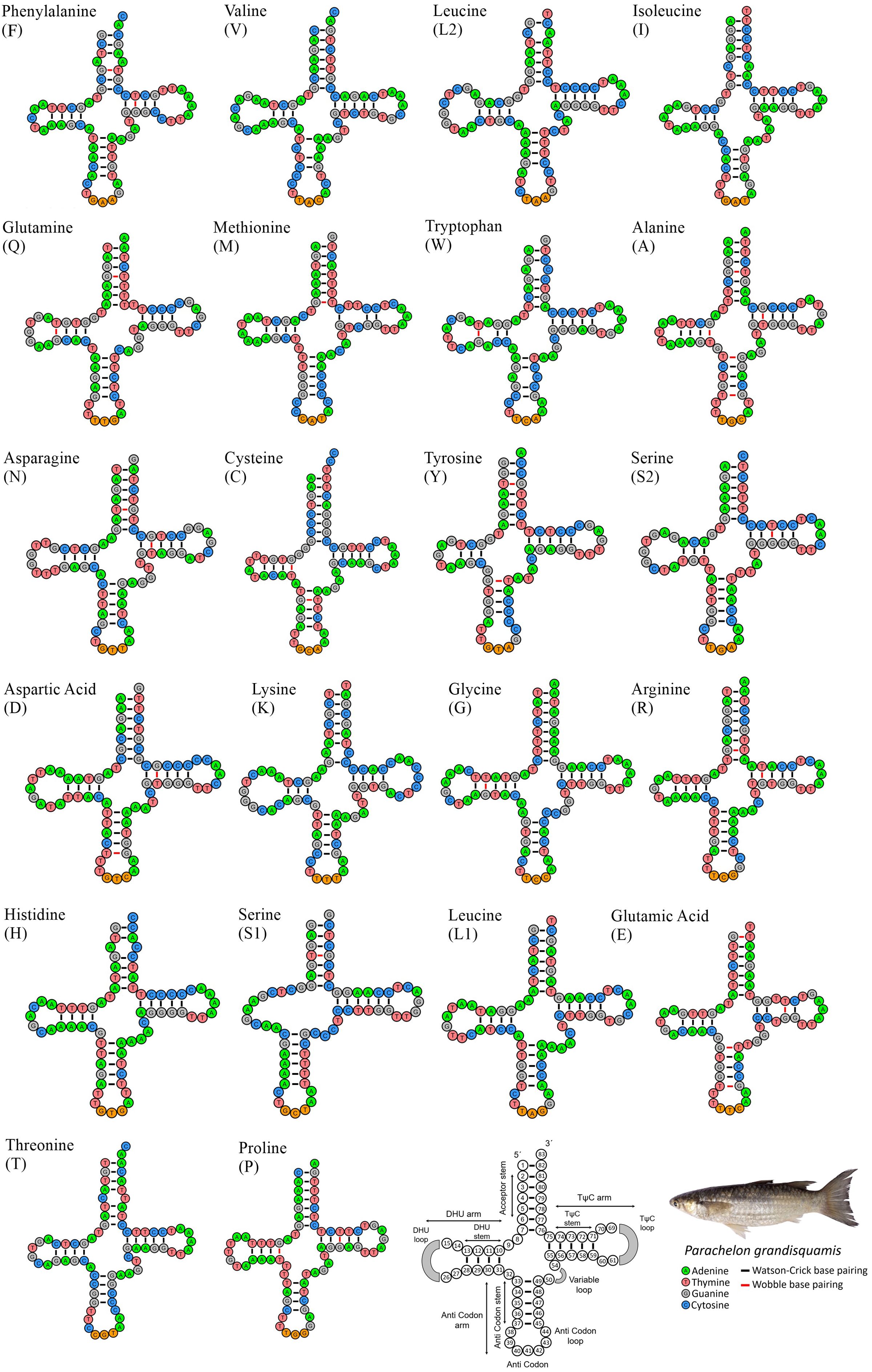
Figure 3. Secondary structures of 22 transfer RNAs (tRNAs) displaying the structural variation in P. grandisquamis. The tRNAs are represented by full names and IUPAC-IUB single letter amino acid codes. Watson-Crick and wobble base pairing are shown by black and red color bars respectively. The last structure shows the nucleotide positions and details of stem-loop of tRNAs.
3.4 Structural features of control region
The total length of control region in P. grandisquamis was 1,103 bp in length, contributing 6.5% of the total mitogenome. The control region of this species showed an AT bias of 57.20%, with an AT-skew of 0.106 and a GC-skew of -0.114 (Table 2). The CR of P. grandisquamis consists of four conserved blocks (CSB-D, CSB-1, CSB-2, and CSB-3), as observed in other fish mitogenomes, including other Mugilids (M. cephalus, Accession No. AP002930 and Moolgarda crenilabis, Accession No. JF911707). Among these conserved blocks, CSB-III is the longest at 31 bp, compared to other regions ranging from 18 bp (CSB-D and CSB-II) to 21 bp (CSB-I) (Figure 4). Comparative analysis of the conserved blocks with other mugilids species demonstrated two groups in CSB-D; the first group with most of species including P. grandisquamis, showed a similarity on conserved nucleotides, however two species (Rhinomugil nasutus and Chaenomugil proboscideus) showed nucleotide similarity as second group. In CSB-I, three groups were observed indicating conserve nucleotides: the first group comprising with four species (Planiliza haematocheilus, Aldrichetta forsteri, R. nasutus, and Dajaus monticola), the second group with two species (M. crenilabis and Osteomugil engeli), and the third group with two species (Planiliza carinata and Planiliza lauvergnii). Similarly, the conserved nucleotides were also found in CSB-II, showing two different groups: the first group comprising six species and the other group with four species. While in CSB-III, three species (P. carinata, P. haematocheilus, and P. lauvergnii) were detected have similar conserved nucleotide, while the other 17 species have different nucleotide patterns, which may be diverged due to the deletion of base pairs in their CRs (Figure 4). In addition, several mugilidis species allowed tandem repeats in the extended termination-associated sequences (ETAS) region, including 2.3 copy of 44 bp consensus sequence in Plicomugil labiosus, 6.8 copies of 79 bp consensus sequence in O. labeo, 2.6 copies of 44 bp consensus sequence in C. proboscideus, and 15.6 copies of 22 bp and 17.4 copies of 22 bp consensus sequences in Minimugil cascasia (Figure 4).
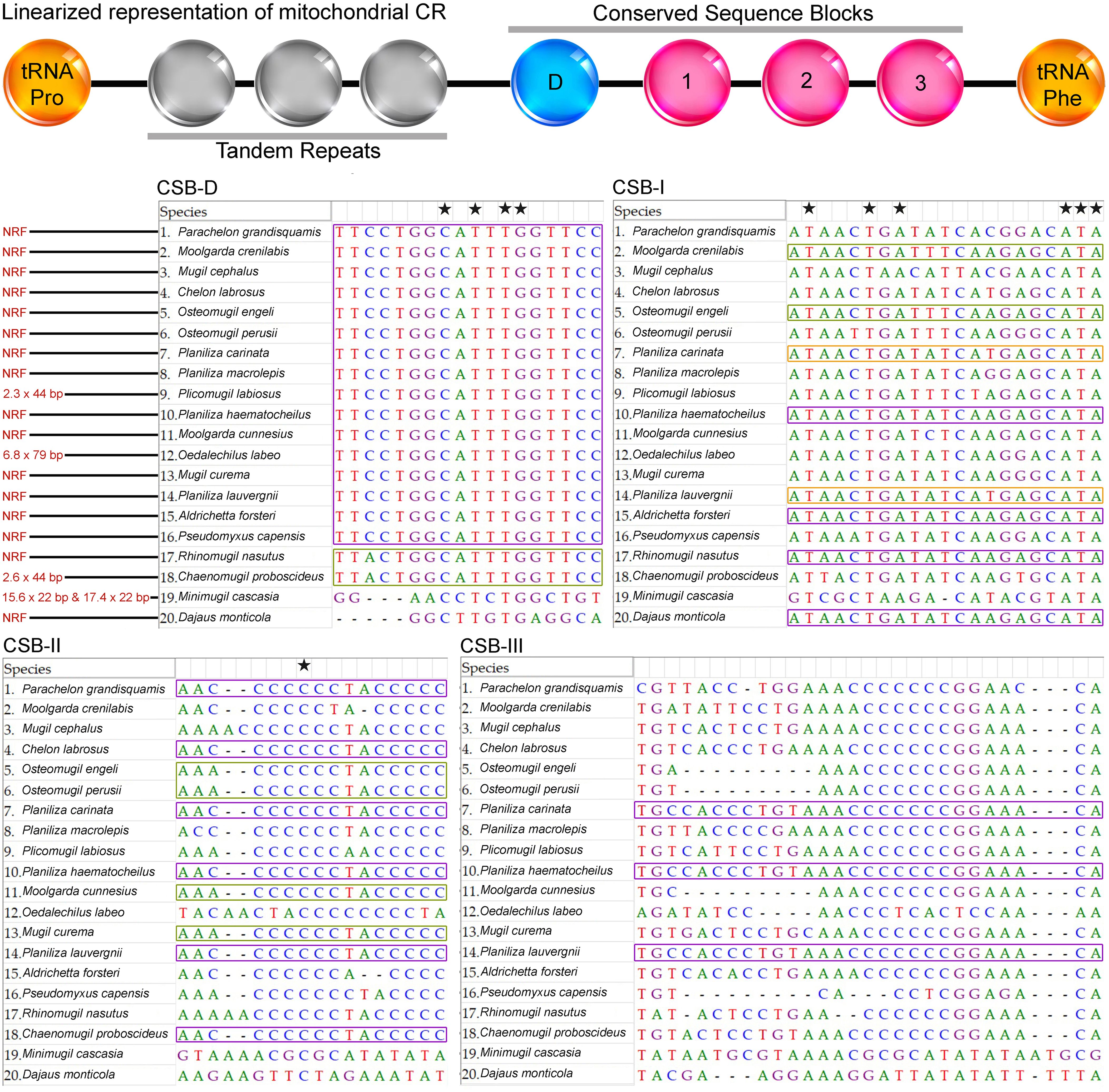
Figure 4. Schematic representation of the different conserved blocks in the control region of P. grandisquamis and other Mugilids species. General linearized representation is showed in the top. The conserved nucleotides pattern in multiple species are marked by different color boxes. Highly conserved bases are denoted by black star across all mugilids.
3.5 Phylogenetic relationship of mullets
A mitogenome-based phylogenetic analysis was performed using 13 PCGs from P. grandisquamis and other related species within the Mugilidae family. The BA phylogenetic tree clearly resolved the species into distinct classifications with high posterior probability support. P. grandisquamis was found to be closely related to species in the genera Chelon and Planiliza (Figure 5). Additionally, the phylogenetic tree confirmed the matrilineal relationships of 10 monotypic species, including P. grandisquamis. The analysis also addressed taxonomic placement of ambiguous species within the Mugilidae family, such as M. cascasia (formerly Sicamugil cascasia), D. monticola (formerly Agonostomus monticola), M. crenilabis (formerly Crenimugil crenilabis), P. labiosus (formerly Oedalechilus labiosus), R. nasutus (formerly Squalomugil nasutus), Pseudomyxus capensis (formerly Myxus capensis), two species of Osteomugil (formerly under the genus Moolgarda), and four species of Planiliza (formerly under the genus Liza) (Figure 5). Notably, the cladistic analysis revealed a non-monophyletic pattern for the genus Moolgarda, with M. crenilabis and M. cunnesius showing distinct lineages, suggesting the need for further investigation.
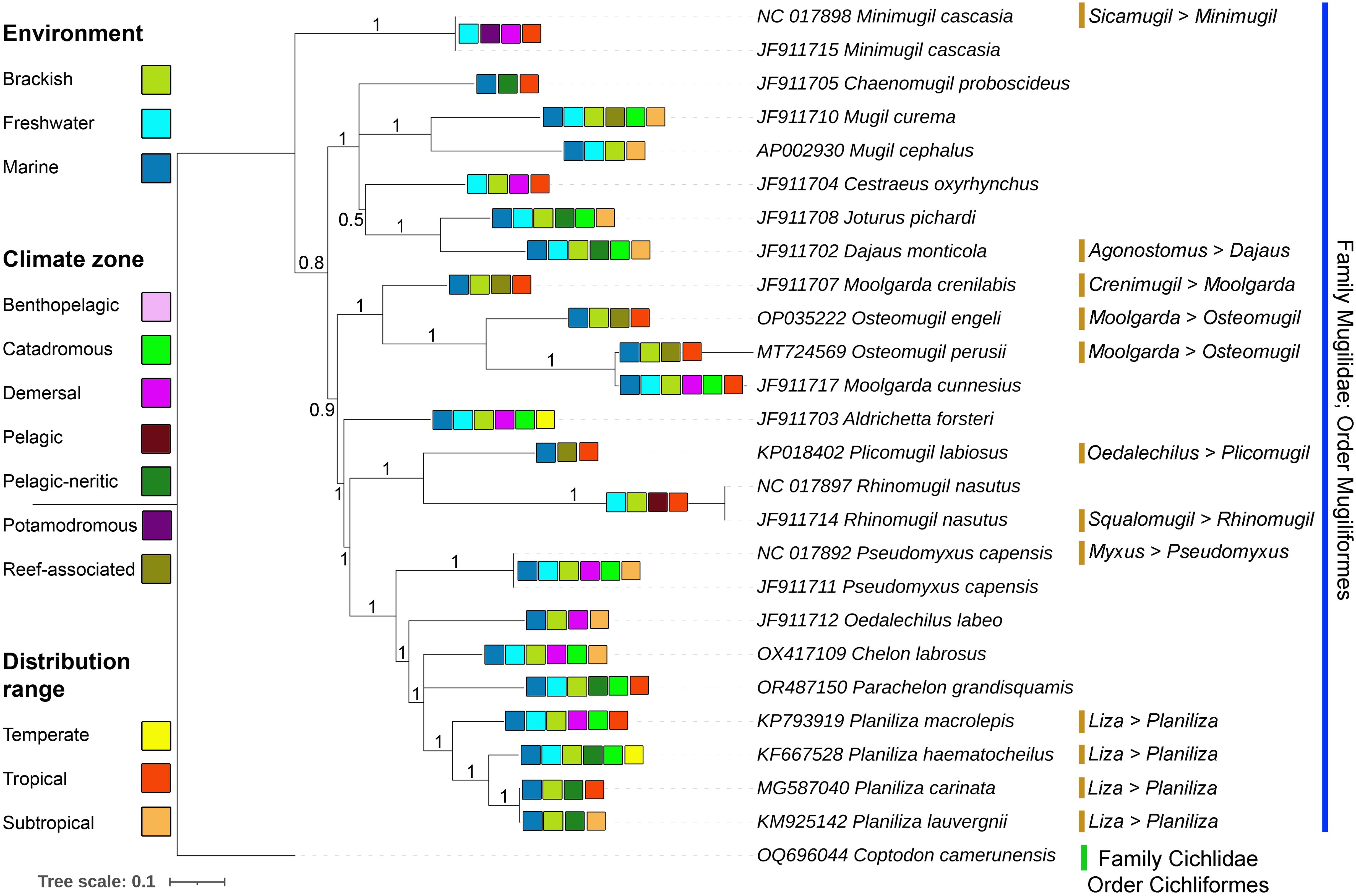
Figure 5. The Bayesian (BA) phylogeny constructed by the concatenated 13 PCGs clearly describe the placement of P. grandisquamis and other mugilids species with high posterior probability supports in each node. The cladogram infers the insights into the revised taxonomical classification and cladistic pattern of mugilids. The environment, climate zone, and distribution range of each species is remarked by different color boxes representing to their distinctive clade.
4 Discussion
4.1 Mitogenomic signature of large-scaled mullet
The mitochondrial genome of P. grandisquamis comprises 37 genes, with 28 genes located on the heavy strand and nine on the light strand. The nucleotide composition of the genome exhibits an A-T bias, which is consistent with the hydrophobic nature of mitochondrial proteins (Naylor et al., 1995). The gene arrangement and strand organization of P. grandisquamis align with those observed in other teleost mitogenomes (Miya et al., 2003, 2005). Our findings suggest that the mitochondrial genome of P. grandisquamis is conserved relative to other species within the Mugilidae family (Miya et al., 2001). Given the general conservation of mitochondrial genomes, gene rearrangements are an important focus in organismal systematics (Gong et al., 2020; Zhang et al., 2020). Therefore, understanding gene arrangement and nucleotide composition across species is essential for refining species classification. The arrangement of mitochondrial genes can influence various aspects of fish physiology, molecular mechanisms, life histories, and genomic evolutionary processes (Montaña-Lozano et al., 2022).
In PCGs, most genes use ATG as the initiation codon, with the exception of COI, which terminates with either complete or incomplete stop codons. This pattern of initiation and termination codon usage in COI is consistent with that observed in other mugilid species (Miya et al., 2001; Shen et al., 2016a, 2016). In this study, amino acids with neutral hydropathy characteristics were found to be more abundant than those with hydrophobic or hydrophilic properties. This observation aligns with findings from previous studies on other teleost species (Wang et al., 2022a). The hydropathy characteristics of amino acids play a crucial role in the evolution of mitochondrial proteins, which are essential for cellular respiration and energy production that are vital for the survival and adaptation of organisms in dynamic environments (Berthelot et al., 2019). Given the frequent fluctuations in salinity, temperature, and oxygen levels experienced by marine fishes, particularly in the Atlantic Ocean (Rutterford et al., 2023), detailed studies on hydropathy-related modifications in mitochondrial proteins could offer valuable insights into their role in optimizing protein conformation and function under such environmental stressors. These adaptations may enhance thermal tolerance and hypoxia resilience, enabling marine species to survive and thrive in highly variable and challenging habitats.
Codon usage was further assessed using RSCU values, revealing that most RSCU values deviated from ‘1’, indicating varying levels of codon bias for different amino acids. Codons for leucine (Leu) and serine (Ser), which have six codon combinations each, were the most frequently used, whereas methionine (Met) and tryptophan (Trp), each represented by a single codon (ATG and TGG, respectively), were less prevalent. Further, the Ka/Ks ratio, a well-established metric for assessing selective pressure and evolutionary associations at the molecular level (Yang and Nielsen, 2000; Zhao et al., 2022). Most PCGs exhibited Ka/Ks ratios less than ‘1’, suggesting strong negative selection in P. grandisquamis and its relatives within the Mugilidae family, with mutations predominantly replaced by synonymous substitutions. This pattern indicates the role of natural selection in reducing deleterious mutations with adverse selective coefficients, positioning with universal patterns detected in other teleosts (Kundu et al., 2024). However, three PCGs (ND4L, ND5, and ND6) showed Ka/Ks ratios greater than ‘1’, indicating potential positive selection. These genes may be more sensitive to genetic mutations, potentially influenced by environmental adaptations or new functional developments (Liao et al., 2024). The Ka/Ks ratio analysis provides valuable insights into how natural selection shapes speciation and evolutionary trajectories in mugilids across diverse aquatic environments. Previous studies have reported that NADH dehydrogenase genes have evolved under positive selection in anadromous salmonids and other deep-sea fishes, suggesting their adaptation to diverse ecosystems influenced by environmental factors such as salinity, temperature, low oxygen levels, darkness, and high-water pressure (Wang et al., 2022b; Shen et al., 2019). However, the specific roles of these genes in mullets remain unclear. The incorporation of this novel mitogenome data of P. grandisquamis into global databases, along with a comparative analysis of substitution patterns with other mugilids, provides baseline information to better understand the environmental adaptations of mullets. This foundational data is crucial for monitoring their protein-level evolutionary dynamics and adaptive strategies.
Additionally, the rRNAs in P. grandisquamis are located on the heavy strand and are separated by tRNA-Val, a pattern consistent with other fish species (Satoh et al., 2006; Liao et al., 2024). Ribosomes, as conserved ribonucleoproteins, play a crucial role in translating genetic information from mRNA into proteins. The structural association of rRNA genes, including their conserved loops, delivers valuable insights into the catalytic developments that are essential for protein synthesis (Lehman, 2004; Satoh et al., 2006). Our analysis also reveals that most tRNAs exhibit a cloverleaf secondary structure, with the exception of tRNA-Ser1. This cloverleaf pattern is common in the mitogenomes of many bony fish, although the absence of the DHU arm in some tRNAs can serve as a distinguishing feature (Liao et al., 2024). The tRNA molecules function as adaptors that translate genetic information into proteins by delivering amino acids during translation. The proper arrangement of tRNAs, particularly in the WANCY region, is crucial for effective mitochondrial gene expression (Cantatore et al., 1987; Ponce et al., 2008). Thus, the analysis of rRNAs and tRNAs in P. grandisquamis, especially through the evaluation of secondary structures, provides important insights into genetic mechanisms and mitochondrial function.
In the mitogenome of P. grandisquamis, the pronounced AT bias in the CR is consistent with observations in other fish species, which typically exhibit a preference for adenine (A) and thymine (T) bases (Miya et al., 2001, 2003, 2005). The CR is particularly significant due to its dynamic nature, as it is the most variable region in the mitochondrial genome. Notably, the repeat-rich ETAS region within the CR is characterized by specific motifs likely forming stable hairpin loops. These structures act as sequence-specific signals to regulate mitochondrial DNA replication termination (Satoh et al., 2016). Understanding the complex mechanisms controlling the CR—such as random loss, dimer formation of mitogenomes, non-random loss, and genomic rearrangement through double replications—is essential for grasping the structural diversity of mitogenomes and the evolutionary dynamics of mitochondrial genomes (Kundu et al., 2024). Thus, our analysis of the CR, including the identification of polymorphic nucleotides in conserved blocks, provides valuable insights for developing distinct molecular markers for species identification and understanding population structure.
The present mitogenome-based phylogenetic assessment provides valuable insights into the evolutionary relationships of P. grandisquamis, particularly in relation to other monotypic species within the Mugilidae family. This analysis supports the revision of classifications and highlights the non-monophyletic clustering of the Moolgarda genus within the Mugilidae family. Molecular phylogenetics offers robust, independent insights into species evolutionary relationships based on maternal lineage (Caldara et al., 1996; Heras et al., 2009; Liu et al., 2010). Integrating multi-dimensional research, including mitogenomic data alongside morphological information, is essential for elucidating ancestral and descendant lineages and resolving evolutionary patterns that have long been debated among ichthyologists due to morphological contradictions (Harrison et al., 2007; Durand et al., 2012; Rajan et al., 2023). Given the species richness and broad distribution of mugilids across diverse aquatic environments, it is crucial to expand mitogenomic research to enhance our understanding of their speciation and adaptation processes.
4.2 Future perspectives for conservation framework of mullets
The large-scale mullet, P. grandisquamis demonstrates considerable versatility and adaptability across various habitats within the coastal ecosystems of the eastern Atlantic region. However, this adaptability may render it susceptible to genetic mutations and increased heterozygosity in response to environmental changes (Martinez et al., 2018). Coastal and riverine habitat alterations often negatively impact fish populations, potentially creating physical barriers to migration, which can lead to population fragmentation and disruptions in gene flow, thereby increasing the risk of inbreeding in restricted ecosystems (Pimentel et al., 2020; Ovidio et al., 2020). This fragmentation can reduce genetic diversity, threaten population viability, and heighten the risk of local extinction (Rourke et al., 2019). Therefore, integrating traditional species knowledge with molecular data is crucial for assessing genetic variation, population structure, and connectivity within specific ecosystems. Mitogenome-based studies are essential for observing adaptive evolution in Parachelon and other mugilids, which are currently underrepresented. The mitogenome characterization provided here serves as a foundational baseline for monitoring the genetic diversity of this migratory fish in the eastern Atlantic. Our findings highlight the vulnerability of P. grandisquamis to positive selection pressures and unique structural variations in its mitochondrial genome. Notably, mitochondrial PCGs such as ND4L, ND5, and ND6 exhibit heightened sensitivity to gene mutations, offering a basis for further research into the adaptive evolution of this endemic and monotypic species. Understanding how specific genes influence physiological adaptation is crucial for evaluating their adaptability to environmental changes and climate variability (Wang et al., 2022b; Shen et al., 2019). Given that P. grandisquamis is typically catadromous, with a tendency to migrate into freshwater systems, this migratory behavior may have a significant impact on mitochondrial gene evolution, as reflected in their Ka/Ks ratios. These insights will be instrumental in developing effective management strategies and conducting population genetic analyses of this species in near future. Additionally, given the restricted distribution of P. grandisquamis in the eastern Atlantic, this mitogenomic data will aid in elucidating lineage diversification patterns through time tree analyses incorporating multiple mitogenomic datasets from the Mugiliformes lineage.
5 Conclusion
In this study, mitochondrial genome sequence analysis was performed to investigate the monotypic P. grandisquamis, which is endemic to the eastern Atlantic. The complete mitochondrial genome provided detailed insights into the genetic configuration and matrilineal evolutionary patterns of P. grandisquamis, confirming its evolutionary relationships with other mugilids. Given the potential conservation implications for mugilids in the eastern Atlantic, this research underscores the necessity of a thorough understanding of both mitogenomic and demographic information within marine ecosystems. Overall, the analysis of the mitochondrial genome and genetic characteristics of the large-scaled mullet provides valuable insights for future advancements in single nucleotide polymorphism (SNP)-based species discrimination, particularly within the Mugilidae family. Additionally, it offers a better understanding of the regulation of mitochondrial genes and potential environmental adaptations of this catadromous species in the eastern Atlantic Ocean.
Data availability statement
The mitogenome of the study species Parachelon grandisquamis (GenBank Accession Number: OR487150) is now publicly accessible at https://www.ncbi.nlm.nih.gov/nuccore/OR487150.
Ethics statement
The animal study was approved by Pukyong National University Animal Care and Use Committee (PKNUIACUC-2022-72). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
T-HY: Conceptualization, Formal Analysis, Funding acquisition, Validation, Writing – original draft. H-EK: Formal Analysis, Investigation, Software, Validation, Writing – original draft. SA: Data curation, Methodology, Software, Visualization, Writing – original draft. AW: Data curation, Methodology, Software, Visualization, Writing – original draft. H-WK: Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – review & editing. SK: Conceptualization, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by Global-Learning & Academic research institution for Master’s, Ph.D. students, and Postdocs (LAMP) Program of the National Research Foundation of Korea (NRF) grant funded by the Ministry of Education (No. RS-2023-00301914). This research was also supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2021R1A6A1A03039211).
Acknowledgments
The authors would like to thank to Fantong Zealous Gietbong from the Ministry of Livestock, Fisheries and Animal Industries (MINEPIA), Yaounde, Cameroon, for invaluable assistance in conducting sampling.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2024.1484198/full#supplementary-material
References
Aurelle D., Barthelemy R. M., Quignard J. P., Trabelsi M., Faure E. (2008). Molecular phylogeny of mugilidae (Teleostei: Perciformes). Open J. Mar. Sci. 2, 29–37. doi: 10.2174/1874450800802010029
Bernt M., Braband A., Schierwater B., Stadler P. F. (2013). Genetic aspects of mitochondrial genome evolution. Mol. Phylogenet. Evol. 69, 328–338. doi: 10.1016/j.ympev.2012.10.020
Berthelot C., Clarke J., Desvignes T., William Detrich H. III, Flicek P., Peck L. S., et al. (2019). Adaptation of proteins to the cold in Antarctic fish: A role for methionine? Genome Biol. Evol. 11, 220–231. doi: 10.1093/gbe/evy262
Blel H., Chatti N., Besbes R., Farjallah S., Elouaer A., Guerbej H., et al. (2008). Phylogenetic relationships in grey mullets (Mugilidae) in a Tunisian lagoon. Aquac. Res. 39, 268–275. doi: 10.1111/j.1365-2109.2007.01893.x
Caldara F., Bargelloni L., Ostellari L., Penzo E., Colombo L., Patarnello T. (1996). Molecular phylogeny of grey mullets based on mitochondrial DNA sequence analysis: Evidence of a differential rate of evolution at the intrafamily level. Mol. Phylogenet. Evol. 6, 416–424. doi: 10.1006/mpev.1996.0090
Cantatore P., Gadaleta M. N., Roberti M., Saccone C., Wilson A. C. (1987). Duplication and remoulding of tRNA genes during the evolutionary rearrangement of mitochondrial genomes. Nature 329, 853–855. doi: 10.1038/329853a0
Chan P. P., Lin B. Y., Mak A. J., Lowe T. M. (2021). tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096. doi: 10.1093/nar/gkab688
Chen W. J., Bonillo C., Lecointre G. (2003). Repeatability of clades as a criterion of reliability: A case study for molecular phylogeny of acanthomorph (Teleostei) with larger number of taxa. Mol. Phylogenet. Evol. 26, 262–288. doi: 10.1016/S1055-7903(02)00371-8
Chen W. J., Mayden R. L. (2010). A phylogenomic perspective on the new era of ichthyology. BioScience 60, 421–432. doi: 10.1525/bio.2010.60.6.6
Chen W. J., Ruiz-Carus R., Orti G. (2007). Relationships among four genera of mojarras (Teleostei: Perciformes: Gerreidae) from the western Atlantic and their tentative placement among percomorph fishes. J. Fish Biol. 70, 202–218. doi: 10.1111/j.1095-8649.2007.01395.x
Crosetti D., Nelson W. S., Avise J. C. (1994). Pronounced genetic structure of mitochondrial DNA among populations of the circumglobally distributed grey mullet (Mugil cephalus Linnaeus). J. Fish Biol. 44, 47–58. doi: 10.1111/j.1095-8649.1994.tb01584.x
Darriba D., Taboada G. L., Doallo R., Posada D. (2012). JModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 9, 772. doi: 10.1038/nmeth.2109
Durand J. D., Shen K. N., Chen W. J., Jamandre B. W., Blel H., Diop K., et al. (2012). Systematics of the grey mullets (Teleostei: Mugiliformes: Mugilidae): Molecular phylogenetic evidence challenges two centuries of morphology-based taxonomy. Mol. Phylogenet. Evol. 64, 73–92. doi: 10.1016/j.ympev.2012.03.006
Erguden D., Gurlek M., Yaglioglu D., Turan C. (2010). Genetic identification and taxonomic relationship of Mediterranean mugilid species based on mitochondrial 16S rDNA sequence data. J. Anim. Vet. Adv. 9, 336–341. doi: 10.3923/javaa.2010.336.341
Fraga E., Schneider H., Nirchio M., Santa-Brigida E., Rodrigues-Filho L. F., Sampaio I. (2007). Molecular phylogenetic analyses of mullets (Mugilidae, Mugiliformes) based on two mitochondrial genes. J. Appl. Ichthyol. 23, 598–604. doi: 10.1111/j.1439-0426.2007.00911.x
(2024).Eschmeyer’s catalog of fishes: genera, species, references. Available online at: http://researcharchive.calacademy.org/research/ichthyology/catalog/fishcatmain.asp (Accessed 20 July 2024).
(2024).FishBase. Available online at: http://www.fishbase.org (Accessed 20 July 2024).
Ghasemzadeh J. (1998). Phylogeny and systematics of Indo-Pacific mullets (Teleostei: Mugilidae) with special reference to the mullets of Australia. D. Phil. Thesis. (Sydney, Australia: Macquarie University).
Ghasemzadeh J., Ivantsoff W., Aarn (2004). Historical overview of mugilid systematics, with descriptions of Paramugil (Teleostei: Mugiliformes: Mugilidae), new genus. Aqua: J. Ichth. Aquat. Biol. 8, 9–22.
Gong L., Lu X., Luo H., Zhang Y., Shi W., Liu L., et al. (2020). Novel gene rearrangement pattern in Cynoglossus melampetalus mitochondrial genome: New gene order in genus Cynoglossus (Pleuronectiformes: Cynoglossidae). Int. J. Biol. Macromol. 149, 1232–1240. doi: 10.1016/j.ijbiomac.2020.02.017
Harrison I. J. (2008). “Mugilidae,” in The fresh and brackish water fishes of Lower Guinea, West-Central Africa. Eds. Stiassny M. L. J., Teugels G. G., Hopkins C. D. (Institut de Recherche pour le Développement, Paris), 450–471.
Harrison I. J. (2016). “Order mugiliformes. Mugilidae,” in The living marine resources of the Eastern Central Atlantic. Volume 3 Bony fishes part 1 (Elopiformes to Scorpaeniformes). Eds. Carpenter, De Angelis (Food and Agriculture Organization, Rome), Pp. 2077–2113.
Harrison I. J., Howes G. J. (1991). The pharyngobranchial organ of mugilid fishes: Its structure, variability, ontogeny, possible function, and taxonomic utility. Bull. Br. Mus. Nat. Hist. Zool. 57, 111–132.
Harrison I. J., Nirchio M., Oliveira C., Ron E., Gaviria J. (2007). A new species of mullet (Teleostei: Mugilidae) from Venezuela, with a discussion on the taxonomy of Mugil gaimardianus. J. Fish Biol. 71, 76–97. doi: 10.1111/j.1095-8649.2007.01520.x
Heras S., Roldán M. I., Gonzalez Castro M. (2009). Molecular phylogeny of Mugilidae fishes revised. Rev. Fish Biol. Fish. 19, 217–231. doi: 10.1007/s11160-008-9100-3
Iwasaki W., Fukunaga T., Isagozawa R., Yamada K., Maeda Y., Satoh T. P., et al. (2013). MitoFish and MitoAnnotator: A mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol. Bio. Evol. 30, 2531–2540. doi: 10.1093/molbev/mst141
Jamandre B. W., Durand J. D., Tzeng W. N. (2009). Phylogeography of the flathead mullet Mugil cephalus in the Northwest Pacific inferred from the mtDNA control region. J. Fish Biol. 75, 393–407. doi: 10.1111/j.1095-8649.2009.02332.x
Ke H. M., Lin W. W., Kao H. W. (2009). Genetic diversity and differentiation of grey mullet (Mugil cephalus) in the coastal waters of Taiwan. Zool. Sci. 26, 421–428. doi: 10.2108/zsj.26.421
Kumar S., Stecher G., Li M., Knyaz C., Tamura K. (2018). MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Bio. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kundu S., Chandra K., Tyagi K., Pakrashi A., Kumar V. (2019). DNA barcoding of freshwater fishes from Brahmaputra River in Eastern Himalaya biodiversity hotspot. Mitochondrial DNA B. Resour. 4, 2411–2419. doi: 10.1080/23802359.2019.1637290
Kundu S., Kang H. E., Kim A. R., Lee S. R., Kim E. B., Amin M. H. F., et al. (2024). Mitogenomic characterization and phylogenetic placement of African Hind, Cephalopholis taeniops: Shedding light on the evolution of groupers (Serranidae: Epinephelinae). Int. J. Mol. Sci. 25, 1822. doi: 10.3390/ijms25031822
Lanfear R., Frandsen P. B., Wright A. M., Senfeld T., Calcott B. (2016). PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Bio. Evol. 34, 772–773. doi: 10.1093/molbev/msw260
Laslett D., Canbäck B. (2008). ARWEN, a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175. doi: 10.1093/bioinformatics/btm573
Lee S. C., Chang J. T., Tsu Y. Y. (1995). Genetic relationships of four Taiwan mullets (Pisces: Perciformes: Mugilidae). J. Fish Biol. 46, 159–162. doi: 10.1111/j.1095-8649.1995.tb05955.x
Lehman N. (2004). Assessing the likelihood of recurrence during RNA evolution in vitro. Artif. Life. 10, 1–22. doi: 10.1162/106454604322875887
Letunic I., Bork P. (2007). Interactive Tree of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. doi: 10.1093/bioinformatics/btl529
Li B., Dettaï A., Cruaud C., Couloux A., Desoutter-Meniger M., Lecointre G. (2009). RNF213, a new nuclear marker for acanthomorph phylogeny. Mol. Phylogenet. Evol. 50, 345–363. doi: 10.1016/j.ympev.2008.11.013
Liao X., Shih Y., Jia C., Gao T. (2024). Complete mitochondrial genome of four Peristediidae fish species: Genome characterization and phylogenetic analysis. Genes 15, 557. doi: 10.3390/genes15050557
Liu J. Y., Brown C. L., Yang T. B. (2010). Phylogenetic relationships of mullets (Mugilidae) in China Seas based on partial sequences of two mitochondrial genes. Biochem. Syst. Ecol. 38, 647–655. doi: 10.1016/j.bse.2010.01.007
Livi S., Sola L., Crosetti D. (2011). Phylogeographic relationships among worldwide populations of the cosmopolitan marine species, the striped gray mullet (Mugil cephalus), investigated by partial cytochrome b gene sequences. Biochem. Syst. Ecol. 39, 121–131. doi: 10.1016/j.bse.2011.01.006
Mabuchi K., Miya M., Azuma Y., Nishida M. (2007). Independent evolution of the specialized pharyngeal jaw apparatus in cichlid and labrid fishes. BMC Evol. Biol. 7, 10. doi: 10.1186/1471-2148-7-10
Martinez A. S., Willoughby J. R., Christie M. R. (2018). Genetic diversity in fishes is influenced by habitat type and life-history variation. Ecol. Evol. 8, 12022–12031. doi: 10.1002/ece3.4661
Miya M., Kawaguchi A., Nishida M. (2001). Mitogenomic exploration of higher teleostean phylogenies: A case study for moderate-scale evolutionary genomics with 38 newly determined complete mitochondrial DNA sequences. Mol. Bio. Evol. 18, 1993–2009. doi: 10.1093/oxfordjournals.molbev.a003741
Miya M., Satoh T. P., Nishida M. (2005). The phylogenetic position of toadfishes (order Batrachoidiformes) in the higher ray-finned fish as inferred from partitioned Bayesian analysis of 102 whole mitochondrial genome sequences. Biol. J. Linn. Soc 85, 289–306. doi: 10.1111/j.1095-8312.2005.00483.x
Miya M., Takeshima H., Endo H., Ishiguro N. B., Inoue J. G., Mukai T., et al. (2003). Major patterns of higher teleostean phylogenies: A new perspective based on 100 complete mitochondrial DNA sequences. Mol. Phylogenet. Evol. 26, 121–138. doi: 10.1016/S1055-7903(02)00332-9
Montaña-Lozano P., Moreno-Carmona M., Ochoa-Capera M., Medina N. S., Boore J. L., Prada C. F. (2022). Comparative genomic analysis of vertebrate mitochondrial genomes reveals a differential rate of rearrangements between taxonomic classes. Sci. Rep. 12, 5479. doi: 10.1038/s41598-022-09512-2
Naylor G. J. P., Collins T., Brown W. M. (1995). Hydrophobicity and phylogeny. Nature 373, 555–556. doi: 10.1038/373565b0
Ovidio M., Sonny D., Watthez Q., Goffaux D., Detrait O., Orban P., et al. (2020). Evaluation of the performance of successive multispecies improved fishways to reconnect a rehabilitated river. Wetl. Ecol. Manage. 28, 641–654. doi: 10.1007/s11273-020-09737-w
Papasotiropoulos V., Klossa-Kilia E., Alahiotis S., Kilias G. (2007). Molecular phylogeny of grey mullets (Teleostei: Mugilidae) in Greece. Evidence from sequence analysis of mtDNA segments. Biochem. Genet. 45, 623–636. doi: 10.1007/s10528-007-9101-2
Perna N. T., Kocher T. D. (1995). Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–359. doi: 10.1007/BF00186547
Pimentel J. S. M., Ludwig S., Resende L. C., Brandão-Diaz P. F. P., Pereira A. H., de Abreu N. L., et al. (2020). Genetic evaluation of migratory fish: Implications for conservation and stocking programs. Ecol. Evol. 10, 10314–10324. doi: 10.1002/ece3.6231
Ponce M., Infante C., Jiménez-Cantizano R. M., Pérez L., ManChado M. (2008). Complete mitochondrial genome of the blackspot seabream, Pagellus bogaraveo (Perciformes: Sparidae), with high levels of length heteroplasmy in the WANCY region. Gene 409, 44–52. doi: 10.1016/j.gene.2007.11.004
Rajan R., Durand J. D., Thomas L., Sidharthan A., Rahman M. A. U., Xavier B., et al. (2023). Barcoding mullets (Mugilidae): Genetic characterization of exploited species in Southern Peninsular India. Diversity 15, 1193. doi: 10.3390/d15121193
Rocha-Olivares A., Garber N. M., Stuck K. C. (2000). High genetic diversity, large inter-oceanic divergence and historical demography of the striped mullet. J. Fish Biol. 57, 1134–1149. doi: 10.1111/j.1095-8649.2000.tb00476.x
Ronquist F., Huelsenbeck J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Rossi A. R., Capula M., Crosetti D., Campton D. E., Sola L. (1998). Genetic divergence and phylogenetic inferences in five species of Mugilidae (Pisces: Perciformes). Mar. Biol. 131, 213–218. doi: 10.1007/s002270050313
Rourke M. L., Robinson W., Baumgartner L. J., Doyle J., Growns I., Thiem J. D. (2019). Sequential fishways reconnect a coastal river reflecting restored migratory pathways for an entire fish community. Restor. Ecol. 27, 399–407. doi: 10.1111/rec.12886
Rozas J., Ferrer-Mata A., Sánchez-DelBarrio J. C., Guirao-Rico S., Librado P., Ramos-Onsins S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Bio. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Rutterford L. A., Simpson S. D., Bogstad B., Devine J. A., Genner M. J. (2023). Sea temperature is the primary driver of recent and predicted fish community structure across Northeast Atlantic shelf seas. Glob. Chang. Biol. 29, 2510–2521. doi: 10.1111/gcb.16633
Satoh T., Miya M., Endo H., Nishida M. (2006). Round and pointed-head grenadier fishes (Actinopterygii: Gadiformes) represent a single sister group: Evidence from the complete mitochondrial genome sequences. Mol. Phylogenet. Evol. 40, 129–138. doi: 10.1016/j.ympev.2006.02.014
Satoh T. P., Miya M., Mabuchi K., Nishida M. (2016). Structure and variation of the mitochondrial genome of fishes. BMC Genom. 17, 719. doi: 10.1186/s12864-016-3054-y
Schultz L. P. (1946). A revision of the genera of mullets, fishes of the family Mugilidae, with descriptions of three new genera. Proc. U. S. Natl. Mus. 96, 377–395. doi: 10.5479/si.00963801.96-3204.377
Semina A. V., Polyakova N. E., Makhotkin M. A., Brykov V. A. (2007). Mitochondrial DNA divergence and phylogenetic relationships in mullets (Pisces: Mugilidae) of the Sea of Japan and the Sea of Azov revealed by PCR–RFLP-analysis. Russ. J. Mar. Biol. 33, 187–192. doi: 10.1134/S1063074007030078
Senou H. (1988). Phylogenetic interrelationships of the mullets (Pisces: mugilidae). D. Phil. Thesis. (Tokyo, Japan: Tokyo University).
Setiamarga D. H. E., Miya M., Yamanoue Y., Mabuchi K., Satoh T. P., Inoue J. G., et al. (2008). Interrelationships of Atherinomorpha (medakas, flyingfishes, killifishes, silversides, and their relatives): The first evidence based on whole mitogenome sequences. Mol. Phylogenet. Evol. 49, 598–605. doi: 10.1016/j.ympev.2008.08.008
Shen K. N., Chen C. H., Hsiao C. D. (2016a). Next generation sequencing yields the complete mitochondrial genome of the hornlip mullet Plicomugil labiosus (Teleostei: Mugilidae). Mitochondrial DNA A. DNA Mapp. Seq. Anal. 27, 2192–2193. doi: 10.3109/19401736.2014.982621
Shen K. N., Jamandre B. W., Hsu C. C., Tzeng W. N., Durand J. D. (2011). Plio-Pleistocene Sea level and temperature fluctuations in the northwestern Pacific promoted speciation in the globally-distributed flathead mullet Mugil cephalus. BMC Evol. Biol. 11, 83. doi: 10.1186/1471-2148-11-83
Shen X., Pu Z., Chen X., Murphy R. W., Shen Y. (2019). Convergent evolution of mitochondrial genes in deep-sea fishes. Front. Genet. 10. doi: 10.3389/fgene.2019.00925
Shen K. N., Tsai S. Y., Chen C. H., Hsiao C. D., Durand J. D. (2016b). Next generation sequencing yields the complete mitochondrial genome of the largescale mullet, Liza macrolepis (Teleostei: Mugilidae). Mitochondrial DNA A. DNA Mapp. Seq. Anal. 27, 4232–4233. doi: 10.3109/19401736.2015.1022755
Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F., Higgins D. G. (1997). The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. doi: 10.1093/nar/25.24.4876
Thomson J. M. (1954). The Mugilidae of Australia and adjacent seas. Aust. J. Mar. Freshw. Res. 5, 70–131. doi: 10.1071/MF9540070
Trape S., Harrison I. J., Diouf P. S., Durand J. D. (2012). Redescription of Liza bandialensis (Teleostei: Mugilidae) with an identification key to mullet species of Eastern Central Atlantic. C. R. Biol. 335, 120–128. doi: 10.1016/j.crvi.2011.12.003
Turan C., Caliskan M., Kucuktas H. (2005). Phylogenetic relationships of nine mullet species (Mugilidae) in the Mediterranean Sea. Hydrobiologia 532, 45–51. doi: 10.1007/s10750-004-8772-5
Vences M., Miralles A., Brouillet S., Ducasse J., Fedosov A., Kharchev V., et al. (2021). iTaxoTools 0.1: Kickstarting a specimen-based software toolkit for taxonomists. Megataxa 6, 77–92. doi: 10.11646/megataxa.6.2.1
Wang Y., Xiong F., Song Z. (2022b). Molecular phylogeny and adaptive mitochondrial DNA evolution of salmonids (Pisces: Salmonidae). Front. Genet. 13. doi: 10.3389/fgene.2022.903240
Wang C., Ye P., Liu M., Zhang Y., Feng H., Liu J., et al. (2022a). Comparative analysis of four complete mitochondrial genomes of epinephelidae (Perciformes). Genes 13, 660. doi: 10.3390/genes13040660
Ward R. D., Zemlak T. S., Innes B. H., Last P. R., Hebert P. D. (2005). DNA barcoding Australia’s fish species. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 360, 1847–1857. doi: 10.1098/rstb.2005.1716
Xia R., Durand J. D., Fu C. (2016). Multilocus resolution of Mugilidae phylogeny (Teleostei: Mugiliformes): Implications for the family’s taxonomy. Mol. Phylogenet. Evol. 96, 161–177. doi: 10.1016/j.ympev.2015.12.010
Yamanoue Y., Miya M., Matsuura K., Yagishita N., Mabuchi K., Sakai H., et al. (2007). Phylogenetic position of tetraodontiform fishes within the higher teleosts: Bayesian inferences based on 44 whole mitochondrial genome sequences. Mol. Phylogenet. Evol. 45, 89–101. doi: 10.1016/j.ympev.2007.03.008
Yang Z. H., Nielsen R. (2000). Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Bio. Evol. 17, 32–43. doi: 10.1093/oxfordjournals.molbev.a026236
Zhang J., Kan X., Miao G., Hu S., Sun Q., Tian W. (2020). qMGR: A new approach for quantifying mitochondrial genome rearrangement. Mitochondrion 52, 20–23. doi: 10.1016/j.mito.2020.02.004
Keywords: Atlantic Ocean, mugilids, endemic species, mitochondrial genome, evolution, conservation
Citation: Yoon T-H, Kang H-E, Aini S, Wujdi A, Kim H-W and Kundu S (2024) Mitogenomic analysis reveals the phylogenetic placement of monotypic Parachelon grandisquamis and distinctive structural features of control regions in mullets. Front. Mar. Sci. 11:1484198. doi: 10.3389/fmars.2024.1484198
Received: 21 August 2024; Accepted: 29 November 2024;
Published: 19 December 2024.
Edited by:
Ana Laura Ibañez, Autonomous Metropolitan University, MexicoReviewed by:
Jiao Cheng, Chinese Academy of Sciences (CAS), ChinaKhaled Mohammed Geba, Menoufia University, Egypt
Copyright © 2024 Yoon, Kang, Aini, Wujdi, Kim and Kundu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hyun-Woo Kim, a2ltaHdAcGtudS5hYy5rcg==; Shantanu Kundu, c2hhbnRhbnUxOTg0QHBrbnUuYWMua3I=; c2hhbnRhbnUxOTg0QGdtYWlsLmNvbQ==