 Anastasiia Barilo
Anastasiia Barilo Aschwin Engelen
Aschwin Engelen Susanne Wilken
Susanne Wilken Harro Bouwmeester
Harro Bouwmeester Gerard Muyzer
Gerard Muyzer
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 24 October 2024
Sec. Microbial Symbioses
Volume 11 - 2024 | https://doi.org/10.3389/fmars.2024.1445441
Caulerpa is a genus of green macroalgae that lives in tropical and subtropical coastal waters. It is an intriguing organism because, despite having plant-like structures, it is one giant cell – which, next to multiple nuclei, chloroplasts, and mitochondria, also contains endo- and epiphytic bacteria. The role of these bacteria is unknown, but they might impact the growth and development of the host, adaptation to environmental parameters, and, hence, the ecological success of these algae. We hypothesised that increased sulphide concentrations would trigger a significant shift in the microbial community composition associated with C. prolifera rhizoids, favouring sulphide-oxidizing bacteria. To test this hypothesis, we conducted a mesocosm experiment incubating C. prolifera in sediments with different sulphide concentrations and analysed the algal photosynthesis, growth, and microbiome composition. While photosynthesis was not affected, the Caulerpa weight-based growth rate decreased linearly with increasing sulphide concentration. To analyse the microbiome, we extracted DNA and RNA from the fronds, rhizoids, and the accompanying sediments and performed 16S amplicon sequencing. The microbiome of the fronds was unaffected in both the DNA and RNA samples. However, an increase in sulphide concentration coincided with a decrease in the relative abundance of sulphate-reducing bacteria associated with Caulerpa rhizoids, particularly from the family Desulfocapsaceae. In the RNA samples, potential sulphide oxidisers of the rhizoid-associated members of the Beggiatoaceae were detected. Our results suggest that the rhizobiome of Caulerpa plays a significant role in its adaptation to sulphide-rich environments, offering new insights into the complex interactions within marine holobionts.
The ocean holds a vast sulphur reservoir with sulphate concentrations around 28 mM (Wasmund et al., 2017). Sulphate-reducing bacteria utilize this sulphate as a terminal electron acceptor to degrade organic matter, producing sulphide (Kasten and Jørgensen, 2000). These microorganisms facilitate up to 29% of organic matter degradation at the seafloor, playing a particularly crucial role in areas rich in organic material (Wasmund et al., 2017). Marine sediments are, therefore, often rich in hydrogen sulphide, a toxic gas that primarily affects mitochondria via inhibition of the cytochrome c oxidase (Paul et al., 2021). In seagrasses (Dooley et al., 2013) and cyanobacteria (Oren et al., 1979), sulphide has been shown to affect the photosynthetic activity and shut down photosystem II, and other studies suggest that these effects are consistent with those in land plants (Gupta et al., 2014; Rolando et al., 2022). Sulphide can also increase free radical production, causing oxidative stress (Joyner-Matos et al., 2006).
Benthic marine organisms, therefore, often need to cope with increased sulphide levels, and various adaptations have evolved to overcome this stress. Invertebrates, for example, have a sulphide-insensitive cytochrome c oxidase or recruit sulphide-oxidizing bacteria (Grieshaber and Völkel, 1998). Deep-sea clams and worms often harbour sulphide-oxidizing bacteria in their tissues that obtain energy from sulphide oxidation (Sogin et al., 2020). Saltmarsh plants that are known to occupy areas with high rates of sulphate reduction (Bahr et al., 2005) can harbour sulphide-oxidizing bacteria in their rhizosphere, especially under elevated sulphide concentrations (Rolando et al., 2022) and employ oxygen transport through aerenchyma to aerate the rhizosphere both for chemical oxidation (Maricle and Lee, 2007) and to provide beneficial conditions for sulphide oxidising bacteria (Martin et al., 2019). Seagrasses - marine flowering plants whose distribution is restricted by high sulphide concentrations (Terrados et al., 1999; Dooley et al., 2013) employ similar mechanisms. A recent study on seagrasses (Crump et al., 2018) showed the presence of sulphate-reducing and sulphide-oxidizing bacteria associated with the roots and the expression of genes involved in these processes. Hence, microbial sulphur oxidation is an important mechanism for detoxifying hydrogen sulphide for many marine organisms.
While sulphide toxicity can be lethal and inhibiting for many organisms, others do well in sulphide-rich environments. Caulerpa prolifera, for example - a green unicellular macroalga that morphologically resembles a multicellular organism with rhizoids (‘roots’) and fronds (‘leaves’) is highly sulphide tolerant (Holmer et al., 2009). It harbours endo- and epiphytic bacteria and is regarded as a holobiont (Aires et al., 2013, 2015; Morrissey et al., 2019). The role of these bacteria is unknown, but they might play a role in the adaptation to environmental conditions. C. prolifera is native to Mediterranean areas and was introduced to the Portuguese South coast in the 19th century (Cunha et al., 2013). For 60 years, it remained undetected and was thought to be extinct; however, in the past ten years, it has started to spread across the Ria Formosa lagoon in the Algarve. Although it is not considered invasive, it can be opportunistic (Parreira et al., 2021); it is often found in sulphide-rich sediments and is known for its fast and efficient nutrient uptake (Bernardeau-Esteller et al., 2023) and for its ability to change the composition and biochemistry of the sediments by increasing sediment organic content and inducing sulphate reduction (Holmer et al., 2009). In this respect, it might outcompete other macrophytes, like seagrasses.
While C. prolifera is associated with high sulphide concentrations (Holmer et al., 2009), the effect of sulphide on the alga is unknown. In the present study, we assessed the impact of sulphide on the growth and photosynthesis of the alga and its microbiome. As microbial oxidation of sulphide is a known mechanism to combat sulphide toxicity in other marine organisms (Grieshaber and Völkel, 1998; Sogin et al., 2020; Rolando et al., 2022; Crump et al., 2018), we hypothesised that changes in the microbiome associated with C. prolifera rhizoids, known as the ‘rhizobiome,’ would promote sulphide-oxidizing bacteria in response to increased sulphide concentrations.
To determine the sulphide concentration in the natural sediments, we sampled the pore water from four types of sediments in the Ria Formosa (Faro, Portugal): (i) bare sediments, (ii) sediments with Caulerpa prolifera, (iii) sediments with the seagrass Cymodocea nodosa, and (iv) sediments with a mixture of C. prolifera and C. nodosa. We sampled three locations for each sediment type and took four or five samples from each location, i.e., 14-15 samples per sediment type (Supplementary Table S1). The water samples were directly mixed with a 10% (w/v) zinc acetate solution to fix the sulphide. Subsequently, the sulphide concentration was measured using the methylene blue method (Trüper and Schlegel, 1964), and the significance of the results was analysed using nested ANOVA.
The mesocosm experiments were performed at the Ramalhete Marine Station of the Centre for Marine Sciences (CCMAR) in Faro (Portugal) in August and September 2020. Caulerpa prolifera specimens maintained in an outdoor tank at the station were collected and incubated in 5 L PET bottles (“mesocosm”) filled with 500 ml 0.7% (w/v) agar containing 0 µM (control), 100 µM (low), 150 µM (medium), 300 µM (high), or 1000 µM (very high) sodium sulphide (Figure 1). These concentrations were chosen based on previous values recorded in areas with declining seagrass populations (Dooley et al., 2013). The mesocosms were then filled with sediments taken from a local marine pond. The sediment was homogenised under running seawater to remove the inherent sulphide before adding 1500 ml sediment to the mesocosms.
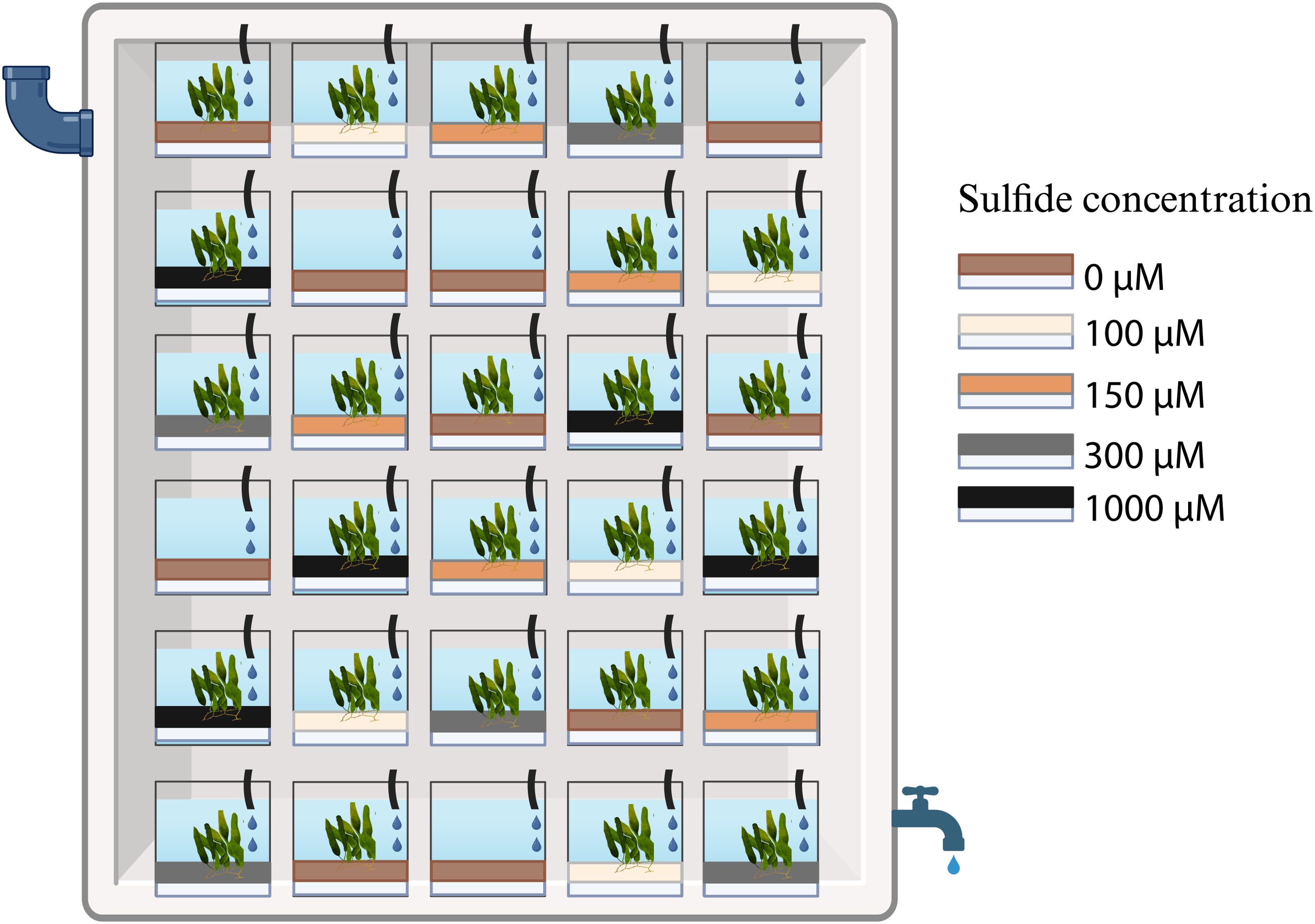
Figure 1. Mesocosm experiment set up: mesocosms with four sulphide concentrations of the agar plug (white) and the control (0 µM), with sediments and Caulerpa on top of the sulphide-containing agar; in addition, five control mesocosms with sediment but without Caulerpa were added to the experiment. All mesocosms were placed randomly in a flow-through system, with five replicates per treatment. Created with BioRender.com.
In addition to the mesocosms with Caulerpa, five mesocosms with sediments but without Caulerpa and sulphide were set up as controls. Every treatment was represented by five replicates, resulting in a total of 30 mesocosms (Figure 1). The mesocosms were kept in a 500 L container with a continuous water flow-through, maintaining an average temperature of 24°C. The average daylight intensity in the setup was 96 µmol m-2 s-1; the mesocosms were placed at random positions that were changed weekly during the experiment. The weight, the number of fronds, and the total leaf length of each Caulerpa specimen were determined at the beginning and the end of the experiment. The photosynthetic yield as a proxy for Caulerpa’s health and performance was measured with a JUNIOR-PAM chlorophyll Fluorometer (Walz, Germany) after a dark adaptation for 60 seconds at standard settings. At the end of the experiment, the fronds, rhizoids, and sediments were collected from each mesocosm and stored in DNA/RNA Shield™ (Zymo Research, United States) for the microbiome analysis. The sulphide levels in the mesocosms were measured at the end of the first, second and fourth week of the experiment using the methylene blue method (Trüper and Schlegel, 1964). Samples that exceeded the spectrophotometer readings were removed.
The growth rate was calculated based on wet weight, average leaf length, and the number of fronds using the following formula (W1-W0)/t where W1 is the weight (or length) at the end of the experiment, W0 – at the beginning of the experiment, and t is the number of days of the experiment (30 days). ANOVA based on the linear model (lm, base R) was used in RStudio to compare weight- and leave-based growth rates and the number of new fronds grown during the experiment and to test whether sulphide treatment influenced these parameters. Linear mixed models (lmm, lmerTest package, Kuznetsova et al., 2017) were used to determine the effect of sulphide on the photosynthetic yield measurements. Homogeneity and normality assumptions of ANOVA were checked visually with Levene’s test and qqplot (car package, Fox and Weisberg, 2019), respectively. The measured sulphide values were log-transformed and analysed by ANOVA.
DNA and RNA were extracted from 79 samples using the ZymoBiomics DNA/RNA kit (Zymo Research, United States). The samples included fronds, rhizoids, and sediments collected from the five experimental sulphide levels (0 µM, 100 µM, 150 µM, 300 µM, 1000 µM) (five replicates per treatment per sample type) and four samples of the control sediments without Caulerpa or sulphide addition. The DNA samples were subsequently used in a two-stage PCR protocol (Naqib et al., 2018) to amplify the V5-V7 region of the 16S rRNA gene. The first stage PCR was done with the chloroplast-avoiding primers CS1_799F - CS2-1193R (Beckers et al., 2016) using one ng of DNA per 10 µL reaction. Subsequently, a second-stage PCR was done with the bar-coded primers (Naqib et al., 2018). A mock community (MSA-1002, ATCC) was used as a positive control, and molecular biology-grade water was used as a negative control.
Despite the DNAse treatment in the ZymoBiomics kit, all RNA samples were checked for DNA contamination with the general 16S primers 515F-806R. The samples that showed a positive PCR product were treated with a TURBO DNA-free kit before transcription into cDNA using SuperScript™ III Reverse Transcriptase (Invitrogen, United States). The cDNA was then used as a template in the PCR, as described above. A total of 147 samples were sent off for paired-end Illumina MiSeq sequencing at the Research Resources Centre of the University of Illinois at Chicago, USA.
The sequencing facility generated and demultiplexed paired-end sequence reads of 150 bp. QIIME2 version 2022.8 (Bolyen et al., 2019) was used to perform quality control and to produce an ASV abundance table and a taxonomic classification. The amplicon sequencing variants (ASVs) were generated using the DADA2-denoise algorithm in QIIME2 (Callahan et al., 2016). The SILVA 16S rRNA database (version 138.1, Quast et al., 2013) was used for reference sequences and Naive Bayes classifier training. Positive and negative controls were verified and checked for biases after QIIME2 analysis but removed from further analysis.
Subsequently, the alpha- and beta-diversity and the taxonomic composition of the microbiomes were analysed with the software package phyloseq in RStudio (version 1.46; McMurdie and Holmes, 2013). For this, the QIIME2-derived taxonomy table, the ASV table and a table with metadata were imported into RStudio and converted to phyloseq objects. Seven samples were removed because of a low number of sequence reads or other problems. Before all analyses except the alpha-diversity analysis, the data were filtered to retain only ASVs with at least one count in at least 20% of the samples.
To determine alpha diversity, non-filtered data were rarefied to 3354 reads per sample (Supplementary Table S2), and Shannon diversity index values were calculated using the vegan package (version 2.6-4 Oksanen et al., 2022). Normality and equality of variances of these indices across samples were assessed with Shapiro-Wilk’s and Levene’s tests. The significance of the diversity differences was tested using ANOVA and Tukey HSD tests if all assumptions were met or Kruskal-Wallis (KW) and Dunn tests if the assumptions were not met.
The filtered data were transformed for the beta diversity analysis with centred log-ratio (clr) transformation (microbiome package; Lahti et al., 2017). The homogeneity of variances was checked with a combination of visual and statistical assessment via the betadisper test (vegan package version 2.6-4; Oksanen et al., 2022). An Aitchison distance matrix was calculated as it is known to be more robust than the widely used Bray-Curtis dissimilarity statistic (Gloor et al., 2017). Adonis2 (vegan package version 2.6-4; Oksanen et al., 2022) was used to perform permutational multivariate analysis of variance (PERMANOVA) with 9999 permutations, three fixed factors were investigated: (i) sulphide concentration (five levels), (ii) DNA or RNA (two levels) and (iii) sample type (three levels). The P-values were corrected according to the Benjamin-Hochberg method; Beta diversity was visualised with redundancy analysis and constrained analysis of principal coordinates (CAP) ordination plots. To further investigate the compositional difference between various sulphide concentrations, PERMANOVA was repeated for each sample type (fronds, rhizoids, sediments) with two fixed factors, i.e., (i) sulphide concentration with five levels and (ii) DNA and RNA with two levels.
The relative abundance of the different taxa was plotted in MicrobiomeAnalyst version 2.0 (Lu et al., 2023). To establish which taxa differ significantly between the treatments, an analysis of the compositions of the microbiomes was performed with package ANCOMBC (Lin and Peddada, 2020). Pairwise comparisons for each sulphide concentration were done on the filtered dataset. Analysis was followed by selecting ASVs that significantly differed in any of these comparisons. The clr-transformed abundances of these ASVs in the rhizoid and sediment samples were then z-centred per taxon and plotted as a heatmap using the ComplexHeatmap package in R. Hierarchical clustering analysis of ASVs was performed using Euclidian distance and average linkage. The significance of the clusters was assessed with the sigclust package (Huang et al., 2015). The enrichment analysis of ASVs in each significant cluster was performed with FuncAssociate 3.0 (Berriz et al., 2003).
To further investigate the compositional differences between treatments, ‘key’ bacterial families for each sulphide treatment were explored for all sample types using a so-called ‘indicator species analysis’ with the “indicspecies” R-package (v 1.7.6) based on relative abundances (Cáceres and Legendre, 2009). Following the results of indicator species analysis, ASVs grouped to key families from sediments and Caulerpa rhizoids were plotted on a heatmap. Ungrouped ASVs from key abundant families from Caulerpa rhizoids were clustered and plotted on a heatmap followed by enrichment analysis (Berriz et al., 2003).
Natural sediments with Caulerpa had a 1.9-fold higher sulphide concentration than sediments with the seagrass C. nodosa and a 3-fold higher concentration than non-vegetated sediments (Nested ANOVA, F3 = 4.32, p < 0.01, Figure 2). The concentrations in the field ranged from 232 µM to 1600 µM for Caulerpa-vegetated sediments, 51 µM to 870 µM for sediments with both Caulerpa and seagrass, 32 µM to 492 µM for empty sediments, and 30 µM to 529 µM for sediments with only seagrasses.
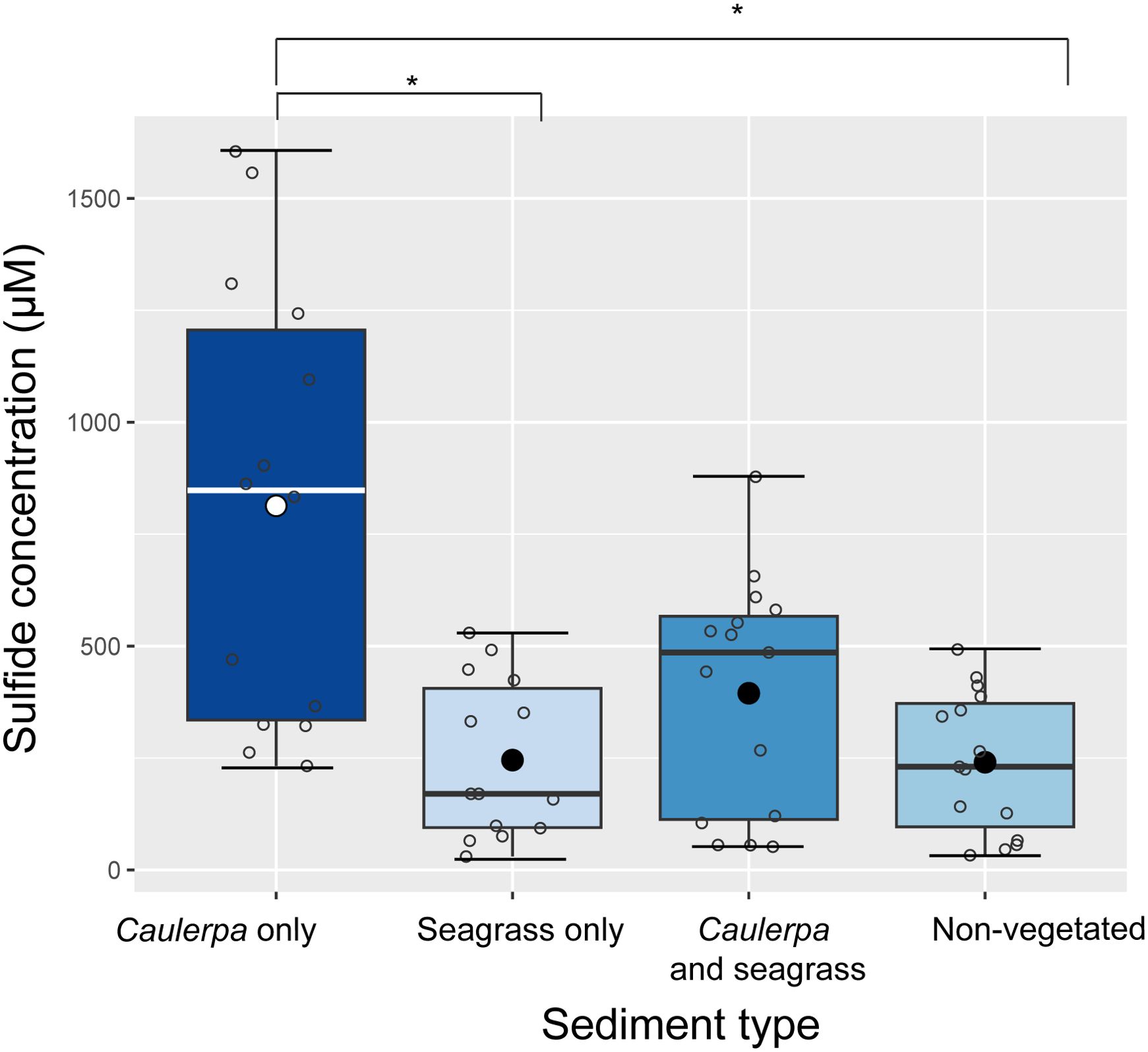
Figure 2. Sulphide concentrations in the pore water of natural sediments with and without macrophytes. Three areas per treatment were sampled, with 14-15 samples from each area (see Supplementary Table S1). The mean is indicated with a black dot; open circles represent individual samples; error bars represent the standard deviation; the line represents the median; the boxplot borders represent the 25th and 75th percentile. Nested ANOVA, F3 = 4.32, p < 0.01; TukeyHSD *p<0.01.
A total of 35 sulphide samples were collected from the mesocosms. A significant effect of both time (ANOVA, F1 = 4.77, p < 0.05) and sulphide addition (ANOVA, F2 = 10.65, p < 0.01) was observed in the mesocosms treated with 0, 100, and 150 µM of sulphide. The sulphide concentration increased steadily (Supplementary Figure S1), indicating a gradual sulphide release from the agar plugs. However, due to the limited number of samples and high variability in the measured concentrations, the added sulphide concentrations were used for all subsequent analyses instead of the measured concentrations.
The weight-based growth rate of Caulerpa decreased linearly with increasing sulphide concentrations (ANOVA, F1 = 7.82, p = 0.01, Figure 3), showing a 3-fold decrease between the 0 µM and the highest (1000 µM) sulphide treatment. However, sulphide did not influence the frond area growth rate, the number of new fronds, or the photosynthetic yield of Caulerpa (Supplementary Figure S2).
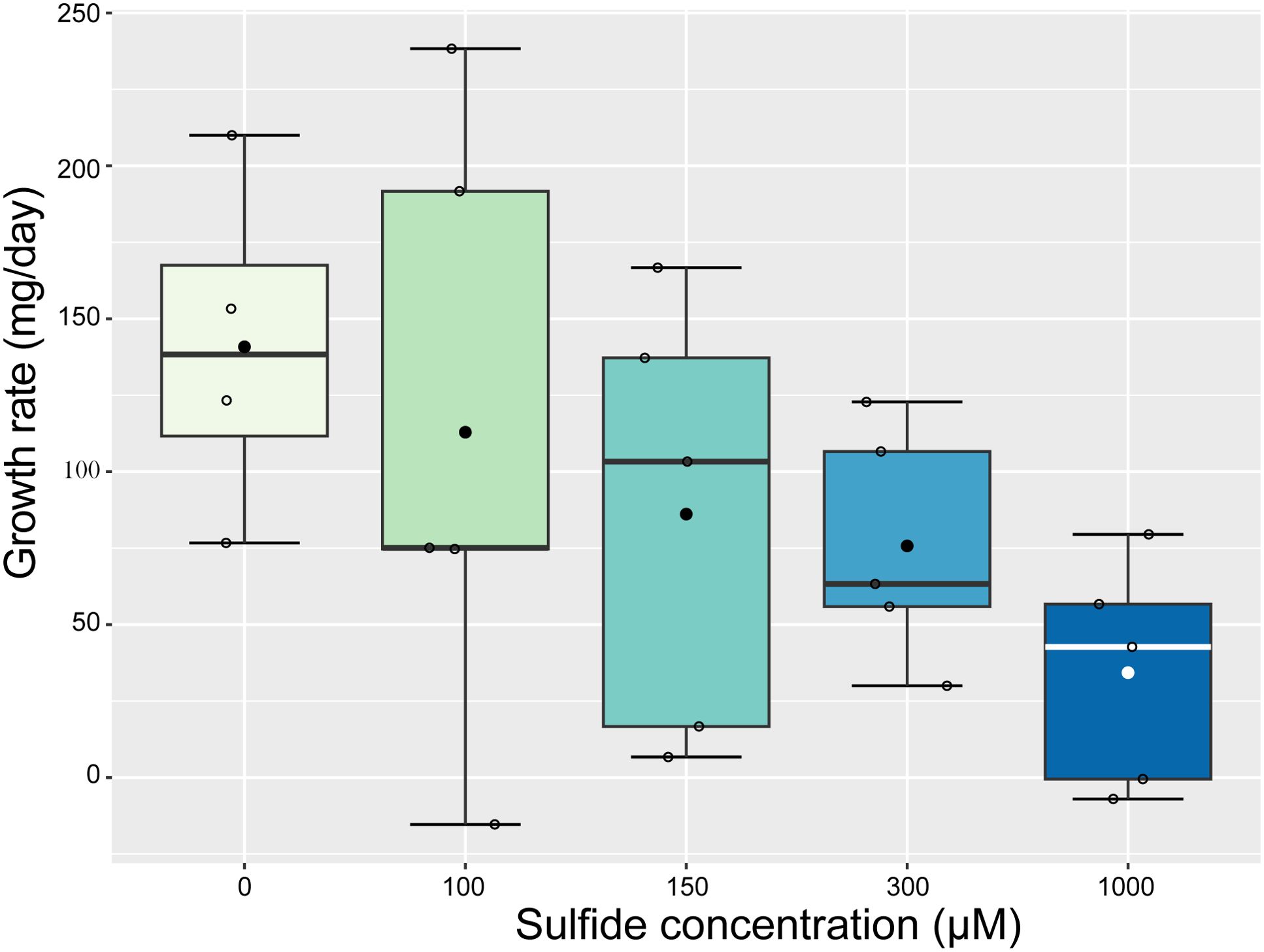
Figure 3. A significant linear decrease in Caulerpa weight-based growth rate with increased sulphide concentrations (ANOVA, F1 = 7.82, p = 0.01). The mean is indicated with a dot; open circles represent samples; error bars represent standard deviation; the line represents the median; and the boxplot borders represent the 25th and 75th percentile.
We obtained sequences for 72 DNA and 75 RNA samples. 65,090 and 65,076 ASVs were retrieved from the 1,129,072 and 703,575 reads of the DNA and RNA samples, respectively. The relative abundance of the phyla showed differences among the different sample types (i.e., fronds, rhizoids, and sediments) and between the DNA and RNA samples (Figure 4; Supplementary Table S3). Only ten phyla were present in the RNA samples, all represented among the 18 phyla found in the DNA samples. Proteobacteria represented the most abundant phylum in the DNA and RNA of the frond samples (Figure 4). In the rhizoid samples, Proteobacteria and Desulfobacterota were the most abundant taxa at all sulphide concentrations for both the DNA and RNA samples, with a decrease in the relative abundance of Desulfobacterota in the sulphide treatments. The relative abundance of Desulfobacterota decreased from 82% and 88% in the RNA and DNA samples from the control, respectively, to 47% and 45% in the highest sulphide treatment. Proteobacteria increased from 13% in the RNA and 4% in the DNA of the control to 40% and 44%, respectively, in the highest sulphide treatment. The sediment samples followed a similar pattern, with Desulfobacterota and Proteobacteria as the most abundant phyla and Desulfobacterota relative abundances declining by 20% (Supplementary Table S3) in the sulphide treatments compared to the control. The most abundant families in the fronds and rhizoids in the control treatment were Rhodobacteraceae (49% DNA and 28% RNA) and Desulfocapsaceae (67% DNA, 38% RNA) respectively (Supplementary Tables S4, S5). In the highest sulphide concentration, the most abundant families were Rhodobacteraceae (59% DNA, 27% RNA) in the fronds and a mix of Desulfosarcinaceae (11% DNA, 18% RNA), Desulfocapsaceae (21% DNA, 11% RNA), Rhodobacteraceae (17% DNA, 2% RNA) and Beggiatoaceae (0.5% DNA, 13% RNA) in the rhizoids (Supplementary Tables S4, S5).
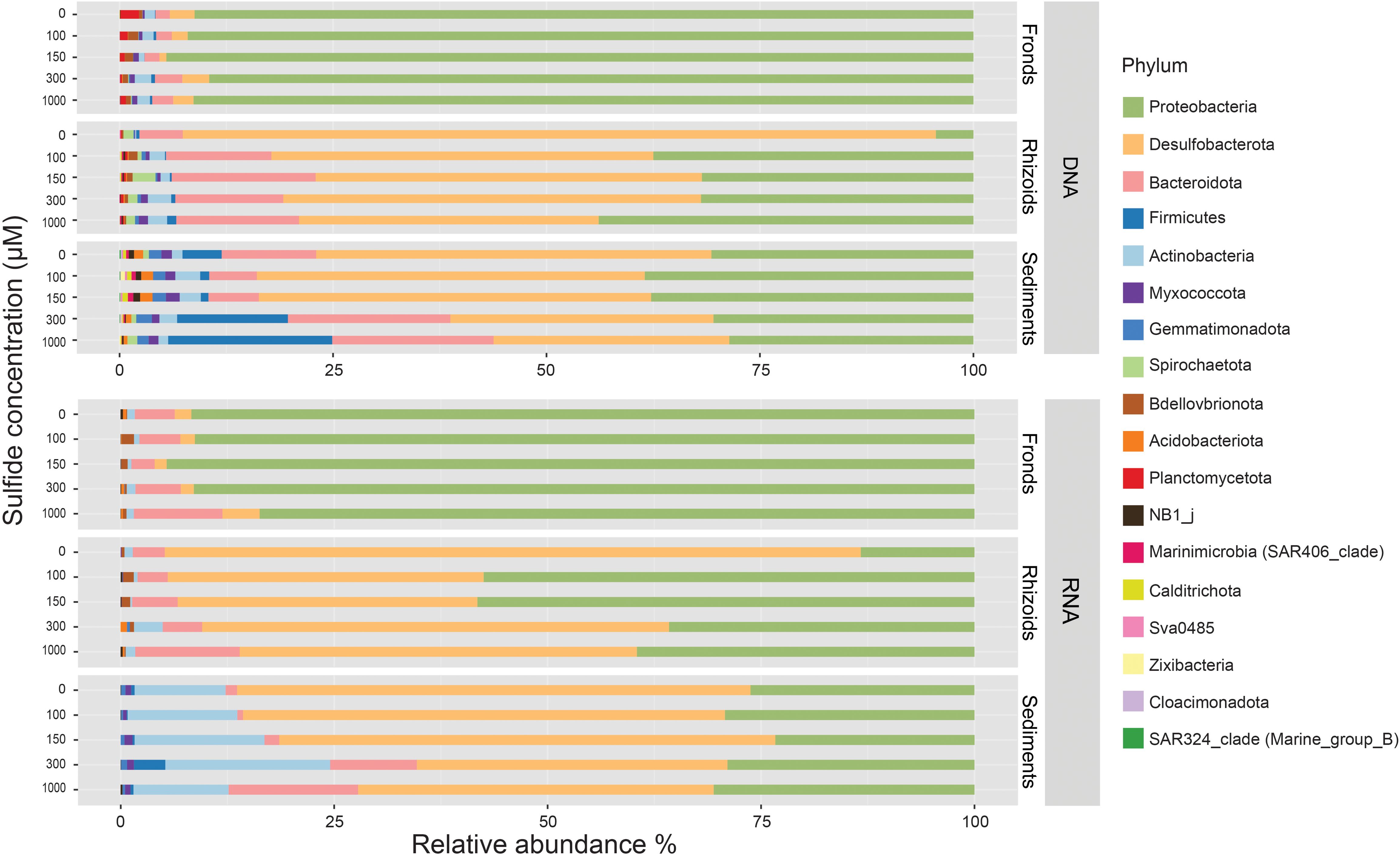
Figure 4. Average relative abundances (in %) of bacterial taxa at the phylum level associated with fronds and rhizoids of Caulerpa prolifera and in the sediment incubated with different sulphide concentrations in DNA and RNA samples (see Supplementary Table S7 for details of all samples).
While the sulphide treatment did not affect the Shannon diversity (richness and evenness) index, it was significantly higher in the rhizoids and sediments compared to the fronds and in sediments compared to the rhizoids (Supplementary Figure S3). In the rhizoids and sediments, it was also higher for the DNA samples than for the RNA samples (Supplementary Figure S4).
Differences in the microbiome were primarily influenced by sample type (Figure 5; Table 1). The microbiomes from the different sample types also showed distinct responses to sulphide concentrations (Table 1). DNA and RNA samples responded similarly to the sulphide treatment (Figure 6; Table 1).
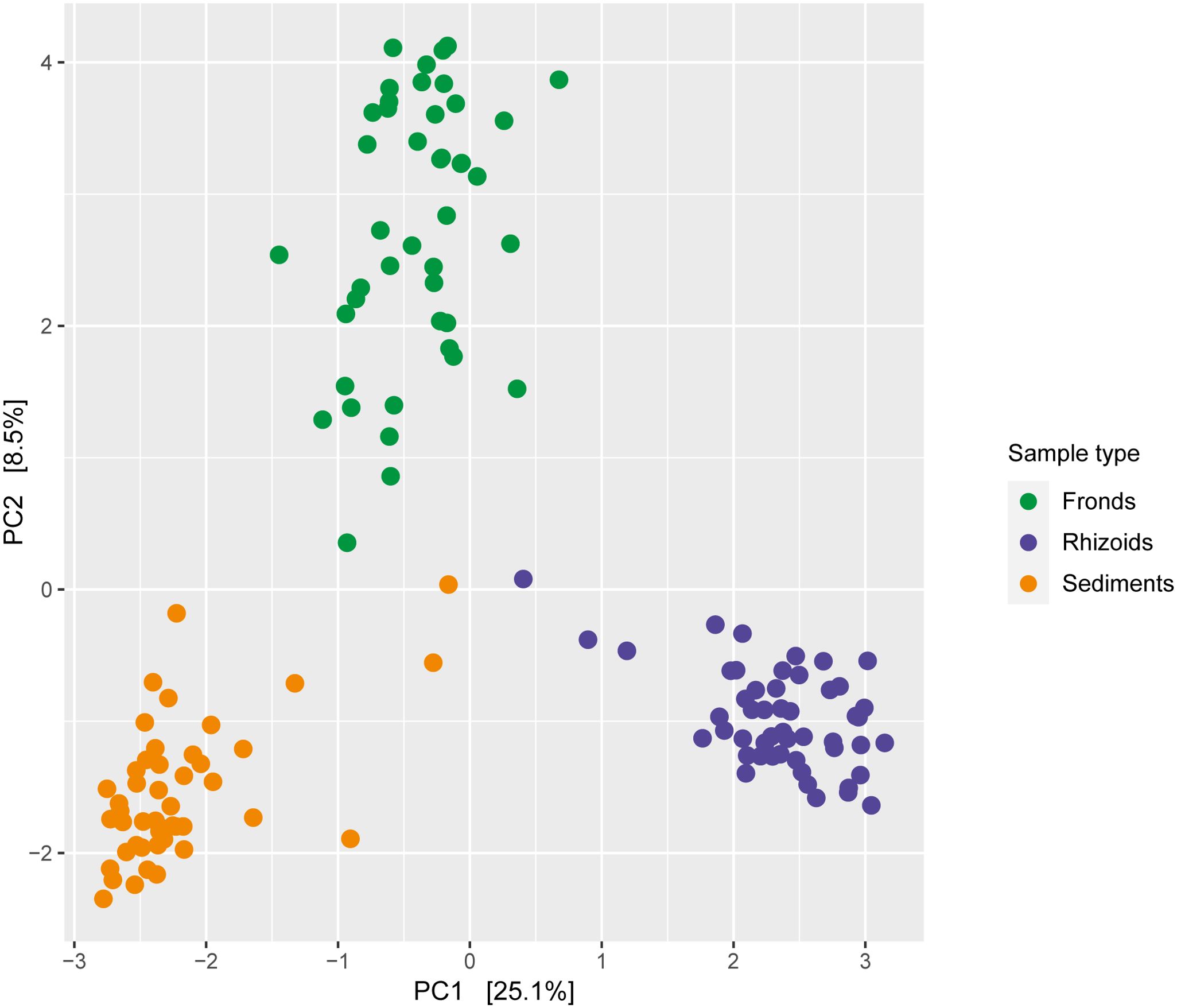
Figure 5. Redundancy analysis of bacteria associated with Caulerpa prolifera fronds (n=45), rhizoids (n=44) and sediments (n=51), based on Euclidian distances. The DNA and RNA samples from experiments with different sulphide concentrations were combined.
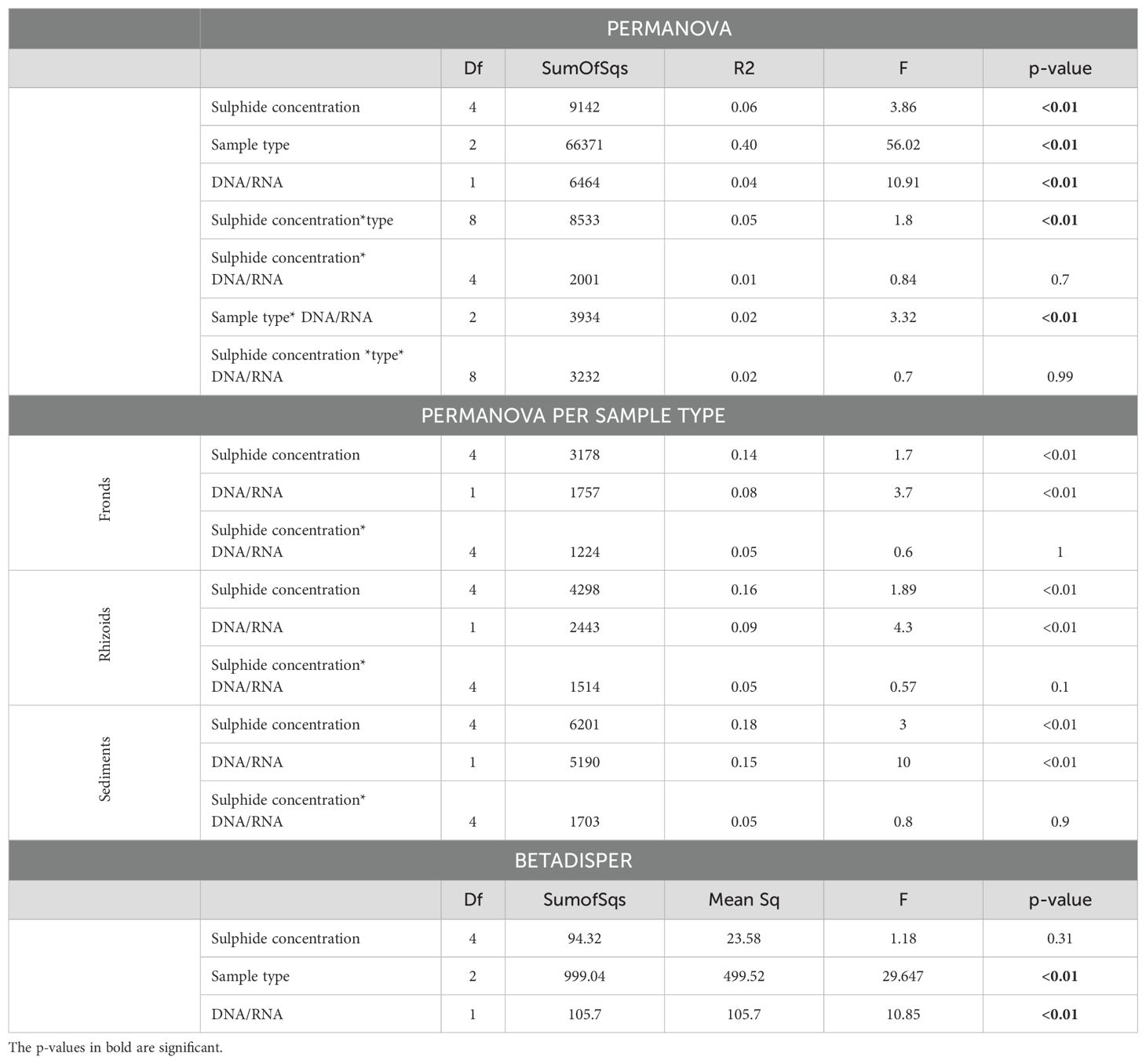
Table 1. PERMANOVA and BETADISPER analysis of microbial communities associated with Caulerpa prolifera (i) sample types (fronds, rhizoids or sediments), (ii) DNA/RNA and (iii) sulphide concentrations.
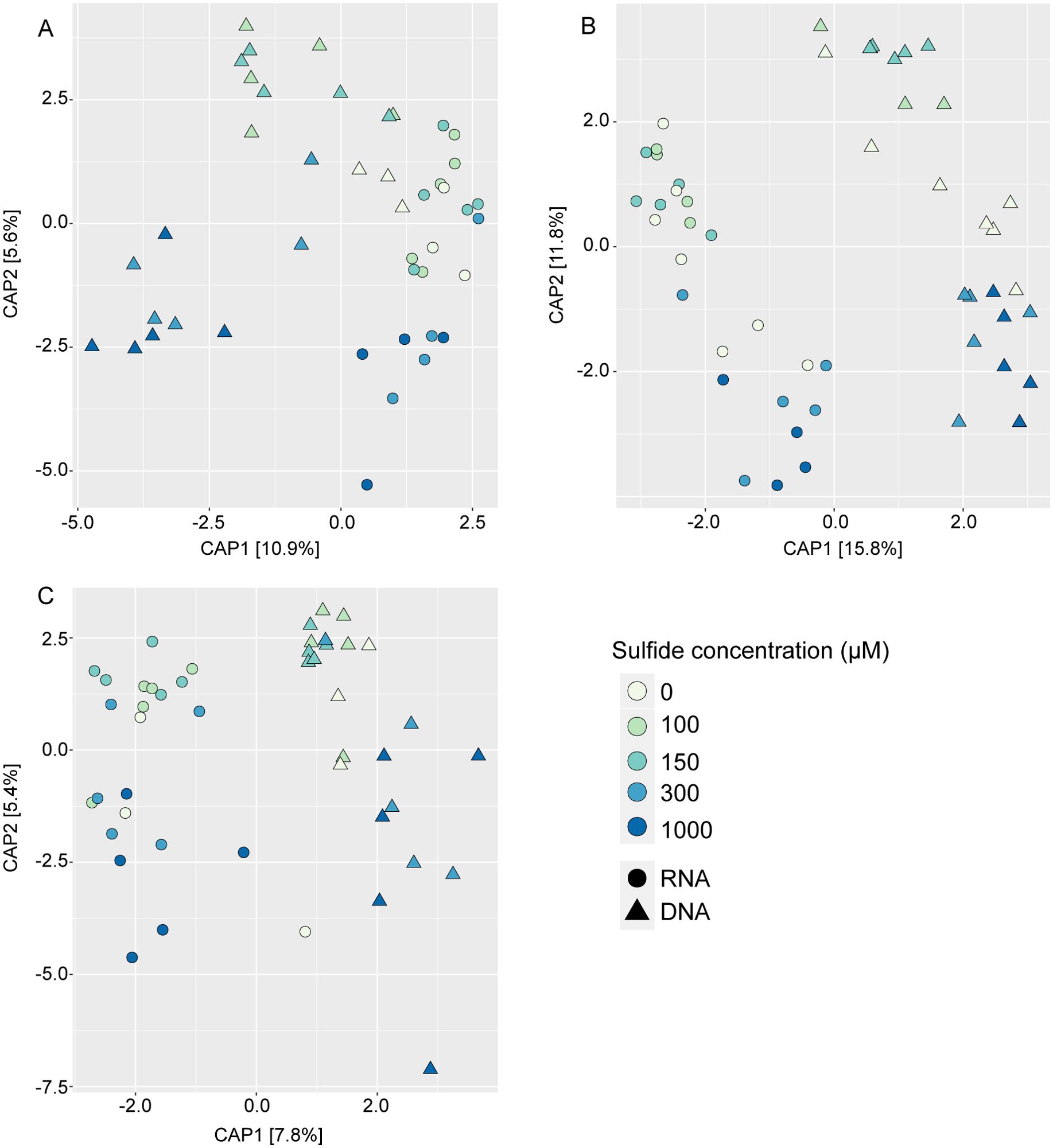
Figure 6. Constrained analysis of principal coordinates of bacteria associated with Caulerpa prolifera rhizoids (A), fronds (B) and sediments (C) at different experimental sulphide concentrations. The ordinations are based on an Euclidian distance matrix. Triangles and circles represent DNA and RNA samples, respectively.
Compositional analysis using ANCOMBC showed the presence of 66 ASVs associated with frond, rhizoid, and sediment samples at different sulphide concentrations. Hierarchical clustering and enrichment analyses showed that the Rhodobacteraceae family was significantly enriched in the cluster associated with the Caulerpa fronds (Supplementary Figure S6). For the sediments and rhizoid clusters, the analysis did not show any enriched families or genera. To further investigate the rhizoids-associated changes we proceeded with the indicator species analysis.
Indicator species analysis identified the sulphate reducer families Desulfocapsaceae and Desulfobulbaceae as indicator taxa for the Caulerpa rhizoids across all sulphide concentrations in DNA and RNA samples (Figure 7; Supplementary Table S6). In contrast, sulphide oxidising Beggiatoaceae was predominantly present in the RNA samples of the 100 µM and 150 µM sulphide treatments. The relative abundance of the Beggiatoaceae in the RNA samples was up to a maximum of 49% for the 100 µM sulphide treatment, whereas the maximum in the DNA samples was less than 1% (Supplementary Tables S4, S5). The sulphate reducer family Desulfosarcinaceae was an indicator taxon in the DNA and RNA samples of the sediment for all sulphide treatments except the highest concentration of 1000 µM. In addition, the sulphide oxidising families Woeseiaceae and Thiotrichacea were indicator taxa in the sediments but not in the rhizoids (Figure 7). For the fronds, abundant families like Rhodobacteraceae and Hyphomonadaceae were indicator taxa for all experimental treatments, suggesting no effect of sulphide on the fronds-associated microbial communities (Supplementary Table S6).

Figure 7. Indicator species analysis results showing key bacterial taxa for Caulerpa prolifera rhizoids and sediments incubated at different sulphide concentrations. Only families with a relative abundance >1% were included. Taxa in red are sulphide-oxidizing bacteria, taxa in blue are sulphate-reducing bacteria, and taxa in green contain members that belong to the sulphate reducers but can oxidise sulphide, such as the “cable” bacteria.
To further investigate the impact of sulphide on the ‘indicator’ families in the rhizoids, these families were clustered at the ASV level, followed by an enrichment analysis. While the use of ASVs is considered accurate and superior to OTU clustering (Weinroth et al., 2022), it could introduce bias by overinflating the diversity because of the possible presence of multiple copies of the 16S rRNA gene in the same bacterium (Schloss, 2021). We, therefore, visualised the ASVs merged at the family level on a heatmap (Supplementary Figure S5) to investigate whether the patterns of both analyses corresponded, which they did. Therefore, we further focused on the ASV-based results.
The enrichment analysis identified six significant clusters (Figure 8). The relative abundance of the indicator taxa followed roughly the same pattern in the DNA and RNA samples, although it was much more pronounced in the former. Two members of the family Desulfocapsaceae were enriched in two clusters; Desulforhophalus sp., in cluster one, was abundant in the 0 µM control but only present at low relative abundance in the sulphide treatments. In contrast, Desulfopila in cluster three was abundant at the highest sulphide concentrations but low in the 0, 100, and 150 µM sulphide treatments (Figure 8). Five ASVs affiliated with the Desulfobulbaceae family (cluster four) were mainly abundant at the two highest sulphide concentrations. Ca. Electrothrix in cluster six did not follow a clear pattern but seemed most abundant in the 100 µM and 150 µM sulphide treatments. Beggiatoaceae was enriched in cluster six; clusters two and five had no enriched species or families.
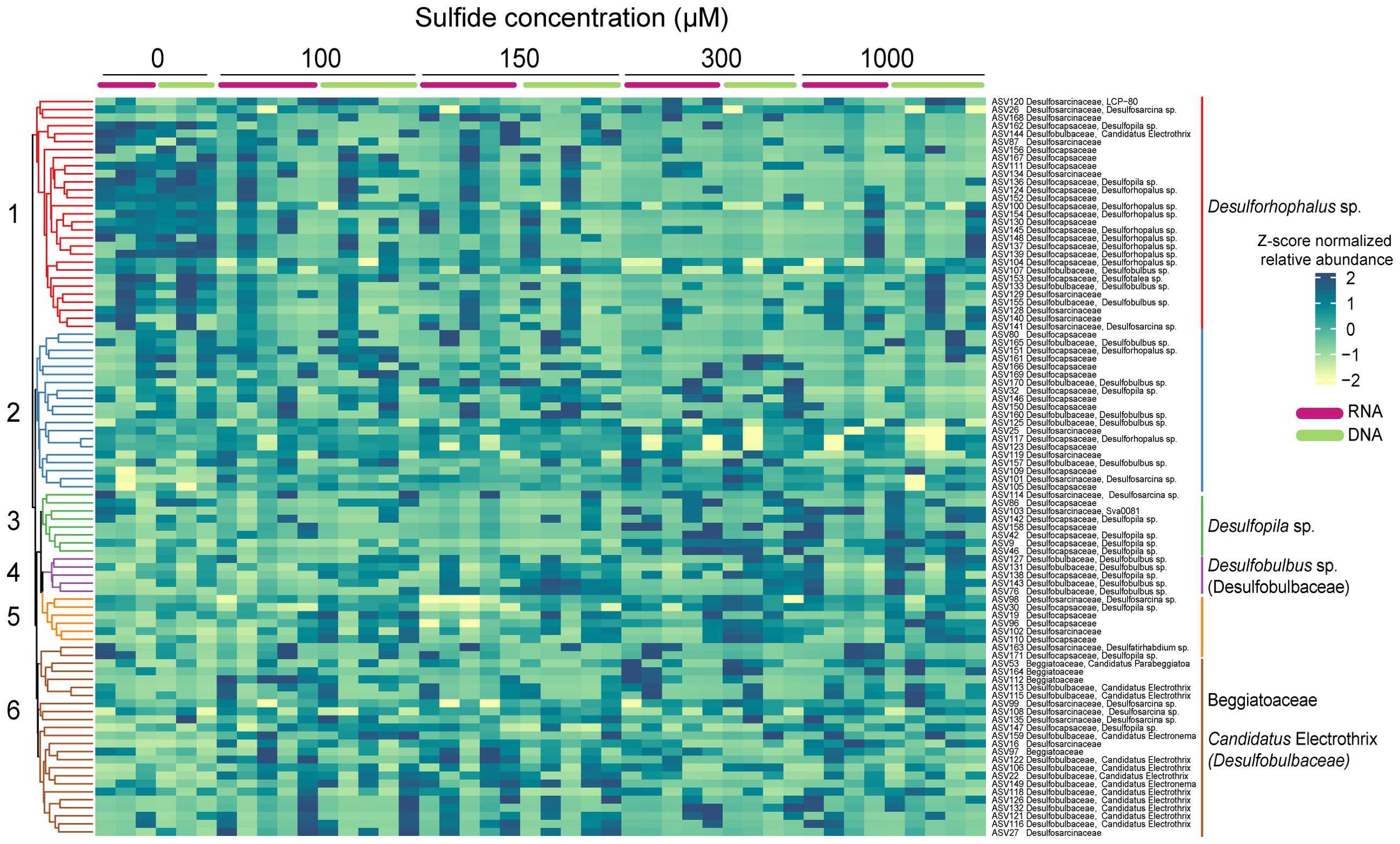
Figure 8. Heatmap of the Euclidian distance, clustering method of average linkage representing the clr-transformed abundances of ASVs belonging to the highly abundant indicator families in Caulerpa prolifera rhizoids DNA and RNA samples incubated at different sulphide concentrations. Significant clusters are highlighted with different colours.
Our measurements of sulphide in the field samples corroborated the findings of Holmer et al. (2009), confirming that sediments inhabited by Caulerpa exhibit elevated sulphide concentrations. In our mesocosm experiment, however, we observed a decline in the weight-based growth rate of Caulerpa in response to higher sulphide levels, indicating that the alga displays a certain sensitivity to sulphide despite continuing to grow. Therefore, while Caulerpa is associated with increased sulphide concentrations in the sediment, its growth is adversely impacted by these elevated sulphide levels. The sulphide increase, however, did not affect the photosynthesis of Caulerpa. While, to our knowledge, no other studies looked at the impact of sulphide on the photosynthesis in Caulerpa, sulphide is known to affect the photosynthesis and survival of seagrass seedlings negatively (Dooley et al., 2013) and seagrass growth (Calleja et al., 2007). So, the Caulerpa above-ground parts are less affected by sulphide than those in seagrasses.
The microbial communities in the rhizoids and sediments displayed a higher diversity than in the fronds. Furthermore, the DNA samples showed a higher diversity than the RNA samples which has been found by others (Gaidos et al., 2011; Bai et al., 2023) and could be due to the presence of both dead and alive bacteria (Laroche et al., 2017). It is known that relic DNA contributes to a large portion of the DNA pool, buffering the dynamics of diversity metrics (Lennon et al., 2018), while RNA might indicate the activity of certain bacteria (Chiba et al., 2022), which might change more rapidly under fluctuating environmental conditions. We suggest that similar results of alpha diversity measurements at different sulphide concentrations are attributed to composition changes, with certain bacteria groups replacing others, while the diversity value stays the same. Previous studies have shown that the fronds and rhizoids of Caulerpa exhibit a distinct microbial community (Morrissey et al., 2019). In our dataset, the sample type (i.e., fronds, rhizoids, sediments) also explained a significant portion (40%) of microbial community composition differentiation. Change in sulphide concentration explained 6% of the overall variation, and each sample type separately - 14, 16, and 18% (Table 1) of the variation in the community composition in the fronds, rhizoids, and sediments. In both analyses a large portion of the variation remained unexplained, which has been seen previously for Caulerpa species (Morrissey et al., 2019), and may indicate bacterial recruitment that is independent of sulphide effects (Morrissey et al., 2019; Kopprio et al., 2021).
The frond-associated microbiome, or ‘phylobiome’, remained unaffected by elevated sulphide concentrations. Both ANCOMBC and Indicator species analysis showed a high abundance of members of the Rhodobacteraceae that remained almost unchanged at the different sulphide treatments. This is not unexpected because sulphide will be chemically oxidised in the seawater without affecting the phylobiome. Members of the Rhodobacteraceae are known to be associated with various Caulerpa species (Aires et al., 2013; Liang et al., 2019; Kopprio et al., 2021) as well as other macroalgae (Dogs et al., 2017; Serebryakova et al., 2018) and seagrasses (Korlević et al., 2021). They might offer functions like vitamin B12 production (Dogs et al., 2017), nitrogen fixation (Chisholm et al., 1996), degradation of polysaccharides from algae, phytohormone production, and promotion of morphogenesis (Kopprio et al., 2021).
Both ANCOMBC and Indicator species analysis showed a dominance of Desulfosarcinaceae and the genus Sva0081 that belongs to the same family in sediments of the control sample without sulphide and in the 100 µM and 150 µM sulphide treatments. Sva0081 bacteria are uncultured sulphate-reducing bacteria in coastal sediments (Dyksma et al., 2018b; Coskun et al., 2019) and seagrass meadows (Zhang et al., 2020; de la Garza Varela et al., 2023). We found no difference in the bacterial composition in sediments of control samples with or without Caulerpa, suggesting that Caulerpa does not actively change the composition of the surrounding sediment microbial communities over a relatively short period of one month.
Caulerpa samples had a completely different microbial community composition than the sediment samples with rhizoid-associated ASVs distinctively clustering together and separated from the sediment-associated ASVs (Supplementary Figure S6). While the ANCOMBC analysis identified several Desulfocapsaceae and Desulfopila ASVs as differentially abundant across sulphide levels, cluster analysis did not resolve these effects. However, Indicator species analysis identified several key bacterial families associated with the rhizoids, all belonging to sulphur-cycling bacteria, and confirmed changes in their relative abundance in response to sulphide. Caulerpa-associated samples had abundant sulphate-reducing taxa; however, their taxonomic classification differed from that of the sediment-associated microbial community. Members of the sulphate-reducing family Desulfocapsaceae were associated with the rhizoids but not with the sediments, suggesting a potential selection by the alga. Our result complements the results of Korlević et al. (2021), who found Desulfocapsaceae associated with the invasive species Caulerpa cylindracea. Another study identified the sulfate-reducing family Desulfobacteraceae in the microbiome of Caulerpa racemosa (Aires et al., 2013), which was also present in our samples, although not abundant. Desulforhopalus sp. might play an important role as a sulphate reducer in the Caulerpa rhizoids (Bahr et al., 2005), as it has also been detected in the rhizobiome of the seagrass Posidonia oceanica (García-Martínez et al., 2009). This species followed a clear pattern in the rhizoids: abundant in the control without sulphide and decreasing in the sulphide treatments in both the DNA and RNA samples. Desulfopila sp., unlike Desulforhophalus, followed an opposite pattern, i.e., an increasing relative abundance with an increase in sulphide. Known initially as a sulphate reducer (Suzuki et al., 2007), recent research suggested that it might perform sulphur disproportionation in which both sulphate and sulphide are produced (Ward et al., 2021). These bacteria are likely to thrive in environments with active sulphate reduction, as these conditions provide intermediate sulphur compounds, such as sulphite, that can serve as a substrate for sulphur disproportionation (Alain et al., 2022). Additionally, the anoxic conditions characteristic of sulphate-reducing environments are conducive to the metabolic activities of sulphur-disproportionating bacteria. Desulfopila has also been detected in marine sediments (Colin et al., 2013), seagrasses (Zhou et al., 2021), and salt marsh plants (Rolando et al., 2022).
Desulfobulbaceae was an indicator taxon in the rhizoid RNA samples, with a high relative abundance of over 8%, and present at all sulphide concentrations. It is a family of sulphate reducers (Dyksma et al., 2018a; Vigneron et al., 2018) that also includes the so-called ‘cable bacteria’ that can oxidise sulphide by transporting electrons over centimetres from the anoxic depth to the oxic sediment surface (Trojan et al., 2016; Sandfeld et al., 2020). Desulfobulbus sp. followed a pattern similar to Desulfopila, i.e., increasing in relative abundance with the highest sulphide concentrations (i.e., 300 µM and 1000 µM). Some Desulfobulbus members are also known to be able to perform sulphur disproportionation and sulphate reduction (Ward et al., 2021; El Houari et al., 2017); these organisms have also been found in seagrasses (Zhou et al., 2021). Candidatus Electrothrix did not follow a specific pattern but seemed abundant in the 100 µM and 150 µM sulphide treatments and was absent in the no-sulphide control. It is known for its association with the rhizoids of seagrasses (Martin et al., 2020; Scholz et al., 2021), salt marsh plants (Scholz et al., 2021), and mangrove sediments (Scholz et al., 2021), potentially protecting against sulphide. Finally, members of the sulphide-oxidizing family Beggiatoaceae were associated with the Caulerpa rhizoids in the lower sulphide treatments but only in the active (RNA) bacterial community. Their presence as an indicator taxon at the intermediate (100 µM and 150 µM) sulphide concentrations suggests a potential role in sulphide detoxification. Their relative abundance seems to drop at the highest sulphide treatments, which could be evidence of sulphide inhibition, as previously described for this family (Dunker et al., 2011; Malkin et al., 2022). Members of this family have also been observed in seagrass rhizospheres (Fuggle et al., 2023).
We observed that the Beggiatoaceae family was detectable only at the RNA level, exhibiting high abundances of over 40% for some treatments while remaining below 1% at the DNA level. We did not detect such dramatic differences in bacteria associated with the Caulerpa fronds, where the relatively abundant taxa constituted >1% of the amplicon abundance in both the DNA and RNA samples. We propose two possible explanations for such discrepancies in the rhizoid-associated microbiome. First, high RNA abundance does not always translate into a high growth rate (Blazewicz et al., 2013). Hence, in our case, a high abundance in the RNA samples coupled with a low abundance in the DNA samples could mean an increase of activity of certain groups like the Beggiatoaceae that are present at low numbers but become active under specific conditions. It is important to note that dormant cells can also exhibit high RNA levels (Blazewicz et al., 2013). While it is theoretically possible that dormant Beggiatoaceae associated with Caulerpa are not active at the time of sampling (Blazewicz et al., 2013), our introduced range of sulphide concentration and the high Beggiatoaceae abundance in the RNA samples, specifically in the moderate sulphide concentrations, suggested otherwise. Second, some of the discrepancy could come from preservation, extraction and sequencing biases (Schirmer et al., 2015; Brauer and Bengtsson, 2022). However, we argue that this cannot fully explain the variation, because we don’t see dramatic differences between RNA and DNA samples in the frond-associated microbiome.
We, therefore, suggest that most of the variation between the DNA and RNA-based taxa abundances comes from a change of activity in the Beggiatoaceae, and its abundance increases as a response to sulphide.
Our findings confirm that C. prolifera is tolerant to sulphide; it inhabits areas with sulphide levels over 1000 µM that are considered extremely high for other organisms like seagrasses, for which the tolerance range is between 10-30 μM (Calleja et al., 2007; Holmer et al., 2009) and even lethal at levels higher than 680 μM (Dooley et al., 2013).
We have shown that Caulerpa prolifera has a high diversity of sulphate-reducing and sulphide-oxidizing bacteria, unique to its rhizoids and not observed in the sediments. As discussed above, all the indicator taxa we have found have been previously recorded in seagrasses and salt marsh plants; however, they are in much lower numbers. Although it is difficult to compare relative amplicon abundances with other studies due to the differences in primer regions (Na et al., 2023), we estimated a relative abundance of 88% of Desulfobacterota, with 66% belonging to the family Desulfocapsaceae in the control treatment (DNA samples), which is much higher than the relative abundances of sulphate reducers previously described for seagrasses and salt marsh plants. While seagrasses are known to host a mix of sulphate-reducing bacteria in their root microbiomes (Crump et al., 2018), the percentage of sulphate reducers found by Crump et al. (2018) - only 16% - was much lower than we identified in Caulerpa. A study on salt marsh plants estimated 7-15% of Desulfobacterota in sediments associated with Spartina alterniflora and Spartina mariqueter (Zheng et al., 2017). We only see a somewhat comparable high relative abundance of sulphur bacteria in another Caulerpa species: similar epiphytic bacteria composition as to our study has been recorded in invasive Caulerpa cylindracea thalli, with Desulfobacterota contributing 25% to the relative abundance of associated bacteria, with the large portion of it being Desulfocapsaceae family and Desulfobulbus, Desulfopila, Desulforhopalus species that we also find in our study (Korlević et al., 2021). The fact that this study only investigated epiphytic communities suggests that the total relative abundances might be higher.
High numbers of sulphate reducers in Caulerpa rhizoids recorded in our study strongly suggest the alga’s ability to change the sediment biochemical properties through its associated bacteria, which has been previously recorded in Spartina alterniflora – an invasive salt marsh plant (Zheng et al., 2017; Zhao et al., 2022) that is known to increase sulphide content of the soil (Zhao et al., 2022). It is possible that, like S. alterniflora, Caulerpa favours the activity of sulphate-reducing bacteria (Zheng et al., 2017), thus having a competitive advantage over sulphide-sensitive macrophytes.
In the study by Crump et al. (2018), sulphide-oxidising taxa (Sedimenticola, Arcobacter, Thiomicrospira, Sulfuromonas) associated with seagrass roots averaged 11%, while we found up to 47% relative abundance of Beggiatoaceae in Caulerpa rhizoids (RNA samples only, 150 µM treatment). This, coupled with the presence of cable bacteria, which are associated with the roots of freshwater and marine plants (Scholz et al., 2021), suggests that bacterial sulphide oxidation could occur around Caulerpa rhizoids at elevated sulphide concentrations.
As hypothesised, we observed a shift in sulphur-cycling bacteria within the rhizobiome of Caulerpa prolifera under sulphide stress, characterised by a decrease in sulphate-reducing bacteria and an increase in sulphide-oxidizing bacteria. This shift may help protect the alga from sulphide toxicity.
The data presented in the study are deposited in the NCBI Sequence Read Archive (SRA) repository, accession number PRJNA1136013.
AB: Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. AE: Conceptualization, Supervision, Writing – review & editing, Formal analysis. SW: Formal analysis, Supervision, Writing – review & editing. HB: Supervision, Writing – review & editing. GM: Supervision, Writing – review & editing, Conceptualization, Funding acquisition, Project administration.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Financial support was obtained from the Research Priority Area ‘Systems Biology of the University of Amsterdam and the Royal Netherlands Academy of Arts and Sciences (KNAW) Ecology Fund. This study received Portuguese national funds from FCT -Foundation for Science and Technology through project UIDB/04326/2020 and AE through contract CEECINST/00114/2018.
We want to thank João Reis for helping us set up and run the experiment at the Ramalhete marine station and guiding us through the research station facilities and Evelien Jongepier for helping with the data analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2024.1445441/full#supplementary-material
Aires T., Moalic Y., Serrao E. A., Arnaud-Haond S. (2015). Hologenome theory supported by co-occurrence networks of species-specific bacterial communities in siphonous algae (Caulerpa). Edited by J. Marchesi. FEMS Microbiol. Ecol. 91, fiv067. doi: 10.1093/femsec/fiv067
Aires T., Serrão E. A., Kendrick G., Duarte C. M., Arnaud-Haond S. (2013). Invasion is a community affair: Clandestine followers in the bacterial community associated to green algae, Caulerpa racemosa, track the invasion source. PloS One 8, e68429. doi: 10.1371/journal.pone.0068429
Alain K., Aronson H. S., Allioux M., Yvenou S., Amend J. P. (2022). Sulfur disproportionation is exergonic in the vicinity of marine hydrothermal vents. Environ. Microbiol. 24, 2210–2219. doi: 10.1111/1462-2920.15975
Bahr M., Crump B. C., Klepac-Ceraj V., Teske A., Sogin M. L., Hobbie J. E. (2005). Molecular characterization of sulfate-reducing bacteria in a New England salt marsh. Environ. Microbiol. 7, 1175–1185. doi: 10.1111/j.1462-2920.2005.00796.x
Bai X., Dinkla I. J. T., Muyzer G. (2023). Shedding light on the total and active core microbiomes in slow sand filters for drinking water production. Water Res. 243, 120404. doi: 10.1016/j.watres.2023.120404
Beckers B., Op De Beeck M., Thijs S., Truyens S., Weyens N., Boerjan W., et al. (2016). Performance of 16S rDNA primer pairs in the study of rhizosphere and endosphere bacterial microbiomes in metabarcoding studies. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00650
Bernardeau-Esteller J., Sandoval-Gil J. M., Belando M. D., Ramos-Segura A., García-Muñoz R., Marín-Guirao L., et al. (2023). The role of Cymodocea nodosa and Caulerpa prolifera meadows as nitrogen sinks in temperate coastal lagoons. Diversity 15, 172. doi: 10.3390/d15020172
Berriz G. F., King O. D., Bryant B., Sander C., Roth F. P. (2003). Characterizing gene sets with FuncAssociate. Bioinformatics 19, 2502–2504. doi: 10.1093/bioinformatics/btg363
Blazewicz S. J., Barnard R. L., Daly R. A., Firestone M. K. (2013). Evaluating rRNA as an indicator of microbial activity in environmental communities: limitations and uses. ISME J. 7, 2061–2068. doi: 10.1038/ismej.2013.102
Bolyen E., Rideout J. R., Dillon M. R., Bokulich N. A., Abnet C. C., Al-Ghalith G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Brauer A., Bengtsson M. M. (2022). DNA extraction bias is more pronounced for microbial eukaryotes than for prokaryotes. Microbiol. Open 11, e1323. doi: 10.1002/mbo3.1323
Cáceres M. D., Legendre P. (2009). Associations between species and groups of sites: indices and statistical inference. Ecology 90, 3566–3574. doi: 10.1890/08-1823.1
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., Holmes S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Calleja M. L., Marbà N., Duarte C. M. (2007). The relationship between seagrass (Posidonia oceanica) decline and sulfide porewater concentration in carbonate sediments. Estuar. Coast. Shelf Sci. 73, 583–588. doi: 10.1016/j.ecss.2007.02.016
Chiba A., Seki M., Suzuki Y., Kinjo Y., Mizunoe Y., Sugimoto S. (2022). Staphylococcus aureus utilizes environmental RNA as a building material in specific polysaccharide-dependent biofilms. NPJ Biofilms Microbiomes 8, 1–10. doi: 10.1038/s41522-022-00278-z
Chisholm J. R. M., Dauga C., Elisabeth A., Grimont P., Jaubert J. (1996). ‘Roots’ in mixotrophic algae. Nature 381, 382. doi: 10.1038/381382a0
Colin Y., Goñi-Urriza M., Caumette P., Guyoneaud R. (2013). Combination of high throughput cultivation and dsrA sequencing for assessment of sulfate-reducing bacteria diversity in sediments. FEMS Microbiol. Ecol. 83, 26–37. doi: 10.1111/j.1574-6941.2012.01452.x
Coskun Ö. K., Özen V., Wankel S. D., Orsi W. D. (2019). Quantifying population-specific growth in benthic bacterial communities under low oxygen using H218O. ISME J. 13, 1546–1559. doi: 10.1038/s41396-019-0373-4
Crump B. C., Wojahn J. M., Tomas F., Mueller R. S. (2018). Metatranscriptomics and amplicon sequencing reveal mutualisms in seagrass microbiomes. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.00388
Cunha A. H., Varela-Álvarez E., Paulo D., Sousa I., Serrao E. (2013). The rediscovery of Caulerpa prolifera in Ria Formosa, Portugal, 60 years after the previous record. Cah. Biol. Mar. 54, 359–364.
de la Garza Varela A., Aguirre-Macedo M. L., García-Maldonado J. Q. (2023). Changes in the rhizosphere prokaryotic community structure of Halodule wrightii monospecific stands associated to submarine groundwater discharges in a karstic costal area. Microorganisms 11, 494. doi: 10.3390/microorganisms11020494
Dogs M., Wemheuer B., Wolter L., Bergen N., Daniel R., Simon M., et al. (2017). Rhodobacteraceae on the marine brown alga Fucus spiralis are abundant and show physiological adaptation to an epiphytic lifestyle. Syst. Appl. Microbiol. 40, 370–382. doi: 10.1016/j.syapm.2017.05.006
Dooley F. D., Wyllie-Echeverria S., Roth M. B., Ward P. D. (2013). Tolerance and response of Zostera marina seedlings to hydrogen sulfide. Aquat. Bot. 105, 7–10. doi: 10.1016/j.aquabot.2012.10.007
Dunker R., Roamily H., Kamp A., Josen B. B. (2011). Motility patterns of filamentous sulfur bacteria, Beggiatoa spp. FEMS Microbiol. Ecol. 77, 176–185. doi: 10.1111/j.1574-6941.2011.01099.x
Dyksma S., Lenk S., Sawicka J. E., Mußmann M. (2018b). Uncultured Gammaproteobacteria and Desulfobacteraceae account for major acetate assimilation in a coastal marine sediment. Front. Microbiol. 9, 3124. doi: 10.3389/fmicb.2018.03124
Dyksma S., Pjevac P., Ovanesov K., Mußmann M. (2018a). Evidence for H2 consumption by uncultured Desulfobacterales in coastal sediments. Environ. Microbiol. 20 (2), 450–461. doi: 10.1111/1462-2920.13880
El Houari A., Ranchou-Peyruse M., Ranchou-Peyruse A., Dakdaki A., Guignard M., Idouhammou L., et al. (2017). Desulfobulbus oligotrophicus sp. nov., a sulfate-reducing and propionate-oxidizing bacterium isolated from a municipal anaerobic sewage sludge digester. Int. J. Syst. Evol. Microbiol. 67, 275–281. doi: 10.1099/ijsem.0.001615
Fuggle R. E., Gribben P. E., Marzinelli E. M. (2023). Experimental evidence root-associated microbes mediate seagrass response to environmental stress. J. Ecol. 111, 1079–1093. doi: 10.1111/1365-2745.14081
Gaidos E., Rusch A., Ilardo M. (2011). Ribosomal tag pyrosequencing of DNA and RNA from benthic coral reef microbiota: community spatial structure, rare members and nitrogen-cycling guilds. Environ. Microbiol. 13, 1138–1152. doi: 10.1111/j.1462-2920.2010.02392.x
García-Martínez M., López-López A., Calleja M. L., Marbà N., Duarte C. M. (2009). Bacterial community dynamics in a seagrass (Posidonia oceanica) meadow sediment. Estuaries Coasts 32, 276–286. doi: 10.1007/s12237-008-9115-y
Gloor G. B., Macklaim J. M., Pawlowsky-Glahn V., Egozcue J. J. (2017). Microbiome datasets are compositional: and this is not optional. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.02224
Grieshaber M. K., Völkel S. (1998). Animal adaptations for tolerance and exploitation of poisonous sulfide. Annu. Rev. Physiol. 60, 33–53. doi: 10.1146/annurev.physiol.60.1.33
Gupta E., Dooley F. D., Ward P. D. (2014). Evolutionary legacy response observed in algae and bryophytes following hydrogen sulfide administration. Toxicol. Environ. Chem. 96, 442–450. doi: 10.1080/02772248.2014.944353
Holmer M., Marbà N., Lamote M., Duarte C. M. (2009). Deterioration of sediment quality in seagrass meadows (Posidonia oceanica) invaded by macroalgae (Caulerpa sp.). Estuaries Coasts 32, 456–466. doi: 10.1007/s12237-009-9133-4
Huang H., Liu Y., Yuan M., Marron J. S. (2015). Statistical significance of clustering using soft thresholding. J. Comput. Graph. Stat. 24, 975–993. doi: 10.1080/10618600.2014.948179
Joyner-Matos J., Downs C. A., Julian D. (2006). Increased expression of stress proteins in the surf clam Donax variabilis following hydrogen sulfide exposure. Comp. Biochem. Physiol. A: Mol. Integr. Physiol. 145, 245–257. doi: 10.1016/j.cbpa.2006.06.033
Kasten S., Jørgensen B. B. (2000). “Sulfate reduction in marine sediments,” in Mar. Geochem. Eds. Schulz H. D., Zabel M. (Springer Berlin Heidelberg, Berlin, Heidelberg), 263–281. doi: 10.1007/978-3-662-04242-7_8
Kopprio G. A., Luyen N. D., Cuong L. H., Duc T. M., Fricke A., Kunzmann A., et al. (2021). Insights into the bacterial community composition of farmed Caulerpa lentillifera: a comparison between contrasting health states. Microbiol. Open 10, e1253. doi: 10.1002/mbo3.1253
Korlević M., Markovski M., Zhao Z., Herndl G. J., Najdek M. (2021). Seasonal dynamics of epiphytic microbial communities on marine macrophyte surfaces. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.671342
Kuznetsova A., Brockhoff P. B., Christensen R. H. B. (2017). lmerTest package: tests in linear mixed effects models. J. Stat. Software 82, 1–26. doi: 10.18637/jss.v082.i13
Lahti L., Shetty S., et al. (2017). Tools for microbiome analysis in R. Microbiome package version 1.23.1. URL. Available online at: http://microbiome.github.com/microbiome. (accessed June 1, 2024).
Laroche O., Wood S. A., Tremblay L. A., Lear G., Ellis J. I., Pochon X. (2017). Metabarcoding monitoring analysis: the pros and cons of using co-extracted environmental DNA and RNA data to assess offshore oil production impacts on benthic communities. PeerJ 5, e3347. doi: 10.7717/peerj.3347
Lennon J. T., Muscarella M. E., Placella S. A., Lehmkuhl B. K. (2018). How, when, and where relic DNA affects microbial diversity. mBio 9, e00637-18. doi: 10.1128/mBio.00637-18
Liang Z., Liu F., Wang W., Zhang P., Sun X., Wang F., et al. (2019). High-throughput sequencing revealed differences of microbial community structure and diversity between healthy and diseased Caulerpa lentillifera. BMC Microbiol. 19, 225. doi: 10.1186/s12866-019-1605-5
Lin H., Peddada S. D. (2020). Analysis of compositions of microbiomes with bias correction. Nat. Commun. 11, 3514. doi: 10.1038/s41467-020-17041-7
Lu Y., Zhou G., Ewald J., Pang Z., Shiri T., Xia J. (2023). MicrobiomeAnalyst 2.0: comprehensive statistical, functional and integrative analysis of microbiome data. Nucleic Acids Res. 51, W310–W318. doi: 10.1093/nar/gkad407
Malkin S. Y., Liau P., Kim C., Hantsoo K. G., Gomes M. L., Song B. (2022). Contrasting controls on seasonal and spatial distribution of marine cable bacteria (Candidatus Electrothrix) and Beggiatoaceae in seasonally hypoxic Chesapeake Bay. Limnol. Oceanogr. 67, 1357–1373. doi: 10.1002/lno.12087
Maricle B. R., Lee R. W. (2007). Root respiration and oxygen flux in salt marsh grasses from different elevational zones. Mar. Biol. 151, 413–423. doi: 10.1007/s00227-006-0493-z
Martin B. C., Bougoure J., Ryan M. H., Bennett W. W., Colmer T. D., Joyce N. K., et al. (2019). Oxygen loss from seagrass roots coincides with colonisation of sulphide-oxidising cable bacteria and reduces sulphide stress. ISME J. 13, 707–719. doi: 10.1038/s41396-018-0308-5
Martin B. C., Sanchez Alarcon M., Gleeson D., Middleton J. A., Fraser M. W., Ryan M. H., et al. (2020). Root microbiomes as indicators of seagrass health. FEMS Microbiol. Ecol. 96, fiz201. doi: 10.1093/femsec/fiz201
McMurdie P. J., Holmes S. (2013). phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8, e61217. doi: 10.1371/journal.pone.0061217
Morrissey K. L., Çavaş L., Willems A., De Clerck O. (2019). Disentangling the influence of environment, host specificity and thallus differentiation on bacterial communities in siphonous green seaweeds. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.00717
Na H. S., Song Y., Yu Y., Chung J. (2023). Comparative analysis of primers used for 16S rRNA gene sequencing in oral microbiome studies. Methods Protoc. 6, 71. doi: 10.3390/mps6040071
Naqib A., Poggi S., Wang W., Hyde M., Kunstman K., Green S. J. (2018). “Making and sequencing heavily multiplexed, high-throughput 16S ribosomal RNA gene amplicon libraries using a flexible, two-stage PCR protocol,” in Gene Expression Analysis: Methods and Protocols. Eds. Raghavachari N., Garcia-Reyero N. (Springer, New York, NY), 149–169. doi: 10.1007/978-1-4939-7834-2_7
Oksanen J., Simpson G., Blanchet F., Kindt R., Legendre P., Minchin P., et al. (2022). vegan: Community Ecology Package. R package version 2.6-4. Available online at: https://CRAN.R-project.org/package=vegan. (accessed June 1, 2024).
Oren A., Padan E., Malkin S. (1979). Sulfide inhibition of photosystem II in cyanobacteria (blue-green algae) and tobacco chloroplasts. Biochim. Biophys. Acta 546, 270–279. doi: 10.1016/0005-2728(79)90045-8
Parreira F., Martínez-Crego B., Lourenço Afonso C. M., MaChado M., Oliveira F., dos Santos Gonçalves J. M., et al. (2021). Biodiversity consequences of Caulerpa prolifera takeover of a coastal lagoon. Estuar. Coast. Shelf Sci. 255, 107344. doi: 10.1016/j.ecss.2021.107344
Paul B. D., Snyder S. H., Kashfi K. (2021). Effects of hydrogen sulfide on mitochondrial function and cellular bioenergetics. Redox Biol. 38, 101772. doi: 10.1016/j.redox.2020.101772
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Rolando J. L., Kolton M., Song T., Kostka J. E. (2022). The core root microbiome of Spartina alterniflora is predominated by sulfur-oxidizing and sulfate-reducing bacteria in Georgia salt marshes, USA. Microbiome 10, 37. doi: 10.1186/s40168-021-01187-7
Sandfeld T., Marzocchi U., Petro C., Schramm A., Risgaard-Petersen N. (2020). Electrogenic sulfide oxidation mediated by cable bacteria stimulates sulfate reduction in freshwater sediments. ISME J. 14, 1233–1246. doi: 10.1038/s41396-020-0607-5
Schirmer M., Ijaz U. Z., D’Amore R., Hall N., Sloan W. T., Quince C. (2015). Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 43, e37. doi: 10.1093/nar/gku1341
Schloss P. D. (2021). Amplicon sequence variants artificially split bacterial genomes into separate clusters. mSphere 6, e00191-21. doi: 10.1128/msphere.00191-21
Scholz V. V., Martin B. C., Meyer R., Schramm A., Fraser M. W., Nielsen L. P., et al. (2021). Cable bacteria at oxygen-releasing roots of aquatic plants: a widespread and diverse plant–microbe association. New Phytol. 232, 2138–2151. doi: 10.1111/nph.17415
Serebryakova A., Aires T., Viard F., Serrão E. A., Engelen A. H. (2018). Summer shifts of bacterial communities associated with the invasive brown seaweed Sargassum muticum are location and tissue dependent. PloS One 13, e0206734. doi: 10.1371/journal.pone.0206734
Sogin E. M., Leisch N., Dubilier N. (2020). Chemosynthetic symbioses. Curr. Biol. 30, R1137–R1142. doi: 10.1016/j.cub.2020.07.050
Suzuki D., Ueki A., Amaishi A., Ueki K. (2007). Desulfopila aestuarii gen. nov., sp. nov., a Gram-negative, rod-like, sulfate-reducing bacterium isolated from an estuarine sediment in Japan. Int. J. Syst. Evol. Microbiol. 57, 520–526. doi: 10.1099/ijs.0.64600-0
Terrados J., Duarte C. M., Kamp-Nielsen L., Agawin N. S. R., Gacia E., Lacap D., et al. (1999). Are seagrass growth and survival constrained by the reducing conditions of the sediment? Aquat. Bot. 65, 175–197. doi: 10.1016/S0304-3770(99)00039-X
Trojan D., Schreiber L., Bjerg J. T., Bøggild A., Yang T., Kjeldsen K. U., et al. (2016). A taxonomic framework for cable bacteria and proposal of the candidate genera Electrothrix and Electronema. Syst. Appl. Microbiol. 39, 297–306. doi: 10.1016/j.syapm.2016.05.006
Trüper H. G., Schlegel H. G. (1964). Sulphur metabolism in Thiorhodaceae I. Quantitative measurements on growing cells of Chromatium okenii. Antonie Van Leeuwenhoek 30, 225–238. doi: 10.1007/BF02046728
Vigneron A., Cruaud P., Alsop E., de Rezende J. R., Head I. M., Tsesmetzis N. (2018). Beyond the tip of the iceberg; a new view of the diversity of sulfite- and sulfate-reducing microorganisms. ISME J. 12, 2096–2099. doi: 10.1038/s41396-018-0155-4
Ward L. M., Bertran E., Johnston D. T. (2021). Expanded genomic sampling refines current understanding of the distribution and evolution of sulfur metabolisms in the Desulfobulbales. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.666052
Wasmund K., Mußmann M., Loy A. (2017). The life sulfuric: microbial ecology of sulfur cycling in marine sediments. Environ. Microbiol. Rep. 9, 323–344. doi: 10.1111/1758-2229.12538
Weinroth M. D., Belk A. D., Dean C., Noyes N., Dittoe D. K., Rothrock M. J. Jr., et al. (2022). Considerations and best practices in animal science 16S ribosomal RNA gene sequencing microbiome studies. J. Anim. Sci. 100, skab346. doi: 10.1093/jas/skab346
Zhang X., Zhao C., Yu S., Jiang Z., Liu S., Wu Y., et al. (2020). Rhizosphere microbial community structure is selected by habitat but not plant species in two tropical seagrass beds. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.00161
Zhao Z., Cheng L., He C., Wang F., Liu J., Li Y., et al. (2022). Spartina alterniflora invaded coastal wetlands by raising soil sulfur contents: a meta-analysis. Water 14, 1633. doi: 10.3390/w14101633
Zheng Y., Bu N. S., Long X. E., Sun J., He C. Q., Liu X. Y., et al. (2017). Sulfate reducer and sulfur oxidizer respond differentially to the invasion of Spartina alterniflora in estuarine salt marsh of China. Ecol. Eng. 99, 182–190. doi: 10.1016/j.ecoleng.2016.11.031
Keywords: Caulerpa, holobiont, microbiome, sulphate reduction, sulphide, sulphide oxidation
Citation: Barilo A, Engelen A, Wilken S, Bouwmeester H and Muyzer G (2024) Shifts in sulphur-cycling bacteria in the rhizobiome support the adaptation of Caulerpa prolifera to elevated sulphide levels. Front. Mar. Sci. 11:1445441. doi: 10.3389/fmars.2024.1445441
Received: 07 June 2024; Accepted: 03 October 2024;
Published: 24 October 2024.
Edited by:
Sébastien Duperron, Muséum National d’Histoire Naturelle, FranceReviewed by:
Maxim Rubin-Blum, Israel Oceanographic and Limnological Research (IOLR), IsraelCopyright © 2024 Barilo, Engelen, Wilken, Bouwmeester and Muyzer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gerard Muyzer, Zy5tdWlqemVyQHV2YS5ubA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.