 Yanbin Tang
Yanbin Tang Rongliang Zhang
Rongliang Zhang Qinghe Liu1,2
Qinghe Liu1,2 Lu Shou
Lu Shou Yibo Liao
Yibo Liao- 1Key Laboratory of Marine Ecosystem Dynamics, Second Institute of Oceanography, Ministry of Natural Resources, Hangzhou, China
- 2Observation and Research Station of Yangtze River Delta Marine Ecosystems, Ministry of Natural Resources, Zhoushan, China
- 3School of Marine Sciences, Ningbo University, Ningbo, China
- 4Key Laboratory of Ocean Space Resource Management Technology, Ministry of Natural Resources, Hangzhou, China
The distinctive tectonic settings and hydrological environments of the marginal seas in the Northwestern Pacific render them a focal point for investigating marine speciation and population diversity. In this study, we focused on understanding the phylogeography and demographic history of Nerita yoldii, a species commonly found in rocky intertidal areas. We examined the COI gene sequences from nine populations of the Northwestern Pacific. The analysis revealed four lineages comprising 35 haplotypes, with the majority of variation found within populations. Approximate Bayesian computation analysis supported the scenario that lineages B and C diverged first, and subsequently, lineage A and D originated from lineage C. Mismatch distribution analysis and neutrality tests suggested demographic expansion of lineage C primarily during the Pleistocene epoch of the Quaternary period, with expansion time estimated at approximately 38,000 years ago. Artificial rocky structures along the northern Changjiang coast, combined with global warming, could possibly facilitate the northward migration of N. yoldii larvae in recent decades. This has resulted in a shift in the species’ northern boundary. This study not only unveils the historical, present, and future impacts of climate and environmental changes on intertidal macrobenthos but also furnishes biological evidence crucial for comprehending related geological historical events. Moreover, it offers insights into the future trajectory of intertidal macrobenthos development and serves as a foundation for guiding future biological conservation efforts.
1 Introduction
The Northwestern Pacific (NWP) region is a significant marine ecosystem known for its diverse biodiversity (Kerswell, 2006). Coastal biogeographic patterns within this area are shaped by multiple factors, including historical occurrences, coastal currents, freshwater influxes, available habitats, and climatic conditions (Dong et al., 2012; Hu et al., 2015; Wang et al., 2015; Ni et al., 2017; Zhong et al., 2020). The distribution of numerous marine populations across the NWP region has been profoundly influenced by climatic shifts during the late Pleistocene ice age (Coyer et al., 2004; Zhang et al., 2019), as species responded to oscillations between glacial and interglacial epochs (Provan and Bennett, 2008). This corresponds to the “expansion-contraction” (EC) model proposed by Provan and Bennett (2008), which posits that southern populations harbor greater genetic diversity and distinct genetic variability compared to their northern counterparts (Taberlet et al., 1998). Consequently, the identification of refugee-seeking populations and recolonization pathways can provide invaluable insights into the conservation of local genetic diversity (Zhang et al., 2019).
In recent decades, ocean warming attributed to global climate change has precipitated the northward migration of the geographic range of numerous marine species in the Northern Hemisphere (Zhang et al., 2019). Previous investigations have shown that escalating sea surface temperatures trigger northward proliferation of certain intertidal organisms such as Crassostrea sikamea and Littoraria sinensis (Hu and Dong, 2022). These findings underscore the importance of biogeographic surveys aimed at cataloging the population distributions of economically and ecologically significant gastropod species.
Nerita yoldii Recluz, 1841 is an endemic species of the family Neritidae (Gasteropoda: Neritimorpha) in East Asia. The reproductive period of N. yoldii in the NWP is from April to August, and the pelagic larval duration lasts for about one month (Hu and Dong, 2022). It is widely distributed along the coastal areas of the NWP region, particularly within the intertidal zone of the lithofacies. N. yoldii can tolerate a wide salinity range and prefer temperature higher than 10 °C (Jiang et al., 2024). During the 1960s, its northern distribution boundary was demarcated at the Shengsi Islands, Zhejiang Province (30°34 ‘-30°52’ N) (Tchang et al., 1963). Subsequently, by the 1980s, it had transcended the Yangtze River Estuary barrier and ventured northward to the Liyashan Oyster Reef (32°08 ‘N) in Jiangsu Province (Quan et al., 2012). Since then, N. yoldii has exhibited rapid northward expansion along artificial rocky coasts, being initially documented at Zhonganpeng (32°51 ‘N) in January 2016 (Wang et al., 2018). Previous research and fossil evidence provide insights into the evolutionary history of Nerita, suggesting that its ancestors emerged as early as the Eocene epoch, approximately 55.2 million years ago (Mya), in the Tethys Sea and the present-day Indo-Western Pacific region (Frey and Vermeij, 2008). N. yoldii originated during the Miocene epoch.
The Changjiang Estuary has traditionally been considered the northern boundary for N. yoldii (Tchang et al., 1963) and several other rocky shore species (Wang et al., 2015), primarily due to unsuitable substratum conditions and significant freshwater discharge (Ni et al., 2017). However, recent observations have documented northward range expansions of N. yoldii and some other intertidal species specifically on artificial rocky shores (Dong et al., 2016; Wang et al., 2018). The northward migration of the N. yoldii population has been attributed to the warming of winter temperatures and the construction of artificial dams on the north bank of the Yangtze River Estuary (Wang et al., 2020). However, these studies lacked comprehensive coverage of the geographical range of the population and omitted the phylogenetic examination of the southern coastal population, which is crucial for inferring population structure across the entire NWP distribution.
Phylogenetic analysis employing molecular markers represents a robust method for unraveling the historical dynamics of populations and species (Avise, 2004). Previous phylogeographical studies focusing on the NWP where sea level fluctuations have led to successive exposure and inundation of continental shelves have elucidated distinctive patterns in response to glacial impacts on its biota, differing notably from findings in other regions (Xu et al., 2009; Ni et al., 2014; Zhang et al., 2024). The consistent findings of demographic expansions during the Pleistocene epoch in previous studies underscore the significant role of glacial events in shaping the demographic dynamics of marine populations (Guo et al., 2015; Wang et al., 2018). Notably, divergent intraspecific lineages have been identified across various seas within the NWP, with overlapping distributions observed in adjacent regions (Dong et al., 2016). Such patterns suggest that each sea within the NWP likely functioned as an independent refugium during periods of glaciation (Ni et al., 2014). However, despite these advancements, the complex environmental dynamics and unique tectonic framework of the NWP present ongoing challenges in fully comprehending phylogeographic divergence. Consequently, the factors and processes underlying community genetics in this region remain predominantly unexplored.
The primary objectives of this study were as follows: (1) to scrutinize the genetic structure of the N. yoldii population in the NWP region; (2) to assess the impact of glacial-interglacial environmental fluctuations on the origin, dispersion, and evolutionary trajectory of this species; and (3) to provide evidence for the examination of biodiversity in tropical and subtropical waters.
2 Materials and methods
2.1 Sample collection and sampling area
From November 2020 to November 2022, 164 specimens of N. yoldii were collected from nine intertidal sites along the NWP coastline and muscle tissues were preserved in anhydrous ethanol. Detailed information regarding the collection dates and locations is presented in Table 1, and the distribution of the sampling stations is illustrated in Figure 1B. The sampling region encompassed three marginal seas within the NWP region. Namely, the Yellow Sea (NT), East China Sea (DYS, SS, NSD, LK, PT, XM), and South China Sea (SZ, ZJ) were included. Concurrently, the South China Sea experienced a decline in sea levels by approximately 100-120 meters, transforming it into a semi-enclosed sea, primarily linked to the Pacific Ocean via the Bashi Strait (Ni et al., 2014). The depth of the NWP increases significantly from north to south. The Yellow Sea has an average depth of only 44 m, while the East China Sea reaches around 350 m, and the South China Sea exceeds 1,200 m. The SS location marks the northern distributional limit of N. yoldii within the NWP since the 1960s (Tchang et al., 1963). SS situated at the mouth of the Changjiang, the third-largest river globally, with an average annual discharge ranging from 8-9×1011 m3. Changjiang exerts significant influence on various ecological and environmental parameters within the East China Sea, separating the North Pacific Temperate Biotic Region (characterized by cold temperate fauna) from the Indo-West Pacific Warm-water Biotic Region (characterized by tropical/subtropical fauna) (Ni et al., 2017). The annual sea surface temperature north of the Changjiang Estuary ranges from 14°C to 19°C, while the sea surface temperature south of the Changjiang Estuary varies between 19°C and 25°C. Consequently, this biogeographic boundary along the NWP coastline has historically restricted the northward dispersion of N. yoldii, although recent decades have witnessed its extension beyond traditional confines (Wang et al., 2022). Notably, investigations in January 2016 (Wang et al., 2018) indicate that the NT location approaches its current northern boundary at approximately 32°51′N.
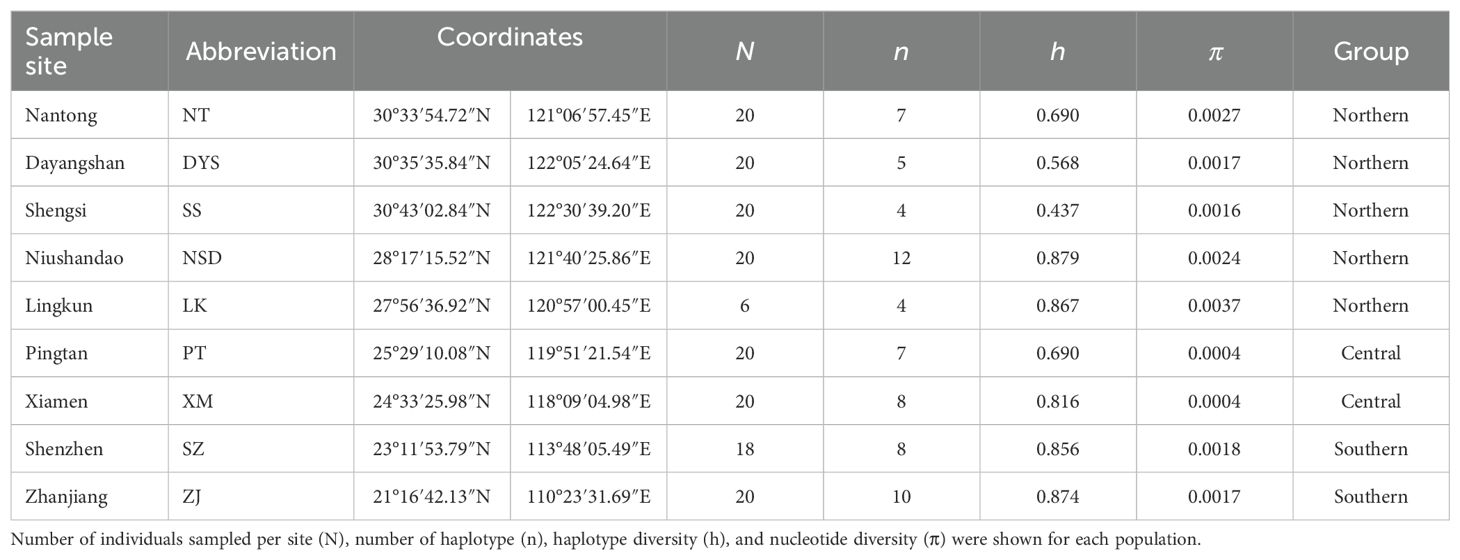
Table 1. Sampling sites and diversity indices for nine populations of Nerita yoldii.
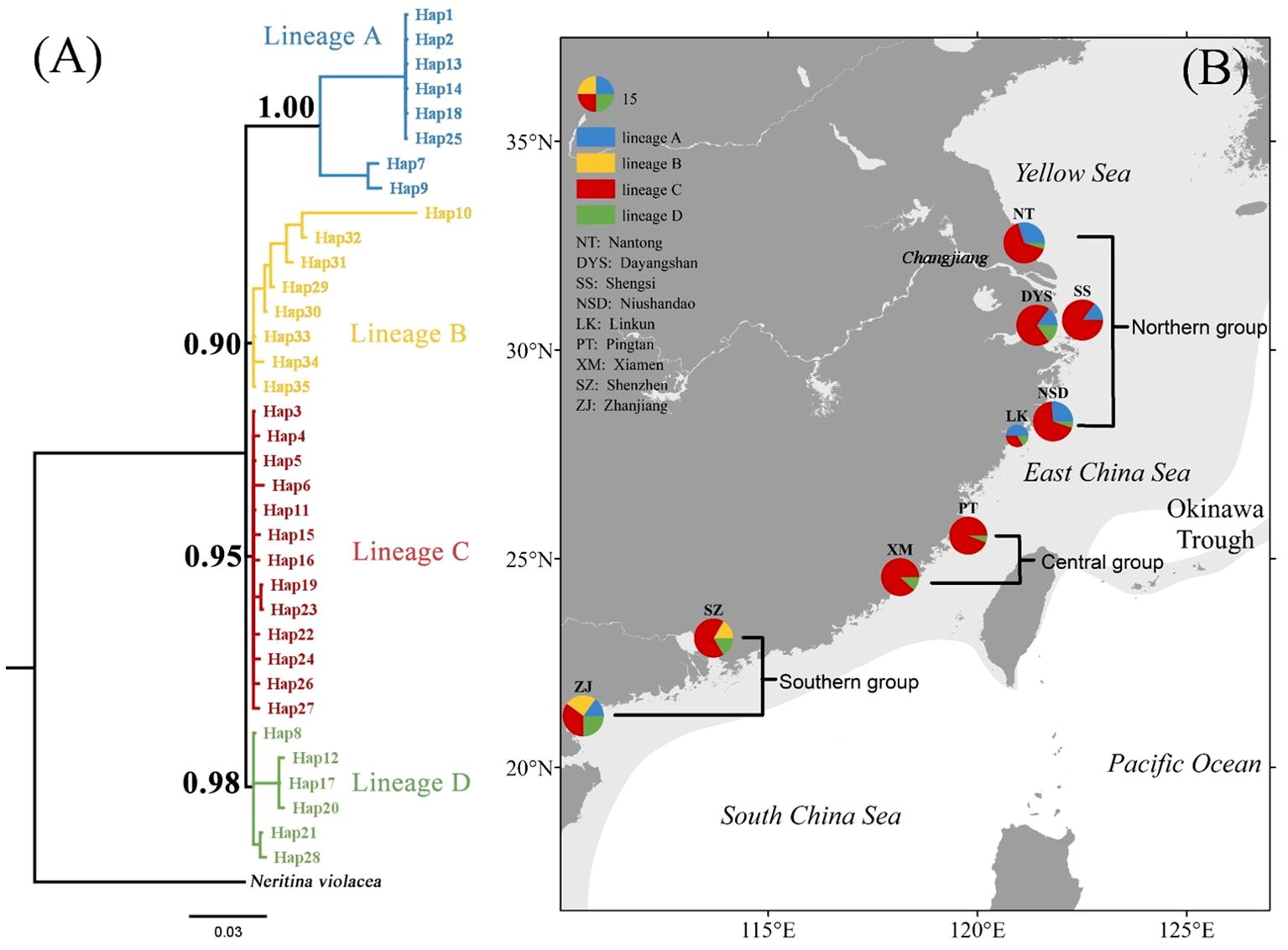
Figure 1. Bayesian tree with sequence of Neritina violacea used as outgroup showing (A) four lineages of Nerita yoldii and (B) their spatial distribution, the posterior probabilities (only values higher than 0.9 are shown) are indicated at the nodes. Map of East Asia showing the sampling sites of Nerita yoldii, dark-grey areas indicate current mainland and island configurations, and light-grey areas indicate palaeogeographic land of East Asia in the early Pleistocene (Osozawa et al., 2012).
2.2 DNA extraction and sequencing
The samples were subjected to DNA extraction using a Tiangen DP304 blood/cell/tissue genomic DNA kit (Tiangen, Beijing, China). Polymerase chain reaction (PCR) amplification was used to obtain COI gene sequences for phylogenetic analysis. The mitochondrial DNA (mtDNA) COI region was amplified and sequenced using the primer pairs LCO1490-GTCAACAAATCATAAAGATATTGG and HCO2198-TAAACTTCAGGGTGACCAAAAAATCA (Folmer et al., 1994). PCR amplification conditions were as follows: an initial denaturation at 94°C for 3 minutes; 35 cycles of denaturation at 94°C for 45 seconds, annealing at 50°C for 45 seconds, and extension at 72°C for 1 minute; and a final extension at 72°C for 10 minutes. Subsequently, the unpurified PCR samples were subjected to electrophoresis on a 1.5% agarose gel at 120V for approximately 30 minutes. A BigDye Terminator v3.1 sequencing kit (ABI, U.S.) was used for PCR. The PCR products underwent purification via the ethanol precipitation method, followed by drying and storage at 4°C away from light. Sequencing was conducted using the Sanger method (double-stranded DNA chain termination technique), followed by capillary electrophoresis to acquire the DNA base sequence. The dried samples were dissolved in 10 µL of formamide, denatured in a PCR apparatus, and sequenced using the 3730XL platform (Thermo Fisher Scientific, U.S.). The initial sequence assembly was conducted using SeqMan software v.7.1.0, and alignment and trimming were performed using BioEdit v.7.0.9 (Hall, 1999). All sequences were deposited in GeBank, with accession number PP436912-PP437075.
2.3 Phylogenetic analysis
The number of haplotypes for each locality was determined using DnaSP v.5.10. Additionally, the number of polymorphic sites, haplotype diversity, and nucleotide diversity were estimated using Arlequin v3.11 (Excoffier et al., 2005). Bayesian analysis was conducted on all N. yoldii specimens, alongside GenBank sequences of Neritina violacea (accession numbers MF688020), utilizing MrBayes v3.2.4 under the F81 model (Huelsenbeck and Ronquist, 2001), four Markov chains were applied for 1,000,000 generations, sampled every 1000 generations, and the first 25% discarded as the burn-in, respectively. A network depicting the relationships among the haplotypes was constructed using TCS (Clement et al., 2000).
2.4 Population analysis
A simulated annealing approach implemented in SAMOVA v. 2.0 was used to test varying numbers of groups (K) to identify phylogeographically homogeneous populations that were maximally differentiated from each other (Dupanloup et al., 2002). The analysis was run for 2 to 8 groups, and the grouping associated with the highest FCT value was selected for further analysis. Analysis of molecular variance (AMOVA) was conducted for COI data using Arlequin v3.11, with 1,000 permutations to explore the partitioning of genetic diversity among populations (Excoffier et al., 1992). Arlequin v3.11 was also employed to compute the pairwise genetic differentiation (FST) among populations and groups identified by SAMOVA (Excoffier et al., 2010). To investigate the relationship between genetic and geographical distances, Mantel’s test with 10,000 randomizations for isolation-by-distance (IBD) (Slatkin, 1993) was performed between the linearized FST (FST/[1-FST]) and geographical distances (shortest marine connection measured in km with ArcMap v10.3) using IBD v1.52 (Bohonak, 2002) to ascertain whether this pattern conformed to the expectation that genetic similarity decreases with geographical distance (Novembre et al., 2008). Linearized genetic differentiation (FST/[1-FST]) values were plotted against geographical distance using Origin software to visualize the relationship between genetic and geographical distances.
2.5 Historical demography analysis
Tajima’s D neutrality test and Fu’s FS neutrality test (Fu and Li, 1993) were conducted using DnaSP 5, with the p-value serving as the criterion for significance. These tests are based on the assumption that mutations occur in each position of the alignment with equal probability consistently over time. Values greater than zero indicate bottleneck effects and/or non-directional selection, whereas values significantly lower than zero indicate population expansion and/or directional selection. Mismatch distribution analysis of the mtDNA sequence of each lineage was performed (Slatkin, 1995). The historical population sizes were estimated using extended Bayesian skyline plots approach employed with the HKY substitution site model using a strict molecular clock with rate 0.0023 (Frey and Vermeij, 2008) and running 1.0 × 106 Markov Chain Monte Carlo (MCMC) simulations in the software BEAST 2.7 (Drummond and Rambaut, 2007; Heled and Drummond, 2008). The convergence of the MCMC runs was assessed using effective sample size (ESS) values for the parameters. The MCMC conditions were adjusted to achieve ESS values greater than 200 for all parameters (Supplementary Table S1). The results were checked with Tracer 1.7 (Rambaut et al., 2018). The population expansion time (t) of N. yoldii was calculated using the formula t = τ/2u (Rogers, 1995), where u = μkg; μ is the nucleotide substitution rate, which is reported to be 0.0023 substitutions/site/myr for the COI gene in genus Nerita (Frey and Vermeij, 2008); k is the length of COI sequence; and g is the generation time of N. yoldii. A generation time of 3 years was set based on studies of the life history of its sister species, N. japonica (Nakano and Nagoshi, 1980). DIY-ABC v2.1.0 was utilized to gain further insights into the timing and divergence history of N. yoldii based on COI gene sequences (Cornuet et al., 2008; Cabrera and Palsbøll, 2017). We tested five plausible divergence scenarios (Supplementary Figure 1) for the four lineages identified by the Bayesian tree. In all scenarios, lineage B and C were the first to diverge. The scenarios modeled the possible hypotheses regarding the origins of lineages A and D, which could either have originated from lineage C or resulted from an admixture between lineage B and C.
3 Results
3.1 Genetic variations
The alignment of the 557 bp COI gene fragment was analyzed in 164 individuals, revealing 384 variable sites and 35 unique haplotypes (Hap1–Hap35). The Bayesian tree for N. yoldii based on the COI gene fragment indicated four lineages: A, B, C, and D, and revealed a polytomy among the lineages (Figure 1A). The haplotype network diagram (Figure 2) aligned closely with the Bayesian tree, indicating that lineages B, C, and D were interconnected, while lineage A was distinct from lineages B and D. Lineage C exhibited a star-like topology with Hap5 as the central haplotype. Populations NSD and ZJ exhibited the highest haplotype diversity, whereas populations SS and LK exhibited only four haplotypes. Hap5 was the most widespread haplotype being found in all populations. The average haplotype diversity (h) across all populations was 0.742, and nucleotide diversity (π) was 0.0018. The NSD population displayed the highest haplotype diversity (h = 0.874), whereas the SS population exhibited the lowest (h = 0.437). The highest nucleotide diversity was observed in LK (π = 0.0037), while PT and XM had the lowest (π = 0.0004).
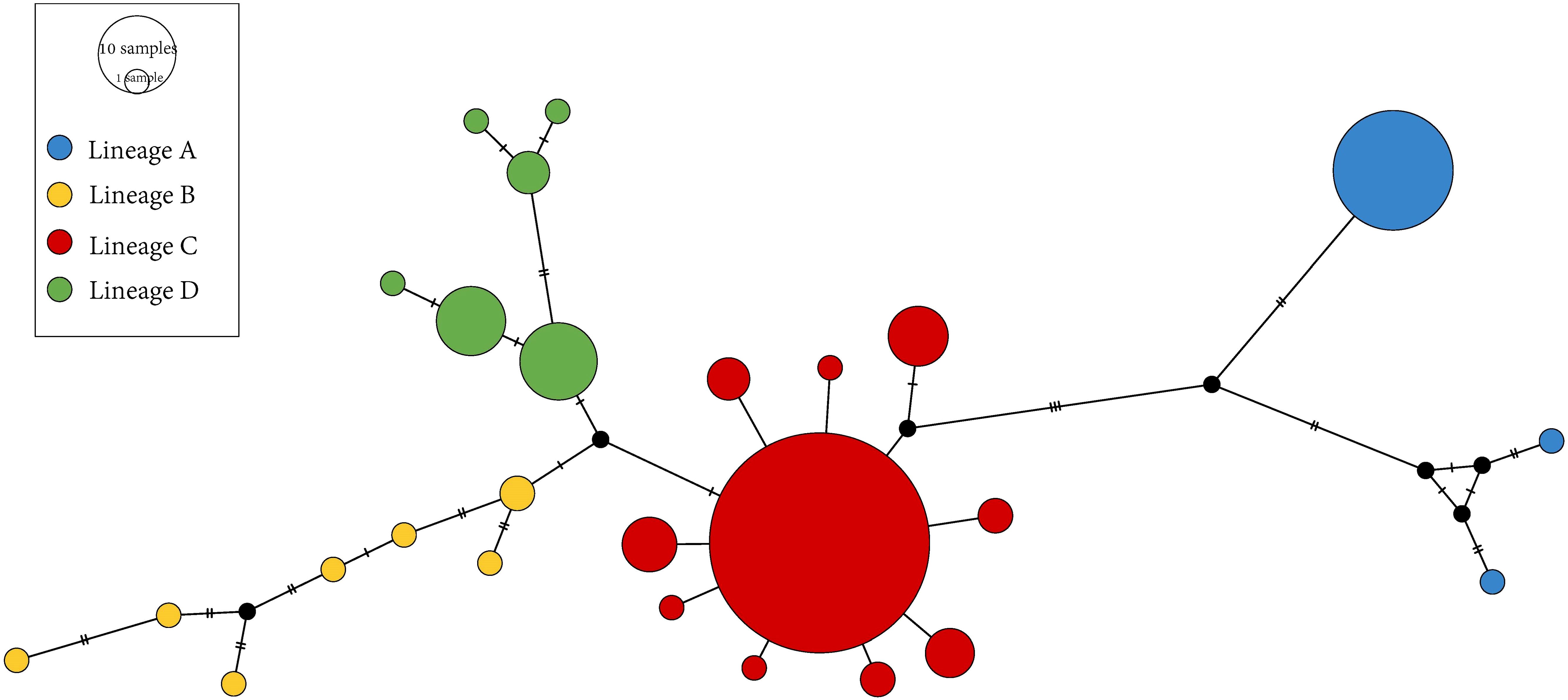
Figure 2. Haplotype network of Nerita yoldii. The size of the circles is proportional to the haplotype frequency, and the different colors of the haplotypes refer to their lineages.
3.2 Phylogenetic structure
According to the SAMOVA, FCT was maximized (FCT = 0.437, P= 0.036) when all populations were divided into three groups: northern (NT, DYS, SS, NSD, and LK), central (PT and XM), and southern (SZ and ZJ). AMOVA indicated that 91.71% of the haplotype variation occurred within populations, while 7.13% occurred among groups and 1.16% among populations within groups (Table 2). Pairwise FST comparisons revealed significant differentiation, with SZ and ZJ exhibiting significant differentiation from the other populations (Table 3). However, the Mantel test indicated no significant relationship between the linearized FST values and geographical distance across the nine populations (Figure 3). The genetic differentiations among groups were all significant, with FST values between the northern and central group higher than those between the northern and southern group (Table 4).
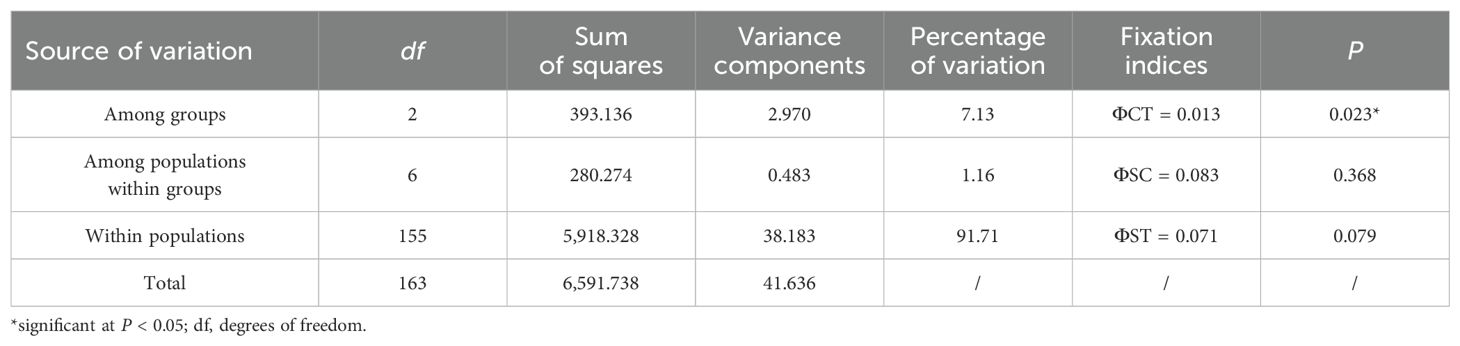
Table 2. Analysis of molecular variation results for samples of Nerita yoldii at different levels.
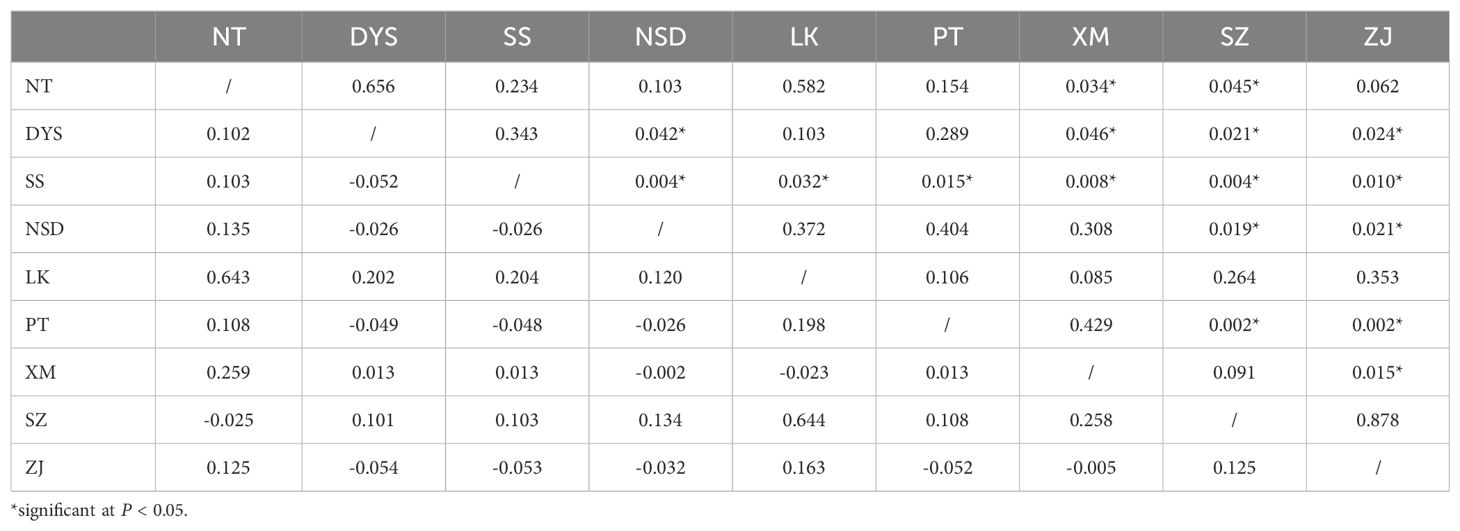
Table 3. Pairwise FST values (below diagonal)) among Nerita yoldii populations and associated P-values (above diagonal).
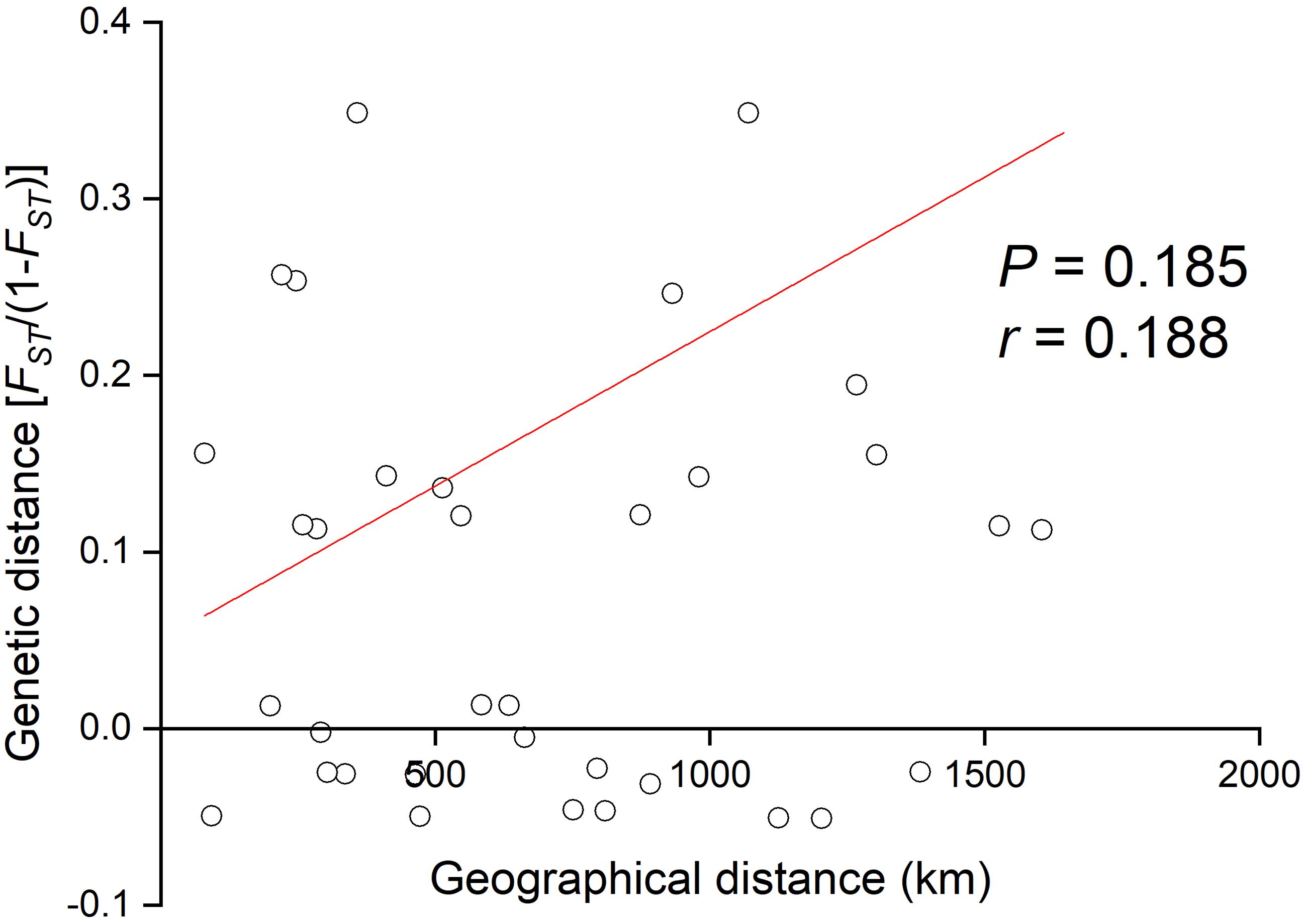
Figure 3. Isolation by distance plot for all Nerita yoldii populations.
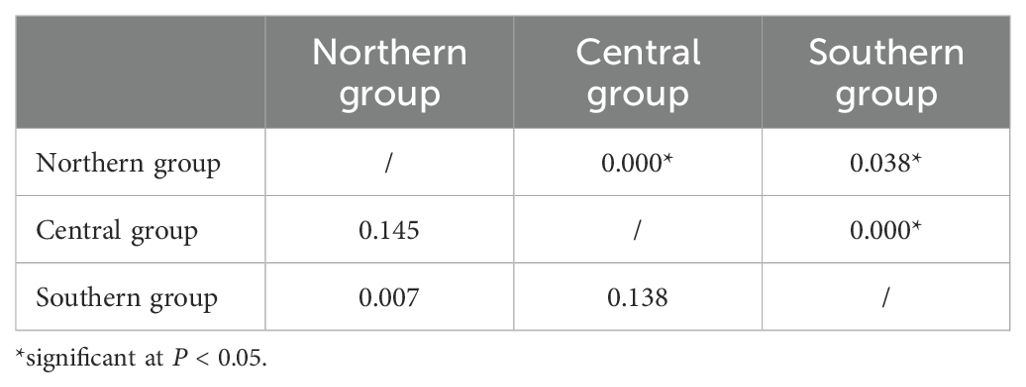
Table 4. Pairwise FST values (below diagonal)) among Nerita yoldii groups and associated P-values (above diagonal).
3.3 Historical demography
The mismatch distribution and neutral tests of the COI gene were used to detect population expansion. Both Tajima’s D and Fu’s FS in the lineage C were negative and statistically significant, indicating demographic expansion. Only the mismatch distribution of lineage C was a unimodal Poisson distribution, confirming demographic expansion in its history. For the other three lineages, there were no explicit signals of population expansion, as neither neutral tests were not significant or the observed mismatch distribution did not identify unimodal (Table 5; Supplementary Figure S4). Reconstruction of population sizes over time indicated a historical population expansion in lineage C, whereas the other three lineages remained relatively stable (Figure 4). Demographic expansion of the lineageC occurred during the late Pleistocene of the Quaternary period with expansion time of 0.038 Mya. The expansion time predate the last glacial maximum (LGM), which occurred approximately 0.020 Mya. Notably, the expansion time occurred during a period characterized by dramatic sea level fluctuations in the late Pleistocene. Approximate Bayesian computation (ABC) analysis supported scenario 1 as the most probable scenario, with posterior probabilities of 0.250 and 0.260 for the direct estimate approach and logistic regression approach, respectively (Supplementary Figure S2). Model testing analysis demonstrated that summary statistics derived from observed data fell within the range of simulated datasets from the posterior predictive distribution based on scenario 1 (Supplementary Figure S3), indicating robust model performance. Scenario 1 posits that ancient populations diverged into lineages B and C at time t3, and lineage A and D was originated from lineage C at time t2 and t1, respectively. Median divergence time were estimated at 73,700 (10,500-324,000), 155,000 (68,600-435,000), and 255,000 (208,000-493,000) generations for t1, t2, and t3, respectively. Assuming a generation time of 3 years for N. yoldii (Nakano and Nagoshi, 1980), these divergence time correspond to approximately 0.221 (0.032-0.927) Mya, 0.465 (0.206-1.305) Mya, and 0.765 (0.206-1.305) Mya for t1, t2, and t3, respectively.
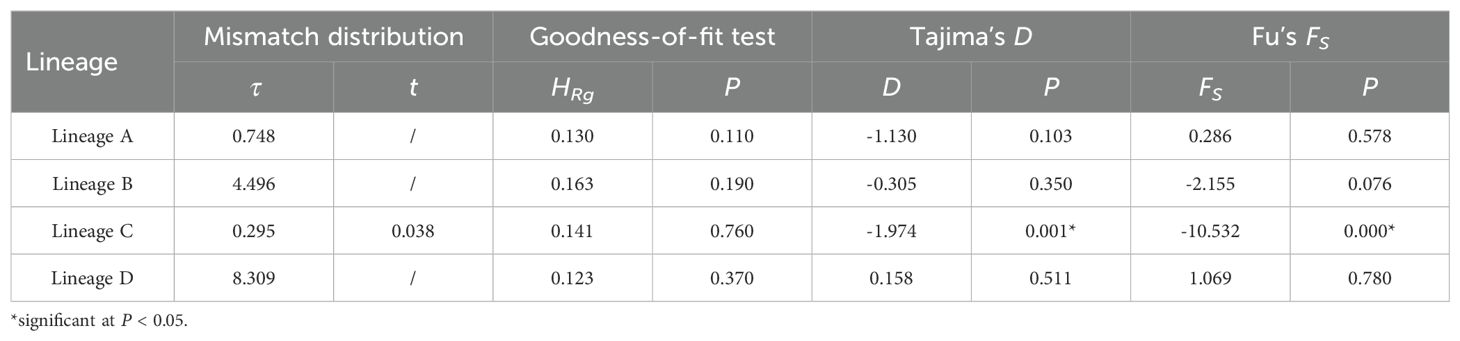
Table 5. Estimates of mismatch distribution, raggedness index, and neutral tests (Tajima’s D and Fu’s FS) for each Nerita yoldii lineage.
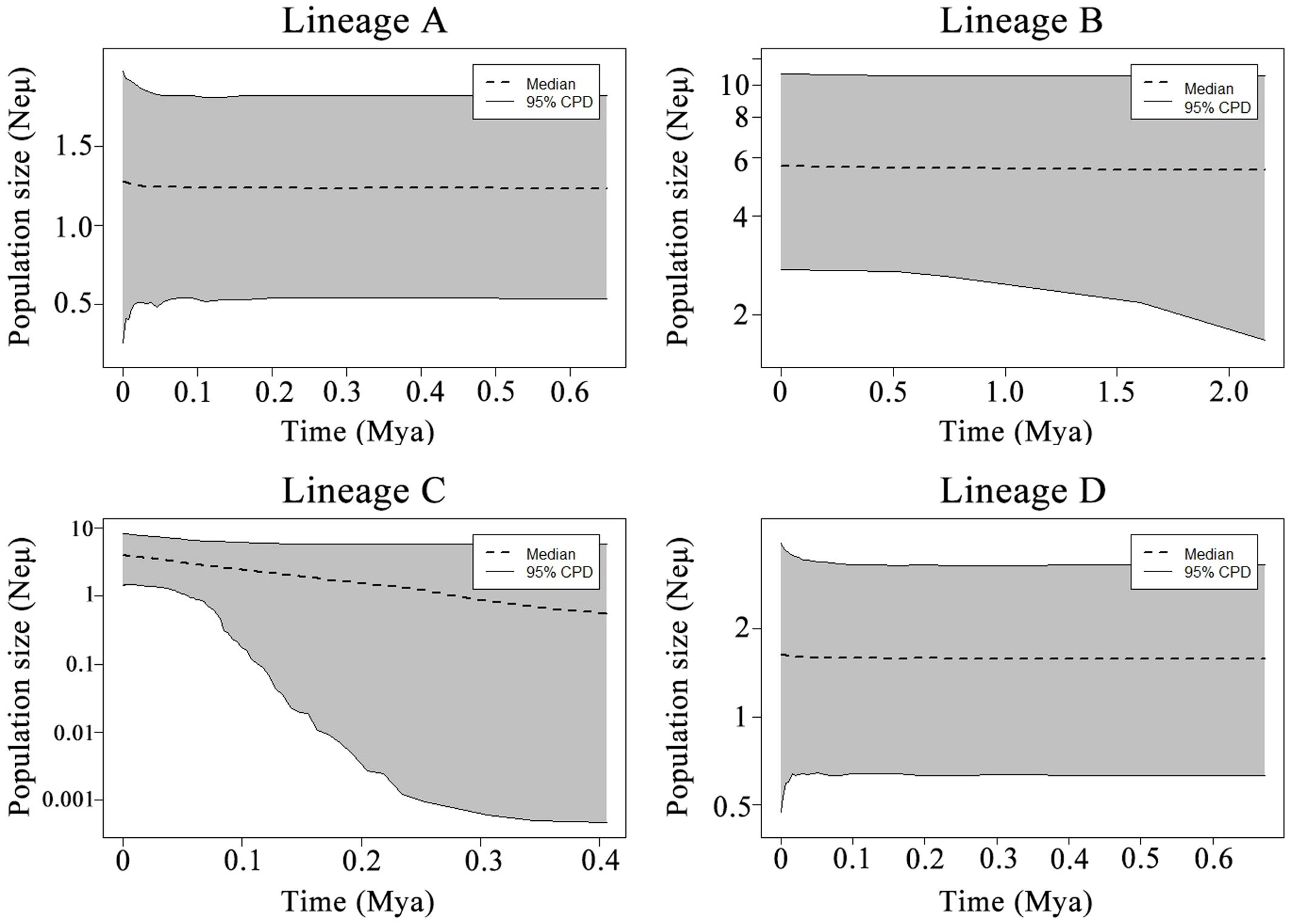
Figure 4. Demographic history of the four lineages of Nerita yoldii estimated using extended Bayesian skyline plots.
4 Discussion
4.1 Different levels of genetic diversity and possible explanations
The haplotype diversity and nucleotide diversity of N. yoldii from the NWP region were comparatively lower than those of N. scabricosta (h = 0.91, π = 0.005) and N. funiculata (h = 0.99, π = 0.008) from the temperate and subtropical East Pacific regions, respectively (Frey and Vermeij, 2008). N. yoldii originated approximately 10.3 Mya during the Miocene epoch (Frey and Vermeij, 2008), This relatively shorter evolutionary history compared to other Nerita species may have constrained the accumulation of genetic variation in N. yoldii. Another possible explanation is that following geographic isolation from the Pacific Ocean during the Pleistocene epoch, small founder populations may have emerged within each lineage of N. yoldii. Subsequent genetic drift within these isolated populations could have led to the reduction of intrapopulation genetic diversity (Hu et al., 2015). Rare variants in small populations are predicted to be eliminated by genetic drift. Recent population isolation and fragmentation during the Pleistocene glacial age may have increased the role of genetic drift in COI variation in N. yoldii (Luo et al., 2012). The dispersal pattern of Nerita’s ancestors from tropical to temperate seas on a large geographical scale is uncommon among gastropods and typically results in low speciation and high extinction rates (Williams, 2007). Additionally, the frequent cycles of glacial and interglacial periods during the Quaternary period likely led to unstable population sizes and low genetic diversity in N. yoldii populations. Despite these factors, only 1.16% of the variation was found among the populations of N. yoldii in this study (Table 2), suggesting limited genetic differentiation among the populations of this species.
The distribution of COI haplotypes of N. yoldii exhibited a clear geographical pattern (Figure 1B), with significant differences among the northern, central and southern groups. Studies of larval diffusion simulations of N. yoldii along the coast of Jiangsu Province have revealed that larvae dispersed as far north as the Jiaodong Peninsula and as far south as the Yangtze River Estuary (Wang et al., 2020). However, there was little gene exchange between the northern and southern groups likely because of significant distances and temperature differences.
4.2 Historical demography and post-glaciation dispersal
Mismatch analysis and neutral tests suggested that the lineage C of N. yoldii in China underwent expansion event. The expansion of the lineage C is estimated to have occurred approximately 0.038 Mya. The population expansion observed in our study occurred prior to the LGM. Traditionally, the LGM has been considered a pivotal factor driving the evolution and genetic structure of marine organisms (Hewitt, 2004). However, recent studies have highlighted the profound impact of Pleistocene glacial-interglacial cycles, including dramatic sea level fluctuations occurring 30-450 thousand years ago, which predate the LGM (Waelbroeck et al., 2002). These cycles have been shown to have substantial influences on population expansions in several species within the Northwestern Pacific (NWP) region (Ni et al., 2014). Despite the absence of a definitive explanation, substantial evidence of pre-LGM expansion has been documented in various marine realms, including the NWP (Marko et al., 2010), collectively emphasizing the importance of pre-LGM events in shaping the demographic trajectories of marine populations. During the Pleistocene glacial period, sea levels fluctuated due to the melting of glaciers, resulting in significant changes in coastal habitats (Wang, 1999). As a result, some marine species appear to have undergone dramatic demographic size changes during the Pleistocene when the sea level fluctuated (Ni et al., 2014). Intertidal species, such as N. yoldii likely experienced demographic expansion and rapidly occupied new habitats as the sea levels rose.
Our data suggested that the lineage divergence of N. yoldii in the NWP likely resulted from recurrent sea area isolation and connection during the mid to late Pleistocene. According to approximate Bayesian Computation (ABC), lineage A and D originated approximately 0.221 and 0.451 Mya, highlighting the significant influence of sea level changes. Lineage A and D are believed to have originated from lineage C during periods of low sea levels, which could have caused isolation between lineages (Waelbroeck et al., 2002). Therefore, the recurrent separations of these marginal seas during glacial-interglacial oscillations are considered significant drivers that influence the abundance and genetic structure of marine taxa (Hu et al., 2011; Cheang et al., 2012; Ni et al., 2014). The glacial period led to a decrease in sea level, causing the marginal sea of China to shrink to approximately 66% of its present size (Waelbroeck et al., 2002). Even when sea levels were low, there were still refugia in the NWP (Lee et al., 2012), in addition to the Okinawa Trough. The ancestors of lineages A and D were likely separated from lineage C during these periods of sea-level decline. These refugia provided suitable environments but restricted their dispersal, leading to relatively stable historical population sizes for lineages A and D compared to the more widespread lineage C. As a result, neither the mismatch distribution nor the extended Bayesian skyline plots indicated demographic expansion in the histories of lineages A and D. Various geological formations, such as lowland plains along the northwest coast of the Pacific Ocean and mountain ranges, including the Changshan Mountains of Vietnam and the Middle Cordillera Mountains of the Philippines, serve as barriers to coastal dispersal. Moreover, the South China Sea became largely landlocked to the south, west, and north, with its east coast blocked by the Sunda Isthmus (Wang, 1999). The only connection to the Pacific Ocean is through the Bashi Channel between Taiwan and the Philippines. These geographical features likely contributed to the isolation of populations in marginal seas from those in the open ocean, leading to genetic differentiation and historical expansion events following rising sea levels.
4.3 Driving factors of phylogeographical patterns
Previous studies have suggested that artificial structures can facilitate the invasion and dispersal of species in lithofacies intertidal zones (Dong et al., 2012; Adams et al., 2014). The haplotype diversity observed in the three populations from the northern group (NT, DYS, and SS) was low. This low diversity can be attributed to several factors including the geographical location of these populations and recent environmental changes. The Yangtze River Estuary, where these populations were sampled, has traditionally been regarded as the northern boundary of the distribution of N. yoldii (Wang et al., 2018). The haplotype diversity of these marginal populations was relatively low compared with that of populations in other regions. Climate change and the construction of artificial hard-bottom structures north of the Yangtze River Estuary have contributed to the expansion of the northern boundary of the distribution of N. yoldii (Wang et al., 2020). These changes may have affected the genetic diversity of populations in this region. Lineage B is predominantly distributed in the South China Sea. Analyses including mismatch distribution and extended Bayesian skyline plots indicate that its historical population size remained relatively stable. The South China Sea, being larger in area compared to the East China Sea, and maintaining connectivity with the Pacific Ocean during Pleistocene sea level decreases due to its deep waters (Wang, 1999), experienced lesser impact from Pleistocene sea level fluctuations. Consequently, ecosystems in the South China Sea were less affected by these fluctuations. The unique haplotype (Hap 23) observed in the NT population from the north of the Yangtze River Estuary suggests that this population has undergone in situ differentiation and is genetically distinct from other populations.
The impacts of climate change, including factors such as rainfall, drought, floods, and typhoons, as well as changes in seabed substrate hydrodynamics and water depth, can significantly influence species dispersal at various spatial scales (Pyron and Covich, 2003). Environmental changes can affect population reproduction, spawning, and larval dispersal dynamics (Lord et al., 2012). Specifically, changes in sea surface salinity, temperature, and other hydrological factors caused by diluted water in the Yangtze Estuary may act as potential barriers to larval spread and gene exchange among species along the estuary (Cheang et al., 2008; Ni et al., 2017). However, the findings of this study on the geographical distribution of Nerita indicate that the low-salinity area of the Yangtze River Estuary may not necessarily hinder the larval diffusion of Nerita. Interestingly, the lack of a hard substrate on the north coast of the Yangtze River and the temperature difference between the Yellow Sea and the East China Sea may be important factors limiting the further spread of N. yoldii to the north (Liu, 2013; Wang et al., 2018). As offshore water temperatures continue to rise in China, the population of N. yoldii will continue to extend northward, potentially forming a new distribution pattern. However, it is important to note that this study included only one mitochondrial gene sampled from nine populations. Therefore, the genetic diversity and faunal composition of Nerita populations distributed along the extensive coastline of the country may not be fully captured. Future studies should incorporate a larger number of samples and additional genetic markers, such as microsatellites and SNPs, to provide a more comprehensive understanding of the phylogenetic relationships among the N. yoldii lineages.
5 Conclusions
Our results identified four distinct lineages (A, B, C, and D) of N.yoldii in the Northwestern Pacific (NWP) region. Lineages A and D are believed to have originated from lineage C during the late Pleistocene, a period marked by recurrent separations of the marginal seas. We identified a demographic expansion event of lineage C during the Pleistocene epoch, highlighting the dynamic history of N. yoldii populations in response to environmental changes. The nine populations could be divided into three distinct groups based on geographic differentiations in genetic diversity distribution patterns, shaped by both historical and ecological factors. Our findings support the historical glaciation hypothesis, which suggests that past climatic events played a significant role in shaping the present-day phylogeographical structure of N. yoldii in the NWP region. Over the past century, human activities, such as the construction of artificial rock facies along the coast of China, coupled with global-warming-induced reductions in water temperature differences between the Yellow Sea and East China Sea, may have influenced the dispersal patterns of N. yoldii larvae. This has led to the northward spread of N. yoldii across the Yangtze River Estuary, resulting in the expansion of its northern distribution boundary. Overall, our study contributes to advancing our understanding of the historical processes influencing coastal species such as N. yoldii and sheds light on the complex interactions among the various factors driving biodiversity in the NWP region.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, PP436912-PP437075.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
YT: Writing – original draft, Investigation, Software. RZ: Investigation, Writing – original draft. QL: Software, Writing – original draft. LS: Conceptualization, Writing – review & editing. YL: Conceptualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Key Research and Development Program of China (Grant No. 2021YFC3101702), the Scientific Research Fund of the Second Institute of Oceanography, MNR (Grant No. JG2312), and the Project of Long-term Observation and Research Plan in the Changjiang Estuary and Adjacent East China Sea (LORCE) (Grant No. SZ2001).
Acknowledgments
We thank Hanren Liu for helping us to collect and sequence the samples.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2024.1396411/full#supplementary-material.
References
Adams T. P., Miller R. G., Aleynik D., Burrows M. T. (2014). Offshore marine renewable energy devices as stepping stones across biogeographical boundaries. J. Appl. Ecol. 51, 330–338. doi: 10.1111/1365-2664.12207
Avise J. C. (2004). What is the field of biogeography, and where is it going? Taxon 53, 893–898. doi: 10.2307/4135555
Bohonak A. J. (2002). IBD (isolation by distance): A program for analyses of isolation by distance. J. Hered. 93, 153–154. doi: 10.1093/jhered/93.2.153
Cabrera A. A., Palsbøll P. J. (2017). Inferring past demographic changes from contemporary genetic data: A simulation-based evaluation of the ABC methods implemented in DIYABC. Mol. Ecol. Resour. 17, e94–e110. doi: 10.1111/1755-0998.12696
Cheang C. C., Chu K. H., Ang P. O. (2008). Morphological and genetic variation in the populations of Sargassum hemiphyllum (Phaeophyceae) in the northwestern Pacific. J. Phycol. 44, 855–865. doi: 10.1111/j.1529-8817.2008.00532.x
Cheang C. C., Tsang L. M., Ng W. C., Williams G. A., Chu K. H., Chan B. K. K. (2012). Phylogeography of the cold-water barnacle Chthamalus challengeri in the north-western Pacific: Effect of past population expansion and contemporary gene flow. J. Biogeogr 39, 1819–1835. doi: 10.1111/j.1365-2699.2012.02742.x
Clement M., Posada D., Crandall K. A. (2000). TCS: A computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659. doi: 10.1046/j.1365-294X.2000.01020.x
Cornuet J. M., Santos F., Beaumont M. A., Robert C. P., Marin J. M., Balding D. J., et al. (2008). Inferring population history with DIY ABC: A user-friendly approach to approximate Bayesian computation. Bioinformatics 24, 2713–2719. doi: 10.1093/bioinformatics/btn514
Coyer J. A., Hoarau G., Stam W. T., Olsen J. L. (2004). Geographically specific heteroplasmy of mitochondrial DNA in the seaweed, Fucus serratus (Heterokontophyta: Phaeophyceae, Fucales). Mol. Ecol. 13, 1323–1326. doi: 10.1111/j.1365-294X.2004.02128.x
Dong Y. W., Huang X. W., Wang W., Li Y., Wang J. (2016). The marine “great wall” of China: Local- and broad-scale ecological impacts of coastal infrastructure on intertidal macrobenthic communities. Divers. Distrib. 22, 731–744. doi: 10.1111/ddi.12443
Dong Y. W., Wang H. S., Han G. D., Ke C. H., Zhan X., Nakano T., et al. (2012). The impact of Yangtze river discharge, ocean currents and historical events on the biogeographic pattern of Cellana toreuma along the China coast. PloS One 7, e36178. doi: 10.1371/journal.pone.0036178
Drummond A. J., Rambaut A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 1–8. doi: 10.1186/1471-2148-7-214
Dupanloup I., Schneider S., Excoffier L. (2002). A simulated annealing approach to define the genetic structure of populations. Mol. Ecol. 11, 2571–2581. doi: 10.1046/j.1365-294X.2002.01650.x
Excoffier L., Lischer H. E. L. (2010). Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Excoffier L., Laval G., Schneider S. (2005). Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinforma. 1, 117693430500100. doi: 10.1177/117693430500100003
Excoffier L., Smouse P. E., Quattro J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 131, 479–491. doi: 10.1093/genetics/131.2.479
Folmer O., Black M., Hoeh W., Lutz R., Vrijenhoek R. (1994). DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299.
Frey M. A., Vermeij G. J. (2008). Molecular phylogenies and historical biogeography of a circumtropical group of gastropods (Genus: Nerita): Implications for regional diversity patterns in the marine tropics. Mol. Phylogenet. Evol. 48, 1067–1086. doi: 10.1016/j.ympev.2008.05.009
Fu Y. X., Li W. H. (1993). Statistical tests of neutrality of mutations. Genetics 133, 693–709. doi: 10.1093/genetics/133.3.693
Guo X., Zhao D., Jung D., Li Q., Kong L. F., Ni G., et al. (2015). Phylogeography of the rock shell Thais clavigera (Mollusca): Evidence for long-distance dispersal in the northwestern Pacific. PloS One 10, 1–16. doi: 10.1371/journal.pone.0129715
Hall T. A. (1999). BioEdit: a user friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleid Acids Symp. Ser., 95–98. doi: 10.1021/bk-1999-0734.ch008
Heled J., Drummond A. J. (2008). Bayesian inference of population size history from multiple loci. BMC Evol. Biol. 8, 1–15. doi: 10.1186/1471-2148-8-289
Hewitt G. M. (2004). Genetic consequences of climatic oscillations in the Quaternary. Philos. Trans. R. Soc B Biol. Sci. 359, 183–195. doi: 10.1098/rstb.2003.1388
Hu L.s., Dong Y.w. (2022). Northward shift of a biogeographical barrier on China’s coast. Divers. Distrib. 28, 318–330. doi: 10.1111/ddi.13463
Hu Z. M., Li J. J., Sun Z. M., Oak J. H., Zhang J., Fresia P., et al. (2015). Phylogeographic structure and deep lineage diversification of the red alga Chondrus ocellatus Holmes in the Northwest Pacific. Mol. Ecol. 24, 5020–5033. doi: 10.1111/mec.13367
Hu Z. M., Uwai S., Yu S. H., Komatsu T., Ajisaka T., Duan D. L. (2011). Phylogeographic heterogeneity of the brown macroalga Sargassum horneri (Fucaceae) in the northwestern Pacific in relation to late Pleistocene glaciation and tectonic configurations. Mol. Ecol. 20, 3894–3909. doi: 10.1111/j.1365-294X.2011.05220.x
Huelsenbeck J. P., Ronquist F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Jiang S., Li Z., Li J., Xu K., Ye Y. (2024). Analysis of Genetic Diversity and Structure of Eight Populations of Nerita yoldii along the Coast of China Based on Mitochondrial COI Gene. Animals 14, 4510–4526. doi: 10.3390/ani14050718
Kerswell A. P. (2006). Global biodiversity patterns of benthic marine algae. Ecology 87, 2479–2488. doi: 10.1890/0012-9658(2006)87[2479:GBPOBM]2.0.CO;2
Lee K. M., Yang E. C., Coyer J. A., Zuccarello G. C., Wang W. L., Choi C. G., et al. (2012). Phylogeography of the seaweed Ishige okamurae (Phaeophyceae): Evidence for glacial refugia in the northwest Pacific region. Mar. Biol. 159, 1021–1028. doi: 10.1007/s00227-012-1882-0
Liu J. Y. (2013). Status of marine biodiversity of the China seas. PloS One 8, e50719. doi: 10.1371/journal.pone.0050719
Lord C., Lorion J., Dettai A., Watanabe S., Tsukamoto K., Cruaud C., et al. (2012). From endemism to widespread distribution: Phylogeography of three amphidromous Sicyopterus species (Teleostei: Gobioidei: Sicydiinae). Mar. Ecol. Prog. Ser. 455, 269–285. doi: 10.3354/meps09617
Luo M. F., Pan H. J., Liu Z. J., Li M. (2012). Balancing selection and genetic drift at major histocompatibility complex class II genes in isolated populations of golden snub-nosed monkey (Rhinopithecus roxellana). BMC Evol. Biol. 12, 1–5. doi: 10.1186/1471-2148-12-207
Marko P. B., Hoffman J. M., Emme S. A., McGovern T. M., Keever C. C., Nicole Cox L. (2010). The “expansion-Contraction” model of Pleistocene biogeography: Rocky shores suffer a sea change? Mol. Ecol. 19, 146–169. doi: 10.1111/j.1365-294X.2009.04417.x
Nakano D., Nagoshi M. (1980). Growth and age structure of Heminerita japonica (Dunker), population of the Shima Peninsula, Japan (Prosobranchia). Japasese J. Malacol. 39, 101–108. doi: 10.18941/venusjjm.39.2_101
Ni G., Kern E., Dong Y. W., Li Q., Park J. K. (2017). More than meets the eye: The barrier effect of the Yangtze River outflow. Mol. Ecol. 26, 4591–4602. doi: 10.1111/mec.14235
Ni G., Li Q., Kong L., Yu H. (2014). Comparative phylogeography in marginal seas of the northwestern Pacific. Mol. Ecol. 23, 534–548. doi: 10.1111/mec.12620
Novembre J., Johnson T., Bryc K., Kutalik Z., Boyko A. R., Auton A., et al. (2008). Genes mirror geography within Europe. Nature 456, 98–101. doi: 10.1038/nature07331
Osozawa S., Shinjo R., Armid A., Watanabe Y., Horiguchi T., Wakabayashi J. (2012). Palaeogeographic reconstruction of the 1.55 Ma synchronous isolation of the Ryukyu Islands, Japan, and Taiwan and inflow of the Kuroshio warm current. Int. Geol. Rev. 54, 1369–1388. doi: 10.1080/00206814.2011.639954
Provan J., Bennett K. D. (2008). Phylogeographic insights into cryptic glacial refugia. Trends Ecol. Evol. 23, 564–571. doi: 10.1016/j.tree.2008.06.010
Pyron M., Covich A. P. (2003). Migration patterns, densities, and growth of Neritina punctulata snails in Rio Espiritu Santo and Rio Mameyes, Northeastern Puerto Rico. Caribb. J. Sci. 39, 338–347. doi: 10.1016/S0006-3207(02)00136-2
Quan W., An C., Ma C., Huang H., Chen W., Wang Y., et al. (2012). Biodiversity and community structure of benthic macroinvertebrates on the Xiaomiaohong oyster reef in Jiangsu province, China. Oceanol. Limnol. Sin. 43, 992–1000.
Rambaut A., Drummond A. J., Xie D., Baele G., Suchard M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Rogers A. R. (1995). Genetic evidence for a Pleistocene population explosion. Evol. (N. Y). 49, 608. doi: 10.2307/2410314
Slatkin M. (1993). Isolation by distance in equilibrium and non-eequilibrium populations. Evol. (N. Y). 47, 264–279. doi: 10.1111/j.1558-5646.1993.tb01215.x
Slatkin M. (1995). A measure of population subdivision based on microsatellite allele frequencies. Genetics 139, 457–462. doi: 10.1093/genetics/139.1.457
Taberlet P., Fumagalli L., Wust-Saucy A. G., Cosson J. F. (1998). Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol. 7, 453–464. doi: 10.1046/j.1365-294x.1998.00289.x
Tchang S., Tsi C., Zhang F., Ma S. (1963). A preliminary study of the demarcation of marine molluscan faunal regions of China and its adjacent waters. Oceanol. Limnol. Sin., 124–138.
Waelbroeck C., Labeyrie L., Michel E., Duplessy J. C., McManus J. F., Lambeck K., et al. (2002). Sea-level and deep water temperature changes derived from benthic foraminifera isotopic records. Quat. Sci. Rev. 21, 295–305. doi: 10.1016/S0277-3791(01)00101-9
Wang J., Cheng Z. Y., Dong Y. W. (2022). Demographic, physiological and genetic factors linked to the poleward range expansion of the snail Nerita yoldii along the shoreline of China. Mol. Ecol. 31, 4510–4526. doi: 10.1111/mec.16610
Wang J., Tsang L. M., Dong Y. W. (2015). Causations of phylogeographic barrier of some rocky shore species along the Chinese coastline. BMC Evol. Biol. 15, 1–15. doi: 10.1186/s12862-015-0387-0
Wang J., Yan H. Y., Cheng Z. Y., Huang X. W., Wang W., Ding M. W., et al. (2018). Recent northward range extension of Nerita yoldii (gastropoda: neritidae) on artificial rocky shores in China. J. Molluscan Stud. 84, 345–353. doi: 10.1093/mollus/eyy042
Wang P. (1999). Response of Western Pacific marginal seas to glacial cycles: Paleoceanographic and sedimentological features. Mar. Geol. 156, 5–39. doi: 10.1016/S0025-3227(98)00172-8
Wang W., Wang J., Choi F. M. P., Ding P., Li X. X., Han G. D., et al. (2020). Global warming and artificial shorelines reshape seashore biogeography. Glob. Ecol. Biogeogr. 29, 220–231. doi: 10.1111/geb.13019
Williams S. T. (2007). Origins and diversification of Indo-West Pacific marine fauna: Evolutionary history and biogeography of turban shells (Gastropoda, Turbinidae). Biol. J. Linn. Soc 92, 573–592. doi: 10.1111/j.1095-8312.2007.00854.x
Xu J., Chan T. Y., Tsang L. M., Chu K. H. (2009). Phylogeography of the mitten crab Eriocheir sensu stricto in East Asia: Pleistocene isolation, population expansion and secondary contact. Mol. Phylogenet. Evol. 52, 45–56. doi: 10.1016/j.ympev.2009.02.007
Zhang J., Yao J., Hu Z. M., Jueterbock A., Yotsukura N., Krupnova T. N., et al. (2019). Phylogeographic diversification and postglacial range dynamics shed light on the conservation of the kelp Saccharina japonica. Evol. Appl. 12, 791–803. doi: 10.1111/eva.12756
Zhang Q., Sun C. H., Huang J. L., Lao Y. L., Chang X. Y., Cao J. N. (2024). Genetic diversity of Hapalogenys analis in the northwest Pacific assessed using dd-RAD sequencing. Front. Ecol. Evol. 12. doi: 10.3389/fevo.2024.1345739
Keywords: Nerita yoldii, COI, Northwestern Pacific, phylogeography, biogeography
Citation: Tang Y, Zhang R, Liu Q, Shou L and Liao Y (2024) Phylogeographical analysis of Nerita yoldii revealed its geographical distribution pattern and drivers of population divergence in the Northwestern Pacific region. Front. Mar. Sci. 11:1396411. doi: 10.3389/fmars.2024.1396411
Received: 05 March 2024; Accepted: 05 November 2024;
Published: 22 November 2024.
Edited by:
Pilar A. Haye, Catholic University of the North, ChileReviewed by:
Hung-Du Lin, National Tainan First Senior High School, TaiwanDon Colgan, Australian Museum, Australia
Victor Piñeros, University of Guadalajara, Mexico
Copyright © 2024 Tang, Zhang, Liu, Shou and Liao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yibo Liao, bGlhb3liQHNpby5vcmcuY24=