 Yusi Zheng
Yusi Zheng Pei Wang1,2†
Pei Wang1,2† Dahui Yu
Dahui Yu Sen Zhao
Sen Zhao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 17 April 2024
Sec. Marine Fisheries, Aquaculture and Living Resources
Volume 11 - 2024 | https://doi.org/10.3389/fmars.2024.1362078
This article is part of the Research TopicBlue Foods Security and SustainabilityView all 15 articles
Background: The marine pearl culture industry is a key industry in the Beibu Gulf of China that achieves large-scale pearl production by artificial nucleus insertion in pearls. High-quality pearls can produced by xenotransplantation, but allotransplantation or xenotransplantation can lead to various immune responses, resulting in nucleus rejection or even the recipient shell death and thereby causing significant losses in pearl production.
Methods: Few studies have investigated the immune defenses of oysters related to allografts and xenografts. In this study, transcriptomic comparisons of allograft and xenograft Pinctada fucata haemocytes were conducted to identify genes associated with immune responses.
Results: A total of 33.11 Gbp of clean reads were generated from five P. fucata haemocytes. De-novo assembly of quality-filtered reads generated a total of 26,526 unigenes, with 22,002 known genes and 4,524 predicted novel genes. In addition, 34,904 novel transcripts were detected, with 15,620 novel alternative splicing isoforms of known protein coding genes and 4,605 belonging to novel protein coding genes, with the remaining 14,679 comprising long non-coding RNA transcripts. Functional enrichment analysis of immune-related differentially expressed genes (DEGs) using the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases revealed 36–44 significantly enriched GO terms and 34 significantly enriched KEGG pathways. Ten DEGs were subjected to validation of expression levels using RT-q PCR analysis, revealing generally consistent values as the high-throughput sequencing data.
Conclusion: Oyster haemocytes were comprehensively evaluated in this study using transcriptomic comparisons and with a focus on immune-related functional genes and pathways. The results revealed numerous DEGs related to immune function that can serve as the basis for subsequent immune response analysis of allotransplantation and xenotransplantation.
The pearl oyster Pinctada fucata martensii is an economically important bivalve that is farmed for marine pearl production globally, with most Chinese marine pearls produced from this species (Zhang et al., 2022). The production of seawater pearls in China is primarily based on artificial cultivation. Specifically, nucleus transplantation is conducted with a mantle graft originating from a donor oyster, along with a shell bead nucleus, into the “pearl sac” of a recipient oyster. The quality of artificial beads consequently depends on the state of the recipient oyster and the choice of mantle graft. To achieve optimal results and produce smooth and round pearls after artificial nucleus insertion, the metabolism of the recipient oyster must remain in a low state, and the gonad must be generated without gametes (Arnaud-Haond et al., 2007; Fang et al., 2008; Inoue et al., 2010; Southgate and Lucas, 2011; Wei et al., 2017; Wang et al., 2024). Moreover, the small mantle graft of the recipient oyster is the primary factor that determines the formation of the pearl sac (Wei et al., 2017), with some studies detecting DNA from the donor oyster mantle in the pearl sac (Wang et al., 2024). Transplantation can either include allogeneic or xenogeneic insertions. Allogenic insertions are primarily conducted in China and produces small pearls with poor coloration, and average economic value. Previous studies demonstrated that xenografts did not significantly affect the formation of pearl sacs and subsequent nucleus retention but did influence pearl color, complexion, shape, nacre deposition, and nacre weight (McGinty et al., 2010). Consequently, xenografts hold potential in enhancing pearl quality attributes such as size, underscoring the importance of donor oysters in achieving desirable pearl growth, color, and surface characteristics (Fukushima et al., 2014). Thus, the study demonstrated the potential for xenografts to improve pearl quality characteristics like pearl size, while emphasizing a role of donor oysters in achieving ideal pearl growth, color, and surface complexion. However, the low output from such methods requires further optimization and improvement.
Allograft and xenograft transplantation have been evaluated in China in recent years. For example, transcriptomic sequencing has been used to evaluate immune system response mechanisms after xenotransplantation and allotransplantation (Wei et al., 2017). These methods have also been applied to investigations of freshwater pearl oysters (Zhang et al., 2016). RNA-seq based transcriptomic analysis is a highly effective approach for investigating the immune responses at the genomic and transcriptomic levels. Such analyses have been widely used in shellfish life science research, including in the investigation of Perumytilus purpuratus (Briones et al., 2018), Patinopecten yessoensis (Zhou et al., 2019), Hyriopsis cumingii (Zhang et al., 2016), P. fucata (Lu et al., 2022), Mytilus galloprovincialis (Dong et al., 2022), and Argopecten irradians (Dong et al., 2022). Numerous immune-related genes have been discovered and intensively studied in bivalves, including the toll-like receptor (Wei et al., 2017), C type lectin (He et al., 2020), HSP70 (Wei et al., 2017), and Interleukin (IL)-17 (Zhang et al., 2016).
Here, a transcriptomic analysis of P. fucata haemocytes after transplantation was conducted to better understand the molecular mechanisms related to P. fucata immune responses after allotransplantation and xenotransplantation. The resulting analyses providing insights into the immune defense mechanisms associated with recipient oyster transplants, help promote the mitigation of host oyster immune rejection of grafts, and inform the improved efficiency of pearl production.
Healthy 1.5-year-old P. fucata (shell lengths: 4.5-5.5 cm, weights: 42-58 g) were obtained from the pearl oyster culture station of the South China Sea Fisheries Research Institute in Xincun Village, Hainan Province, China. Donor P. maxima (shell lengths: 8-15 cm, weights: 100-200 g) and Pteria penguin (shell lengths: 10-15 cm, weights: 200-300 g) were selected from wild populations in Hainan. Oysters were cultured in oxygenated seawater at 25°C and were fed Chlorella vulgaris. After temporary breeding for a week, professional technicians performed nucleus transplantation. The donors (P. fucata, P. maxima, and P. penguin) were first sacrificed and strips of mantle tissue were excised from the midventral regions of the mantle and then thoroughly cleaned in sterile seawater. Samples were then sectioned into small grafts (about 3×3 mm) and inserted into pearl sacs. Transplanted host oysters were placed in temporary culture ponds to breed, with four experimental groups and one control group established for analyses. The experimental groups were respectively implanted with P. fucata, P. maxima, and P. penguin mantle slices in the P. fucata group with nucleus insertion. Another group was implanted with P. fucata mantle slices without nucleus insertion. A control group was established comprising P. fucata without any implants. Host oysters were referred to as Pf_Pf, Pf_Pm, and Pf_Pp, respectively, after mantle allograft and xenograft surgery. The shellfish group that had only undergone surgical treatment without nucleus insertion was referred to as Mock, while the untreated control group was referred to as CK.
At 192 hours post-transplantation, whole hemolymph samples were collected from the adductor muscles using 1 mL syringes (1 mL withdrawn per oyster and samples from 10 oysters were pooled together). Hemocytes were then harvested via centrifugation at 4,000 g for 10 minutes, the supernatant was discarded and 1 mL of Trizol (TransGen Biotech, China) was added, followed by mixing with the hemocytes. The samples were then immediately frozen in liquid nitrogen and stored at -70°C.
Total RNA was extracted from the experimental and control group samples following the manufacturer’s instructions. RNA concentration and integrity of the RNA were determined using a Lunatic high-throughput microfluidic spectrophotometer (Unchained Labs, USA). cDNA libraries were then sequenced using the BGISEQ-500RS RNA-Seq platform.
Library construction and RNA-Seq were conducted at BGI Genomics (Shenzhen, China) following typical protocols. To ensure the reliability of the analyses, the resulting raw data were filtered using the SOAPnuke software (Chen et al., 2018) program to remove data containing adaptors, poly-N’s, and reads of low quality. The resulting clean reads were used in the subsequent analyses. After comparing the transcriptomic data against the reference genome, transcribed regions not originally annotated were observed to identify potentially novel genes for the species. The new genes were then compared against several databases, with the confidence in annotations following order of: Nt <KOG <Pfam <Swiss Prot <KO <GO <Nr (Ai et al., 2016).
To determine physiological differences of P. fucata with different mantle inserts, the gene expression levels of each sample were calculated using the RSEM (Reads Per Kilobase Million) and FPKM (Fragments Per Kilobase Million) methods, followed by correction for sequencing depth. Unigenes meeting the criteria of p < 0.005 and |log2(fold change)| > 2 were considered differentially expressed genes (DEGs). DEGs were selected for further analyzed using the hypergeometric distribution principle and subjected to GO and KEGG enrichment analysis. GO enrichment was used to primarily analyze the functional classification of major biological features related to DEGs. In addition, KEGG pathway enrichment was performed to identify the primary biochemical metabolic and signal transduction pathways that DEGs were involved in. The statistical threshold for GO or KEGG enrichment was set at p < 0.05.
To evaluate the accuracy of the transcriptomic analyses, 10 DEGs were selected for real-time quantitative PCR (RT-qPCR) analysis. The sequences of the10 genes were used for primer design (Table 1) using 18S rRNA genes as internal reference genes. The RNA samples used for RT-qPCR amplifications were the same as those used for constructing the RNA-Seq libraries described above. Total RNA was reverse transcribed using the EasyScript® All-in-One First-Strand cDNA Synthesis SuperMix for qPCR (TransGen Biotech China). Each reaction contained 1 μL reverse and forward primers each, 10 μL of SYBR Green Mix (TransGen, Beijing, China), and 2 μL of 1:8 diluted cDNA, with RNase-free water added to achieve a final reaction volume of 20 μL. The qPCR cycling program including 1 cycle of 10 min at 95°C, followed by 40 cycles of 10 s at 95°C, 15 s at 55°C, and 15 s at 72°C. The mRNA relative expression levels were calculated using the 2^−ΔΔCt method, and each experiment was performed in triplicate. Data were analyzed using the Least Significant Difference (LSD) method in the SPSS software program (version 22), with statistically significant differences identified at p < 0.05.

Table 1 RT-qPCR primer sequence information.
The transcriptomes from four experimental and control samples yielded a total of 33.11 Gbp of clean data (Table 2). The clean reads data can be downloaded from NCBI under accession code PRJNA1068836. Over 88% of each sample exhibited >Q30 quality scores. The GC contents for the CK, Mock, Pf_Pf, Pf_Pm, and Pf_Pp libraries ranged from 39.43% to 39.88%. In addition, between 64.03 to 66.09% of the clean reads from each group aligned to the P. fucata genome (Table 2). Further, between 37.16% and 39.6% of the reads for the four experimental and control samples uniquely mapped to the genome (Table 2).
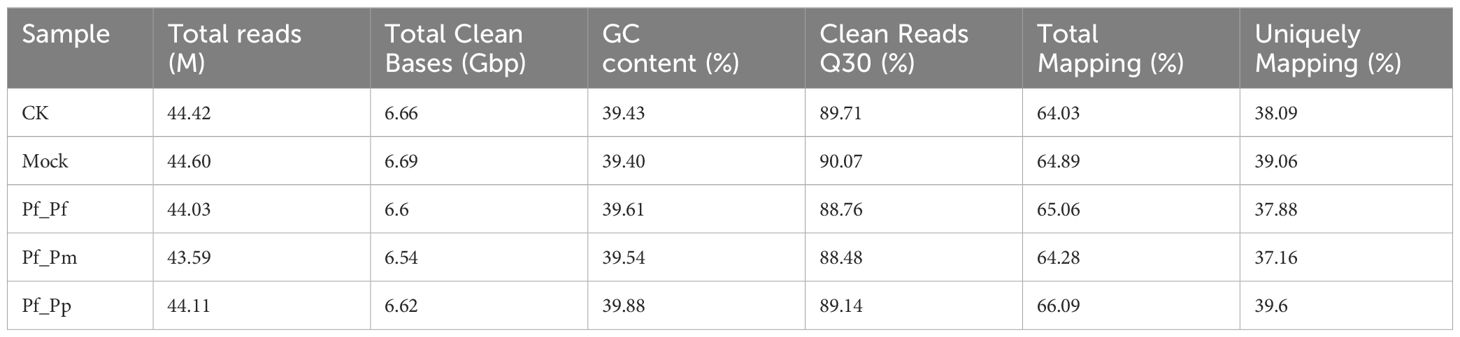
Table 2 Transcriptomic sequencing data statistics.
In addition, A total of 26,526 unigenes were identified among the expressed genes, with 22,002 known unigenes and 4,524 predicted novel genes. A total of 34,904 novel transcripts were also detected, of which 15,620 belonged to novel alternative splicing isoforms of known protein coding genes and 4,605 belonged to novel protein coding genes, with the remaining 14,679 comprising long non-coding RNA transcripts.
The CK vs Pf_Pm comparison comprised the largest number of differential genes, with 2,786 DEGs, of which 886 were up-regulated and 1,900 were down-regulated (Figure 1, Table 3). The DEGs were annotation based on GO classifications, revealing functions primarily related to biological processes, cellular components, and molecular functions, with each group comprising 36 to 44 sub-categories (Figure 2).
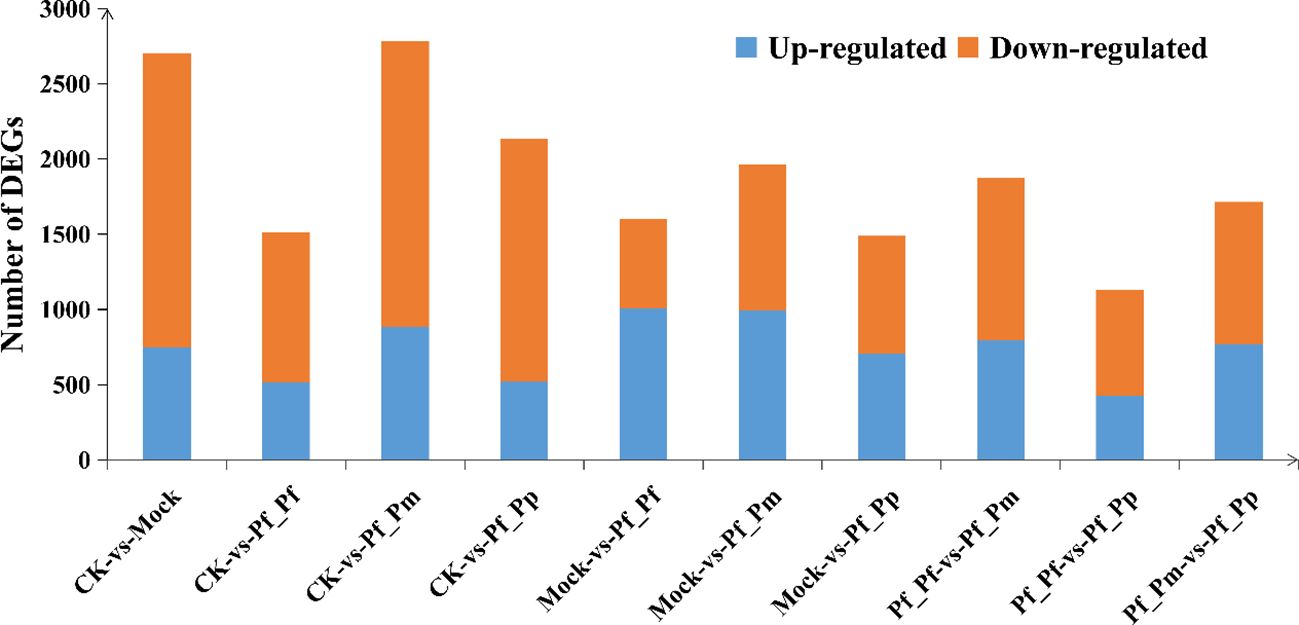
Figure 1 Up- and down-regulated DEGs of P. fucata in comparisons between post-transgraft groups.
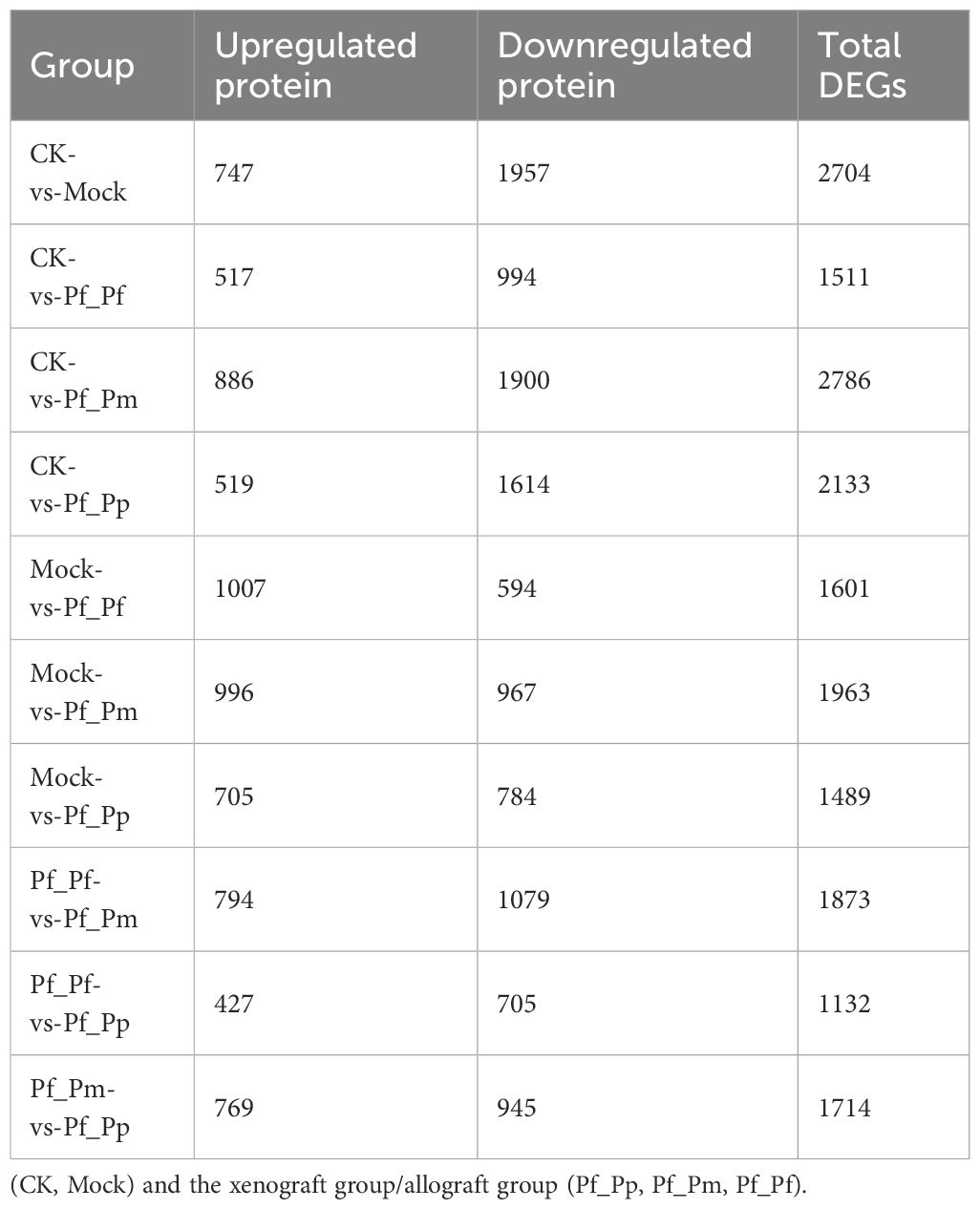
Table 3 Comparison of the number of DEGs in the serum between the control group.
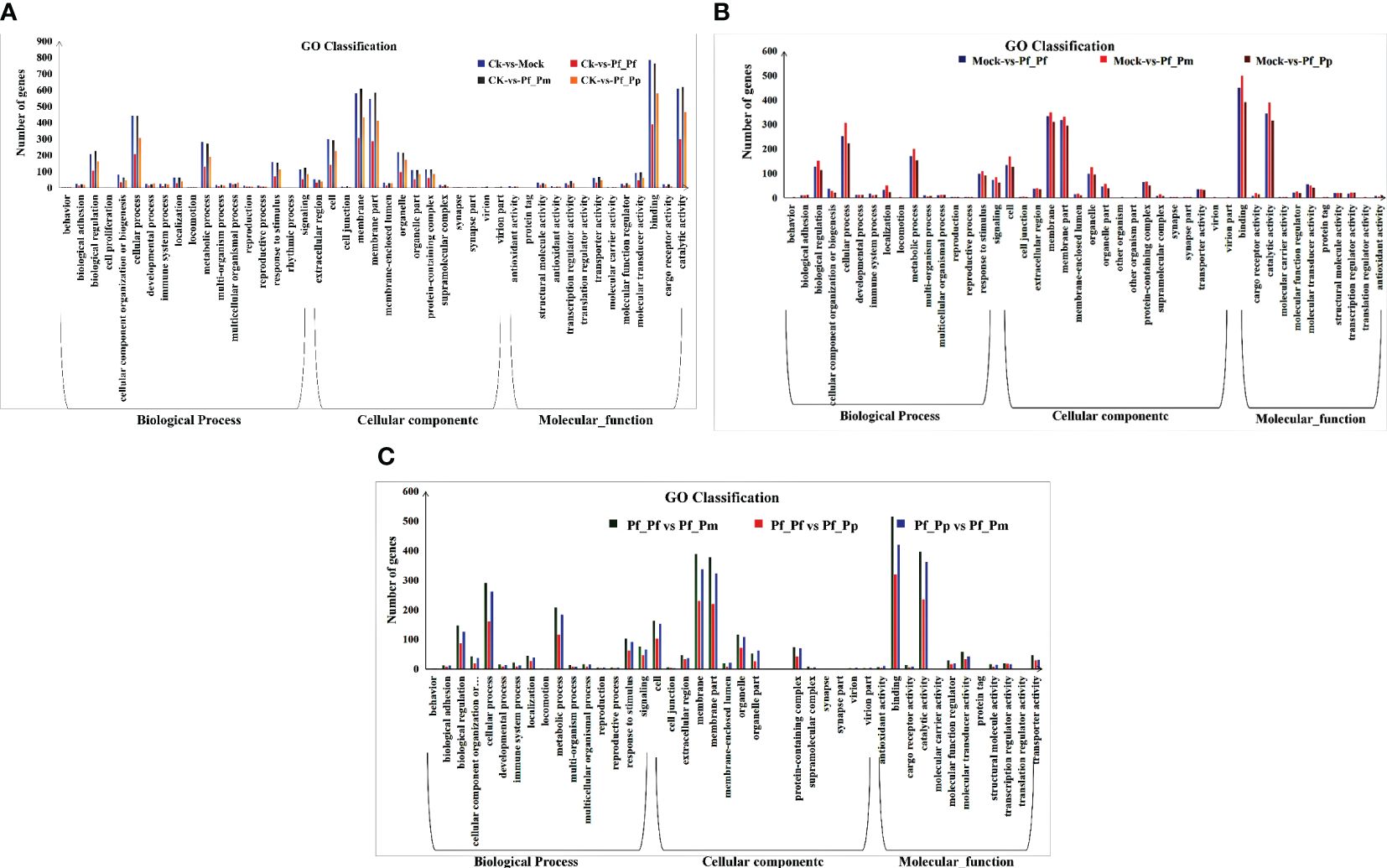
Figure 2 Functional classification of DEGs. (A) GO enrichment analysis of DEGs between the control and nucleus insertion groups. (B) GO Enrichment analysis of DEGs between the non-inserted and different insertion group. (C) GO enrichment analysis of DEGs between the allograft and xenograft groups.
Considering the DEGs annotated within the biological processes group, the sub-categories of “cell movement process,” “metabolic process,” and “biological regulation” were the most abundant. Within the cellular component group, most differential genes were annotated to the “membrane” and “membrane part” sub-categories. Lastly, the “binding” and “catalytic activity” sub-categories were particularly prominent among the molecular function group.
DEGs classified to KEGG pathways related to signal transduction pathways comprised 398, 213, 427, 341, 268, 293, 269, 290, 188, and 253 pathways in the CK-vs-Mock, CK-vs-Pf_Pf, CK-vs-Pf_Pm, CK-vs-Pf_Pp, Mock-vs-Pf_Pf, Mock-vs-Pf_Pm, Mock-vs-Pf_Pp, Pf_Pf-vs-Pf_Pm, Pf_Pf-vs-Pf_Pp, and Pf_Pm-vs-Pf_Pp group comparisons. A total of 26 KEGG pathways were enriched among the DEGs (Figure 3), including the NOD-like receptor signaling pathway, the cytosolic DNA-sensing pathway, the Toll and Imd signaling pathway, the C-type lectin receptor signaling pathway, and the chemokine signaling pathway. Notably, the cytosolic DNA-sensing pathway had the highest enrichment of DEGs in the CK-vs-Mock group.

Figure 3 The KEGG pathways enriched among DEGs. (A) Ck-vs-Mock, (B) CK-vs-Pf_Pf, (C) CK-vs-Pf_Pm, and (D) CK-vs-Pf_Pp comparisons. The number inside represents the number of DEGs enriched.
DEGs among all samples were compared, revealing 19,012 DEGs common to all samples, with the other different modules comprising unique genes for each sample (Figure 4). KEGG enrichment analysis revealed that the enriched pathways (p < 0.05) including a cytosolute DNA sensing pathway, in addition to pathways related to long-term inhibition, cholesterol metabolism, the PPAR signaling pathway, apoptosis, and terpenoid skeleton biosynthesis. Notably, long-term inhibition was represented in each differential comparison and comprised genes encoding cGMP-dependent protein kinase 1(PRKG1), ryanodine receptor 1 (RYR1), cytosolic phospholipase A2 (PLA2), and phosphatidylinositol phospholipase C and beta (PLCB). In addition, other DEGs were identified, such as those encoding chitinase, perilipin-2 (PLIN2), calpain-5, neurexin, very low-density lipoprotein receptor, microphthalmia-associated transcription factor, and Man 1. The above genes exhibited higher expression in the recipient shellfish but lower expression in the control group. In addition, the mucin-2 and tubulin beta genes were highly expressed in the control group but lowly expressed in the surgical group.

Figure 4 Venn diagram showing overlap in DEGs among treatment groups.
To further validate the gene expression of DEGs, 10 immune-related DEGs were randomly selected for RT-qPCR validation (Figure 5), followed by comparison of fold-changes detected by qRT-PCR to the RNA-Seq expression levels. Most of the RT-qPCR results were consistent with the high-throughput sequencing data. Thus, the RNA-Seq data were appropriate for inference of differential gene expression.
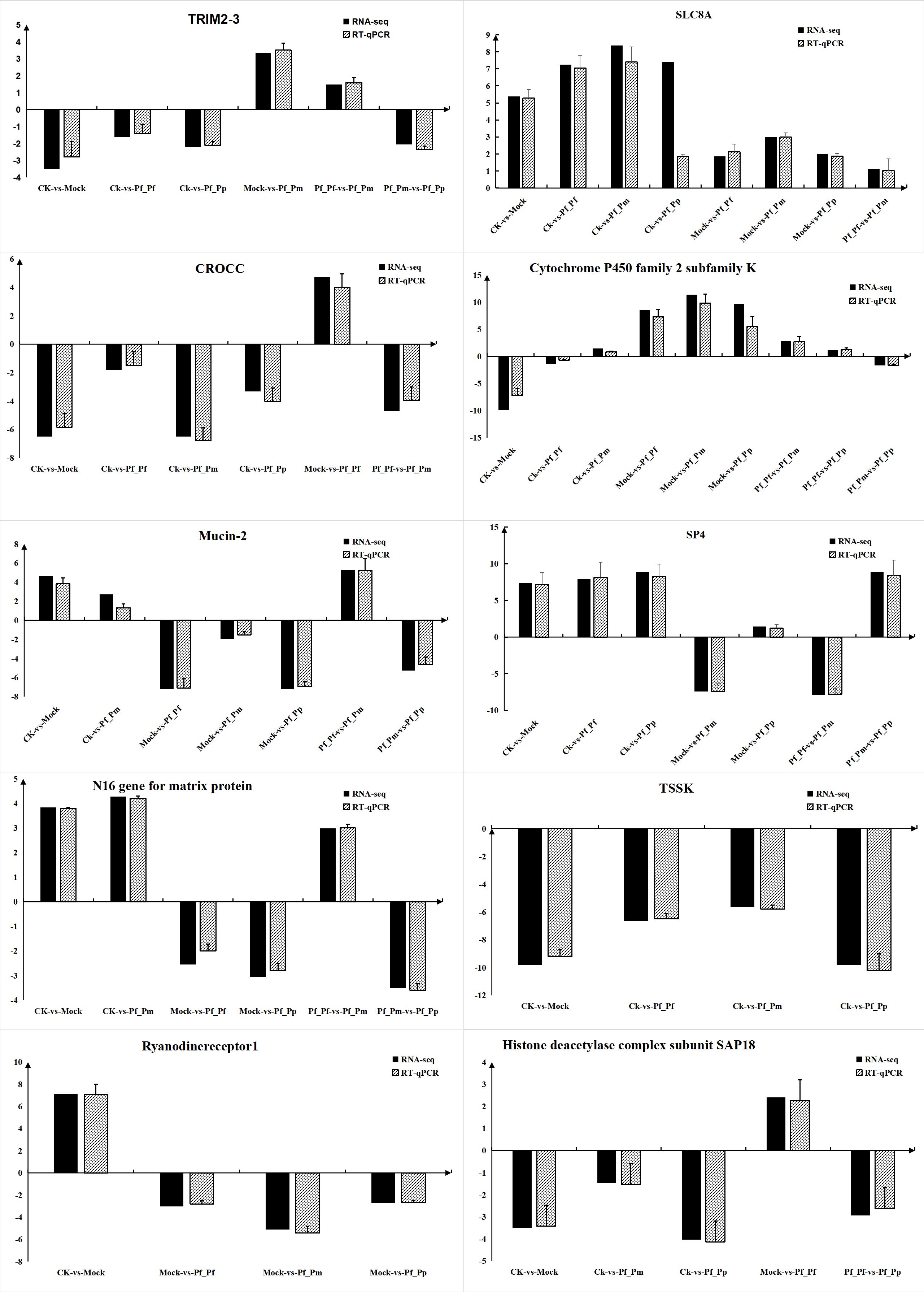
Figure 5 RT-qPCR validation of the expression levels for 10 DEGs. RNA-seq represents the increase or decrease multiple of the gene in the transcriptome data relative to a control group.RT-qPCR means using RT-qPCR technology to verify the quantitative test of the experimental group and the control group, and then calculating the multiple of the increase or decrease of the gene in the experimental group relative to the control group.
Mantle allograft surgery in pearl aquaculture has been successfully employed without immunosuppressants throughout the last century (Zhang et al., 2016). However, little is known of the immune responses generated after mantle tissue transplantation. The seventh day after mantle tissue implantation comprises the pearl sac formation period and an inflammatory milieu has been suggested in donor mantle tissues during this time. The inflammatory microenvironment leads to allograft rejection and also induces allograft tolerance in invertebrates (Land, 2012). Transcriptomic analyses of haemocytes at 192 h were consequently conducted in this study after allograft and xenograft transplantation to identify genes and important pathways involved in development and gametogenesis, along with evaluation of their differential expression between experimental and control groups. These findings provide a basis for the subsequent immune responses analysis of allotransplantation and xenotransplantation.
NOD-like receptors (NLRs) comprise a subgroup of cytosolic pattern recognition receptors (PRRs) that have recently been suggested to play new roles in antiviral innate immune signaling pathways (Zheng, 2021). The NLR protein family comprises 22 members that contain a common structural of an N-terminal effector domain, a central NACHT domain, and a C-terminal leucine-rich repeat sequence (LRR) (Zheng, 2021). The N-terminal effector domain participates in signal transduction, while the NACHT domain interacts with ATP/GTPase-specific P-rings, Mg2+ binding sites, and five other motifs to perform nucleotide binding functions based on self-oligomerization and ATPase activity (Koonin and Aravind, 2000). In addition, LRRs are responsible for pathogen-associated molecular patterns (PAMPs) recognition (Kanneganti et al., 2007). In this study, 86 DEGs were enriched in NOD-like receptors in the CK-vs-Pf-Pm comparison, followed by 67 DEGs in the CK-vs-Pf-Pp comparison, and relatively fewer in the CK-vs-Mock comparison. We speculate that these trends may be related to the degree of immune response produced by allotransplantation and xenotransplantation.
The cytosolic DNA-sensing pathway is primarily responsible for detecting foreign DNA derived from invading microorganisms or host cells and generating an innate immune response. The first identified cytosolic DNA sensor is DAI that activates the IRF and NF-kappaB transcription factors, to produce type I interferons and other cytokines. The second cytoplasmic DNA sensor is AIM2. After sensing DNA, AIM2 interacts with the assembly of the inflammasome, eventually leading to interleukin maturation (Yanai et al., 2009; Huijser et al., 2022). In the control and surgical groups comparison, a higher expression of cGAMP synthase (MB21D1) was detected in the control group and less expressed in the recipient shell. The cGAMP synthetase gene acts as an intracellular pattern recognition receptor (PRR) that senses cytosolic pathogen DNA and subsequently produces the second messenger cGAMP to initiate the TMEM173/STING pathway to produce interferon (IFN) that causes the overall immune response (Liang et al., 2014). Transcriptomic analyses revealed that its expression was down-regulation in the recipient shell, indicating a immune rejection effect. However, this hypothesis requires additional confirmation, including via the role of MB21D1 in the body during immune rejection reaction.
Type C lectin receptors (CLRs) contain one or more type C lectin-like domains (CTLDs) (Viswambari et al., 2010) and are involved in immune recognition reactions of some cells as pattern recognition receptors for pathogen-derived ligands. Dectin-1 is a CLR example that can recognize the fungal-derived ligandsβ-Glucan and high-mannose carbohydrates (Kato et al., 2006). After ligand binding, C-type lectins stimulate intracellular signaling cascades, include the production of various factors, and trigger immune responses against pathogens (Mentrup et al., 2022). The C-type lectin receptor signaling pathways appeared in each group of comparison between allogeneic and xenogeneic transplantation in this study, indicating that the C-type lectin receptor signaling pathway is an important pathway in the immune response of interspecific transplantation, with the abundant immune genes identified in the study deserving further investigation.
Immune genes all exhibited different expression patterns in individual fractions, suggesting that these genes may play important roles in immune processes. N16 is an active protein in P. fucata and can be inhibited to prevent the occurrence of osteoclasts (Lin et al., 2020). N16 may also be a membrane protein-like component of the nacre layer that involved in both crystal formation and the formation of Water Insoluble Organic Matrix (WISM) as a microfibrillar matrix, similar to the role of High Glycine/Tyrosine Proteins (FGTPs), and implicating the specificity of N16 in pearl formation and specific expression only in the mantle (Samata et al., 1999). N16 was distributed in almost every component, relative to that above the control group, consistent with the results of the study.
TSSK proteins establish relationships with two T6SS subcomplexes through direct interactions with TssL, Hcp, and TssC (Zoued et al., 2013). Interestingly, TSSK exhibited down-regulation relative to the control group. However, the roles of TSSK molecules remain unclear and further studies are needed. In addition, Ryanodinereceptor 1 (RYR1) is a skeletal muscle sarcoplasmic reticulum (SR), required for excitation-contraction coupling (EC coupling) of Ca2+ release channels in the sarcoplasmic terminal pool (O'connor et al., 2023). In this study, RYR1 was up-regulation in the CK-vs-Mock comparison, while the Pf_Pf, Pf_Pm, and Pf_Pp comparisons exhibited lower expression of RYR1 than in Mock. RYR1 may be consequently related to pearl sac formation from mantle fragments.
SAP18 was originally identified by immunopurification of sin3-related proteins and is a component of the Sin3-HDAC complex that can enhances sin3-mediated transcriptional inhibition. Specifically, SAP18 is a protein-protein adapter linking the transcription factor Gli, which is a transcriptional repressor fused to [Su (fu)] and the Sin3-HDAC complex (Cheng and Bishop, 2002). Overall, these proteins were down-regulation among the treatment groups, indicating that SAP18 may be involved in immune transplantation, although its mechanistic role remains unclear. CROCC plays important roles in tumors and the involvement of cytokine and cancer-related gene expression (Xu et al., 2019). CROCC was down-regulated as SAP18. A previous study (Xu et al., 2019) observed the up-regulation of miR-33 that could potentially suppress cell proliferation, migration, invasion, and epithelial–mesenchymal transition (EMT) in Gallbladder cancer (GBC) through down-regulation of CROCC. These results suggest that CROCC may modulate immune mechanisms by down-regulation after transplantation.
Cytochrome P450 isoenzymes (CYPs) are a hemoglobin superfamily and terminal oxidases of mixed functional oxidase systems on endoplasmic reticulum membranes. CYPs play critical roles in metabolism of many harmful substances (Muntane et al., 1995; Nelson, 2009), and modulation of CYP pathways can ultimately trigger immunosuppression of immune cells. In this study, CYPs were down-regulated in the Pf_Pf, Pf_Pm, and Pf_Pp groups relative to the control group. Consequently, the involvement of CYPs in the immune response mechanisms of organisms may indicate the generation of immune refection. MUC2 is one the most abundant gastrointestinal gel-forming mucins and exhibits constitutive expression throughout the gastrointestinal tract (Yamashita and Melo, 2018). The MUC2 mucus barrier acts as the first defense against direct contact between intestinal bacteria and colon epithelial cells (Yao et al., 2021). During the development of ulcerative colitis (UC), bacterial factors associated with the MUC2 mucus barrier play important roles in responses to altered dietary patterns, dysfunction of the MUC2 mucus barrier, stimulation of contact with colon epithelial cells, and responses to mucosal and submucosal inflammation (Yao et al., 2021).
Sp4 is a member of the Sp1-like transcription factor family and is primarily expressed in neurons, where it is associated with various neuronal processes, including signal transduction and energy production, and conditions like bipolar disorder (Sheehan et al., 2019). Current research of SP4 focuses on controlling various neuronal processes (Sheehan et al., 2019; Zhang H. et al., 2020), with few studies investigating the direction of immune rejection. Thus, further studies in this area may help clarify its mechanism of action.
Transcripts encoding tripartite motif containing 23 (TRIM23) is an ubiquitin ligase belonging to the tripartite motif (TRIM) family (Bu et al., 2020). TRIM proteins with E3 ubiquitin ligase activity play important roles in virus infection in vertebrates and invertebrates (Zhang R. et al., 2020). Current understanding of TRIM23 primarily focuses on its antiviral immune defense mechanisms, although its precise role in the immune mechanisms remains unclear and requires further study.
The sodium/calcium exchanger or NCX (SLC8A) family is primarily expressed in excitable tissues including muscle and heart tissue, because their rapid and massive transport of Ca2+ is important in muscle and heart contractions (Brini and Carafoli, 2011). All the components of this family were up-regulated, with little observed differences in each group. SLC8A is primarily expressed in tissues like the heart, with the blood cells in the samples we collected primarily coming from hearts. These reasons might consequently explain their consistent up-regulation.
Here, a transcriptomic analysis was conducted to identify host defense gene activities against allograft and xenograft transplantation in P.fucata during pearl cultivation. Specifically, immune-related pathways and genes were identified and discussed. These results provide a theoretical basis and framework to further understand the role of P.fucata in immune defense systems, thereby helping to reduce host immunological rejection to transplantation.
Raw sequence reads generated during the current study were deposited in the Sequence Read Archive of the National Center for Biotechnology Information (NCBI), accession number: PRJNA1068836.
This study adhered to the guidelines set forth by the Ministry of Science and Technology of the People's Republic of China in their "Guidelines for the Care and Use of Experimental Animals" (approval number: 2006-398) and received approval from the Animal Research and Ethics Committee of Beibu gulf University for all experimental animal manipulations.
YZ: Investigation, Writing – original draft. PW: Writing – review & editing. YG: Writing – review & editing. LB: Writing – review & editing. DY: Supervision, Writing – review & editing. SZ: Funding acquisition, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Natural Science Foundation of Guangxi Zhuang Autonomous Region of China (Grant No. 2021GXNSFAA220031 and 2021GXNSFAA075008) and the Natural Science Foundation of China (Grant No. 31860734).
We thank LetPub (www.letpub.com) for linguistic assistance and pre-submission expert review.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2024.1362078/full#supplementary-material
Ai Y., Zhang Q., Wang W., Zhang C., Cao Z., Bao M., et al. (2016). Transcriptomic analysis of differentially expressed genes during flower organ development in genetic male sterile and male fertile Tagetes erecta by digital gene-expression profiling. PloS One 11, e0150892. doi: 10.1371/journal.pone.0150892
Arnaud-Haond S., Goyard E., Vonau V., Herbaut C., Prou J., Saulnier D. (2007). Pearl formation: persistence of the graft during the entire process of biomineralization. Mar. Biotechnol. 9, 113–116. doi: 10.1007/s10126-006-6033-5
Brini M., Carafoli E. (2011). The plasma membrane Ca2+ ATPase and the plasma membrane sodium calcium exchanger cooperate in the regulation of cell calcium. Cold Spring Harbor Perspect. Biol. 3, a004168. doi: 10.1101/cshperspect.a004168
Briones C., Nuñez J. J., Pére Z. M., Espinoza-Rojas D., Molina-Quiroz C., Guiñez R. (2018). De novo male gonad transcriptome draft for the marine mussel Perumytilus purpuratus with a focus on its reproductive-related proteins. J. Genomics 6, 127. doi: 10.7150/jgen.27864
Bu N., Dong Z., Zhang L., Zhu W., Zheng S. (2020). CircPVT1 regulates cell proliferation, apoptosis, and glycolysis in hepatocellular carcinoma via miR-377/TRIM23 axis. Cancer Manage. Res. 12, 12945–12956. doi: 10.2147/CMAR.S280478
Chen Y., Chen Y., Shi C., Huang Z., Zhang Y., Li S., et al. (2018). SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, 1–6. doi: 10.1093/gigascience/gix120
Cheng S. Y., Bishop J. M. (2002). Suppressor of Fused represses Gli-mediated transcription by recruiting the SAP18-mSin3 corepressor complex. Proc. Natl. Acad. Sci. United States America 99, 5442–5447. doi: 10.1073/pnas.082096999
Dong C., Wu H., Zheng G., Peng J., Guo M., Tan Z. (2022). Transcriptome analysis reveals MAPK/AMPK as a key regulator of the inflammatory response in PST detoxification in Mytilus galloprovincialis and Argopecten irradians. Toxins 14, 516. doi: 10.3390/toxins14080516
Fang Z., Feng Q. L., Chi Y. Z., Xie L., Zhang R. (2008). Investigation of cell proliferation and differentiation in the mantle of Pinctada fucata (bivalve, Mollusca). Mar. Biol. 153, 745–754. doi: 10.1007/s00227-007-0851-5
Fukushima E., Iwai T., Miura C., Celino F. T., Urasaki S., Miura T. (2014). A xenograft mantle transplantation technique for producing a novel pearl in an akoya oyster host. Mar. Biotechnol. 16, 10–16. doi: 10.1007/s10126-013-9525-0
He J., Shen C., Liang H., Fang X., Lu J. (2020). Antimicrobial properties and immune-related gene expression of a C-type lectin isolated from Pinctada fucata martensii. Fish Shellfish Immunol. 105, 330–340. doi: 10.1016/j.fsi.2020.07.017
Huijser E., Bodewes I. L., Lourens M. S., Van H. C. G., Van d. B. T. P. P., Grashof D. G. B., et al. (2022). Hyperresponsive cytosolic DNA-sensing pathway in monocytes from primary Sjgren's syndrome. Rheumatology 61, 3491–3496. doi: 10.1093/rheumatology/keac016
Inoue N., Ishibashi R., Ishikawa T., Atsumi T., Aoki H., Komaru A. (2010). Gene expression patterns and pearl formation in the Japanese pearl oyster (Pinctada fucata): comparison of gene expression patterns between the pear sac and mantle tissues. Aquaculture 308, 68–74. doi: 10.1016/j.aquaculture.2010.06.036
Kanneganti T. D., LamkanfiM, Núñez G. (2007). Intracellular NOD-like receptors in host defense and disease. Immunity 27, 549–559. doi: 10.1016/j.immuni.2007.10.002
Kato Y., Adachi Y., Ohno N. (2006). Contribution of N-linked oligosaccharides to the expression and functions of β-glucan receptor, Dectin-1. Biol. Pharm. Bull. 29, 1580–1586. doi: 10.1248/bpb.29.1580
Koonin E. V., Aravind L. (2000). The NACHT family–a new group of predicted NTPases implicated in apoptosis and MHC transcription activation. Trends Biochem. Sci. 25, 223–224. doi: 10.1016/S0968-0004(00)01577-2
Land W. G. (2012). Emerging role of innate immunity in organ transplantation part III: the quest for transplant tolerance via prevention of oxidative allograft injury and its consequences. Transplant. Rev. 26, 88–102. doi: 10.1016/j.trre.2011.07.001
Liang Q., Seo G. J., Choi Y. J., Ge J., Rodgers M. A., Shi M., et al. (2014). Autophagy side of MB21D1/cGAS DNA sensor. Autophagy 10, 1146–1147. doi: 10.4161/auto.28769
Lin J. B., Wu H., Liu Y. L., Shaw P. C., Li P. B. (2020). N16 suppresses RANKL-mediated osteoclatogenesis by down-regulating RANK expression. Int. J. Biol. Macromolecules 151, 1154–1162. doi: 10.1016/j.ijbiomac.2019.10.159
Lu X., Zhang M., Yang S., Deng Y., Jiao Y. (2022). Transcriptome analysis reveals the diverse response of pearl oyster Pinctada fucata martensii after different PAMP stimulation. Fish Shellfish Immunol. 131, 881–890. doi: 10.1016/j.fsi.2022.10.058
McGinty E. L., Evans B. S., Taylor J. U. U., Jerry D. R. (2010). Xenografts and pearl production in two pearl oyster species, P. maxima and P. margaritifera: Effect on pearl quality and a key to understanding genetic contribution. Aquaculture 302, 175–181. doi: 10.1016/j.aquaculture.2010.02.023
Mentrup T., Stumpff-niggemann A. Y., Leinung N., Schlosser C., Schubert K., Wehner R., et al. (2022). Phagosomal signalling of the C-type lectin receptor Dectin-1 is terminated by intramembrane proteolysis. Nat. Commun. 13, 1880. doi: 10.1038/s41467-022-29474-3
Muntane J., Ourlin J. C., Domergue J., Maurel P. (1995). Differential effects of cytokines on the inducible expression of CYP1A1, CYP1A2, and CYP3A4 in human hepatocytes in primary culture. Hepatology 22, 1143–1153. doi: 10.1002/hep.1840220420
Nelson D. R. (2009). The cytochrome p450 homepage. Hum. Genomics 4, 1–7. doi: 10.1186/1479-7364-4-1-59
O'connor T. N., Van Den Bersselaar L. R., Chen Y. S., Nicolau S., Simon B., Huseth A., et al (2023). RYR-1-related diseases international research workshop: From mechanisms to treatments Pittsburgh, PA, USA, 21-22 July 2022. J. Neuromuscular Dis. 10, 135–154. doi: 10.3233/JND-221609
Samata T., Hayashi N., Kono M., Hasegawa K., Horita C., Akera S. (1999). A new matrix protein family related to the nacreous layer formation of Pinctada fucata. FEBS Lett. 462, 225–229. doi: 10.1016/S0014-5793(99)01387-3
Sheehan K., Lee J., Chong J., Zavala K., Sharma M., Philipsen S., et al. (2019). Transcription factor Sp4 is required for hyperalgesic state persistence. PloS One 14, 1–24. doi: 10.1371/journal.pone.0211349
Viswambari D. R., Basilrose M. R., Mercy P. D. (2010). Prospect for lectins in arthropods. Ital. J. Zoology 77, 254–260. doi: 10.1080/11250003.2010.492794
Wang P., Guo Y., Li S., Zheng Y., Li T., Zhao S., et al. (2024). Comparative proteomics reveal the humoral immune rejection of pearl oyster Pinctada fucata to xenograft from Pinctada maxima. Aquaculture 582, 740515. doi: 10.1016/j.aquaculture.2023.740515
Wei J., Liu B., Fan S., et al. (2017). Differentially expressed immune-related genes in hemocytes of the pearl oyster Pinctada fucata against allograft identified by transcriptome analysis. Fish Shellfish Immunol. 62, 247–256. doi: 10.1016/j.fsi.2017.01.025
Xu G., Wei X., Tu Q., Zhou C. (2019). Up-regulated microRNA-33b inhibits epithelial-mesenchymal transition in gallbladder cancer through downregulating CROCC. Bioscience Rep. 40, BSR20190108. doi: 10.1042/BSR20190108
Yamashita M. S. A., Melo E. O. (2018). Mucin 2 (MUC2) promoter characterization: an overview. Cell Tissue Res. 374, 455–463. doi: 10.1007/s00441-018-2916-9
Yanai H., Savitsky D., Tamura T., Taniguchi T. (2009). Regulation of the cytosolic DNA-sensing system in innate immunity: a current view. Curr. Opin. Immunol. 21, 17–22. doi: 10.1016/j.coi.2009.01.005
Yao D., Dai W., Dong M., Dai C., Wu S. (2021). MUC2 and related bacterial factors: therapeutic targets for ulcerative colitis. Ebio Med. 74, 103751. doi: 10.1016/j.ebiom.2021.103751
Zhang H., Lu J., Wu S. (2020). Sp4 controls constitutive expression of neuronal serine racemase and NF-E2-related factor-2 mediates its induction by valproic acid. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 1863, 194597. doi: 10.1016/j.bbagrm.2020.194597
Zhang M., Lu J., Liang H., Zhang B., Liang B., Zou H. (2022). The succinylome of Pinctada fucata martensii implicates lysine succinylation in the allograft-induced stress response. Fish Shellfish Immunol. 127, 585–593. doi: 10.1016/j.fsi.2022.07.009
Zhang R., Dai X., Cao X., Zhang C., Wang K., Huang X., et al. (2020). Trim23 promotes WSSV replication though negative regulation of antimicrobial peptides expression in Macrobrachium nipponense. Mol. Immunol. 124, 172–179. doi: 10.1016/j.molimm.2020.06.007
Zhang R., Wang M., Xia N., Yu S., Chen Y., Wang N. (2016). Cloning and analysis of gene expression of interleukin-17 homolog in triangle-shell pearl mussel, Hyriopsis cumingii, during pearl sac formation. Fish Shellfish Immunol. 52, 151–156. doi: 10.1016/j.fsi.2016.03.027
Zheng C. F. (2021). The emerging roles of NOD-like receptors in antiviral innate immune signaling pathways. Int. J. Biol. Macromolecules 169, 407–413. doi: 10.1016/j.ijbiomac.2020.12.127
Zhou L., Liu Z., Dong Y., Sun X., Wu B., Yu T., et al. (2019). Transcriptomics analysis revealing candidate genes and networks for sex differentiation of yesso scallop (Patinopecten yessoensis). BMC Genomics 20, 1–15. doi: 10.1186/s12864-019-6021-6
Zoued A., Durand E., Bebeacua C., Brunet Y. R., Douzi B., Cambillau C., et al. (2013). TssK is a trimeric cytoplasmic protein interacting with components of both phage-like and membrane anchoring complexes of the type VI secretion system. J. Biol. Chem. 288, 27031–27041. doi: 10.1074/jbc.M113.499772
Keywords: Pinctada fucata, transcriptome, allograft, xenograft, immunological response
Citation: Zheng Y, Wang P, Guo Y, Bai L, Yu D and Zhao S (2024) Comparative transcriptome analysis reveals immune-related genes involved in allograft and xenograft transplantation in Pinctada fucata. Front. Mar. Sci. 11:1362078. doi: 10.3389/fmars.2024.1362078
Received: 27 December 2023; Accepted: 02 April 2024;
Published: 17 April 2024.
Edited by:
Tangtian He, Hong Kong Polytechnic University, Hong Kong SAR, ChinaReviewed by:
Michio Suzuki, The University of Tokyo, JapanCopyright © 2024 Zheng, Wang, Guo, Bai, Yu and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sen Zhao, emhhb3NlbkBiYmd1LmVkdS5jbg==; Dahui Yu, cGVhcmx5ZGhAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.