 Giulia Trapella1,2†
Giulia Trapella1,2† Daniel Scicchitano1,2†Lucia Foresto1,2Andrea Nicoló Dell’Acqua1,2Elena Radaelli1,2
Daniel Scicchitano1,2†Lucia Foresto1,2Andrea Nicoló Dell’Acqua1,2Elena Radaelli1,2 Silvia Turroni1
Silvia Turroni1 Simone Rampelli1,2Cinzia Corinaldesi3
Simone Rampelli1,2Cinzia Corinaldesi3 Giorgia Palladino1,2
Giorgia Palladino1,2 Marco Candela1,2*
Marco Candela1,2*- 1Unit of Microbiome Science and Biotechnology, Department of Pharmacy and Biotechnology (FaBiT), Alma Mater Studiorum – University of Bologna, Bologna, Italy
- 2Fano Marine Center, The Inter-Institute Center for Research on Marine Biodiversity, Resources and Biotechnologies, Fano, Italy
- 3Department of Materials, Environmental Sciences and Urban Planning, Polytechnic University of Marche, Ancona, Italy
Introduction: In our paper, we explored the impact of different anthropogenic stressors, namely, mussel farming, methane extraction platforms, and summer mass tourism, on the epipelagic microbiomes of the North-Western Adriatic Sea.
Methods: By 16S rRNA gene sequencing, we provided the epipelagic microbiome layout from selected sites corresponding to each of the considered impacts. As an unimpacted reference, we used already published 16S rRNA sequencing data.
Results: According to our findings, each one of the anthropogenic stressors resulted in a peculiar increase of specific epipelagic microbial components, including copiotrophic R-strategists and host-restricted bacteria, as well as some pathobiome components, the latter being detected exclusively in impacted sites. Particularly, potentially harmful pathogenic species such as Legionella impletisoli and Staphylococcus epidermidis have been detected in proximity to the mussel farms, and Escherichia coli and Campylobacter ureolyticus were present close to the methane extraction platform and at the summer mass tourism site, respectively. Particularly, C. ureolyticus is an emerging human gastrointestinal pathogen, capable of destroying intestinal microvilli.
Discussion: In addition to providing evidence supporting the existence of recognizable and impact-driven fingerprints on the epipelagic marine microbiome peculiar to the different anthropogenic stressors, our findings also raise concern about the ecological relevance of the observed changes, in terms of possible loss of ecosystem services and also for the potential release of pathogenic microorganisms in the environment.
1 Introduction
It is now a matter of fact that the epipelagic microbiome plays a central role in primary productivity, the global biogeochemical cycling, and the functioning of food webs of marine ecosystems (Arrigo, 2005; Luna, 2015; Dang and Lovell, 2016). In a recent publication (Scicchitano et al., 2022), the structure and function of the epipelagic microbiome of the North-Western Adriatic Sea (Mediterranean Sea) have been described, showing a well-balanced microbial ecosystem structured for the provision of key ecosystem services. Indeed, by applying 16S rRNA next-generation sequencing and a network-based approach on epipelagic samples collected from a 130-km2 area located 13.5 km away from the Emilia-Romagna coast, the authors reported that the epipelagic layer of the North-Western Adriatic Sea was dominated by Synechococcus-like Cyanobacteria Subsection I, a photosynthetic primary producer (Scanlan and West, 2002; Flombaum et al., 2013), and by Flavobacteriales, Oceanospirillales and Rhodobacterales, copiotrophic microbiome components playing an important role in the cycling of the dissolved organic matter (DOM) (Lauro et al., 2009). Conversely, cosmopolitan marine heterotrophs, such as SAR11 clade and Cellvibrionales, represented only minor components of the epipelagic microbiome, as typically dominated more oligotrophic waters (Giovannoni et al., 2005; Salter et al., 2015; Liao et al., 2020).
The North-Western Adriatic Sea is characterized by shallow waters, with a maximum depth of approximately 40 m. The ecosystem productivity in the coastal area is mainly sustained by nutrient inputs, especially from the Po River (Findlay et al., 1990; Grilli et al., 2020), with two main currents dominating the circulation in the Adriatic Sea, namely, the Western Adriatic Current (WAC), flowing toward the southeast along the Western Italian coast, and the East Adriatic Current (EAC), flowing from the northwest along the eastern Croatian coast (Findlay et al., 1990; Grilli et al., 2020).
Its peculiar geochemical and hydrodynamic features make the overexploited North-Western Adriatic Sea a remarkable model of a marine system dynamically influenced by complex anthropogenic impacts (Danovaro, 2003; Zuccato et al., 2005; Zuccato et al., 2006; Corinaldesi et al., 2022). Indeed, in the last decades, the Adriatic Sea has been increasingly threatened by several anthropogenic stressors (e.g., hydrocarbon extraction, over-fishing, aquaculture, marine traffic, plastic contamination, and tourism), and, recently, it has been identified as one of the areas in the Mediterranean Sea most worthy of protection (Bastari et al., 2016). In this scenario, the assessment of the impact of these different threats on the epipelagic ecosystems of the Adriatic Sea is becoming particularly relevant (Orel et al., 2022) in terms of impaired ecological functions and also for possible concerns regarding human and animal health. Indeed, the changes induced in the structure and dynamics of the marine pathobiome (Naidoo and Olaniran, 2014; Buccheri et al., 2019; Numberger et al., 2019) represent a global risk for the spread and consolidation of infectious diseases resulting from exposure to contaminated waters and/or the consumption of contaminated seafood as defined in the One Health concept (Orel et al., 2022).
In our work, three different impact sources have been considered in the North-Western Adriatic Sea, namely, i) mussel farming, where the North-Western Adriatic Sea accounts for 50% of the Italian mussel production (Prioli, 2006); ii) methane extraction platforms, with 82 platforms installed since 1960 in the Adriatic Sea (Colaleo et al., 2022), 50 of which occupy the area between Rimini and Ravenna (http://ytaa.miesbcn.com/work/220, 2016 Edition, Ghiselli, and Melandri); iii) summer mass tourism, where the Emilia-Romagna coast represents one of the most important touristic hotspots of the Mediterranean Sea, where the summer tourism peak has been associated with serious threats to the coastal ecosystems (Andolina et al., 2021). Seawater samples were collected in an area of approximately 585 km2 in the North-Western Adriatic Sea during the summer of 2021 (September), including a mussel farm located offshore Cesenatico, a methane extraction plant (“Azalea”) located offshore Rimini, and a coastal site in Riccione, one of the main Italian sites for mass summer tourism in the North-Western Adriatic Sea since 1960 (Istat Annual report on the tourist movement and hotel and complementary consistency in Emilia romagna, 2020). Epipelagic microbiomes from these impacted sites were assessed by 16S rRNA next-generation sequencing and compared with those from a relatively unimpacted reference site from Scicchitano et al. (2022). Findings reported in this study provide glimpses into possible risks from the overexploitation of marine resources, posing significant threats to the health of fragile marine ecosystems such as those of the Adriatic Sea.
2 Materials and methods
2.1 Study areas and sampling collection
The present study was conducted in September 2021 in four different sites located offshore a coast trait encompassing Cesenatico to Riccione (Emilia Romagna, Italy). A total of 44 seawater samples were collected using a Niskin bottle from three sites subject to different anthropogenic impacts. In particular, 12 samples were collected near a mussel farm located offshore Cesenatico at a depth of 3 m. Eleven samples were collected in a concentric area around the Azalea methane extraction platform off the Rimini coast at a depth of 10 m. Twenty-one samples were collected close to the Riccione coast, which is subject to mass tourism especially during summer, at a depth of 2.5 m close to the artificial structures, referred to as WMesh, originally designed to prevent coastal erosion (Palladino et al., 2022a).
Finally, 16S rRNA sequencing data from Scicchitano et al. (2022), including 19 epipelagic samples from a relatively unimpacted offshore area of 130 km2 at a depth of 10 m, have been used as a control. Geographic coordinates, water depths, and distance from the coast for each sample are reported in Supplementary Table S1. Immediately after collection, 2 L of seawater was poured into a previously sterilized plastic bottle. Samples were stored in the dark until arrival at the laboratory. Seawater samples were pre-filtered through a 1.2-μm cellulose mixed ester polycarbonate filter to remove large, suspended particles (Chen et al., 2019) and then filtered through 0.22-μm 47-mm-diameter cellulose mixed ester pore-size filters (MF-Millipore, Merck Millipore, Billerica, MA, USA) through vacuum filtration system (Behzad et al., 2022) under laminar flow hood. Filters were stored in sterile Eppendorf at −80°C until processed. The environmental parameters (temperature, pH, and concentration of chlorophyll a, dissolved inorganic carbon, phosphate, nitrate, and ammonium) of the study sites at the time of sample collection (September 2021) were retrieved using the monthly data present within the Copernicus database (https://www.copernicus.eu/en, retrieved 5 January, 2024). Specifically, for the extraction site, the mussel farming site, and the coastal tourism site, a single point was considered, while for the pristine site, a mean of the data in the whole sampling area was obtained.
2.2 DNA extraction, 16S rRNA gene amplification, and sequencing
Extraction of the total microbial DNA from water samples was performed from the entire membrane filters using the DNAeasy PowerWater extraction kit (QIAGEN, Hilden, Germany) following the manufacturer’s instructions (Scicchitano et al., 2022). Extracted DNA was then quantified using NanoDrop ND-1000 (NanoDrop Technologies, Wilmington, DE, USA) and stored at −20°C until further processing. The V3–V4 hypervariable region of the 16S rRNA gene was PCR amplified in a 50 µL reaction containing 25 ng of microbial DNA, 2X KAPA HiFi HotStart ReadyMix (Roche, Basel, Switzerland), and 200 nmol/L of 341F and 785R primers carrying Illumina overhang sequencing adapter (Klindworth et al., 2013). The thermal cycle consisted of 3 min at 95°C, 25 cycles of 30 s at 95°C, 30 s at 55°C and 30 s at 72°C, and a final elongation step of 5 min at 72°C (Palladino et al., 2022b). PCR products were purified using Agencourt AMPure XP magnetic beads (Beckman Coulter, Brea, CA, USA). Indexed libraries were prepared using limited-cycle PCR with Nextera technology and cleaned up using Agencourt AMPure XP magnetic beads (Beckman Coulter, Brea, CA, USA). Libraries were normalized to 4 nM and pooled. The sample pool was denatured with 0.2 N NaOH and diluted to a final concentration of 4.5 pM with a 20% PhiX control. Sequencing was performed on an Illumina MiSeq platform using a 2 × 250 bp paired-end protocol, according to the manufacturer’s instructions (Illumina, San Diego, CA, USA).
2.3 Bioinformatics and biostatistics
The QGIS software (https://qgis.org/it/site/) was used to construct the maps of the study area. Raw sequencing outputs for a total of 63 samples were processed using a pipeline combining PANDAseq (Masella et al., 2012) and QIIME2 (Bolyen et al., 2019). High-quality reads (min/max length = 350/550 bp) were retained using the “fastq filters” function of Usearch11 (Edgar, 2010). Specifically, reads with an expected error per base E = 0.03 (i.e., 3 expected errors every 100 bases) were discarded, based on the phred Q score probabilities. The resulting reads from the length and quality filtering were binned into amplicon sequence variants (ASVs) using DADA2 (Callahan et al., 2016). The taxonomy was assigned using the hybrid method combining VSEARCH and q2 classifier trained on the Silva database release 138.1 (Bokulich et al., 2018) against the SILVA database (2022, v138.1) (Quast et al., 2012). All the sequences assigned to eukaryotes (i.e., chloroplasts and mitochondria) or unassigned were discarded. Sequencing reads were deposited in ENA (project number PRJEB69608).
All statistical analyses were performed using the R software (R Core Team; www.r-project.org—last access: March 2021), v. 4.1.2, with the package “vegan” (https://cran.r-project.org/web/packages/vegan/index.html) and Made4 (Culhane et al., 2005). Beta diversity was estimated by computing Weighted UniFrac distance, and the data separation in the principal coordinate analysis (PCoA) was tested using a permutation test with pseudo-F ratios (function “adonis” in the vegan package). Wilcoxon rank-sum test and Kruskal–Wallis test were used to assess significant differences in alpha diversity and taxon relative abundance between groups. p-Values were corrected for multiple testing with the “p.adjust” function in R, with a false discovery rate (FDR) ≤0.05 considered statistically significant.
Sequences corresponding to the ASVs assigned to Leptothrichiaceae_sp, Legionellaceae, Campylobacteraceae, Staphylococcaceae, and Enterobacteriaceae were retrieved and uploaded to the Silva online tool for alignment, classification, and tree service (SINA v1.2.12, https://www.arb-silva.de/aligner/). The default parameters were used, and for the search and classify section, the 10 closest neighbors with a minimum identity of 97% were evaluated. For the construction of the phylogenetic trees, the “compute tree” option was selected using the RAxML program and GTR model including the neighbor sequences. The tree was exported in newick format and uploaded on iTOL for graphical representation (Letunic and Bork, 2021).
For the microbiome network construction, bacterial co-abundance groups were obtained by computing the association among the bacterial families using the Kendall correlation test. The Wiggum plot network structure was created using Cytoscape (http://www.cytoscape.org/). Circle sizes were proportional to families’ abundance or over-abundance, and connections between nodes were represented as “red line” or “dashed gray line” for positive or negative correlation, respectively. Over-abundance values were calculated using the ratio between the mean relative abundance in a specific site and the average relative abundance in the whole dataset of the study (meanArea/meanTot). Hub nodes, total cohesion, negative/positive cohesion ratio, and modularity were calculated on the overall structure of the network. Specifically, hub nodes were identified by looking at the combination of the highest values of closeness centrality, betweenness centrality, and degree retrieved from Cytoscape as previously described (Agler et al., 2016). Cohesion and modularity were calculated using the “igraph” R package as proposed by Hernandez et al. (2021).
3 Results
3.1 Impact of different anthropogenic pollution sources (mussel farming, methane extraction, and coastal tourism) on the epipelagic microbiome
In this study, we characterized the epipelagic microbiome by 16S rRNA gene sequencing from a total of 63 water samples collected in the North-Western Adriatic Sea, including i) 12 samples from the mussel farming-impacted site, ii) 11 samples from the methane extraction-impacted site, and iii) 21 samples from the tourism-impacted site. As a control, 16S rRNA sequencing data from Scicchitano et al. (2022) were used, representing 19 epipelagic samples from a relatively unimpacted control area. In Figure 1, we provide an overview of the samples collected from each impacted site and the relatively unimpacted control. Environmental parameters at the time of sampling for the impacted sites and for the reference control are reported in Table 1. Overall, 44 samples have been newly sequenced, providing a total of 588,896 high-quality reads (13,884 ± 3,638) corresponding to 3,408 total ASVs.

Figure 1 Geolocation of sampling sites. General distribution of sampling sites within the study area with a focus on the coastal tourism and extraction platform sites (platform CH4), where the distance between sampling points is on a finer scale.
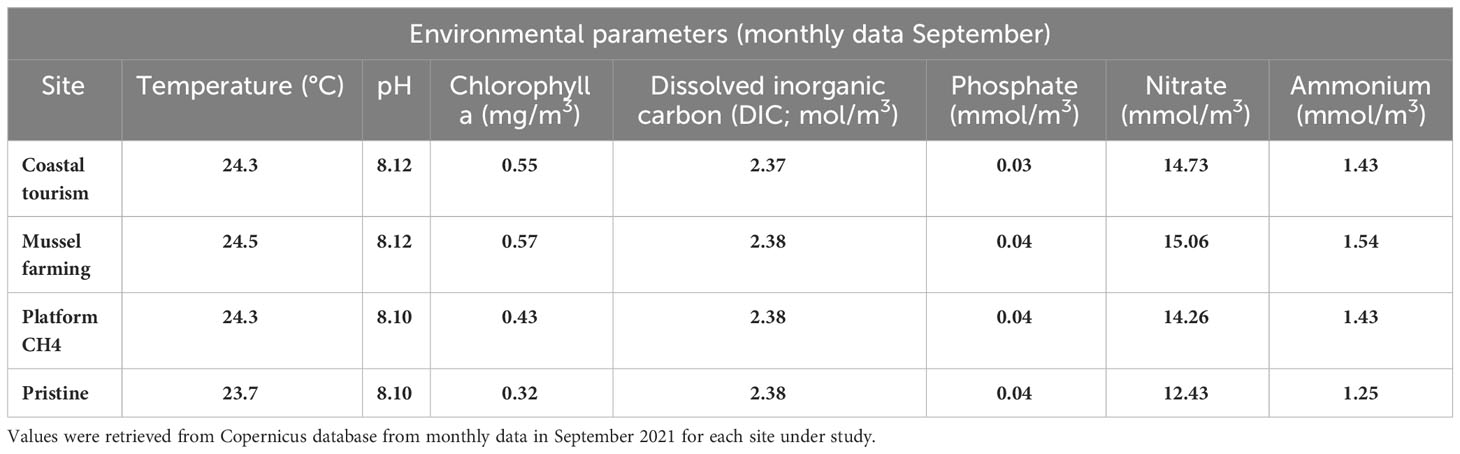
Table 1 Environmental parameters at the time of sample collection.
According to our findings, the main phyla representing the epipelagic microbiome were Proteobacteria (mean r.ab ± SD; 53.1% ± 11.7%), Bacteroidota (17.6% ± 7.5%), Cyanobacteria (8.7% ± 8.7%), Actinobacteriota (5.2% ± 8.7%), Planctomycetota (4.5% ± 4.7%), and Verrucomicrobia (7.3% ± 5.1%) (Supplementary Figure S1), with site-specific declinations in terms of relative abundances, as noticeable at the corresponding family level (Figure 2; Supplementary Figure S2). Focusing on the phylogenetic resolution at the ASV level, the PCoA plot of the microbiomes’ compositional structure showed sharp segregation between the epipelagic microbiomes at the control and impacted sites (Figure 3A). More specifically, according to our data, each impacted site showed a peculiar ASV-level compositional profile of the epipelagic microbiome, well segregating from the unimpacted control and the other impacted sites. Noticeably, when accounting for the alpha-diversity distribution (Figure 3B), the methane extraction-impacted site showed an overall lower diversity compared to the other sites (particularly, tourism-impacted site and control site). To highlight the site-specific impact on the epipelagic microbiome in terms of compositional diversity, the boxplots of the microbial families showing a significantly different distribution between each of the impacted sites and unimpacted reference control are provided in Supplementary Figure S3. According to our findings, each of the impacted sites showed a specific layout of significantly increased or depleted bacterial taxa compared to the pristine control. In particular, the mussel farming-impacted site was characterized by a higher abundance of Hyphomonadaceae (mean r.ab variation + 5.17%), Nannocystaceae (+0.70%), Saprospiraceae (+5.29%), Oleiphilaceae (+0.30%), family AB1 of Rickettsiales (+0.64%), Alteromonadaceae (+2.97%), Ruminococcaceae (+0.05%), Leptothrichiaceae (+0.12%), and Pseudoalteromonadaceae (+6.48%) while being depleted in Puniceicoccaceae (−2.83%), Cyanobiaceae (−13.07%), SAR11 (−0.22%), and SAR116 clades (−4.35%). Focusing on the methane extraction impact, the epipelagic microbiome in the proximity of the extraction platform was enriched in Pseudoalteromonadaceae (+12.40%), Actinomarinaceae (+6.61%), Vibrionaceae (+13.62%), Enterobacteriaceae (+0.18%), Staphylococcaceae (+0.07%), Alteromonadaceae (+2.19%), Rhodobacteraceae (+3.84%), Bifidobacteriaceae (+0.04%), and Bacillaceae (+0.56%) while depleted in Rhodospirillales (Aegean 169 marine group) (−2.39%), Cyanobiaceae (−9.15%), Flavobacteriaceae (−3.16%), PS1 clade of Parvibaculales (−0.89%), Puniceicoccaceae (−2.64%), SAR11 (−3.00%), and NS9 marine group of Flavobacteriales (−1.48%). Finally, the coastal tourism-impacted site was characterized by a significant increase of Rhodobacteraceae (+7.52%), Cryomorphaceae (+3.96%), Rubritaleaceae (+3.02%), Methylophilaceae (+0.62%), DEV007 of Verrucomicrobiales (+4.07%), and Alteromonadaceae (+0.67%), with a depletion in SAR11 clades (−1.04%), Puniceicoccaceae (−3.12%), Actinomarinaceae (−0.75%), and Pirellulaceae (−1.83%). In order to explore the variation of the epipelagic microbiome network structure at the impacted and control sites, the overall network of the epipelagic microbiome was first obtained, and then its declination at the different impacted and reference sites was assessed. For the construction of the microbiome network, co-abundance associations between microbial families have been computed, obtaining six Co-Abundance Groups (CAGs), namely, Vibrionaceae CAG, Halieaceae CAG, Saprospiraceae CAG, Rhodobacteraceae CAG, Cyanobiaceae CAG, and Clade_I CAG. The detailed composition of each CAG is provided in the Supplementary Table S2. The Wiggum plot showing the compositional relationships between the microbial network components is provided in Supplementary Figure S4. According to our findings, the obtained epipelagic microbiome network structure showed modularity of 0.197, a ratio between negative to positive cohesions of 0.913, and a total cohesion of 0.42. Finally, Clade_I; SAR116_clade; Puniceicoccaceae; Pirellulaceae, Saprospiraceae; DEV007; Flavobacteriales, f_NS9_marine_group; Hyphomonadaceae; and Cyanobiaceae and Cryomorphaceae resulted as keystone components supporting the whole network topology. The variation of the epipelagic network in the different impacted and at the control sites was then explored. To this aim, site-specific patterns of over-abundance families were computed, and the respective site-specific over-abundant network plots were created (Supplementary Figure S5). According to our findings, each impacted sites correspond to a specific layout of over-abundant families, resulting in recognizable site-specific declination of the epipelagic network structure.
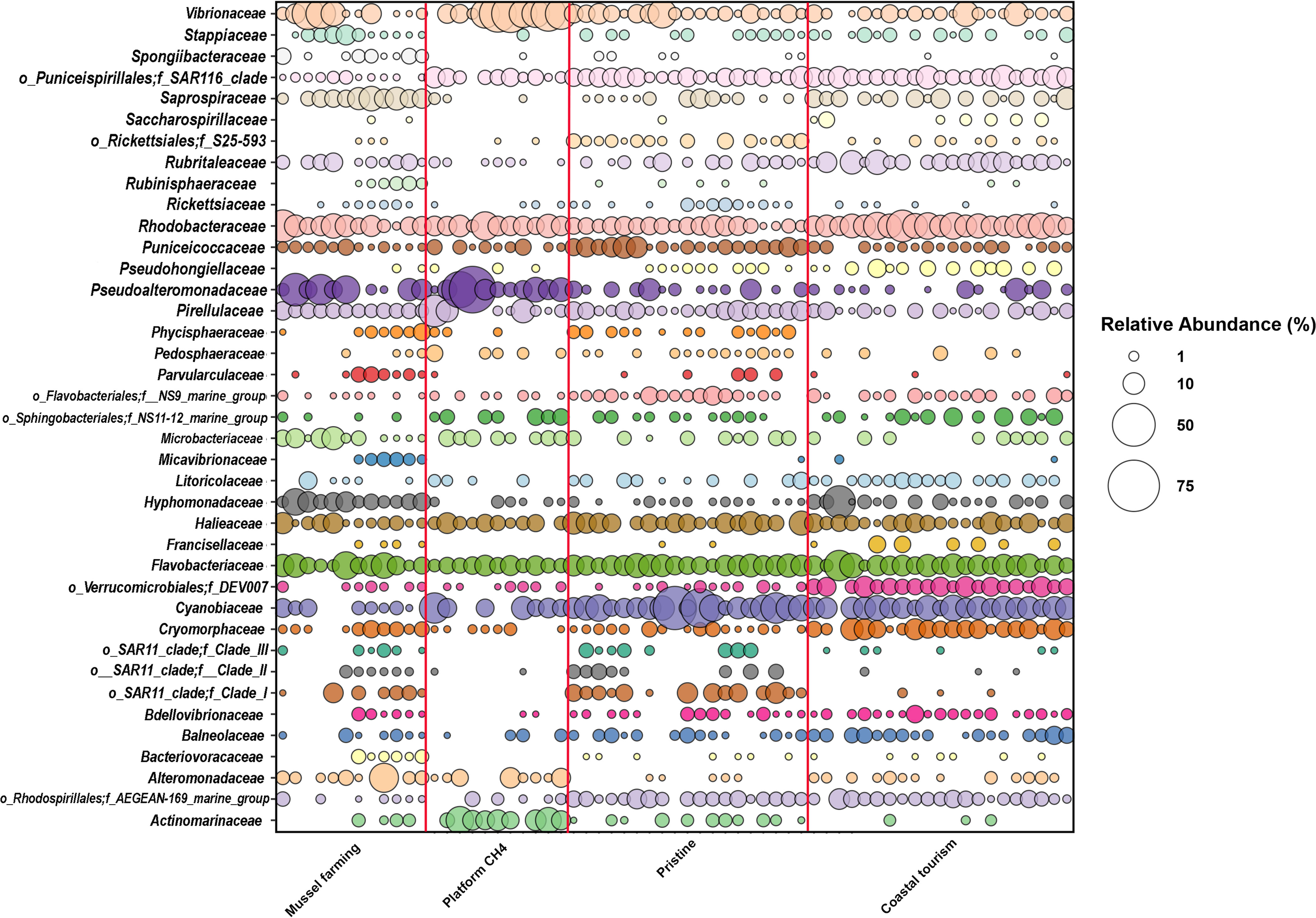
Figure 2 Microbial families’ relative abundance distribution in the study sites. Bubble chart showing the relative abundance of most abundant families (r.ab. ≥ 2.5% in at least two samples) within the four sampling sites (starting from the left: mussel farming, extraction platform, pristine site, and coastal tourism site). Circle sizes correspond to the legend on the right part of the figure.
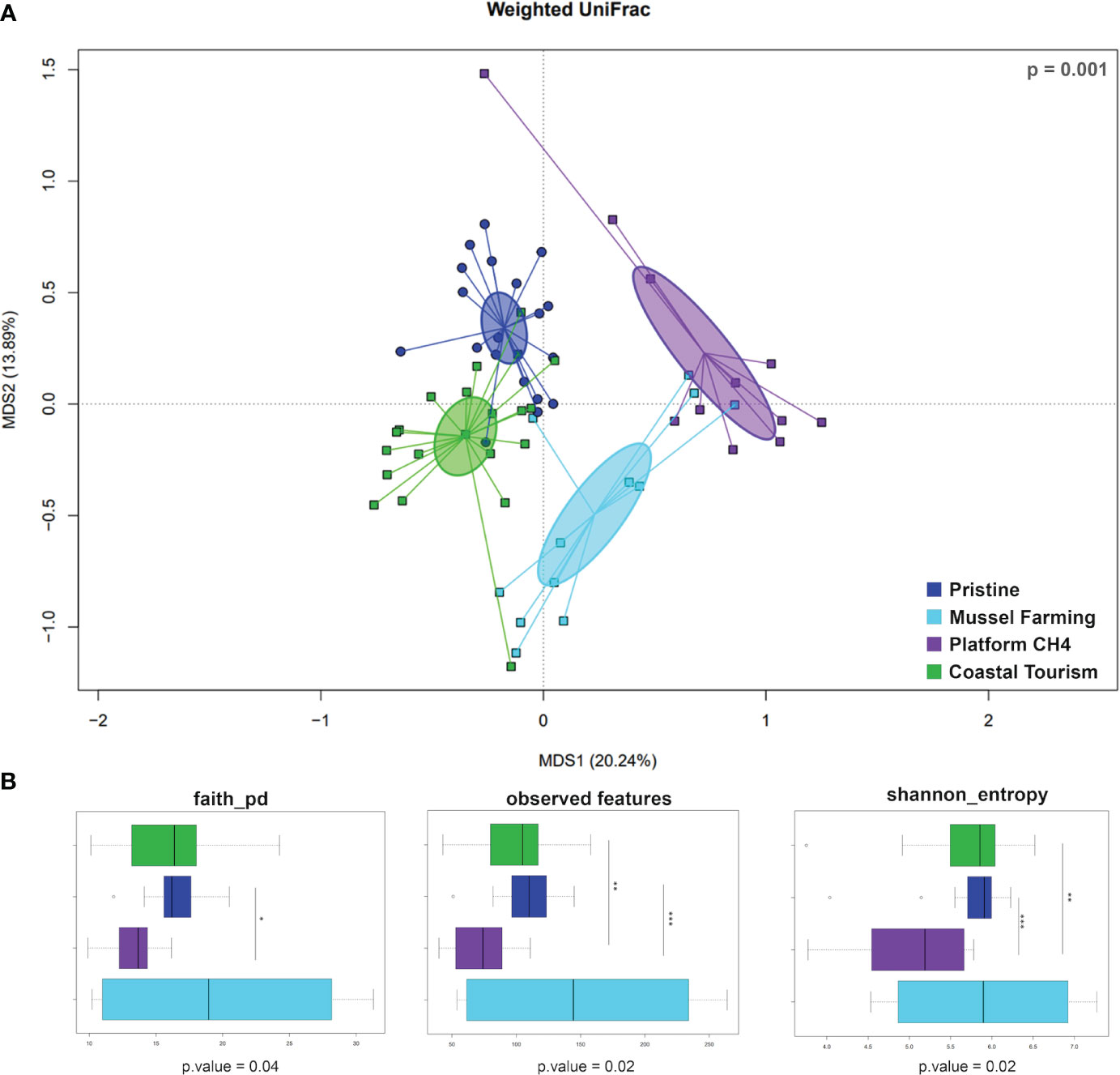
Figure 3 Principal coordinate analysis (PCoA) and alpha-diversity indices of the study sites. (A) PCoA based on weighted UniFrac distances, with color code legend for each site at the bottom-right part of the panel. Study sites are significantly separated (Adonis; p = 0.001). (B) Boxplots of alpha-diversity indices calculated for each study site. The central box represents the distance between the 25th and 75th percentiles, while the median between them is marked with a bold line. Significant variations across study sites are highlighted in the figure (Wilcoxon rank-sum test; p ≤ 0.05*, p ≤ 0.01**, p ≤ 0.001***).
3.2 Variations of the epipelagic pathobiome at the impact sites
In order to characterize the variations of the epipelagic pathobiome structure at the impacted sites, the ASVs assigned to Leptothrichiaceae_sp, Legionellaceae, Campylobacteraceae, Staphylococcaceae, and Enterobacteriaceae have been considered, representing microbial families encompassing well-known pathogens and/or opportunistic pathogens (Palusińska-Szysz and Cendrowska-Pinkosz, 2008; Otto, 2009; O'Donovan et al., 2014; Rock and Donnenberg, 2014; Eisenberg et al., 2016; Sabaté Brescó et al., 2017). Using this approach, we detected 84 epipelagic pathobiome ASVs in our dataset. The Silva classification is provided in Supplementary Table S3. For six of these ASVs, it has been possible to assign the corresponding species using the Silva database, of which one ASV was assigned to Campylobacter ureolyticus and five ASVs were assigned to Leptothrichiaceae_sp. To further improve the species-level assignment of the epipelagic pathobiome members, we implemented the approach reported by Orel et al. (2022). Coherently, when possible, for the ASVs belonging to the abovementioned pathobiont families—and assigned at the genus level on the Silva database—we generated a phylogenetic tree including matching reference species manually curated, allowing the ASV identification based on their clustering close to specific references. We thus obtained a phylogenetic tree for ASVs belonging to Escherichia, Staphylococcus, and Legionella, as shown in Figure 4. According to the trees, one Escherichia ASV was assigned to Escherichia coli, one Staphylococcus ASV to Staphylococcus epidermidis, and one Legionella ASV to Legionella impletisoli. For the ASVs belonging to the possible pathobiome and being identified at the species level, the correspondent distribution in the impacted and control sites was assessed in terms of presence or absence (Supplementary Figure S6). Specifically, the ASV assigned to E. coli was only detected in proximity to the methane extraction platform, whereas the ASVs assigned to Leptothrichiaceae_sp, L. impletisoli, and S. epidermidis were in proximity to mussel farms. Finally, the ASV assigned to C. ureolyticus was only detected at the touristic coastal site.
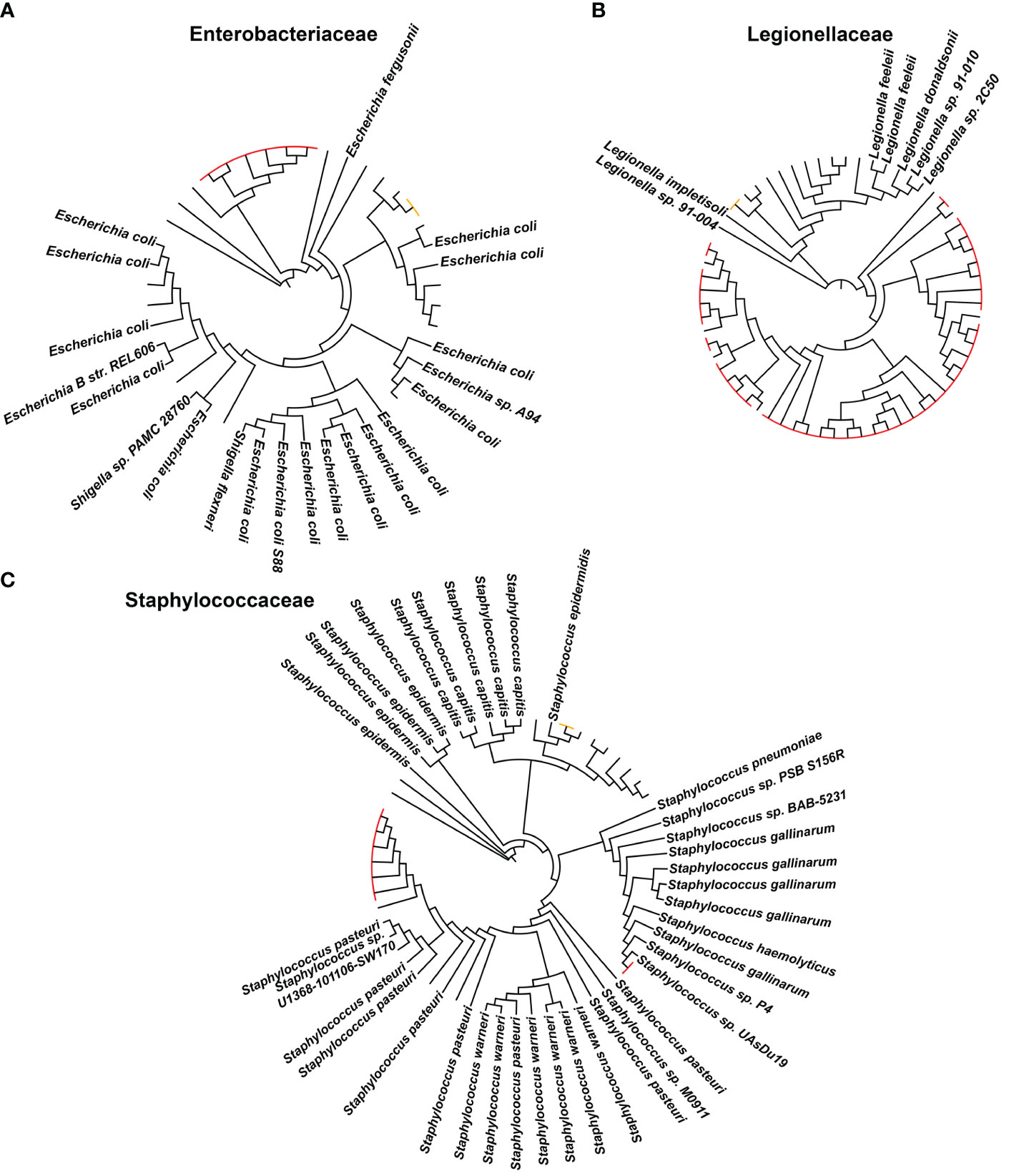
Figure 4 Phylogenetic tree with amplicon sequence variants (ASVs) assigned to (A) Enterococcaceae, (B) Legionellaceae, and (C) Staphylococcaceae families and their neighbors. Within each phylogenetic tree, the taxonomic classification of known species is highlighted with the complete name, while the red-colored labels highlight the ASVs retrieved from our study (the orange ones are the ASVs taken into account for the evaluation of ASV distribution among control and impacted sites).
4 Discussion
In the present work, we explored variations in the marine epipelagic microbiome in response to different sources of anthropogenic stressors, namely, mussel farming, methane extraction activities on offshore platforms, and mass coastal tourism. According to our findings, the three different impacted sites resulted in a well-defined and site-specific fingerprint on the epipelagic microbiome, shaping its general compositional structure at different phylogenetic levels and resulting in site-specific layouts of co-abundant taxa. More specifically, the epipelagic microbiome in the proximity of the mussel farms was enriched in specific carbon oxidizing copiotrophic r-strategists, such as Pseudoalteromonadaceae and Alteromonadaceae (Bowman, 2007; Liu et al., 2011; Ivanova et al., 2014); host-associated microbiome components, such as Ruminococcaceae and Rickettsiales (Darby et al., 2007; Afouda et al., 2019; Klinges et al., 2019); and some potentially pathogenic microorganisms, such as the Leptothrichiaceae (Eisenberg et al., 2016). Not surprisingly, the epipelagic waters in the proximity of the mussel farms were also enriched in known microbial plastisphere components from the marine environment, such as Hypomonadaceae and Saprospiraceae (Dudek and Neuer, 2023) due to the use of plastic socks and other items in this mariculture practice (Skirtun et al., 2022). When the variation of the epipelagic microbiome in the proximity of a methane extraction platform was taken into account, we observed that close to the extraction platform, the epipelagic waters were enriched in specific host-associated microorganisms, such as members of, Vibrionaceae, Bifidobacteriaceae, Bacillaceae, Enterobacteriaceae, and Staphylococcaceae, with the latter four of probable terrestrial origin (Sanders, 2015; Mandic-Mulec et al., 2016; Lugli et al., 2017; Gorrasi et al., 2021; Soto, 2022). Moreover, waters close to the extraction platforms were enriched with specific carbon oxidizing copiotrophic r-strategists, such as Pseudoalteromonadaceae and Alteromonadaceae (Bowman, 2007; Liu et al., 2011; Ivanova et al., 2014), also found to be enriched in the proximity of the mussel farm, and Rhodobacteraceae, a copiotrophic sulfur oxidizer (Sun et al., 2020). Finally, for the touristic impacted site, we observed an increase in a specific set of copiotroph microorganisms, such as Verrucomicrobiales, Alteromonadaceae, and Rhodobacteraceae, possibly due to the proximity with the coast (Liu et al., 2011; Baltar et al., 2018; Sun et al., 2020), as well as the host-associated marine microbiome component (i.e., Rubritalaceae) (Garibay-Valdez et al., 2021).
Although each one of the anthropogenic stressors taken into account resulted in a peculiar increase of specific epipelagic microbial components, all of them shared a common fingerprint in terms of depletion of epipelagic marine microorganisms, such as well-known carbon oxidizing oligotrophic k-strategists (i.e., members of the SAR11 and SAR116 clades) (Choi et al., 2015; Tinta et al., 2015; Hu et al., 2021), phototrophic primary producers (i.e., Cyanobiaceae) (Salazar et al., 2020), and copiotrophic heterotrophs (i.e., Puniceicoccaceae) (Choo et al., 2007).
When exploring changes in the epipelagic network topology at the impacted and control sites, impact-specific changes in the abundance patterns of the six epipelagic microbiome CAGs were observed. More specifically, mussel farming was characterized by an over-abundance of the Saprospiraceae and Clade_I CAGs, while the Vibrionaceae and Rhodobacteraceae CAGs were over-abundant in the extraction platform and coastal tourism-impacted sites, respectively. Finally, all impacted sites were depleted in the Halieaceae CAG with respect to the reference site.
When focusing on the pathobiome components, we detected different potential pathogenic species in the impacted sites. Specifically, Leptothrichiaceae_sp, L. impletisoli, and S. epidermidis were all detected in the proximity of the mussel farm site, whereas E. coli and C. ureolyticus were found in the epipelagic waters in the proximity of the methane extraction platform and at the coastal tourism-impacted site, respectively. While E. coli typically colonizes the human gastrointestinal tract and only a few strains show a pathogenic behavior (Kaper et al., 2004), C. ureolyticus is an emerging gastrointestinal pathogen, capable of causing the destruction of the filamentous microvilli (O'Donovan et al., 2014). In contrast to the impacted sites, no pathogenic microbial species were detected in the control site. Taken together, these findings suggest the release of potentially pathogenic strains in the epipelagic waters from each of the characterized impacted sites, showing a site-specific pathobiome with a recognizable pattern of pathogenic components, possibly representing potential threats to human health.
In conclusion, our findings provide evidence of the existence of identifiable and impact-driven fingerprints on the epipelagic marine microbiome specific to different anthropogenic activities. According to our findings, a specific layout of enriched epipelagic microbiome components, including impact-specific pathogenic species, is associated with each of the described stressors. Conversely, all anthropogenic stressors result in the depletion of characteristic epipelagic microbiome components, such as SAR11 and SAR116 clades and Cyanobiaceae. Even though our findings are still preliminary and need to be confirmed in a wider spatial and temporal context, here, we provide new insights into the potential of the microbiome to provide specific fingerprints for different anthropogenic activities. In addition to providing a new set of indicator bacterial species, specific for the different anthropogenic impact sources, our results also raise the concern of the release of pathogenic microorganisms in the marine ecosystem potentially harmful to human and marine life health.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: Sequencing reads were deposited in ENA (project number PRJEB69608).
Author contributions
GT: Conceptualization, Data curation, Formal Analysis, Methodology, Writing – original draft. DS: Conceptualization, Data curation, Formal Analysis, Methodology, Writing – original draft. LF: Writing – review & editing. AD’A: Writing – review & editing. ER: Writing – review & editing. ST: Writing – review & editing. SR: Writing – review & editing. CC: Writing – review & editing. GP: Methodology, Writing – original draft. MC: Conceptualization, Data curation, Funding acquisition, Methodology, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the “Controlling Microbiomes Circulations for Better Food Systems” (CIRCLES) project, which was funded by the European Union’s Horizon 2020 research and innovation program under grant agreement no. 818290.
Acknowledgments
This study represents partial fulfillment of the requirements for the PhD thesis of Giulia Trapella, Lucia Foresto, Andrea Nicoló Dell’Acqua, and Elena Radaelli in the PhD course of Innovative Technologies and Sustainable Use of Mediterranean Sea Fishery and Biological Resources (FishMed—University of Bologna, Italy). We would like to thank the recreational scuba diving association Blennius (Riccione, Italy), Fondazione Cetacea Onlus (Riccione, Italy), and all the staff and partners of the Sailing for Blue Life Project (Rimini, Italy) for their primary contribution to the sample collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2024.1340088/full#supplementary-material
References
Afouda P., Traore S. I., Dione N., Andrieu C., Tomei E., Richez M., et al. (2019). Description and genomic characterization of Massiliimalia massiliensis gen. nov., sp. nov., and Massiliimalia timonensis gen. nov., sp. nov., two new members of the family Ruminococcaceae isolated from the human gut. Antonie. van Leeuwenhoek. 112, 905–918. doi: 10.1007/s10482-018-01223-x
Agler M. T., Ruhe J., Kroll S., Morhenn C., Kim S. T., Weigel D., et al. (2016). Microbial hub taxa link host and abiotic factors to plant microbiome variation. PloS Biol. 14 (1), e1002352. doi: 10.1371/journal.pbio.1002352
Andolina C., Signa G., Tomasello A., Mazzola A., Vizzini S. (2021). Environmental effects of tourism and its seasonality on Mediterranean islands: the contribution of the Interreg MED BLUEISLANDS project to build up an approach towards sustainable tourism. Environment. Dev. Sustainabil. 23, 8601–8612. doi: 10.1007/s10668-020-00984-8
Arrigo K. R. (2005). Marine microorganisms and global nutrient cycles. Nature 437 (7057), 349–355. doi: 10.1038/nature04159
Istat Annual report on the tourist movement and hotel and complementary consistency in Emilia romagna. Available at: https://statistica.regione.emilia-romagna.it/documentazione/pubblicazioni/documenti_catalogati/rapporto-annuale-turismo-2020.
Baltar F., Gutiérrez-Rodríguez A., Meyer M., Skudelny I., Sander S., Thomson B., et al. (2018). Specific effect of trace metals on marine heterotrophic microbial activity and diversity: Key role of iron and zinc and hydrocarbon-degrading bacteria. Front. Microbiol. 9, 3190. doi: 10.3389/fmicb.2018.03190
Bastari A., Micheli F., Ferretti F., Pusceddu A., Cerrano C. (2016). Large marine protected areas (LMPAs) in the Mediterranean Sea: the opportunity of the Adriatic Sea. Mar. Policy 68, 165–177. doi: 10.1016/j.marpol.2016.03.010
Behzad H., Ohyanagi H., Alharbi B., Ibarra M., Alarawi M., Saito Y., et al. (2022). A cautionary signal from the Red Sea on the impact of increased dust activity on marine microbiota. BMC Genomics 23 (1), 1–12. doi: 10.1186/s12864-022-08485-w
Bokulich N. A., Kaehler B. D., Rideout J. R., Dillon M., Bolyen E., Knight R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6 (1), 1–17. doi: 10.1186/s40168-018-0470-z
Bolyen E., Rideout J. R., Dillon M. R., Bokulich N. A., Abnet C. C., Al-Ghalith G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37 (8), 852–857. doi: 10.1038/s41587-019-0209-9
Bowman J. P. (2007). Bioactive compound synthetic capacity and ecological significance of marine bacterial genus Pseudoalteromonas. Mar. Drugs 5 (4), 220–241. doi: 10.3390/md504220
Buccheri M. A., Salvo E., Coci M., Quero G. M., Zoccarato L., Privitera V., et al. (2019). Investigating microbial indicators of anthropogenic marine pollution by 16S and 18S High-Throughput Sequencing (HTS) library analysis. FEMS Microbiol. Lett. 366 (14), fnz179. doi: 10.1093/femsle/fnz179
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., Holmes S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13 (7), 581–583. doi: 10.1038/nmeth.3869
Chen L., Tsui M. M., Lam J. C., Hu C., Wang Q., Zhou B., et al. (2019). Variation in microbial community structure in surface seawater from Pearl River Delta: Discerning the influencing factors. Sci. Total. Environ. 660, 136–144. doi: 10.1016/j.scitotenv.2018.12.480
Choi D. H., Park K. T., An S. M., Lee K., Cho J. C., Lee J. H., et al. (2015). Pyrosequencing revealed SAR116 clade as dominant dddP-containing bacteria in oligotrophic NW Pacific Ocean. PloS One 10 (1), e0116271. doi: 10.1371/journal.pone.0116271
Choo Y. J., Lee K., Song J., Cho J. C. (2007). Puniceicoccus vermicola gen. nov., sp. nov., a novel marine bacterium, and description of Puniceicoccaceae fam. nov., Puniceicoccales ord. nov., Opitutaceae fam. nov., Opitutales ord. nov. and Opitutae classis nov. in the phylum ‘Verrucomicrobia’. Int. J. Syst. Evol. Microbiol. 57 (3), 532–537. doi: 10.1099/ijs.0.64616-0
Colaleo G., Nardo F., Azzellino A., Vicinanza D. (2022). Decommissioning of offshore platforms in Adriatic Sea: The total removal option from a life cycle assessment perspective. Energies 15 (24), 9325. doi: 10.3390/en15249325
Corinaldesi C., Bianchelli S., Rastelli E., Varrella S., Canensi S., Gambi C., et al. (2022). The paradox of an unpolluted coastal site facing a chronically contaminated industrial area. Front. Mar. Sci. 8, 813887. doi: 10.3389/fmars.2021.813887
Culhane A. C., Thioulouse J., Perrière G., Higgins D. G. (2005). MADE4: an R package for multivariate analysis of gene expression data. Bioinformatics 21 (11), 2789–2790. doi: 10.1093/bioinformatics/bti394
Dang H., Lovell C. R. (2016). Microbial surface colonization and biofilm development in marine environments. Microbiol. Mol. Biol. Rev. 80 (1), 91–138. doi: 10.1128/MMBR.00037-15
Danovaro R. (2003). Pollution threats in the Mediterranean Sea: an overview. Chem. Ecol. 19 (1), 15–32. doi: 10.1080/0275754031000081467
Darby A. C., Cho N. H., Fuxelius H. H., Westberg J., Andersson S. G. (2007). Intracellular pathogens go extreme: genome evolution in the Rickettsiales. Trends Genet. 23 (10), 511–520. doi: 10.1016/j.tig.2007.08.002
Dudek K. L., Neuer S. (2023). Environmental exposure more than plastic composition shapes marine microplastic-associated bacterial communities in Pacific versus Caribbean field incubations. Environ. Microbiol. 25 (12), 2807–2821. doi: 10.1111/1462-2920.16519
Edgar R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26 (19), 2460–2461. doi: 10.1093/bioinformatics/btq461
Eisenberg T., Fawzy A., Nicklas W., Semmler T., Ewers C. (2016). Phylogenetic and comparative genomics of the family Leptotrichiaceae and introduction of a novel fingerprinting MLVA for Streptobacillus moniliformis. BMC Genomics 17 (1), 1–12. doi: 10.1186/s12864-016-3206-0
Findlay R. H., Trexler M. B., Guckert J. B., White D. C. (1990). Laboratory study of disturbance in marine sediments: response of a microbial community. Mar. Ecol. Prog. Series. Oldendorf. 62 (1), 121–133. doi: 10.3354/meps062121
Flombaum P., Gallegos J. L., Gordillo R. A., Rincón J., Zabala L. L., Jiao N., et al. (2013). Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. Proc. Natl. Acad. Sci. 110 (24), 9824–9829. doi: 10.1073/pnas.1307701110
Garibay-Valdez E., Cicala F., Martinez-Porchas M., Gómez-Reyes R., Vargas-Albores F., Gollas-Galván T., et al. (2021). Longitudinal variations in the gastrointestinal microbiome of the white shrimp, Litopenaeus vannamei. PeerJ 9, e11827. doi: 10.7717/peerj.11827
Giovannoni S. J., Tripp H. J., Givan S., Podar M., Vergin K. L., Baptista D., et al. (2005). Genome streamlining in a cosmopolitan oceanic bacterium. science 309 (5738), 1242–1245. doi: 10.1126/science.1114057
Gorrasi S., Pasqualetti M., Franzetti A., Gonzalez-Martinez A., Gonzalez-Lopez J., Muñoz-Palazon B., et al. (2021). Persistence of Enterobacteriaceae drawn into a marine saltern (Saline di Tarquinia, Italy) from the adjacent coastal zone. Water 13 (11), 1443. doi: 10.3390/w13111443
Grilli F., Accoroni S., Acri F., Bernardi Aubry F., Bergami C., Cabrini M., et al. (2020). Seasonal and interannual trends of oceanographic parameters over 40 years in the northern Adriatic Sea in relation to nutrient loadings using the EMODnet chemistry data portal. Water 12 (8), 2280. doi: 10.3390/w12082280
Hernandez D. J., David A. S., Menges E. S., Searcy C. A., Afkhami M. E. (2021). Environmental stress destabilizes microbial networks. ISME J. 15 (6), 1722–1734. doi: 10.1038/s41396-020-00882-x
Hu C., Li X., He M., Jiang P., Long A., Xu J. (2021). Effect of ocean acidification on bacterial metabolic activity and community composition in oligotrophic oceans, inferred from short-term bioassays. Front. Microbiol. 12, 583982. doi: 10.3389/fmicb.2021.583982
Ivanova E. P., Ng H. J., Webb H. K. (2014). The family pseudoalteromonadaceae. Prokaryotes., 575–582. doi: 10.1007/978-3-642-38922-1_229
Kaper J. B., Nataro J. P., Mobley H. L. (2004). Pathogenic escherichia coli. Nat. Rev. Microbiol. 2 (2), 123–140. doi: 10.1038/nrmicro818
Klindworth A., Pruesse E., Schweer T., Peplies J., Quast C., Horn M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41 (1), e1–e1. doi: 10.1093/nar/gks808
Klinges J. G., Rosales S. M., McMinds R., Shaver E. C., Shantz A. A., Peters E. C., et al. (2019). Phylogenetic, genomic, and biogeographic characterization of a novel and ubiquitous marine invertebrate-associated Rickettsiales parasite, Candidatus Aquarickettsia rohweri, gen. nov. sp. nov. ISME J. 13 (12), 2938–2953. doi: 10.1038/s41396-019-0482-0
Lauro F. M., McDougald D., Thomas T., Williams T. J., Egan S., Rice S., et al. (2009). The genomic basis of trophic strategy in marine bacteria. Proc. Natl. Acad. Sci. 106 (37), 15527–15533. doi: 10.1073/pnas.0903507106
Letunic I., Bork P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49 (W1), W293–W296. doi: 10.1093/nar/gkab301
Liao H., Lin X., Li Y., Qu M., Tian Y. (2020). Reclassification of the taxonomic framework of orders cellvibrionales, oceanospirillales, pseudomonadales, and alteromonadales in class gammaproteobacteria through phylogenomic tree analysis. Msystems 5 (5), 10–1128. doi: 10.1128/mSystems.00543-20
Liu M., Dong Y., Zhao Y., Zhang G., Zhang W., Xiao T. (2011). Structures of bacterial communities on the surface of Ulva prolifera and in seawaters in an Ulva blooming region in Jiaozhou Bay, China. World J. Microbiol. Biotechnol. 27, 1703–1712. doi: 10.1007/s11274-010-0627-9
Lugli G. A., Milani C., Turroni F., Duranti S., Mancabelli L., Mangifesta M., et al. (2017). Comparative genomic and phylogenomic analyses of the Bifidobacteriaceae family. BMC Genomics 18 (1), 1–15. doi: 10.1186/s12864-017-3955-4
Luna G. M. (2015). Diversity of marine microbes in a changing Mediterranean Sea. Rendiconti. Lincei. 26, 49–58. doi: 10.1007/s12210-014-0333-x
Mandic-Mulec I., Stefanic P., van Elsas J. D. (2016). Ecology of bacillaceae. Bacterial. Spore.: From. Mol. to Syst., 59–85. doi: 10.1128/9781555819323.ch3
Masella A. P., Bartram A. K., Truszkowski J. M., Brown D. G., Neufeld J. D. (2012). PANDAseq: paired-end assembler for illumina sequences. BMC Bioinf. 13, 1–7. doi: 10.1186/1471-2105-13-31
Naidoo S., Olaniran A. O. (2014). Treated wastewater effluent as a source of microbial pollution of surface water resources. Int. J. Environ. Res. Public Health 11 (1), 249–270. doi: 10.3390/ijerph110100249
Numberger D., Ganzert L., Zoccarato L., Mühldorfer K., Sauer S., Grossart H. P., et al. (2019). Characterization of bacterial communities in wastewater with enhanced taxonomic resolution by full-length 16S rRNA sequencing. Sci. Rep. 9 (1), 9673. doi: 10.1038/s41598-019-46015-z
O'Donovan D., Corcoran G. D., Lucey B., Sleator R. D. (2014). Campylobacter ureolyticus: a portrait of the pathogen. Virulence 5 (4), 498–506. doi: 10.4161/viru.28776
Orel N., Fadeev E., Klun K., Ličer M., Tinta T., Turk V. (2022). Bacterial indicators are ubiquitous members of pelagic microbiome in anthropogenically impacted coastal ecosystem. Front. Microbiol. 12, 765091. doi: 10.3389/fmicb.2021.765091
Otto M. (2009). Staphylococcus epidermidis—the'accidental'pathogen. Nat. Rev. Microbiol. 7 (8), 555–567. doi: 10.1038/nrmicro2182
Palladino G., Caroselli E., Tavella T., D’Amico F., Prada F., Mancuso A., et al. (2022b). Metagenomic shifts in mucus, tissue and skeleton of the coral Balanophyllia europaea living along a natural CO2 gradient. ISME Commun. 2 (1), 65. doi: 10.1038/s43705-022-00165-w
Palladino G., Rampelli S., Galià-Camps C., Scicchitano D., Trapella G., Nanetti E., et al. (2022a). Plasticity of the Anemonia viridis microbiota in response to different levels of combined anthropogenic and environmental stresses. Front. Mar. Sci. 9, 956899. doi: 10.3389/fmars.2022.956899
Palusińska-Szysz M., Cendrowska-Pinkosz M. (2008). Occurrence and pathogenicity of the family of Legionellaceae. Adv. Hygiene. Exp. Med. 62. doi: 10.1007/s00005-009-0035-8
Prioli G. (2006). “Perspectives and problems in italian shellfish farming,” in AQUA2006 International Conference, Florence. (pp. 9–(pp13).
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41 (D1), D590–D596. doi: 10.1093/nar/gks1219
Rock C., Donnenberg M. S. (2014). Human pathogenic enterobacteriaceae, reference module in biomedical sciences. Elsevier. doi: 10.1016/B978-0-12-801238-3.00136-7
Sabaté Brescó M., Harris L. G., Thompson K., Stanic B., Morgenstern M., O'Mahony L., et al. (2017). Pathogenic mechanisms and host interactions in Staphylococcus epidermidis device-related infection. Front. Microbiol. 8, 1401. doi: 10.3389/fmicb.2017.01401
Salazar V. W., Tschoeke D. A., Swings J., Cosenza C. A., Mattoso M., Thompson C. C., et al. (2020). A new genomic taxonomy system for the Synechococcus collective. Environ. Microbiol. 22 (11), 4557–4570. doi: 10.1111/1462-2920.15173
Salter I., Galand P. E., Fagervold S. K., Lebaron P., Obernosterer I., Oliver M. J., et al. (2015). Seasonal dynamics of active SAR11 ecotypes in the oligotrophic Northwest Mediterranean Sea. ISME J. 9, 347–360. doi: 10.1038/ismej.2014.129
Sanders J. G. (2015). Disentangling the coevolutionary histories of animal gut microbiomes. Harvard. Univ.
Scanlan D. J., West N. J. (2002). Molecular ecology of the marine cyanobacterial genera Prochlorococcus and Synechococcus. FEMS Microbiol. Ecol. 40 (1), 1–12. doi: 10.1111/j.1574-6941.2002.tb00930.x
Scicchitano D., Lo Martire M., Palladino G., Nanetti E., Fabbrini M., Dell’Anno A., et al. (2022). Microbiome network in the pelagic and benthic offshore systems of the northern Adriatic Sea (Mediterranean Sea). Sci. Rep. 12 (1), 16670. doi: 10.1038/s41598-022-21182-8
Skirtun M., Sandra M., Strietman W. J., van den Burg S. W., De Raedemaecker F., Devriese L. I. (2022). Plastic pollution pathways from marine aquaculture practices and potential solutions for the North-East Atlantic region. Mar. pollut. Bull. 174, 113178. doi: 10.1016/j.marpolbul.2021.113178
Soto W. (2022). Emerging research topics in the vibrionaceae and the squid–vibrio symbiosis. Microorganisms 10 (10), 1946. doi: 10.3390/microorganisms10101946
Sun F., Wu M., Wang Y., Sun C., Xu Z. (2020). Diversity and potential function of bacterial communities in different upwelling systems. Estuarine. Coast. Shelf. Sci. 237, 106698. doi: 10.1016/j.ecss.2020.106698
Tinta T., Vojvoda J., Mozetič P., Talaber I., Vodopivec M., Malfatti F., et al. (2015). Bacterial community shift is induced by dynamic environmental parameters in a changing coastal ecosystem (northern A driatic, northeastern M editerranean S ea)–a 2-year time-series study. Environ. Microbiol. 17 (10), 3581–3596. doi: 10.1111/1462-2920.12519
Zuccato E., Castiglioni S., Fanelli R. (2005). Identification of the pharmaceuticals for human use contaminating the Italian aquatic environment. J. Hazardous. Mter. 122 (3), 205–209. doi: 10.1016/j.jhazmat.2005.03.001
Keywords: anthropogenic threats, epipelagic ecosystems, marine microbiome, Adriatic Sea, metagenomic next- generation sequencing
Citation: Trapella G, Scicchitano D, Foresto L, Dell’Acqua AN, Radaelli E, Turroni S, Rampelli S, Corinaldesi C, Palladino G and Candela M (2024) Signature of the anthropogenic impacts on the epipelagic microbiome of the North-Western Adriatic Sea (Mediterranean Sea). Front. Mar. Sci. 11:1340088. doi: 10.3389/fmars.2024.1340088
Received: 17 November 2023; Accepted: 15 January 2024;
Published: 02 February 2024.
Edited by:
Hao Zhang, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Yantao Liang, Ocean University of China, ChinaYafen Wang, China University of Geosciences, China
Copyright © 2024 Trapella, Scicchitano, Foresto, Dell’Acqua, Radaelli, Turroni, Rampelli, Corinaldesi, Palladino and Candela. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Candela, bWFyY28uY2FuZGVsYUB1bmliby5pdA==
†These authors have contributed equally to this work and share first authorship