 Matthew R. Jones1*
Matthew R. Jones1* Rosie Chance1
Rosie Chance1 Thomas Bell2
Thomas Bell2 Oban Jones2†David C. Loades1Rebecca May2†Liselotte Tinel1†Katherine Weddell1†
Oban Jones2†David C. Loades1Rebecca May2†Liselotte Tinel1†Katherine Weddell1† Claire Widdicombe2
Claire Widdicombe2 Lucy J. Carpenter1*
Lucy J. Carpenter1*- 1Wolfson Atmospheric Chemistry Laboratory, University of York, York, United Kingdom
- 2Plymouth Marine Laboratory, Prospect Place, Plymouth, United Kingdom
The surface ocean is the main source of iodine to the atmosphere, where it plays a crucial role including in the catalytic removal of tropospheric ozone. The availability of surface oceanic iodine is governed by its biogeochemical cycling, the controls of which are poorly constrained. Here we show a near two-year time series of the primary iodine species, iodide, iodate and dissolved organic iodine (DOI) in inner shelf marine surface waters of the Western English Channel (UK). The median ± standard deviation concentrations between November 2019 and September 2021 (n=76) were: iodide 88 ± 17 nM (range 61-149 nM), iodate 293 ± 28 nM (198-382 nM), DOI 16 ± 16 nM (<0.12-75 nM) and total dissolved iodine (dIT) 399 ± 30 nM (314-477 nM). Though lower than inorganic iodine ion concentrations, DOI was a persistent and non-negligible component of dIT, which is consistent with previous studies in coastal waters. Over the time series, dIT was not conserved and the missing pool of iodine accounted for ~6% of the observed concentration suggesting complex mechanisms governing dIT removal and renewal. The contribution of excess iodine (I*) sourced from the coastal margin towards dIT was generally low (3 ± 29 nM) but exceptional events influenced dIT concentrations by up to ±100 nM. The seasonal variability in iodine speciation was asynchronous with the observed phytoplankton primary productivity. Nevertheless, iodate reduction began as light levels and then biomass increased in spring and iodide attained its peak concentration in mid to late autumn during post-bloom conditions. Dissolved organic iodine was present, but variable, throughout the year. During winter, iodate concentrations increased due to the advection of North Atlantic surface waters. The timing of changes in iodine speciation and the magnitude of I* subsumed by seawater processes supports the paradigm that transformations between iodine species are biologically mediated, though not directly linked.
1 Introduction
Elemental iodine transformations are an important global biogeochemical cycle that influences our environment (Tsunogai and Sase, 1969; Carpenter et al., 2021). Iodine in seawater impacts climate (He et al., 2021; Cuevas et al., 2022) and global health because it contributes to the removal of harmful ozone from the atmospheric boundary layer directly via the reaction of ozone with iodide at the sea surface (Garland et al., 1980) and indirectly via release of iodine in gaseous form to the atmosphere (Carpenter et al., 2013; Carpenter et al., 2021). Photochemical transformations of iodine gases can lead to new particle formation that may act as cloud condensation nuclei (He et al., 2023), thus influencing atmospheric radiative properties (Saiz-Lopez et al., 2012; Gómez Martín et al., 2022). Iodine emissions from seawater are in the form of organic compounds (Lovelock, 1975; Carpenter et al., 2003) and inorganic molecules (Carpenter et al., 2013; MacDonald et al., 2014; Tinel et al., 2020; Carpenter et al., 2021), the latter are most important in terms of emissions (Carpenter et al., 2021). Iodine in the terrestrial environment is predominantly sourced by emissions from coastal seawater (Fuge, 1989; Fuge and Johnson, 2015). These emissions improve regional air quality by removing ground level ozone (Carpenter et al., 2013; Carpenter et al., 2021) and are the main source of iodine – an essential micronutrient (WHO) – to soils and subsequently the food chain (Fuge and Johnson, 2015).
In oceanic seawater, iodine is classified as a biophilic element (Goldschmidt, 1954) and is primarily in the forms of thermodynamically stable iodate (IO3–), iodide (I–; ~25% of the total dissolved iodine (dIT) in surface waters) and iodine associated with dissolved organic material (typically representing ~5% of dIT; Elderfield and Truesdale, 1980; Wong, 1980; Luther et al., 1991; Wong, 1991; Chance et al., 2014; Fuge and Johnson, 2015; Luther, 2023). The central tenet of iodine marine biogeochemistry is that transformations between soluble chemical species are biologically mediated (Tsunogai and Sase, 1969; Amachi et al., 2007), though chemical processes cannot be ruled out (Schnur et al., 2024). This tenet derives from observations of temporal changes in iodine speciation in ocean waters concluding that from spring through to autumn iodide accumulates and iodate is removed, and over winter the reverse occurs (Truesdale, 1978; Jickells et al., 1988; Campos et al., 1996; Tian et al., 1996; Truesdale and Jones, 2000; Truesdale and Upstill-Goddard, 2003; Waite et al., 2006; Chance et al., 2010; Satoh et al., 2019a; Shi et al., 2023). These observations support the observed global ocean iodide-nitrate anticorrelation and the supposition that iodate is either assimilated during primary production and iodide is released, or iodate is enzymatically reduced, possibly by nitrate reductase enzymes (Tsunogai and Sase, 1969; Wong and Zhang, 1992; Campos et al., 1999; Amachi et al., 2005; Amachi et al., 2007).
Microbially forced changes to iodine species have been observed in experimental systems. Iodate reduction occurred during phytoplankton growth (Waite and Truesdale, 2003; Hung et al., 2005; Amachi et al., 2007) and senescence, the latter concurrently with increasing bacterial biomass (Bluhm et al., 2010; Carrano et al., 2020; Hepach et al., 2020). Both, laboratory and field observations indicate a significant lag time between iodate loss and iodide formation (Chance et al., 2007; Bluhm et al., 2010; Chance et al., 2010; Carrano et al., 2020; Hepach et al., 2020). Iodide oxidation may also occur directly by bacteria (iodide oxidation rate, ~75 mM d-1) or be an indirect artefact of bacterial processes [e.g., ammonia oxidation (~2.5 μM d-1), organic acid-mediated hydrogen peroxide production (~14.4 mM d-1) or manganese oxide catalysed superoxide production (Amachi et al., 2005; Li et al., 2012; Li et al., 2014; Hughes et al., 2021)].
Relative to open ocean and outer shelf waters, the concentration of dissolved iodine in coastal inner shelf waters is lower (Truesdale, 1978; Truesdale, 1994; Truesdale and Upstill-Goddard, 2003; Satoh et al., 2019a; Satoh et al., 2023; Shi et al., 2023). The reason for this is not well known (Chance et al., 2014), though a chemical transformation to a particulate fraction may account for the apparent depreciated soluble iodine concentrations (Truesdale, 1994; Truesdale and Upstill-Goddard, 2003; Satoh et al., 2019a; Satoh et al., 2023). Processes directly influencing the iodine cycle in wider coastal environments include interaction with dissolved organic material during its remineralization and sorption to particulate material (Truesdale, 1978; Tian et al., 1996; Satoh et al., 2019a; Shi et al., 2023), the mixing of deeper low oxygen high iodide waters to the surface (Shi et al., 2023), and significant changes in dissolved organic iodine (DOI) concentration (ΔDOI up to 100 nM; Satoh et al., 2019a). In coastal systems, the iodine cycle may also be influenced by higher life forms including macroalgae and invertebrates (e.g., jellyfish) that use organic and inorganic iodine species as antioxidants and signaling molecules (Silverstone et al., 1977; Küpper et al., 1998; Prieto et al., 2010; Cross et al., 2015; Gonzales et al., 2017; Tymon et al., 2017; Carrano et al., 2020; Carrano et al., 2021). Furthermore, inner shelf waters have a greater opportunity for interactions with sediments, which can influence water column iodine speciation (e.g., Ullman and Aller, 1980; Francois, 1987; Farrenkopf and Luther, 2002). When abiotic processes are combined with microbial processes they should meet the observed net rates of coastal in situ iodide oxidation of 0.33-0.52 nM d-1 (Hardisty et al., 2020).
The presence of elevated levels of organic material and nutrients from terrestrial sources is a significant difference between inner shelf and open ocean marine systems (Mendoza and Zika, 2014; Barrón and Duarte, 2015; Carr et al., 2019). The presence of organic material influences DOI formation and iodine geochemistry because DOI can account for up to 40% of dIT in estuarine and coastal environments (Luther et al., 1991; Abdel-Moati, 1999; Cook et al., 2000; Wong and Cheng, 2001a; Satoh et al., 2019a). Though DOI consists of many varied compounds (Schulz et al., 1974; Harvey, 1980; Wong, 1982; Luther et al., 1991; Cook et al., 2000; Moulay, 2013) some may be hypervalent organic iodine (Francois, 1987) while a fraction (pico-molar) is covalently bonded volatile iodine (Lovelock, 1975; Schall and Heumann, 1993; Dembitsky, 2006).
In this paper, we present a near two-year time series detailing the major iodine species in temperate inner shelf marine surface waters of the Western English Channel. These coastal waters extend from the shoreline and outer limit of estuaries to depths of ~30 m, and have near to oceanic salinities. The time series provides insight into the factors controlling iodine speciation at the sea surface and therefore its impacts on the magnitude of ozone deposition, sea-to-air iodine flux, availability of iodine as a nutrient to terrestrial environments and air quality (Fuge, 1989; Carpenter et al., 2013; Fuge and Johnson, 2015; Carpenter et al., 2021). To evaluate the controlling factors, the inner shelf iodine species were compared to those on the outer shelf at the Western Channel Observatory (Station L4). All observations were part of a larger project aiming to constrain the role of the sea-surface microlayer in controlling ozone flux. The data will be used to inform those observations and subsequent research. The novelty of these data is the inclusion of DOI and a quantification of the fraction of excess iodine due to the interaction of the terrestrial marine system. These data also provide vital inner shelf iodine species concentrations to constrain modelling studies, i.e., Sherwen et al. (2019); Wadley et al. (2020) and Pound et al. (2023).
2 Methods
2.1 Location and sampling
From November 2019 through to September 2021 seawater was collected from inner and outer shelf locations in the Western English Channel (Figure 1) by the crew of the Plymouth Marine Laboratory’s (UK) research vessel Plymouth Quest. The samples were taken from the vessel’s clean underway system (inlet depth 3 m). The inner shelf location was ~0.5 km offshore of Rame Head, Cornwall, and in the vicinity of the Penlee Point Atmospheric Observatory, the water column depth was 20-25 m. The inner shelf site was further split into two; an east site (50° 19’ 12” N, 4° 10’ 12” W) and a west site (50° 18’ 32.4” N, 4° 11’ 27.6” W) because sampling was always conducted upwind of the Penlee Point Atmospheric Observatory. The samples were collected approximately weekly (weather permitting), sampling was more intensive in 2021 than in 2020. A total of 76 weeks of samples were collected from these locations. The outer shelf location was the Western Channel Observatory (Station L4; 50°15′ N, 4°13′ W; 52 m depth) situated ~6 km offshore from which 14 samples were collected at monthly intervals during the springs, summers and autumns of 2020 and 2021. Following collection, all seawater was filtered through Whatman fine glass fibre membranes (GF/F; nominal pore size 0.7 μm). Filtered seawater was stored at -20°C in acid-washed plastic containers to preserve iodine speciation (Campos, 1997). Before analysis, samples were thawed overnight at 4°C and in the dark.
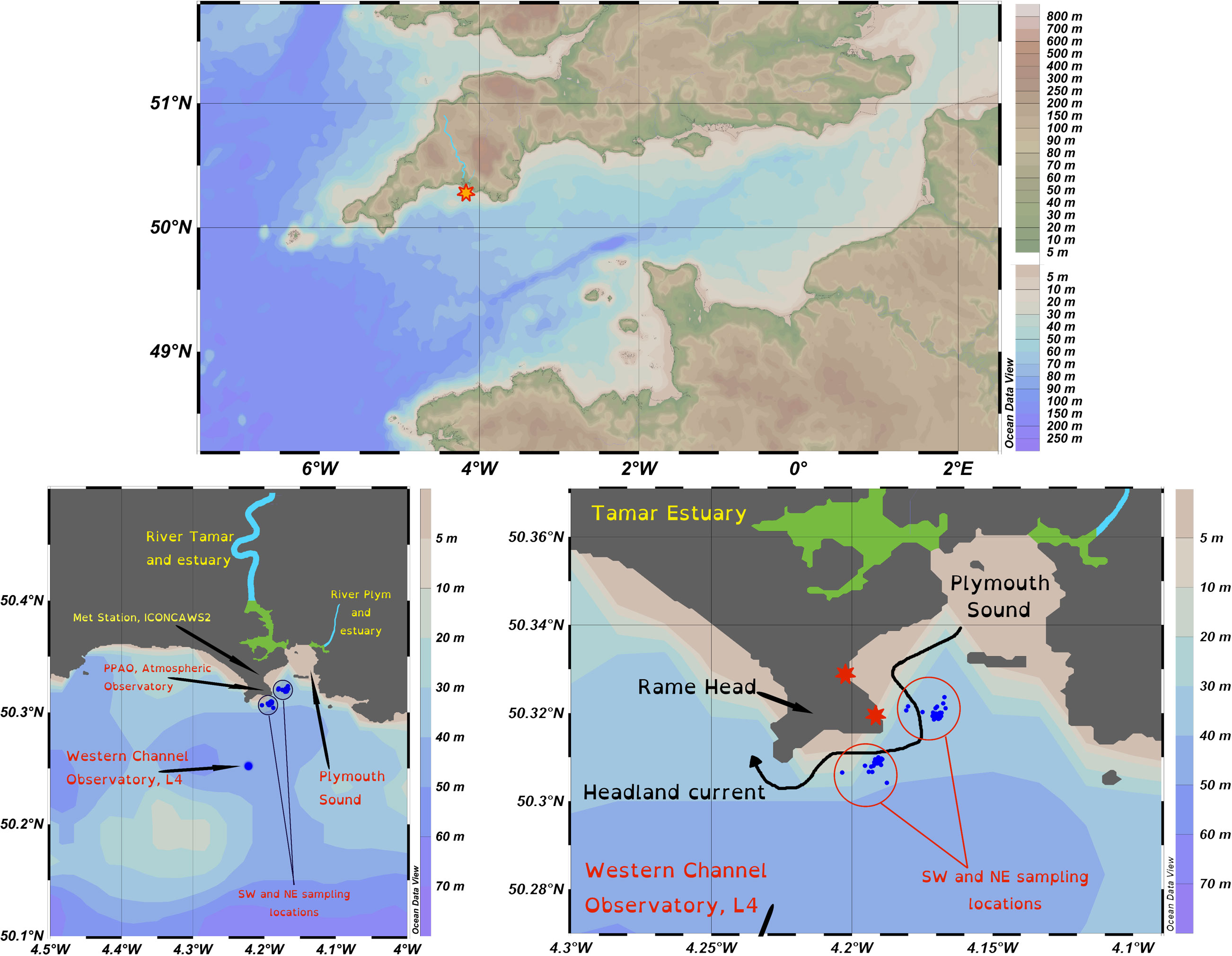
Figure 1 Location map of sampling site(s). Top, English Channel with the UK to the north and France to the south, the Tamar River is highlighted in light blue and the sampling locations by the star. Bottom left, annotated map showing inner and outer shelf sampling locations (SW, NE and L4), and major rivers (light blue) and their estuaries (green) running into Plymouth Sound. Bottom right, inner shelf sampling stations relative to Rame Head, the meteorological stations (red stars), and the path of the Rame Head Current. Map produced in Ocean Data View, Schlitzer, R., Ocean Data View, https://odv.awi.de, 2018.
2.2 Analytical
2.2.1 Iodine species
Iodide was determined spectrophotometrically following an ion chromatography (IC) separation of a 400 μL sample (Jones et al., 2023). Separation and determination were conducted on an Agilent 1100 HPLC with a 1260 series detector. The UV detector monitored absorbance at 226 nm over a 60 mm path length. The IC isocratic mobile phase (0.4 M NaCl) ran at 0.64 mL min-1. The IC guard and analytical columns were Dionex IonPac AS-23 4×50 mm and 4×250 mm, respectively. Once injected, the chromatogram was collected for 16.1 minutes, iodide eluted at c.11 minutes. Gravimetrically prepared stock solutions of potassium iodide (KI; Fisher >99.0%) in 18.1 MΩ deionized water were used to prepare analytical standards. Iodate and DOI were quantified as iodide following chemical amendments before the chromatography (Jones et al., 2023). Hydroxylamine hydrochloride (NH2OH-HCl; Sigma-Aldrich Reagent Plus, 99%) was added to a final concentration of 7 mM to reduce iodate to iodide and enable the measurement of the inorganic iodine fraction (Iinorg = iodide + iodate) and quantification, by difference, of iodate. A second chemical manipulation enabled measurement of the total dissolved iodine fraction (dIT = Iinorg + DOI) and hence quantification, by difference, of DOI (Lister and Rosenblum, 1963; Takayanagi and Wong, 1986; Abdel-Moati, 1999; Jones et al., 2023). For DOI determination, the chemical amendment to the sample was an addition of hypochlorite, in the form of calcium hypochlorite (Ca(ClO)2; Sigma-Aldrich Technical Grade), to a final concentration of 189 μM. After 1 h, the hypochlorite was neutralized through addition of sodium sulfite (Na2SO3; Sigma-Aldrich, min. 98%) to a concentration of 380 μM and then NH2OH-HCl was added to a final concentration of 7 mM. There may be an underestimation of the DOI concentration as the analytical method may not capture all iodine within DOI (Luther and Cole, 1988; Wong and Cheng, 1998; Wong and Cheng, 2001b). The methods were found to yield iodate and DOI measurements in excellent agreement with values obtained using the established methods of spectrophotometry (116 ± 9%, n=103) and UV-degradation (98 ± 4%, n=98) (Jones et al., 2023). All samples, treated and untreated, were measured in triplicate and the standard deviation of those determinations was used to calculate the analytical error. The relative standard deviation for each analysis was typically <2%. Sample concentrations were blank corrected using values derived from the additions of the amendments to deionized H2O. All samples were quantified as iodide, which had a detection limit of 0.12 nM, calculated as the equivalent concentration of 3× the standard deviation of ten repeated 1 nM standards.
2.2.2 Nutrient, physicochemical, dissolved organic carbon and meteorological measurements
Except for the soluble iodine species and dissolved organic carbon, all analytes [nutrients (Supporting Information (SI), Supplementary Figure SI.1) and physicochemical parameters (Supplementary Figure SI.2)] were determined by the Plymouth Marine Laboratory (UK). Nutrient analysis was conducted on a 5-channel segmented flow system (Bran+Luebbe) with quality control procedures implemented. Phosphate was determined as phosphor-molybdenum blue (analytical Λ = 710 nm) (Zhang and Chi, 2002) and silicate as silico-molybdenum blue (Kirkwood, 1989). Nitrate and nitrite ions were determined using the Greiss method; nitrate was reduced on a copper cadmium column in an ammonium chloride solution (Brewer and Riley, 1965; Grasshoff, 1976). Ammonia was determined as indophenol blue (analytical Λ = 650 nm) (Mantoura and Woodward, 1983). Due to it being subject to storage and contamination problems, the ammonium data should be viewed more as trends rather than highly accurate concentrations. Chlorophyll-α (chl-α; mg L-1) concentrations were determined fluorometrically from acetone extractions of the particulate phase captured on GF/F filters and normalized for filtrate volume (Yentsch and Menzel, 1963). An array of electronic sensors (Supplementary Table SI.1), referred to as the CTD, were deployed from the research vessel beginning in January 2020. The calibrated sensors measured seawater conductivity (salinity), temperature, pressure, turbidity, in situ photosynthetically active radiation (PAR), oxygen and fluorescence (a proxy for chl-α). Phytoplankton biomass (mg carbon (C) L-1) on the outer shelf location was taken from the British Oceanographic Data Centre (BODC; Widdicombe and Harbour, 2021). The biomass on the inner shelf was calculated by applying the ratio of the CTD-measured fluorescence from the inner to outer shelf sites as a multiplicative factor to the outer shelf biomass. Dissolved organic carbon (DOC) was calibrated using a total organic carbon standard (TOC standard 50 mg L-1, Sigma-Aldrich). Samples, standards and blanks for DOC analysis were diluted in 18.1 MΩ deionized water, placed in 12 mL vials and covered with tin foil. These samples were acidified with small volumes of 1.2 M HCl and then the DOC content was determined using an Elementar Vario TOC cube. Supporting meteorological data was retrieved from the WeatherUnderground station (ICONCAWS2, Cawsands Bay). The met station was situated ~1 km from the inner shelf sampling locations (Figure 1).
2.2.3 Rationalisation and excess iodine (I*)
The established practice that accounts for the dilution of seawater by freshwater is to normalize analyte concentrations to a salinity of 35 (termed ‘rationalization’, see Equation 1; Truesdale, 1994), this approach assumes that the freshwater end member has a zero iodine concentration. Rationalisation (R) does not provide knowledge of a species/elements behavior in the environment, just a relative abundance to all dissolved salts (Truesdale, 1995).
To evaluate contributions to dIT from estuarine and submarine groundwater discharge, we examined how observed dIT concentrations deviated from a conservative mixing curve (linear model). This model was chosen because most observations suggest that dIT behaves conservatively in estuarine systems (Luther et al., 1991; Abdel-Moati, 1999; Cook et al., 2000; Truesdale et al., 2001; Lin, 2023). Mixing curve end members were defined using the outer shelf average dIT (406.5 ± 15.0, n=6) of the six highest salinity samples (SP = 34.93 ± 0.08, n=6), and an assumed freshwater dIT concentration of 25 nM (Fuge, 1989) and salinity of zero. The freshwater endmember is the average of southern UK surface (x̅=23 nM, n=9) and well waters (x̅=26 nM, n=6). Excess iodine (I*) was calculated as the difference in determined and predicted dIT.
2.2.4 Statistical analysis
Concentrations of all chemical species may exhibit short-term variability, i.e. concentration changes driven by variability on a scale shorter than the approximately weekly sampling resolution. This variability may reflect relatively rapid diel natural processes that may not have been fully captured such as tidal fluctuation, precipitation, photosynthesis and photochemistry or multi-day processes such as phytoplankton bloom progression. To focus on longer-term seasonal changes, the subsequent comparisons of chemical species concentrations, unless explicitly stated, are from a LOESS regression model with a 12-point spline (R-package(s) ggplot2/predictdf or Tidyverse/geom_smooth). The model fitted a localized quadratic curve with a 12-point spline that effectively smoothed short-period natural variability while succinctly capturing mid to longer-period changes. A consequence of comparing LOESS model values is that the relative concentration changes between chemical species are more conservative than were measured. Pearson correlation coefficients were used to assess potential relationships between all biological, physiochemical and iodine species, both rationalized and unrationalised (Data Table 01).
3 Results
Here we focus on the 76 samples collected from the inner shelf waters. The (14) outer shelf samples are used as a comparison to inform the discussion of the inner shelf samples. The similarities, within a 12-point spline 95% confidence limit, and difference of the inner and outer shelf samples are observable in Figure 2; Supplementary Figures SI.1, SI.2.
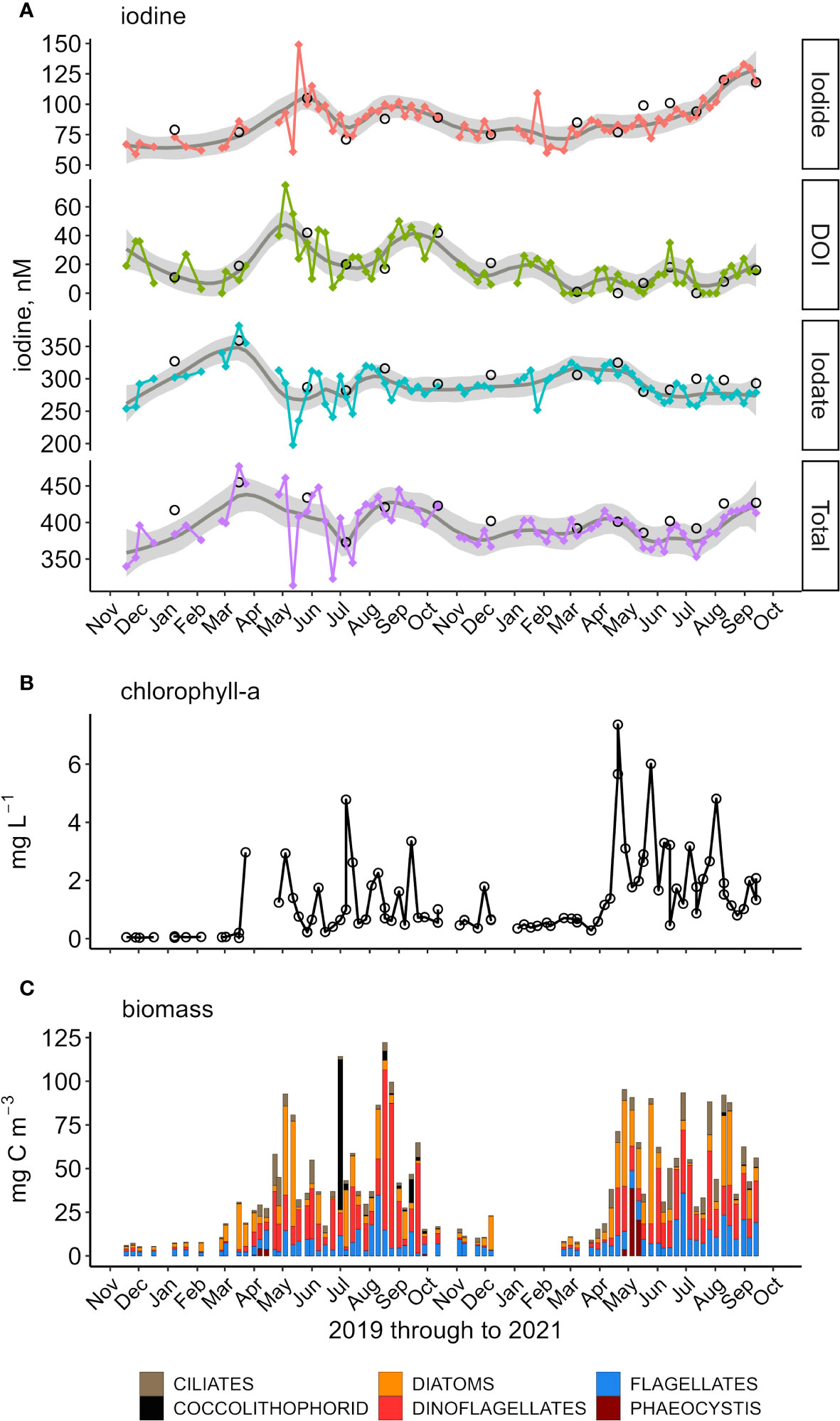
Figure 2 (A) Temporal change of iodine species and the total dissolved iodine in inner shelf waters; DOI, dissolved organic iodine. The grey envelope represents the 95% confidence range over a 12-point LOESS moving average. Large circles, outer shelf. Breaks in the time series data are because no samples were collected for a period of >16 d. (B) Inner shelf chlorophyll-α concentrations and (C) Outer shelf phytoplankton biomass broken down by dominant group.
3.1 Water column characteristics
Within the inner shelf waters, the homogenous temperature distribution with depth indicates that the summer waters were generally well-mixed, i.e., there was no significant temperature step change between the surface and deepest measuring point (Figure 3; Supplementary Figure SI.3). However, there were small differences in salinity, during the 2020-2021 autumn-winter and winter-spring transitions because of a winter pycnocline (δSP ~1) at ~8 m (Figure 3; Supplementary Figure SI.3). Also, in summer 2020 there was higher salinity (SP >35.5) in the near surface waters (<2 m) (Figure 3; Supplementary Figure SI.3). The salinity profiles indicated a small seasonality in the waters to a depth of ~10 m (Figure 3; Supplementary Figure SI.3). Winter salinity generally ranged between 33.2 and 34.5 (two early samples were measured at SP = 32.9) while summer salinity ranged between 34.5 to 35.5 (Figure 3). Lower winter salinities are likely because of higher flows of terrestrially sourced water (river and groundwater) due to greater precipitation (Supplementary Figure SI.4).
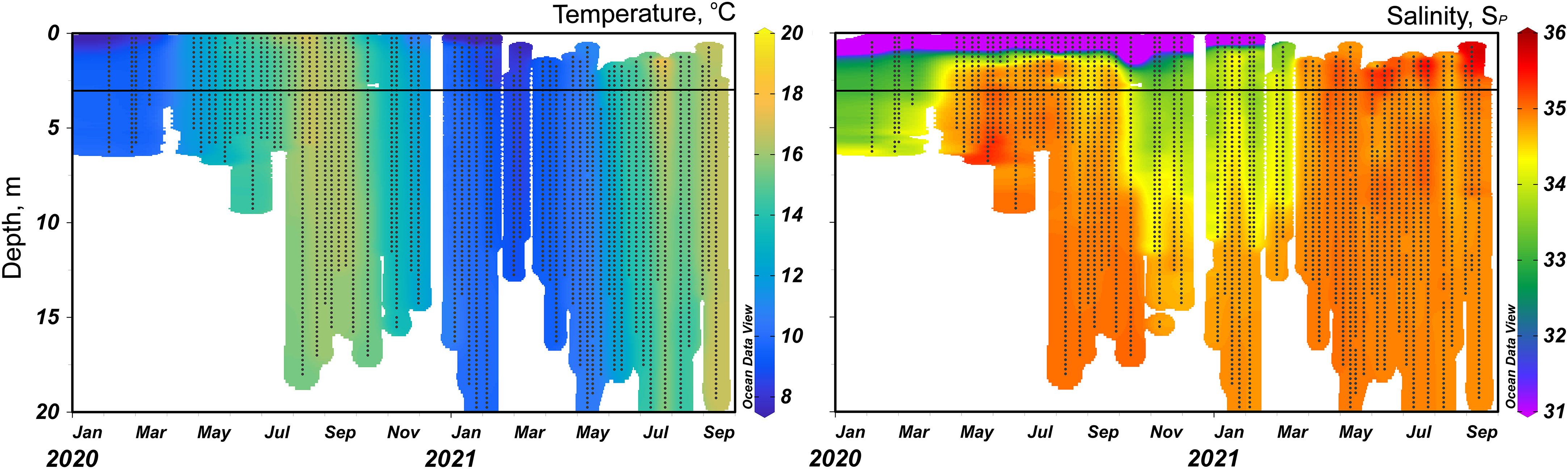
Figure 3 Temperature and salinity profiles of the inner shelf waters, the water column depth is variable but was no more than 20-25 m. The horizontal black line represents the sampling depth. #1.) During winter to summer of 2020 water column measurements were curtailed at ~6 m and full profiles are unavailable. #2.) 2020 abnormally low salinities in waters shallower than 2.5 m is a sampling artefact. #3.) 2021 near surface water values are not available as sampling was not conducted. Profiles produced in Ocean Data View, Schlitzer, R., Ocean Data View, https://odv.awi.de, 2018.
3.2 Dissolved iodine species
3.2.1 Inner shelf total dissolved iodine
Total dissolved iodine (dIT) exhibited a bi-modal seasonal maximum with maxima in the winter/spring and summer/autumn transitions (Table 1; Figures 2A, 4E) – referred to as the early and late season maxima. These maxima were more evident in 2020 than in 2021. The 2020 early season maxima were due to an accumulation over the four months beginning December 2019 where dIT increased by 54 nM up to 440 nM. That increase was entirely removed by July 2020 leaving only 369 nM of dIT. The late season maxima (Table 1), was due to dIT increasing by ~60 nM up to 426 nM from July to late August. High dIT concentrations (>415 nM) remained for 2 months during autumn. A subsequent loss of ~50 nM down to 375 nM dIT occurred between November 2019 and the December 2020 winter minimum. The changes in dIT during 2021 follow a broadly similar pattern to those during 2020, but the maximum concentrations were lower. The 2020 early and late season maxima, 406 and 419 nM, were in early April and mid-September, respectively, with new iodine contributing 21 and 34 nM. Also, the early summer (June) of both years exhibited a small (relative to early and late season maxima) increase in dIT. This early summer maximum in 2020 was incorporated within the spring maximum.
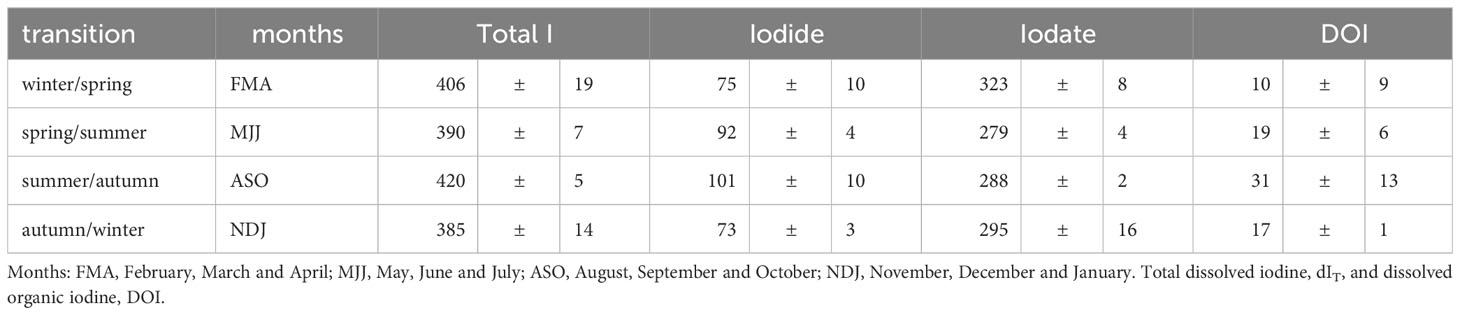
Table 1 Comparison of iodine species concentrations during transitions between the classical season categories.
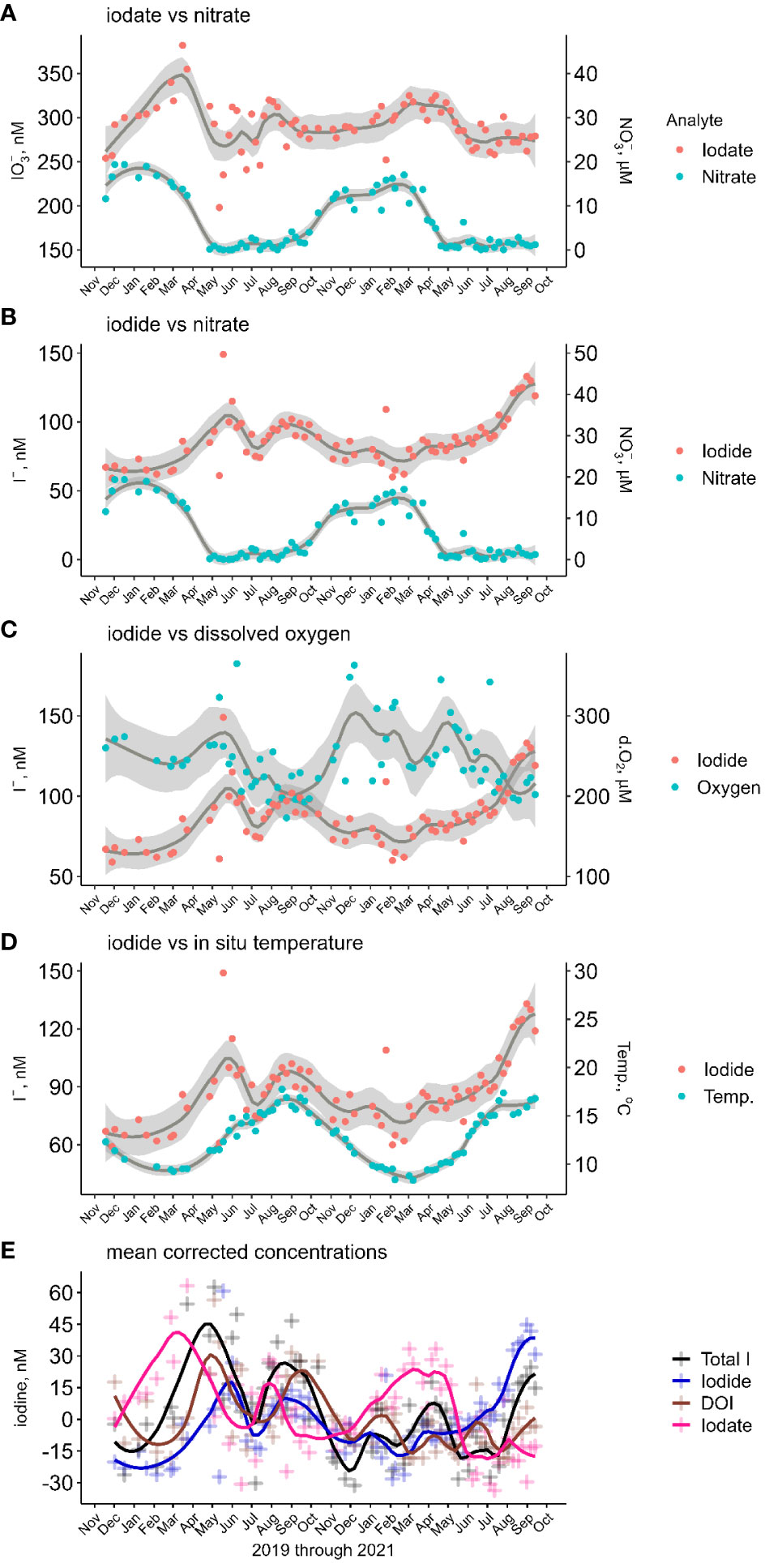
Figure 4 (A–D) Comparison of iodate and iodide to nitrate, dissolved oxygen and seawater temperature in the inner shelf waters. The grey envelopes represent the 95% confidence range over a 12-point LOESS moving average. (E) Temporal change from mean corrected total dissolved iodine and iodine species in inner shelf waters.
3.2.2 Inner shelf iodide
Iodide ranged from 61 to 149 nM (average 88 ± 17 nM), with a seasonality characterized by high concentrations in the summer/autumn transition and low concentrations in the autumn/winter transition (Table 1), albeit with significant temporal differences between years (Figure 2A). In 2020, iodide was represented by a bi-modal distribution model (maxima in May and August) and in 2021 a single maximum (September). During winter 2019/2020 until early March iodide concentrations were relatively invariant at 68 ± 3 nM. Iodide increased at a rate of 1.1 nM d-1 through to the first maximum of 109 nM in late May. The summer iodide minimum (81 nM) in July was followed by a late summer maximum in August (98 nM), this maximum concentration plateaued for ~6 weeks. The year 2020 ended with iodide decreasing to 77 nM (end of November). In contrast, and following the winter minimum of 2021, iodide concentrations gradually increased at ~0.1 nM d-1 between mid-March and July, then more rapidly by ~1 nM d-1 over August. For both years, the lowest iodide concentrations were found in February, 65 nM in 2020 and 71 nM in 2021.
3.2.3 Inner shelf dissolved organic iodine
Dissolved organic iodine represented 5 ± 4% of dIT and had an average concentration of 19 ± 16 nM. In 2020, temporal changes in DOI were broadly similar to those of iodide with bi-modal maxima over spring-summer, while in 2021 changes were more nuanced (Figure 2A). Both years began with decreasing concentrations through to March. Over the first winter, DOI concentrations decreased from 30 to 7 nM, from December 2019 to February 2020. Coincidently, the relative decrease in DOI from January 2021 (20 nM) through to March 2021 (2 nM) was similar to the previous year. The bimodal 2020 DOI profile showed early and late maxima occurring in May and September with >40 nM DOI. From late summer, the DOI concentrations decreased through to December by >30 nM. This was followed by a mid-winter maximum of 20 nM DOI (January 2021). In both 2020 and 2021, DOI concentrations decreased at the beginning of each year as iodate increased.
3.2.4 Inner shelf iodate
Iodate contributed 74 ± 5% of dIT and had an average concentration of 293 ± 27 nM (Table 2). Though a significant contributor of dIT, iodate showed only a single seasonal maximum which occurred in the winter/spring transition (Table 1; Figure 2A) and followed a gradual increase over autumn and winter. In March 2020, iodate peaked at 351 nM following a 64 nM increase (LOESS regression model, actual increase of 95 nM) from November 2019. In March 2021, the iodate maximum of 316 nM followed a smaller increase of 34 nM over the seven months from September 2020. There were inter-annual differences in the changes of iodate concentration following its seasonal maximum (Figure 2A). In the spring of 2020, iodate decreased by 85 nM from 351 to 269 nM according to the LOESS model. These model values do not capture the significantly larger measured decrease of 184 nM observed between the only two iodate outliers (> ± 3σ) of 382 nM on 16/03/2020 and 198 nM on 12/05/2020, representing a rate of iodate decrease of 3.3 nM d-1 between early and late spring. In comparison, the rate of observed iodate decrease in spring 2021 was less rapid at only 1.2 nM d-1; in this year the early spring (March–April) iodate concentrations were reasonably stable (314 ± 1 nM) rather than spiking to >380 nM. In the summer of 2020, iodate increased up to 305 nM in August whereas in the summer of 2021 concentrations remained low (275 ± 2 nM LOESS model vs 275 ± 12 nM measured, n=16).
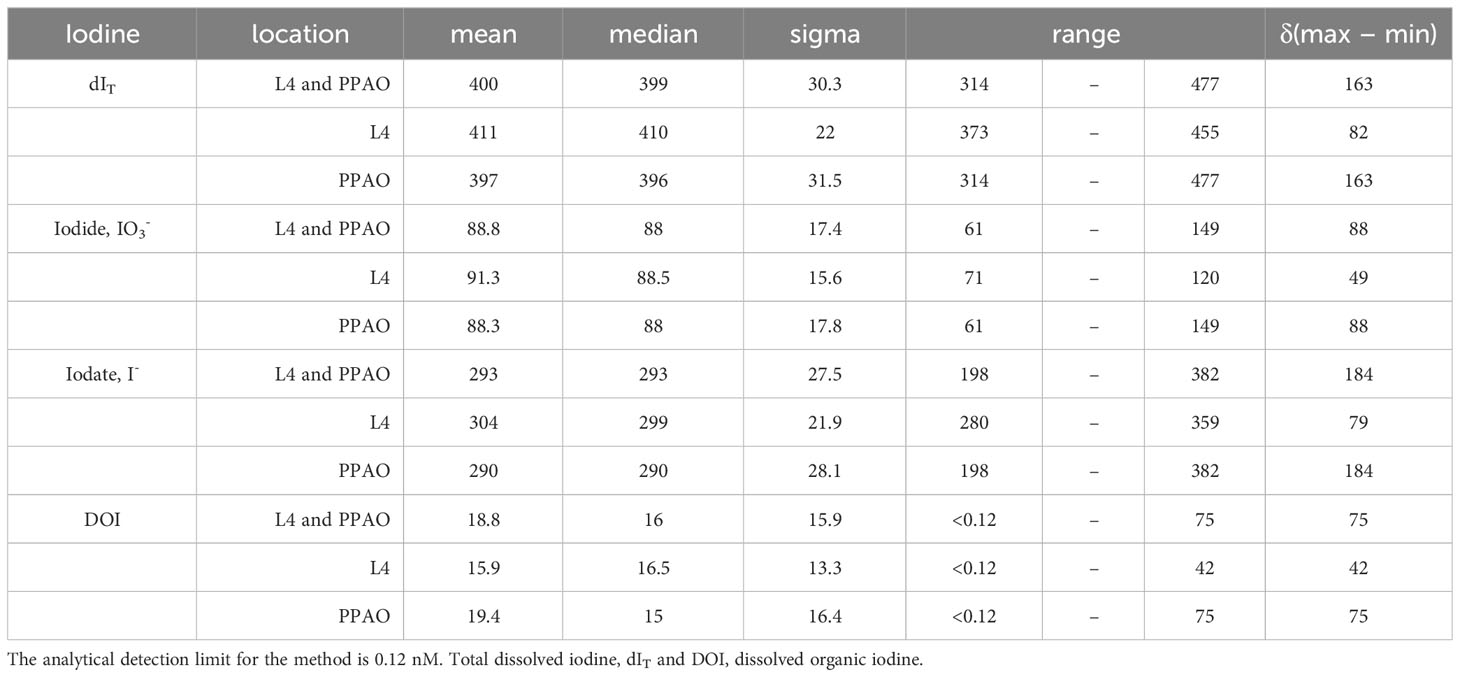
Table 2 Statistical analysis of two years of measured iodine species concentrations (nM) broken down by inner or outer shelf sampling location.
3.3 Chlorophyll-α, biomass, phytoplankton assemblage and nutrients
Chlorophyll-α is an indicator of phytoplankton biomass and reflects the level of primary production. In general, chl-α was low in winter (2019: 0.05 mg L-1; 2020: 0.4 mg L-1) and higher in summer, spiking to >3 mg L-1 (2020) and >5 mg L-1 in 2020 (Figure 2B). Both years saw the first rise in chl-α in late March whereafter concentrations varied by up to 3 mg L-1 as phytoplankton blooms progressed through the summer. Nevertheless, in the summer of 2020 chl-α minimum concentrations were maintained at the relatively low winter 2020 concentrations whereas in 2021 summer chl-α was generally (except June 2002) maintained at >1.5 mg L-1. In the outer shelf waters, four primary phytoplankton groups were present: diatoms, dinoflagellates, flagellates and to some lesser extent ciliates (Figure 2C). During the 2019/2020 winter, the biomass (mg carbon (C) m-3) was approximately half of that during winter 2020/2021. Following the first winter, an early spring (March) diatom bloom preceded the subsequent gradual build-up of biomass through April. The main spring bloom was in May 2020 (>50% diatoms by biomass). In contrast, the May 2021 spring bloom appeared more suddenly, from April onwards. Following the spring bloom, there were differences between years in the progression of phytoplankton and how biomass was maintained within the water column. In 2020, and with one exception, the summer biomass was ~40 mg C m-3 until the late summer bloom (>100 mg C m-3 with >80% dinoflagellates). The exception was a 1-week period where coccolithophores were present in mid-summer (July) and the biomass increased to 114 mg C m-3 (75% coccolithophores). This coccolithophore bloom was present over a large area (Supplementary Figures SI.6, SI.7). In contrast, following the 2021 spring bloom, there was an approximately 5-week periodicity in the arrival of mixed assemblage phytoplankton blooms. This cycling ensured that the median biomass during the summer of 2021 remained higher than that of the summer of 2020, 56 ± 25 vs 42 ± 29 mg C m-3, respectively. In general, concentrations of the major (nitrate, phosphate, silicate) and minor (ammonia, nitrate) nutrients were greater on the inner shelf than on the outer shelf (Supplementary Figure SI.1, compare the position of discrete open circles (outer shelf) relative to connected filled circles (inner shelf)). Nitrate showed a strong winter-to-summer seasonality; winter concentrations were between 15 and 20 μM (outer shelf >5 μM) and summer concentrations were typically <2 μM (outer shelf <0.5 μM). The seasonal cycles for phosphate and silicate were similar to nitrate. The minor nutrients, nitrite and ammonia, behaved similarly within the inner shelf waters, these species exhibited a bi-modal model with high concentrations in February and October (~1.0 μM NH3 and >0.3 μM NO2-). The corresponding minima were in spring early/summer (<0.4 μM NH3 and <0.1 μM NO2-) and mid-winter.
4 Discussion
4.1 Nature of the sampling site
The North Atlantic Ocean is the source water for the Western English Channel (French and British territorial and exclusive economic zone waters). Prevailing winds force Atlantic waters into the Western English Channel and these progress eastwards, water column mixing on the shelf is controlled by tides and weather (Cooper, 1960; Huthnance, 2010). Outer shelf waters develop a thermocline with a typical depth of ~20 m (Supplementary Figure SI.5), this stratification begins in March and ends in October, though severe wind events induce mixing with deep waters (Cooper, 1960; Huthnance, 2010). Seasonal stratification is known to impact the vertical distribution of iodine species, in that development of a thermocline facilitates the accumulation of iodide in the surface layer in association with biological activity, while overturning will dilute this by mixing with deeper, higher iodate/lower iodide waters (Chance et al., 2014; Wadley et al., 2020). These processes will impact the seasonally stratified outer shelf site which supplies the inner shelf, but will not take place at the inner shelf site as it is fully mixed year-round.
The water column at the inner shelf location was within the Rame Head current [Figure 1 and Uncles et al. (2015)]. Even in summer, the inner shelf sampling site does not undergo seasonal thermal stratification as observed on the outer shelf (Uncles et al., 2015). Moreover, the seawater dynamics of Plymouth Sound and within the Rame Head current mean that turbulent mixing processes including seabed friction, tide-, wind- and density-driven flows and the Coriolis Force limit stratification of waters between 0.5 and ~30 m deep on periods longer than a diel cycle (Wright, 1993; Upstill-Goddard, 2006; Bosboom and Stive, 2021). The temperature of the generally well mixed inner shelf water column (Figure 3; Supplementary Figure SI.3) was between 8 and 11°C during winter, 12 to 13°C during spring and autumn, and >16°C during summer.
Relative to the estimated global deep ocean dIT concentration of ~450-480 nM (Tsunogai and Sase, 1969; Barker and Zeitlin, 1972; Elderfield and Truesdale, 1980; Jickells et al., 1988; Nakayama et al., 1989; McTaggart et al., 1994; Campos et al., 1996; Tian et al., 1996; Farrenkopf et al., 1997; Moriyasu et al., 2020; Moriyasu et al., 2023; Schnur et al., 2024) inner shelf waters were depleted (x̅=400 ± 30 nM, range 314-477 nM; Table 2). Nevertheless, the average dIT concentration of both inner and outer shelf seawater was within the range of reported Atlantic surface waters dIT concentrations (≈435 nM, Elderfield and Truesdale (1980) and Truesdale et al. (2000); 390 ± 20 nM, He et al. (2013)). The inner shelf dIT concentration showed a bi-modal seasonal maximum (Figures 2A, 4E). The first maximum was likely caused by winter advection of Atlantic surface waters across the shelf as the spring dIT concentrations (>400 nM) were more similar to surface Atlantic source waters (Elderfield and Truesdale, 1980; Truesdale et al., 2000; He et al., 2013).
Advection and tidal forcing of Western English Channel shelf sea waters into Plymouth Sound results in those waters exiting via a broad headland current that first flows into Cawsand Bay and then around Rame Head (Siddorn et al., 2003; Uncles et al., 2015). The residence time of seawater in Plymouth Sound prior to arriving at the sampling site is ~2.5 days (Uncles et al., 2015). During this period, freshwaters, (e.g., submarine groundwater discharge and estuarine waters – River Plym (minor contribution) and River Tamar (major contribution)) will influence seawater composition. Additions of terrestrial and anthropogenic material within freshwaters entering Plymouth Sound coupled with the residence time are the likely cause of the higher inner shelf nutrient concentrations (Section 3.3; Supplementary Figures SI.1, SI.4) with higher nutrients driving higher primary productivity (Lotze et al., 2006; Canfield et al., 2010).
4.2 Iodine dynamics in a terrestrially influenced inner shelf seawater
4.2.1 Influence of freshwater
Seawater at the outer shelf site is primarily ocean sourced with only episodic summer inputs from the Tamar River (Rees et al., 2009; Supplementary Figure SI.5) while inner shelf waters are continuously modified by terrestrially derived fresh water. In freshwater, the primary iodine species are iodide or DOI (Schwehr and Santschi, 2003; Tang et al., 2013; Zhang et al., 2013). Freshwater chemical species entering the marine system because of submarine groundwater discharge (Tait et al., 2023) or from estuaries may first undergo chemical changes and removal and resuspension cycles due to ion exchange and precipitation, though whether dIT behavior is conservative is under debate (Smith and Butler, 1979; Ullman and Aller, 1980; Takayanagi and Cossa, 1985; Francois, 1987; Luther and Cole, 1988; Abdel-Moati, 1999; Cook et al., 2000; Jones and Tebo, 2021; Lin, 2023). In the case of iodine, estuarine processes may even entirely remove any freshwater iodine such that the only effect of freshwater is expected to be dilution of the seawater concentration, i.e., it can be described by Equation 1 (Takayanagi and Cossa, 1985). However, with no measurement of iodine concentrations in Plymouth Sound’s freshwater sources (two major estuaries plus ground water submarine discharge), we cannot definitively state this.
Calculation of excess iodine (I*) provides a means to estimate additional sources of dIT to the inner shelf, albeit with uncertainty arising from poorly constrained freshwater and marine endmember concentrations. From 73 samples, 34 had negative I* and 39 (53%) had positive I*, therefore there was both a sink and a source of dIT. The average I* contribution was 3 ± 29 nM (n=73) and 60 (82%) samples were within one standard deviation (29 nM) of equality (I* = 0). There were nine samples with I* >25 nM (range, 28-98 nM) and five with I* ranging from -28 to -90 nM indicating that episodic process(es) that influence I* could significantly alter dIT by more than the assumed freshwater concentration (25 nM). Though I* is higher in summer than winter when river flow is correspondingly low (Supplementary Figure SI.8A) there appears to be no relationship with river flow (Supplementary Figure SI.8B), nor is there a strong relationship in I* with the fraction of freshwater in the sample (Supplementary Figure SI.8C) or tide height (Supplementary Figure SI.8D). Nevertheless, iodine concentrations in the water column of an estuary have previously been observed to increase as river flow decreases (Abdel-Moati, 1999). Lower estuarine flow rates result in longer residence times for estuarine waters, and hence, more estuarine processes/sediment pore water interactions. This is important because upper sediment pore water dIT concentrations of the lower Tamar River estuary are generally between 2-5 μM (Upstill-Goddard and Elderfield, 1988) so at low river flow they may be a source of iodine as iodide but not iodate. Conversely, sediment entrapment to the water column that increases suspended load may decrease soluble iodine concentration (Truesdale, 1994; Truesdale and Upstill-Goddard, 2003; Satoh et al., 2019a; Satoh et al., 2023). The low relative mean contribution of I* (3 nM) and the relative consistency of dIT (399 ± 30 nM) indicates that seawater successfully subsumes seawater margin processed dIT contributions.
Rationalized iodine (Section 2.2.3) supported the initial conclusion that dIT was not conserved over seasonal and annual scales at the inner shelf sampling site (Figures 5B, C). This is consistent with the fact these waters show asynchronous cycling of iodate, iodide and DOI (Figures 2A, 4E). Over a year it was anticipated that the dIT concentrations may be conserved because dIT has previously shown conservative properties (Tian et al., 1996). A contributing factor towards the non-conservation of dIT is that the variability in the spatial concentration of dIT in North Atlantic surface waters is also high (390 ± 20 nM He et al., 2013 and outer shelf dIT Figure 2A). In terms of concentration, and once normalized for salinity, the dIT (x̅=413 ± 31 nM) concentrations observed at the inner shelf are closer to the Truesdale et al. (2000) than the He et al. (2013) observations. Importantly, observed and normalized dIT changes over time were similar (Figure 5B) indicating dilution of seawater by freshwater while in Plymouth Sound is not a significant driver of the concentration changes over the seasonal time series. This conclusion is also in agreement with Shi et al. (2023) who did not find a relationship between salinity and dIT over a wider salinity range, SP = 26-31 during a 4.5-year time series in a fjord.
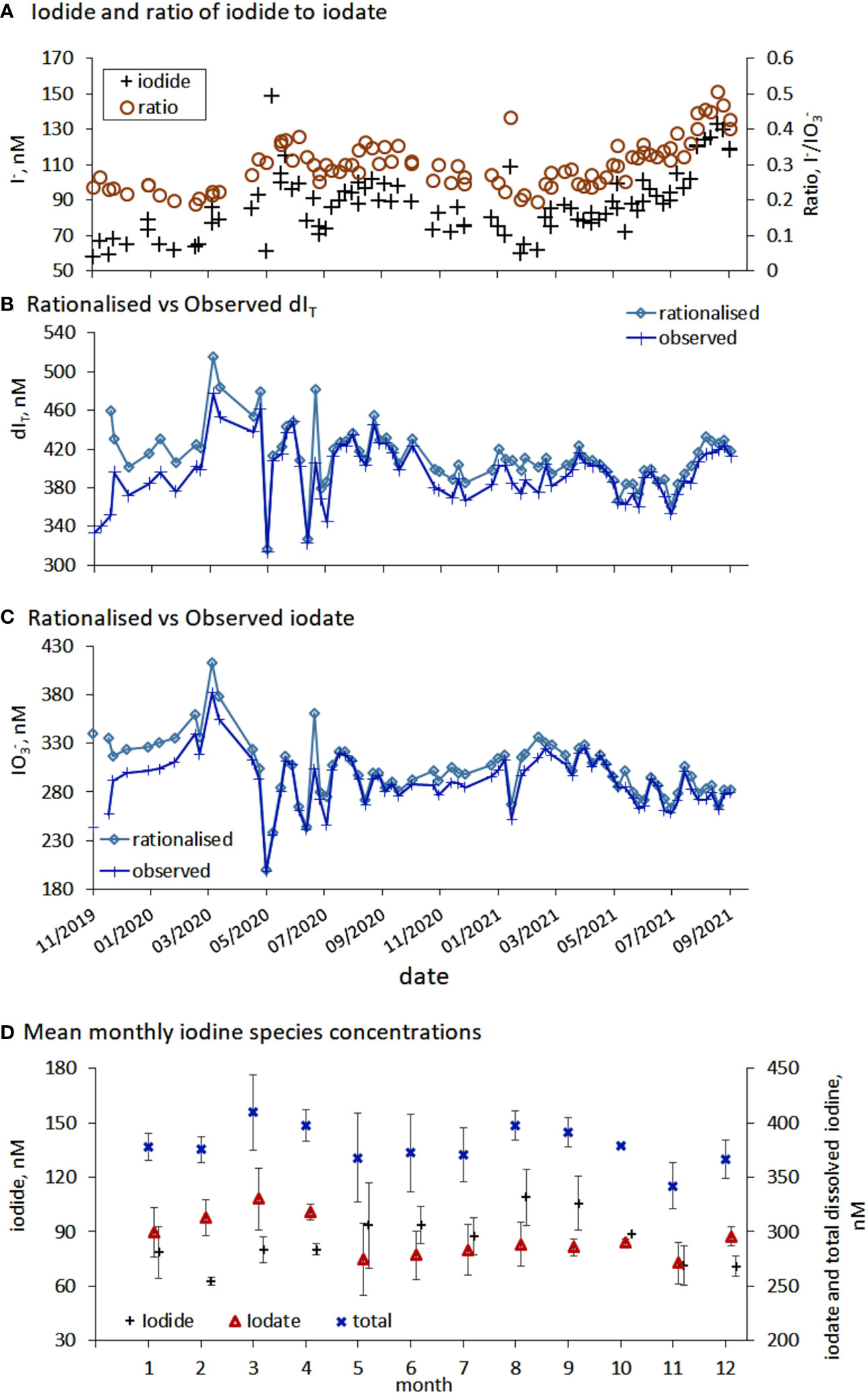
Figure 5 (A) Comparison between the inners shelf iodide concentration and the ratio of iodide to iodate. (B–C) Comparison between temporal changes in rationalised (normalised to a salinity of SP=35) and observed total dissolved iodine (dIT) and iodate. (D) Average calendar (2020 and 2021) iodine concentration for inorganic iodine species (iodide, iodate and total dissolved iodine) in the inner shelf waters, error bars represent the standard deviations of the measurements.
4.2.2 Terrestrial influence on inner shelf seawater
In UK coastal waters, in situ processes have been found to dominate over estuarine inputs for influencing iodine concentrations (Smith and Butler, 1979; Truesdale and Upstill-Goddard, 2003). To test whether inner shelf processes were influencing iodine concentration and speciation we compared 14 paired inner and outer shelf samples, each pair collected within 30 minutes. A Student t-test of those paired samples (Table 3) indicated that outer and inner shelf iodide and DOI were statistically similar but dIT and iodate were significantly lower. The monthly average difference of iodate (20 ± 5 nM or 7%) between the outer and inner shelf was consistent throughout the year except during the transition periods in May and October (Figure 6). Salinity rationalized iodate and dIT are known to decrease towards the coast though a cause for this effect has not yet been identified (Truesdale, 1978; Wong and Zhang, 1992; Truesdale and Jones, 2000; Truesdale and Upstill-Goddard, 2003). Mesoscale processes on the inner shelf that can change as a function of water column depth and influence iodine speciation range from the physical (e.g., mixing of the bottom boundary layer (deepest 10 m) of the water column by interaction with the seabed and wave refraction from the coasts) to chemical (e.g., additions through submarine groundwater discharge, anthropogenic material from point and diffusive sources, natural terrestrial organic and inorganic material and elevated mineral surface areas or suspended load that catalyze reactions). Despite the statistical differences, the 95% confidence level of the inner shelf LOESS model tracks the outer shelf measured dIT and iodine speciation concentrations (Figure 2A). We infer from these similar temporal changes that the inner and outer shelf concentrations were likely driven by the same suite of processes. Superimposed on these, we propose that there are additional process taking place closer to the shore that decreased inner shelf iodate by ~20 nM without increasing iodide or DOI concentrations. An iodate loss rate of ~8 nM d-1 was calculated from the residence time of seawater in Plymouth Sound (~2.5 days) and the 20 nM inner to outer shelf iodate concentration difference (Figure 6). This rate is similar to Truesdale’s (1978) estimated rate of iodate removal, ~16 nM d-1, in the Menai Strait (Wales, UK). In our study, the outer to inner shelf iodide concentration difference (-2 nM or ~2% of the iodide, Table 3) was not significant and therefore the inner shelf waters did not follow the previously observed behavior of iodide increasing shoreward (Truesdale, 1978; Wong and Zhang, 1992; Truesdale and Jones, 2000; Truesdale and Upstill-Goddard, 2003). The inner and outer shelf DOI concentrations of the present study were also very similar (Table 3), though the 1 nM higher inner shelf DOI is ~6% of the total DOI. The asynchronous seasonal concentration changes of iodine species (Figures 2A, 4E) and non-conservation of dIT (Figures 2A, 6B) indicate that there was another pool of (undetected) iodine or an ex situ removal and renewal process that influenced the inner shelf waters.
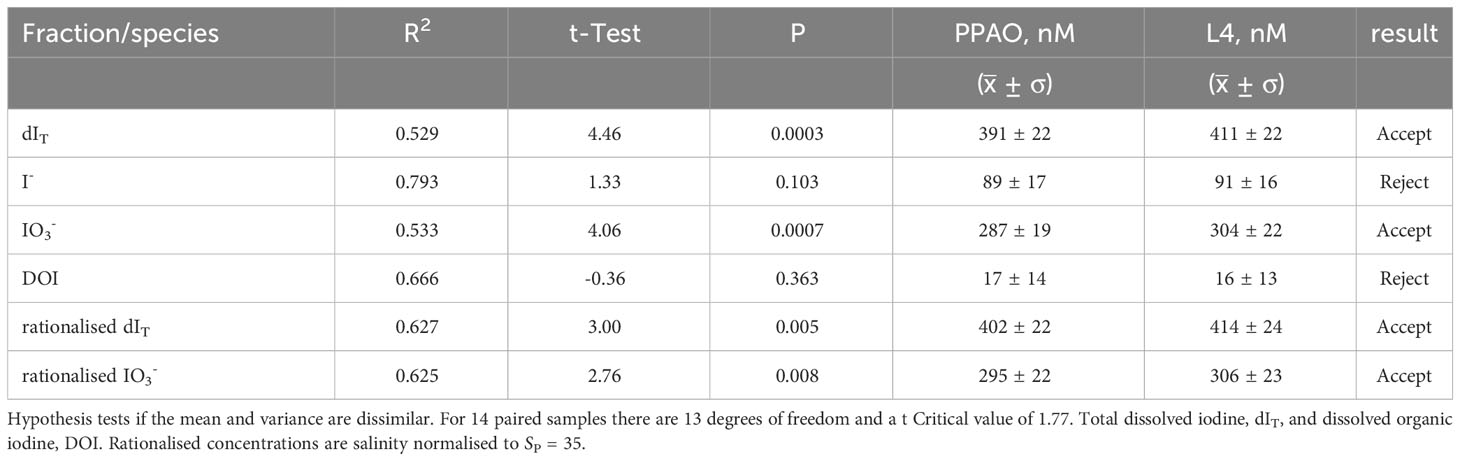
Table 3 Average (± standard deviation), correlation co-efficient (R2) and results of one tail student t-Test for the 14 paired samples that were collected on the same days at the inner and outer shelf location.
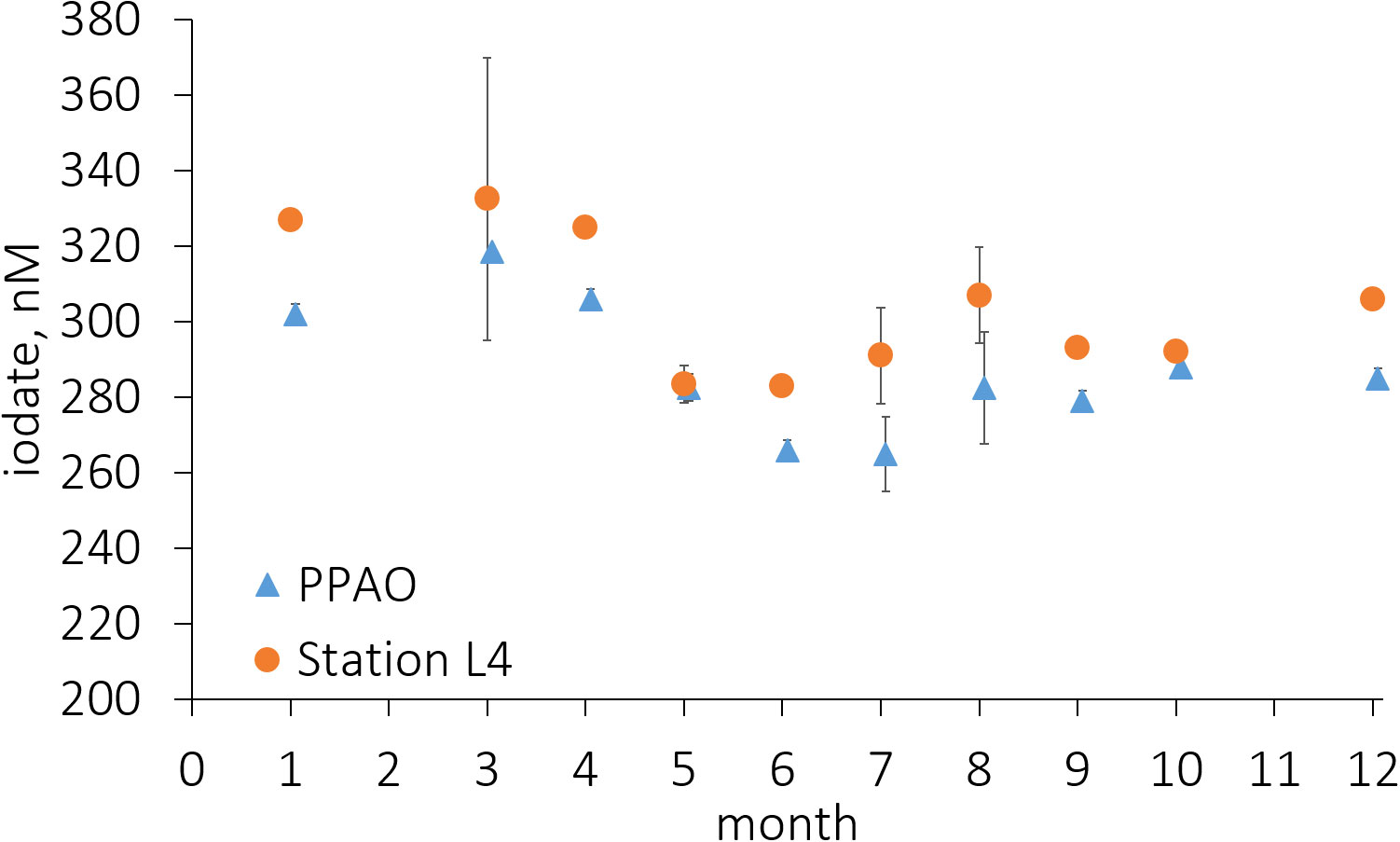
Figure 6 Average collated inner and outer shelf monthly observed coincident mean iodate (IO3-) concentrations. Mean iodate difference, excluding the transition months of May and October, is ~20 nM. Error bars represent the standard deviation of all observations for those calendar months.
4.3 The missing pool of iodine
4.3.1 Biological particulate iodine
Our study aimed to constrain iodine cycling within the soluble fraction but because dIT was not conserved we must consider the possibility that the missing iodine is in the particulate phase. The concentration of total particulate iodine (pIT) in the coastal Pacific and open Atlantic Ocean surface seawaters is ~1 nM (Wong et al., 1976; Satoh et al., 2023). The low pIT concentration suggests that pIT cannot account for the tens of nanomolar dIT inner shelf missing pool of iodine. Though pIT was not measured we can estimate the biological contribution towards pIT. Particulate iodine is split into three pools, biological [pIBio, incorporated within organisms, primarily phytoplankton as the largest biomass in the water column (Sigman and Hain, 2012)], organic (i.e., large amorphous colloids, aggregates and detrital matter) and inorganic (e.g., calcite or ferromanganese particles). The iodine content of the plankton biomass can be estimated from the carbon to iodine ratio of 1:1×10-4 (Elderfield and Truesdale, 1980) and the inner shelf biomass (mg C m-3, Section 2.2.2). Our estimate of pIBio ranged from 0.004 to 0.096 nM, this is 1000× lower than dIT and 10× lower than the expected pIT (Wong et al., 1976; Satoh et al., 2023). Therefore, pIBio is only a small portion of pIT and unlikely to be the missing pool of iodine.
4.3.2 Dissolved/particulate organic iodine
The significant concentration of DOI observed (19 nM, Table 2) is evidence that iodine was present in soluble organic structures. There exist a large variety of possible iodinated low molecular weight organic material (Bichsel and von Gunten, 1999; Wong and Cheng, 2001a; Sherrill et al., 2004; Du et al., 2023). One reason for elevated DOI is that there are higher organic material concentrations in inner shelf waters relative to outer shelf water. This organic material has a different structure and composition strongly influenced by terrestrially derived material (Muller, 2018 and references therein). Inner shelf organic material availability is also influenced seasonally because increased seasonal precipitation flushes material from the terrestrial to the marine environment. However, on the inner shelf, the temporal variation in dissolved organic carbon (DOC) was low and its concentration did not exhibit a strong seasonality (data not shown). A steady-state DOC system has the potential for continuous DOI formation. As well as bacterial processes in estuarine systems that convert DOI to inorganic iodine (Luther et al., 1991) the decomposition of DOI via photochemistry or ozonation produces iodide and iodate (Wong and Cheng, 2001b; Sherrill et al., 2004). Photochemical breakdown of DOI is of the order of hours and ozonation of DOI is of the order of minutes (Wong and Cheng, 2001b; Sherrill et al., 2004). With lower winter solar irradiation and therefore levels of photochemistry this may allow winter DOI to accumulate as observed (Figures 2A, 4E). Organic material is generally also required for the photochemical formation of reactive intermediate species that influence iodine cycling (Liebhafsky, 1934; Hughes et al., 1971; Rush and Bielski, 1985; Neta et al., 1988; Lymar et al., 2000; Wong and Zhang, 2008; Sharpless and Blough, 2014; Hardisty et al., 2020; Luther, 2023).
The iodine species hypoiodous acid and molecular iodine, which oxidize organic carbon, are also the most likely to undertake nucleophilic substitution reactions to form DOI. Hypoiodous acid, though thermodynamically stable at pH = 8 (pK = 10.6; Truesdale and Luther, 1995), forms molecular iodine following its comproportionation with iodide (Luther et al., 1995; Truesdale and Luther, 1995; Bichsel and von Gunten, 1999; Li et al., 2020; Du et al., 2023). Iodate is also an iodinating agent for organic material (Baer et al., 2011; Zhdankin and Muñiz, 2017) and may be significant as a precursor ion for DOI. Iodate sequestration into organic material which then precipitates to sediments because of increasing ionic strength has been suggested as the mechanism for iodate removal in estuarine systems (Cook et al., 2000; Wong and Zhang, 2003). In soils, estuaries and marine systems an organoiodate has been found to form concurrently with the removal of inorganic iodate (Francois, 1987; Luther et al., 1991). When considering whether there is iodide or iodate within organic structures it has been found that iodate forms the more reactive form of DOI in both ex situ seawaters amended with humic acid and porewaters fluxing from sediments (Francois, 1987; Bowley et al., 2016; Satoh and Imai, 2021). In laboratory experiments that had no external forcing, i.e., no advanced oxidation processes such as photochemistry or ozonation, organoiodate had a half-life of circa 10 d (Bowley et al., 2016). As DOI is present, iodine may also be retained in particulate organic carbon and may contribute to the missing pool of iodine.
Alternatively, we must also acknowledge that there may be a portion of the DOI that is not recoverable by the analytical method (Jones et al., 2023). However, a comparison of the concentration recoveries of the chemical digestion method versus a UV digestion showed the chemical digestion method was high (98 ± 4%, n=92; Jones et al. (2023)). Furthermore, the agreement between the measured DOI and the ratio of DOI to inorganic iodine found here and that found by other authors using different digestion techniques suggests that this undetected pool of dissolved iodine may not be significant (Truesdale, 1975; Luther et al., 1991; Wong and Cheng, 1998; Truesdale et al., 2001; Schwehr and Santschi, 2003; Takeda et al., 2016). Any unrecovered organic iodine in the dissolved fraction is likely to be small and not account for the missing pool of iodine.
4.3.3 Sediment-water interactions
As well as a ‘missing iodine’ pool in the water column, we consider mechanisms that might remove (and subsequently renew) dissolved iodine on the inner shelf over seasonal scales. Once iodine species are adsorbed or absorbed by the water column’s suspended load (organic or inorganic) they can be deposited onto the sediment-water interface. At the interface, iodate may be retained or chemically reduced (Francois, 1987; Anschutz et al., 2000). Sediment porosity controls the distance oxygen can travel into sediments and whether redox processes occur at the sediment-water interface or deeper. Plymouth Sound sediments are porous because they have a high percentage of sand (Franco et al., 2017; Johnson et al., 2022). In interstitial porewaters, the iodine species derive from organic material remineralization in oxygenated (Francois, 1987) or anoxic sediments (Ullman and Aller, 1980) or following iodate reduction in hypoxic or suboxic sediments (Chapman and Truesdale, 2011). Solution phase iodine species is scavenged at the oxic interface by ferro-manganese minerals or other reactions relating to soluble phase metal cycling and/or organic material cycling (Harvey, 1980; Ullman and Aller, 1985; Francois, 1987; Luther et al., 1997; Anschutz et al., 2000; Satoh and Imai, 2021). Nevertheless, oxygenated sediments can also be a source of iodide to overlying waters and iodide fluxes increase as oxygen concentrations decrease (Chapman and Truesdale, 2011; Satoh and Imai, 2021; Ooki et al., 2022).
To test if iodide was fluxing from sediments and increasing concentrations in inner shelf waters we considered excess nitrogen (N*) because sediments are a known source of nitrogen. The N* value provides the nitrogen deficit or excess relative to the canonical nitrogen to phosphate Redfield Ratio (RR) of 16 (Equation 2).
If the concentration of seawater nitrogen is lower than that predicted there will be a negative N*, whereas a positive N* value indicates that the seawater is enriched with nitrogen. The N* rationalization is typically used in open ocean settings though it has been used in shelf sea systems (Kim et al., 2011; Moon et al., 2021). The N*-iodide relationship was negative (Supplementary Figure SI.9), so inner shelf waters had higher iodide when nitrogen was deficient. The N*-iodate relationship was positive and this was likely forced by the winter advection of low primary productivity, high nutrient North Atlantic waters combining with excess freshwater bringing terrestrial runoff (Supplementary Figures SI.4, SI.9). Nevertheless, the iodate loss during spring and summer, the accumulation of iodide over summer and autumn and the non-conservation of dIT may be accounted for by iodate deposition to and a slow flux of iodide from the sedimentary system.
4.4 Seasonal controls on dissolved iodine speciation
Within dIT, the relative contribution of iodine species was: iodate 73 ± 6%; iodide 22 ± 4%; and DOI 5 ± 4%. The relative contributions are of a similar magnitude to those observed in northeast Atlantic surface waters, 89% iodate and 12% iodide (He et al., 2013). The late autumn dIT maxima was caused by the gradual build-up of iodide through the warmer summer months, a process observed in the Mediterranean Sea and North Atlantic Fjords (Tian et al., 1996; Shi et al., 2023). The seasonal iodine speciation changes at the inner shelf, which were also observed at the outer shelf (Figure 2), are the first to be reported for UK coastal waters (Truesdale and Upstill-Goddard, 2003) though seasonal changes in iodine speciation have been reported in other locations (Jickells et al., 1988; Campos et al., 1996; Tian et al., 1996; Chance et al., 2010; Shi et al., 2023). Changes in iodine species and dIT mean that the iodide to iodate ratio was not conserved but the iodide/iodate ratio (0.3 ± 0.07 (x̅±σ), range 0.19-0.51) did follow the same temporal pattern as the change in iodide (Results Section 3.1.2 and Figure 5A).
4.4.1 Biological control
The timing of certain changes in iodine speciation (Figures 2, 4) supports the paradigm that transformations between iodine species are biologically mediated. However, the near coincident relationship between seasonal changes in inorganic iodine species and nitrate (Figures 4A, B), a key nutrient for marine biological systems, and inorganic iodine and dissolved oxygen (Figure 4C), the product of photosynthesis, were not statistically significant. Oxygen is an oxidant of iodide, although not thought to be significant in oxygenated systems (Luther et al., 1995), thus the relationship between dissolved oxygen and iodine speciation was likely not causal. In terms of phytoplankton, which are the main controllers of nutrient concentration, laboratory culture experiments have shown iodide production as phytoplankton cultures collapse and bacterial numbers increase (Chance et al., 2007; Bluhm et al., 2010; Hepach et al., 2020). For discrete phytoplankton blooms, a time lag of 60 days between iodate reduction and iodide production was observed in Antarctic coastal waters (Chance et al., 2010) and in culturing studies (Chance et al., 2007; Bluhm et al., 2010; Hepach et al., 2020). A time lag of 60 days was successfully used to constrain an ocean iodine model (Wadley et al., 2020). The lag times from the 2020 iodate LOESS maximum (358 nM, March 2020) and iodide and DOI maxima were 65 and 42 days, respectively (Figures 2A, 4E). However, for 2021, and because iodide concentrations were still increasing in September 2021, the lag between the iodate (314 nM, March) and iodide maxima was a minimum of 190 days. The different phytoplankton succession sequences of summer 2020 and 2021 may explain the different lag phases of the iodine species. In 2020, the outer shelf had an early and late bloom (Figure 2C) with a transient coccolithophore presence in mid-summer (Supplementary Figures SI.6I, SI.7G). In 2021, the bloom cycle occurred at approximately 5-week intervals. With just two discrete blooms in 2020, we propose that a senescence phase evolved between those blooms that allowed for heterotrophic organisms to take advantage of the conditions resulting in a time lag of 60 d, whereas in 2021, the heterotrophic organisms could not establish themselves as the bloom successions cycled too quickly so the time lag, 190 d, arrived towards the end of the growing season.
If dIT is conserved (Tian et al., 1996; Waite et al., 2006) and iodine speciation changes were due to in situ biological conversion between iodide to iodate and vice versa and pIBio was low, then a correlation could be expected between soluble iodine species. Moreover, as DOI may form because of the iodination of organic material (Dunford and Ralston, 1983) a relationship may also exist between DOI and an inorganic iodine species. The linear correlation coefficient between iodate and iodide was R2iodide = 0.05 and between iodate and DOI, R2DOI = 0.07. Adjusting the temporal changes in iodine species concentrations to synchronize based on time lags did not improve correlations; after applying the 2020 time lag of 65 days the correlation coefficients were R2iodide = 0.01 and R2DOI = 0.03. Other than for iodate, there was no observed iodine speciation seasonality with concentration changes related to major nutrient changes (nitrate, phosphate and silicate). That iodine did not follow a nutrient-like profile supports Truesdale and Jones’s (2000) proposal that iodine does not show nutrient-like behavior. Therefore, the pattern of iodine speciation change on the inner and outer shelf of the Western English Chanel is not consistent with a simple biological uptake or a direct iodate to iodide reduction coupled with phytoplankton growth (Waite and Truesdale, 2003; Hung et al., 2005; Amachi et al., 2007). As decreasing concentrations of dIT are not concurrent with increasing biomass then other processes must also be involved in iodine speciation changes and iodine storage.
As well as phytoplankton, higher life forms and the habitats in which they live may influence iodine speciation. The multi-stage growth cycle of jellyfish depends on the availability of iodide and a specific DOI (Silverstone et al., 1977; Prieto et al., 2010). On the inner shelf, elevated DOI is somewhat coincident with elevated iodide (Figure 2A). High DOI concentrations can kinetically protect organo-iodine signaling compounds used by mature jellyfish to induce strobilation in benthic populations (Silverstone et al., 1977; Prieto et al., 2010). Also, cnidarian (jellyfish) control iodine speciation to help relieve oxidative stress (Berking et al., 2005). On the outer shelf, zooplankton pre-empts the winter/spring transition primary production bloom (Smyth et al., 2010) but during initial bloom conditions, zooplankton concentrations are kept low by a significant population of gelatinous planula larvae (jellyfish; Cross et al., 2015). Macroalgae (kelp) also interact with the iodine cycle for the benefit of relieving oxidative stress (Küpper et al., 2008). The Rame Head current flows through Cawsand Bay (Siddorn et al., 2003; Uncles et al., 2015), which is a warm, shallow, lateral bay and a habitat for Laminaria kelp (Teagle and Smale, 2018). Though used to relieve chemical stress the effect of iodine species cycling in this role is highly localized, within 0.1-1.0 m distance, from the kelp (Küpper et al., 1998; Carrano et al., 2021). If Cawsand Bay kelp beds were a strong influence on iodine speciation we would expect similar seasonal cycles yet cycles between 2020 and 2021 were different (Figure 2). Finally, seagrass can produce halocarbons including iodomethane (Weinberg et al., 2013) and the iodine content within the flesh of seagrass fluctuates on a seasonal cycle (Satoh et al., 2019b; Satoh et al., 2020), so it is possible that they may locally influence iodine speciation. However, even though seagrass is present in Plymouth Sound and Cawsand Bay (Howard-Williams, 2022) there is insufficient evidence to assess its effect here.
5 Conclusions
A range of biologically mediated processes coupled with potential sediment interactions and seawater margin processed aqueous iodine may influence the cycling of iodine in the temperate coastal marine environment. Nearly two years of observations of iodine species on the inner and outer shelf of temperate coastal surface waters showed the following; 1.) winter iodate concentrations increased because of renewal through advection/mixing of source waters, 2.) dissolved organic iodine increased during winter, 3.) iodate reduction began as light levels and biomass increased, 4.) there was a significant time lag between iodate reduction and iodide and DOI accumulation, 5.) iodide and DOI accumulated during warmer periods within a well-mixed water column, 6.) the asynchronous progression of iodine species resulted in dIT not being conserved, 7.) the near coincident seasonal concentration changes for iodate and nitrate were not statistically significant, 8.) seawater margin processed aqueous iodine may sometimes be significant but concentrations are subsumed by seawater and 9.) a significant pool of iodine remains unaccounted for and is required to conserve the concentration of dIT in the water column.
There are many processes and systems, which range in scale from the mesoscale to the molecular, that influence iodine species formation and cycling. We were unable to identify a definitive suite of coupled processes that cause iodine species transformations. However, the combination of processes that influenced iodine speciation on a seasonal scale commenced in late spring and ran through to autumn, so were likely biological in origin. The processes driving the iodine cycle may be due to the relationship between phytoplankton and bacterial or archaeal populations or due to seasonal changes in organic material composition. Autochthonous and allochthonous (terrestrial input) DOC are also photochemically labile and this characteristic influences the formation of reactive intermediate species and their subsequent interaction with iodine and the function of organoiodate. This work has shown that our understanding of iodine’s marine biogeochemistry is still insufficient for the requirement to understand the processes controlling oceanic surface iodine speciation and thus how it influences iodine’s role in the chemistry of the atmosphere.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
MJ: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. RC: Conceptualization, Data curation, Formal analysis, Investigation, Writing – review & editing. TB: Methodology, Writing – review & editing. OJ: Investigation, Writing – review & editing. DL: Investigation, Writing – review & editing. RM: Investigation, Writing – review & editing. LT: Conceptualization, Formal analysis, Investigation, Writing – review & editing. KW: Formal analysis, Investigation, Writing – review & editing. CW: Formal analysis, Funding acquisition, Methodology, Writing – review & editing. LC: Conceptualization, Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by a European Research Council (ERC) (project O3-SML; grant agreement no. 833290) grant under the European Union’s Horizon 2020 programme to LC. This work was also supported by the UK Natural Environment Research Council’s National Capability Long-term Single Centre Science Programme, Climate Linked Atlantic Sector Science, grant number NE/R015953/1, and is a contribution to Theme 1.3 - Biological Dynamics.
Acknowledgments
This paper also contributes to the science plan of the Surface Ocean-Lower Atmosphere Study (SOLAS), which is partially supported by the U.S. National Science Foundation (Grant OCE-1840868) via the Scientific Committee on Oceanic Research (SCOR). We are especially grateful to Plymouth Marine Laboratory and the crew of the RV Plymouth Quest, for collecting and filtering seawater samples. We thank the reviewers for their time, insights and comments that helped improve this paper.
Conflict of interest
Author KW is employed by Fidra, though at the time of research, she was with the University of York.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2024.1277595/full#supplementary-material
References
Abdel-Moati M. A. R. (1999). Iodine speciation in the Nile River estuary. Mar. Chem. 65, 211–225. doi: 10.1016/S0304-4203(99)00003-1
Amachi S., Kawaguchi N., Muramatsu Y., Tsuchiya S., Watanabe Y., Shinoyama H., et al. (2007). Dissimilatory iodate reduction by marine Pseudomonas sp. strain SCT. Appl. Environ. Microbiol. 73, 5725–5730. doi: 10.1128/AEM.00241-07
Amachi S., Muramatsu Y., Akiyama Y., Miyazaki K., Yoshiki S., Hanada S., et al. (2005). Isolation of iodide-oxidizing bacteria from iodide-rich natural gas brines and seawaters. Microb. Ecol. 49, 547–557. doi: 10.1007/s00248-004-0056-0
Anschutz P., Sundby B., Lefrançois L., Luther G. W., Mucci A. (2000). Interactions between metal oxides and species of nitrogen and iodine in bioturbated marine sediments. Geochimica Cosmochimica Acta 64, 2751–2763. doi: 10.1016/S0016-7037(00)00400-2
Baer M. D., Pham V.-T., Fulton J. L., Schenter G. K., Balasubramanian M., Mundy C. J. (2011). Is iodate a strongly hydrated cation? J. Phys. Chem. Lett. 2, 2650–2654. doi: 10.1021/jz2011435
Barker D. R., Zeitlin H. (1972). Metal-ion concentrations in sea-surface microlayer and size-separated atmospheric aerosol samples in Hawaii. J. Geophysical Res. 77, 5076–5086. doi: 10.1029/JC077i027p05076
Barrón C., Duarte C. M. (2015). Dissolved organic carbon pools and export from the coastal ocean. Global Biogeochemical Cycles 29, 1725–1738. doi: 10.1002/2014GB005056
Berking S., Czech N., Gerharz M., Herrmann K., Hoffmann U., Raifer H., et al. (2005). A newly discovered oxidant defence system and its involvement in the development of Aurelia aurita (Scyphozoa, Cnidaria): reactive oxygen species and elemental iodine control medusa formation. Int. J. Dev. Biol. 49, 969–976. doi: 10.1387/ijdb.052024sb
Bichsel Y., von Gunten U. (1999). Determination of iodide and iodate by ion chromatography with postcolumn reaction and UV/Visible detection. Anal. Chem. 71, 34–38. doi: 10.1021/ac980658j
Bluhm K., Croot P., Wuttig K., Lochte K. (2010). Transformation of iodate to iodide in marine phytoplankton driven by cell senescence. Aquat. Biol. 11, 1–15. doi: 10.3354/ab00284
Bosboom J., Stive M. (2021) Coastal dynamics (LibreTexts). Available at: https://geo.libretexts.org/Bookshelves/Oceanography/Coastal_Dynamics_(Bosboom_and_Stive (Accessed November 10, 2023).
Bowley H. E., Young S. D., Ander E. L., Crout N. M. J., Watts M. J., Bailey E. H. (2016). Iodine binding to humic acid. Chemosphere 157, 208–214. doi: 10.1016/j.chemosphere.2016.05.028
Brewer P. G., Riley J. P. (1965). The automatic determination of nitrate in sea water. Deep Sea Res. Oceanographic Abstracts 12, 765–772. doi: 10.1016/0011-7471(65)90797-7
Campos M. L. A. M. (1997). New approach to evaluating dissolved iodine speciation in natural waters using cathodic stripping voltammetry and a storage study for preserving iodine species. Mar. Chem. 57, 107–117. doi: 10.1016/S0304-4203(96)00093-X
Campos M., Farrenkopf A. M., Jickells T. D., Luther G. W. (1996). A comparison of dissolved iodine cycling at the Bermuda Atlantic Time-Series station and Hawaii Ocean Time-Series station. Deep-Sea Res. Part II-Topical Stud. Oceanography 43, 455–466. doi: 10.1016/0967-0645(95)00100-X
Campos M., Sanders R., Jickells T. (1999). The dissolved iodate and iodide distribution in the South Atlantic from the Weddell Sea to Brazil. Mar. Chem. 65, 167–175. doi: 10.1016/S0304-4203(98)00094-2
Canfield D. E., Glazer A. N., Falkowski P. G. (2010). The evolution and future of earth’s nitrogen cycle. Science 330, 192–196. doi: 10.1126/science.1186120
Carpenter L. J., Chance R. J., Sherwen T., Adams T. J., Ball S. M., Evans M. J., et al. (2021). Marine iodine emissions in a changing world. Proc. R. Soc. A: Mathematical Phys. Eng. Sci. 477, 20200824. doi: 10.1098/rspa.2020.0824
Carpenter L. J., Liss P. S., Penkett S. A. (2003). Marine organohalogens in the atmosphere over the Atlantic and Southern Oceans. J. Geophysical Research: Atmospheres 108. doi: 10.1029/2002JD002769
Carpenter L. J., MacDonald S. M., Shaw M. D., Kumar R., Saunders R. W., Parthipan R., et al. (2013). Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine. Nat. Geosci 6, 108–111. doi: 10.1038/ngeo1687
Carr N., Davis C. E., Blackbird S., Daniels L. R., Preece C., Woodward M., et al. (2019). Seasonal and spatial variability in the optical characteristics of DOM in a temperate shelf sea. Prog. Oceanography 177, 101929. doi: 10.1016/j.pocean.2018.02.025
Carrano M. W., Carrano C. J., Edwards M. S., Al-Adilah H., Fontana Y., Sayer M. D. J., et al. (2021). Laminaria kelps impact iodine speciation chemistry in coastal seawater. Estuarine Coast. Shelf Sci. 262, 107531. doi: 10.1016/j.ecss.2021.107531
Carrano M. W., Yarimizu K., Gonzales J. L., Cruz-López R., Edwards M. S., Tymon T. M., et al. (2020). The influence of marine algae on iodine speciation in the coastal ocean. Algae 35, 167–176. doi: 10.4490/algae.2020.35.5.25
Chance R., Baker A. R., Carpenter L., Jickells T. D. (2014). The distribution of iodide at the sea surface. Environ. Sci.: Processes Impacts 16, 1841–1859. doi: 10.1039/C4EM00139G
Chance R., Malin G., Jickells T., Baker A. R. (2007). Reduction of iodate to iodide by cold water diatom cultures. Mar. Chem. 105, 169–180. doi: 10.1016/j.marchem.2006.06.008
Chance R., Weston K., Baker A. R., Hughes C., Malin G., Carpenter L., et al. (2010). Seasonal and interannual variation of dissolved iodine speciation at a coastal Antarctic site. Mar. Chem. 118, 171–181. doi: 10.1016/j.marchem.2009.11.009
Chapman P., Truesdale V. W. (2011). Preliminary evidence for iodate reduction in bottom waters of the Gulf of Mexico during an hypoxic event. Aquat Geochem 17, 671–695. doi: 10.1007/s10498-011-9123-6
Cook P. L. M., Carpenter P. D., Butler E. C. V. (2000). Speciation of dissolved iodine in the waters of a humic-rich estuary. Mar. Chem. 69, 179–192. doi: 10.1016/S0304-4203(99)00104-8
Cooper L. H. N. (1960). The water flow into the English Channel from the south-west. J. Mar. Biol. Assoc. United Kingdom 39, 173–208. doi: 10.1017/S0025315400013242
Cross J., Nimmo-Smith W. A. M., Hosegood P. J., Torres R. (2015). The role of advection in the distribution of plankton populations at a moored 1-D coastal observatory. Prog. Oceanography 137, 342–359. doi: 10.1016/j.pocean.2015.04.016
Cuevas C. A., Fernandez R. P., Kinnison D. E., Li Q., Lamarque J.-F., Trabelsi T., et al. (2022). The influence of iodine on the Antarctic stratospheric ozone hole. Proc. Natl. Acad. Sci. 119, e2110864119. doi: 10.1073/pnas.2110864119
Dembitsky V. M. (2006). Biogenic iodine and iodine-containing metabolites. Natural Product Commun. 1, 139–175. doi: 10.1177/1934578X0600100210
Du J., Kim K., Son S., Pan D., Kim S., Choi W. (2023). MnO2-induced oxidation of iodide in frozen solution. Environ. Sci. Technol. 57, 5317–5326. doi: 10.1021/acs.est.3c00604
Dunford H. B., Ralston I. M. (1983). On the mechanism of iodination of tyrosine. Biochem. Biophys. Res. Commun. 116, 639–643. doi: 10.1016/0006-291X(83)90572-7
Elderfield H., Truesdale V. W. (1980). On the biophilic nature of iodine in seawater. Earth Planetary Sci. Lett. 50, 105–114. doi: 10.1016/0012-821X(80)90122-3
Farrenkopf A. M., Luther III, G. W. (2002). Iodine chemistry reflects productivity and denitrification in the Arabian Sea: Evidence for flux of dissolved species from sediments of western India into the OMZ. Deep Sea Res. Part II: Topical Stud. Oceanography 49, 2303–2318. doi: 10.1016/S0967-0645(02)00038-3
Farrenkopf A. M., Luther G. W., Truesdale V. W., van der Weijden C. H. (1997). Sub-surface iodide maxima: evidence for biologically catalyzed redox cycling in Arabian Sea OMZ during the SW intermonsoon. Deep Sea Res. Part II: Topical Stud. Oceanography 44, 1391–1409. doi: 10.1016/S0967-0645(97)00013-1
Franco A., Mazik K., Roberts L. (2017). Whitsand and Looe Bay MCZ and surround subtidal sediment data analysis and reporting. The University of Hull.
Francois R. (1987). The influence of humic substances on the geochemistry of iodine in nearshore and hemipelagic marine sediments. Geochimica Cosmochimica Acta 51, 2417–2427. doi: 10.1016/0016-7037(87)90294-8
Fuge R. (1989). Iodine in waters: possible links with endemic goitre. Appl. Geochemistry 4, 203–208. doi: 10.1016/0883-2927(89)90051-6
Fuge R., Johnson C. C. (2015). Iodine and human health, the role of environmental geochemistry and diet, a review. Appl. Geochemistry 63, 282–302. doi: 10.1016/j.apgeochem.2015.09.013
Garland J. A., Elzerman A. W., Penkett S. A. (1980). The mechanism for dry deposition of ozone to seawater surfaces. J. Geophys. Res.-Oceans Atmos. 85, 7488–7492.
Gómez Martín J. C., Saiz-Lopez A., Cuevas C. A., Baker A. R., Fernández R. P. (2022). On the speciation of iodine in marine aerosol. J. Geophysical Research: Atmospheres 127, e2021JD036081. doi: 10.1029/2021JD036081
Gonzales J., Tymon T., Küpper F. C., Edwards M. S., Carrano C. J. (2017). The potential role of kelp forests on iodine speciation in coastal seawater. PloS One 12, e0180755. doi: 10.1371/journal.pone.0180755
Hardisty D. S., Horner T. J., Wankel S. D., Blusztajn J., Nielsen S. G. (2020). Experimental observations of marine iodide oxidation using a novel sparge-interface MC-ICP-MS technique. Chem. Geology 532, 119360. doi: 10.1016/j.chemgeo.2019.119360
Harvey G. R. (1980). A study of the chemistry of iodine and bromine in marine sediments. Mar. Chem. 8, 327–332. doi: 10.1016/0304-4203(80)90021-3
He P., Hou X., Aldahan A., Possnert G., Yi P. (2013). Iodine isotopes species fingerprinting environmental conditions in surface water along the northeastern Atlantic Ocean. Sci. Rep. 3, 2685. doi: 10.1038/srep02685
He X.-C., Simon M., Iyer S., Xie H.-B., Rörup B., Shen J., et al. (2023). Iodine oxoacids enhance nucleation of sulfuric acid particles in the atmosphere. Science 382, 1308–1314. doi: 10.1126/science.adh2526
He X.-C., Tham Y. J., Dada L., Wang M., Finkenzeller H., Stolzenburg D., et al. (2021). Role of iodine oxoacids in atmospheric aerosol nucleation. Science 371, 589–595. doi: 10.1126/science.abe0298
Hepach H., Hughes C., Hogg K., Collings S., Chance R. (2020). Senescence as the main driver of iodide release from a diverse range of marine phytoplankton. Biogeosciences 17, 2453–2471. doi: 10.5194/bg-17-2453-2020
Howard-Williams E. (2022). Seagrass Natural Capital Assessment: The Plymouth Sound and Estuaries SAC. NECR420. Second edition (UK: Natural England).
Hughes C., Barton E., Hepach H., Chance R., Pickering M. D., Hogg K., et al. (2021). Oxidation of iodide to iodate by cultures of marine ammonia-oxidising bacteria. Mar. Chem. 234, 104000. doi: 10.1016/j.marchem.2021.104000
Hughes M. N., Nicklin H. G., Sackrule W. (1971). The chemistry of peroxonitrites. Part III. The reaction of peroxonitrite with nucleophiles in alkali, and other nitrite producing reactions. J. Chem. Soc A, 3722–3725. doi: 10.1039/J19710003722
Hung C.-C., Wong G. T. F., Dunstan W. M. (2005). Iodate reduction activity in nitrate reductase extracts from marine phytoplankton. Bull. Mar. Sci. 76, 61–72.
Huthnance J. (2010). Temperature and salinity Vol. 39–106. Eds. Buckley P., Connor D., Cook D., Cox M., Dale T., Dye S., et al (London: DEFRA on behalf of the United Kingdom Marine Monitoring and Assessment Strategy (UKMMAS) Community).
ICONCAWS2 (-) WeatherUnderground station ICONCAWS2. Available at: https://www.wunderground.com/dashboard/pws/ICONCAWS2 (Accessed December 12, 2021).
Jickells T. D., Boyd S. S., Knap A. H. (1988). Iodine cycling in the Sargasso Sea and the Bermuda inshore waters. Mar. Chem. 24, 61–82. doi: 10.1016/0304-4203(88)90006-0
Johnson G., Burrows F., Crabtree R., Warner I. (2022). Plymouth Sound & Estuaries SAC Subtidal Sediment Data Analysis Report 2017. (UK: Natural England).
Jones M. R., Chance R., Dadic R., Hannula H.-R., May R., Ward M., et al. (2023). Environmental iodine speciation quantification in seawater and snow using ion exchange chromatography and UV spectrophotometric detection. Analytica Chimica Acta 1239, 340700. doi: 10.1016/j.aca.2022.340700
Jones M. R., Tebo B. M. (2021). Novel manganese cycling at very low ionic strengths in the Columbia River Estuary. Water Res. 207, 117801. doi: 10.1016/j.watres.2021.117801
Kim T.-W., Lee K., Najjar R. G., Jeong H.-D., Jeong H. J. (2011). Increasing N abundance in the Northwestern Pacific Ocean due to atmospheric nitrogen deposition. Science 334, 505–509. doi: 10.1126/science.1206583
Kirkwood D. S. (1989). Simultaneous determination of selected nutrients in seawater. Int. Council Explor. Sea (ICES). Annual Report, 29.
Küpper F. C., Carpenter L. J., McFiggans G. B., Palmer C. J., Waite T. J., Boneberg E.-M., et al. (2008). Iodide accumulation provides kelp with an inorganic antioxidant impacting atmospheric chemistry. PNAS 105, 6954–6958. doi: 10.1073/pnas.0709959105
Küpper F. C., Schweigert N., Ar Gall E., Legendre J.-M., Vilter H., Kloareg B. (1998). Iodine uptake in Laminariales involves extracellular, haloperoxidase-mediated oxidation of iodide. Planta 207, 163–171. doi: 10.1007/s004250050469
Li H.-P., Daniel B., Creeley D., Grandbois R., Zhang S., Xu C., et al. (2014). Superoxide production by a manganese-oxidizing bacterium facilitates iodide oxidation. Appl. Environ. Microbiol. 80, 2693–2699. doi: 10.1128/AEM.00400-14
Li H.-P., Yeager C. M., Brinkmeyer R., Zhang S., Ho Y.-F., Xu C., et al. (2012). Bacterial production of organic acids enhances H2O2-dependent iodide oxidation. Environ. Sci. Technol. 46, 4837–4844. doi: 10.1021/es203683v
Li J., Jiang J., Pang S., Cao Y., Zhou Y., Guan C. (2020). Oxidation of iodide and hypoiodous acid by non-chlorinated water treatment oxidants and formation of iodinated organic compounds: A review. Chem. Eng. J. 386, 123822. doi: 10.1016/j.cej.2019.123822
Liebhafsky H. A. (1934). The catalytic decomposition of hydrogen peroxide by the iodine—iodide couple. IV. The approach to the steady state. J. Am. Chem. Soc 56, 2369–2372. doi: 10.1021/ja01326a043
Lin J. (2023). Dissolved iodine in the changjiang river estuary, China. Water Sci. Technol. 88, 1269–1279. doi: 10.2166/wst.2023.263
Lister M. W., Rosenblum P. (1963). Rates of reaction of hypochlorite ions with sulphite and iodide ions. Can. J. Chem. 41, 3013–3020. doi: 10.1139/v63-442
Lotze H. K., Lenihan H. S., Bourque B. J., Bradbury R. H., Cooke R. G., Kay M. C., et al. (2006). Depletion, degradation, and recovery potential of estuaries and coastal seas. Science 312, 1806–1809. doi: 10.1126/science.1128035
Lovelock J. E. (1975). Natural halocarbons in the air and in the sea. Nature 256, 193–194. doi: 10.1038/256193a0
Luther G. W. (2023). Review on the physical chemistry of iodine transformations in the oceans. Front. Mar. Sci. 10. doi: 10.3389/fmars.2023.1085618
Luther G. W., Cole H. (1988). Iodine speciation in Chesapeake Bay waters. Mar. Chem. 24, 315–325. doi: 10.1016/0304-4203(88)90039-4
Luther G. W., Ferdelman T., Culberson C. H., Kostka J., Wu J. (1991). Iodine chemistry in the water column of the Chesapeake Bay: Evidence for organic iodine forms. Estuarine Coast. Shelf Sci. 32, 267–279. doi: 10.1016/0272-7714(91)90020-C
Luther G. W., Sundby B., Lewis B. L., Brendel P. J., Silverberg N. (1997). Interactions of manganese with the nitrogen cycle: Alternative pathways to dinitrogen. Geochimica Cosmochimica Acta 61, 4043–4052. doi: 10.1016/S0016-7037(97)00239-1
Luther G. W., Wu J., Cullen J. B. (1995). “Redox chemistry of iodine in seawater,” in Aquatic Chemistry Advances in Chemistry (American Chemical Society), 135–155. doi: 10.1021/ba-1995-0244.ch006
Lymar S. V., Schwarz H. A., Czapski G. (2000). Medium effects on reactions of the carbonate radical with thiocyanate, iodide, and ferrocyanide ions. Radiat. Phys. Chem. 59, 387–392. doi: 10.1016/S0969-806X(00)00277-2
MacDonald S. M., Gómez Martín J. C., Chance R., Warriner S., Saiz-Lopez A., Carpenter L. J., et al. (2014). A laboratory characterisation of inorganic iodine emissions from the sea surface: dependence on oceanic variables and parameterisation for global modelling. Atmospheric Chem. Phys. 14, 5841–5852. doi: 10.5194/acp-14-5841-2014
Mantoura R. F. C., Woodward E. M. S. (1983). Optimization of the indophenol blue method for the automated determination of ammonia in estuarine waters. Estuarine Coast. Shelf Sci. 17, 219–224. doi: 10.1016/0272-7714(83)90067-7
McTaggart A. R., Butler E. C. V., Haddad P. R., Middleton J. H. (1994). Iodide and iodate concentrations in eastern Australian subtropical waters, with iodide by ion chromatography. Mar. Chem. 47, 159–172. doi: 10.1016/0304-4203(94)90106-6
Mendoza W. G., Zika R. G. (2014). On the temporal variation of DOM fluorescence on the southwest Florida continental shelf. Prog. Oceanography 120, 189–204. doi: 10.1016/j.pocean.2013.08.010
Moon J.-Y., Lee K., Lim W.-A., Lee E., Dai M., Choi Y.-H., et al. (2021). Anthropogenic nitrogen is changing the East China and Yellow seas from being N deficient to being P deficient. Limnology Oceanography 66, 914–924. doi: 10.1002/lno.11651
Moriyasu R., Bolster K. M., Hardisty D. S., Kadko D. C., Stephens M. P., Moffett J. W. (2023). Meridional survey of the central pacific reveals iodide accumulation in equatorial surface waters and benthic sources in the abyssal plain. Global Biogeochemical Cycles 37, e2021GB007300. doi: 10.1029/2021GB007300
Moriyasu R., Evans N., Bolster K. M., Hardisty D. S., Moffett J. W. (2020). The distribution and redox speciation of iodine in the eastern tropical North Pacific Ocean. Global Biogeochemical Cycles 34, e2019GB006302. doi: 10.1029/2019GB006302
Moulay S. (2013). Molecular iodine/polymer complexes. J. Polymer Eng. 33, 389–443. doi: 10.1515/polyeng-2012-0122
Muller F. L. L. (2018). Exploring the potential role of terrestrially derived humic substances in the marine biogeochemistry of iron. Front. Earth Sci. 6. doi: 10.3389/feart.2018.00159
Nakayama E., Kimoto T., Isshiki K., Sohrin Y., Okazaki S. (1989). Determination and distribution of iodide- and total-iodine in the North Pacific Ocean - by using a new automated electrochemical method. Mar. Chem. 27, 105–116. doi: 10.1016/0304-4203(89)90030-3
Neta P., Huie R. E., Ross A. B. (1988). Rate constants for reactions of inorganic radicals in aqueous solution. J. Phys. Chem. Reference Data 17, 1027–1284. doi: 10.1063/1.555808
Ooki A., Minamikawa K., Meng F., Miyashita N., Hirawake T., Ueno H., et al. (2022). Marine sediment as a likely source of methyl and ethyl iodides in subpolar and polar seas. Commun. Earth Environ. 3, 1–7. doi: 10.1038/s43247-022-00513-7
Pound R. J., Brown L. V., Evans M. J., Carpenter L. J. (2023). An improved estimate of inorganic iodine emissions from the ocean using a coupled surface microlayer box model. EGUsphere 1–40. doi: 10.5194/egusphere-2023-2447
Prieto L., Astorga D., Navarro G., Ruiz J. (2010). Environmental control of phase transition and polyp survival of a massive-outbreaker jellyfish. PloS One 5, e13793. doi: 10.1371/journal.pone.0013793
Rees A. P., Hope S. B., Widdicombe C. E., Dixon J. L., Woodward E. M. S., Fitzsimons M. F. (2009). Alkaline phosphatase activity in the western English Channel: Elevations induced by high summertime rainfall. Estuarine Coast. Shelf Sci. 81, 569–574. doi: 10.1016/j.ecss.2008.12.005
Rush J. D., Bielski B. H. J. (1985). Pulse radiolytic studies of the reactions of HO2/O2- with Fe(II)/Fe(III) ions - the reactivity of HO2/O2- with ferric ions and its implication on the occurrence of the Haber-Weiss reaction. J. Phys. Chem. 89, 5062–5066. doi: 10.1021/j100269a035
Saiz-Lopez A., Plane J. M. C., Baker A. R., Carpenter L. J., von Glasow R., Gómez Martín J. C., et al. (2012). Atmospheric chemistry of iodine. Chem. Rev. 112, 1773–1804. doi: 10.1021/cr200029u
Satoh Y., Imai S. (2021). Flux and pathway of iodine dissolution from brackish lake sediment in the northeast of Japan. Sci. Total Environ. 789, 147942. doi: 10.1016/j.scitotenv.2021.147942
Satoh Y., Otosaka S., Suzuki T., Nakanishi T. (2023). Factors regulating the concentration of particulate iodine in coastal seawater. Limnol. Oceanogr. 68, 1580–1594. doi: 10.1002/lno.12369
Satoh Y., Wada S., Hama T. (2019a). Vertical and seasonal variations of dissolved iodine concentration in coastal seawater on the northwestern Pacific coast of central Japan. Continental Shelf Res. 188, 103966. doi: 10.1016/j.csr.2019.103966
Satoh Y., Wada S., Hisamatsu S. (2019b). Seasonal variations in iodine concentrations in a brown alga (Ecklonia cava Kjellman) and a seagrass (Zostera marina L.) in the northwestern Pacific coast of central Japan. J. Oceanogr 75, 111–117. doi: 10.1007/s10872-018-0479-8
Satoh Y., Wada S., Hisamatsu S. (2020). Relationship between iodine and carbohydrate contents in the seagrass Zostera marina on the northwestern Pacific coast of central Japan. Botanica Marina 63, 273–281. doi: 10.1515/bot-2020-0004
Schall C., Heumann K. G. (1993). GC determination of volatile organoiodine and organobromine compounds in Arctic seawater and air samples. Fresenius J. Anal. Chem. 346, 717–722. doi: 10.1007/BF00321279
Schnur A. A., Sutherland K. M., Hansel C. M., Hardisty D. S. (2024). Rates and pathways of iodine speciation transformations at the Bermuda Atlantic Time Series. Front. Mar. Sci. 10. doi: 10.3389/fmars.2023.1272870
Schulz R. C., Fleischer D., Henglein A., Bössler H. M., Trisnadi J., Tanaka H. (1974). Addition compounds and complexes with polymers and models. Pure Appl. Chem. 38, 227–247. doi: 10.1351/pac197438010227
Schwehr K. A., Santschi P. H. (2003). Sensitive determination of iodine species, including organo-iodine, for freshwater and seawater samples using high performance liquid chromatography and spectrophotometric detection. Analytica Chimica Acta 482, 59–71. doi: 10.1016/S0003-2670(03)00197-1
Sharpless C. M., Blough N. V. (2014). The importance of charge-transfer interactions in determining chromophoric dissolved organic matter (CDOM) optical and photochemical properties. Environ. Sci.: Processes Impacts 16, 654–671. doi: 10.1039/C3EM00573A
Sherrill J., Whitaker B. R., Wong G. T. F. (2004). Effects of ozonation on the speciation of dissolved iodine in artificial seawater. J. Zoo Wildlife Med. 35, 347–355. doi: 10.1638/03-025
Sherwen T., Chance R. J., Tinel L., Ellis D., Evans M. J., Carpenter L. J. (2019). A machine-learning-based global sea-surface iodide distribution. Earth System Sci. Data 11, 1239–1262. doi: 10.5194/essd-11-1239-2019
Shi Q., Kim J. S., Wallace D. W. (2023). Speciation of dissolved inorganic iodine in a coastal fjord: a time-series study from Bedford Basin, Nova Scotia, Canada. Front. Mar. Sci. 10. doi: 10.3389/fmars.2023.1171999
Siddorn J. R., Allen J. I., Uncles R. J. (2003). Heat, salt and tracer transport in the Plymouth Sound coastal region: a 3-D modelling study. J. Mar. Biol. Assoc. United Kingdom 83, 673–682. doi: 10.1017/S002531540300763Xh
Sigman D. M., Hain M. P. (2012). The biological productivity of the ocean | Learn science at scitable. Nat. Educ. Knowledge 10, 21.
Silverstone M., Tosteson T. R., Cutress C. E. (1977). The effect of iodide and various iodocompounds on initiation of strobilation in Aurelia. Gen. Comp. Endocrinol. 32, 108–113. doi: 10.1016/0016-6480(77)90087-9
Smith J. D., Butler E. C. V. (1979). Speciation of dissolved iodine in estuarine waters. Nature 277, 468–469. doi: 10.1038/277468a0
Smyth T. J., Fishwick J. R., AL-Moosawi L., Cummings D. G., Harris C., Kitidis V., et al. (2010). A broad spatio-temporal view of the Western English Channel observatory. J. Plankton Res. 32, 585–601. doi: 10.1093/plankt/fbp128
Tait D. R., Santos I. R., Lamontagne S., Sippo J. Z., McMahon A., Jeffrey L. C., et al. (2023). Submarine groundwater discharge exceeds river inputs as a source of nutrients to the Great Barrier Reef. Environ. Sci. Technol. 57, 15627–15634. doi: 10.1021/acs.est.3c03725
Takayanagi K., Cossa D. (1985). Behaviour of dissolved iodine in the upper St. Lawrence Estuary. Can. J. Earth Sci. 22, 644–646. doi: 10.1139/e85-067
Takayanagi K., Wong G. T. F. (1986). The oxidation of iodide to iodate for the polarographic determination of total iodine in natural waters. Talanta 33, 451–454. doi: 10.1016/0039-9140(86)80115-1
Takeda A., Tsukada H., Takaku Y., Satta N., Baba M., Shibata T., et al. (2016). Determination of iodide, iodate and total iodine in natural water samples by HPLC with amperometric and spectrophotometric detection, and off-line uv irradiation. Analytical Sci. 32, 839–845. doi: 10.2116/analsci.32.839
Tang Q., Xu Q., Zhang F., Huang Y., Liu J., Wang X., et al. (2013). Geochemistry of iodine-rich groundwater in the Taiyuan Basin of central Shanxi Province, North China. J. Geochemical Explor. 135, 117–123. doi: 10.1016/j.gexplo.2012.08.019
Teagle H., Smale D. A. (2018). Climate-driven substitution of habitat-forming species leads to reduced biodiversity within a temperate marine community. Diversity Distributions 24, 1367–1380. doi: 10.1111/ddi.12775
Tian R. C., Marty J. C., Nicolas E., Chiavérini J., Ruiz-Ping D., Pizay M. D. (1996). Iodine speciation: a potential indicator to evaluate new production versus regenerated production. Deep Sea Res. Part I: Oceanographic Res. Papers 43, 723–738. doi: 10.1016/0967-0637(96)00023-4
Tinel L., Adams T. J., Hollis L. D. J., Bridger A. J. M., Chance R. J., Ward M. W., et al. (2020). Influence of the sea surface microlayer on oceanic iodine emissions. Environ. Sci. Technol. 54, 13228–13237. doi: 10.1021/acs.est.0c02736
Truesdale V. W. (1975). ‘Reactive’ and ‘unreactive’ iodine in seawater — A possible indication of an organically bound iodine fraction. Mar. Chem. 3, 111–119. doi: 10.1016/0304-4203(75)90018-3
Truesdale V. W. (1978). Iodine in inshore and off-shore marine waters. Mar. Chem. 6, 1–13. doi: 10.1016/0304-4203(78)90002-6
Truesdale V. W. (1994). Distribution of dissolved iodine in the Irish Sea, a temperate shelf sea. Estuarine Coast. Shelf Sci. 38, 435–446. doi: 10.1006/ecss.1994.1030
Truesdale V. W. (1995). The distribution of dissolved iodine in hebridean waters during mid-winter. Mar. Environ. Res. 40, 277–288. doi: 10.1016/0141-1136(94)00147-H
Truesdale V. W., Bale A. J., Woodward E. M. S. (2000). The meridional distribution of dissolved iodine in near-surface waters of the Atlantic Ocean. Prog. Oceanography 45, 387–400. doi: 10.1016/S0079-6611(00)00009-4
Truesdale V. W., Jones K. (2000). Steady-state mixing of iodine in shelf seas off the British Isles. Continental Shelf Res. 20, 1889–1905. doi: 10.1016/S0278-4343(00)00050-9
Truesdale V. W., Luther G. W. (1995). Molecular iodine reduction by natural and model organic substances in seawater. Aquat Geochem 1, 89–104. doi: 10.1007/BF01025232
Truesdale V. W., Nausch G., Baker A. (2001). The distribution of iodine in the Baltic Sea during summer. Mar. Chem. 74, 87–98. doi: 10.1016/S0304-4203(00)00115-8
Truesdale V. W., Upstill-Goddard R. (2003). Dissolved iodate and total iodine along the British east coast. Estuarine Coast. Shelf Sci. 56, 261–270. doi: 10.1016/S0272-7714(02)00161-0
Tsunogai S., Sase T. (1969). Formation of iodide-iodine in the ocean. Deep Sea Res. Oceanographic Abstracts 16, 489–496. doi: 10.1016/0011-7471(69)90037-0
Tymon T. M., Miller E. P., Gonzales J. L., Raab A., Küpper F. C., Carrano C. J. (2017). Some aspects of the iodine metabolism of the giant kelp Macrocystis pyrifera (phaeophyceae). J. Inorganic Biochem. 177, 82–88. doi: 10.1016/j.jinorgbio.2017.09.003
Ullman W. J., Aller R. C. (1980). Dissolved iodine flux from estuarine sediments and implications for the enrichment of iodine at the sediment water interface. Geochimica Cosmochimica Acta 44, 1177–1184. doi: 10.1016/0016-7037(80)90071-X
Ullman W. J., Aller R. C. (1985). The geochemistry of iodine in near-shore carbonate sediments. Geochimica Cosmochimica Acta 49, 967–978. doi: 10.1016/0016-7037(85)90311-4
Uncles R. J., Stephens J. A., Harris C. (2015). Physical processes in a coupled bay–estuary coastal system: Whitsand Bay and Plymouth Sound. Prog. Oceanography 137, 360–384. doi: 10.1016/j.pocean.2015.04.019
Upstill-Goddard R. C. (2006). Air–sea gas exchange in the coastal zone. Estuarine Coast. Shelf Sci. 70, 388–404. doi: 10.1016/j.ecss.2006.05.043
Upstill-Goddard R. C., Elderfield H. (1988). The role of diagenesis in the estuarine budgets of iodine and bromine. Continental Shelf Res. 8, 405–430. doi: 10.1016/0278-4343(88)90012-X
Wadley M. R., Stevens D. P., Jickells T. D., Hughes C., Chance R., Hepach H., et al. (2020). A global model for iodine speciation in the upper ocean. Global Biogeochemical Cycles 34, e2019GB006467. doi: 10.1029/2019GB006467
Waite T. J., Truesdale V. W. (2003). Iodate reduction by Isochrysis galbana is relatively insensitive to de-activation of nitrate reductase activity—are phytoplankton really responsible for iodate reduction in seawater? Mar. Chem. 81, 137–148. doi: 10.1016/S0304-4203(03)00013-6
Waite T. J., Truesdale V. W., Olafsson J. (2006). The distribution of dissolved inorganic iodine in the seas around Iceland. Mar. Chem. 101, 54–67. doi: 10.1016/j.marchem.2006.01.003
Weinberg I., Bahlmann E., Michaelis W., Seifert R. (2013). Determination of fluxes and isotopic composition of halocarbons from seagrass meadows using a dynamic flux chamber. Atmospheric Environ. 73, 34–40. doi: 10.1016/j.atmosenv.2013.03.006
WHO. Available at: https://www.who.int/health-topics/micronutrients (Accessed January 11, 2024).
Widdicombe C. E., Harbour D. (2021). Phytoplankton taxonomic abundance and biomass time-series at Plymouth Station L4 in the Western English Channel 1992-2020. English Channel Publisher: NERC EDS Br. Oceanographic Data Centre NOC. doi: 10.5285/c9386b5c-b459-782f-e053-6c86abc0d129
Wong G. T. F. (1980). The stability of dissolved inorganic species of iodine in seawater. Mar. Chem. 9, 13–24. doi: 10.1016/0304-4203(80)90003-1
Wong G. T. F. (1982). The stability of molecular iodine in seawater. Mar. Chem. 11, 91–95. doi: 10.1016/0304-4203(82)90051-2
Wong G. T. F., Brewer P. G., Spencer D. W. (1976). The distribution of particulate iodine in the Atlantic Ocean. Earth Planetary Sci. Lett. 32, 441–450. doi: 10.1016/0012-821X(76)90084-4
Wong G. T. F., Cheng X.-H. (1998). Dissolved organic iodine in marine waters: Determination, occurrence and analytical implications. Mar. Chem. 59, 271–281. doi: 10.1016/S0304-4203(97)00078-9
Wong G. T. F., Cheng X.-H. (2001a). Dissolved organic iodine in marine waters: role in the estuarine geochemistry of iodine. J. Environ. Monit. 3, 257–263. doi: 10.1039/B007229J
Wong G. T. F., Cheng X.-H. (2001b). The formation of iodide in inshore waters from the photochemical decomposition of dissolved organic iodine. Mar. Chem. 74, 53–64. doi: 10.1016/S0304-4203(00)00095-5
Wong G. T. F., Zhang L. (1992). Changes in iodine speciation across coastal hydrographic fronts in southeastern United States continental shelf waters. Continental Shelf Res. 12, 717–733. doi: 10.1016/0278-4343(92)90027-H
Wong G. T. F., Zhang L.-S. (2003). Seasonal variations in the speciation of dissolved iodine in the Chesapeake Bay. Estuarine Coast. Shelf Sci. 56, 1093–1106. doi: 10.1016/S0272-7714(02)00310-4
Wong G. T. F., Zhang L.-S. (2008). The kinetics of the reactions between iodide and hydrogen peroxide in seawater. Mar. Chem. 111, 22–29. doi: 10.1016/j.marchem.2007.04.007
Wright L. D. (1993). Micromorphodynamics of the inner continental shelf: A middle atlantic bight case study. J. Coast. Res., 93–124.
Yentsch C. S., Menzel D. W. (1963). A method for the determination of phytoplankton chlorophyll and phaeophytin by fluorescence. Deep-Sea Res. 10, 221–231. doi: 10.1016/0011-7471(63)90358-9
Zhang J.-Z., Chi J. (2002). Automated analysis of nanomolar concentrations of phosphate in natural waters with liquid waveguide. Environ. Sci. Technol. 36, 1048–1053. doi: 10.1021/es011094v
Zhang E., Wang Y., Qian Y., Ma T., Zhang D., Zhan H., et al. (2013). Iodine in groundwater of the North China Plain: Spatial patterns and hydrogeochemical processes of enrichment. J. Geochemical Explor. 135, 40–53. doi: 10.1016/j.gexplo.2012.11.016
Keywords: iodine speciation, biogeochemistry, marine systems, seasonal time series, iodide, iodate
Citation: Jones MR, Chance R, Bell T, Jones O, Loades DC, May R, Tinel L, Weddell K, Widdicombe C and Carpenter LJ (2024) Iodide, iodate & dissolved organic iodine in the temperate coastal ocean. Front. Mar. Sci. 11:1277595. doi: 10.3389/fmars.2024.1277595
Received: 14 August 2023; Accepted: 29 January 2024;
Published: 21 February 2024.
Edited by:
Shigeki Wada, University of Tsukuba, JapanReviewed by:
Yuhi Satoh, Japan Agency for Marine-Earth Science and Technology, JapanAtsushi Ooki, Hokkaido University, Japan
Copyright © 2024 Jones, Chance, Bell, Jones, Loades, May, Tinel, Weddell, Widdicombe and Carpenter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew R. Jones, bWF0dGhld19yX2pvbmVzQHJvY2tldG1haWwuY29t; Lucy J. Carpenter, bHVjeS5jYXJwZW50ZXJAeW9yay5hYy51aw==
†Present addresses: Oban Jones, Bermuda Institute for Ocean Sciences (BIOS), St. George’s, Bermuda
Rebecca May, Bermuda Institute for Ocean Sciences (BIOS), St. George’s, Bermuda
Liselotte Tinel, Institut Mines-Té lé com (IMT) Nord Europe, Université of Lille, Centre for Energy and Environment, Lille, France
Katherine Weddell, Fera Science Ltd, Biotech Campus, York, United Kingdom