 Lili Zhang
Lili Zhang Xinyu Gu
Xinyu Gu Chong Chen
Chong Chen Xing He
Xing He Ying Qi
Ying Qi Jin Sun
Jin Sun- 1Key Laboratory of Evolution & Marine Biodiversity (Ministry of Education) and Institute of Evolution & Marine Biodiversity, Ocean University of China, Qingdao, China
- 2Laoshan Laboratory, Qingdao, China
- 3X-STAR, Japan Agency for Marine-Earth Science and Technology (JAMSTEC), Yokosuka, Japan
Neomphalida is an order of gastropod molluscs with highly diverse morphology and a global distribution across various chemosynthesis-based ecosystems from organic falls to hot vents. The phylogenetic relationships of taxa within this order remain contentious, due to the rarity of material leading to a low taxonomic coverage and few genetic markers used. Neomphalida includes three families—Melanodrymiidae, Neomphalidae, and Peltospiridae—and molecular sequences are especially lacking in Melanodrymiidae. Here, we assembled a total of 11 mitogenomes covering these three families and 14 genus-level groups to reconstruct the most complete phylogeny of Neomphalida to date. Our current result recovered the monophyly of three families with maximum support and a likely interfamilial relationship of (Melanodrymiidae + Neomphalidae) + Peltospiridae. These indicate the possibility of habitat shifting from non-chemosynthetic deep sea to hot vent and then to sunken wood, accompanied by elevated mitogenome rearrangements and amino acid substitution rates in Melanodrymiidae. By mapping species distribution on the phylogeny, our findings suggest a Pacific origin of Neomphalida and multiple historical dispersal events of Peltospiridae to the Indian Ocean and at least once to the Atlantic.
Introduction
Deep-sea chemosynthetic ecosystems are “oases” fueled by bacterial primary production (Van Dover et al., 2001; Dubilier et al., 2008), such as deep-sea hot vents, cold seeps, and organic falls like sunken woods and whale falls (Martin et al., 2008; He et al., 2023). Many species inhabiting these chemosynthesis-based ecosystems are specialists that cannot be found in other habitats (Mills and Harrison, 1998). Hydrothermal vent specialists are especially notable, as vents are often distributed like stepping stones along mid-ocean ridges species with dispersal most likely mediated by currents along the mid-ocean ridges (Tunnicliffe and Mary R. Fowler, 1996; Rogers et al., 2012), although other systems in settings like hot spots and back-arc basins are also present (Breusing et al., 2016; Xu et al., 2021; Brunner et al., 2022).
Neomphalida is an order of gastropod molluscs endemic to chemosynthetic ecosystems, forming the subclass Neomphaliones together with the cocculiniform gastropods in the order Cocculinida (Ponder et al., 2019; Uribe et al., 2022). Neomphalida includes a single superfamily Neomphaloidea with three families: Melanodrymiidae, Neomphalidae, and Peltospiridae. Among these three families, Melanodrymiidae has been found on sunken wood and hydrothermal vents, while living Neomphalidae and Peltospiridae are vent specialists (Mclean, 1989; Heß et al., 2008; Chen et al., 2015b; Chen et al., 2015c; Zhong et al., 2022). One genus, Retiskenea, is known from both recent and fossil hydrocarbon seeps as early as the Early Jurassic with unclear familial affinity, although it has been tentatively assigned to Neomphalidae (Warén and Bouchet, 2001; Kaim et al., 2014; Kaim, 2022). Neomphaloideans are morphologically and ecologically diverse, ranging from filter feeding to grazing to relying on endosymbionts (Sasaki et al., 2010). The latter is seen in the two coiled genera Chrysomallon and Gigantopelta (Chen et al., 2015a; Chen et al., 2015b; Sun et al., 2020), which have independently evolved (Chen et al., 2015c; Zhong et al., 2022) endosymbiosis in an enlarged and modified esophageal gland (Lan et al., 2021; Lan et al., 2022). To better understand the evolution of these traits, a robust phylogeny is required.
Reconstructing the internal phylogenetic relationships among different taxa within Neomphalida has proved challenging with different studies finding disparate topologies. For instance, multi-gene and mitogenome-based phylogenetic studies have placed the Scaly-foot Snail Chrysomallon squamiferum in Peltospiridae, which agrees with its morphology, but it has been recovered nested within Neomphalidae when only the mitochondrial COI gene was used (Chen et al., 2017). Monophyly of the three Neomphalida families was sometimes supported by molecular phylogeny (Heß et al., 2008) but not in others (Aktipis and Giribet, 2010; Chen et al., 2017; Zhong et al., 2022). These studies were performed either using a small number of genes or with a limited taxon coverage. For instance, in the only mitogenome-based molecular phylogeny study focusing on this group, Melanodrymiidae was unrepresented (Zhong et al., 2022). In this study, we expanded the mitogenome sampling to the whole order by adding a total of 11 mitogenomes from all three families. We are the first to include the five mitogenomes from Melanodrymiidae, with phylogenetic analyses performed using various methods, matrix, and models for robustness.
Materials and methods
Data collection and sequencing
All mitochondrial genomes used in this study are listed in Tables 1, 2. Melanodrymia galeronae, Melanodrymia telperion, and Melanodrymia laurelin were collected from inactive sulfide mounds at 9°N on the East Pacific Rise by a manipulator of the human-occupied vehicle (HOV) Alvin during R/V Atlantis cruise AT50-06 (Chen et al., 2024). The specimens were preserved in 80% Ethanol. We assembled further mitogenomes using unassembled reads openly available on the NCBI SRA database (Table 2). These were complemented with published mitogenomes (Zhong et al., 2022) for phylogenetic analyses.
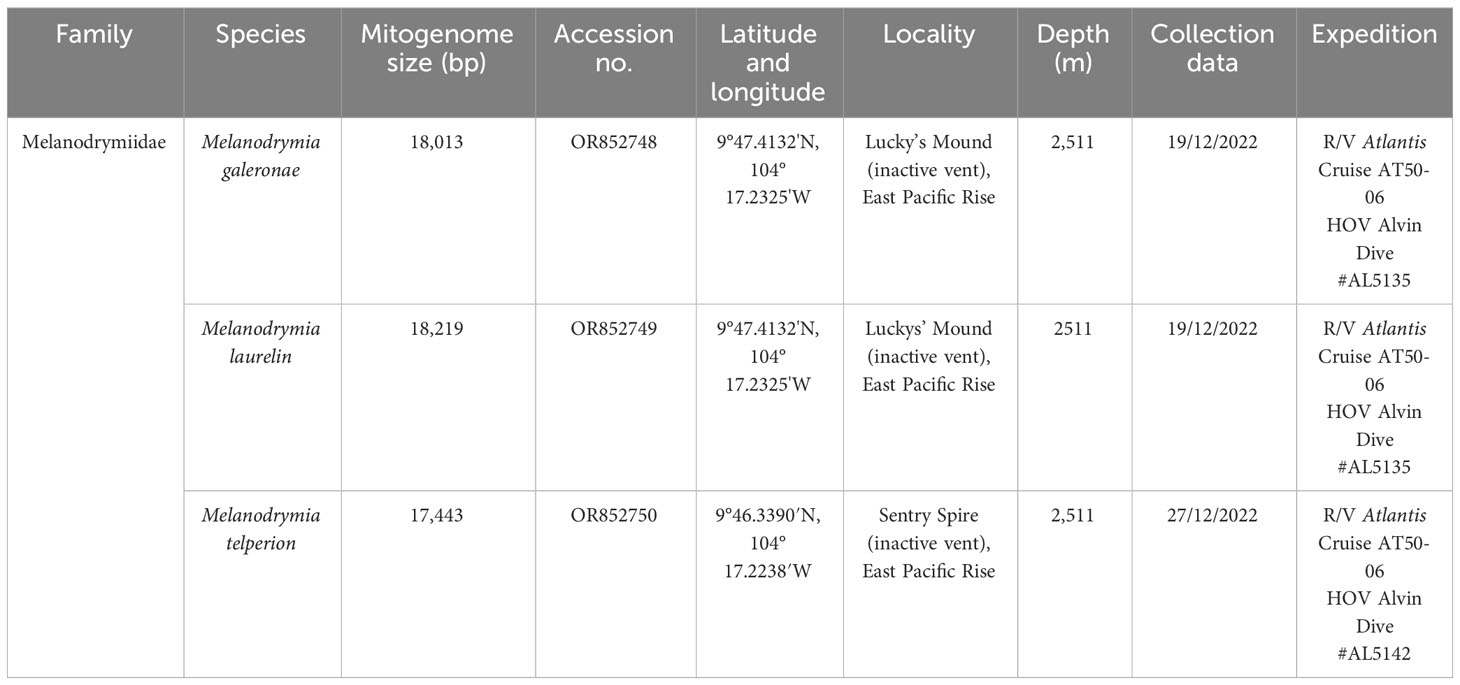
Table 1 Species newly collected with mitogenomes assembled in this study.
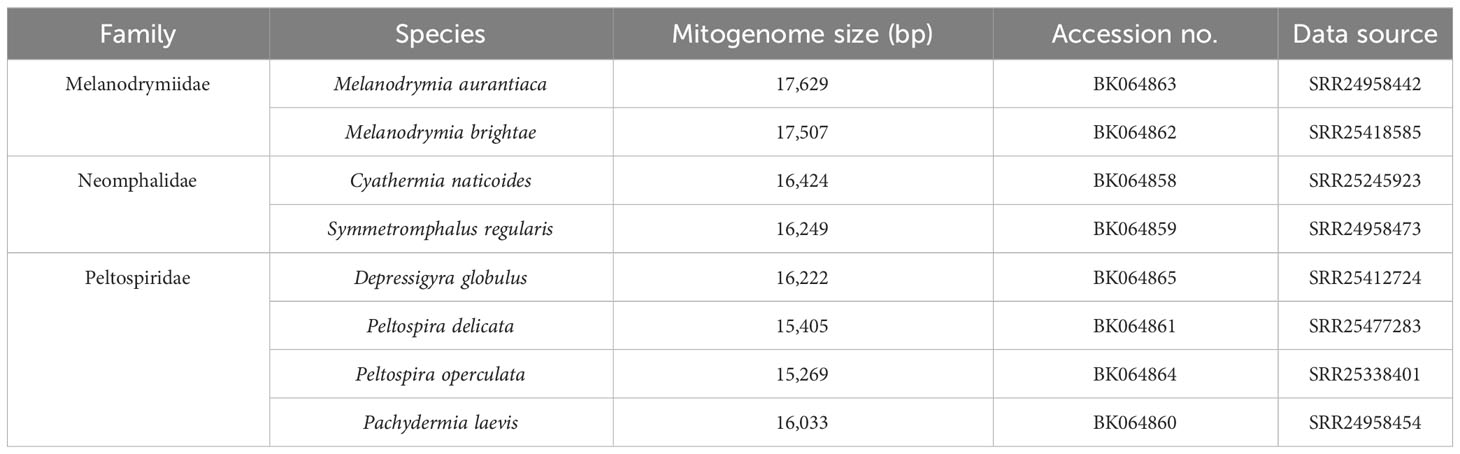
Table 2 New mitochondrial genomes assembled from the raw Illumina sequencing data available on the NCBI SRA database.
Total genomic DNA of the three newly sequenced species were extracted using the SDS method (Phillips and Simon, 1995); then, the DNA libraries were constructed with NEB Next® Ultra™ DNA Library Prep Kit for Illumina (NEB, USA) following the manufacturer’s instructions. These were then sequenced using 150 bp paired-end Illumina sequencing to generate approximately 20 Gb of raw data on a NovaSeq 6000 platform.
Assembly and mitogenome annotation
The raw data were trimmed by Trimmomatic v 0.39 (Bolger et al., 2014) with the default settings. The clean data were used for mitogenome assembly by either Novoplasty v 4.3.1 (Dierckxsens et al., 2017), MEGAHIT v 1.2.9 (Li et al., 2016), or GetOrganelle v 1.7.7.0 (Jin et al., 2020). For Novoplasty, the COI sequence from the closed lineage was used, e.g., within the same genus or family, as the “seed input”.
The complete mitogenome was annotated by the MITOS2 web server (Bernt et al., 2013) with the invertebrate genetic code. Some protein-coding genes were not annotated with the stop codon or with inconsistent sequence length, and these were corrected manually by comparing with published gastropod mitochondrial sequences.
Phylogenetic analysis and conflicting phylogenetic signal test
We used 21 species belonging to Neomphalida, plus four species of its sister-order Cocculinida and two non-Neomphaliones vetigastropods as the outgroup. To overcome the third codon saturation issue during the phylogenetic analyses, A and G in the third codon position were converted to R, while C and T were converted to Y, respectively, a common method used in other mitogenome phylogenetic reconstructions (Lee et al., 2019). Three types of datasets were used in this study [i.e., the amino acid sequences of the 13 protein-coding genes (PCGs), the nucleotide sequences of the 13 PCGs, and the nucleotide sequences of the 13 PCGs + two rRNAs (12S rRNA and 16S rRNA)]. The amino acid sequences of the 13 PCGs were independently aligned by MUSCLE v 3.8.31 (Edgar, 2004) and areas of poor alignments were clipped by trimAl v 1.4.1 (Capella-Gutiérrez et al., 2009) with the setting of “automated1”, respectively. Supermatrix and the corresponding partition file were generated by a python script (Liu et al., 2023). Then, two methods were used in contrasting tree, namely, concatenation and coalescence. For a concatenated method, IQ-TREE v 2.1.3 (Minh et al., 2020) with “-MFP” was used to select the best-fit model for each partition. An additional empirical profile mixture model, C60, was carried out on the amino acid sequences of the 13 PCGs. RAxML v 8.2.12 (Stamatakis, 2014) and PhyloBayes MPI v1.8c (Lartillot et al., 2013) were used to reconstruct the phylogenetic tree with the model of CAT+GTR. ASTRAL - MP v 5.15.5 (Yin et al., 2019) was used for the coalescent method. To test the phylogenetic signals on each partition, different species tree topologies were tested on each partition to check the distribution of the phylogenetic signal of the gene tree based on a previous study (Shen et al., 2017). All the commands used in the current study can be found in the Supplementary Material.
Mitochondrial genome properties
We analyzed the GC content, amino acid substitution rate, nucleic acid substitution rate, and mitogenome size in Melanodrymiidae, Neomphalidae, and Peltospiridae. We calculated the GC content using SeqKit v2.2.0 with the command fx2tab. To measure the evolutionary rates of amino acid and nucleic acid substitutions, we used the tip-to-root distance. We transformed the GC content and amino acid substitution rate using the arcsine square root transformation to fit for downstream t-test statistics. For nucleic acid substitution rate and genome size, we used the formula y = ln(x) to convert the values. We classified the three families into three groups and used Student’s t-test for pairwise comparison to determine if there were any significant differences in their properties. All analyses were conducted in the R package ggplot2 v3.4.4 (Villanueva and Chen, 2019) and violin plot (vioplot) v0.4.0.
The distribution of the 21 species in the three families
Melanodrymia galeronae, Melanodrymia laurelin, and Melanodrymia telperion were collected in this study. For the remaining species, their distribution was surveyed from the original description and subsequent records in the literature. These include the following: Chrysomallon squamiferum (Chen et al., 2015b), Depressigyra globulus (Warén, 2001; Desbruyères et al., 2006), Pachydermia laevis (Kiel, 2004), Lirapex politus (Chen et al., 2017), Dracogyra subfusca (Chen et al., 2017), Gigantopelta aegis (Chen et al., 2015c), Symmetriapelta wareni (Chen and Sigwart, 2023), Symmetriapelta sp. (Zhong et al., 2022), Cyathermia naticoides (Kiel, 2004), Peltospira smaragdina (Kiel, 2004), Nodopelta heminoda (Mclean, 1989), Peltospira operculata (Mclean, 1989), Peltospira delicata (Mclean, 1989), Melanodrymia aurantiaca (Haszprunar, 1989), Melanodrymia brightae (Wareín and Bouchet, 1993), Lamellomphalus manusensis (Zhang and Zhang, 2017), Symmetromphalus regularis (Mclean, 1990), and Neomphalidae gen. et sp. Hatoma sensu Zhong et al., 2022 (Zhong et al., 2022).
Results
A total of 11 mitogenomes were assembled in Neomphalida in this study, and the mitogenomes of Melanodrymia laurelin, Melanodrymia galeronae, Melanodrymia brightae, Melanodrymia aurantiaca, Cyathermia naticoides, and Symmetromphalus regularis are circular. The mitogenomes ranged from 15,269 bp (Peltospira operculata) to 18,219 bp (Melanodrymia laurelin) in length (Tables 1, 2). All mitochondrial genomes contain exclusively 13 protein-coding genes, 22 tRNA genes, and two rRNA genes (12S and 16S), without any gene duplication or deletion.
By applying the concatenated and coalescent methods of phylogenetic tree construction, we obtained a total of 13 phylogenetic trees, comprising two different tree topologies with conflicting phylogenetic signals. In topology 1 (Figure 1A), Melanodrymiidae is sister to (Neomphalidae + Peltospiridae), supported by the following methods: 13PCGs with MFP, C60, RAxML, PB, ASTRAL − MP, and PCGs with BP. In topology 2, however, Peltospiridae was recovered sister to (Neomphalidae + Melanodrymiidae), supported by the following methods: 13PCGs with MFP, RAxML, ASTRAL – MP and PCGs + rRNA with MFP, RAxML, BP, ASTRAL − MP (Figure 1B). The three Neomphalida families were monophyletic with maximal support across all methods, and the whole order Neomphalida.
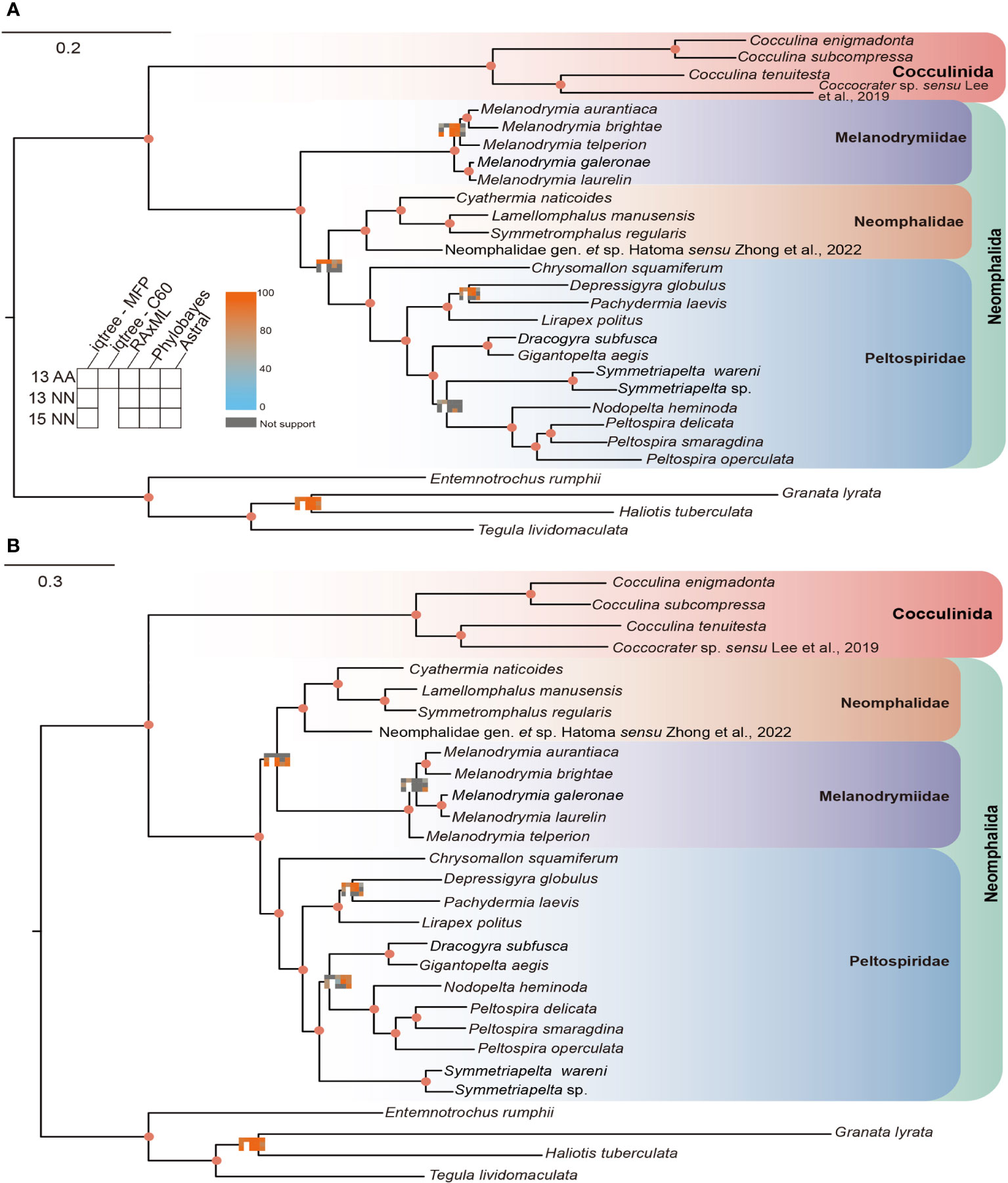
Figure 1 Two topologies obtained by different phylogenetic reconstruction methods used in this study, (A) topology 1 and (B) topology 2. Node supports represented by a matrix consisting of 13 squares that are colored with a continuous scale bar ranging from 0 to 100. Each small square represents a different method of phylogenetic analysis. Gray indicates non-support, while the darker shades of orange and blue indicate higher and lower support, respectively.
To further test the phylogenetic signals of these two topologies on all the partitioned genes, 15 nucleotide sequences of the 13 PCGs + 2 rRNAs and 13 PCGs were separately tested on the two trees. For the 13 PCGs and two rRNAs, the gene tree from 10 of them supported topology 2 and the remaining five supported topology 1. Meanwhile, for the 13 PCGs, seven supported topology 1 and six supported topology 2 (Figure 2).
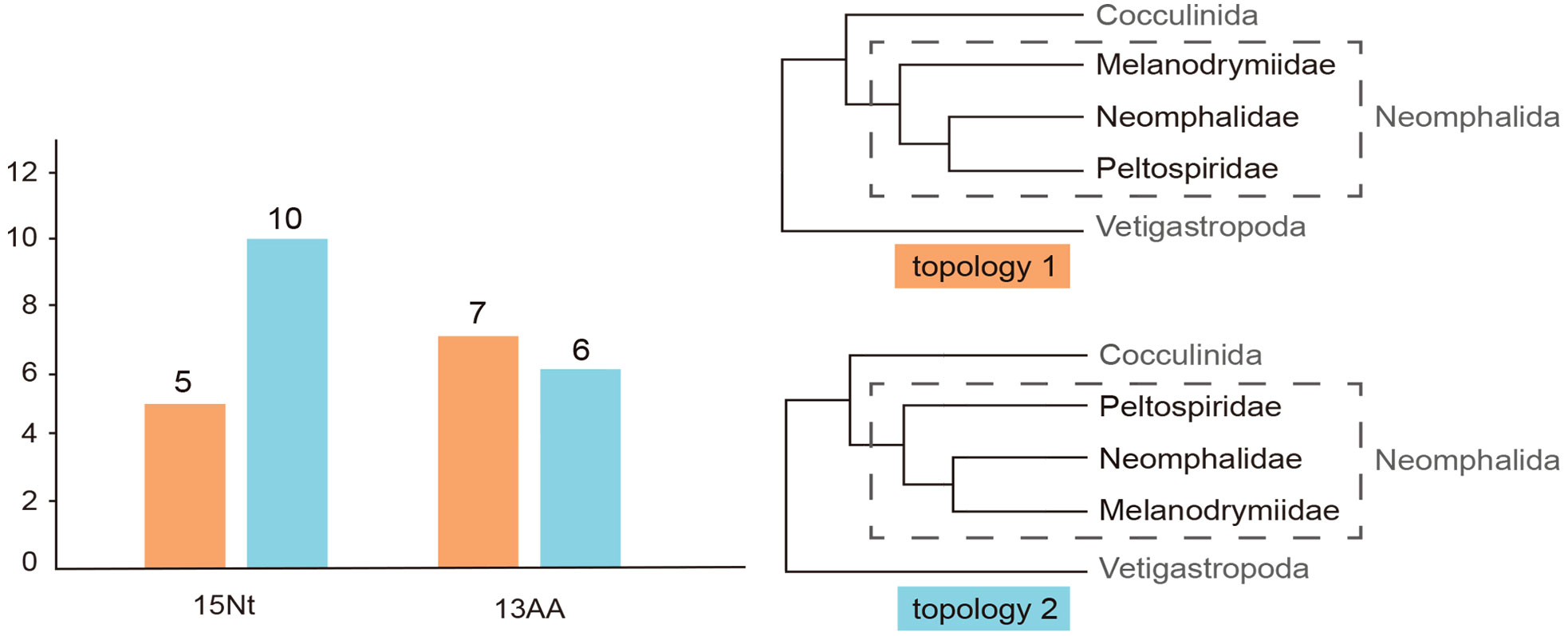
Figure 2 The result of conflicting phylogenetic signal test. Two topologies generated (topology 1 and topology 2), for 13PCGs + 2rRNA and the amino acid sequences of the 13 protein coding genes, 4/7 gene trees support the topology 2. Pink color represents topology 1, while blue represents topology 2.
For intra-family level relationships, the phylogenetic positions were relatively stable in both topologies. The lineage with contentious position was the clade of (Dracogyra + Gigantopelta), which was found to be either sister to ((Nodopelta + Peltospira) + Symmetriapelta) in topology 1 or to (Nodopelta + Peltospira) in topology 2. In addition, the phylogenetic position of Melanodrymia telperion was variable, being either sister to (Melanodrymia aurantiaca +Melanodrymia brightae) or to all other Melanodrymia species included (Figure 1).
The gene order of each mitogenome was analyzed. Overall, the gene order within each family was consistent, and they shared the identical gene order of atp6–atp8–trnD–cox2–nad5–trnH–nad4–nad4L–trnT–trnS2 (Figure 3). Comparing the gene order of each family with the hypothetical ancestral gene order of the whole Gastropoda, the gene order of both Neomphalidae + Peltospiridae was more similar with less translocation and transversion, while the gene order of Melanodrymiidae was more deviated with the translocation of cob–trnE–trnF–trnR–trnC–trnG–cox3–trnK–trnI–nad3–trnS1–nad2. Both Neomphalida and Cocculinida have a shared transversion of atp6–atp8–trnD–cox2 when compared with the hypothetical ancestral gene order.
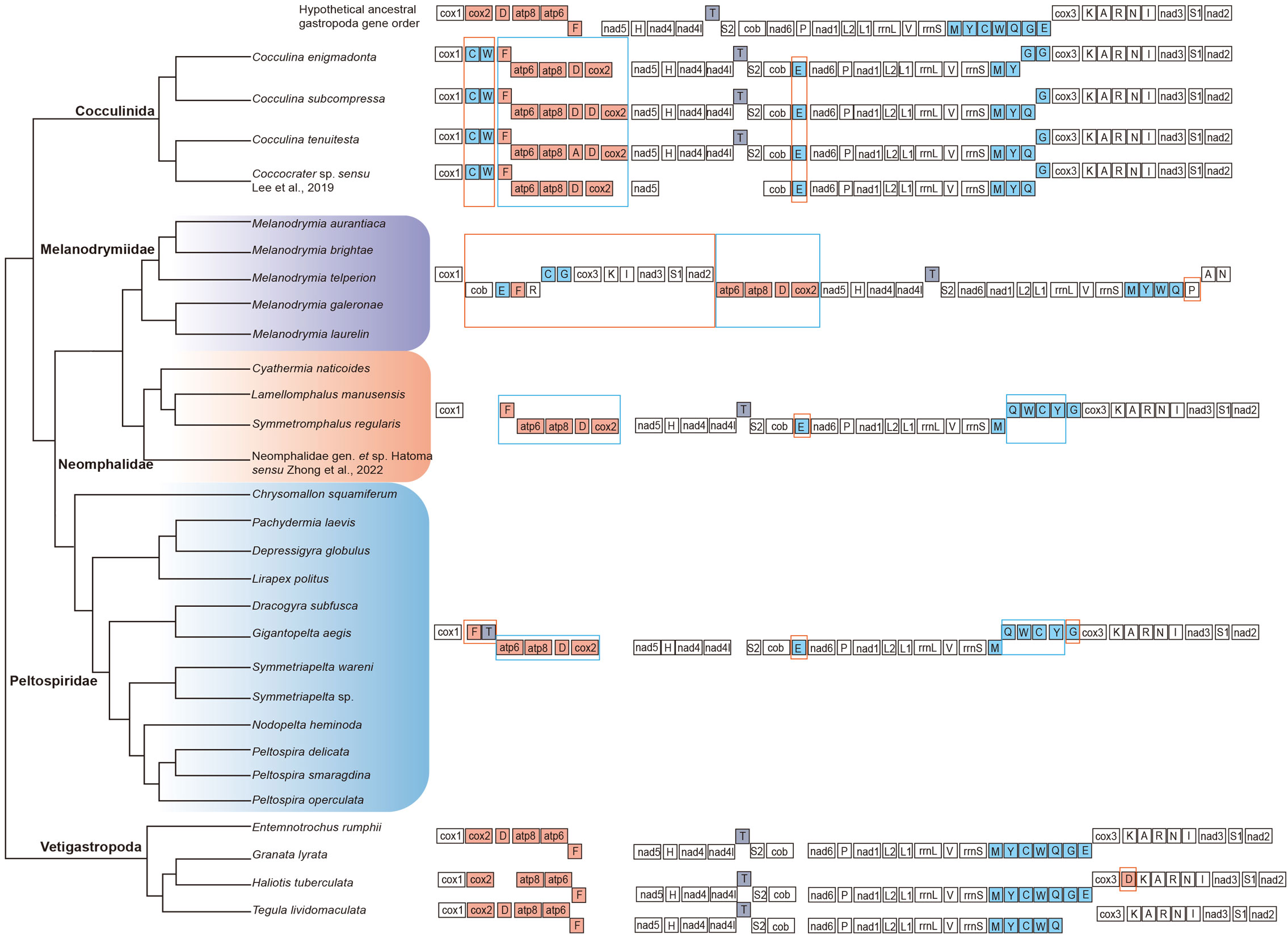
Figure 3 Comparative analysis of mitochondrial gene rearrangements between three families of Neomphalida and the hypothesized ancestor of Gastropoda. Transposition and inversion are indicated with orange and blue boxes, respectively.
The mitogenome GC content, mitogenome size, and substitution rate of the three families were also calculated (Figure 4). Melanodrymiidae had the lowest GC content, which significantly differed from Peltospiridae and Neomphalidae (p < 0.01) (Figure 4A). There is a significant difference in amino acid substitution rates among the three families, with Peltospiridae exhibiting the highest value compared to Neomphalidae (p < 0.01) (Figure 4B). For the nucleotide substitution rate, Neomphalidae has the lowest value compared with other two families and differs significantly from Melanodrymiidae (p < 0.05) (Figure 4C). Melanodrymiidae was found to have the largest mitogenome size among the three families (p < 0.05) (Figure 4D). Among the three families, Peltospiridae showed a much wider range of GC content and substitution rates of nucleotide and amino acids. For the GC content, C. squamiferum had the highest (33.84%), while S. wareni had the lowest (27.81%). For the substitution rates of nucleotide and amino acids, G. aegis showed the lowest substitution rates (1.03 and 0.59, respectively), while P. operculata had the highest rates (1.13 and 0.74, respectively).
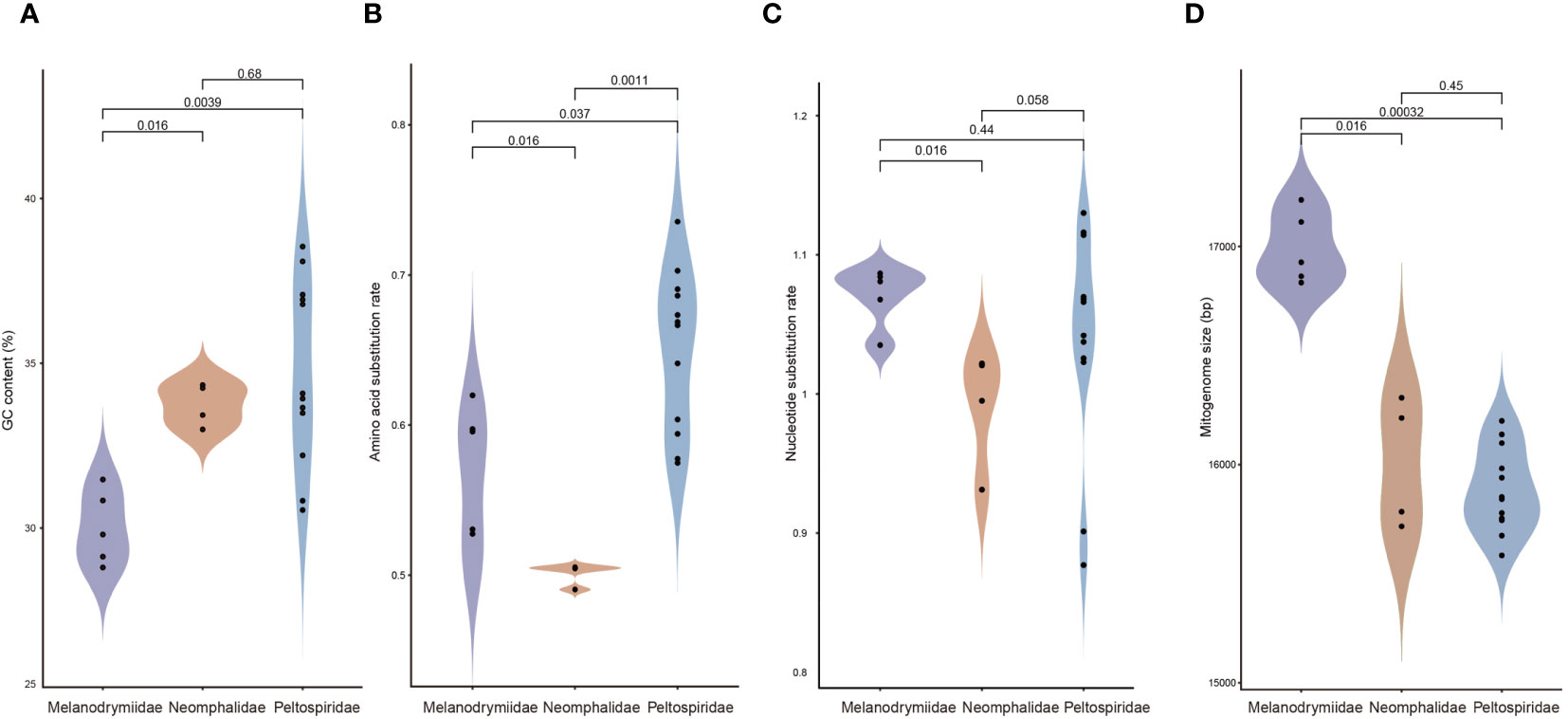
Figure 4 Four violin plots comparing (A) GC content, evolutionary rates including (B) amino acid substitution rate and (C) nucleotide substitution rate, and (D) mitogenome size in Melanodrymiidae, Neomphalidae, and Peltospiridae. Student’s t-tests with p-values were used to evaluate the statistical significance of differences between each group. p-values below 0.05 are considered to indicate statistically significant differences.
Discussion
As one of the six subclasses in Gastropoda, Neomphaliones is relatively understudied compared to the other five, largely due to sample availability since species in this clade are mainly from deep-sea chemosynthetic ecosystems. By including 11 new mitogenomes and a total of 21 mitogenomes covering all three known families in Neomophlida, we tested their phylogenetic positions with a combination of different methods and matrixes, gene order analysis, GC content, and evolutionary rate analysis. As of now, 26 genera are included in Neomphalida, and our analyses cover 13 genera plus an undescribed genus (Neomphalidae gen. et sp. Hatoma sensu Zhong et al., 2022).
Our phylogenetic analysis has recovered the monophyly of the three Neomphalida families across all methods, indicating a good consistency between molecular phylogeny and morphology at the family level. This result is in line with a former phylogenetic tree analysis (Heß et al., 2008) but with better support on the nodes and improved taxon coverage. Nevertheless, the relationships among these three families remain unsettled even with complete mitogenome sequencing. By checking the phylogenetic signals on each partition of both nucleotide and amino acid sequences of our two conflicting topologies recovered, we show that 10 nucleotide partitions support topology 2 versus five supporting topology 1, while seven amino acid partitions supported topology 1 versus six supporting topology 2. There shall be very little effect of the third codon saturations on the molecular phylogeny, since the third codons was replaced by degenerated nucleotides. It is possible that the phylogenetic signal in the nucleotide data is lost in the amino acid dataset after translation for Neomphalida, and overall, it seems that topology 2 is better supported by our results. To further resolve the phylogenetic positions of these three families, a more comprehensive study using transcriptome or genome sequencing is needed. However, these methods require high-quality samples, which are usually lacking for many small and rare species in this order. In addition, more taxon sampling is also required, especially sunken wood melanodrymiid species like Leptogyra and Leptogyropsis with some plesiomorphic characters (Heß et al., 2008).
Our topology 2 is different from a former study (Heß et al., 2008), which included only 11 species with two partial genes (COI and histone H3). Based on their phylogenetic result and that some melanodrymiids show plesiomorphic characters inhabit the likely ancestral sunken wood habitat, the authors deduced a scenario that sunken wood could have served as an ecological stepping stone to hot vent. From our phylogenetic results, it seems equally likely that the subclass Neomphaliones diverged from non-chemosynthetic deep seafloor according to habitats, first splitting into the sunken-wood lineage (i.e., Cocculinida) and the hot-vent lineage (i.e., Neomphalida). Then, there could have been a secondary habitat shift from hydrothermal vents to sunken wood in Melanodrymiidae. This secondary habitat shift could be a plausible reason of the elevated nucleotide and amino acid substitution rate and the deviated mitogenome gene order in Melanodrymiidae when compared to Neomphalidae. This habitat transition scenario is complex and also differs from other deep-sea lineages, such as bathymodioline mussels, which took “wooden steps” to the vents and seeps (Lorion et al., 2013)—warranting future phylogenetic analyses using genomic-level data to test.
Species in Peltospiridae have a higher amino acid substitution rate (p < 0.05) and nucleotide substitution rate (though not very significant) comparing with the other two families, which may be related to its wider species distributions. By mapping the species distribution in combination with the phylogenetic analysis, it is clear that the Neomphalida species included are mostly from the eastern Pacific, especially Melanodrymiidae and Neomphalidae (Figure 5) (Mclean, 1981). This is consistent with the distribution of known species of these two families, with Neomphalidae limited to the Pacific and Melanodrymiidae being largely Pacific except four species of Leptogyra from the Atlantic (Bush, 1897; Warén and Bouchet, 2001). For Peltospiridae, species in this family are also mostly found in the Pacific, but with species also distributing in the Indian Ocean, Atlantic Ocean, and Southern Ocean (East Scotia Ridge). Our tree suggests that peltospirids have colonized the Indian Ocean hot vents at least three times in historical dispersal. The earliest event was the clade leading up to Chrysomallon (Zhong et al., 2022), followed by the (Gigantopelta aegis + Dracogyra subfusca) clade and then Lirapex politus, in agreement with previous COI analyses (Chen et al., 2017). These three Indian Ocean colonization events may be linked to major tectonic events in the Indian Ocean (McElhinny, 1970), but the limited fossil records in the whole order means that it is difficult to carry out accurate molecular clock analysis with sufficient calibration nodes. Our results, combined with overall distribution pattern of Neomphalida, point to an eastern Pacific origin of this group.
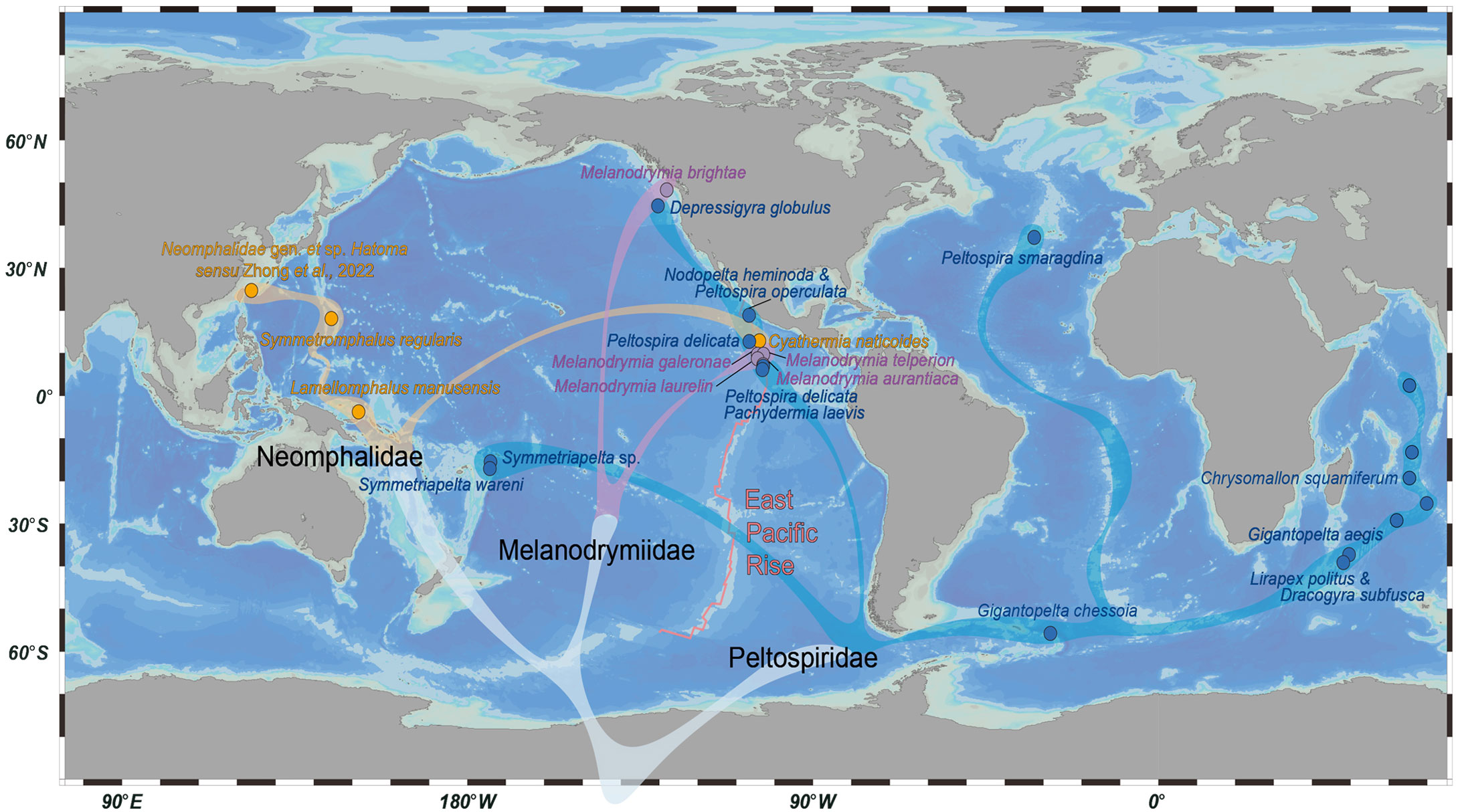
Figure 5 Overview map of the distribution pattern of the 21 Neomphalida species included in this study, across the three families. Dots indicate representative localities of each species or sites of collection for the newly collected species in this study. Species belonging to the same family are indicated by the same color. The tree in gray below is a stylized representation of their phylogenetic relationship, as deduced from their mitogenomes.
Data availability statement
The data presented in the study are deposited in the NCBI repository (https://www.ncbi.nlm.nih.gov/), under BioProject number PRJNA1048888.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study. No experiment on live animals were carried out in this study.
Author contributions
LZ: Data curation, Formal analysis, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. XG: Methodology, Software, Visualization, Writing – review & editing. CC: Data curation, Methodology, Visualization, Writing – review & editing. XH: Data curation, Methodology, Writing – review & editing. YQ: Methodology, Software, Writing – review & editing. JS: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Science and Technology Innovation Project of Laoshan Laboratory (LSKJ202203104), National Natural Science Foundation of China (42176110), the Fundamental Research Funds for the Central Universities (202172002 and 202241002), and the Young Taishan Scholars Program of Shandong Province (tsqn202103036). The collection of newly sequenced Melanodrymia specimens were funded by the U.S. National Science Foundation grants OCE 1947735 and OCE 2152453 to Lauren S. Mullineaux and Stace E. Beaulieu.
Acknowledgments
We thank the captain, crew, and on-board scientists of R/V Atlantis cruise AT50-06 (chief scientist: Shawn Arellano, Western Washington University) for their support of the scientific activities; which we also extend to the HOV Alvin team. Stace E. Beaulieu, Michael Meneses, and Lauren Mullineaux (Woods Hole Oceanographic Institution) are gratefully acknowledged for collecting, sorting, and providing the gastropod specimens for study. The bioinformatics analysis was performed in the high-performance cluster IEMB-1, Ocean University of China. Comments from the three reviewers improved an earlier version of this paper.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1341869/full#supplementary-material
References
Aktipis S. W., Giribet G. (2010). A phylogeny of Vetigastropoda and other “archaeogastropods”: re-organizing old gastropod clades. Invertebrate. Biol. 129 (3), 220–240. doi: 10.1111/j.1744-7410.2010.00198.x
Bernt M., Donath A., Jühling F., Externbrink F., Florentz C., Fritzsch G., et al. (2013). MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69 (2), 313–319. doi: 10.1016/j.ympev.2012.08.023
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Breusing C., Biastoch A., Drews A., Metaxas A., Jollivet D., Vrijenhoek R. C., et al. (2016). Biophysical and population genetic models predict the presence of “Phantom” stepping stones connecting mid-atlantic ridge vent ecosystems. Curr. Biol. 26 (17), 2257–2267. doi: 10.1016/j.cub.2016.06.062
Brunner O., Chen C., Giguère T., Kawagucci S., Tunnicliffe V., Watanabe H. K., et al. (2022). Species assemblage networks identify regional connectivity pathways among hydrothermal vents in the Northwest Pacific. Ecol. Evol. 12 (12), e9612. doi: 10.1002/ece3.9612
Bush K. J. (1897). Revision of the marine gastropods referred to Cyclostrema, Adeorbis, Vitrinella, and related genera; with descriptions of some new genera and species belonging to the Atlantic fauna of America. Trans. ConnecticutAcademy. Arts. Sci. 10, 97–144.
Capella-Gutiérrez S., Silla-Martínez J. M., Gabaldón T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25 (15), 1972–1973. doi: 10.1093/bioinformatics/btp348
Chen C., Copley J. T., Linse K., Rogers A. D., Sigwart J. (2015a). How the mollusc got its scales: convergent evolution of the molluscan scleritome. Biol. J. Linn. Soc. 114 (4), 949–954. doi: 10.1111/bij.12462
Chen C., Li Y., Sun J., Beaulieu S. E., Mullineaux L. S. (2024). Two new melanodrymiid snails from the East Pacific Rise indicate the potential role of inactive vents as evolutionary stepping-stones. Systemat. Biodivers. 22 (1), 2294014. doi: 10.1080/14772000.2023.2294014
Chen C., Linse K., Copley J. T., Rogers A. D. (2015b). The ‘scaly-foot gastropod’: a new genus and species of hydrothermal vent-endemic gastropod (Neomphalina: Peltospiridae) from the Indian Ocean. J. Molluscan. Stud. 81 (3), 322–334. doi: 10.1093/mollus/eyv013
Chen C., Linse K., Roterman C. N., Copley J. T., Rogers A. D. (2015c). A new genus of large hydrothermal vent-endemic gastropod (Neomphalina: Peltospiridae). Zool. J. Linn. Soc. 175 (2), 319–335. doi: 10.1111/zoj.12279
Chen C., Sigwart J. (2023). The lost vent gastropod species of Lothar A. Beck. Zootaxa 5270, 401–436. doi: 10.11646/zootaxa.5270.3.2
Chen C., Zhou Y., Wang C., Copley J. (2017). Two new hot-vent peltospirid snails (Gastropoda: neomphalina) from longqi hydrothermal field, southwest Indian ridge. Front. Mar. Sci. 4. doi: 10.3389/fmars.2017.00392
Desbruyères D., Segonzac M., Bright M. (2006). Handbook of Deep-sea Hydrothermal Vents, Second Completely Revised Edition. Denisia. 18, 1–544.
Dierckxsens N., Mardulyn P., Smits G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45 (4), e18. doi: 10.1093/nar/gkw955
Dubilier N., Bergin C., Lott C. (2008). Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat. Rev. Microbiol. 6 (10), 725–740. doi: 10.1038/nrmicro1992
Edgar R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32 (5), 1792–1797. doi: 10.1093/nar/gkh340
Haszprunar G. (1989). The anatomy of Melanodrymia aurantiaca Hickman a coiled archaeogastropod from the east pacific hydrothermal vents (Mollusca, gastropoda). Acta Zool. 70 (3), 175–186. doi: 10.1111/j.1463-6395.1989.tb01068.x
He X., Xu T., Chen C., Liu X., Li Y.-X., Zhong Z., et al. (2023). Same (sea) bed different dreams: Biological community structure of the Haima seep reveals distinct biogeographic affinities. Innovation Geosci. 1 (2), 100019. doi: 10.59717/j.xinn-geo.2023.100019
Heß M., Beck F., Gensler H., Kano Y., Kiel S., Haszprunar G. (2008). Microanatomy, shell structre and molecular phylogeny of Leptogyra, Xyleptogyra and Leptogyropsis (Gastropoda: Neomphalida: Melanodrymiidae) from sunken wood. J. Molluscan. Stud. 74 (4), 383–401. doi: 10.1093/mollus/eyn030
Jin J. J., Yu W. B., Yang J. B., Song Y., dePamphilis C. W., Yi T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21 (1), 241. doi: 10.1186/s13059-020-02154-5
Kaim A. (2022). “A review of gastropods at ancient hydrocarbon seeps,” in Ancient hydrocarbon seeps. Eds. Kaim A., Cochran J. K., Landman N. H. (Cham: Springer International Publishing), 323–374.
Kaim A., Jenkins R., Tanabe K., Kiel S. (2014). Mollusks from late Mesozoic seep deposits, chiefly in California. Zootaxa 3861, 401–440. doi: 10.11646/zootaxa.3861.5.1
Kiel S. (2004). Shell structures of selected gastropods from hydrothermal vents and seeps. Malacologia 46, 169–183.
Lan Y., Sun J., Chen C., Sun Y., Zhou Y., Yang Y., et al. (2021). Hologenome analysis reveals dual symbiosis in the deep-sea hydrothermal vent snail Gigantopelta aegis. Nat. Commun. 12 (1), 1165. doi: 10.1038/s41467-021-21450-7
Lan Y., Sun J., Chen C., Wang H., Xiao Y., Perez M., et al. (2022). Endosymbiont population genomics sheds light on transmission mode, partner specificity, and stability of the scaly-foot snail holobiont. ISME. J. 16 (9), 2132–2143. doi: 10.1038/s41396-022-01261-4
Lartillot N., Rodrigue N., Stubbs D., Richer J. (2013). PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 62 (4), 611–615. doi: 10.1093/sysbio/syt022
Lee H., Chen W.-J., Puillandre N., Aznar-Cormano L., Tsai M.-H., Samadi S. (2019). Incorporation of deep-sea and small-sized species provides new insights into gastropods phylogeny. Mol. Phylogenet. Evol. 135, 136–147. doi: 10.1016/j.ympev.2019.03.003
Li D., Luo R., Liu C. M., Leung C. M., Ting H. F., Sadakane K., et al. (2016). MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11. doi: 10.1016/j.ymeth.2016.02.020
Liu X., Sigwart J. D., Sun J. (2023). Phylogenomic analyses shed light on the relationships of chiton superfamilies and shell-eye evolution. Mar. Life Sci. Technol. 5 (4), 525–537. doi: 10.1007/s42995-023-00207-9
Lorion J., Kiel S., Faure B., Kawato M., Ho S. Y., Marshall B., et al. (2013). Adaptive radiation of chemosymbiotic deep-sea mussels. Proc. Biol. Sci. 280 (1770), 20131243. doi: 10.1098/rspb.2013.1243
Martin W., Baross J., Kelley D., Russell M. J. (2008). Hydrothermal vents and the origin of life. Nat. Rev. Microbiol. 6 (11), 805–814. doi: 10.1038/nrmicro1991
McElhinny M. W. (1970). Formation of the Indian ocean. Nature 228 (5275), 977–979. doi: 10.1038/228977a0
Mclean J. H. (1981). The Galapagos rift limpet Neomphalus relevance to understanding the evolution of a major Paleozoic-Mesozoic radiation. Science 21, 291–336.
Mclean J. H. (1989). New archaeogastropod limpets from hydrothermal vents: new family Peltospiridae, new superfamily Peltospiracea. Zool. Scripta. 18 (1), 49–66. doi: 10.1111/j.1463-6409.1989.tb00123.x
Mclean J. H. (1990). A new genus and species of neomphalid limpet from the Mariana vents with a review of current understanding of relationships amoàng neomphalacea and Peltospiracea. Nautilus. 104, 77–86.
Mills R., Harrison K. (1998). Modern ocean floor processes and the geological record. Geol. Soc. Spec. Pub. 148, 17–28.
Minh B. Q., Schmidt H. A., Chernomor O., Schrempf D., Woodhams M. D., von Haeseler A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37 (5), 1530–1534. doi: 10.1093/molbev/msaa015
Phillips A. J., Simon C. (1995). Simple, efficient, and nondestructive DNA extraction protocol for arthropods. Ann. Entomol. Soc. America 88 (3), 281–283. doi: 10.1093/aesa/88.3.281
Ponder W. F., Lindberg D. R., Ponder J. M. (2019). Biology and evolution of the mollusca, volume 2 (Boca Raton: CRC Press).
Rogers A. D., Tyler P. A., Connelly D. P., Copley J. T., James R., Larter R. D., et al. (2012). The discovery of new deep-sea hydrothermal vent communities in the southern ocean and implications for biogeography. PloS Biol. 10 (1), e1001234. doi: 10.1371/journal.pbio.1001234
Sasaki T., Warén A., Kano Y., Okutani T., Fujikura K. (2010). “Gastropods from recent hot vents and cold seeps: systematics, diversity and life strategies,” in The vent and seep biota: aspects from microbes to ecosystems. Ed. Kiel S. (Dordrecht: Springer Netherlands), 169–254.
Shen X.-X., Hittinger C. T., Rokas A. (2017). Contentious relationships in phylogenomic studies can be driven by a handful of genes. Nat. Ecol. Evol. 1 (5), 126. doi: 10.1038/s41559-017-0126
Stamatakis A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30 (9), 1312–1313. doi: 10.1093/bioinformatics/btu033
Sun J., Chen C., Miyamoto N., Li R., Sigwart J. D., Xu T., et al. (2020). The Scaly-foot Snail genome and implications for the origins of biomineralised armour. Nat. Commun. 11 (1), 1657. doi: 10.1038/s41467-020-15522-3
Tunnicliffe V., Mary R. Fowler C. (1996). Influence of sea-floor spreading on the global hydrothermal vent fauna. Nature 379 (6565), 531–533. doi: 10.1038/379531a0
Uribe J. E., González V. L., Irisarri I., Kano Y., Herbert D. G., Strong E. E., et al. (2022). A phylogenomic backbone for gastropod molluscs. Syst. Biol. 71 (6), 1271–1280. doi: 10.1093/sysbio/syac045
Van Dover C. L., Humphris S. E., Fornari D., Cavanaugh C. M., Collier R., Goffredi S. K., et al. (2001). Biogeography and ecological setting of Indian ocean hydrothermal vents. Science 294 (5543), 818–823. doi: 10.1126/science.1064574
Villanueva R. A. M., Chen Z. J. (2019). ggplot2: elegant graphics for data analysis (2nd ed.). Measurement.: Interdiscip. Res. Perspect. 17 (3), 160–167. doi: 10.1080/15366367.2019.1565254
Wareín A., Bouchet P. (1993). New records, species, genera, and a new family of gastropods from hydrothermal vents and hydrocarbon seeps. Zool. Scripta. 22 (1), 1–90. doi: 10.1111/j.1463-6409.1993.tb00342.x
Warén A. (2001). Gastropoda and Monoplacophora from hydrothermal vents and seeps; new taxa and records. Veliger. 44, 116–231.
Warén A., Bouchet P. (2001). Gastropoda and Monoplacophora from hydrothermal vents and seeps; New taxa and records. Veliger 44, 116–231.
Xu T., Wang Y., Sun J., Chen C., Watanabe H. K., Chen J., et al. (2021). Hidden historical habitat-linked population divergence and contemporary gene flow of a deep-sea patellogastropod limpet. Mol. Biol. Evol. 38 (12), 5640–5654. doi: 10.1093/molbev/msab278
Yin J., Zhang C., Mirarab S. (2019). ASTRAL-MP: scaling ASTRAL to very large datasets using randomization and parallelization. Bioinformatics 35 (20), 3961–3969. doi: 10.1093/bioinformatics/btz211
Zhang S., Zhang S. (2017). A new genus and species of Neomphalidae from a hydrothermal vent of the Manus Back-Arc Basin, western Pacific (Gastropoda: Neomphalina). Nautilus. 131, 76–86.
Keywords: Neomphalida, mitogenome, hot vent, sunken wood, phylogeny
Citation: Zhang L, Gu X, Chen C, He X, Qi Y and Sun J (2024) Mitogenome-based phylogeny of the gastropod order Neomphalida points to multiple habitat shifts and a Pacific origin. Front. Mar. Sci. 10:1341869. doi: 10.3389/fmars.2023.1341869
Received: 21 November 2023; Accepted: 26 December 2023;
Published: 16 January 2024.
Edited by:
Min Hui, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Shao’E Sun, Chinese Academy of Sciences (CAS), ChinaWong Yue Him, Shenzhen University, China
Yadong Zhou, Ministry of Natural Resources, China
Copyright © 2024 Zhang, Gu, Chen, He, Qi and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin Sun, amluX3N1bkBvdWMuZWR1LmNu