 Xue Kong1,2,3
Xue Kong1,2,3 Wei Wang1,2Sunan Chen1,2
Wei Wang1,2Sunan Chen1,2 Ning Mao1,2Yuefeng Cai1,2,3Yanan Li4Sunan Xia1,2
Ning Mao1,2Yuefeng Cai1,2,3Yanan Li4Sunan Xia1,2 Haibin Zhang5
Haibin Zhang5 Xin Shen1,2,3*
Xin Shen1,2,3*- 1School of Marine Science and Fisheries, Jiangsu Ocean University, Lianyungang, China
- 2Jiangsu Key Laboratory of Marine Bioresources and Environment/Jiangsu Key Laboratory of Marine Biotechnology, Jiangsu Ocean University, Lianyungang, China
- 3Co-Innovation Center of Jiangsu Marine Bio-Industry Technology, Jiangsu Ocean University, Lianyungang, China
- 4College of Animal Sciences and Technology, Zhongkai University of Agriculture and Engineering, Guangzhou, China
- 5Institute of Deep-sea Science and Engineering, Chinese Academy of Sciences, Sanya, China
Introduction: The deep-sea environment is always characterized by high hydrostatic pressure, fluctuating temperatures, heavy metals, darkness, and others. The Vesicomyidae inhabit cold seep zones, hydrothermal vents, and other chemically reduced environments.
Methods: To enhance the understanding of the adaptation mechanisms of clams in extreme environments, a comprehensive proteomic study was conducted on the cold seep clam Archivesica marissinica and shallow water clam Ruditapes philippinarum.
Results: A total of 4,557 proteins were identified from the comparative groups. The Gene Ontology results indicated that the differentially expressed proteins (DEP) for the comparative group Rpgill vs. Amgill were enriched in the nitrogen compound metabolic process, and others. The comparative analysis for Amfoot vs. Amgill and Ammantle vs. Amgill revealed significant enrichment of the differential proteins that were involved in metal ion transport, divalent inorganic cation transport, and so on. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis for the comparative group Rpgill vs. Amgill was significantly enriched in the regulation of actin cytoskeleton, lysosome, and others. The proteins that exhibited differential expression in the cold seep clam’ different tissues were also enriched in important pathways, such as lysosome, fatty acid degradation, nitrogen metabolism.
Discussion: The further analysis identified crucial response proteins involved in various biological pathways. For example, the pattern recognition receptors, such as galectin and peptidoglycan recognition protein, participated in recognition of symbiotic microorganisms. The lysosome pathway members, such as cathepsin and saposins, were engaged in the degradation process of symbiont proteins during symbiont digestion. Profilin and gelsolin from actin cytoskeleton pathway might be pressure-related proteins. Furthermore, carbonic anhydrases from nitrogen metabolism KEGG pathway provide inorganic carbon for symbiotic bacteria. Additionally, Mn superoxide dismutase plays a role in the scavenging of superoxide anion radicals and antioxidant activity. Then, arginine kinases facilitate the low temperature adaptation of deep-sea shellfish with its cold adaptation characters. These findings offer novel perspectives on the proteins that are implicated in A. marisica’s response to cold seep environments, thereby contributing to the understanding of deep-sea biological adaptation and the preservation of deep-sea ecosystems.
Introduction
Due to its high hydrostatic pressure, year-round low temperature, and lack of light, the deep sea has traditionally been regarded as a “life desert”. However, with the advancement of sea surveys, it has become evident that deep sea ecosystems have remarkable diversity, which is concentrated around hydrothermal vents, whale falls, seamounts, and cold seeps (Vrijenhoek, 2010). Hydrothermal vents, for instance, are characterized by the formation of hot, reducing, metal- and sulfur-rich fluids, and they are commonly found in areas of volcanic activity, tectonic plate separation zones, oceanic basins, and hotspots (Beaulieu et al., 2013; Galley et al., 2020). Cold seeps are areas where methane-rich fluid is emitted, which are typically found in highly productive or tectonically active continental margins and tectonic plate convergence zones (Jean-paul et al., 2009). Deep-sea ecosystems containing macrofaunal invertebrates are known for their high biomass but low species diversity (Levin, 2005; Govenar, 2010). Various species, such as deep-sea mussels, tube worms, vesicomyid clams, and crustacea, have been discovered in these environments (Dubilier et al., 2008). Deep-sea invertebrates possess the capacity for filter feeding, yet they predominantly acquire their nutrients through a symbiotic association with chemosynthetic bacteria. This symbiotic relationship involves the utilization of reduced chemical compounds, such as H2S, H2, or CH4, as electron donors, and O2 as electron acceptors, thereby generating substantial energy that is necessary for carbon fixation (Ling et al., 2020). Consequently, deep-sea invertebrates and their symbiotic bacteria engage in a mutualistic relationship.
Deep-sea ecosystems are characterized by arduous environmental conditions, such as low temperature, poor food distribution, and high pressure (McClain et al., 2012; Jones et al., 2017). Thus, diverse technological approaches have been employed to investigate the adaptive mechanisms of deep-sea organisms. Through the sequencing of the genomes of Archivesica marissinica and its bacterial symbiont, Ip et al. discovered an expansion in the gene families that was related to cellular processes that potentially facilitate chemoautotrophy, apoptosis repression, and gas transportation. Additionally, a reduction in the gene families associated with bacterial recognition was observed, indicating a reliance on host transcriptional regulation and a mutually beneficial metabolic interdependence between the host and symbiont (Ip et al., 2021). Moreover, Sun et al. (Sun et al., 2022) utilized 16S ribosomal DNA and metatranscriptome sequencing in Gigantidas platifrons and revealed a significant upregulation of quinone oxidoreductase in vent mussels, indicating a collaborative effort among mussel hosts, endosymbionts, and epibionts to facilitate sulfide detoxification, thereby enabling adaptation to challenging H2S-rich environment. Penhallurick et al. (Penhallurick and Ichiye, 2021) conducted a comparative analysis of dihydrofolate reductase (DHFR) from two piezophiles at varying depths, and revealed that Moritella DHFR exhibits favorable adaptability to high-pressure environments due to its enhanced compressibility, which effectively prevents water infiltration into internal cavities. Additionally, many others deep-sea organisms have been studied, such as tubeworms (Li et al., 2018; Li et al., 2019), Rimicaris sp.(Zhang et al., 2017), Shinkaia crosnieri (Cheng et al., 2019), hadal amphipod (Lan et al., 2017), and hadal sea cucumber (Liu et al., 2021), among others.
The field of proteomics involves the examination of the protein composition of cells, tissues, or entire organisms, and the investigation of their expression characteristics and regulatory principles (Pandey and Mann, 2000). Similar to the transcriptome, the proteome refers to the complete collection of proteins that are actively expressed within a cell or organism during a specific moment in time (Cai et al., 2022). It encompasses the molecular composition of cellular organelles and protein interactions, and plays a crucial role in determining the cellular phenotype. This is due to the fact that proteins, as functional units, interact with various molecules such as metabolites, lipids, and nucleotides. Consequently, the proteome holds greater significance in relation to the cellular phenotype compared to the genome or transcriptome (Crick, 1970; Spirin and Mirny, 2003). Therefore, in the field of deep-sea biology, proteomics has emerged as a prominent tool. For instance, Yan et al. used liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) to examine the ocular composition of the prevailing hadal endemic snailfish species, Pseudoliparis swirei, inhabiting the Mariana Trench. The investigation identified that rhodopsin exhibited sensitivity toward low-intensity blue light, alongside a notable enrichment of proteins associated with visual phototransduction (Yan et al., 2021). To investigate the response of respiratory trees in the sea cucumber Apostichopus japonicus, Huo et al. performed a comparative quantitative proteomics study using the iTRAQ technique. Results showed that sea cucumber used different coping strategies to different environmental stresses, and the interaction between stressors had a superimposed effect at the proteome level (Huo et al., 2019). Kwan et al. applied LC−MS/MS to identify and quantify the protein expression changes of Abyssorchomene distinctus after in situ copper exposure, and notably, certain differentially expressed proteins exhibited a dose-dependent relationship with copper ion concentration and demonstrated a remarkable level of sensitivity. These findings suggest the potential utility of these proteins as biomarkers for the evaluation of deep-sea mining endeavors in the future (Kwan et al., 2019). Therefore, proteomics research has been conducted on various marine organisms, including barnacle (Yan et al., 2020), deep-sea mussel (Ponnudurai et al., 2020; Shi et al., 2022), deep sea copepods (Renz et al., 2021), deep sea isopods (Paulus et al., 2022), and so on.
The Vesicomyidae clam species inhabit cold seep zones, hydrothermal vents, and other chemically reduced environments. These clams primarily depend on symbiotic bacteria within their gill tissue to synthesize essential nutrients for their survival. In turn, they provide essential nutrient sources and habitats for these bacteria (Girguis et al., 2008; Krylova and Sahling, 2010). Transcriptome and genome sequencing on Vesicomyid clams has been conducted (Yi et al., 2019; Ip et al., 2021). As previous studies have not focused on changes in protein levels between cold seep clam and shallow water clam, a comprehensive proteomic study was conducted on Archivesica marissinica and Ruditapes philippinarum (mainly gill tissues of shallow shellfish and deep-sea clams). Four-dimensional Label Free Liquid Chromatography-tandem Mass Spectrometry (LC-MS/MS) was used to obtain the proteome data. Differentially regulated proteins in various tissues of the cold seep clam or clams residing in different habitats were analyzed using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). Then, the reliability of proteomic data was confirmed through Quantitative Real-time Polymerase Chain Reaction (qPCR). The outcomes of these analyses may provide insights into the molecular mechanisms underlying the response of A. marissinica to symbiotic bacteria, low temperature, high hydrostatic pressure, and others. This, in turn, could enhance our comprehension of the potential adaptive strategies employed by deep-sea organisms in cold-seep habitats.
Materials and methods
Sample collection and species identification
Deep-sea cold-seep clams A. marissinica were collected from the Haima cold seep zone in the South China Sea. The depth was 1361m. When three clams were collected on deck, they were washed three times with sterile water, and then dissected into adductor muscle, gill, mantle, and foot. The average shell length of cold sea clams is 128.75 ± 5.23 mm. Liquid nitrogen was used to store samples for RNA extraction, and 95% ethanol was used to store DNA samples. The shallow water shellfish R. philippinarum were obtained from Haizhou Bay (Jiangsu Province, China). R. philippinarum live in the intertidal zone of Haizhou Bay and sink into the sea during rising tide. The annual average surface temperature of the seawater in Haizhou Bay is about 16°. The average shell length of the shallow water shellfish R. philippinarum is 37.8 ± 0.88 mm. In order to study the environmental adaptation mechanism (mainly symbiosis mechanism and pressure) of deep-sea clams, the gill tissues of shallow shellfish were used for comparative analysis. The shallow water samples (three clams) were processed using the same methods as those for the deep-sea shellfish.
To identify the species, the mitochondrial COI genes of both deep-sea and shallow water organisms were amplified. DNA extractions were performed using marine animal tissue genomic DNA extraction kit (Code No. DP324-03, TIANGEN, Beijing, China). A polymerase chain reaction (PCR) kit was used to amplify the target fragments (Code No. RR001Q, Takara, Beijing, China). The COI-F and COI-R primers employed were L1490 (GGTCAACAAATCATAAAGATATTGG) and H2198 (TAAACTTCAGGGTGACCA AAAAATCA), respectively.
Protein extraction, digestion, and peptide desalination
About 100 mg of tissue was cut and placed in a homogenizer with 500-1000 μL of sodium dodecyl sulfate pyrolysis solution (4% (w/v) sodium dodecyl sulfate, 100mM Tris-HCl with pH 7.6, 0.1 M dithiothreitol). Samples were slowly homogenized on ice for 5 min and boiled at 100 ° for 3 min. Afterwards, the homogenate was centrifuged at 10, 000g for 10 minutes, and the supernatant was collected. The Bicinchoninic Acid method was used for protein quantification and the detailed method was based on the instructions (Order No. C503021, Shanghai Sangon Biotech Co., Ltd, Shanghai, China).
The protein extract (50 μg of protein) was digested with trypsin. Dithiothreitol (Item No. D9760, Sigma-Aldrich, St. Louis, MO, United States) was added to the solution at a final concentration of 10 mM. A 30 minute incubation was performed on the mixture at 30°C. Then, it was cooled to ambient temperature and iodoacetamide (Item No. I1149, Sigma-Aldrich) was added (final concentration of 40 mM). The solution was incubated at room temperature for 20 min (in the dark). Trypsin (Cat No. VA9000, Promega, Madison, WI, United States) was added and the mass ratio of trypsin to protein was 1:100. The mixture was incubated at 37 ° for 6 hours, and then the same volume (1:100) of trypsin was added to the protein solution again. The digestion time was extended for a further 6-10 h.
A C18 cartridge (Cat No. 89851, Thermo Fisher Scientific) was used to desalt the peptide solution. The desalting column was first optimized. Then, about 100 μL of methanol was added to the desalting column, and then centrifuged at 1509 g for 1 min. About 100 μL of 70% acetonitrile (v/v, 0.1% formic acid) was added to the desalting column, and then centrifuged at 1509 g for 1 min. Subsequently, about 200 μL of 0.1% formic acid (purity, >99.9%) was added to the desalting column, and then centrifuged at 1509 g for 1 min. The peptide solution was then added to the optimized column. About 1 μL of formic acid was added to the trypsin digestion, and the pH of the digestion was kept at 2-3. The digestion was added to the desalting column, and then it was centrifuged at 750 g for 10 min, and the liquid was discarded. Next, about 200 μL of 0.1% formic acid was added to the desalting column, and then it was centrifuged at 1509 g for 1 min. The liquid was discarded and the procedure was repeated once. The desalting column was transferred to a new clean centrifuge tube. About 100 μL of 70% acetonitrile (v/v, 0.1% formic acid) was added to the desalting column, and then centrifuged at 1509 g for 1 min. The elution was collected and the operation was repeated for 2-3 times. The elution was lyophilized under a vacuum, and about 40 μL of 0.1% formic acid was used to dissolve the dried powder. OD280 was used for peptide quantification (NanoDrop 2000, Thermo Fisher Scientific).
Four-dimensional Label Free Liquid Chromatography-tandem Mass Spectrometry analysis
The sample was analyzed using High-Performance Liquid Chromatography (NanoElute) with a nanoliter flow rate. Buffer A contained 0.1% formic acid, and buffer B contained 0.1% formic acid (84% acetonitrile). The chromatographic column was balanced with 95% buffer A. The sample was loaded from the autosampler onto the sample loading column (Cat no. 164941, Thermo Scientific Acclaim PepMap100), and separated using the analytical column (Cat no. ES900, Thermo Scientific EASY column). The flow rate was 300 nL/min. After chromatographic separation, the sample was analyzed by mass spectrometry with timsTOF Pro mass spectrometer (timsTOF pro, German Bruker Daltonics, Bremen, Germany). A 1.5 kV ion source voltage and positive ion detection mode was used. Both mass spectrometry (MS) and MS/MS used TOF for detection and analysis. The scanning range of mass spectrum ranged from 100 to 1700 m/z. The data acquisition mode was parallel cumulative serial fragmentation (PASEF) acquisition method, and the parention was collected in the PASEF mode for 10 times after a primary mass spectrum was collected. The window time of one cycle was 1.17 seconds. The charge number of secondary spectrum was in the range of 0-5. The dynamic elimination time of tandem mass spectrometry scanning was set to 24 s to avoid repeated scanning of parention.
Protein identification and quantification
The original mass spectrometry data was RAW file. MaxQuant software (version no. 1.6.14) (Cox, 2008) was used for database identification and quantification. Relevant parameters and descriptions were as follows: enzyme, trypsin; max missed cleavages, 2; main search, 6 ppm; first search, 20 ppm; MS/MS tolerance, 20 ppm; fixed modifications, carbamidomethyl (C); variable modifications, oxidation (M); database, Amav1.0_pep.fasta (Ip et al., 2021); database pattern, reverse; include contaminants, true; protein false discovery rate (FDR), ≤0.01; peptide FDR, ≤0.01; peptides used for protein quantification, razor and unique peptides; time window (match between runs), 2 min; protein quantification, label-free quantitation; minimum ratio count, 1.
Protein cluster, subcellular localization, protein domain, Gene Ontology, Kyoto Encyclopedia of Genes and Genomes and protein interaction analyses
Protein cluster analysis
Initially, the quantitative data for the target protein set underwent normalization to the (-1,1) interval. Subsequently, the complexheatmap R package, version 3.4, was employed to perform two-dimensional classification of samples and protein expression utilizing the Euclidean distance algorithm and Average link connection method. The resulting output consisted of hierarchical clustering heat maps.
Subcellular localization analysis
The CELLO tool (http://cello.life.nctu.edu.tw/) was used to employ multi-class support vector machine (SVM) machine learning approach to predict subcellular localization. This approach models protein sequence data using known subcellular location information from public databases, thereby predicting the subcellular location information for the protein of interest.
Protein domain analysis
The analysis of protein domains involved the utilization of the Pfam database. To characterize the function of a given sequence, a scanning algorithm from the InterPro database was executed using the InterProScan software package. This process provided domain annotation information for the target protein sequence within the Pfam database.
Gene Ontology function annotation
The target protein set was annotated using Blast2GO, which involved a four-step process consisting of sequence alignment (Blast), GO item extraction (Mapping), GO annotation, and InterProScan supplementary annotation.
Kyoto Encyclopedia of Genes and Genomes pathway annotation
Annotating target proteins with KEGG pathways was carried out using the KEGG Automatic Annotation Server software.
Enrichment analysis
To compare the distribution of each GO classification (or KEGG pathway, or Domain) in the goal protein set and the total protein set, Fisher’s Exact Test was employed. Furthermore, an enrichment analysis of GO (or KEGG pathway, or Domain) annotation was conducted on the target protein set.
Analysis of protein-protein interaction network
The IntAct (http://www.ebi.ac.uk/intact/main.xhtml) or STRING (http://string-db.org/) database were used to find the direct and indirect interaction relationships between target proteins. STRING was used to produce and analyze the interaction network.
Real-time Polymerase chain reaction validation
To authenticate the differential expression of significant genes encoding the proteins, real-time fluorescence quantitative PCR was conducted. The TRIZOL reagent (Cat no.TR201-50, JIANSHIBIO, China) was employed to extract total RNA from the gill and adductor muscle of A. marissinica, following the manufacturer’s guidelines. The initial strand of complementary DNA (cDNA) was synthesized as a template using reverse transcriptase (Cat no.KR118-02, TIANGEN Biotech(Beijing) Co., Ltd.). The messenger RNA (mRNA) expression level was assessed using SYBR Green real-time PCR and ABI StepOne Plus real-time PCR system (Applied Biosystems, Waltham, MA, USA), with β-actin serving as the internal reference gene for standardization purposes. The primers (Supplementary Table S1) were designed based on sequence information obtained from the transcriptome database(Ip et al., 2021), with selection based on optimal performance. The amplification mixture (20 μL), was comprised of 10 μL of TOROGreen® qPCR Master Mix (Cat no. QST-100, Toroivd), 2 μL of diluted cDNA template, 4 μL (2 μM) of forward primer, and 4 μL (2 μM) of reverse primer. The thermal cycle were: 60 s at 95°C, followed by 15 s at 95°C, 15 s at 58°C, and 45 s at 72°C, with a total of 40 cycles. The amplification products’ specificity was confirmed through dissolution curve analysis. The 2−△△CT method was utilized to compare and analyze the mRNA expression of chosen genes. Statistical analysis was conducted using SPSS26 software (IBM Corp., Armonk, NY, USA), and all data were presented as mean ± standard deviation. The statistical significance of each gene was assessed through one-way analysis of variance and t-test, with P <0.05 indicating a significant difference.
Results
Species identification
The sequencing of the COI products of the deep-sea and shallow water shellfish was conducted. The results were subsequently subjected to analysis using the National Center for Biotechnology Information website (https://www.ncbi.nlm.nih.gov/). Among the deep-sea shellfish, Calyptogena marissinica (also known as A. marissinica) exhibited the highest degree of similarity to the COI sequence, with a 100% match. Similarly, the species of shallow water shellfish was confirmed to be R. philippinarum, with a similarity of 100%. Further information regarding the specific sequences can be found in Supplementary Table S2.
Overview of the proteome data
Protein extraction was performed on the tissues of A. marissinica, specifically the gill (Amgill), adductor muscle (Ammuscle), mantle (Ammantle), and foot (Amfoot), and the gill of R. philippinarum (Rpgill). The resulting extracted proteins met the necessary criteria in terms of mass concentration and total mass, ensuring their suitability for subsequent proteomic analysis. A total of 1,254,723 spectrums were acquired, of which 193,436 spectrums were successfully matched to the A. marissinica peptide database(Ip et al., 2021). The number of peptides obtained and the number of unique peptides were 30,073 and 29,081, respectively. The identified proteins and quantified proteins were 4,587 and 4,576, respectively (Table 1). A quantified protein refers to a protein that has quantitative information available in at least half of the biological replicates within a given group. The maximum and minimum numbers of proteins that were identified in a single sample were 4,105 and 3,175, respectively. The mean quantity of proteins detected in a single sample was 3,795. The Venn diagram showed that the proteins that were shared among the samples within the group exceeded 3,000 (excluding the Ammuscle group), while proteins that were shared between the groups amounted to 3,227 (refer to Figure 1).
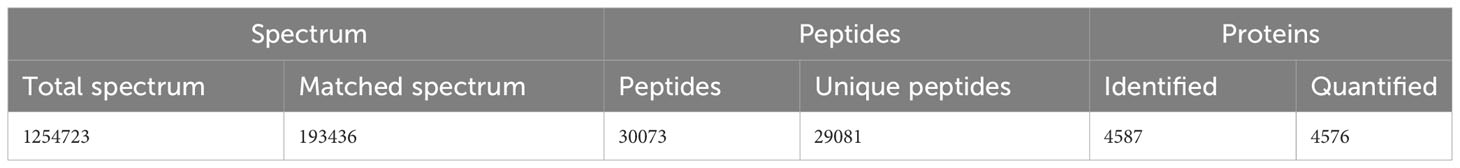
Table 1 The results of the spectra, peptides, and proteins.
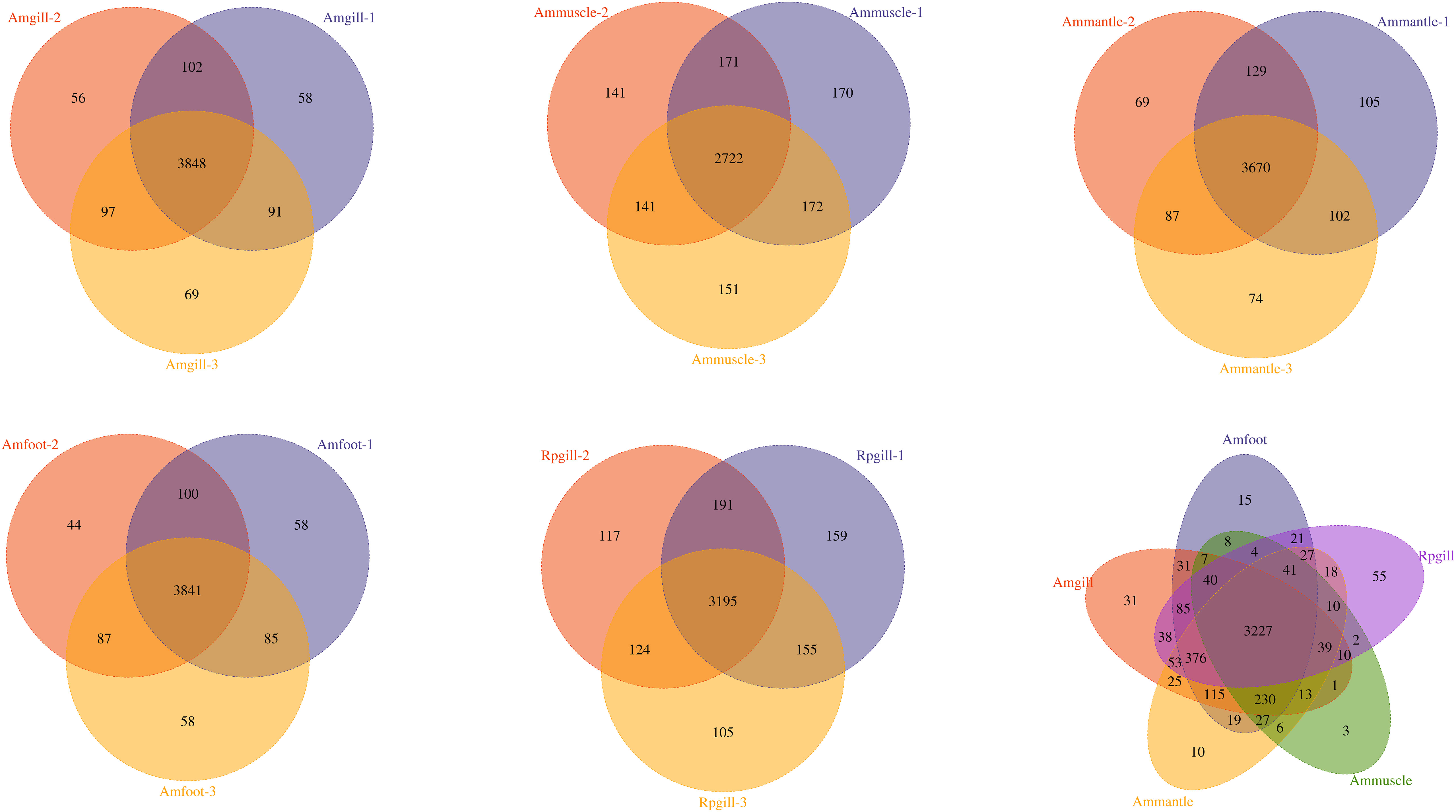
Figure 1 The Venn diagram of the overlapping proteins within the groups and between the groups Notes: abbreviations, Amgill, Archivesica marissinica gill; Ammuscle, A. marissinica adductor muscle; Ammantle, A. marissinica mantle; Amfoot, A. marissinica foot; and Rpgill, Ruditapes philippinarum gill.
Global view of the differential protein expression patterns
To investigate the differential expression of the proteins across the distinct groups, additional differential screening was performed on the experimental data. The amount of differentially expressed proteins between the compared groups was determined based on a fold change threshold (>2.0), indicating upregulation exceeding 2.0 or downregulation below 0.5, with a significance level of P <0.05. A total of 6,352 proteins with differential expression were identified, comprising 2,609 upregulated and 3,743 downregulated proteins.
The amount of differentially expressed proteins that were upregulated and downregulated by more than ten times were 1,244 and 508, respectively. In the “presence or absence” difference comparison, the number of proteins were obtained using the screening criteria that they were present two or more times in a group of samples, and all the information in the other group were null values. The amount of 1,940 differential proteins were identified, of which 482 proteins were upregulated and 1458 proteins were downregulated. The results also showed that there were relatively more down-regulated proteins in the “presence or absence” difference comparison (Table 2).
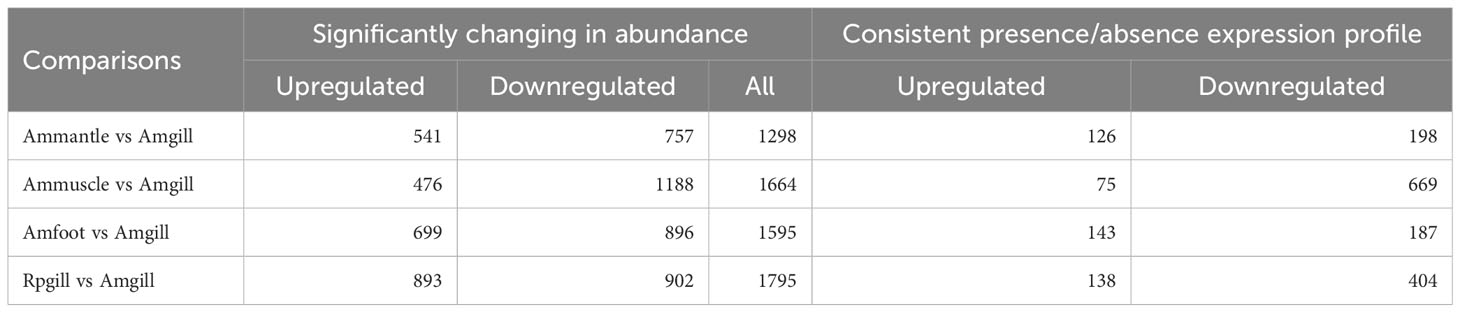
Table 2 Statistics of quantitative differential proteins.
Differential protein expression analysis
A hierarchical cluster analysis was conducted on the proteins that exhibited differential expression (Figure 2). The findings demonstrated that these differentially expressed proteins could be used to distinguish between the comparative groups, thereby suggesting the potential for further analysis of these proteins.
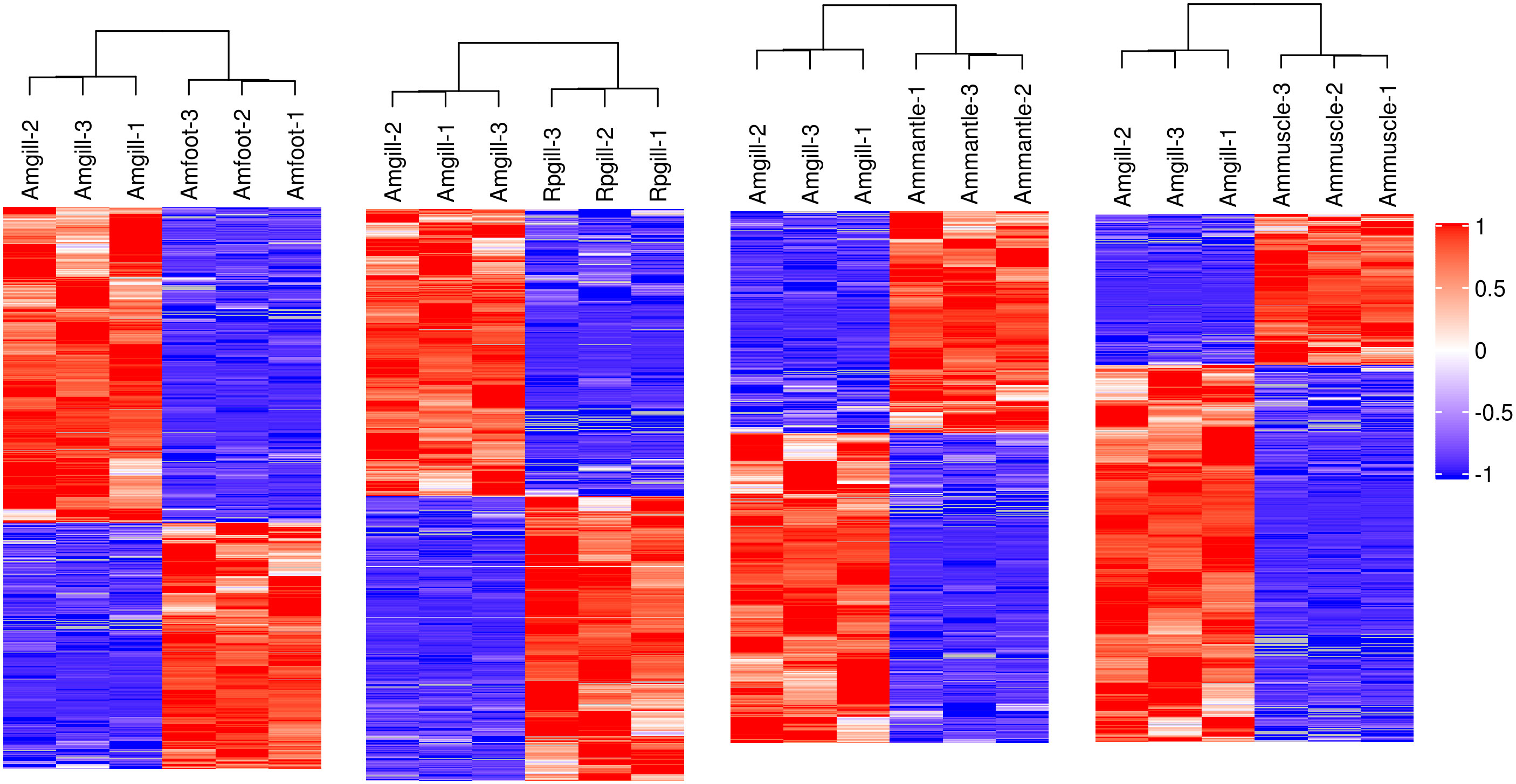
Figure 2 Hierarchical cluster analysis of the differentially expressed proteins Notes: each column represents one sample of a group, and each row represents a protein with significant differential expression. Significantly different protein expression levels in different samples are displayed in different colors on the heat map. Red represents a significantly upregulated protein, blue represents a significantly downregulated protein, and the gray portion represents no protein quantitative information.
Gills’ differential regulated proteins between cold seep and shallow water clams
The gill tissues of cold seep clam and shallow water shellfish were subjected to a comparative analysis. The results in Table 2 indicate that a total of 1,795 proteins exhibited differential expression, with 893 being upregulated and 902 being downregulated. Further information regarding the differential proteins (Rpgill vs. Amgill) can be found in Supplementary Table S3. Additionally, subcellular localization and domain analysis were performed on these differential proteins. Figure 3A reveals that the majority of differential proteins from comparative group Rpgill vs. Amgill were predominantly localized in the nuclear compartment (1020/2935, 34.75%) and cytoplasmic region (947/2935, 32.26%). The results of the domain analysis indicated that the majority of differentially expressed proteins possessed the following domains: RNA recognition motif, protein kinase domain, WD domain (G-beta repeat), Ras family, and immunoglobulin I-set domain, with the latter being the most prevalent (top 5; Figure 3B). To further investigate the enrichment patterns of these domains in the differentially expressed proteins and identify any significant enrichments, Fisher’s Exact Test was employed for domain enrichment analysis. The functional domain enrichment analysis identified the PDZ domain (also referred to as DHR or GLGF), AAA domain (specifically the dynein-related subfamily), papain family cysteine protease, the hydrolytic ATP binding site of the dynein motor region D1, and the dynein heavy chain with region D6 of the dynein motor as the top five domains with the highest significance (Figure 3C).
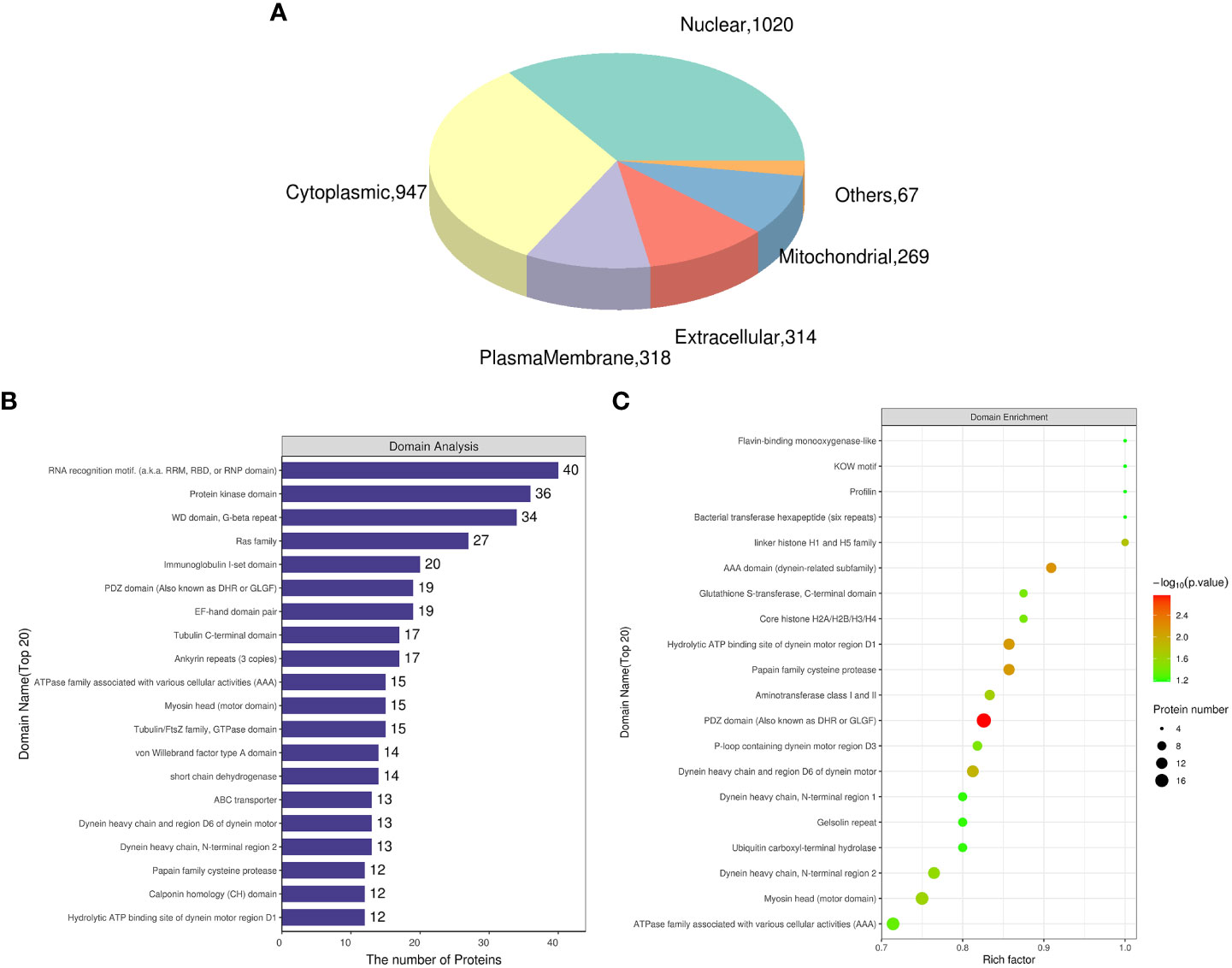
Figure 3 Subcellular localization and domain analysis of the differential proteins for Ruditapes philippinarum gill (Rpgill) vs. Archivesica marissinica gill (Amgill) Notes: (A), subcellular localization of the differential proteins; (B), domain analysis of the differential proteins; (C), domain enrichment analysis of the differential proteins.
Differentially regulated proteins between cold seep clam tissues
This study also involved a comparative analysis of the gill, mantle, muscle, and foot of the cold seep clam A. marissinica. A total of 4,557 differentially expressed proteins were identified in the comparative groups, namely Amfoot vs. Amgill, Ammuscle vs. Amgill, and Ammantle vs. Amgill. Among these, 1,716 proteins were upregulated, while 2,841 proteins were downregulated (Table 2). Detailed information regarding the differential proteins in each comparison (Amfoot vs. Amgill, Ammuscle vs. Amgill, and Ammantle vs. Amgill) can be found in Supplementary Tables S4–S6. Additionally, subcellular localization and domain analysis were performed on the differential proteins.
Based on the data presented in Supplementary Figure S1, A1, it is evident that most of differential proteins between Amfoot and Amgill were primarily located in the nuclear (816/2415, 33.78%) and cytoplasmic (720/2415, 29.81%) regions. This pattern was also observed in the comparative groups Ammuscle vs. Amgill and Ammantle vs. Amgill. Furthermore, the domain analysis of the Amfoot vs. Amgill comparison revealed that the most prevalent domains among the differential proteins were the immunoglobulin I-set domain, RNA recognition motif, protein kinase domain, fibronectin type III domain, and Ras family (top 5; Supplementary Figure S1, B1). The comparative groups Ammuscle vs. Amgill and Ammantle vs. Amgill exhibited similar findings in domain analysis, which were similar to those of the comparative group Amfoot vs. Amgill (Supplementary Figures S1, B2, S1, B3).
In the functional domain enrichment analysis of Amfoot vs. Amgill, the domains that exhibited the highest significance (top 5) were the immunoglobulin I-set domain, papain family cysteine protease, collagen triple helix repeat, mitochondria-eating protein, and immunoglobulin domain (Supplementary Figure S1, C1). Similarly, in the comparison of Ammuscle vs. Amgill, the domains with the highest significance (top 5) were the RNA recognition motif, papain family cysteine protease, immunoglobulin I-set domain, myosin head (motor domain), and calponin family repeat (Supplementary Figure S1, C2). The domains exhibiting the greatest significance between Ammantle and Amgill (top 5) were the immunoglobulin I-set domain, papain family cysteine protease domain, C1q domain, von Willebrand factor type A domain, and tektin family (Supplementary Figure S1, C3).
Gene Ontology Enrichment and Kyoto Encyclopedia of Genes and Genomes pathways analyses of differentially expressed proteins between cold seep clams’ gills and shallow water clams’ gills
To comprehend the role, distribution, and biochemical pathways of differentially expressed proteins in cold seep/shallow water organisms, the proteins were subjected to annotation using GO and KEGG pathway enrichment analyses. As depicted in Figure 4A, the first three terms of biological processes included cellular process, metabolic process, and localization. Similarly, the first three terms of molecular functions included catalytic activity, binding, and structural molecule activity. The first three terms of cellular components are cell, cell part, and membrane.
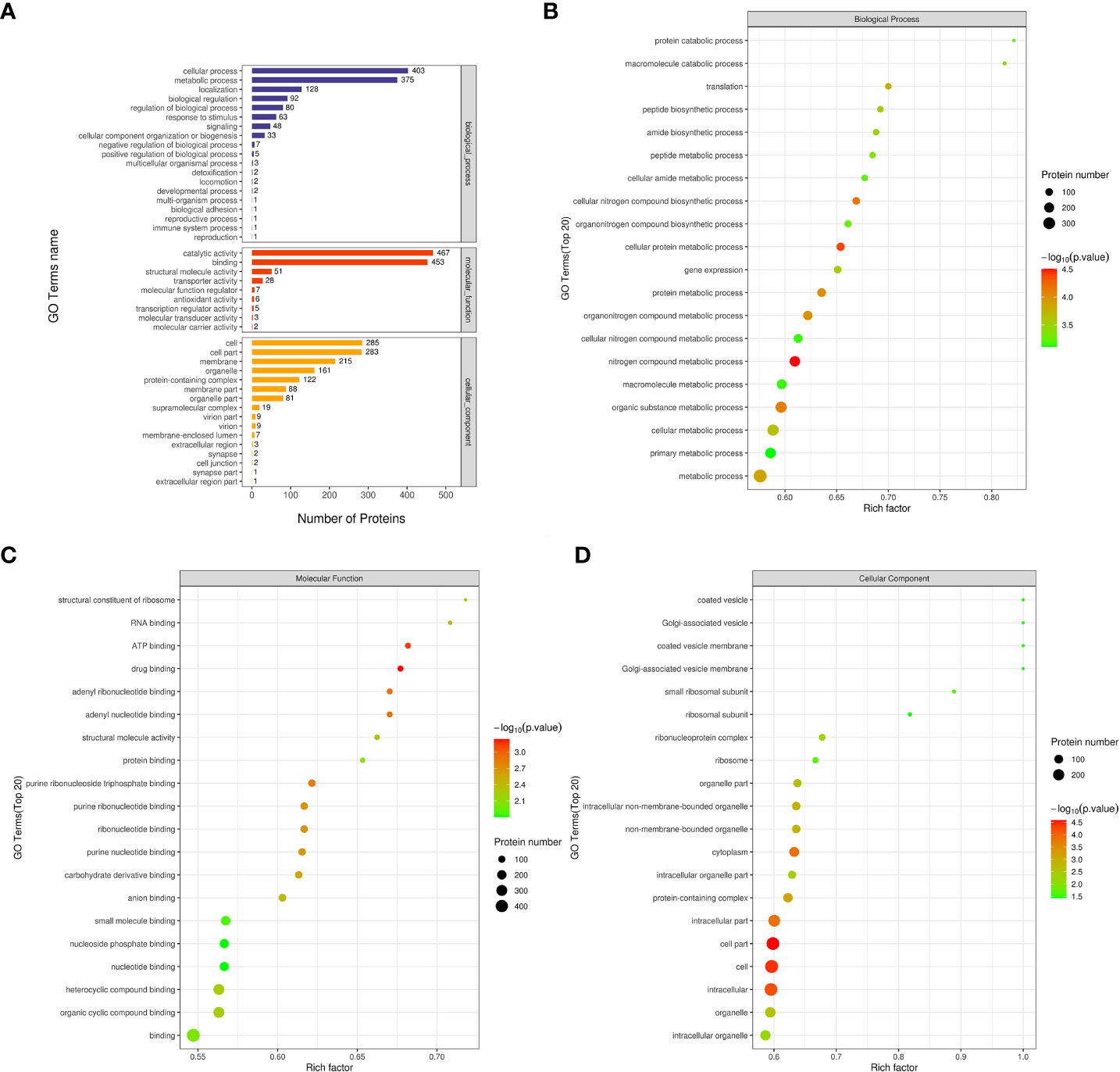
Figure 4 GO analysis related maps of differentially expressed proteins from Ruditapes philippinarum gill (Rpgill) vs. Archivesica marissinica gill (Amgill) Notes: (A), GO annotation statistical map; (B), biological process enrichment bubble map; (C), molecular function enrichment bubble map; (D), cellular component enrichment bubble map.
To identify GO terms that were significantly enriched, Fisher’s Exact Test was employed to conduct a GO functional enrichment analysis on proteins that exhibited differential expression. The results of the analysis, as depicted in Figure 4B, revealed notable enrichment of differential proteins in crucial biological processes including nitrogen compound metabolic process, cellular protein metabolic process, cellular nitrogen compound biosynthetic process, organic substance metabolic process, protein metabolic process, and others. The analysis revealed significant enrichment of molecular functions related to drug binding, ATP binding, adenyl nucleotide binding, adenyl ribonucleotide binding, and purine ribonucleoside triphosphate binding, as indicated by the top five results presented in Figure 4C. Similarly, the differential proteins exhibited significant enrichment in cellular components, such as cell part, cell, intracellular region, cytoplasm, and intracellular part, as shown in Figure 4D.
Differentially expressed proteins from the comparative group Rpgill vs. Amgill were annotated for the KEGG pathway. Simultaneously, the protein count corresponding to the KEGG pathway was determined, and the initial pathways with the greatest number of differentially expressed proteins were identified as ribosome, phagosome, lysosome, biosynthesis of cofactors, and spliceosome (Figure 5A). The Fisher’s exact test method was employed to conduct KEGG pathway enrichment analysis of the differentially expressed proteins in the comparative group. The results indicated that significant changes occurred in important pathways such as biosynthesis of cofactors, spliceosome, regulation of actin cytoskeleton, hedgehog signaling pathway, and lysosome (top 5; Figure 5B).
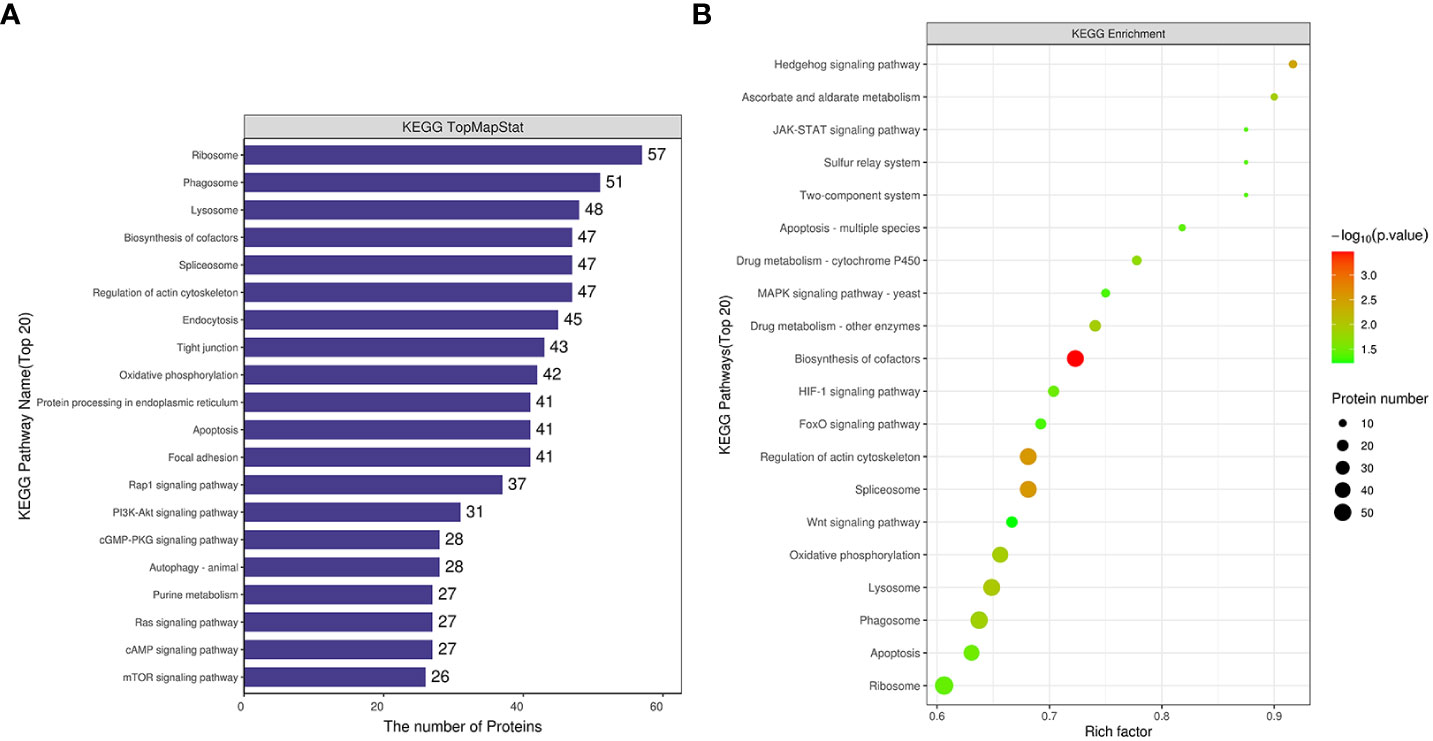
Figure 5 Kyoto Encyclopedia of Genes and Genomes pathway analysis of differentially expressed proteins for Rpgill vs. Amgill Note: KEGG pathway annotation statistical map (A), KEGG pathway enrichment bubble map (B).
Gene Ontology Enrichment and Kyoto Encyclopedia of Genes and Genomes pathways analyses of differentially expressed proteins between cold seep clam tissues
The first three GO terms of the biological process of the comparative groups Amfoot vs. Amgill, Ammuscle vs. Amgill, and Ammantle vs. Amgill were cellular process, metabolic process, and localization. It is similar to those of Rpgill vs. Amgill. The first three terms associated with molecular function in the comparative groups Amfoot vs. Amgill, Ammuscle vs. Amgill, and Ammantle vs. Amgill were catalytic activity, binding, and structural molecule actibvity/transporter activity. The first three terms associated with cellular components in the comparative groups Amfoot vs. Amgill, Ammuscle vs. Amgill, and Ammantle vs. Amgill were cell, membrane, and cell part. (Supplementary Figures S2, A1–A3).
The biological process enrichment bubble map comparing Amfoot and Amgill revealed significant enrichment of differential proteins involved in metal ion transport, divalent inorganic cation transport, calcium ion transport, divalent metal ion transport and response to chemical (top 5; Supplementary Figure S2, B1). Biological process enrichment bubble map from Ammuscle vs. Amgill showed that differential proteins were significantly enriched in protein-containing complex subunit organization, regulation of actin polymerization/depolymerization, actin filament polymerization, regulation of actin filament polymerization and actin polymerization/depolymerization (top 5; Supplementary Figure S2, B2). Biological process enrichment bubble map from Ammantle vs. Amgill showed that differential proteins were significantly enriched in divalent metal ion transport, calcium ion transport, divalent inorganic cation transport, metal ion transport and cellular amino acid catabolic process (top 5; Supplementary Figure S2, B3). This result was similar to the biological process result for Amfoot vs. Amgill.
The molecular functions observed in the comparison between Amfoot and Amgill were found to be significantly enriched in oxidoreductase activity, acting on peroxide as acceptor, peroxidase activity, heme binding and tetrapyrrole binding (top 5; Supplementary Figure S2, C1). The molecular functions identified in the comparison between Ammuscle and Amgill were significantly enriched in the structural constituent of ribosome, structural molecule activity, peptidase activity, actin binding and acting on L-amino acid peptides (top 5; Supplementary Figure S2, C2). Molecular function from Ammantle vs. Amgill was significantly enriched in active transmembrane transporter activity, calcium ion binding, transmembrane transporter activity, symporter activity and oxidoreductase activity (acting on peroxide as acceptor) (top 5; Supplementary Figure S2, C3).
The cellular component enrichment bubble map revealed that there were significant differences in the proteins enriched in the cytoskeleton, supramolecular fiber, supramolecular complex, supramolecular polymer, and polymeric cytoskeletal fiber between Amfoot and Amgill (top 5; Supplementary Figure S2, D1). Additionally, the proteins enriched in the ribonucleoprotein complex, ribosome, peptidase complex, proteasome complex, and endopeptidase complex showed significant differences between Ammuscle and Amgill (top 5; Supplementary Figure S2, D2). Differential proteins for Ammantle vs. Amgill were significantly enriched in the COPI vesicle coat, myosin complex, dynactin complex, Golgi subcompartment, and Golgi membrane (top 5;Supplementary Figure S2, D3).
Proteins that were differentially expressed for Amfoot and Amgill were annotated for the KEGG pathway. Among these, the pathways with the most differentially expressed proteins were lysosome, phagosome, focal adhesion, apoptosis, and biosynthesis of cofactors (Supplementary Figure S3, A1). The KEGG pathway enrichment bubble map revealed significant changes in important pathways, including methane metabolism, lysosome, glutathione metabolism, purine metabolism, and drug metabolism-cytochrome P450, among others (Supplementary Figure S3, B1).
Proteins that exhibited differential expression for the comparative groups Ammuscle and Amgill were annotated for the KEGG pathway. Among these, the pathways with the greatest number of differentially expressed proteins included ribosome, lysosome, biosynthesis of cofactors, spliceosome, and regulation of actin cytoskeleton (Supplementary Figure S3, A2). The KEGG pathway enrichment bubble map revealed significant alterations in key pathways such as ribosome, proteasome, lysosome, glycolysis/gluconeogenesis, fatty acid degradation, and others (Supplementary Figure S3, B2).
Differentially expressed proteins in the comparative group Ammantle and Amgill for the KEGG pathway were annotated. Among these, the pathways with the highest number of differentially expressed proteins included lysosome, focal adhesion, phagosome, rap1 signaling pathway, and apoptosis (Supplementary Figure S3, A3). The KEGG pathway enrichment bubble map revealed significant changes in important pathways such as peroxisome, lysosome, ECM-receptor interaction, nitrogen metabolism, and fatty acid degradation, among others (Supplementary Figure S3, B3).
The presence of the lysosome pathway was observed in all four comparative groups, while fatty acid degradation was detected in two comparative groups (Ammuscle vs. Amgill and Ammantle vs. Amgill). Subsequently, the interactions between the proteins that were involved in the major signaling pathways (top 5) were determined, and the results were shown in Figure 6, Supplementary Figures S4–S6. Proteins involved in KEGG pathway (top 5) and their protein expression level for comparative groups Rpgill vs. Amgil, Amfoot vs. Amgill, Ammuscle vs. Amgill and Ammantle vs. Amgil were listed in Supplementary Tables S7–S10. Some of the important proteins and their protein expression levels from comparative groups were listed in Table 3. The important proteins includes galectin, peptidoglycan recognition protein (immune recognition), carbonic anhydrase (nitrogen metabolism), Mn-superoxide dismutase, extracellular superoxide dismutase [Cu-Zn], intracellular CuZn superoxide dismutase (antioxidant response), arginine kinase (energy metabolism), cathepsins, saposins (maintenance of the symbiotic relationship, Supplementary Table S7), profilin, gelsolin (pressure related protein, Supplementary Table S7) and so on.
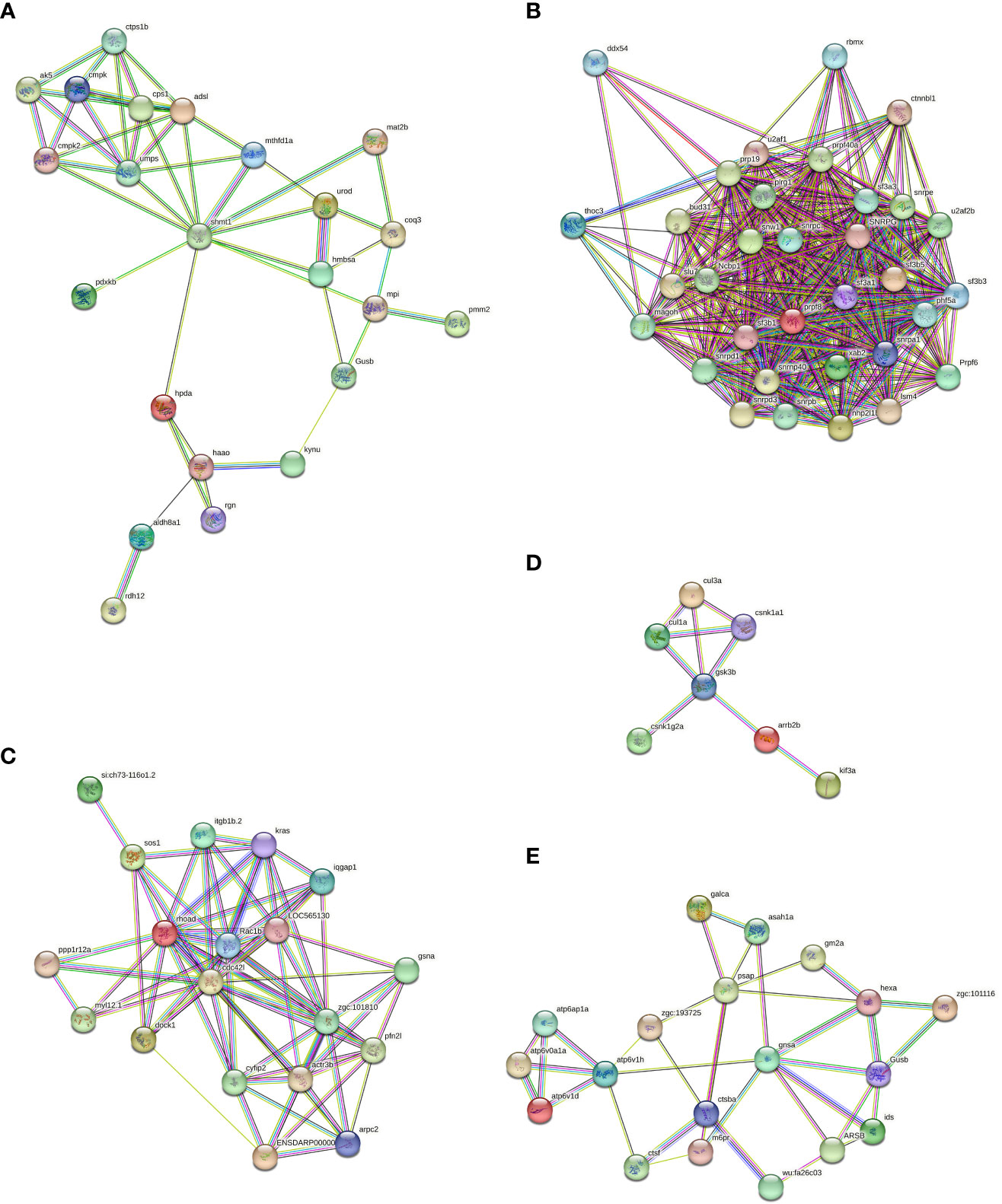
Figure 6 Protein-protein interaction diagrams of which involved in the biosynthesis of cofactors (A), spliceosome (B), regulation of actin cytoskeleton (C), hedgehog signaling pathway (D) and lysosome signaling pathway (E) from Rpgill vs. Amgill Note: protein full name and protein abbreviation. A, 3-hydroxyanthranilate 3,4-dioxygenase: haao; 4-hydroxyphenylpyruvate dioxygenase: hpda; adenylate kinase: ak5; adenylosuccinate lyase:adsl; aldehyde dehydrogenase (NAD+): aldh8a1; beta-glucuronidase:Gusb; carbamoyl-phosphate synthase:cps1;gluconolactonase:rgn; glycine hydroxymethyltransferase: shmt1; hydroxymethylbilane synthase:hmbsa;kynureninase: kynu; mannose-6-phosphate isomerase:mpi; methylenetetrahydrofolate dehydrogenase (NADP+):mthfd1;nucleoside-diphosphate kinase: cmp; phosphomannomutase:pmm2; polyprenyldihydroxybenzoate methyltransferase:coq3; pyridoxine kinase:pdxkb; retinol dehydrogenase 12:rdh12;S-adenosylmethionine synthetase:mat2b; UMP-CMP kinase:cmpk; UMP-CMP kinase 2, mitochondrial: cmpk2; uridine monophosphate synthetase:umps; uroporphyrinogen decarboxylase: urod. B, ATP-dependent RNA helicase: ddx54; beta-catenin-like protein 1: ctnnbl1; bud site selection protein 31: bud31; heterogeneous nuclear ribonucleoprotein G: rbmx; nuclear cap-binding protein subunit 1: Ncbp1; PHD finger-like domain-containing protein 5A: phf5a;pleiotropic regulator 1: plrg1; pre-mRNA-processing factor 19: prp19;pre-mRNA-processing factor 40: prpf40a; pre-mRNA-processing factor 6: Prpf6; pre-mRNA-processing factor 8: prpf8;pre-mRNA-processing factor SLU7: slu7; pre-mRNA-splicing factor SYF1: xab2; protein mago nashi: magoh; Prp8 binding protein:snrnp40; small nuclear ribonucleoprotein B and B:snrpb; small nuclear ribonucleoprotein D1: snrpd1; small nuclear ribonucleoprotein D3: snrpd3; small nuclear ribonucleoprotein E:snrpe; small nuclear ribonucleoprotein G: SNRPG;SNW domain-containing protein 1: snw1; splicing factor 3A subunit 1: sf3a1; splicing factor 3A subunit 3:sf3a3;splicing factor 3B subunit 1:sf3b1;splicing factor 3B subunit 3:sf3b3;splicing factor 3B subunit 5: sf3b5; splicing factor U2AF 65 kDa subunit: u2af2b; THO complex subunit 3: thoc3; U1 small nuclear ribonucleoprotein C: snrpc; U2 small nuclear ribonucleoprotein A: snrpa1; U4/U6 small nuclear ribonucleoprotein SNU13: nhp2l1b; U6 snRNA-associated Sm-like protein LSm4: lsm4. C, actin related protein 2/3 complex, subunit 2: arpc2; actin related protein 2/3 complex, subunit 4: ENSDARP00000115888; actin-related protein 2: zgc:101810; actin-related protein 3: actr3b; cell division control protein 42: cdc42l; cytoplasmic FMR1 interacting protein: cyfip2; dedicator of cytokinesis protein 1: dock1; gelsolin: gsna; GTPase KRas: kras; integrin beta 1: itgb1b.2; myosin regulatory light chain 12: myl12.1; paxillin: LOC565130; profilin:pfn2l; protein phosphatase 1 regulatory subunit 12A: ppp1r12a; Ras GTPase-activating-like protein IQGAP1: iqgap1; Ras homolog gene family, member A: rhoad; Ras-related C3 botulinum toxin substrate 1: Rac1b; Ras-related protein M-Ras: si:ch73-116o1.2; son of sevenless: sos1. D, beta-arrestin: arrb2b; casein kinase 1, alpha: csnk1a1; casein kinase 1, gamma: csnk1g2a; cullin 1: cul1a; cullin 3: cul3a; glycogen synthase kinase 3 beta: gsk3b; kinesin family member 3A: kif3a. E, acid ceramidase: asah1a; alpha-L-fucosidase: zgc:101116; arylsulfatase B: ARSB; beta-glucuronidase: Gusb; cathepsin B: ctsba; cathepsin F: ctsf; cathepsin L: wu:fa26c03; cation-dependent mannose-6-phosphate receptor: m6pr; galactosylceramidase: galca; ganglioside GM2 activator: gm2a; hexosaminidase: hexa; iduronate 2-sulfatase: ids; N-acetylglucosamine-6-sulfatase: gnsa; Niemann-Pick C2 protein: zgc:193725; saposin: psap; V-type H+-transporting ATPase S1 subunit: atp6ap1a; V-type H+-transporting ATPase subunit a: atp6v0a1a; V-type H+-transporting ATPase subunit d: atp6v1d; V-type H+-transporting ATPase subunit H: atp6v1h.
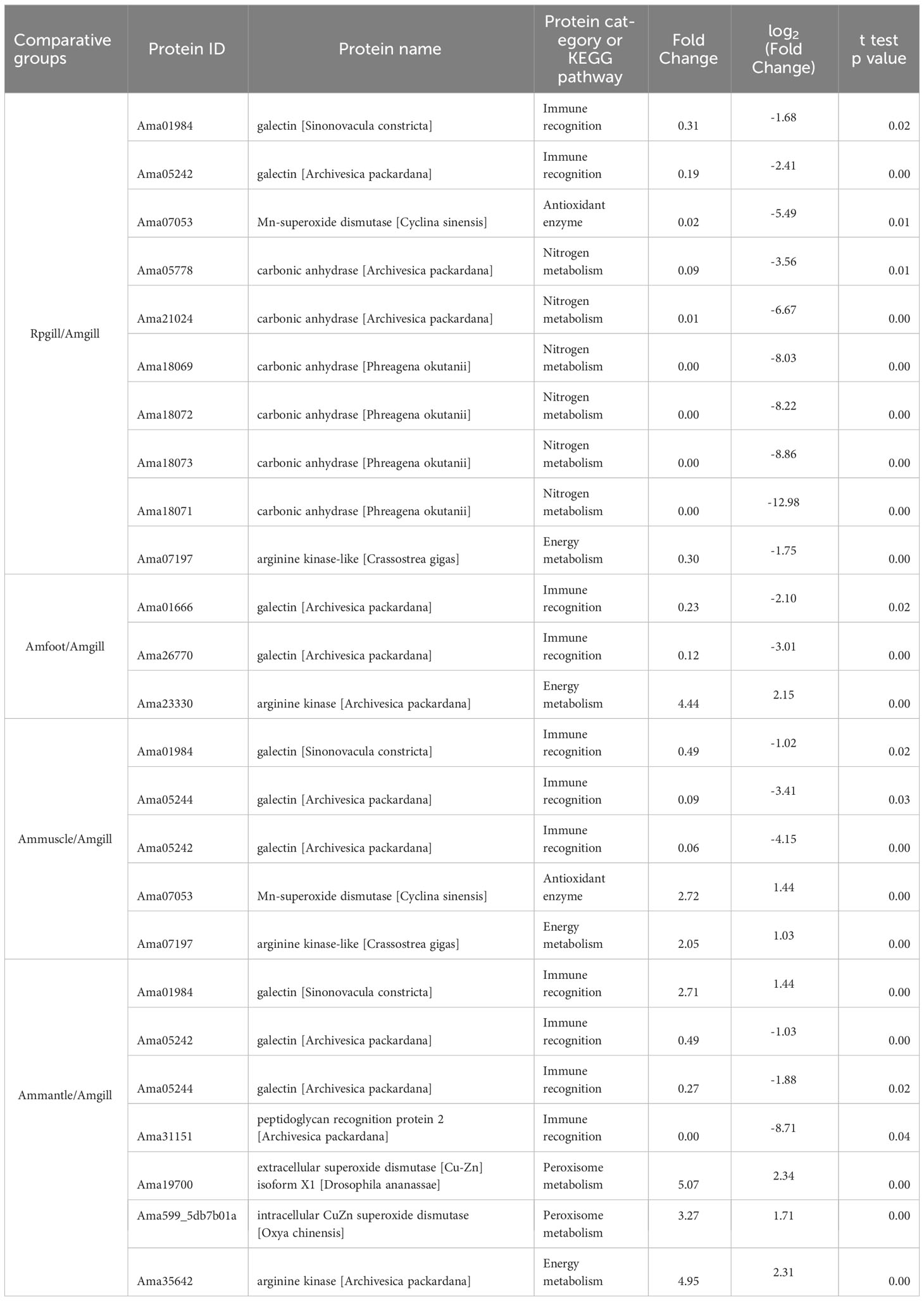
Table 3 Important proteins and their protein expression levels from comparative groups.
mRNA- and protein-level expression of differentially expressed proteins for validation
To investigate the mRNA levels of the identified proteins, seven differentially expressed proteins from comparative group Ammuscle vs. Amgill, namely Wilms’ tumor 1-associated protein, arginine kinase-like, major egg antigen, SPARC-like, fermitin family homolog 2-like, heat shock protein 90, and laminin receptor, were chosen for the mRNA-levels analysis. The qPCR results indicated that Wilms’ tumor 1-associated protein, arginine kinase-like, major egg antigen, SPARC-like and fermitin family homolog 2-like were upregulated in Ammuscle (Ammuscle vs. Amgill), whereas the two proteins heat shock protein 90 and laminin receptor were downregulated in Ammuscle (Ammuscle vs. Amgill). The mRNA expression levels of the differentially expressed proteins were statistically different among the two tissues (p <0.05; Figure 7). When compared with the protein expression levels, the mRNA expression exhibited similar patterns with the formers.
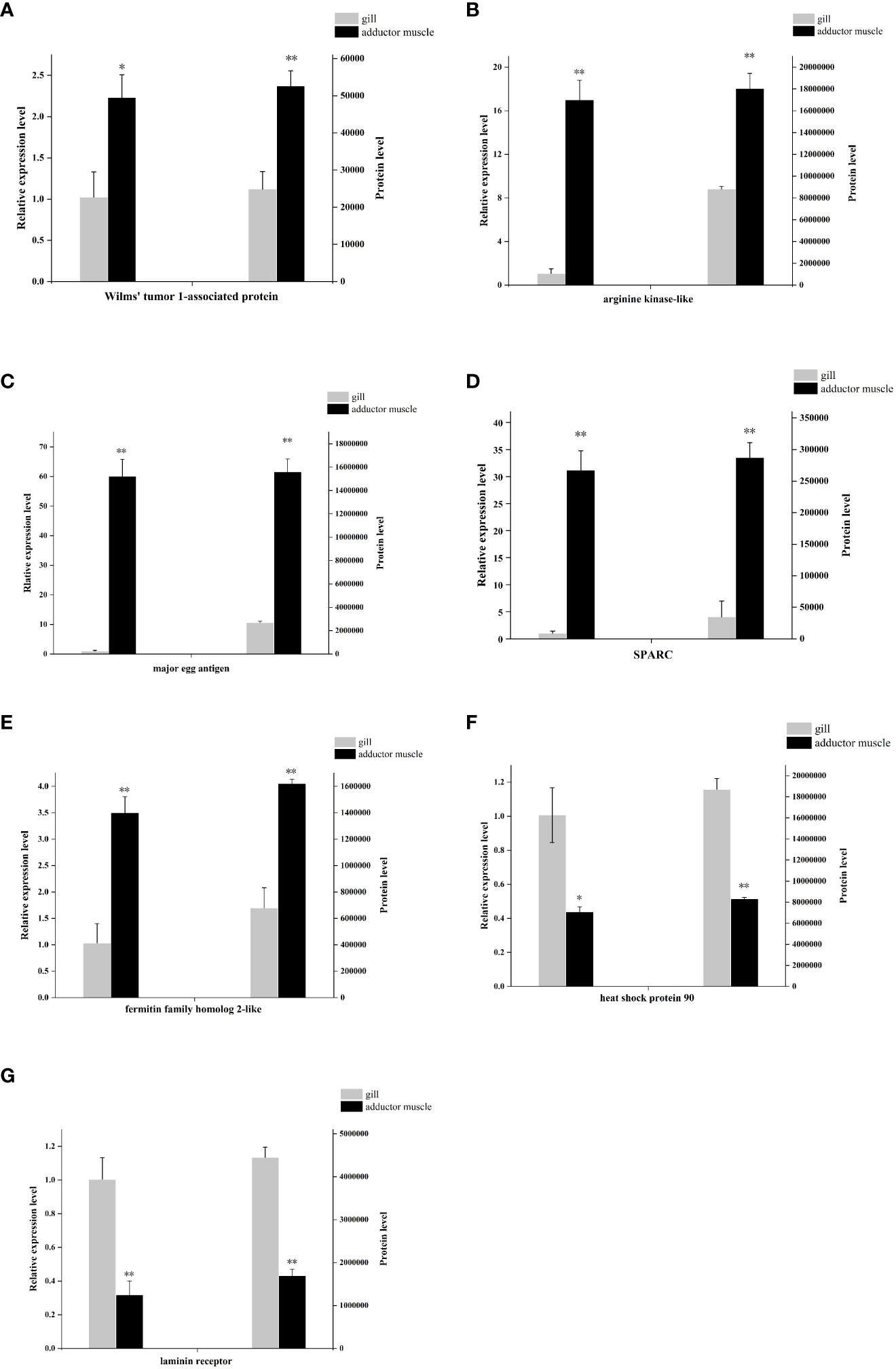
Figure 7 mRNA- and protein-level expression of 7 differentially expressed proteins Note: (A), Wilms’ tumor 1-associated protein; (B), arginine kinase-like; (C), major egg antigen; (D), SPARC-like; (E), fermitin family homolog 2-like; (F), heat shock protein 90; (G), laminin receptor. “**” indicate p < 0.01, “*” indicate p < 0.05.
Discussion
In this study, a total of 4557 proteins were identified from cold seep clam A. marissinica and shallow water clam R. philippinarum. The GO, KEGG, and protein interaction analyses of differential expressed proteins were conducted, and the qPCR validation of partial proteins were processed to verify the reliability of proteome data. Considering the differences between shallow-water and cold seep zone environments, as well as the differences between shallow sea and cold seep shellfish, researchers will discuss immune recognition (establishment and maintenance of symbiotic relationships), pressure related proteins, nitrogen metabolism, antioxidant response, and energy metabolism (cold-adaptation), and so on.
Host proteins related symbiotic relationship
The mutualistic relationship between deep sea shellfish and symbiotic bacteria have been demonstrated in previous research(Yue et al., 2015; Sun et al., 2017). These studies have suggested that pattern recognition receptors may play a crucial role in facilitating symbiotic relationships among deep-sea organisms, specifically by facilitating the colonization of related symbiotic microorganisms. Our study further supported these findings as we observed high expression levels of mostly galectin and peptidoglycan recognition protein in the gill tissue (Table 3). After the stable establishment of the symbiotic relationship, it enters the maintenance stage of the symbiotic relationship, which mainly involves the treatment of the symbiotic bacteria after apoptosis. In the KEGG pathway analysis conducted on the four comparative groups, particular attention was given to the lysosomal signaling pathway. The main members of the signaling pathway are cathepsin B, cathepsin C, cathepsin D, cathepsin F, cathepsin L, cathepsin X, saposin, and others. They were highly expressed in Vesicomyidae clam gill tissue (except for a transcript from cathepsin L, Ama38338, which was low expressed in A. marissinica gill tissue; Supplementary Tables S7-S10, lysosome pathway). Cathepsins, including cysteine cathepsins, were previously thought to exclusively engage in terminal protein degradation in the context of necrotic and autophagic cell death(Turk et al., 2012). Saposins, on the other hand, activate multiple lysosomal hydrolases that play a role in the metabolic processes of diverse sphingolipids (Kishimoto et al., 1992; Popovic et al., 2012). Cathepsin and saposin have both been identified as potential facilitators in the degradation process of symbiont proteins during symbiont digestion. A similar occurrence was observed in the deep sea mussel Bathymodiolus azoricus, where the levels of cathepsin and saposin in the gill tissue were found to be 80-fold and 13-fold higher, respectively, in comparison to gill tissue devoid of symbiotic bacteria(Ponnudurai et al., 2017). Moreover, five glycosidases, namely lysosomal alpha-glucosidase, lysosomal alpha-mannosidase, beta-mannosidase, alpha-L-fucosidase, and galactocerebrosidase, were identified and predominantly localized in the gills (Supplementary Tables S7-S10, lysosome pathway). These enzymes potentially participate in the degradation of polysaccharides present on the bacterial cell surface(Davies and Henrissat, 1995; Ponnudurai et al., 2017).
Pressure related proteins-regulation of actin cytoskeleton
In contrast to organisms inhabiting shallow waters, deep-sea organisms have adapted to thrive in environments characterized by elevated hydrostatic pressure. It has been observed that the hydrostatic pressure rises by one standard atmosphere for every 10.06 m of water depth (Somero, 1992). Therefore, marine organisms that live in deep-sea environments must have mechanisms to withstand such high hydrostatic pressure. Studies have indicated that the trimethylamine N-oxide (TMAO) content in deep-sea fish increases proportionally with water depth, and this high TMAO concentration serves various purposes, such as reducing osmotic regulation costs, enhancing buoyancy, and mitigating protein destabilization caused by pressure (Kelly and Yancey, 1999). In our study, the proteins involved in the regulation of actin cytoskeleton KEGG pathway were found to be enriched in the comparative group Rpgill vs. Amgill. Within this pathway, it was observed that actin related proteins, myosin, most of ras related proteins, and cdc42 exhibited low expression in the Amgill tissue (Rpgill vs. Amgill). However, profilin and gelsolin exhibited high expression in the Amgill tissue (Rpgill vs. Amgill) (Figure 6C, Supplementary Table S7). These findings suggest that profilin and gelsolin may have significant roles in actin organization and cell motility, as suggested by previous researchers (Krebs, 2004; Pimm et al., 2020). It is hypothesized that deep-sea shellfish cells respond to pressure by undergoing a process of “rounding up” or depolymerizing actin cytoskeletal proteins. This phenomenon has been documented in previous studies on cell-pressure experiments (Crenshaw et al., 1996; Tokuda et al., 2009). The GO analysis also found that differential proteins from Ammuscle vs. Amgill were significantly enriched in actin related GO terms (Supplementary Figure S2, B2). These GO terms includes regulation of actin polymerization or depolymerization, actin filament polymerization, regulation of actin filament polymerization, actin polymerization or depolymerization, and so on. This observation can be attributed to the abundance of actin in the muscle tissue.
Host nitrogen metabolism provides Ci for the symbiotic bacteria
It is well-established that clam species belonging to the Vesicomyidae family, found in vent and seep ecosystems, possess symbionts in their gills(Vrijenhoek, 2010; Dover et al., 2011). These symbionts play a crucial role in providing the clams with necessary nutrients (Kuwahara et al., 2007; Newton et al., 2007). In the case of Calyptogena clams, the symbionts act as primary producers and utilize sulfur oxidation to fix inorganic carbon(Ci) (Childress et al., 1991; Childress et al., 1993). The majority of Ci, such as CO2 and HCO- 3, exists in the form of HCO- 3 in seawater (Siegenthaler and Sarmiento, 1993). However, Ci in this form cannot permeate the cell membrane(Geers and Gros, 2000). Thus, the mechanism by which Ci is transferred from the external water to symbionts is a topic of interest. It has been observed that carbonic anhydrase (CA) can facilitate a reversible reaction: CO2 + H2O↔ HCO- 3 + H+ (Supuran and De Simone, 2015; Hongo et al., 2016). Moreover, in Calyptogena clams, the CA activity was measured in various tissues including the gills, foot, and mantle, revealing that the gills exhibited the highest level of activity(Kochevar and Childress, 1996). Subsequently, researchers found that host CA (Calyptogena clams) facilitated a chemical reaction involving the conversion of CO2 and HCO- 3 (transfer HCO- 3 into CO2) in the clam gills, which provided intracellular CO2 for symbiotic bacteria residing in the symbiont (Hongo et al., 2013; Hongo et al., 2016). The proteins derived from Ammantle vs. Amgill exhibited a significant enrichment in the nitrogen metabolism KEGG signaling pathway, as indicated by Supplementary Figure S3, B3; Supplementary Table S10. The main members of this pathway were CAs, with the majority of their transcripts displaying high expression levels in Amgill (Ammantle/Amgill), as shown in Supplementary Table S10 (Nitrogen metabolism). Similarly, it was observed that the differential proteins from Rpgill vs. Amgill were enriched in the nitrogen compound metabolic process (GO term) and cellular nitrogen compound biosynthetic process (GO term), as depicted in Figure 4B. The gill tissue of deep-sea mollusks demonstrated a notable upregulation of CA expression (Rpgill/Amgill), as indicated in Table 3. Therefore, carbonic anhydrase from A. marissinica might also play a similar function and provide a carbon source for symbiotic bacteria.
Antioxidant response-superoxide dismutase
The deep-sea environment, characterized by low temperatures and high pressures, has the potential to exert a substantial impact on various cellular aspects, such as the integrity of cells, the fluidity of membranes, macromolecular interactions, and the utilization of energy. Consequently, this influence may disrupt cell function, resulting in metabolic disorders and redox imbalances (De Maayer et al., 2014; Xiao and Zhang, 2014). Specifically, the detrimental effects of low temperature and high pressure on the cell membrane can lead to electron leakage, whereby leaked electrons react with oxygen to produce reactive oxygen species (ROS) (Green and Paget, 2004). The decrease in temperature has the potential to enhance the solubility of oxygen, consequently increasing the ROS content, which may lead to oxidative damage. The excessive accumulation of ROS can cause severe harm to biological macromolecules, including DNA and proteins, and may even result in cellular death (Sharma et al., 2012; Dumanović et al., 2021; Sachdev et al., 2021). Thus, the combination of low temperature and high pressure in deep-sea environments disrupts the redox equilibrium of organisms, leading to the generation of a substantial quantity of ROS that causes significant physiological harm. Consequently, organisms must confront the challenge of oxidative damage by employing antioxidant systems (Xiao and Zhang, 2014).
Superoxide dismutase (SOD) is an important antioxidant enzyme that directly acts on superoxide anion radicals, and it converts them into water and hydrogen peroxide. SOD serves as the primary defense mechanism in organisms’ antioxidant defense system (Policar et al., 2022). In the comparative analysis between Rpgill and Amgill, there was a significant upregulation of the antioxidant protein Mn superoxide dismutase in the gill tissue of deep-sea mollusks (Amgill) (Table 3). This finding suggests a potential role of Mn superoxide dismutase in the scavenging of superoxide anion radicals. Li et al. (2019) discovered that the sea cucumber MnSOD derived from the Mariana Trench exhibits a low Km value, along with resistance to heavy metals, chemical reagents, and potent protein denaturants. These characteristics prove advantageous in the preservation of redox equilibrium within the highly challenging deep-sea environments. Additionally, our findings indicated that both extracellular CuZn superoxide dismutase and intracellular CuZn superoxide dismutase it the Ammantle vs. Amgill comparative group exhibited enrichment in the Peroxisome KEGG signaling pathway (Supplementary Table S10). However, these enzymes displayed notably diminished expression levels in the gill tissue of deep-sea molluscs (Amgill), as evidenced by Table 3. Furthermore, Mn-superoxide dismutase originating from Ammuscle vs. Amgill also exhibited reduced expression in the gill tissue of A. marissinica (Amgill), as indicated in Table 3. Therefore, we speculate that symbiotic bacteria in the gill tissue of Vesicomyidae clams is thiotropic γ-protobacteria, which might consume more oxygen and produce less ROS in the gill tissue(Newton et al., 2007; Vrijenhoek, 2010). Nevertheless, it should be noted that different types of SOD in different tissues play different roles in antioxidant activity in cold seep clam.
Cold adaptation-arginine kinase
The deep-sea environment exhibits significant temperature variations, with hydrothermal vents reaching temperatures as high as 350 ° and cold seep zones experiencing temperatures as low as 2-10 ° (Martin et al., 2008; Feng et al., 2018). Thus, deep-sea organisms need to have mechanisms to adapt to these extreme temperature conditions, particularly in low-temperature habitats. When exposed to low temperatures, organisms often experience reduced metabolic rates compared with warm-water species(Peck, 2016). Consequently, it is important to investigate how these organisms manage their energy consumption in response to low temperatures. Research has demonstrated that shallow-water invertebrates possess the ability to utilize proteins or enzymes with cold-adapted characteristics to adapt to low temperature environments. Examples of these proteins and enzymes include cold shock protein, isocitric acid dehydrogenase, arginine kinase(AK, EC 2.7.3.3), trypsin, and others(Stark et al., 2022). Among these, AK plays a crucial role in the energy metabolism process. It facilitates the reversible exchange of the gamma-phosphoryl group between ATP and arginine, resulting in the production of ADP, phosphorylated guanidine, and energy(Uda et al., 2006). Researchers found that the catalytic efficiency of arginine kinases with two domains, derived from cold seep clams, exhibited a threefold increase at a temperature of 10 ° when compared with that at to 25 ° (Suzuki et al., 2012). This finding suggests a potential mechanism by which cold seep shellfish can adapt to low-temperature surroundings. Moreover, researchers revealed that domain 2 of cold seep clam two-domain AKs may be subject to more substantial evolutionary selection pressure than domain 1 (Kong et al., 2018). This is supported by the presence of a greater number of positive selective sites in domain 2. Additionally, the proteomic analysis in this study demonstrated that deep-sea shellfish exhibit significantly elevated expression of AK in gill tissue (Amgill) when compared with that in shallow sea shellfish, as indicated in Table 3. These findings collectively suggested that AKs may play crucial roles in cold-adaptation of deep-sea shellfish.
Conclusion
Transcriptome and genome technologies have been widely employed in research on deep-sea organisms. However, there have been limited investigations into proteomic analysis of deep-sea organisms inhabiting extreme environments. This study conducted a comprehensive examination of differentially regulated proteins in the cold seep clam A. marisinica and the shallow water clam R. filippinarum, providing a global perspective. This study identified crucial response proteins involved in various biological pathways. The identification of several noteworthy KEGG pathways, such as “regulation of actin cytoskeleton,” “lysosome,” “peroxisome,” and “nitrogen metabolism,” suggests some survival strategies employed by A. marisinica in extreme environments. These findings offer novel perspectives on the proteins implicated in A. marisica’s response to cold seep environments, thereby contributing to the understanding of deep-sea biological adaptation and the preservation of deep-sea ecosystems.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The animal study was approved by An Institutional Animal Care and Use Committee, Jiangsu Ocean University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
XK: Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing, Formal Analysis, Visualization, Funding acquisition. WW: Formal Analysis, Validation, Writing – review & editing, Investigation. SC: Writing – review & editing, Investigation. NM: Writing – review & editing, Investigation. YC: Writing – review & editing, Investigation. YL: Writing – review & editing, Investigation. SX: Writing – review & editing, Investigation. HZ: Investigation, Resources, Writing – review & editing, Funding acquisition. XS: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Natural Science Foundation of Jiangsu Province (Grant No. BK20210927), National Nature Science Foundation of China (Grant No. 42376139), Strategic Priority Research Program of the Chinese Academy of Sciences (CAS) (XDA22050303), Natural Science Foundation of the Jiangsu Higher Education Institutions of China (Grant No. 21KJB170022), Open Project of Jiangsu Institute of Marine Resources Development (Grant No. JSIMR202204), Scientific Research Foundation of Jiangsu Ocean University (Grant No. KQ19043), A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Acknowledgments
Samples were collected on board of “Explorer 1” scientific research ship of Institute of Deep-sea Science and Engineering (Chinese Academy of Sciences) by using “Deep-sea Warrior” manned submersible, we thank the crew for their help during the cruise.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1294736/full#supplementary-material
Supplementary Figure 1 | Subcellular localization and domain analysis of the differential proteins for Amfoot vs. Amgill (A1, B1, C1), Ammuscle vs. Amgill (A2, B2, C2), and Ammantle vs. Amgill (A3, B3, C3) Notes: A1-3, subcellular localization of the differential proteins; B1-3, domain analysis of the differential proteins; C1-3, domain enrichment analysis of the differential proteins.
Supplementary Figure 2 | The Gene Ontology analysis related maps of the differentially expressed proteins for Amfoot vs. Amgill (A1, B1, C1, D1), Ammuscle vs. Amgill (A2, B2, C2, D2) and Ammantle vs. Amgill (A3, B3, C3, D3) Note: GO annotation statistical map (A1, A2, A3), Biological Process enrichment bubble map (B1, B2, B3), Molecular Function enrichment bubble map (C1, C2, C3), Cellular Component enrichment bubble map (D1, D2, D3).
Supplementary Figure 3 | KEGG pathway analysis of differentially expressed proteins from Amfoot vs. Amgill (A1, B1), Ammuscle vs. Amgill (A2, B2) and Ammantle vs. Amgill (A3, B3) Note: KEGG pathway annotation statistical map (A1, A2, A3), KEGG pathway enrichment bubble map (B1, B2, B3).
Supplementary Figure 4 | Protein-protein interaction diagrams of which involved in the methane metabolism (A), lysosome (B), glutathione metabolism (C), purine metabolism (D) and drug metabolism-cytochrome P450 signaling pathway (E) from Amfoot vs. Amgill Note: protein full name and protein abbreviation. A, 6-phosphofructokinase 1: pfkma; D-3-phosphoglycerate dehydrogenase: phgdh; enolase: eno4; fructose-1,6-bisphosphatase I: tigara; fructose-bisphosphate aldolase, class I: aldoaa; glycine hydroxymethyltransferase: shmt1; phosphoserine phosphatase: psph; S-(hydroxymethyl) glutathione dehydrogenase: adh5; S-formylglutathione hydrolase: esd. B, acid ceramidase: asah1a; alpha-L-fucosidase: zgc:101116; arylsulfatase B: ARSB; cathepsin B: ctsba; cathepsin C: ctsc; cathepsin D:ctsd; cathepsin F: ctsf; cathepsin L: wu:fa26c03; cation-dependent mannose-6-phosphate receptor: m6pr; CD63 antigen: cd63; galactosylceramidase: galca; ganglioside GM2 activator: gm2a; hexosaminidase: hexa; lysosome membrane protein 2: LOC564077; Niemann-Pick C2 protein: zgc:193725; N-sulfoglucosamine sulfohydrolase: sgsh; saposin: psap; V-type H+-transporting ATPase S1 subunit: atp6ap1a; V-type H+-transporting ATPase subunit a: atp6v0a1a; V-type H+-transporting ATPase subunit H: atp6v1h. C, 6-phosphogluconate dehydrogenase: pgd; cytosol aminopeptidase: lap3; glucose-6-phosphate 1-dehydrogenase: g6pd; glutathione peroxidase: gpx1b; glutathione S-transferase: gstm; glutathione-specific gamma-glutamylcyclotransferase: chac1; isocitrate dehydrogenase: idh3a; peroxiredoxin 6: prdx6. D, 5-hydroxyisourate hydrolase: urah; adenine phosphoribosyltransferase: aprt;adenosine kinase: adka;adenylosuccinate lyase: adsl; ADP-sugar pyrophosphatase: nudt5; amidophosphoribosyltransferase:ppat; GMP reductase: gmpr; hypoxanthine phosphoribosyltransferase: prtfdc1; IMP dehydrogenase: impdh2; nucleoside-diphosphate kinase: cmpk; phosphoribosylaminoimidazole carboxylase: paics; urate oxidase: uox; xanthine dehydrogenase: xdh. E, S-(hydroxymethyl)glutathione dehydrogenase: ADH5; glutathione S-transferase: GSTO1; prostaglandin-H2 D-isomerase: HPGDS.
Supplementary Figure 5 | Protein-protein interaction diagrams of which involved in the ribosome (A), proteasome (B), lysosome (C), glycolysis/gluconeogenesis (D) and fatty acid degradation signaling pathway (E) from Ammuscle vs. Amgill Note: protein full name and protein abbreviation. A, large subunit ribosomal protein L38e: rpl38; small subunit ribosomal protein S14e: rps14; large subunit ribosomal protein L3e: rpl3; small subunit ribosomal protein S9e: rps9; small subunit ribosomal protein SAe: rpsa; small subunit ribosomal protein S15Ae: rps15a; large subunit ribosomal protein L17: mrpl17; large subunit ribosomal protein L10e: rpl10; small subunit ribosomal protein S2e: rps2; smallsubunit ribosomal protein S3e: rps3; large subunit ribosomal protein L31e: rpl31; large subunit ribosomal protein L35e: rpl35; large subunit ribosomal protein L21e: rpl21; small subunit ribosomal protein S3Ae: rps3a; large subunit ribosomal protein L44e: rpl36a; small subunit ribosomal protein S4e: rps4x; largesubunit ribosomal protein L21: mrpl21; large subunit ribosomal protein L10Ae: rpl10a; large subunit ribosomal protein L13Ae: rpl13a; large subunit ribosomal protein L19: mrpl19; large subunit ribosomal protein L17e: rpl17; ubiquitin-small subunit ribosomal protein S27Ae: rps27a; small subunit ribosomal protein S10: mrps10; small subunit ribosomal protein S30e: faua; small subunit ribosomal protein S12e: rps12; small subunit ribosomal protein S21e: rps21; small subunit ribosomal protein S28e: rps28; large subunit ribosomal protein L13: mrpl13; large subunit ribosomal protein L37e: rpl37; large subunit ribosomal protein L32: mrpl32; large subunit ribosomal protein L23e: rpl23; largesubunit ribosomal protein L13e: rpl13; large subunit ribosomal protein L2: MRPL2; large subunit ribosomal protein L18Ae: rpl18a; large subunit ribosomal protein L6e: rpl6; large subunit ribosomal protein LP2: rplp2l; small subunit ribosomal protein S10e: rps10; large subunit ribosomal protein L32e: rpl32; small subunit ribosomal protein S26e: rps26; large subunit ribosomal protein L16: mrpl16; large subunit ribosomal protein L15e: rpl15; small subunit ribosomal protein S13e: rps13; small subunit ribosomal protein S18e: rps18; large subunit ribosomal protein L12e: rpl12; small subunit ribosomal protein S16e: rps16; large subunit ribosomal protein L36e: rpl36; large subunit ribosomal protein L4e: rpl4; small subunit ribosomal protein S17e: rps17; smallsubunit ribosomal protein S15e: rps15; small subunit ribosomal protein S6e: rps6; large subunit ribosomal protein L26e: rpl26; large subunit ribosomal protein L7e: rpl7; large subunit ribosomal protein L5e: rpl5a; large subunit ribosomal protein L7Ae: rpl7a; large subunit ribosomal protein L10: mrpl10; small subunit ribosomal protein S8e: rps8a; large subunit ribosomal protein L27e: rpl27; large subunit ribosomal protein L37Ae: RPL37A; large subunit ribosomal protein L35Ae: rpl35a; large subunit ribosomal protein L8e: rpl8; large subunit ribosomal protein LP1: rplp1; large subunit ribosomal protein L30e: rpl30; large subunit ribosomal protein L11e: rpl11; large subunit ribosomal protein L19e: rpl19; small subunit ribosomal protein S27e: rps27.1; small subunit ribosomal protein S24e: rps24; large subunit ribosomal protein L23Ae: rpl23a; large subunit ribosomal protein L20: mrpl20; small subunit ribosomal protein S11e: rps11. B, 26S proteasome regulatory subunit N1: psmd2; 26S proteasome regulatory subunit N10: psmd4b; 26S proteasome regulatory subunit N11: psmd14; 26S proteasome regulatory subunit N12: psmd8; 26S proteasome regulatory subunit N3: psmd3; 26S proteasome regulatory subunit N5: psmd12; 26S proteasome regulatory subunit N7: psmd6; 26S proteasome regulatory subunit N8: psmd7; 26S proteasome regulatory subunit T1: psmc2; 26S proteasome regulatory subunit T2: psmc1a; 26S proteasome regulatory subunit T3: psmc4; 26S proteasome regulatory subunit T4: psmc6; 26S proteasome regulatory subunit T5: psmc3; 26S proteasome regulatory subunit T6: psmc5. C, acid ceramidase: asah1a; alpha-L-fucosidase: zgc:101116; alpha-N-acetylglucosaminidase: NAGPA; AP-1 complex subunit beta-1: ap1b1; AP-1 complex subunit gamma-1: ap1g2; AP-1 complex subunit mu: ap1m2; AP-3 complex subunit beta: AP3B2; arylsulfatase B: ARSB; beta-galactosidase: GLB1L2; beta-glucuronidase: Gusb; cathepsin B: ctsba; cathepsin C: ctsc; cathepsin D: ctsd; cathepsin F: ctsf; cathepsin L: wu:fa26c03; cation-dependent mannose-6-phosphate receptor: m6pr; CD63 antigen: cd63; clathrin heavy chain: cltca; deoxyribonuclease II: DNASE2B; galactosylceramidase: galca; ganglioside GM2 activator: gm2a; glucosylceramidase: gba; hexosaminidase: hexa; iduronate 2-sulfatase: ids; legumain: LOC569153; lysosome membrane protein 2: LOC564077; Niemann-Pick C1 protein: npc1; Niemann-Pick C2 protein: zgc:193725; saposin: psap; V-type H+-transporting ATPase S1 subunit: atp6ap1a; V-type H+-transporting ATPase subunit a: atp6v0a1a; V-type H+-transporting ATPase subunit d: atp6v1d. D, 6-phosphofructokinase 1: pfkma; ADP-dependent glucokinase: adpgk2; aldehyde dehydrogenase (NAD+): aldh8a1; dihydrolipoamide dehydrogenase: dldh; enolase: eno4; fructose-bisphosphate aldolase, class I: aldoaa; glucose-6-phosphate isomerase: gpia; hexokinase: hkdc1; phosphoglycerate kinase: pgk1; pyruvate dehydrogenase E1 component alpha subunit: pdha1a; pyruvate dehydrogenase E2 component (dihydrolipoamide acetyltransferase): dbt; pyruvate kinase: pklr. E, 3-hydroxyacyl-CoA dehydrogenase: ehhadh; acetyl-CoA acyltransferase: acaa2; acetyl-CoA acyltransferase 1: acaa1; acyl-CoA dehydrogenase: acad9; acyl-CoA oxidase: acox1; aldehyde dehydrogenase (NAD+): aldh8a1; carnitine O-palmitoyltransferase 2: cpt2; long-chain acyl-CoA synthetase: acsl4a.
Supplementary Figure 6 | Protein-protein interaction diagrams of which involved in the peroxisome (A), lysosome (B), ECM-receptor interaction (C), nitrogen metabolism (D) and fatty acid degradation signaling pathway (E) from Ammantle vs. Amgill Note: protein full name and protein abbreviation. A, acetyl-CoA acyltransferase 1: acaa1; acyl-CoA oxidase: acox1; carnitine O-octanoyltransferase: crot; catalase: cat; long-chain acyl-CoA synthetase: acsl4a; peroxin-19: pex19; peroxiredoxin 1: prdx1; peroxisomal membrane protein 2: pxmp2; superoxide dismutase, Cu-Zn family: sod1. B, acid ceramidase: asah1a; arylsulfatase B: ARSB; cathepsin B: ctsba; cathepsin C: ctsc; cathepsin D: ctsd; cathepsin F: ctsf; cathepsin L: wu:fa26c03; cation-dependent mannose-6-phosphate receptor: m6pr; CD63 antigen: cd63; ganglioside GM2 activator: gm2a; lysosome membrane protein 2: LOC564077; Niemann-Pick C2 protein: zgc:193725; N-sulfoglucosamine sulfohydrolase: sgsh; saposin: psap. C, collagen type II alpha: col2a1b; collagen type IV alpha: col4a5; collagen type VI alpha: col6a2; integrin beta 1: itgb1b.2; laminin, alpha 3/5: tmem5; laminin, beta 1: gyltl1b; laminin, gamma 1: lamc1. D, glutamate dehydrogenase (NAD(P)+): Gdh; glutamate synthase (NADH): CG9674. E, 3-hydroxyacyl-CoA dehydrogenase: ehhadh; acetyl-CoA acyltransferase: acaa2; acetyl-CoA acyltransferase 1: acaa1; acyl-CoA dehydrogenase: acad9; acyl-CoA oxidase: acox1; aldehyde dehydrogenase (NAD+): aldh8a1; carnitine O-palmitoyltransferase 2: cpt2; long-chain acyl-CoA synthetase: acsl4a.
References
Beaulieu S. E., Baker E. T., German C. R., Maffei A. (2013). An authoritative global database for active submarine hydrothermal vent fields. Geochemistry Geophysics Geosystems 14 (11), 4892–4905. doi: 10.1002/2013GC004998
Cai Z., Poulos R. C., Liu J., Zhong Q. (2022). Machine learning for multi-omics data integration in cancer. iScience 25 (2), 103798. doi: 10.1016/j.isci.2022.103798
Cheng J., Hui M., Sha Z. (2019). Transcriptomic analysis reveals insights into deep-sea adaptations of the dominant species, Shinkaia crosnieri (Crustacea: Decapoda: Anomura), inhabiting both hydrothermal vents and cold seeps. BMC Genomics 20 (1), 388–398. doi: 10.1186/s12864-019-5753-7
Childress J. J., Fisher C. R., Favuzzi J. A., Arp A. J., Oros D. R. (1993). The role of a zinc-based, serum-borne sulfide-binding component in the uptake and transport of dissolved sulfide by the chemoautotrophic symbiont-containing clam Calyptogena Clongata. J. Exp. Biol. 179 (1), 131–158. doi: 10.1242/jeb.179.1.131
Childress J. J., Fisher C. R., Favuzzi J. A., Sanders N. K. (1991). Sulfide and carbon dioxide uptake by the hydrothermal vent clam, calyptogena magnifica, and its chemoautotrophic symbionts. Physiol. Zoology 64 (6), 1444–1470. doi: 10.2307/30158224
Crenshaw H. C., Allen J. A., Skeen V., Harris A., Salmon E. D. (1996). Hydrostatic pressure has different effects on the assembly of tubulin, actin, myosin II, vinculin, talin, vimentin, and cytokeratin in mammalian tissue cells. Exp. Cell Res. 227 (2), 285–297. doi: 10.1006/excr.1996.0278
Crick F. (1970). Central dogma of molecular biology. Nature 227 (5258), 561–563. doi: 10.1038/227561a0
Davies G., Henrissat B. (1995). Structures and mechanisms of glycosyl hydrolases. Structure 3 (9), 853–859. doi: 10.1016/s0969-2126(01)00220-9
De Maayer P., Anderson D., Cary C., Cowan D. A. (2014). Some like it cold: understanding the survival strategies of psychrophiles. EMBO Repots 15 (5), 508–517. doi: 10.1002/embr.201338170
Dover C. V., Smith C. R., Ardron J., Arnaud S., Beaudoin Y., Bezaury J. (2011). Environmental Management of Deep-Sea Chemosynthetic Ecosystems: Justification of and Considerations for a Spatially Based Approach. Ifremer (Jamaica), 1–75.
Dubilier N., Bergin C., Lott C. (2008). Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat. Rev. Microbiol. 6 (10), 725–740. doi: 10.1038/nrmicro1992
Dumanović J., Nepovimova E., Natić M., Kuča K., Jaćević V. (2021). The significance of reactive oxygen species and antioxidant defense system in plants: A concise overview. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.552969
Feng D., Qiu J.-W., Hu Y., Peckmann J., Guan H., Tong H., et al. (2018). Cold seep systems in the South China Sea: An overview. J. Asian Earth Sci. 168, 3–16. doi: 10.1016/j.jseaes.2018.09.021
Galley C. G., Jamieson J. W., Lelièvre P. G., Farquharson C. G., Parianos J. M. (2020). Magnetic imaging of subseafloor hydrothermal fluid circulation pathways. Sci. Adv. 6 (44), 5–10. doi: 10.1126/sciadv.abc6844
Geers C., Gros G. (2000). Carbon dioxide transport and carbonic anhydrase in blood and muscle. Physiol. Rev. 80 (2), 681–715. doi: 10.1152/physrev.2000.80.2.681
Girguis P. R., Newton Irene L. G., Cavanaugh C. M. (2008). Comparative genomics of vesicomyid clam (Bivalvia: Mollusca) chemosynthetic symbionts. BMC Genomics 9 (1), 585–595. doi: 10.1186/1471-2164-9-585
Govenar B. (2010). “Shaping vent and seep communities: habitat provision and modification by foundation species,” in The Vent and Seep Biota: Aspects from Microbes to Ecosystems. Ed. Kiel S. (Dordrecht: Springer Netherlands), 403–432.
Green J., Paget M. S. (2004). Bacterial redox sensors. Nat. Rev. Microbiol. 2 (12), 954–966. doi: 10.1038/nrmicro1022
Hongo Y., Ikuta T., Takaki Y., Shimamura S., Shigenobu S., Maruyama T., et al. (2016). Expression of genes involved in the uptake of inorganic carbon in the gill of a deep-sea vesicomyid clam harboring intracellular thioautotrophic bacteria. Gene 585 (2), 228–240. doi: 10.1016/j.gene.2016.03.033
Hongo Y., Nakamura Y., Shimamura S., Takaki Y., Uematsu K., Toyofuku T., et al. (2013). Exclusive localization of carbonic anhydrase in bacteriocytes of the deep-sea clam Calyptogena okutanii with thioautotrophic symbiotic bacteria. J. Exp. Biol. 216 (Pt 23), 4403–4414. doi: 10.1242/jeb.092809
Huo D., Sun L., Zhang L., Ru X., Liu S., Yang X., et al. (2019). Global-warming-caused changes of temperature and oxygen alter the proteomic profile of sea cucumber Apostichopus japonicus. J. Proteomics 193, 27–43. doi: 10.1016/j.jprot.2018.12.020
Ip J. C., Xu T., Sun J., Li R., Chen C., Lan Y., et al. (2021). Host-endosymbiont genome integration in a deep-sea chemosymbiotic clam. Mol. Biol. Evol. 38 (2), 502–518. doi: 10.1093/molbev/msaa241
Jean-paul F., Graham W., Boetius A., Ceramicola S., Dupré S., Mascle J., et al. (2009). Structure and drivers of cold seep ecosystems. Oceanography (Washington D.C.) 22, 92–109. doi: 10.5670/oceanog.2009.11
Jones D. O., Kaiser S., Sweetman A. K., Smith C. R., Menot L., Vink A., et al. (2017). Biological responses to disturbance from simulated deep-sea polymetallic nodule mining. PloS One 12 (2), e0171750. doi: 10.1371/journal.pone.0171750
Kelly R. H., Yancey P. H. (1999). High contents of trimethylamine oxide correlating with depth in deep-sea teleost fishes, skates, and decapod crustaceans. Biol. Bull. 196 (1), 18–25. doi: 10.2307/1543162
Kishimoto Y., Hiraiwa M., O’Brien J. S. (1992). Saposins: structure, function, distribution, and molecular genetics. J. Lipid Res. 33 (9), 1255–1267. doi: 10.1016/S0022-2275(20)40540-1
Kochevar R. E., Childress J. J. (1996). Carbonic anhydrase in deep-sea chemoautotrophic symbioses. Mar. Biol. 125 (2), 375–383. doi: 10.1007/BF00346318
Kong X., Liu H., Zhang H. (2018). Positive selection adaptation of two-domain arginine kinase (AK) from cold seep Vesicomyidae clams. Mol. Biol. Rep. 45 (5), 1527–1532. doi: 10.1007/s11033-018-4227-3
Krebs J. (2004). “Calcium-binding proteins: cytosolic (Annexins, gelsolins, C2-domain proteins,” in Encyclopedia of Biological Chemistry. Eds. Lennarz W. J., Lane M. D. (New York: Elsevier), 287–293.
Krylova E. M., Sahling H. (2010). Vesicomyidae (bivalvia): current taxonomy and distribution. PloS One 5 (4), 9957. doi: 10.1371/journal.pone.0009957
Kuwahara H., Yoshida T., Takaki Y., Shimamura S., Nishi S., Harada M., et al. (2007). Reduced genome of the thioautotrophic intracellular symbiont in a deep-sea clam, calyptogena okutanii. Curr. Biol. 17 (10), 881–886. doi: 10.1016/j.cub.2007.04.039
Kwan Y. H., Zhang D., Mestre N. C., Wong W. C., Wang X., Lu B., et al. (2019). Comparative proteomics on deep-sea amphipods after in situ copper exposure. Environ. Sci. Technol. 53 (23), 13981–13991. doi: 10.1021/acs.est.9b04503
Lan Y., Sun J., Tian R., Bartlett D. H., Li R., Wong Y. H., et al. (2017). Molecular adaptation in the world’s deepest-living animal: Insights from transcriptome sequencing of the hadal amphipod Hirondellea gigas. Mol. Ecol. 26 (14), 3732–3743. doi: 10.1111/mec.14149
Levin L. A. (2005). Ecology of cold seep sediments: Interactions of fauna with flow, chemistry and microbes. Oceanography Mar. Biol. 43, 1–46. doi: 10.1201/9781420037449-3
Li Y., Liles M. R., Halanych K. M. (2018). Endosymbiont genomes yield clues of tubeworm success. ISME J. 12 (11), 2785–2795. doi: 10.1038/s41396-018-0220-z
Li Y., Tassia M. G., Waits D. S., Bogantes V. E., David K. T., Halanych K. M. (2019). Genomic adaptations to chemosymbiosis in the deep-sea seep-dwelling tubeworm Lamellibrachia luymesi. BMC Biol. 17 (1), 019–0713. doi: 10.1186/s12915-019-0713-x
Ling J., Guan H., Liu L., Tao J., Li J., Dong J., et al. (2020). The diversity, composition, and putative functions of gill-associated bacteria of bathymodiolin mussel and vesicomyid clam from haima cold seep, south China sea. Microorganisms 8 (11), 1699–1712. doi: 10.3390/microorganisms8111699
Liu R., Liu J., Zhang H. (2021). Positive selection analysis reveals the deep-sea adaptation of a hadal sea cucumber (Paelopatides sp.) to the Mariana Trench. J. Oceanology Limnology 39 (1), 266–281. doi: 10.1007/s00343-020-0241-0
Martin W., Baross J., Kelley D., Russell M. J. (2008). Hydrothermal vents and the origin of life. Nat. Rev. Microbiol. 6 (11), 805–814. doi: 10.1038/nrmicro1991
McClain C. R., Allen A. P., Tittensor D. P., Rex M. A. (2012). Energetics of life on the deep seafloor. Proc. Natl. Acad. Sci. U.S.A. 109 (38), 15366–15371. doi: 10.1073/pnas.1208976109
Newton I. L., Woyke T., Auchtung T. A., Dilly G. F., Dutton R. J., Fisher M. C., et al. (2007). The Calyptogena magnifica chemoautotrophic symbiont genome. Science 315 (5814), 998–1000. doi: 10.1126/science.1138438
Pandey A., Mann M. (2000). Proteomics to study genes and genomes. Nature 405 (6788), 837–846. doi: 10.1038/35015709
Paulus E., Brix S., Siebert A., Martínez Arbizu P., Rossel S., Peters J., et al. (2022). Recent speciation and hybridization in Icelandic deep-sea isopods: An integrative approach using genomics and proteomics. Mol. Ecol. 31 (1), 313–330. doi: 10.1111/mec.16234
Peck L. S. (2016). A cold limit to adaptation in the sea. Trends Ecol. Evol. 31 (1), 13–26. doi: 10.1016/j.tree.2015.09.014
Penhallurick R. W., Ichiye T. (2021). Pressure adaptations in deep-sea moritella dihydrofolate reductases: compressibility versus stability. Biol. (Basel) 10 (11), 1211–1220. doi: 10.3390/biology10111211
Pimm M. L., Hotaling J., Henty-Ridilla J. L. (2020). “Chapter Five - Profilin choreographs actin and microtubules in cells and cancer,” in International Review of Cell and Molecular Biology. Eds. Thomas C., Galluzzi L. (London & New York: Academic Press), 155–204.
Policar C., Bouvet J., Bertrand H. C., Delsuc N. (2022). SOD mimics: From the tool box of the chemists to cellular studies. Curr. Opin. Chem. Biol. 67, 102–109. doi: 10.1016/j.cbpa.2021.102109
Ponnudurai R., Heiden S. E., Sayavedra L., Hinzke T., Kleiner M., Hentschker C., et al. (2020). Comparative proteomics of related symbiotic mussel species reveals high variability of host-symbiont interactions. ISME J. 14 (2), 649–656. doi: 10.1038/s41396-019-0517-6
Ponnudurai R., Kleiner M., Sayavedra L., Petersen J. M., Moche M., Otto A., et al. (2017). Metabolic and physiological interdependencies in the Bathymodiolus azoricus symbiosis. ISME J. 11 (2), 463–477. doi: 10.1038/ismej.2016.124
Popovic K., Holyoake J., Pomès R., Privé G. G. (2012). Structure of saposin A lipoprotein discs. Proc. Natl. Acad. Sci. U.S.A. 109 (8), 2908–2912. doi: 10.1073/pnas.1115743109
Renz J., Markhaseva E. L., Laakmann S., Rossel S., Martinez Arbizu P., Peters J. (2021). Proteomic fingerprinting facilitates biodiversity assessments in understudied ecosystems: A case study on integrated taxonomy of deep sea copepods. Mol. Ecol. Resour. 21 (6), 1936–1951. doi: 10.1111/1755-0998.13405
Sachdev S., Ansari S. A., Ansari M. I., Fujita M., Hasanuzzaman M. J. A. (2021). Abiotic stress and reactive oxygen species: Generation, signaling, and defense mechanisms. Antioxidants (Basel) 10, 2, 277–290. doi: 10.3390/antiox10020277
Sharma P., Jha A. B., Dubey R. S., Pessarakli M. (2012). Reactive oxygen species, oxidative damage, and antioxidative defense mechanism in plants under stressful conditions. J. Bot. 2012, 217–247. doi: 10.1155/2012/217037
Shi Y., Yao G., Zhang H., Jia H., Xiong P., He M. (2022). Proteome and transcriptome analysis of gonads reveals intersex in gigantidas haimaensis. BMC Genomics 23 (1), 174–184. doi: 10.1186/s12864-022-08407-w
Siegenthaler U., Sarmiento J. L. (1993). Atmospheric carbon dioxide and the ocean. Nature 365 (6442), 119–125. doi: 10.1038/365119a0
Somero G. N. (1992). Adaptations to high hydrostatic pressure. Annu. Rev. Physiol. 54, 557–577. doi: 10.1146/annurev.ph.54.030192.003013
Spirin V., Mirny L. A. (2003). Protein complexes and functional modules in molecular networks. Proc. Natl. Acad. Sci. U.S.A. 100 (21), 12123–12128. doi: 10.1073/pnas.2032324100
Stark C., Bautista-Leung T., Siegfried J., Herschlag D. (2022). Systematic investigation of the link between enzyme catalysis and cold adaptation. Elife 11, e72884. doi: 10.7554/eLife.72884
Sun J., Zhang Y., Xu T., Mu H., Lan Y., Fields C. J., et al. (2017). Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat. Ecol. Evol. 1 (5), 017–0121. doi: 10.1038/s41559-017-0121
Sun Y., Wang M., Zhong Z., Chen H., Wang H., Zhou L., et al. (2022). Adaption to hydrogen sulfide-rich environments: Strategies for active detoxification in deep-sea symbiotic mussels, Gigantidas platifrons. Sci. Total Environ. 804, 150–159. doi: 10.1016/j.scitotenv.2021.150054
Supuran C. T., De Simone G. (2015). “Chapter 1 - Carbonic Anhydrases: An Overview,” in Carbonic Anhydrases as Biocatalysts. Eds. Supuran C. T., De Simone G. (Amsterdam: Elsevier), 3–13.
Suzuki T., Yamamoto K., Tada H., Uda K. (2012). Cold-adapted features of arginine kinase from the deep-sea clam Calyptogena kaikoi. Mar. Biotechnol. 14 (3), 294–303. doi: 10.1007/s10126-011-9411-6
Tokuda S., Miyazaki H., Nakajima K., Yamada T., Marunaka Y. (2009). Hydrostatic pressure regulates tight junctions, actin cytoskeleton and transcellular ion transport. Biochem. Biophys. Res. Commun. 390 (4), 1315–1321. doi: 10.1016/j.bbrc.2009.10.144
Turk V., Stoka V., Vasiljeva O., Renko M., Sun T., Turk B., et al. (2012). Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim. Biophys. Acta 1824 (1), 68–88. doi: 10.1016/j.bbapap.2011.10.002
Uda K., Fujimoto N., Akiyama Y., Mizuta K., Tanaka K., Ellington W. R., et al. (2006). Evolution of the arginine kinase gene family. Comp. Biochem. Physiol. Part D Genomics Proteomics 1 (2), 209–218. doi: 10.1016/j.cbd.2005.10.007
Vrijenhoek R. C. (2010). Genetics and evolution of deep-sea chemosynthetic bacteria and their invertebrate hosts. (Berlin: Springer Netherlands).
Xiao X., Zhang Y. (2014). Life in extreme environments: Approaches to study life-environment co-evolutionary strategies. Sci. China Earth Sci. 57 (5), 869–877. doi: 10.1007/s11430-014-4858-8
Yan G., Lian C. A., Lan Y., Qian P. Y., He L. (2021). Insights into the vision of the hadal snailfish Pseudoliparis swirei through proteomic analysis of the eye. Proteomics 21 (19), e2100118. doi: 10.1002/pmic.202100118
Yan G., Sun J., Wang Z., Qian P. Y., He L. (2020). Insights into the Synthesis, Secretion and Curing of Barnacle Cyprid Adhesive via Transcriptomic and Proteomic Analyses of the Cement Gland. Mar. Drugs 18 (4), 1–10. doi: 10.3390/md18040186
Yi L., Sun J., Zhang W., Xu T., Zhang Y., Chen C., et al. (2019). Host–symbiont interactions in deep-sea chemosymbiotic Vesicomyid clams: insights from transcriptome sequencing. Front. Mar. Sci. 6. doi: 10.3389/fmars.2019.00680
Yue H. W., Sun J., He L. S., Chen L. G., Qiu J. W., Qian P. Y. (2015). High-throughput transcriptome sequencing of the cold seep mussel Bathymodiolus platifrons. Sci. Rep. 5, 165–171. doi: 10.1038/srep16597
Keywords: Vesicomyidae clam, cold seep zone, comparative proteomics, adaptation mechanism, deep-sea ecosystems
Citation: Kong X, Wang W, Chen S, Mao N, Cai Y, Li Y, Xia S, Zhang H and Shen X (2024) Comparative proteomic analysis of cold seep clam Archivesica marissinica and shallow water shellfish Ruditapes philippinarum provides insights into the adaptation mechanisms of deep-sea mollusks. Front. Mar. Sci. 10:1294736. doi: 10.3389/fmars.2023.1294736
Received: 18 September 2023; Accepted: 19 December 2023;
Published: 09 January 2024.
Edited by:
Crisalejandra Rivera-Perez, Centro de Investigación Biológica del Noroeste (CIBNOR), MexicoReviewed by:
Miguel Tripp, Centro de Investigación Biológica del Noroeste (CIBNOR), MexicoWendy Espinosa De Aquino, Universidad Autónoma de Baja California Sur, Mexico
Copyright © 2024 Kong, Wang, Chen, Mao, Cai, Li, Xia, Zhang and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Shen, c2hlbnRoaW5AMTYzLmNvbQ==