 Ginevra Lilli
Ginevra Lilli Charlotte Sirot2
Charlotte Sirot2 Isabelle F. George
Isabelle F. George
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 05 December 2023
Sec. Marine Biology
Volume 10 - 2023 | https://doi.org/10.3389/fmars.2023.1286706
This article is part of the Research TopicMediterranean Coastal Fish Biology and EcologyView all 6 articles
The gut microbiome holds an important role in the health and homeostasis of fishes. However, despite the large diversity and distribution of this vertebrate group, only the intestinal microbiome of a limited number of freshwater and marine fish species has been well characterized to date. In this study, we characterize the gut mucosal microbial communities of three commercially valuable Scorpaena spp. (n=125) by using a comprehensive comparative dataset including 16S rRNA gene amplicon data from four different locations in the Mediterranean Sea. We report that the geographical origin of the individuals influences the diversity and the composition of the gut microbial communities more than the host’s phylogenetic relatedness in this fish group. Moreover, we observe a positive correlation between the composition of the gut microbiota and the phylogenetic distance between the hosts (i.e. phylosymbiosis). Finally, the core microbiota of each species is described both regionally and across the Mediterranean Sea. Only a few bacterial genera appear to be residents of the scorpionfishes’ gut microbiota across the Mediterranean Sea: Photobacterium, Enterovibrio, Vibrio, Shewanella, Epulopiscium, Clostridium sensu stricto 1 and Rombutsia in S. notata, Clostridium sensu stricto 1, Cetobacterium and Rombutsia in S. porcus, and only Clostridium sensu stricto 1 in S. scrofa. This study highlights the importance of investigating the gut microbiome across a species’ geographical range and it suggests this as a general procedure to better characterize the gut microbial ecology of each fish species.
The microbial consortia living in the body niches of fishes, especially in the intestinal system, have stimulated particular attention in recent years for their symbiotic role in their farmed and wild hosts (Tarnecki et al., 2017; Butt and Volkoff, 2019). Understanding the contribution of the intestinal communities to fish health and metabolism can inform the development of rearing enhancement measures in aquaculture such as the administration of probiotics and prebiotics (Chauhan and Singh, 2019). In farmed and wild fishes, increased susceptibility to pathogens and reduced absorption of nutrients due to structural alteration of the intestinal communities (i.e., microbial dysbiosis) can be better explained by identifying the host-related and environmental factors that affect these communities. Despite these applications, studies of wild fish gut microbiota have predominantly focused on a limited number of teleost fishes (Nayak, 2010; Egerton et al., 2018) and gut microbiota is well characterized for only a handful of freshwater and marine fish species (Gajardo et al., 2016). As a consequence, phenomena like phylosymbiosis, which is the subject of considerable study in other animal clades (e.g., mammals, insects) (Lim and Bordenstein, 2020; Mallott and Amato, 2021) have not been explored in detail for fishes. Phylosymbiosis is an eco-evolutionary pattern that is evident when the composition of the host-associated microbiota reflects the host’s phylogenesis (Brooks et al., 2017). Hence, the composition of the microbial communities of phylogenetically closer hosts will be more similar than that of phylogenetically distant ones. This pattern is promoted both by host-microbe co-diversification and by ecological selection driven by host’s genetics and behavior (Kohl, 2020; Lim and Bordenstein, 2020). Although the diversity and structure of the microbiome associated with fish’s gut has been reported to vary depending on the fish species (Tarnecki et al., 2017), it is still a matter of debate whether this variation is driven by the phylogenetic relationships existing among the hosts (i.e. phylosymbiosis). In carnivorous Sparidae for example, distantly related host species were found to harbor similar gut microbiota and this convergence was explained by their comparable gut morphology and dietary habits (Escalas et al., 2021). Conversely, evidence of phylosymbiosis has been found in the microbiome of different body locations of multiple North Pacific fishes (Minich et al., 2022). Furthermore, the latter study showed that the hosts’ evolutionary history was more evident in the microbial communities present in the intestinal mucosa (hindgut) than in other body sites.
Factors other than host’s taxonomy and phylogeny also affect the structure and diversity of the gut microbiota in fishes. Generally, diet and trophic level are the main factors influencing the composition of the gut microbial communities of both marine and freshwater fishes (Tarnecki et al., 2017; Egerton et al., 2018; Butt and Volkoff, 2019). However, the gut microbiota of fishes can also be affected indirectly by several other environmental variables. Temperature, by influencing the nutrient load and the resources available in the water column, indirectly acts on the gut microbiota of organisms feeding in this ecosystem (Butt and Volkoff, 2019). Temperature and salinity, by influencing the bacterioplankton communities ingested by fish during osmoregulation, may influence the fish gut microbiota (Sullam et al., 2012). Finally, the different habitats of the host (i.e. freshwater vs marine; captive vs wild; degraded habitat vs intact) are also relevant in determining the dominant bacterial taxa in their gut communities (Dulski et al., 2020; Clever et al., 2022). In contrast, the effect of geographic location is not well defined yet, due to its strong dependency on the species investigated. For example, geographic location was reported to explain the dissimilarity of the gut microbial communities found in individuals of gizzard shad (Dorosoma cepedianum, Rafinesque, 1820) (Ye et al., 2014), but it was found to have no such effect for other species both from freshwaters and in the marine environment (Hypophthalmichthys molitrix, Gasterosteus aculeatus, Sparus aurata, Dicentrarchus labrax and Gadus morhua) (Ye et al., 2014; Smith et al., 2015; Nikouli et al., 2018; Riiser et al., 2019). Spatial heterogeneity of the environment is known to influence parameters such as the biomass, the body conditions and the dietary habits of marine fishes (Ordines et al., 2009). However, further evidence is needed to demonstrate its effect on the intestinal microbial communities of wild fishes.
The black scorpionfish (Scorpaena porcus, Linnaeus, 1758), the small red scorpionfish (Scorpaena notata, Rafinesque 1810) and the red scorpionfish (Scorpaena scrofa, Linnaeus, 1758) are common species, widely distributed in the Mediterranean Sea and represent commercially valuable resources (Harmelin-Vivien et al., 1989). These species have similar habitat preferences (occurring primarily in rocky bottom and seagrass beds) (Ordines et al., 2009; Özgül et al., 2019), foraging behavior (all three species are ambush predators with a sit and wait hunting strategy) (Harmelin-Vivien et al., 1989) and occupy relatively similar trophic levels (S. scrofa = 4.3 ± 0.5, S. notata = 3.7 ± 0.2, S. porcus = 3.9 ± 0.2; source: https://www.fishbase.se/). However, a difference in the relative importance of some specific diet items exploited by the three species exists, with S. scrofa reported to consume fishes more often and in higher quantities that the other two species, which appear to rely mainly on Crustacea (Table 1).
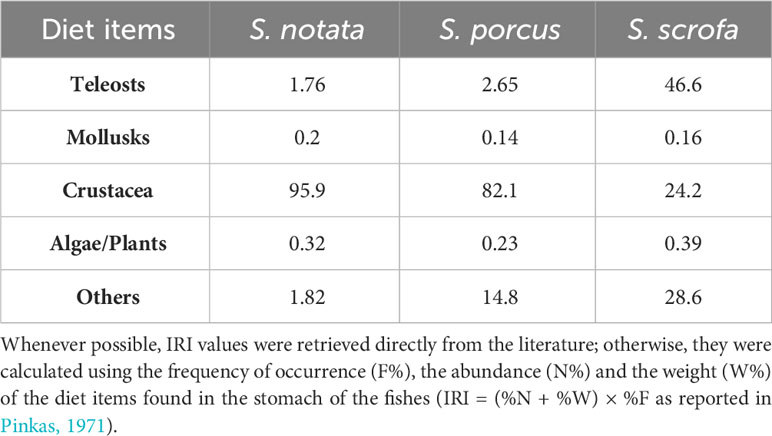
Table 1 Average IRI (Index of Relative Importance) values for the diet items exploited by S. scrofa, S. notata and S. porcus based on data from the literature (Harmelin-Vivien et al., 1989; Bradai and Bouain, 1990; Cabiddu and Pedoni, 2010; Castriota et al., 2012 Shahrani, 2015; Rafrafi-Nouira et al., 2016; Ferri and Matić-Skoko, 2021; Šantić et al., 2021).
This group of species is potentially a good model to investigate phylosymbiosis as they share a recent common ancestor (~49 MYA) (Kumar et al., 2017): in fact, the signal of phylosymbiosis is observed to weaken when the evolutionary history is too extended (inter-clade) and to strengthen when the species investigated share a recent common ancestor (intra-clade) (Groussin et al., 2017). Moreover, the pair-wise phylogenetic distances between these three species are uneven. When considering the 16S mitochondrial gene of Scorpaena species living in the Mediterranean Sea, S. porcus and S. notata form one of the two lineages, while S. scrofa belongs to the other lineage shared with S. elongata and S. maderiensis (Turan et al., 2009). Finally, the gut microbiota of these three species is under studied: to the best of our knowledge, the gut microbial communities of only 12 individuals (4 S. notata, 4 S. porcus and 4 S. scrofa) have been described to date (Ruiz-Rodríguez et al., 2020; Kormas et al., 2022).
In light of this, the purpose of our study was threefold: first, to investigate the effect of host’s phylogeny on the composition and diversity of the gut microbiota of the three species and consider the evidence for phylosymbiosis. Second, to investigate the effect of geographic location on the gut microbiota of scorpionfish in the North-Western Mediterranean, by extending our sampling to different locations and including data retrieved from the literature in this study. Lastly, it aims to enrich the literature about wild fish microbiota by characterizing the poorly described gastrointestinal communities of three species of Mediterranean scorpionfish (Scorpaena notata, Scorpaena porcus and Scorpaena scrofa).
Samples were collected between the 16th of July and the 9th of September 2021. The three geographical regions selected for this study (Figure S1) are all proximal to fully protected marine areas (Réserve marine de Cerbère-Banyuls; Réserve de Carry-Le-Rouet in the marine Park of Côte Bleue; Réserve des Iles-des-Moines and Bruzzi in the marine Park of Bonifacio). They differ in terms of marine habitat types: the first is characterized by a corraligenous biocenosis while the latter two are characterized by a biocenosis of Posidonia oceanica (Figure S2). In addition, the region of Carry-le-Rouet is the most heavily impacted by human activities due to its proximity to the largest harbor in France, the Port of Marseilles-Fos. For simplicity in this study the region of Banyuls-sur-mer will be referred to as “BA”, that of Carry-le-Rouet as “CR” and that of Bonifacio as “BO”.
In BO, specimens of Scorpaena notata (n=11), Scorpaena porcus (n=26) and Scorpaena scrofa (n=10) were collected at a maximum of 50m depth with gillnets by artisanal fishermen. The same fishing method was applied to collect individuals of Scorpaena notata from BA (n=36) and CR (n=33) (Figure S1). Scorpaena notata is the most frequently captured species among the three studied (Harmelin-Vivien et al., 1989) and therefore, it was the only species for which a sufficient number of specimens could be collected from each geographic location. In each geographic location, fishes were caught from a set of specific fishing locations characterized by similar bathymetry and sea bottom (polygon layers in Figure S2). Some fishing locations were treated as a single location for the subsequent analysis because, due to their close proximity, it is likely that the fishing vessel would have passed through them during a single haul (Figure S2; Table S1). The ethical review and approval was not necessary for this study because the fish individuals were collected already dead from the artisanal fishermen.
All 116 individual fish were identified using the meristic characters normally used to distinguish scorpaeniformes (Fischer, 1987), then the total length of each individual (i.e. from the tip of the snout to the tip of the longer lobe of the caudal fin) was recorded. The size range of the collected specimens differed between species and between geographic locations. The mean total length of the specimens collected in BO was 19.7 cm (sd=3.34 cm) for S. porcus, 21.8 cm (sd=4.79 cm) for S. scrofa and 13.1 cm (sd=1.56 cm) for S. notata. The average size of S. notata was 14.7 cm in BA (sd = 3.01 cm) and 12.94 cm in CR (sd = 0.98 cm).
Fishes were kept on ice immediately post capture and dissected within a maximum of three hours from their collection. To avoid contamination of the samples by the skin microbiota, the body surface of the fishes was cleaned with 70% ethanol. Then the middle and posterior portion of the intestine (midgut and hindgut) of each individual were extracted and the intestinal content (digesta) was removed by rinsing with PBS 1x in order to keep only the bacterial community adherent to the intestinal wall: this community is known to be autochthonous and less influenced by the diet and the environment compared to the one living in the intestinal content (Tarnecki et al., 2017). The intestinal wall samples were placed in sterile 50-ml collection tubes and completely immersed in RNAlater (Sigma-Aldrich solutions) up to five times their volume. They were stored at 4°C overnight, then at -20°C for the rest of the field campaign (~55 days maximum) and eventually at -80°C until DNA extraction (November 2021).
Bacterial DNA was extracted from ~350 mg of the intestinal samples (midgut and hindgut). First, the intestinal segment was split longitudinally and one of the two halves was used for the extraction of DNA. The residual RNALater was removed by centrifugation step at 7000g and 4°C. Samples were centrifuged for 15 minutes, then rinsed with sterile PBS 1x and centrifuged again for 15 minutes. DNA was isolated following the procedure for pathogen detection provided by the QIAMP Fast Stool Mini Prep kit (Cat. No. 51604, QIAGEN) with a modification at the initial step of the protocol: the first reagent of the kit was added with 250 mg of 1-mm zirconium beads to the sample and the mix was vortexed horizontally at 2700 rpm for 30 minutes; after the removal of the beads through a quick centrifugation step (1 minute at 1000 g), the samples were processed following the manufacturer’s instructions. Negative controls were included in the DNA extraction procedure to check for cross and reagent contamination. The final DNA purity (A260/A280 and A260/A230) was measured with a UV-vis spectrophotometer Nanodrop (Thermofisher Scientific) and DNA concentration by fluorometry with Qubit (Thermofisher Scientific). DNA was stored at -20°C until library preparation and sequencing.
Generation of sequencing libraries and amplicon sequencing were performed in three runs at StarSEQ GmbH (Mainz, Germany). The V4 region of the 16S rRNA gene was amplified using the primers 515F (5′-GTGYCAGCMGCCGCGGTA-3′) and 806bR (5′-GGACTACNVGGGTWTCTAAT-3′) (Apprill et al., 2015; Parada et al., 2016); the sequencing was performed applying the MiSeq Illumina technology to generate paired-end 300-bp reads. Positive (a defined amount of DNA of bacteria and fungi) and negative controls were included in the sequencing runs by the sequencing company to detect the success of the process and the cross contamination among the samples, respectively.
Only two other studies investigated the gut microbiota of Mediterranean scorpionfishes in the past using the 16S rRNA sequencing technique (Ruiz-Rodríguez et al., 2020; Kormas et al., 2022). The amplicon data they reported were integrated with those obtained in our study to extend our dataset. The study by Kormas et al. (2022) reported data from the mucosal gut microbiota of four Scorpanea porcus and four Scorpanea scrofa sampled in the Greek Island of Gyaros (GY), while the study by Ruiz-Rodríguez et al. (2020) reported that of four individuals of Scorpaena notata sampled in BA. The raw FASTQ files from both studies were downloaded from the NCBI Sequence Read Archive (SRA) using the publicly available accession numbers.
The 16S rRNA gene sequencing data obtained from the three sequencing runs and those retrieved from the literature were processed independently using the dada2 pipeline (Callahan et al., 2016). Due to the drastically lower quality of the reverse reads obtained from the sequencing runs, only the forward reads of the samples collected in this study were processed (as explained in Dacey and Chain, 2021).
The dada2 “filterAndTrim” function was used to: remove the reads with Ns (maxN=0); trim the first 10bp of the raw reads to eliminate the primers sequences (trimLeft = 10); trim the end of the reads at a specific base pair positions where the quality of the majority of the reads dropped under Q=30 (in this case: truncLen = 260bp); filter out the reads belonging to the PhiX bacteriophage(Mukherjee et al., 2015) (rm.phix=TRUE); select only the reads with a number of error equal or lower than 2 (maxEE= 2) and truncate them at the first instance of a quality score equal to or lower than 2 (truncQ=2; the value 2 is used by Illumina as read end quality indicator). After dereplicating, the Amplicon Sequence Variants (ASVs) were inferred by the dada2 algorithm (Divisive Amplicon Denoising Algorithm).
Contaminant reads found in the negative controls of the sequencing runs were removed from the ASVs tables.
The ASVs tables obtained (three tables from this study, one table for the reads from Kormas et al. (2022) and one for Ruiz-Rodríguez et al. (2020) were merged together (using the dada2 mergeSequenceTables() function) before assigning the taxonomy that was performed with the naive Bayesian RDP classifier implemented in dada2 by using the SILVA rRNA database (release v138) (Quast et al., 2013). Each ASV was assigned at the taxonomical level of Kingdom, Phylum, Class, Order, Family and Genus. A maximum likelihood tree was generated using the DECIPHER package in R (Wright et al., 2012) and FastTree (Price et al., 2009).
A few following filtration steps were performed on the ASVs table: the ASVs assigned to Mitochondria, Chloroplasts and Archaea and those unclassified at the Class level were removed from the dataset; in addition, the samples with less than 10,000 reads were removed from the dataset. Among all the samples, three out of the four BA samples of S. notata from the study by Ruiz-Rodríguez et al. (2020) were excluded from further analyses due to their low number of reads.
The final dataset (Extended-dataset) comprised 125 samples belonging to S. porcus, S. notata and S. scrofa from four different locations (BA; CR; BO; GY) and three different studies (this study, Kormas et al., 2022 and Ruiz-Rodríguez et al., 2020). Before starting to analyze the bacterial communities of the scorpionfish, two additional adjustments were made on the Extended-dataset. First, it was used to generate a reduced dataset (Species-dataset) which comprised only the samples belonging to the three different species of scorpionfish collected in Corsica (BO) (N=47 samples): this Species-dataset was created for investigating the effect of host’s phylogeny on the gut microbiota of scorpionfishes at a regional scale. Secondly, all the unique sequences (ASVs) present in the Extended-dataset were agglomerated at the Genus level to override the differences among the three studies in: the section of the intestine analyzed (midgut and hindgut in this study and Ruiz-Rodríguez et al., 2020, and only midgut in Kormas et al., 2022), the DNA extraction method implemented, and the region of the 16S rRNA gene amplified (V4 region for this study and Ruiz-Rodríguez et al., 2020; V3-V4 for the study by Kormas et al., 2022). The Extended-dataset was then used to generalize the effect of host’s species on a broader geographical scale, and to determine the effect of geographic location on the gut microbiota of the Mediterranean scorpionfishes. All statistical analyses were implemented in R studio (R Core Team, 2021) on the two datasets in parallel.
The two datasets were normalized by rarefaction: the Species-dataset was rarefied to 20,000 reads per sample, and the Extended-dataset was rarefied to a lower number of reads per sample (18,025) to avoid the removal of one of the four S. porcus samples from GY.
The alpha diversity of the bacterial communities was assessed in the rarefied datasets using the total number of ASVs (or genera) observed in the samples (Observed index), the evenness and richness of ASVs (or genera) (Shannon’s diversity index), and the phylogenetic diversity of ASVs (or genera) (Faith’s diversity index).
In order to determine the level of variation in bacterial communities diversity across Scorpaena species and geographic locations, a one-way ANOVA (or Kruskal-Wallis test, if the requirement of normal distribution and/or homogeneity of variance of the data were not met) was performed on the alpha diversity indices; in case of a significant outcome, the Tukey’s HSD test (or the non-parametric Dunn’s test) was then performed for multiple pairwise post-hoc comparisons among the groups (Scorpaena species or geographic locations). Spearman’s rank correlations were calculated to examine the relationship between fish size (i.e. total length) and the alpha diversity indices.
A compositional approach was implemented as suggested in Gloor et al., 2017 to calculate the inter-individual dissimilarities in terms of gut microbiota composition (beta diversity): the zeros in the unrarefied ASV tables were replaced with near-zero counts by the cmultRepl() function of the R package “zComposition” through the Bayesian multiplicative treatment (Palarea-Albaladejo and Martín-Fernández, 2015). Then, the ASV tables were transformed by the centered log-ratio transformation (clr) and pairwise dissimilarities were calculated among the Scorpaena individuals using the Aitchinson’s distance (Aitchison et al., 2000).
To test whether the inter-individual bacterial dissimilarity was greater between the groups (i.e. the three Scorpaena species and the different geographic locations) than within them, the PERMDISP test (Anderson, 2001) through the betadispers() function from the vegan package in R (Oksanen et al., 2013) was performed. In case of absence of homogeneity of multivariate variances (i.e. significant PERMDISP test) or unbalanced sample size, the multivariate Welch MANOVA (Wd Test) (Hamidi et al., 2019) – performed by the MicEco package in R (Russel, 2020) – was used to test the effect of variables (i.e. host’s species and geographic locations) on the bacterial composition. Otherwise, the permutational multivariate analysis of variance (PERMANOVA) (Anderson, 2014) was used. In case of significant outcome, the pairwise.adonis() function from the pairwiseAdonis package in R (Martinez Arbizu, 2020) was applied to perform a multilevel pairwise comparison of the composition of the bacterial community in each category (i.e. the host species and the geographic locations).
Because the samples for this study were collected at different locations along the coast in BA, CR and BO, the effect of fishing location on the composition of bacterial community was investigated for the two datasets. Additionally, given the size range displayed by the samples collected in this study, the effect of fish size on the gut microbiota composition was also tested for both datasets. The specimens not collected for this study were excluded from these additional analyses.
Due to the difference in sample size both in the Species-dataset (S. notata =11, S. scrofa = 10 and S. porcus= 26) and in the Extended-dataset (BA = 37, CR = 33, BO = 11, GY=8) the above mentioned statistical analyses were repeated on sub-datasets created by the random combination of samples evenly distributed across the categories analyzed (10 samples for each Species, 11 samples for each geographic location of S. notata and four samples of S. porcus and of S. scrofa for each geographic location). The statistical analyses on alpha (one-way ANOVA or Kruskal-Wallis test) and beta (PERMANOVA or Welch MANOVA) diversity indexes were performed on 1000 different sub-datasets, and the results were compared with those obtained using the full datasets.
Finally, to highlight which specific taxa (ASVs or genera) were significantly different in the gut microbiome of the three species in BO and of each species in the different geographic locations, the microbiome differential abundance method DESeq2 (Differential Expression analysis for Sequence count data) (Love et al., 2014) was implemented. In order to confirm the taxa detected by DESeq2, ANCOM II (Mandal et al., 2015) was implemented as suggested in Nearing et al. (2022). Although having less statistical power, this second method is more conservative and has a lower false discovery rate (FDR) than DESeq2; the taxa unanimously detected by both methods were eventually selected as those differentiating the different fish species and geographic locations.
Phylogenetic information about the three scorpionfish species were gathered from the study by Turan et al., 2009 which compared the mitochondrial 16S rDNA sequence across different species of Scorpaena in the Mediterranean sea. Total nucleotide distances for S. notata, S. porcus and S. scrofa were used to generate a genetic dissimilarity matrix of the individuals collected in BO only.
The Mantel test from the vegan package in R (Oksanen et al., 2013) was applied to detect the presence of a positive or negative relationship between the host phylogenetic dissimilarity matrix and the bacterial dissimilarity matrix generated by the Aitchinson’s distance on the Species-dataset. This is recognized as the most appropriate test for the detection of phylosymbiosis (Lim and Bordenstein, 2020).
The core microbiota is defined as the set of bacterial taxa that are characteristic of a host’s species or an environment (Neu et al., 2021). The frequency of appearance of a taxon in a sample pool (i.e. occurrence) and its relative abundance in the bacterial community were combined to identify the core microbiota of Mediterranean scorpionfish. Using the Species-dataset, the core microbiota was identified in order to determine which bacterial taxa were common and unique to each scorpionfish species. In addition, the core microbiota of S. notata, S. porcus and S. scrofa were also identified using the Extended-dataset because the definition of the symbiotic core microbiota of a host species is considered more accurate when analyzed across a spatial gradient (Neu et al., 2021). These core bacterial taxa, found for each species in the different geographic locations, were then combined to detect those common in the genus Scorpaena and those unique to each species across the Mediterranean Sea.
The identification of the core microbiota was performed using the core_members() function of the R package microbiome (Lahti, 2023) on the rarefied datasets agglomerated at the Genus level. For each Scorpaena species, the bacterial genera occurring in at least 75% of individuals and displaying a relative abundance in the community higher than 0.01% were considered members of the core microbiota.
In order to understand the contribution of the core members to the whole bacterial community, the average relative abundance of the core taxa was calculated across the samples of the same group (i.e. host’s species or geographic locations); finally, the taxa shared by all groups (i.e. host’s species or geographic locations) included in the analysis were labelled as “Shared by all”, those shared by two out of the three groups of the analyses as “Shared”, and those unique to every group as “Unique”, all the rest was labelled as “Not core” and its relative abundance was also calculated.
The three Illumina MiSeq sequencing runs of the 16S rRNA gene (V4 region) produced together 9,375,424 uncontaminated reads with a median number of reads per sample equal to 68,840. The dada2 pipeline generated a total of 14,188 ASVs that were merged with the additional 546 ASVs obtained for the data retrieved from the literature. Then, 18.3% of the ASVs were removed by the filtration steps resulting in a final dataset of 12,042 ASVs (included in 1,202 different bacterial Genera). Three S. notata samples from the study by Ruiz-Rodríguez et al. (2020) were discarded after filtering to a minimum of 10,000 reads (Figure S3) per sample. Three additional samples (one individual of S. porcus collected in BO and two individuals of S. notata collected in BA and in CR) were also removed from the Species-dataset after rarefying to 20,000 reads.
The fish specimens included in this study were observed to vary in their total length (see Methods section). Among the three species collected in BO, S. notata displayed significantly lower sizes compared to the other two species (TukeyHSD test, P-value < 0.0001 for both comparisons). The individuals of S.notata collected in BA were significantly bigger than those collected in CR and BO (Dunn’s test: P-value = 0.001 between BA and BO; P-value < 0.0001 between BA and CR).
In light of this, the relationship between the fishes’ size and the alpha diversity of their gut microbiota was investigated prior to checking for the effect of other factors: a not significant Spearman’s correlation of Shannon (Figures S4 and S5) and Faith indices and total number of Observed ASVs was found (data available upon request).
The inter-individual dissimilarity of the gut microbiota composition (beta diversity, Figure 1A) was calculated by the Aitchinson’s distance on the clr transformed ASVs counts. The dissimilarity of the bacterial gut communities of the three Scorpaena species distributed homogenously (PERMDISP test = 0.12). However, the uneven samples size of the three fish categories in the Species-dataset justified the application of the Welch MANOVA test. This analysis revealed the influence of host species on the gut microbiota composition in Scorpaena fishes (WdTest; W*d statistic = 1.29, P value = 0.006); however, the W*d statistics reported by the test suggests that other variables should be taken into account to fully understand the reason behind this difference. Therefore, we tested for the effect of the size (i.e. total length) of the fish but this did not help to explain better the variability of the gut microbiota composition (PERMANOVA on the total length of the fishes of each species: S. notata, P-value = 0.68; S. porcus, P-value = 0.91; S. scrofa, P-value = 0.89). On the contrary, a weak effect of fishing location on the gut microbiota composition of S. porcus was observed (WdTest; W*d statistic = 1.19, P -value = 0.003).
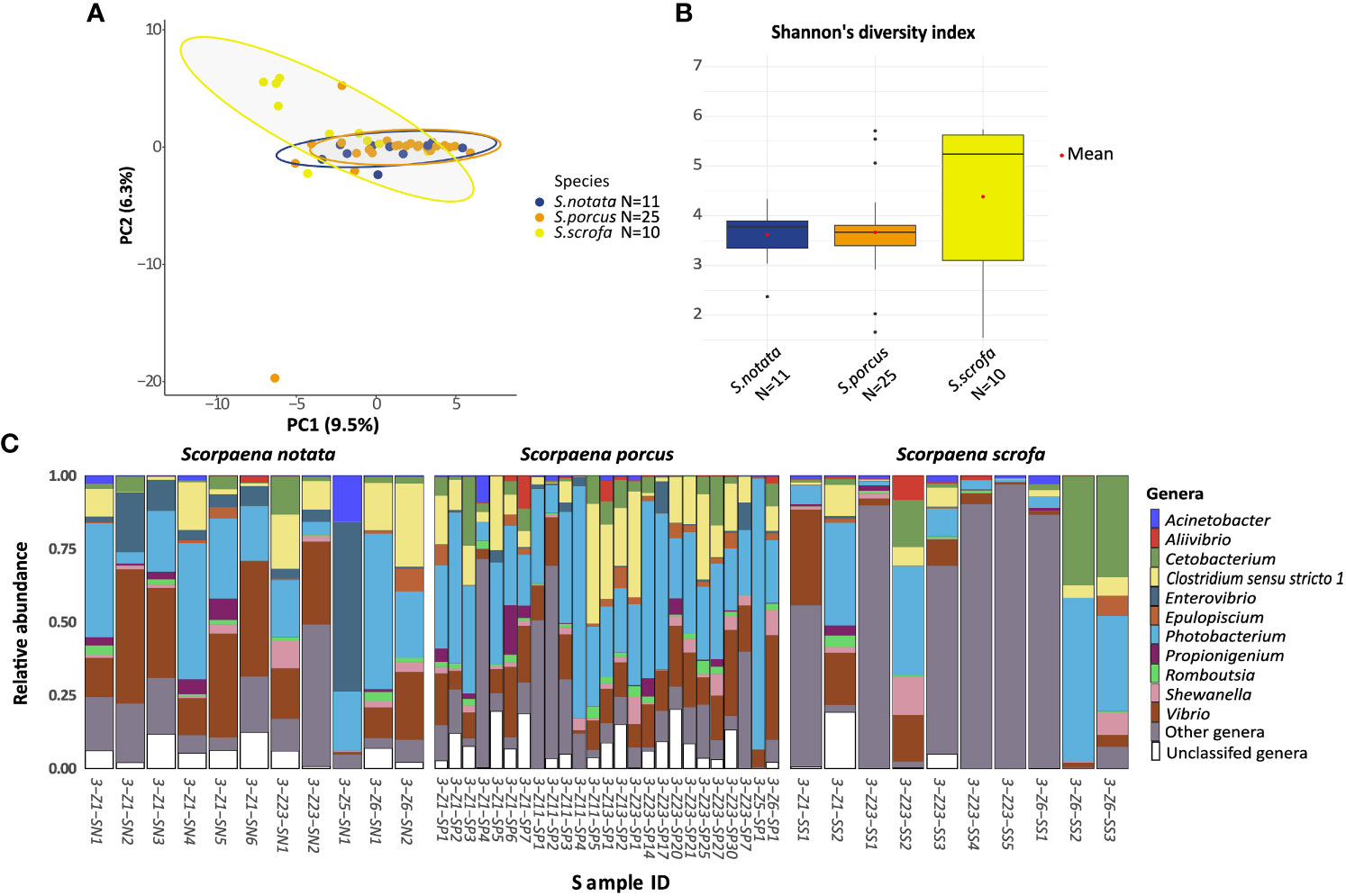
Figure 1 Analysis of Alpha and beta bacterial diversity in the Species-dataset. The analyses were performed at the ASV level. (A) Principal Component Analysis ordination representing the gut bacterial communities of specimens from the three species of Scorpaena collected in BO. The distances were calculated using the Aitchinson’s distance. (B) Median and variation in Shannon alpha diversity index of the bacterial gut communities of the three species in BO. Differences in the Shannon index of the gut bacterial communities of the three species were not significant according to the Kruskal-Wallis test (P-value =0.45); (C) barplot of the 11 most abundant Genera in the three fish species samples from BO. “Other genera” represents ASVs classified at the genus level but not belonging to the top 11 genera; “Unclassified genera” represents all the ASVs that could not be classified at the Genus level. Sample IDs include the geographic location (3=Bonifacio) (see Table S1), the Fishing location (e.g. Z1), the species (i.e. SN, S. notata; SP, S. porcus; SS, S. scrofa) and the specimen number.
The pairwise analysis on the three host species revealed that only one species was characterized by a gut microbiota composition significantly different from the other two: Scorpaena scrofa (pairwiseAdonis; P-value = 0.02 for the comparison with S. porcus; P-value = 0.003 for the comparison with S. notata; Figure 1A). The gut bacterial communities belonging to this species seemed to be the most diverse ones in terms of richness and evenness (Shannon alpha diversity index, Figure 1B), phylogenetic distance of the ASVs (Faith’s index) and total number of ASVs observed in the community (Figure S6). However, the greater alpha bacterial diversity in S. scrofa could not be confirmed statistically (Kruskal-Wallis test on the three indexes: P-value > 0.05; see Figures 1 and S6 caption for exact P-values).
DESeq2 and ANCOM II were then applied to determine which bacterial genera significantly differed in the gut microbiota of the three scorpionfish species. Only two taxa were found by this analysis (DESeq2 and ANCOM II: adj. P-value < 0.05): the genus Enterovibrio, which was more abundant in the gut microbiota of S. notata (10.2% of the gut bacterial community) and S. porcus (1.5%) than in the one of S. scrofa (0.1%) and Aliivibrio which was more abundant in the gut microbiota of. S. porcus (where it accounted for 1.1% of the gut bacterial community) (Figure 1C).
A positive correlation (Mantel test. r = 0.26, P-value = 0.009) was found between the dissimilarity matrix generated using the total genetic distances of the mitochondrial 16S rDNA and the bacterial dissimilarity matrix produced by the application of the Aitchinson’s distance on the clr transformed ASVs counts. When comparing pairs of individuals of the three species, a weak but positive correlation between the two matrices was visible (Figure 2); however, the pairwise bacterial dissimilarities values dispersed largely along the y-axis, confirming again that the host species is not the only factor explaining the variability in the gut microbiota composition.
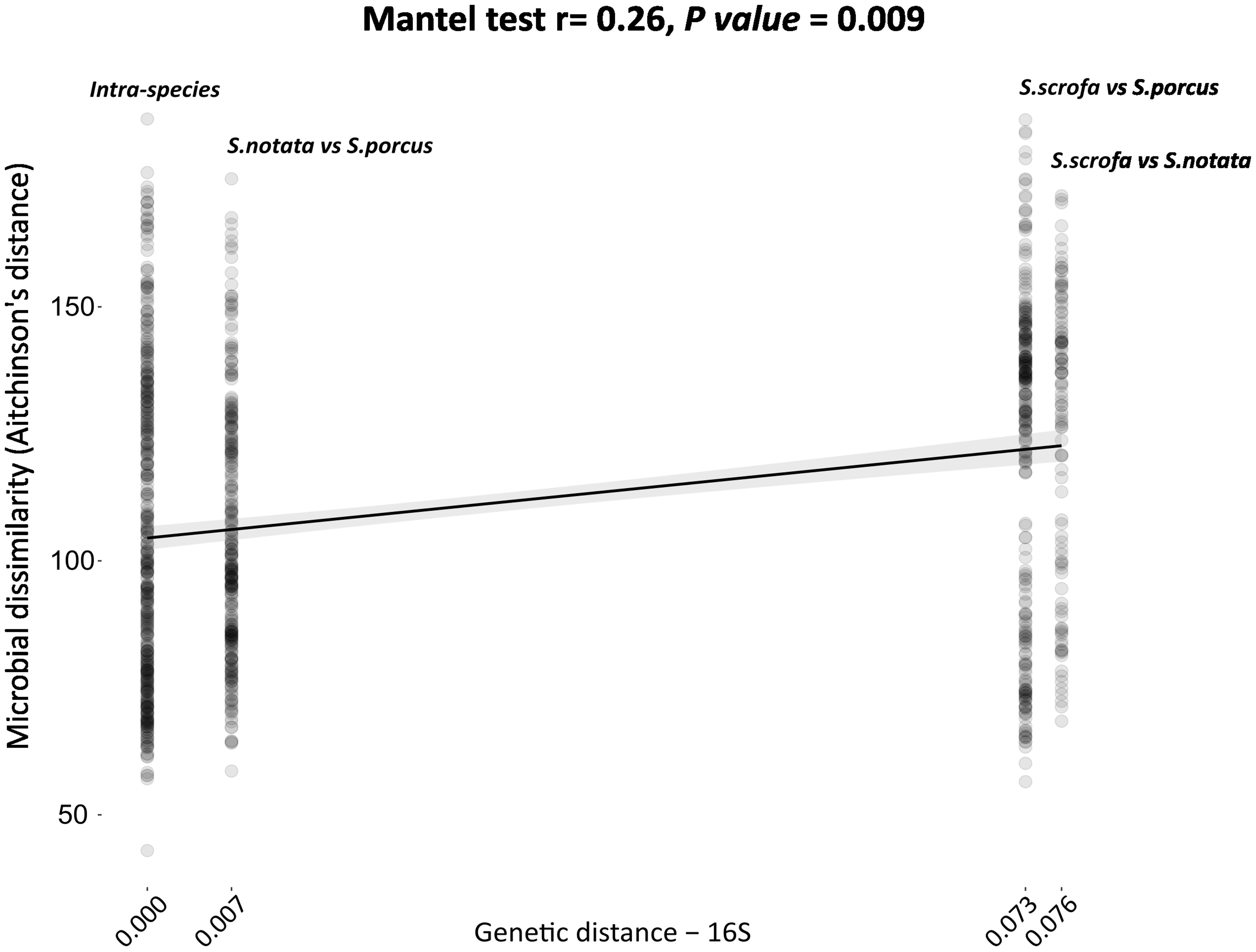
Figure 2 Phylogenetic distance of the three species of the genus Scorpaena (x-axis) compared to the inter-individual bacterial dissimilarity (beta-diversity) calculated by the application of the Aitchinson’s distances (y-axis). Each point in the plot represents a pair of individuals. The comparison was performed using the Mantel test. Genetic divergence among the three species was based on the total genetic distances reported in Turan et al., 2009 (Turan, Gunduz, and Yaglioglu, 2009) for the 16S rRNA mitochondrial gene.
All the individuals collected for this study in BA, CR and BO (n=116 samples) and those already investigated in the literature (n=9: 1 S. notata specimen from BA, 4 S. porcus and 4 S. scrofa from GY) were included in the Extended-dataset (n=125). This dataset was used to generalize the effect of host species through a broader geographical range and to investigate the variation of the gut microbiota of the three Scorpaena species across the Mediterranean sea. The ASVs in this dataset were agglomerated to the Genus level prior to performing the diversity analyses (see Methods section).
In contrast to the observation in BO (at the ASVs and Genus level, respectively in Figures 1B and S7A), a significant effect of the host species on the levels of richness and evenness (Shannon’s index), phylogenetic diversity (Faith’s index) and total number of observed genera of the gut microbiota was found when testing the Extended-dataset (Kruskal-Wallis: Shannon’s index, P-value = 0.007; Faith’s index, P-value = 0.01; Observed ASVs, P-value = 0.03). The gut microbiota of Scorpaena scrofa had a significantly higher median Shannon’s index among the three species and it was significantly different from that of S. notata (Dunn’s test; P-value = 0.01) (Figure S7B).
By analyzing the alpha diversity of the bacterial communities of each species across a geographical range, a significant difference was observed among the individuals of S. notata collected in BA, CR and BO (Figure 3) (Kruskal-Wallis: Shannon’s index, P-value = 0.001; Faith’s and Observed ASVs, P-value < 0.001). More specifically, the specimens from BA showed lower levels of the alpha diversity indexes than those collected in CR (Dunn’s test: Shannon’s, P-values = 0.003; Faith’s and Observed ASVs, P-value < 0.001) and in BO (Dunn’s test: Shannon’s, P-values = 0.02; Faith’s and Observed ASVs, P-value < 0.001). Differently, geographic location had no significant effect on the alpha diversity values of the S. porcus and S. scrofa microbiota (Figure 3. Mann-Whitney test for S. porcus: Shannon’s, P-values = 0.3; Faith’s, P-values = 0.051 and Observed ASVs, P-value = 0.07; Mann-Whitney test for S. scrofa: P-value > 0.1 for the three indexes).
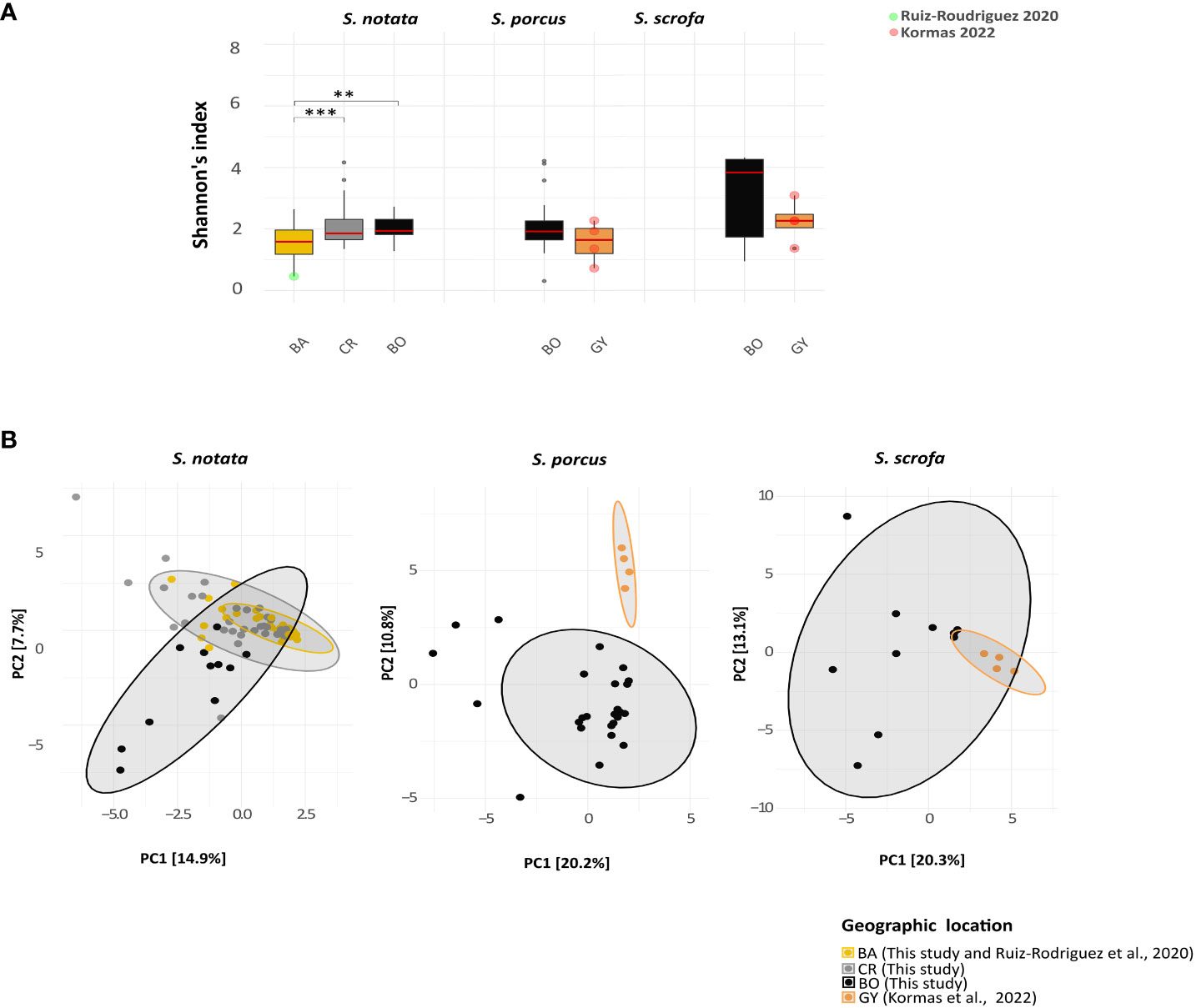
Figure 3 Analysis of Alpha and beta bacterial diversity in the Extended-dataset. The analyses were performed at the genus level. (A) Boxplots representing the Shannon alpha diversity index compared among the four different geographic locations. Only significant comparisons (according to the Dunn’s test for notata and the Mann-Whitny test for porcus and scrofa) are labeled: ** P-value < 0.05; *** P-value < 0.01 ). (B) Principal Component Analysis ordination representing the gut bacterial communities of the individuals of the three Scorpaena species from the four different geographic locations. The distances were calculated using the Aitchinson’s distance.
Due to the effect of geographic location on the alpha diversity observed for the individuals of S. notata, it was not possible to confirm the effect of host species on the alpha diversity of the gut microbiota in the extended dataset (Figure S7B). In fact, to disentangle the effect of host species from that of geographic location on the alpha diversity, similar values of alpha diversity should have been observed among the individuals of each species from the different geographic locations.
The composition of the bacterial communities (beta diversity, Aitchinson’s distance, Figure 3) differed more between the samples from the same species collected in the different locations of the Mediterranean (WdTest: S. notata, W*d statistic = 3.20, P-value = 0.001; S. porcus, W*d statistic = 5.43, P-value = 0.001; S. scrofa, W*d statistic = 3.33, P-value = 0.003) than between the different species in each location (WdTest: BO, P-value = 0.07; PERMANOVA: GY, P-value = 0.19). Among the samples of S. notata collected from the three geographic locations, BO samples separated from CR and BA samples in the PC ordination space (Figure 3). A pairwise analysis was performed to highlight whether the gut microbiota composition of the former location significantly differed from that of the latter ones. Significant differences were actually observed for every pair of geographic locations tested (pairwiseAdonis; Bonferroni adjusted P-value = 0.003) implying a general effect of this factor.
Although the individuals of S. notata collected from BA in this study displayed significantly larger sizes than those from CR and BO (as reported in the methods section), this factor did not explain the inter-individual dissimilarity of the bacterial communities (PERMANOVA, P-value = 0.57).
Additionally, the location where the fishes were collected (fishing locations) explained the variance among the composition of the bacterial communities for the individuals of S. notata from BA (WdTest: BA, W*d statistic = 0.98, P-value = 0.04); and for the individuals of S. porcus in BO (WdTest, W*d statistic = 1.43, P-value = 0.001). However, the pairwise comparisons performed to detect which fishing locations significantly differed in BA and BO for the two species respectively, revealed that this effect was not generalizable. In fact, the significancy reported by the Welch MANOVA for S. notata in BA and for S. porcus in BO was driven by the dissimilarity among the samples collected only from two fishing locations (S. notata, pairwiseAdonis between fishing location 4 and 11 in BA: Bonferroni adjusted P-value = 0.03; S.porcus, pairwiseAdonis between fishing location 2-3 and 11 in BO: Bonferroni adjusted P-value = 0.02).
DeSEQ2 and ANCOM II were implemented on each species from the Extended-dataset parallelly to determine the bacterial genera mainly responsible for the variation in the gut microbiota composition across the geographical range. The relative abundance of 12 genera was found to be significantly different among the specimens of S. notata collected in BA, CR and BO (Figure S8). BA and BO displayed the largest number of differently abundant genera (8). The genus Acinetobacter was more abundant in the fish collected in BO and CR than in BA; on the other hand fish from BA were enriched in the genera Moritella and Catenococcus. However, these discriminant taxa were generally not highly abundant in the communities (mean rel. abund. = 1% for Acinetobacter in the individuals of CR and BO, and 0.1% for Moritella and 0.5% for Catenococcus in the individuals from BA).
A few bacterial genera were also detected to be differently abundant in the individuals of S. porcus and S. scrofa from BO and from GY (Figure 4). The gut microbiota of the scorpionfishes from GY were indeed characterized by a higher abundance of Diaphorobacter, Cutibacterium, Pelomonas and Cloacibacterium. Differently, in BO, the genera Aliivibrio and Enterovibrio were found to be more abundant in S. porcus and Photobacterium in S. scrofa. Some of these taxa accounted for a large portion of the whole bacterial community: for example, the two genera Diaphorobacter and Cutibacterium made up to 19% of the whole gut bacterial community of the individuals of S. scrofa from the GY while Photobacterium – alone – represented 18.6% of the gut bacterial community of those from BO.
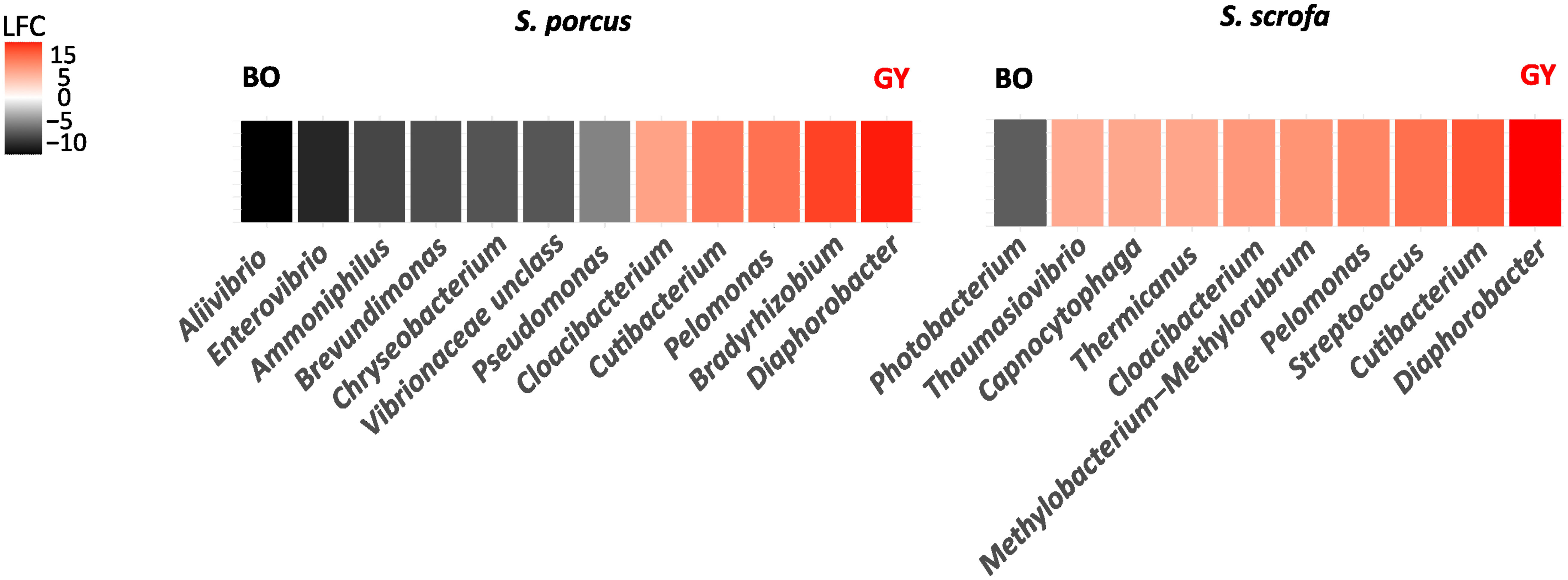
Figure 4 Bacterial genera differently abundant in the gut microbiota of S. porcus and S. scrofa collected for this study in Bonifacio (BO) and for the study of Kormas et al. (2022) in the island of Gyaros (GY). DeSEQ2 and ANCOM II were implemented to determine the bacterial genera whose abundance was significantly different between the individuals from BO and GY (adj.P-value < 0.05). A negative/positive change on a Log2Fold scale (LFC) indicates that the taxa are more abundant in the reference/compared group, respectively.
The different sample size among the three species investigated appeared to not influence the outcome of the statistical tests performed to assess the effect of the host species or of the geographic location. Indeed, the test repeated on the 1000 different sub-datasets displaying even sample size across the three phylogenetic and geographical categories provided results that confirmed those obtained using the full dataset.
In BO, S. scrofa displayed a smaller number of total core genera (n =7) than the other two species (n=20 for S. notata and n=17 for S. porcus) (Figure 5). In addition, the relative abundance of the core microbiota was significantly lower in S. scrofa (45.3%) than in S. notata (90.3%) and in S. porcus (85.2%) (Dunn’s test, P-value = 0.01 for both comparisons) (Figure 5 and Table S2).
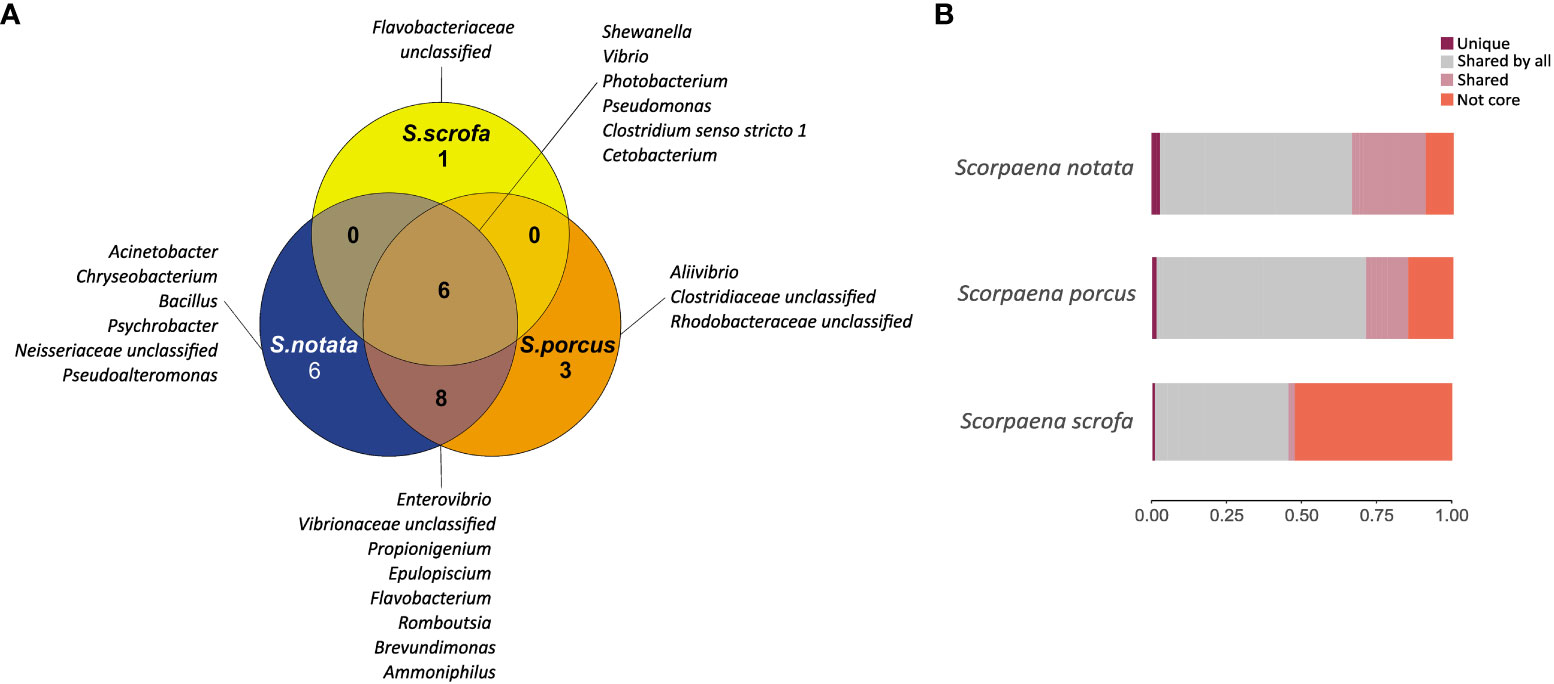
Figure 5 (A) Venn diagram reporting the number and the name of the genera shared and unique in the three species of Scorpaena in BO; (B) Barplot representing the relative abundances of the bacterial genera found in the gut microbiota of the three scorpionfish species in BO: relative abundance of the genera found uniquely in the species (dark red), of the genera shared among the three species (grey), of the genera shared between two of the three species (pink); and relative abundance of all the other bacterial genera not included in the core microbiota (orange).
A small number of core genera were unique to each fish species: Acinetobacter, Chryseobacterium, Bacillus, Psychrobacter, Pseudoalteromonas and one unclassified genus belonging to the Neisseriaceae family were found solely in the core microbiota of S. notata and represented 3.4% of the whole gut bacterial community (Figure 5 and Table S2); likewise Aliivibrio and two bacterial genera belonging to the family of Clostridiaceae and Rhodobacteraceae were found only in the core microbiota of S. porcus (1.7% of the whole community) while one genera included in the Flavobacteriaceae family was found only in the one of S. scrofa (0.7% of the whole community).
In contrast, six bacterial genera belonging to the Classes of Gammaproteobacteria, Clostridia and Fusobacteria were common among the three species. The most abundant core genus shared by the three species was Photobacterium (25.1% of the community in S. notata, 33.8% in S. porcus and 18.6% in S. scrofa; Table S2) followed by Shewanella, Vibrio, Pseudomonas, Clostridium sensu stricto 1, Cetobacterium. Interestingly Cetobacterium was the second more abundant core taxon in the gut microbiota of S. scrofa (9.5% of the whole community), while it was less represented in the bacterial communities of the other two species (4.7% in S. porcus and 2.8% in S. notata) (Table S2).
To better describe the core microbiota of the three Scorpaena spp., each species was analyzed in the different geographic locations separately. Then, the bacterial genera commonly found for each species across the Mediterranean Sea were selected as those characterizing the core microbiota. After expanding the analysis to a broader geographical range, the number of core bacterial genera characterizing each of the three Scorpaena species appeared to be drastically reduced (Figure 6 compared to Figures 5A and S9). In addition, all the bacterial genera that were found to be exclusive of each Scorpaena species in BO (Figure 5) were not recorded in the core microbiota by the extended geographic analysis anymore (Figure 6).
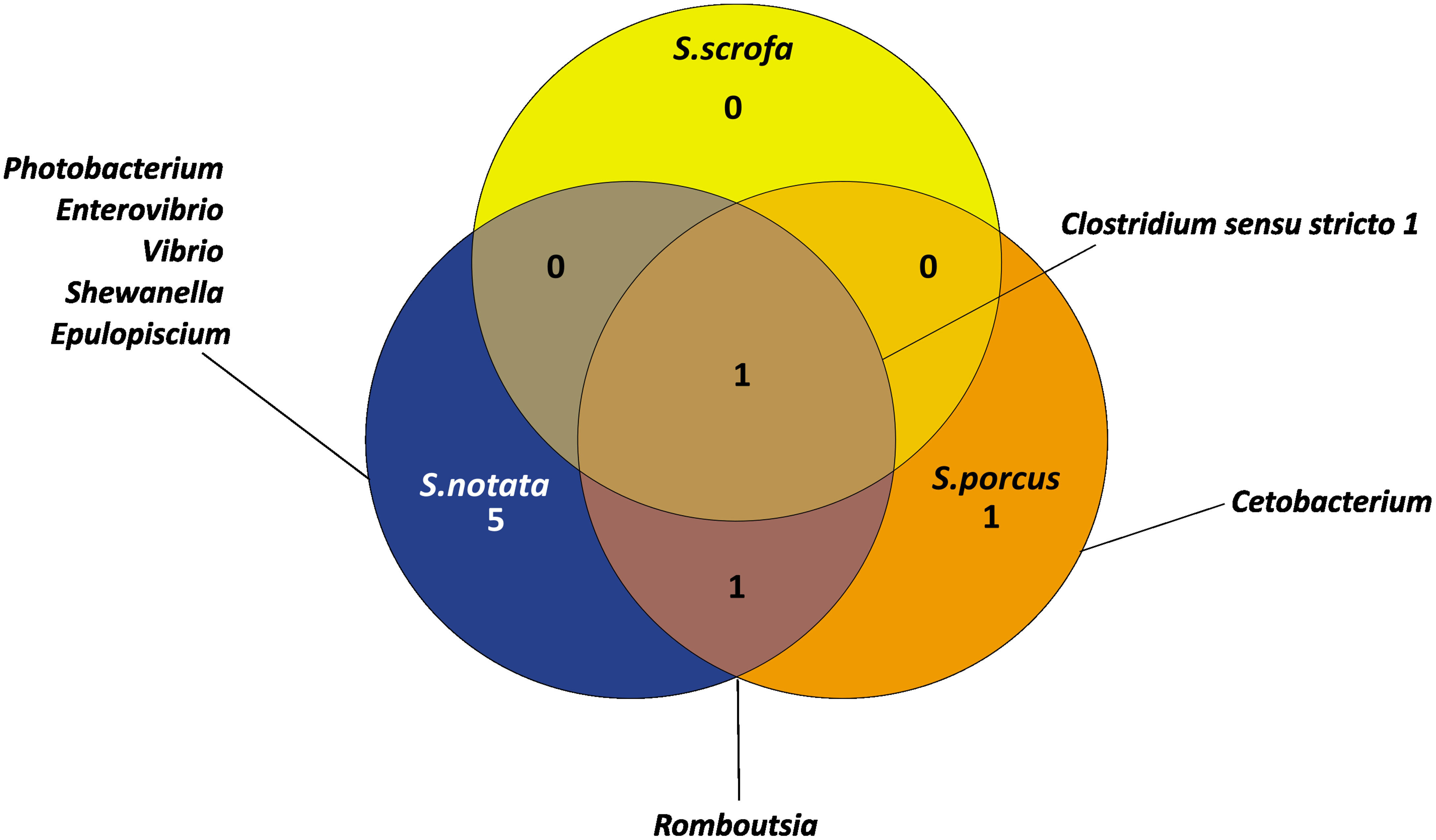
Figure 6 Venn diagram representing the number and the name of bacterial genera shared and unique for the three species of Scorpaena in the Mediterranean Sea. The core microbiota of each species analyzed separately in the different geographical locations (BA, CR, BO, GY) was integrated to obtain the unique and shared bacterial genera in Scorpaena spp.
The core microbiota of S. notata across the Mediterranean Sea was characterized by seven bacterial genera (Figure 6): Photobacterium, Enterovibrio, Vibrio, Shewanella, Epulopiscium, Clostridium sensu stricto 1 and Rombutsia. It represented a great proportion of the total microbiota: 80.7% of the whole bacterial community of the individuals from BA, for 72.8% of the BO ones and for 59.5% of the CR ones (Table S3).
The core microbiota of S. porcus across the Mediterranean Sea included only three genera: Clostridium sensu stricto 1, Cetobacterium and Rombutsia (Figure 6). All together they represented 52.5% of the whole bacteria community of the samples from GY and 19.3% of those from BO (Table S4). Finally, the core microbiota of S. scrofa was reduced only to Clostridium sensu stricto 1 (Figure 6). This genus accounted for 3.8% and 2.3% of the whole bacterial community in the samples collected in BO and in the GY island, respectively (Table S5).
The majority of the genera recorded as the core microbiota of S. notata by the extended analysis were previously identified as genera characterizing the whole Scorpaena genus when the investigation was limited to BO (Figure 5); eventually, the only bacterial genus found to characterize the Scorpaena genus across the Mediterranean Sea was Clostridium sensu stricto 1.
In this study the bacterial communities associated with the mucosa of the mid and lower intestinal tract of three species of commercially important scorpionfishes (Scorpaena scrofa, Scorpaena notata and Scorpaena porcus) were investigated. By using a comprehensive comparative dataset including 16S rRNA gene amplicon data from four different locations in the Mediterranean Sea (n=125), it was possible to describe an important influence of the geographic location on the composition of the gut microbiota of the three species. In addition, the presence of a distinct microbiota in the individuals of S. scrofa from that of the other two species highlighted the existence of a relation between the composition of the gut microbiota and the phylogenetic relation between the hosts (i.e. phylosymbiosis). Finally, this study is the first to define the bacterial taxa that characterize the core microbiota of Mediterranean scorpionfishes across a broad geographical range.
Genetic diversity of the host has been defined as one of the most influential factors shaping the structure and composition of the gut bacterial communities (Huang et al., 2020). However, relatively few studies have investigated the variation of the gut microbiota among species belonging to the same fish genus and sharing very similar habitat, diet and behavior. When analyzed at the ASVs level in this study, the composition of the gut bacterial communities was observed to vary depending on the host species, with the gut bacterial community of S. scrofa differing from the other two species (Figure 1A). However, the low amount of variance explained by this variable and the absence of a significant effect of the size of the fish entails that other factors, not included in the analyses, may drive this diversity. A large inter-individual variability was found for each of the three species: the effect of genotype (individual effect) on the microbiota associated with the gut mucosa has, indeed, already been reported for other fishes (Tarnecki et al., 2017). However, additional life history information (i.e. age, sex) could be included in future analyses to have a full overview of the causes leading to the changes in the composition of the autochthonous gut bacterial community of Scorpaena spp.
The presence of different enterotypes was also investigated looking at the phylogenetic distances in the Scorpaena genus. A positive correlation was found between the bacterial community dissimilarity among the three species and their phylogenetic distance based on the 16S rDNA (Figure 2) (Turan et al., 2009). Of course, the robustness of this results could be further strengthened by the inclusion of additional species belonging to the Scorpaena genus and Scorpaenidae family, however the presence of phylosymbiosis in this fish genus can already be discussed. As reported in Lim and Bordenstein (2020) phylosymbiosis can be caused by deterministic and/or stochastic forces: among the former, there is the possibility that bacterial organisms colonize hosts with a specific background or traits (e.g gut pH, gut morphology). Differently, stochastic forces that drive phylosymbiosis of the gut mucosa refer to random events such the ecological drift in bacterial communities, the change in host’s geographical range and the ecological and dietary niche variation among hosts. Niche variation across the host lineages seems to be the determinant that, most likely, explains the presence of phylosymbiosis in the Scorpaena genus. The three species included in this study are closely related, they occupy the same type of habitat and rely mostly on the same resources and for this reason the interspecific competition is elevated; however, although their fundamental niches are highly overlapping, the width of the realized niches is reduced at night when the prey diversity is at its maximum allowing the coexistence of Scorpaena spp. feeding on different preys belonging to the same groups (crustaceans, chiefly prawns and brachyurans) (Harmelin-Vivien et al., 1989). Besides, S. scrofa exploits more frequently higher quantities of fish in the diet (Table 1) (Harmelin-Vivien et al., 1989; Bradai and Bouain, 1990; Cabiddu and Pedoni, 2010; Castriota et al., 2012; Shahrani, 2015; Rafrafi-Nouira et al., 2016; Šantić et al., 2021; Ferri and Matić-Skoko, 2021). Therefore, a different exposition to prey-associated bacterial communities, due to the variation in the dietary niche, may explain the development of distinct bacterial communities in the three Scorpaena species. Above all, the genus Enterovibrio appeared to be the main taxon differently represented in the gut bacterial community of the three Scorpaena species in BO (Figure 1C). Indeed Enterovibrio was underrepresented in S. scrofa while it displayed the highest relative abundance in S. notata. This genus is found mainly in the gut of herbivorous species (Egerton et al., 2018) but it is also reported to produce chitinase (therefore essential to digest crustaceans) (Ray et al., 2012).
Generally speaking, the composition of the fish microbiota associated with different body niches is known to be correlated with the phylogenetic distance between host species (Chiarello et al., 2018; Minich et al., 2022; Sylvain et al., 2022). Nevertheless, phylosymbiosis has been mostly observed between fish species belonging to different genera or families. In contrast, the results reported in this study highlight the existence of phylosymbiosis even for species within the same fish genus. Future studies should investigate whether this eco-evolutionary pattern is also observed for the microbial communities associated with the external body surfaces of scorpionfishes, such as the gills and skin. The bacterial communities inhabiting these body niches are known to be influenced by host genetics (Wang et al., 2010; Berggren et al., 2022), so the existence of a relationship with the evolutionary history of the hosts is foreseeable. However, it is important to consider that both environmental conditions and the composition of the bacterioplankton communities have a greater influence on the microbiota associated with the skin and the gills than on that associated with the intestinal mucosa (Minich et al., 2020; Sylvain et al., 2020). Consequently, future studies exploring the relationship between the host’s phylogeny and the skin and gills microbiota composition should be designed specifically to minimize the effect of these external variables.
The Mediterranean Sea hosts a wide variety of habitats, resulting in a great variability in the abundance, the biomass and the biodiversity of fish communities (Coll et al., 2010) and in fish’s body conditions (Ordines et al., 2009; Ferri and Matić-Skoko, 2021). The possibility that the microbiota associated with the body niches of fishes also vary geographically was already investigated in the past without reaching a unanimous conclusion (Ye et al., 2014; Riiser et al., 2019; Nikouli et al., 2020).
In this study the specimens of S. notata collected in the three different geographic locations (BA, CR, BO) displayed a different diversity and composition of the gut mucosa bacterial community (Figures 3A, B). Similarly, the composition of the gut microbiota of the specimens of S. porcus and S. scrofa significantly differed between the individuals collected in BO (our study) and in GY (study of Kormas et al., 2022) (Figure 3). However, the overall diversity did not change spatially for these two species (Figure 3).
Overall, the inclusion of multiple geographic locations in the analysis revealed that geographical location had a stronger influence than the host species on the structure of the bacterial community living on the gut mucosa of Mediterranean scorpionfishes (higher W*d statistic values of the Welch MANOVA, see Results section). The analysis of the bacterial communities at the Genus level also contributed to reducing the differences previously observed among the three species in BO. The already weak effect of the host species on the gut microbiota composition, observed by analyzing the communities at the ASVs level (Figure 1A), disappeared completely when the same test was performed at the Genus level.
Although it is important to point out that for the species S. porcus and S. scrofa, a likely “study” effect can also justify part of the variance observed across the bacterial communities, analyzing the communities at the Genus level should have limited this effect.
The variability of the gut microbiota composition found for the three different species across the different geographical locations can be first explained from an environmental perspective. The temperatures and the salinity of the water registered by the Argo floats (Wong et al., 2020) in the period of sampling in the different location were indeed different: during the months of July and August 2021, the water was warmer in BO (24°C) than in BA (22°C) and CR (21°C). When the Greek samples were collected (July 2018 and June 2019) the water in GY displayed higher levels of salinity and temperature (39.2 psu-practical salinity units; 25°C) than in BO (38 psu; 24°C) (Wong et al., 2020). Both temperature and salinity are described to indirectly affect the composition of the fish gut bacterial community by altering the nutrients load and the composition of the free-living bacterial community living in the water column (Butt and Volkoff, 2019).
The significantly lower values of alpha diversity reported for the gut microbiota of the S. notata collected in BA compared to the other two locations (Figure 3) may be explained by differences in the seabed type at the three geographic locations. Indeed, the sea bottom of the three locations is characterized by different abundance levels of Posidonia oceanica: this plant is almost absent in BA, mixed with rocks and coralligenous species in BO and prevalent in CR (Figure S2). S. notata is reported to be the most abundant species found in the Posidonia oceanica meadows (Harmelin-Vivien et al., 1989), and its feeding potential correlates positively with the abundance of different algal and plant species in the sea bottom (Ordines et al., 2009). In addition, Posidonia oceanica holds important functions that could influence the stability of the gut microbiota such as sustaining biodiversity and food-web, recycling nutrients and absorbing pollutants (Lepoint et al., 2002; Houngnandan, 2020). In light of this, it is possible that a higher abundance of this plant may benefit the gut microbiota of S. notata.
Nevertheless, the autochthonous gut microbiota, investigated in this study, is known to be more influenced by the host related factors (e.g. species, population, genotype, and ontogeny) than by environmental variables (Tarnecki et al., 2017). Therefore it is possible to consider that the differences observed in terms of microbiota composition across the geographical range of the three species may also be a consequence of the genetic divergence between their populations (Smith et al., 2015). However, to be able to disentangle the environmental and the population effect, a genetic analysis of the populations of S. notata, S. porcus and S. scrofa in the Mediterranean is needed. In practical, this can be implemented by the combined analysis of mitochondrial DNA sequence markers and microsatellites to define the existing haplogroups for the three species in the Mediterranean Sea (Gandolfi et al., 2017).
In BO, the bacterial communities characterizing the intestinal walls of Scorpaena spp. appeared to be mainly composed of members of the Gammaproteobacteria, Clostridia and Fusobacteriia Class. Six bacterial genera were found to be shared in the core microbiota of the three Scorpaena species: Photobacterium, Vibrio, Shewanella, Pseudomonas (all belonging to Gammaproteobacteria), Clostridium sensu stricto 1 (included in Clostridia) and Cetobacterium (Fusobacteria) (Figure 5). These bacterial taxa have already been described to have a mutualistic relation with marine fishes. Clostridium sensu stricto 1 contributes to the supplementation of fatty acids and vitamins, the degradation of cellulose and in general to host digestion (Huang et al., 2020). Shewanella is one of the taxa which characterize the gut microbiota of piscivorous/carnivorous fishes (Huang et al., 2020). Photobacterium is ubiquitous in carnivorous, omnivorous and herbivorous fishes (Egerton et al., 2018; Burtseva et al., 2021) and together with Pseudomonas it has been found to produce chitinases and to be essential for the digestion of crustaceans in the diet (Egerton et al., 2018; Burtseva et al., 2021). Vibrio is known to be widespread in the marine environment and was also reported in the intestine of marine fishes where some Vibrio strains possess cellulase and amylase activity (Ray et al., 2012; Liu et al., 2016). In addition, Vibrio and Photobacterium, which were the two most abundant core genera in the gut of Scorpaena porcus and Scorpaena notata (Table S2), have been described as important taxa for the Scorpaenidae family in a study targeting a large range of fish species (Huang et al., 2020).
The lower relative abundance and number of genera displayed in the core microbiota of S. scrofa compared to the other two species (Figure 5A, B) can be traced back to a different foraging strategy among the three species. By being less selective compared to the other two species (mainly feeding on Crustacea) (Table 1), the individuals of S. scorfa may ingest a large variety of preys (and prey-associated bacteria) that can influence the structure of the intestinal mucosa microbiota (Butt and Volkoff, 2019). This can result in a higher inter-individual variability of the gut microbiota composition in this species (also possible to observe in Figure 1A) which does not allow the bacterial taxa to satisfy the thresholds imposed for the definition of the core microbiota in this study (i.e. minimum occurrence across the samples and relative abundance of a taxon in the community).
The core microbiota of each Scorpaena species from BO was also characterized by a set of exclusive genera that defined their enterotype (Figure 5). In S. notata, some of these exclusive genera are known to hold beneficial functions for the host: for example the genus Acinetobacter is known to possess lipase activity which improves the absorption of lipids in the GI tract (Ray et al., 2012), and the genus Bacillus is reported for its probiotic potential and production of natural antimicrobial compounds in the intestine of fishes (Santos et al., 2021).
Extending the investigation of the core microbiota to a larger geographical scale (BA, CR and GY) contributed to define the core bacterial genera associated with the three Scorpaena across the Mediterranean Sea (Figure 6). Among all, Clostridium sensu stricto 1 turned out to be the only bacterial genus shared by the individuals of the three species in every geographic location considered. In addition, it also displayed high relative abundances in the bacterial communities. Hence, being aware of the threshold applied in this study to define the core microbiota (i.e. minimum occurrence = 75% of the individuals and minimum relative abundance in the community = 0.01%, see Methods section for details), Clostridium sensu stricto 1 can be considered a resident of the gut microbiota of the three species.
It was interesting to observe that the number of bacterial taxa characterizing the core gut microbiota of the three species was drastically reduced by the analysis across the Mediterranean sea. Several bacterial genera that were recorded with high relative abundance in each species regionally were not recorded as core taxa across the Mediterranean Sea anymore (e.g. for S. porcus and S. scrofa: Photobacterium in BO and Diaphorobacter, Bradyrhizobium and Streptococcus in GY). This could have been easily anticipated by the strong inter-individual dissimilarity observed among the samples from different geographic locations (Figure 3).
A combined effect of environmental factors and functional role of the core microbiota might explain these dissimilarities in the core microbiota of the three species across the Mediterranean Sea. A fish population is exposed throughout life and generations to bacterial taxa present in the environment. Among these, the ones that hold important functions for the host (i.e. metabolism and homeostasis) are selected by host-microbe or microbe-microbe interactions in the gut community and become part of the core microbiota (Lemanceau et al., 2017). This would make the core microbiota taxonomically different for the same species across a geographical range, although preserving its functionality. The highly abundant bacterial genera found exclusively in the species from GY are known for their functionality in the intestine of fishes: for example, Bradyrhizobium and Diaphorobacter have been described for being capable of fixing nitrogen and to metabolize complex and potentially toxic organic compounds (Klankeo et al., 2009; Vaninsberghe et al., 2015). Nevertheless, to determine whether the taxonomical differences of the core microbiota are linked with differences in its functionality, analyses of shotgun metagenomic (i.e. potential functionality) or metatranscriptomics (i.e. actual functionality) data are required.
This study adds a tile to the mosaic of knowledge of bacterial symbiosis in wild fishes; it does so by characterizing the gut microbiota of three species of Scorpaena widely distributed in the Mediterranean Sea. The importance of S. porcus, S. notata and S. scrofa for the local fisheries and the possibility for Scorpaena scrofa to become an aquaculture reared species in a close future justifies the choice of these species for the study of their gut microbiota. The three species share a small core microbiota when analyzed regionally (i.e. the study region of Bonifacio) which is characterized by six bacterial genera known to hold important functions for the metabolism and the homeostasis of the host. However, the core microbiota of the three species investigated throughout the Mediterranean Sea appear to be characterized only by Clostridium senso stricto I.
Although the composition of the bacterial communities (beta-diversity) was found to change according to the host species and to the host phylogenetic relatedness (i.e. phylosymbiosis), other host’s related factors (e.g. genotype, sex, age, etc.) should be taken into account in future to fully understand the variability of the gut microbiota.
The importance of analyzing the gut microbiota of fish species through a spatial range was highlighted in this study. This approach allows to determine whether populations of the same species display different gut bacterial communities. Studying the overall diversity and composition of the gut microbiota of a fish species across its area of distribution may contribute – combined with other parameters such as the biomass, the body conditions and the dietary habits – to the identification of populations that can resist and resile differently to environmental perturbations (e.g. alien species, climate warming, or pollution) and that need different management plans in case of intense environmental and anthropogenic stress. Moreover, this practice allows to characterize more precisely the composition of the autochthonous communities living in the gut of fish species and to determine the bacteria lineages that are resident and associated to a fish species. In this study, this practice revealed that the core microbiota associated with the gut mucosa of scorpionfishes is not stable across the Mediterranean Sea, being more dependent on the fish population than on the species. In future, a function-oriented investigation of the core microbiota of Mediterranean scorpionfishes across a geographical range should determine the type of relationship existing between scorpionfishes and the bacterial genera associated with their gut mucosa.
The sequence data controls (negative and positive) produced and analyzed in this study have been deposited in the NCBI Sequences read archive (SRA, https://www.ncbi.nlm.nih.gov/sra) and are publicly available under the accession number PRJNA954707. Metadata and the SRA accession numbers generated for each of the 116 samples are provided in Table S6. The codes used to perform the raw sequences preprocessing and statistical analyses are publicly available in the Github repository https://github.com/ginevralilli93/MedScorpaena_GutMicrobiota.
Ethical approval for using animals: the ethical review and approval was not necessary for this study because the fish individuals were collected already dead from the artisanal fishermen.
GL: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Validation, Visualization, Writing – original draft, Writing – review & editing. CS: Conceptualization, Formal analysis, Funding acquisition, Project administration, Resources, Validation, Writing – review & editing. HC: Resources, Validation, Writing – review & editing. CG: Formal analysis, Funding acquisition, Validation, Writing – review & editing. DB: Formal analysis, Funding acquisition, Validation, Writing – review & editing. IG: Conceptualization, Formal analysis, Funding acquisition, Supervision, Writing – review & editing, Validation.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by the grant BiodivERsA2018-A-517 through the project METRODIVER (https://metrodiver2020.wixsite.com/website).
A special thank goes to the artisanal fishermen that kindly provided us with the fish samples and to the French students that enormously helped in the field.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1286706/full#supplementary-material
Aitchison J., Barceló-Vidal C., Martín-Fernández J. A., Pawlowsky-Glahn V. (2000). Logratio analysis and compositional distance. Math. Geol. 32 (3), 271–275. doi: 10.1023/A:1007529726302
Anderson M. J. (2001). A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26 (1), 32–46. doi: 10.1046/j.1442-9993.2001.01070.x
Anderson M. J. (2014). Permutational Multivariate Analysis of Variance (PERMANOVA) (Wiley StatsRef: Statistics Reference Online), 1–15.
Apprill A., Mcnally S., Parsons R., Weber L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microbial Ecol. 75 (2), 129–137. doi: 10.3354/ame01753
Berggren H., Tibblin P., Yıldırım Y., Broman E., Larsson P., Lundin D., et al (2022). Fish skin microbiomes are highly variable among individuals and populations but not within individuals. Front. Microbiol. 12
Bradai M. N., Bouain A. (1990). Regime alimentaire de Scorpaena porcus et de S. scrofa (Teleostei, Scorpaenidae) du Golfe de Gabes, Tunisie. Cybium 14 (3), 207–216. doi: 10.26028/cybium/1990-143-002
Brooks A. W., Kohl K. D., Brucker R. M., van Opstal E. J., Bordenstein S. R. (2017). Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PloS Biol. 15 (1), 1–29. doi: 10.1371/journal.pbio.1002587
Burtseva O., Kublanovskaya A., Fedorenko T., Lobakova E., Chekanov K. (2021). Gut microbiome of the White Sea fish revealed by 16S rRNA metabarcoding. Aquaculture 533, 736175. doi: 10.1016/j.aquaculture.2020.736175
Butt R. L., Volkoff H. (2019). Gut microbiota and energy homeostasis in fish. Front. Endocrinol. 10, 6–8. doi: 10.3389/fendo.2019.00009
Cabiddu S., Pedoni C. (2010). Trophic habits of red scorpionfish Scorpaena scrofa (osteichthyes, scorpaeniformes) in the central western mediterranean. Rapp. Commun. Int. Mer Médit. 39, 464.
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., Holmes S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13 (7), 581–583. doi: 10.1038/nmeth.3869
Castriota L., Falautano M., Finoia M. G., Consoli P., Pedà C., Esposito V., et al. (2012). Trophic relationships among scorpaeniform fishes associated with gas platforms. Helgoland Mar. Res. 66 (3), 401–411. doi: 10.1007/s10152-011-0281-0
Chauhan A., Singh R. (2019). Probiotics in aquaculture: a promising emerging alternative approach. Symbiosis 77 (2), 99–113. doi: 10.1007/s13199-018-0580-1
Chiarello M., Auguet J. C., Bettarel Y., Bouvier C., Claverie T., Graham N. A. J., et al. (2018). Skin microbiome of coral reef fish is highly variable and driven by host phylogeny and diet. Microbiome 6, (147). doi: 10.1186/s40168-018-0530-4
Clever F., Sourisse J. M., Preziosi R. F., Eisen J. A., Guerra E. C. R., Scott J. J., et al. (2022). The gut microbiome variability of a butterflyfish increases on severely degraded Caribbean reefs. Commun. Biol. 5 (1), 770. doi: 10.1038/s42003-022-03679-0
Coll M., Piroddi C., Steenbeek J., Kaschner K., Lasram F. B. R., Aguzzi J. (2010). The biodiversity of the mediterranean sea: estimates, patterns, and threats. PloS One 5 (8), e11842. doi: 10.1371/journal.pone.0011842
Dacey D. P., Chain F. J. J. (2021). Concatenation of paired-end reads improves taxonomic classification of amplicons for profiling microbial communities. BMC Bioinf. 22 (1), 1–25. doi: 10.1186/s12859-021-04410-2
Dulski T., Kozłowski K., Ciesielski S. (2020). Habitat and seasonality shape the structure of tench (Tinca tinca L.) gut microbiome. Sci. Rep. 10 (1), 1–11. doi: 10.1038/s41598-020-61351-1
Egerton S., Culloty S., Whooley J., Stanton C., Ross R. P. (2018). The gut microbiota of marine fish. Front. Microbiol. 9, 1–17. doi: 10.3389/fmicb.2018.00873
Escalas A., Auguet J. C., Avouac A., Seguin R., Gradel A., Borrossi L., et al. (2021). Ecological specialization within a carnivorous fish family is supported by a herbivorous microbiome shaped by a combination of gut traits and specific diet. Front. Mar. Sci. 8, 622883. doi: 10.3389/fmars.2021.622883
Ferri J., Matić-Skoko S. (2021). The spatial heterogeneity of the black scorpionfish, Scorpaena porcus (Scorpaenidae): Differences in length, dietary and age compositions. Appl. Sci. (Switzerland) 11 (24). doi: 10.3390/app112411919
Fischer W. (1987). Méditerranée et mer Noire. Zone de pêche 37. Fiches FAO d’identification Des Espèces Pour Les Besoins de la Pêche, (Food and Agriculture Organization of the United Nations) Vol. 2. 761–1530.
Gajardo K., Rodiles A., Kortner T. M., Krogdahl Å., Bakke A. M., Merrifield D. L., et al. (2016). A high-resolution map of the gut microbiota in Atlantic salmon (Salmo salar): A basis for comparative gut microbial research. Sci. Rep. 6, 30893. doi: 10.1038/srep30893
Gandolfi A., Ferrari C., Crestanello B., Girardi M., Lucentini L., Meraner A. (2017). Population genetics of pike, genus Esox (Actinopterygii, Esocidae), in Northern Italy: evidence for mosaic distribution of native, exotic and introgressed populations. Hydrobiologia 794 (1), 73–92. doi: 10.1007/s10750-016-3083-1
Gloor G. B., Macklaim J. M., Pawlowsky-Glahn V., Egozcue J. J. (2017). Microbiome datasets are compositional: And this is not optional. Front. Microbiol. 8, 1–6. doi: 10.3389/fmicb.2017.02224
Groussin M., Mazel F., Sanders J. G., Smillie C. S., Lavergne S., Thuiller W., et al. (2017). Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nat. Commun. 8, 14319. doi: 10.1038/ncomms14319
Hamidi B., Wallace K., Vasu C., Alekseyenko A. V. (2019). W d∗ -test: Robust distance-based multivariate analysis of variance. Microbiome 7 (1), 1–9. doi: 10.1186/s40168-019-0659-9
Harmelin-Vivien M. L., Kaim-Malka R. A., Ledoyer M., Jacob-Abraham S. S. (1989). Food partitioning among scorpaenid fishes in Mediterranean seagrass beds. J. Fish Biol. 34 (5), 715–734. doi: 10.1111/j.1095-8649.1989.tb03352.x
Houngnandan F. (2020). Impact des pressions anthropiques et de l’environnement sur les herbiers de Posidonia oceanica en Mediterranee Française. Dissertation. (Université de Montpellier)
Huang Q., Sham R. C., Deng Y., Mao Y., Wang C., Zhang T., et al. (2020). Diversity of gut microbiomes in marine fishes is shaped by host-related factors. Mol. Ecol. 29 (24), 5019–5034. doi: 10.1111/mec.15699
Klankeo P., Nopcharoenkul W., Pinyakong O. (2009). Two novel pyrene-degrading Diaphorobacter sp. and Pseudoxanthomonas sp. isolated from soil. JBIOSC 108 (6), 488–495. doi: 10.1016/j.jbiosc.2009.05.016
Kohl K. D. (2020). Ecological and evolutionary mechanisms underlying patterns of phylosymbiosis in host-associated microbial communities. Philos. Trans. R. Soc. B.: Biol. Sci. 375 (1798), 20190251. doi: 10.1098/rstb.2019.0251
Kormas K., Nikouli E., Kousteni V., Damalas D. (2022). Midgut bacterial microbiota of 12 fish species from a marine protected area in the aegean sea (Greece). Microbial Ecol. doi: 10.1007/s00248-022-02154-x
Kumar S., Stecher G., Suleski M., Blair Hedges S. (2017). TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34 (7), 1812–1819. doi: 10.1093/molbev/msx116
Lahti L. (2023) microbiome R package. Available at: http://microbiome.github.io.
Lemanceau P., Blouin M., Muller D., Moënne-loccoz Y. (2017). Let the core microbiota be functional. Trends Plant Sci. 22 (7), 583–595. doi: 10.1016/j.tplants.2017.04.008
Lepoint G., Defawe O., Gobert S., Dauby P., Bouquegneau J. M. (2002). Experimental evidence for N recycling in the leaves of the seagrass Posidonia oceanica. J. Sea Res. 48 (3), 173–179. doi: 10.1016/S1385-1101(02)00164-8
Lim S. J., Bordenstein S. R. (2020). An introduction to phylosymbiosis. Proc. R. Soc. B. 287 (1922). doi: 10.1098/rspb.2019.2900
Liu H., Guo X., Gooneratne R., Lai R., Zeng C., Zhan F., et al. (2016). The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Sci. Rep. 6, 24340. doi: 10.1038/srep24340
Love M. I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 1–21. doi: 10.1186/s13059-014-0550-8
Mallott E. K., Amato K. R. (2021). Host specificity of the gut microbiome. Nat. Rev. Microbiol. 19 (10), 639–653. doi: 10.1038/s41579-021-00562-3
Mandal S., Van Treuren W., White R. A., Eggesbø M., Knight R., Peddada S. D. (2015). Analysis of composition of microbiomes: a novel method for studying microbial composition. Microbial Ecol. Health Dis. 26 (1), 27663. doi: 10.3402/mehd.v26.27663
Martinez Arbizu P. (2020). pairwiseAdonis: Pairwise multilevel comparison using adonis (R package version 0.4.1). Available at: https://github.com/pmartinezarbizu/pairwiseAdonis.
Martín-Fernández J. A., Hron K., Templ M., Filzmoser P., Palarea-Albaladejo J. (2015). Bayesian-multiplicative treatment of count zeros in compositional data sets. Stat. Model. 15 (2), 134–158. doi: 10.1177/1471082X14535524
Minich J. J., Härer A., Vechinski J., Frable B. W., Skelton Z. R., Kunselman E., et al. (2022). Host biology, ecology and the environment influence microbial biomass and diversity in 101 marine fish species. Nat. Commun. 13 (1), 1–19. doi: 10.1038/s41467-022-34557-2
Minich J. J., Poore G. D., Jantawongsri K., Johnston C., Bowie K., Knight R., et al. (2020). Microbial ecology of atlantic salmon (Salmo salar) hatcheries: impacts of the built environment on fish mucosal microbiota. Appl. Environ. Microbiol. 86 (12), e00411–e00420. doi: 10.1128/AEM.00411-20
Mukherjee S., Huntemann M., Ivanova N., Kyrpides N. C., Pati A. (2015). Large-scale contamination of microbial isolate genomes by illumina Phix control. Standards Genomic Sci. 10, 1–4. doi: 10.1186/1944-3277-10-18
Nayak S. K. (2010). Role of gastrointestinal microbiota in fish. Aquacult. Res. 41 (11), 1553–1573. doi: 10.1111/j.1365-2109.2010.02546.x
Nearing J. T., Douglas G. M., Hayes M. G., MacDonald J., Desai D. K., Allward N., et al. (2022). Microbiome differential abundance methods produce different results across 38 datasets. Nat. Commun. 13 (1), 1–16. doi: 10.1038/s41467-022-28034-z
Neu A. T., Allen E. E., Roy K. (2021). Defining and quantifying the core microbiome: Challenges and prospects. Proc. Natl. Acad. Sci. United States America 118 (51), e2104429118. doi: 10.1073/pnas.2104429118
Nikouli E., Meziti A., Antonopoulou E., Mente E., Kormas K. A. (2018). Gut Bacterial Communities in Geographically Distant Populations of Farmed Sea Bream (Sparus aurata) and Sea Bass (Dicentrarchus labrax). Microorganisms 6 (3), 92. doi: 10.3390/microorganisms6030092
Nikouli E., Meziti A., Smeti E., Antonopoulou E., Mente E., Kormas K. A. (2020). Gut microbiota of five sympatrically farmed marine fish species in the aegean sea. Microbial Ecol. 81 (2), 460–470. doi: 10.1007/s00248-020-01580-z
Oksanen J., Blanchet F. G., Kindt R., Legendre P., Minchin P. R. (2013) vegan: R package for community ecologists: popular ordination methods, ecological null models and diversity analysis. Available at: https://github.com/vegandevs/vegan.
Ordines F., Quetglas A., Massutí E., Moranta J. (2009). Habitat preferences and life history of the red scorpion fish, Scorpaena notata, in the Mediterranean. Estuarine Coast. Shelf Sci. 85 (4), 537–546. doi: 10.1016/j.ecss.2009.09.020
Özgül A., Lök A., Tansel Tanrıkul T., Alós J. (2019). Home range and residency of Scorpaena porcus and Scorpaena scrofa in artificial reefs revealed by fine-scale acoustic tracking. Fisheries Res. 210, 22–30. doi: 10.1016/j.fishres.2018.10.008
Palarea-Albaladejo J., Martín-Fernández J. A. (2015). zCompositions — R package for multivariate imputation of left-censored data under a compositional approach. Chemometrics Intelligent Lab. Syst. 143, 85–96. doi: 10.1016/j.chemolab.2015.02.019
Parada A. E., Needham D. M., Fuhrman J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18 (5), 1403–1414. doi: 10.1111/1462-2920.13023
Pinkas L. (1971). Food habits of albacore, bluefin tuna, and bonito in california waters. Fish Bull. 152.
Price M. N., Dehal P. S., Arkin A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26 (7), 1641–1650. doi: 10.1093/molbev/msp077
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Rafrafi-Nouira S., El Kamel-Moutalibi O., Boumaïza M., Reynaud C., Capapé C. (2016). Food and feeding habits of black scorpionfish, Scorpaena porcus (Osteichthyes: Scorpaenidae) from the northern coast of Tunisia (Central Mediterranean). J. Ichthyol. 56 (1), 107–123. doi: 10.1134/S0032945216010112
Ray A. K., Ghosh K., Ringø E. (2012). Enzyme-producing bacteria isolated from fish gut: A review. Aquacult. Nutr. 18 (5), 465–492. doi: 10.1111/j.1365-2095.2012.00943.x
R Core Team (2021). R: A Language and Environment for Statistical Computing (Vienna, Austria: R Foundation for Statistical Computing). Available at: https://www.R-project.org/.
Riiser E. S., Haverkamp T. H. A., Varadharajan S., Borgan Ø., Jakobsen K. S., Jentoft S., et al. (2019). Switching on the light: using metagenomic shotgun sequencing to characterize the intestinal microbiome of Atlantic cod. Environ. Microbiol. 21 (7), 2576–2594. doi: 10.1111/1462-2920.14652
Ruiz-Rodríguez M., Scheifler M., Sanchez-Brosseau S., Magnanou E., West N., Suzuki M., et al. (2020). Host species and body site explain the variation in the microbiota associated to wild sympatric mediterranean teleost fishes. Microbial Ecol. 80 (1), 212–222. doi: 10.1007/s00248-020-01484-y
Russel J. (2020). MicEco: Various functions for analysis of microbial community data (Russel88/MicEco: V0.9.19 (v0.9.19). Zenodo).
Šantić M., Pallaoro A., Rađa B. (2021). Feeding habits of Scorpaena notata (Scorpaenidae) from eastern Adriatic Sea Regular paper Feeding habits of Scorpaena notata (Scorpaenidae) from eastern Adriatic Sea. Cybium 45 (3), 217–224. doi: 10.26028/cybium/2021-453-006
Santos R. A., Teles A. O., Pousão P., Russell F., Maria J., Enes P., et al. (2021). Isolation and characterization of fish − Gut bacillus spp. as source of natural antimicrobial compounds to fight aquaculture bacterial diseases. Mar. Biotechnol. 23, 276–293. doi: 10.1007/s10126-021-10022-x
Shahrani S. E. (2015). Food and feeding habits of Scorpaena scrofa (Linnaeus 1758) in the western part of Libya. Lybian J. Mar. Sci. 14, 5–15.
Smith C. C. R., Snowberg L. K., Gregory Caporaso J., Knight R., Bolnick D. I. (2015). Dietary input of microbes and host genetic variation shape among-population differences in stickleback gut microbiota. ISME J. 9 (11), 2515–2526. doi: 10.1038/ismej.2015.64
Sullam K. E., Essinger S. D., Lozupone C. A., O’Connor M. P., Rosen G. L., Knight R., et al. (2012). Environmental and ecological factors that shape the gut bacterial communities of fish: A meta-analysis. Mol. Ecol. 21 (13), 3363–3378. doi: 10.1111/j.1365-294X.2012.05552.x
Sylvain F. É., Holland A., Bouslama S., Audet-Gilbert É., Lavoie C., Luis Val A., et al. (2020). Fish skin and gut microbiomes show contrasting signatures of host species and habitat. Appl. Environ. Microbiol. 86 (16), 1–15. doi: 10.1128/AEM.00789-20
Sylvain F. É., Normandeau E., Holland A., Luis Val A., Derome N. (2022). Genomics of Serrasalmidae teleosts through the lens of microbiome fingerprinting. Mol. Ecol. 31 (18), 4656–4671. doi: 10.1111/mec.16574
Tarnecki A. M., Burgos F. A., Ray C. L., Arias C. R. (2017). Fish intestinal microbiome: diversity and symbiosis unravelled by metagenomics. J. Appl. Microbiol. 123 (1), 2–17. doi: 10.1111/jam.13415
Turan C., Gunduz I., Yaglioglu D. (2009). Systematics of scorpaeniformes species in the mediterranean sea inferred from mitochondrial 16S rDNA sequence and morphological data. Folia Biol. (Kraków) 57, 219–226. doi: 10.3409/fb57_1-2.219-226
Vaninsberghe D., Maas K. R., Cardenas E., Strachan C. R., Hallam S. J., Mohn W. W. (2015). Non-symbiotic Bradyrhizobium ecotypes dominate North American forest soils. ISME J. 9, 2435–2441. doi: 10.1038/ismej.2015.54
Wang W., Zhou Z., He S., Liu Y., Cao Y., Shi P., et al. (2010). Identification of the adherent microbiota on the gills and skin of poly-cultured gibel carp (Carassius auratus gibelio) and bluntnose black bream (Megalobrama amblycephala Yih). Aquacult. Res. 41, e72–e83. doi: 10.1111/j.1365-2109.2009.02459.x
Wong A. P. S., Wijffels S. E., Riser S. C., Pouliquen S., Hosoda S., Roemmich D., et al (2020). Argo data 1999–2019: two million temperature-salinity profiles and subsurface velocity observations from a global array of profiling floats. Front. Mar. Sci. 7, 1–23. doi: 10.3389/fmars.2020.00700
Wright E. S., Yilmaz L. S., Noguera D. R. (2012). DECIPHER, a search-based approach to chimera identification for 16S rRNA sequences. Appl. Environ. Microbiol. 78 (3), 717. doi: 10.1128/AEM.06516-11
Keywords: fish gut microbiome, phylosymbiosis, Mediterranean Sea, core microbiota, spatial variation, Scorpaena
Citation: Lilli G, Sirot C, Campbell H, Brophy D, Graham CT and George IF (2023) Geographic origin and host’s phylogeny are predictors of the gut mucosal microbiota diversity and composition in Mediterranean scorpionfishes (Scorpaena spp.). Front. Mar. Sci. 10:1286706. doi: 10.3389/fmars.2023.1286706
Received: 31 August 2023; Accepted: 20 November 2023;
Published: 05 December 2023.
Edited by:
Francesco Tiralongo, University of Catania, ItalyReviewed by:
Giacomo Zaccone, University of Messina, ItalyCopyright © 2023 Lilli, Sirot, Campbell, Brophy, Graham and George. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ginevra Lilli, TGlsbGkuR2luZXZyYUB1bGIuYmU=
†ORCID: Ginevra Lilli, orcid.org/0009-0006-7089-9287
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.