 Iulia-Mădălina Ştreangă
Iulia-Mădălina Ştreangă Daniel J. Repeta
Daniel J. Repeta Jerzy S. Blusztajn3,4
Jerzy S. Blusztajn3,4 Tristan J. Horner
Tristan J. Horner- 1Department of Marine Chemistry and Geochemistry, Woods Hole Oceanographic Institution, Woods Hole, MA, United States
- 2Department of Earth, Atmospheric and Planetary Sciences, Massachusetts Institute of Technology, Cambridge, MA, United States
- 3NIRVANA Labs, Woods Hole Oceanographic Institution, Woods Hole, MA, United States
- 4Department of Geology and Geophysics, Woods Hole Oceanographic Institution, Woods Hole, MA, United States
Iodine intersects with the marine biogeochemical cycles of several major elements and can influence air quality through reactions with tropospheric ozone. Iodine is also an element of interest in paleoclimatology, whereby iodine-to-calcium ratios in marine carbonates are widely used as a proxy for past ocean redox state. While inorganic iodine in seawater is found predominantly in its reduced and oxidized anionic forms, iodide (I−) and iodate (IO3−), the rates, mechanisms and intermediate species by which iodine cycles between these inorganic pools are poorly understood. Here, we address these issues by characterizing the speciation, composition and cycling of iodine in the upper 1,000 m of the water column at Station ALOHA in the subtropical North Pacific Ocean. We first obtained high-precision profiles of iodine speciation using isotope dilution and anion exchange chromatography, with measurements performed using inductively coupled plasma mass spectrometry (ICP-MS). These profiles indicate an apparent iodine deficit in surface waters approaching 8% of the predicted total, which we ascribe partly to the existence of dissolved organic iodine that is not resolved during chromatography. To test this, we passed large volumes of seawater through solid phase extraction columns and analyzed the eluent using high-performance liquid chromatography ICP-MS. These analyses reveal a significant pool of dissolved organic iodine in open ocean seawater, the concentration and complexity of which diminish with increasing water depth. Finally, we analyzed the rates of IO3− formation using shipboard incubations of surface seawater amended with 129I−. These experiments suggest that intermediate iodine species oxidize to IO3− much faster than I− does, and that rates of IO3− formation are dependent on the presence of particles, but not light levels. Our study documents the dynamics of iodine cycling in the subtropical ocean, highlighting the critical role of intermediates in mediating redox transformations between the major inorganic iodine species.
1 Introduction
Understanding marine iodine geochemistry is imperative due to its integral role in numerous environmental processes. The ocean is the major source of iodine to the atmosphere via both biological and non-biological pathways (Fuge and Johnson, 2015). Once released, photochemically active iodocarbons have tropospheric lifetimes ranging from a few minutes to a few days (Solomon et al., 1994; Carpenter et al., 2021). Upon photolysis and oxidation, these organic halogens form IO radicals, a major source of particles that can act as cloud condensation nuclei and influence cloud properties (Bluhm et al., 2010). Furthermore, dissolved iodide reacts with tropospheric ozone at the surface ocean, fueling a catalytic pathway (Chance et al., 2014) that accounts for approximately 15% tropospheric ozone loss globally (Sherwen et al., 2016a; Sherwen et al., 2016b). Iodine is also an element of interest in paleoclimatology, with iodine-to-calcium ratios in marine carbonates being widely interpreted as proxies for the oxygenation state of the ocean through geological time (e.g. Lu et al., 2018; Lu et al., 2020; Sun et al., 2023). In seawater, iodine is primarily found in its reduced and oxidized anionic forms, iodide (I−) and iodate (IO3−), respectively. Measurements of total iodine concentrations across the world’s oceans reveal relatively constant values between 450–500 nM. Despite being thermodynamically unstable in oxygenated waters, I− can be the dominant iodine species in coastal and low latitude regions, with concentrations as high as 300 nM (e.g., Chance et al., 2014; Sherwen et al., 2019). At latitudes above 40° north and south and deeper in the water column, IO3− is the dominant species (Chance et al., 2014; Chance et al., 2020). Unraveling the underlying reasons for this distinctive speciation pattern, particularly in the surface ocean, necessitates a deeper understanding of the processes governing iodine transformations.
One of the central challenges to understanding iodine biogeochemistry lies in elucidating the rates, mechanisms and intermediates involved in the transformation of iodine between its reduced and oxidized forms. It is generally thought that I− oxidation to IO3− is much slower than the reverse transformation at both high and low oxygen concentrations (Hardisty et al., 2021); however, I− oxidation is a highly uncertain process (Carpenter et al., 2021). Previous studies have reported I− oxidation rates ranging between 1.5–8.2 nM yr–1 (He et al., 2013) and 560 nM yr–1 in oxic waters (Campos et al., 1996), mostly determined by mass balance approaches. Abiotic I− oxidation is thought to be too slow to account for the more rapid I− oxidation rates reported to date. Instead, I− oxidation is thought to be mediated by particles, such as reactive Mn oxides, by reactive oxygen species produced photochemically, or via direct or indirect biological processes (e.g., Amachi, 2008; Bluhm et al., 2010; Carpenter et al., 2021; Luther, 2023). Given that there is a six-electron transfer between I− and IO3−, it is unlikely that I− oxidation leads directly to IO3− production. Instead, several intermediate iodine species likely mediate this transformation, such as molecular iodine (I2), hypoiodous acid (HOI) and iodite (IO2-; Luther, 2023; Figure 1). Both HOI and I2 can react with organic matter to form iodine-bearing organic compounds (Luther, 2023), which have yet to be chemically characterized. The existence of these compounds has been documented in coastal areas (Luther et al., 1991), but they are widely considered to be negligible in the open ocean (Edwards and Truesdale, 1997; Wadley et al., 2020). A mismatch observed between the amounts of IO3− reduced and I− produced during incubation studies has led to the suggestion that dissolved organic iodine (DOI) can be synthesized by a wide range of microalgae and cyanobacteria (Chance et al., 2007; van Bergeijk et al., 2016; Hepach et al., 2020).
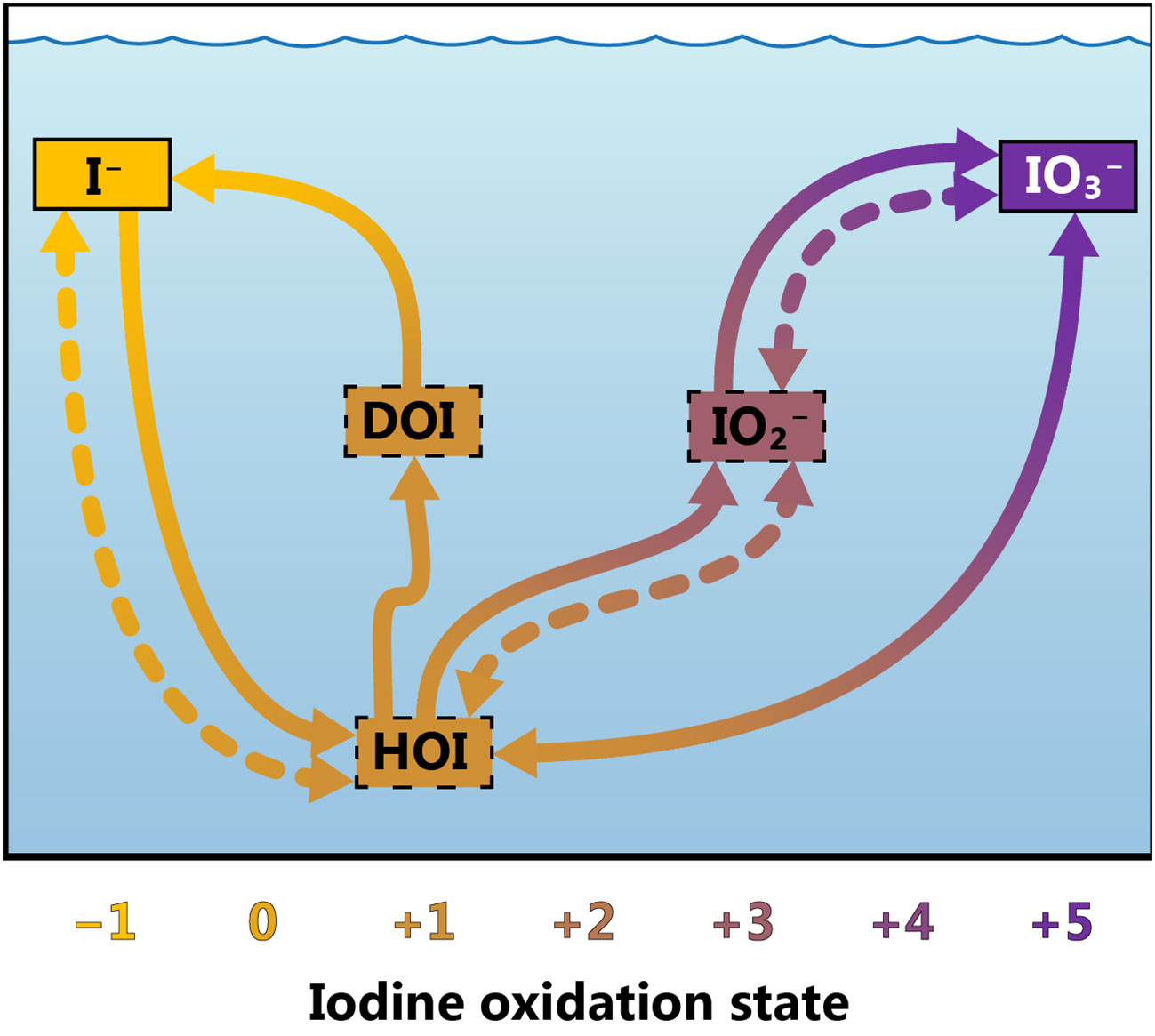
Figure 1 Conceptual model of iodine cycling at Station ALOHA. Arrows represent transformations between various iodine species. Abiotic processes are represented as dotted lines and biotic or biologically mediated processes with solid lines. The solid bounding boxes around iodide and iodate indicate that these species are resolved by our ion-exchange chemistry, while the dotted bounding boxes around the other species imply that they are not resolved by our method. Iodide (I−), iodate (IO3−), iodite (IO2−), hypoiodous acid (HOI) and dissolved organic iodine (DOI) have been included, while molecular iodine, I2 (oxidation state 0), has been omitted. Dissolved organic iodine has been assigned an oxidation state of +1, although its precise composition is unknown.
Conventional methods lack the precision required to discern subtle variations in iodine speciation during an experiment, which can be particularly significant in the case of slow processes, such as I− oxidation. The most common method used for measuring I− concentrations is cathodic stripping square wave voltammetry (Luther et al., 1988a; Chance et al., 2014; Moriyasu et al., 2020; Moriyasu et al., 2023), while IO3− is usually quantified using differential pulse polarography (Herring and Liss, 1974) and spectrophotometric methods (Wong and Brewer, 1974; Rue et al., 1997). In most studies, only I− or IO3− is measured, alongside total inorganic iodine, and the difference between the two is used to calculate the abundance of the other species (Liss et al., 1973; Takayanagi and Wong, 1986; Jickells et al., 1988; Truesdale and Jones, 2000). These conventional approaches for measuring iodine speciation achieve a typical precision of between 5 and 10% (Campos et al., 1996), which are unlikely to resolve the small changes in iodine speciation that are believed to occur over short timescales. Thus, incubation experiments are particularly suitable for studying iodine cycling over short timescales. While a new method using ion exchange chromatography coupled to spectrophotometric iodine detection can achieve a precision as good as 2–4% (Jones et al., 2023), any concentration-based approach to studying iodine redox transformations is fundamentally limited by the difficulty in constraining the specific reaction pathway involved in the formation of a given iodine species (Figure 1).
To address these challenges, we leveraged a recently developed method using 129I, a long-lived radioactive isotope of iodine (half-life of 15.7 Myr), as a tracer to study iodine redox transformations in seawater. In the context of our experiments, the natural abundance of 129I in the open ocean is negligible, with 129I/127I ratios varying between 10-11 and 10-6 (Qi et al., 2023). By coupling isotope dilution to chromatographic separation of iodine measurement by ICP-MS, this method, adapted from Hardisty et al. (2020), offers unprecedented precision for measuring iodine speciation and transformation rates. Short incubation experiments can be conducted to study the rates and mechanisms of these reactions, on timescales relevant to biological processes occurring in the ocean. Likewise, recent developments in high-pressure liquid chromatography ICP-MS (HPLC-ICP-MS) and electrospray ionization MS have enabled the isolation and quantification of low-abundance organic ligands in seawater (Boiteau et al., 2013; Boiteau and Repeta, 2015; Li et al., 2021). These methods can be used to test for the existence of DOI.
In this study, we investigated the rates, mechanisms and intermediates governing iodine redox transformations in the subtropical North Pacific Ocean near Station ALOHA. We tackled these issues in four steps. First, we report profiles of iodine speciation measured using isotope dilution ICP-MS and compare our results with the pioneering work of Campos et al. (1996). Second, we apply a novel HPLC-ICP-MS method to investigate the existence of DOI in open ocean seawater samples. Third, we conduct short-term 129I-amended incubation experiments to constrain rates of IO3− formation. Finally, we use the same series of experiments to investigate the roles of particles and light on the formation of IO3−. This research advances our understanding of iodine cycling in seawater and the role of particles and organics in sustaining it.
2 Methods
2.1 Sample collection
Samples were collected onboard the R/V Kilo Moana between 22 July and 5 August 2021 (Cruise ID KM2112). Sampling occurred at and around Station ALOHA (Figure 2), as part of the Simons Collaboration on Ocean Processes and Ecology (SCOPE) Particles and Growth in the Oceanic Nutricline (PARAGON) cruise. The profile samples were collected in 60 mL high density polyethylene (HDPE) bottles that were cleaned in several steps. First, bottles were soaked in a 1% Citranox© solution for one week, before rinsing five times with deionized water. Second, bottles were soaked in a 10% (by volume) solution of hydrochloric acid (HCl) for one week, then rinsed three times with 18.2 MΩ cm–2-grade water from a Milli-Q system (MilliporeSigma©). For all profile samples, 60 mL of seawater were filtered directly from the Go-Flo bottle using a 0.2 μm Pall© Acropak-200 Supor filter. Information about the time and location at which the three CTD profiles were collected is summarized in Table 1. Profile 2 was collected with a conventional rosette, whereas Profiles 1 and 3 were collected with a trace-metal–clean rosette. The samples were refrigerated immediately after collection. Spiking and chromatographic separation of iodine was performed within 14 months of collection using 20 mL of seawater from each profile sample. Iodine speciation in filtered seawater is stable for at least a year if samples are stored refrigerated and sealed (Campos, 1997).
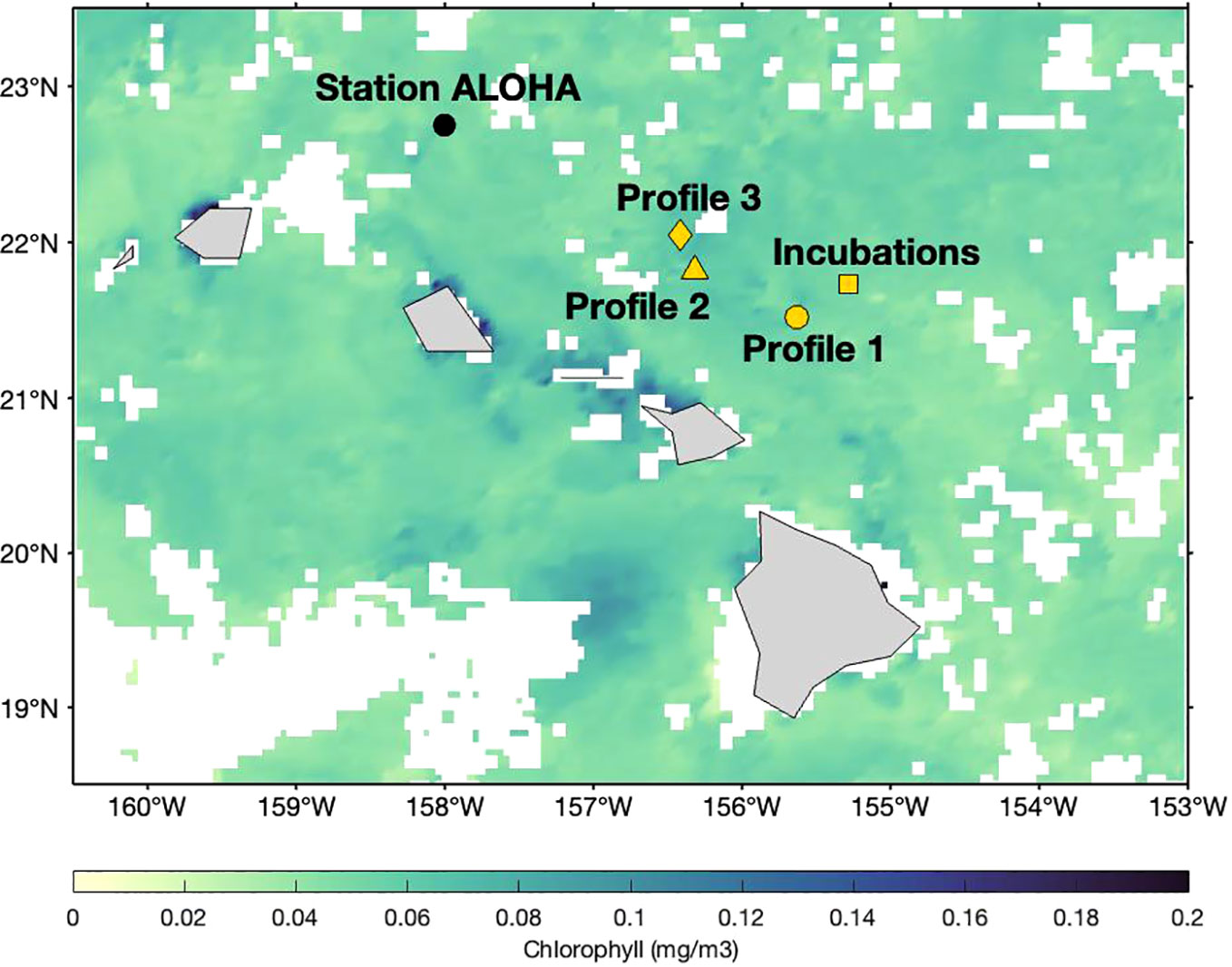
Figure 2 Locations of deployments relative to Station ALOHA: Profile 1 (filled circle), Profile 2 (filled triangle), Profile 3 (filled diamond) and location corresponding to the collection of incubation samples (filled square). The surface chlorophyll data is the 8-day composite of Stage Level 3 NASA Aqua-MODIS Ocean Color data product between 28 July–4 August 2021.
Table 1 Summary of deployment information for the profile and incubation samples.
The shipboard incubations were conducted in triplicate over a 60-hour period, with surface water collected with a conventional rosette from a fourth CTD deployment (Table 1; Figure 2). All 60 mL incubation samples were collected into polycarbonate bottles cleaned using the same protocol as for HDPE bottles. Polycarbonate bottles were placed in an on-deck incubator that was continuously flushed with surface seawater to maintain in situ temperature. Four different conditions were tested (Table 1). Prior to the start of the incubation period, each 60 mL sample was individually spiked with 45 µL of a 129I working solution. At each of the six sampling timepoints, an entire sample volume was sacrificed for all corresponding triplicates under the four conditions. The added spike amount was ≈ 45 nM 129I, approximately 10% of the previously determined in situ total 127I concentrations. The working solution was prepared by adding 1.366 mL of an Eckert & Ziegler© stock spike solution, with a total activity of 18,500 Bq (0.5 μCi) 129I in the form of sodium iodide, to 48.634 mL of 18.2 MΩ H2O. Triplicates were incubated and processed every 12 hours. At each sampling point, the unfiltered samples were filtered through a 0.2 µm filter, collected and stored at 4°C. The filtered samples were not filtered again prior to refrigeration. Laboratory analysis on these samples was conducted between 14–18 months after collection.
2.2 Determination of stock spike solution composition and speciation
In order to accurately use 129I for isotope dilution ICP-MS, the 129I/127I ratio and total iodine concentration of the spike solution needed to be determined. This was achieved by preparing a set of 10 standards with spike-to-standard volumetric mixing ratios between 0.03 and 1. These were prepared by mixing a SPEX CertiPrep© 1,000 μg mL−1 127 I− standard solution, the stock spike solution and 18.2 MΩ H2O in various known proportions, and measuring using a quadrupole ICP-MS (Thermo Scientific© iCap-Q) at the Woods Hole Oceanographic Institution (WHOI) Plasma Facility. The theoretical 129I/127I ratio of each mixture was calculated using Equation 1:
where is the volumetric fraction of the certified 127I standard solution in the prepared standard solution; is the iodine isotopic ratio of the stock spike solution; is the total iodine concentration of the spike solution in nmol mL-1 and is the total iodine concentration of the certified 127I standard solution. We then calculated the 129I/127I of the spike by finding the composition that minimized the root-mean-square error between theoretical and measured 129I/127I for the 10 mixtures. This exercise revealed that the best-fit spike composition was = 3.76 and = 626 nmol mL−1. The spike composition was also determined using MC-ICP-MS, yielding a 129I/127I of 3.58, which is ≈ 5% lower than that determined using ICP-MS. The MC-ICP-MS value was obtained without standard addition, via a direct analysis of the spike itself using the wet-sparge sample introduction method (Section 2.5.). The slight difference in spike composition between the two measurement approaches likely derives from differences in the sample introduction method (liquid versus wet sparge) and from instrument-specific considerations (e.g., collision cell, plasma conditions, lens stack). For consistency between approaches, a spike composition of 3.76 was assumed for all samples measured using the ICP-MS and a value of 3.58 for those measured using MC-ICP-MS.
2.3 Calculation of spike amounts to be added to profile samples
Optimal spiking for isotope dilution is achieved when the 129I/127I ratio of the target mixture is close to the geometric mean of the natural and spike compositions (Stracke et al., 2014). This raises two issues for iodine. First, in most natural seawater, 129I is essentially absent compared to 127I (e.g., 0.03 fM in the subtropical Pacific Ocean; Qi et al., 2023). This means that the optimal 129I/127I ratio would be so low as to be unamenable to analysis by ICP-MS. Second, the concentration of different inorganic iodine species varies with depth. Thus, optimal spiking requires prior knowledge of the iodine distribution. To work around these issues, we estimated likely in situ [IO3–] and [I–] from literature data and aimed for a target 129I/127I ratio of between 0.5–1.0 for both iodine fractions. This wider target ensured that there was sufficient 129I for analysis in each sample, and likewise reduced the relative magnitude of the interference from isobaric xenon-129 on m/z 129.
Iodine species separation was achieved in two steps: first by eluting I−, followed by separating IO3− after reducing it to I− and similarly eluting it. Therefore, each 20 mL seawater sample was spiked twice, before and after the isolation of the I− fraction. Table 2 summarizes the targeted concentrations and the amounts of 129I working solution which were added to the profile samples collected from various depths. The approximate I− and IO3− in situ concentrations were estimated from the results reported in Campos et al. (1996) for Station ALOHA during the summer. Procedural blanks were monitored by processing 20 mL aliquots of 18.2 MΩ H2O alongside profile samples. Blanks were spiked with the same amount of 129I as seawater samples below 500 m depth. No procedural blanks were processed alongside the incubation experiment samples, since this correction would affect all samples equally and would not change the overall trends.
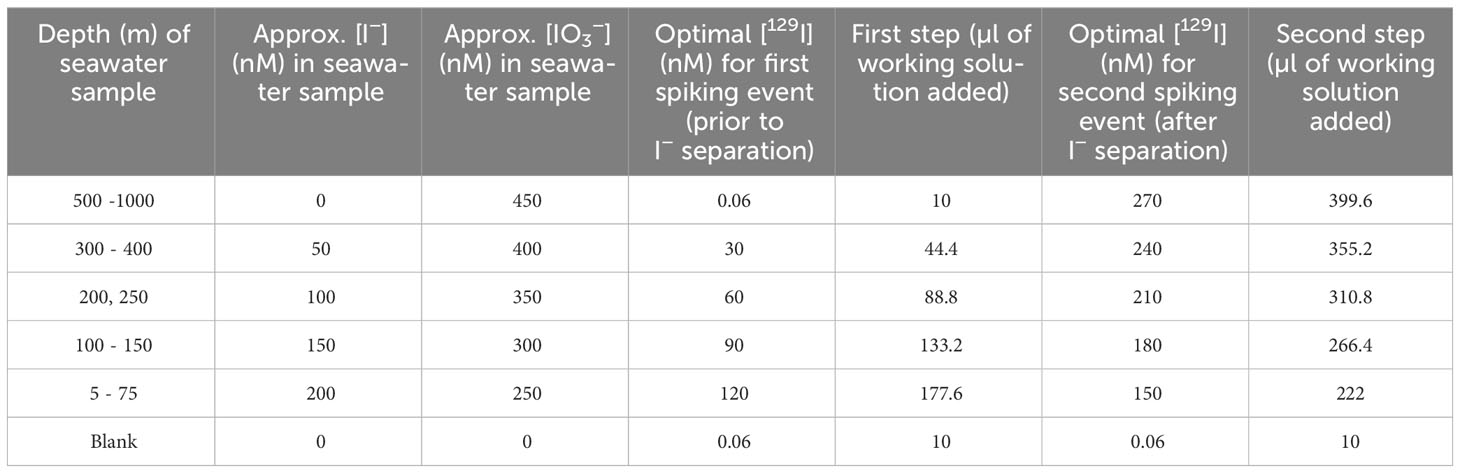
Table 2 Determination of optimal volume of working solution to be added to profile samples in the first and the second steps, based on approximate I− and IO3− in situ concentrations at each depth.
2.4 Chromatographic separation of reduced and oxidized iodine species
Sample processing took place within Class 100 laminar flow workbenches situated in the NIRVANA Labs (Non-traditional Isotope Research on Various Advanced Novel Applications). We used a two-step chromatographic protocol to separate and isolate the major inorganic forms of iodine in seawater. This protocol was based on the method described by Hardisty et al. (2020). Briefly, separation was performed by packing acid-cleaned 5 mm (inner diameter) quartz-glass columns with 0.5 mL of AG-1X8 anion-exchange resin (BioRad©). The resin was batch cleaned with 6 M reagent-grade HCl (Fischer Scientific© ACS plus) and thoroughly rinsed with 18.2 MΩ H2O prior to loading onto the columns. Due to initially very high iodine concentrations in our procedural blanks, we introduced a cleaning protocol which followed the first step of the chromatographic procedure, substituting seawater sample with 18.2 MΩ H2O.
After rinsing the resin and loading 20 mL seawater sample, the oxidized iodine fraction was collected, spiked, refrigerated overnight and further processed the following day. Simultaneous with the spike addition, 500 µL of 1.2 M HCl and 80 µL of 0.3 M sodium bisulfite (NaHSO3, Acros© ACS-grade) were added to the IO3− fraction to completely reduce it to I−. The final I− fraction was eluted following the addition of 15 mL of a mixed 2 M nitric acid (HNO3; Fisher Scientific© certified ACS plus) and 18% tetramethylammonium hydroxide solution (TMAH, Alfa Aesar© 25% w/w aq. Solution Electronic Grade) and refrigerated. The role of TMAH was to keep iodine stable in solution and prevent its volatilization (Hardisty et al., 2020). The second step of the separation involved rinsing the loaded resin again and loading the oxidized fraction (containing the reduced IO3−) onto the columns. The final IO3− fraction was then eluted (as I−) with 15 mL of the 2 M HNO3 and 18% TMAH solution, collected and refrigerated. The processed samples were analyzed via mass spectrometry within weeks of separation.
The chromatographic separation of the profile samples differed slightly from that of the incubation samples. Since the incubation samples had already been amended with 129I−, they were not spiked again in the laboratory prior to the separation of reduced and oxidized iodine. The entire volume (60 mL) of the incubation experiments was loaded onto the columns to maximize the quantity of 129I for measurement via MC-ICP-MS. This difference in processed sample volumes necessitated an increase in the amounts of HCl and NaHSO3 added to the incubation samples prior to the second step in the separation (1 mL HCl and 160 µL NaHSO3, respectively).
2.5 Instrumental analyses and data reduction
All spiked seawater samples were analyzed at the WHOI Plasma Facility. Seawater profile samples were analyzed using a quadrupole ICP-MS operated in Kinetic Energy Discrimination mode with helium as the collision cell gas. Solutions were prepared in 2 mL polypropylene vials soaked in a 50% (by volume) solution of concentrated reagent-grade HCl. The vials contained 100 µL of the collected final fraction (in 2 M HNO3 and 18% TMAH), 20 µL of a 10 µg mL−1 Tellurium (Te, Assurance© 1000 mg L−1 Te) standard solution (internal standard) and 1.88 mL 18.2 MΩ H2O. To account for instrument memory, a solution containing only 2 M HNO3, 18% TMAH and Te was analyzed between each sample. Likewise, the wash solution used between samples contained 2 M HNO3 and 18% TMAH, but no Te.
Data processing comprised several steps, namely: removing xenon (Xe) interferences on m/z 129, noting that Xe is present in trace amounts in the argon carrier gas; correcting for instrumental mass fractionation; isolating memory effects; and quantifying procedural blanks. The process started by assigning an initial value for the instrumental mass bias correction factor, f (in this case, 6). This value was used together with raw 131Xe/126Te to calculate true (solar) fractionation-corrected 131Xe/126Te ratios, which were then used to determine the contribution of 126Te to the m/z 126 ion beam for each sample and blank. This allowed us to calculate raw Xe-corrected 126Te/125Te ratios, which could then be used to re-assess f according to Equation 2. The process was iterated three times to ensure convergence of the final f value. The next step involved correcting for the contribution of 129Xe to the 129 ion beam in a similar fashion, in order to obtain 129Xe-corrected 129I counts. The preceding blank in the instrumental run sequence was subtracted from every sample to correct for instrument memory. The resultant 129I/127I ratios were then corrected for instrumental mass fractionation. These corrected values were then used, together with the stock spike solution isotopic composition and the known volumes of added working solution, to calculate each sample’s initial 127I concentration in nM.
The incubation experiments were analyzed using a ThermoFinnigan© Neptune MC-ICP-MS, also situated in the WHOI Plasma Facility, according to the gas-based sparge approach described in Hardisty et al. (2020). This method involves introducing iodine into the instrument as a gas, by reacting it with a strong oxidizing agent, in this case concentrated HNO3. For all measurements, 4 mL of sample (in 2 M HNO3 and 18% TMAH) were combined with 2 mL of 18.2 MΩ H2O, and 4 mL of concentrated HNO3 were injected through a syringe to volatilize the iodine to I2 gas (Hardisty et al., 2020). To minimize the risk of extinguishing the plasma, the tubing connecting the sparge vial with the sample to the Neptune was disconnected upon HNO3 addition and reconnected after the first few seconds of reaction.
Corrections similar to those described above for the profile samples were applied in MATLAB (Mathworks©) to account for background iodine contributions, Xe interference and instrumental mass fractionation. Final values for 129I/127I ratios were weighted using an integration-by-integration approach based on the intensities of the 129- and 127-iodine beams (Hardisty et al., 2020).
2.6 Separation and analysis of DOI
Polycarbonate bottles, cleaned following the protocol described above, were used to collect 4L samples at various depths from the CTD27 cast (using the conventional rosette; Table 1; Figure 2). These samples were first filtered through 0.2 μm Pall Acropak-200 Supor filters before being extracted on Bond-Elut ENV (1 g, 6 mL, Agilent©) solid phase extraction (SPE) columns, by pumping each sample at a rate of 20 mL min−1. Before sample collection, the SPE columns were conditioned with three column volumes of methanol, followed by three column volumes of 18.2 MΩ H2O. The SPE columns were frozen at −20°C after extraction and shipped frozen to WHOI at the end of the cruise. Approximately one year after the cruise, the SPE columns were thawed, rinsed with 6 mL of 18.2 MΩ H2O to remove salts, and eluted with 6 mL of distilled methanol (Optima, Fischer Scientific©). To concentrate the DOI compounds in a final volume of 500 μL, the methanol extracts were placed in a vacuum centrifuge (SpeedVac, Thermo Scientific©) for 3.5 hours at 35°C.
The samples were analyzed via HPLC-ICP-MS, which allows the separation of iodine-bearing organic compounds in each sample based on the compounds’ polarity. A 20 μL sample aliquot was combined with 5 μL of a cobalamin solution (used as internal standard) and 25 μL of 18.2 MΩ H2O. A 10 μL aliquot of this solution was loaded onto a C18 column (0.5 x 150 mm, 3.5 μm particle size, Agilent©) connected to a Dionex Ultimate 3000 LC system (Thermo Scientific©). The LC system flow was directed into an iCAP Q (Thermo Scientific©) quadrupole MS at a rate of 20 μL min−1 using the instrument’s loading pump module. After a 10 minute initial hold at 95/5% A/B (A = 5mM ammonium formate solution in 18.2 MΩ H2O, B = 5mM ammonium formate solution in distilled methanol), the DOI compounds were separated using a 30-minute gradient to 5/95% A/B, followed by a 10-minute isocratic hold at 5/95% A/B. Helium was used as the collision gas and oxygen was introduced at 25 mL min−1 throughout the analysis, to prevent carbon deposits from forming on the ICP-MS interface. The intensity of the 127I ion beam was monitored in Kinetic Energy Discrimination mode using an integration time of 0.01 seconds.
3 Results
3.1 In situ measurements
3.1.1 Inorganic speciation
The concentrations of reduced, oxidized, total, and salinity-predicted iodine for the three vertical profiles are shown in Figure 3. All associated data are provided in Supplementary Table 1. Iodide and IO3− concentrations were determined using isotope dilution ICP-MS. Total iodine concentrations were calculated as the sum of I− and IO3−. The salinity-predicted iodine concentrations were estimated assuming a total iodine:salinity ratio of 15., which is the arithmetic mean of the seven samples measured at or below 500 m. In situ salinity was then used to predict total iodine at each depth, and any excess (or deficit) in iodine was quantified by comparing the salinity-predicted value to the measured total.
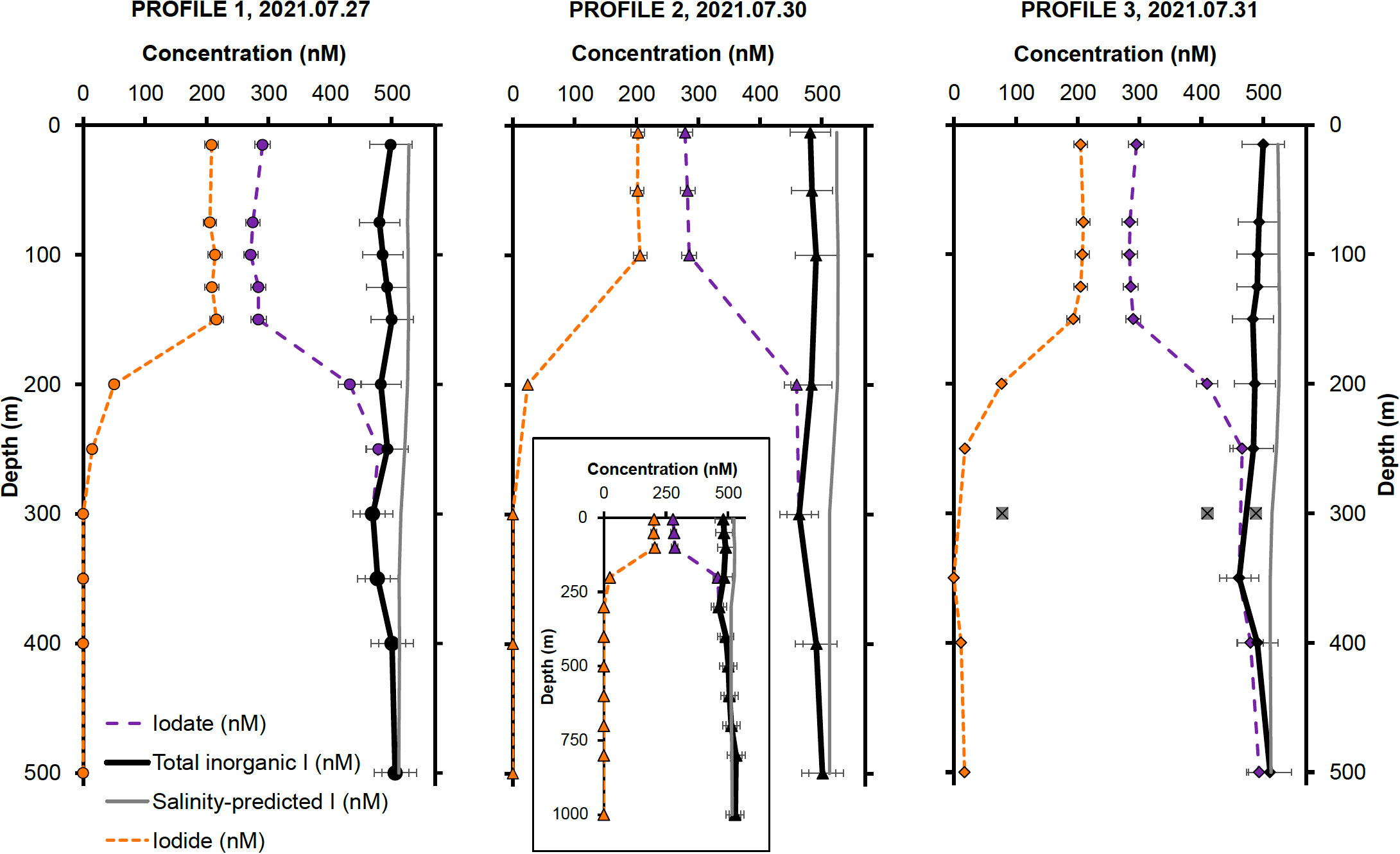
Figure 3 Inorganic iodine speciation and concentrations for Profile 1, Profile 2 and Profile 3, collected on July 27, 30 and 31, respectively. The outliers at 300 m in Profile 3 are most probably due to contamination during shipboard sampling or from bottles triggering at a different depth. Error bars represent two relative standard deviations from the arithmetic mean of all measurements for a particular iodine species (I−, IO3−) in the first 150 m of the water column. The inset in the Profile 2 panel shows iodine speciation concentrations in nM over the whole depth profile (1000 m). The symbols in each panel match the profile collection sites shown in Figure 2.
In addition to the samples, the I− and IO3− concentrations for two 18.2 MΩ H2O blanks and one 800 m sample (from Profile 2), which was analyzed twice, were processed to quantify contamination during processing. The 800 m sample was assumed to have negligible I− concentration, as expected of deep-water samples. The average of the two I− concentrations measured in this sample (4.5 nM) was used to calculate sample processing blanks and was subtracted from all I− measurements. Negative values obtained for all but one 18.2 MΩ H2O I− and IO3− blanks could not be used to determine process blanks. The limit of detection, calculated as 3 standard deviations of all I− and IO3 process blanks (excluding IO3− concentrations measured in the 800 m sample), was determined as 10.5 nM 3 SD, (n = 6). All I− concentrations below this value were considered below detection and assigned a value of zero.
Iodide concentrations are highest in the first 150 m of the water column, remaining relatively constant at 206 ± 11 nM ( ± 2 SD, n = 13). Below that, I− concentrations decrease rapidly to negligible values deeper than 300 m. The distribution of IO3− shows the opposite pattern, with lower concentrations of 284 ± 12 nM ( ± 2 SD, n = 13) in the first 150 m, followed by an increase to concentrations around 482 ± 60 nM ( ± 2 SD, n = 19) below this depth. As previously mentioned, it is expected for IO3− to dominate iodine speciation deeper in the water column, due to its thermodynamic stability under oxygenated conditions. These inorganic iodine concentration patterns agree well with previously reported measurements at Station ALOHA, which show high surface I− and corresponding low surface IO3− concentrations at low latitudes, characteristic of the Atlantic (Truesdale et al., 2000; Campos et al., 1999), Pacific (Tsunogai and Henmi, 1971) and Indian oceans (Chance et al., 2020) tropical waters. There is good agreement between the I− and IO3− distributions in all three CTD profiles, as shown in Figure 3.
We estimate the precision of our iodine speciation measurements using the profile data. To do so, we assumed that I− and IO3− were invariant over the top 150 m, and calculated the RSD of all I− and IO3− measurements in this depth range. Using this method, our measurement precision is estimated as ±4.3% for IO3−, ± 5.3% for I−, and ±6.8% (2 RSD) for total iodine concentrations.
A comparison of total iodine with salinity-predicted iodine reveals resolvable apparent iodine deficits near the sea surface. This deficit may signify a potential loss of iodine from surface waters, either to the atmosphere or deeper ocean, or it might denote an ‘apparent deficit,’ whereby certain iodine species are unresolved by our chromatographic methods. We discuss possible explanations in more detail in Section 4.3. Regardless of the cause, this apparent iodine deficit is lowest for Profile 3 (24 ± 0.5 nM in surface waters; ± 2 SD, n = 7) and highest for Profile 2 (43 ± 0.5 nM at the surface; ± 2 SD, n = 7). The apparent deficit constitutes 6% and 7% of salinity-predicted total iodine in the top 100 m for Profile 3 and Profile 1 respectively, and 8% for Profile 2.
3.1.2 Dissolved organic iodine
The chromatograms shown in Figure 4 correspond to samples collected at multiple depths. Each of the well-defined peaks represents one or several discrete DOI compounds, which elute at different times during the HPLC-ICP-MS analysis as a function of their polarities. More polar compounds elute early, while non-polar compounds elute toward the end of the analysis. Several such peaks of different intensities are observed in the 5 m sample. Fewer discrete peaks are present as depth increases, with three large peaks seen in the 200 m sample, but only one dominant peak in the 500 m and 1000 m samples. A major feature of the 5 m chromatogram is the presence of a large unresolved complex mixture, which appears as a rise in the baseline in Figure 4 and decreases in abundance deeper in the water column, as seen in the other chromatograms. The unresolved complex mixture represents DOI compounds that are present in seawater, but are not well resolved by the chromatographic methods used here. More unresolved compounds are present in the upper 200 m of the water column than at depth.
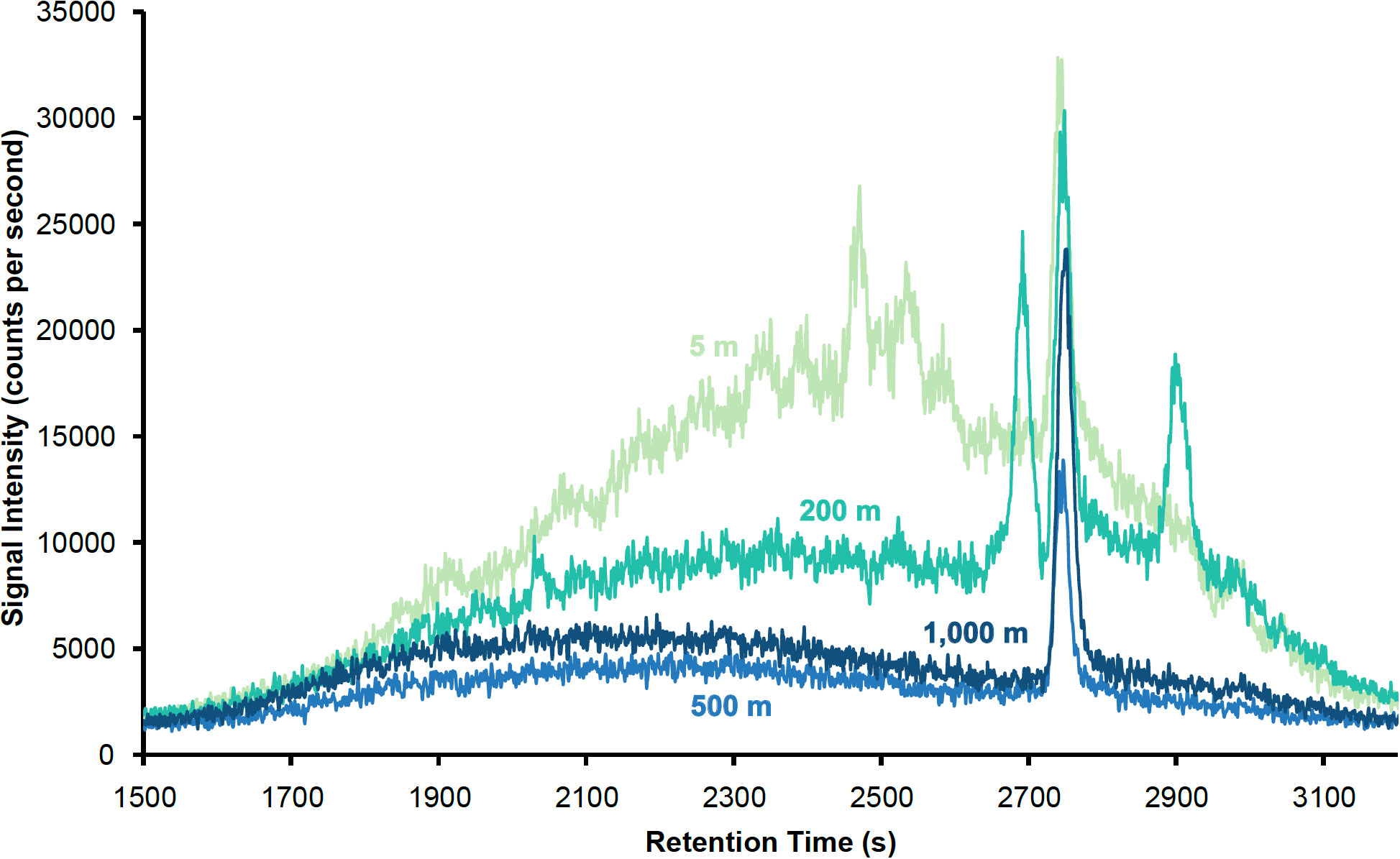
Figure 4 127I HPLC-ICP-MS chromatograms of samples collected from the CTD27 cast at 5, 200, 500 and 1000 m.
3.2 Rate experiments
We observe significant and systematic changes in (129I/127I)iodate during our 60-hour incubation experiments (Figure 5). Since some IO3− is present in the 129I− spike (Hardisty et al., 2020), all experiments start with (129I/127I)iodate ≈ 0.04. This value represents a mixture between IO3− added in the spike (129I/127I ≈ 3.6; Section 2.2.) and background IO3− present in seawater (129I/127I ≈ 0). Additionally, incubation samples were not homogenized prior to the start of the experiments, as each of the 60 mL bottles was filled and spiked separately. Therefore, the 129I/127I ratios at t=0 varied between individual samples, and this variation is a source of uncertainty between triplicates analyses. To calculate the precision of individual (129I/127I)iodate values, we performed a bootstrapped uncertainty estimation. Here we randomly selected three of the t0 samples and calculated their mean 129I/127I ratio, repeating the random selection 100,000 times. The initial IO3− isotopic ratio for all four experiments, (129I/127I)iodate = 0.0360, was assumed to be the mean ratio from the 100,000 iterations (dotted lines in Figure 5). The measurement uncertainty was calculated as two standard deviations of the bootstrapped means ( ± 0.00302; 2 SD; gray shaded areas in Figure 5), equivalent to an uncertainty of 8.3% (2 RSD). The 129I/127I ratio in the I− fraction for the rate experiments was also measured. If we apply the same uncertainty (8.3%) to the measured (129I/127I)iodide, no significant changes in the I− isotopic ratios are observed in any of the experiments (data shown in Supplementary Table 2). Therefore, our discussion is focused on investigating trends in (129I/127I)iodate.
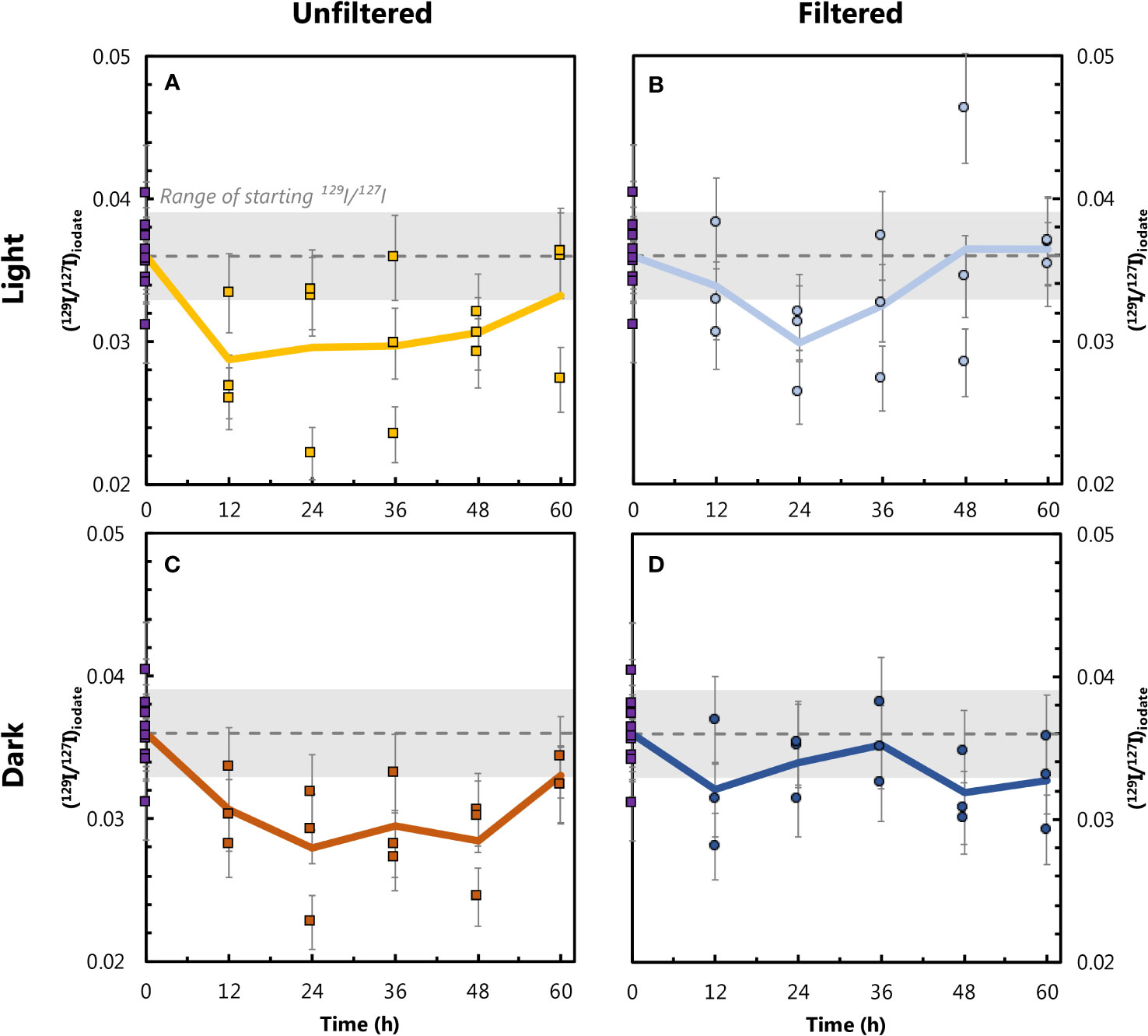
Figure 5 Changes in (129I/127I)iodate over the course of the incubation experiments for the unfiltered–light (A), filtered–light (B), unfiltered–dark (C) and filtered–dark (D) conditions. The dotted lines correspond to the average isotopic ratio at t=0 (0.036), generated via bootstrapping as described in the main text. The gray shaded areas correspond to two standard deviations resulting from the bootstrapping analysis. Error bars for triplicate data points represent 8.3% measurement precision uncertainty. The thick lines represent arithmetic means of triplicate measurements throughout the experiments.
Variations in (129I/127I)iodate across the four experiments ranged from 0.0280 to 0.0365, equivalent to relative changes from the initial value of 0.0360 of -22.3 and +1.3%, respectively (Figure 5; Table 3). Of the 60 time course measurements, 51 fall below the initial (129I/127I)iodate, although only the two unfiltered experiments remain outside the initial value during the 60-hour interval. Both unfiltered experiments have mean (129I/127I)iodate=0.030 between t=12 hours and t=60 hours, which is 16-17% lower than (129I/127I)iodate at t=0. In contrast, the filtered–dark experiment has a decrease of only 8% to 129I/127I=0.033, while the filtered–light experiment decreases by 6% to 129I/127I=0.034 over the last 48 hours. These smaller changes for the filtered experiments are within measurement uncertainty of the initial value. Beyond 48 hours, both unfiltered experiments show a slight increase in (129I/127I)iodate, with the final samples at 60 hours back to within uncertainty of the initial composition.
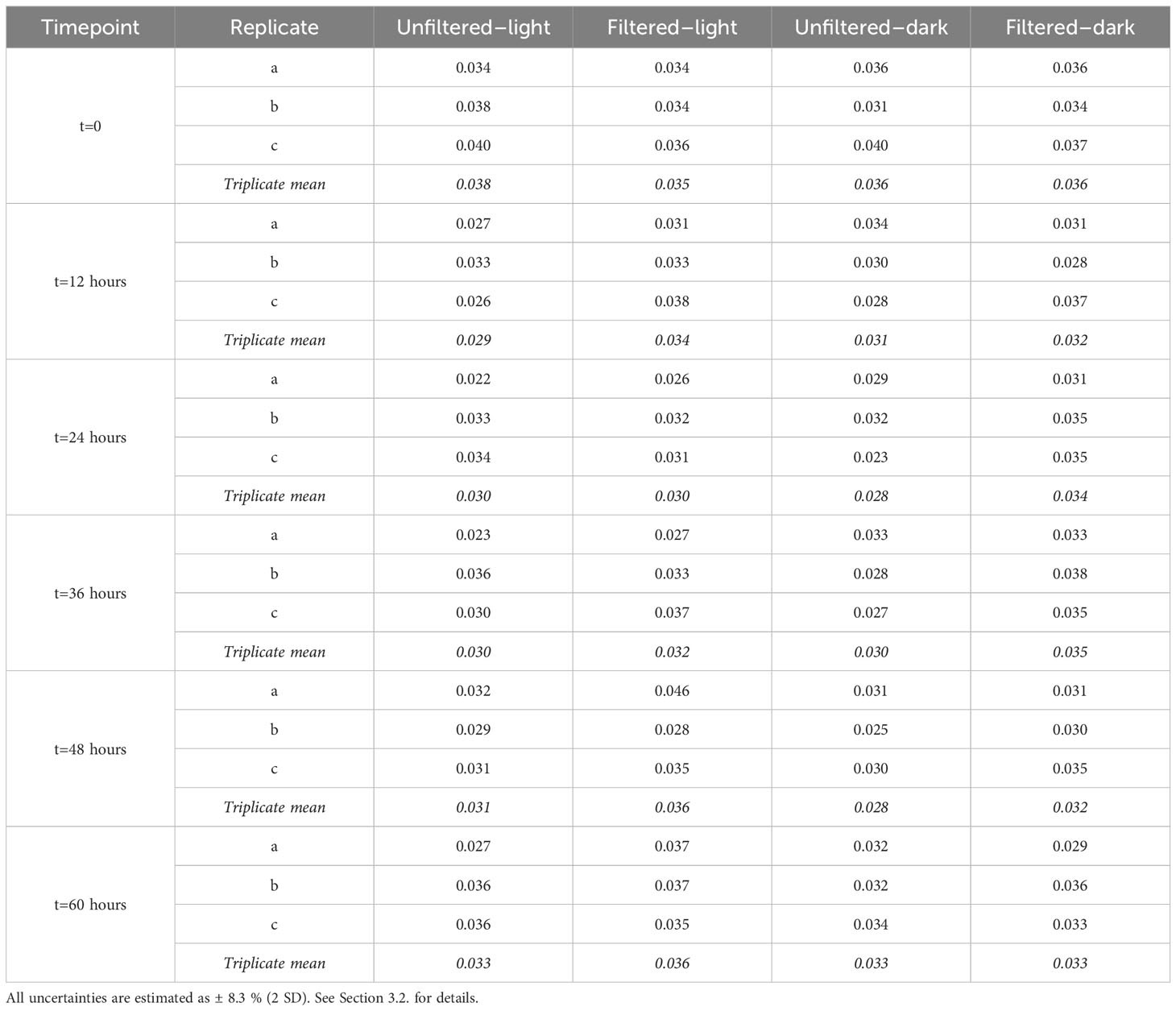
Table 3 Replicate measurements of iodate 129I/127I at each time point and arithmetic means of replicates for the four incubation experiments.
4 Discussion
4.1 Comparison with historical data
The concentration profiles chosen for comparison with our dataset are the June 1994 measurements reported in the seasonal study that Campos et al. (1996) conducted at the Hawaii Ocean Time series (HOT), centered around Station ALOHA. These profiles were selected because they were also sampled during summer, and they covered a depth range similar to the one used for our measurements. In their study, concentrations of I−, IO3− and total iodine were determined in the upper 500 m of the water column using voltammetry and differential pulse polarography (Takayanagi and Wong, 1986; Luther et al., 1988a), with higher measurement resolution in the top 250 m compared to the rest of the profiles (Figure 6A). For a more direct comparison between our study and that of Campos et al. (1996), a composite upper 500 m profile was created by averaging iodine concentrations from Profiles 1, 2 and 3 from this study at each depth (Figure 6B). Profile 2 values were used at depths 5 and 50 m.
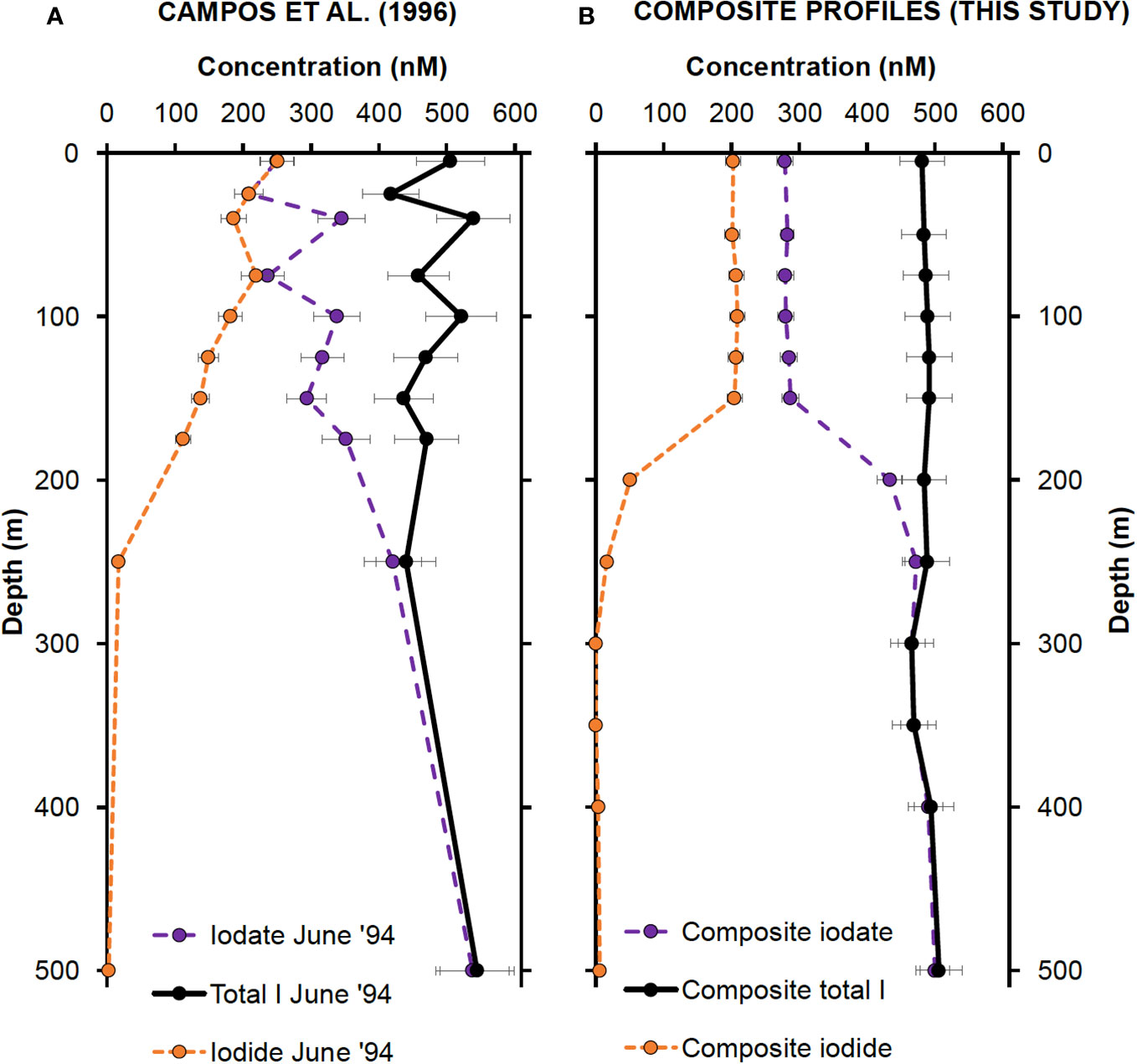
Figure 6 (A) Inorganic iodine speciation measurements made in June 1994 by Campos et al., 1996. The data were extracted graphically using the WebPlotDigitizer software (https://automeris.io/WebPlotDigitizer/). Error bars represent 10% uncertainty estimates. (B) Composite upper 500 m profiles created by averaging iodine concentrations from Profiles 1, 2 and 3 (this study) at each depth. Profile 2 values were used at depths 5 and 50 m. Error bars correspond to precision previously estimated for iodide, iodate and total iodine (5.3%, 4.3% and 6.8%, respectively).
The general shape of the profiles reported by Campos et al. (1996) for summer 1994 is similar to the results reported here, showing the characteristic decrease in I− and increase in IO3− with increasing water depth. However, one difference between the two studies is the rate of change in iodine concentrations between surface and deeper waters. Our results indicate a rapid change in iodine concentrations between 150 and 200 m depth, whereas Campos et al. (1996) record a more gradual decline in I− and increase in IO3− from the surface down to 250 m depth. Their results also show greater variability in iodine speciation in the upper 200 m, with IO3− concentrations changing by up to 140 nM between 25 and 40 m, for instance. The lack of variability in surface iodine concentrations reported in this study can mostly be attributed to the increased precision of our isotope dilution method compared to the conventional methods used by Campos et al. (1996). We believe that the smaller variations in the upper water column are indicative of our newer measurements being more oceanographically consistent.
Despite these differences in precision, however, there is good agreement between iodine concentrations measured almost 30 years apart at Station ALOHA. For instance, I− concentrations in the upper 100 m average 215 ± 52 nM in 1994 and 206 ± 11 nM for the three profiles analyzed in this study. This overall similarity increases our confidence in affirming that the results we report here are representative of general conditions around Station ALOHA. The long residence time of surface water at Station ALOHA (approximately 10 years) led Campos et al. (1996) to conclude that IO3− reduction by phytoplankton is the main source of I− to the euphotic zone. Thus, they invoked a correspondingly rapid rate of I− oxidation (560 nM/year) to explain why I− concentrations remain constant and do not approach total iodine concentrations over the residence time of the water.
It is also noteworthy that Campos et al. (1996) did not detect any evidence for iodine depletion at the time of their measurements. In contrast, we observe deficits in the top 100 m that constitute up to 8% of salinity-predicted total iodine. Our observations are consistent with data reported by Elderfield and Truesdale (1980), who noted that total iodine depletions of up to 13% are common in stratified tropical waters. To discern whether these deficits indicate actual losses of iodine to the atmosphere or deeper water, or if they represent apparent deficits due to organic iodine species that are unresolved during chromatography, we performed a direct examination of the characteristics of DOI, discussed next.
4.2 Identification of significant open-ocean DOI
In order to gain a more complete understanding of iodine cycling at Station ALOHA, we collected samples for DOI analysis. These samples provide insight into a less studied dimension of iodine cycling in the region, but also more generally in the open ocean, where it is widely believed that DOI does not constitute a significant component of the iodine pool (Luther et al., 1991; Wadley et al., 2020).
The dominant peak observed at all depths in Figure 4 is a common feature of all four chromatograms, indicating that the DOI compound(s) it corresponds to are found throughout the upper 1000 m of the water column at Station ALOHA, at higher concentration compared to the other DOI compounds resolved by this chromatographic separation. The overall decrease in the amount of unresolvable complex DOI compounds with increasing water depth suggests that the DOI fraction at Station ALOHA is more abundant and increasingly complex closer to the surface.
These results confirm the presence of a DOI fraction in the euphotic zone at Station ALOHA. Although identification of the individual compounds that comprise this fraction will have to await further processing, this analysis is the first step toward the characterization of DOI in this region. While it is challenging to provide absolute concentrations with the method as implemented here, a semi-quantitative analysis indicates that the most abundant discrete peaks correspond to compounds found in concentrations on the order of pM in seawater. However, only a fraction of the dissolved organic matter present in seawater might be retained by the SPE column, therefore these estimates are a lower bound on DOI concentrations.
4.3 Iodate formation rates during the incubation experiments
The results from the incubation experiments can be used to provide insight into the factors which determine the rate of IO3− formation in surface waters at Station ALOHA. Campos et al. (1996) assumed that horizontal advection processes in the region do not play an important role in iodine cycling, although they considered mixing out of the euphotic zone as a sink for I−. Assuming I− oxidation is a first-order reaction, the authors calculated the rate constant of this reaction to be 5.15 year−1. In this and the next sections, we use the results from the incubation experiments to revisit the magnitude of the contribution of in situ I− oxidation to the total sink of I− at Station ALOHA.
The goal of the incubation experiments was to observe and quantify the rate of IO3− formation. This was achieved by labeling the I− pool, as this is believed to be the primary substrate for IO3− formation. Were we to observe IO3− formation from I− oxidation over the course of the experiments, we would expect to see an increase in (129I/127I)iodate. Our experimental approach is analogous to how oxidation kinetics for other elements have been investigated in the past (for instance, Moffett, 1994).
We observe systematic differences in (129I/127I)iodate over the course of the experiments. Moreover, inter-experiment variations indicate that certain treatments affected IO3− formation rates. In the case of the filtered–light and filtered–dark experiments, the arithmetic means of the triplicates of most time points are within (or very close to) uncertainty of the starting composition, indicating no significant change in (129I/127I)iodate over the course of these incubations. However, the arithmetic means of the triplicates from the unfiltered experiments dip below the initial value after t=0. The largest decline in (129I/127I)iodate occurs in the first 12 hours of the unfiltered–light and unfiltered–dark incubations. For the unfiltered–light experiment, average (129I/127I)iodate at t=12 hours is 0.029, which is 19% lower than (129I/127I)iodate at t=0 (0.036). For the unfiltered–dark experiment, average (129I/127I)iodate decreases by 14% between t=0 and t=12 hours, from 0.036 to 0.031. This initial fast decline is followed by a period between t=12 hours and t=48 hours, during which (129I/127I)iodate shows little variation for both unfiltered experiments, remaining well below the initial isotopic composition. The unfiltered experiments show a slight increase in (129I/127I)iodate at t=60 hours, with the triplicates from both experiments averaging 0.033 (8% below the initial value). While it is not clear whether this trend would have continued beyond the final time point, it is plausible that this slight increase in (129I/127I)iodate reflects the oxidation of some spike-derived 129I− through to 129IO3−.
Iodate 129I/127I shows the most significant changes in experiments where particles were present. These particles are most likely a mixture of abiotic and biotic components, and they can have multiple roles in the formation of IO3−. For instance, abiotic particles containing iodine might dissolve during the experiments, adding 127I to the dissolved IO3− pool and causing the (129I/127I)iodate to decrease. Likewise, 127I could be added to the IO3− fraction through biotic particles, such as from cell lysis. We deem both possibilities unlikely. First, we are not aware of any abiotic particles that contain significant amounts of iodine. Second, seawater at Station ALOHA typically contains< 2 μg L−1 of organic carbon in living biomass (Karl and Church, 2014). If considering an iodine:carbon ratio of 10−4 for organic matter (Elderfield and Truesdale, 1980), complete oxidation of this material would add less than 17 pM to the IO3− pool. Microorganisms such as ammonia-oxidizing bacteria have been shown to have a catalytic role in IO3− formation, promoting I− oxidation to IO3− (Hughes et al., 2021). However, substantial ammonium oxidation rates have only been detected in the lower euphotic zone at Station ALOHA, below the primary nitrite maximum (Dore and Karl, 1996). This suggests that ammonia-oxidizing bacteria do not play a significant role in I− oxidation in surface waters at Station ALOHA. Due to a lack of culture-based studies focused on I− oxidation, it is unclear which other microbial groups are able to oxidize significant amounts of I− to IO3− in the ocean, although partial I− oxidation to small molecular weight organics has been previously documented for various classes of phytoplankton (Manley and de la Cuesta, 1997). Given that there are no obvious differences between the unfiltered–dark and unfiltered–light experiments, it is reasonable to infer that photoautotrophs do not contribute significantly to IO3− formation, at least over the course of our experiments.
The decrease in (129I/127I)iodate observed at the beginning of the unfiltered incubations indicates a small but rapid gain of 127I in the IO3− pool. The gain of 127I must originate from a pool of iodine that does not contain spike-derived 129I and is not resolved by anion-exchange chromatography. This pool likely contains iodine with oxidation states intermediate between that of I− (-1) and IO3− (+5; Figure 1). As noted in Section 1, several intermediate species are known to mediate I− oxidation to IO3− and would have existed in our incubation samples at the time of spiking. Our analyses do not allow us to confidently identify these intermediates, but we suspect that DOI and HOI are major components of this pool. Indeed, our results in Section 3.1.2. confirm the existence of a significant pool of DOI at Station ALOHA. Thus, for (129I/127I)iodate to initially decline in the unfiltered experiments, the transformation between intermediates and IO3− must be much faster than the net oxidation of I− to IO3−. Similarly, intermediates oxidation must be faster than the oxidation of I− into intermediates, assuming that I− must first pass through intermediates before being oxidized to IO3−.
In this context, it is reasonable to assume that the apparent iodine deficits inferred from our total iodine calculations can be accounted for by this pool of intermediates. The variability in apparent iodine deficits implied by our measurements suggests that changes in the speciation and distribution of intermediate iodine compounds can occur on small spatial and temporal scales, indicating a higher reactivity of this pool compared to I− and IO3−. As mentioned above, the largest changes in (129I/127I)iodate are recorded between t=0 and t=12 hours. Over this interval, the rapid decline in (129I/127I)iodate can be attributed to the transformation of intermediates into IO3−, and the measured isotopic ratios at the two initial time points can be used to estimate rate constants for this reaction.
Several studies have documented that the oxidized fraction separated during the second step of the chromatography (Section 2.4.) includes some DOI (Hou et al., 1999; Hardisty et al., 2020). At Station ALOHA, dissolved organic carbon concentrations average 56 μM in the first 1000 m of the water column (Zigah et al., 2017). Considering our sample size of 20 mL (for the profile samples) and an iodine:carbon ratio of 10-4 as before, this would correspond to approximately 5.6 nM added DOI, which is below our detection limit of 10.5 nM (Section 3.1.1.). Moreover, were the DOI and IO3− pools to be analyzed together, our results would not imply apparent iodine deficits in the surface, nor would they show a change in (129I/127I)iodate during the incubation experiments. Therefore, we believe that no significant amounts of DOI were resolved by our inorganic chromatographic separation.
4.4 Calculation of oxidation rates
The surface seawater used for the incubation experiments was collected from a fourth CTD cast (Table 1; Figure 2). Nevertheless, Profile 1 was the most proximal to this fourth cast, both chronologically and geographically. Therefore, for the purpose of estimating the rate of intermediates oxidation to IO3−, we considered the seawater 127IO3− concentration prior to spiking equal to that measured at 15 m depth in the TM5 profile (291 nM). The (129I/127I)iodate at t=0 was set to 0.036, as calculated from the bootstrapping analysis detailed in Section 3.2. Using this initial isotopic ratio, concentrations of 127IO3− and 129IO3− immediately after spike addition were determined. The (129I/127I)iodate values at t=12 hours were set to 0.029 and 0.031 for the unfiltered–light and unfiltered–dark experiments respectively (average of triplicates, Table 3). These calculations assume that the pool of intermediates is initially free of 129I and that there is no formation of 129IO3− during the first 12 hours. Given the uncertainty regarding the nature of the oxidation reactions, we calculated rate constants assuming zero, first and second order kinetics.
The reaction rate constants for the oxidation of intermediates to IO3− in the unfiltered experiments are summarized in Table 4. These rate constants were calculated as a function of the product (IO3−) and represent minimum gross rates at which IO3− is being generated from the oxidation of spike-free intermediates in surface waters. Our calculations do not account for possible concomitant reduction of IO3− to DOI, so these numbers represent net, rather than gross, oxidation rates over the first 12 hours. These rates correspond to a consumption of around 70 nM of intermediates during the initial 12 hours, which is around two times larger than our estimates for the apparent iodine deficits in any of the CTD profiles discussed in this study (the largest being 43 ± 0.5 nM at the surface for Profile 2). Considering zero-order kinetics, we estimate that between 34,000 (for the unfiltered–dark conditions) and 50,000 (for the unfiltered–light experiment) nM of intermediates oxidize to IO3− every year. For such high oxidation rates to be sustained, the pool of intermediates in seawater must be rapidly replenished, but these experiments do not shed light on this process.
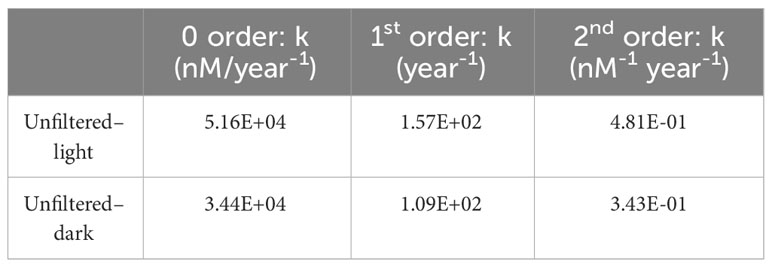
Table 4 Reaction rate constants for the oxidation of iodine intermediates into iodate, determined as a function of iodate concentrations and isotopic ratios in the unfiltered experiments for various reaction kinetics.
Our calculated rates of intermediates oxidation at Station ALOHA are extremely rapid relative to rates of I− oxidation to IO3−. Since (129I/127I)iodate ratios decrease systematically during the experiments, but do not increase significantly until t=60 hours, we infer that net I− to IO3− oxidation rates at Station ALOHA are slower than the rate of oxidation of intermediates to IO3−. Unfortunately, given the large uncertainty in reaction kinetics and the lack of constraints on the abundance and isotopic evolution of the intermediates, it is not possible to obtain more precise estimates at this time. The first order rate constant reported by Campos et al. (1996) of 5.15 year−1 is orders of magnitude smaller than our first order estimates for the oxidation of intermediates. However, the overall reaction rates also depend on the concentrations of the reactants. The I− pool is much larger than the pool of intermediates in seawater; in the absence of more information about the in situ formation of intermediate iodine species, we would therefore expect I− oxidation to be the dominant source of IO3− over longer time scales, as opposed to oxidation of intermediates, due to the fast exhaustion of this latter pool.
The slight increase in (129I/127I)iodate in the last 12 hours of the unfiltered experiments may be evidence of some 129I− being completely oxidized to IO3−. The trend can be seen in both light and dark unfiltered treatments; however, this trend remains tentative and longer-term incubation studies will be required to assess whether oxidation continues beyond this time.
We propose a conceptual model of iodine cycling in the oligotrophic ocean, schematically represented in Figure 1. Although we can estimate the contribution of only one of these arrows to total iodine cycling, this model could be referred to for future work aimed at disentangling the complex interactions between the numerous iodine species co-existing in the ocean. More information about the abundance and identity of DOI and iodine intermediates are needed in order to better constrain the variables included in this model and to confidently determine rates of I− oxidation in the surface ocean.
5 Conclusions
The results reported in this study offer an updated perspective on iodine speciation and cycling at Station ALOHA. Our methodological approaches include the application of isotope dilution and mass spectrometry to measure concentrations and isotopic ratios of various iodine pools with increased precision compared to historical speciation measurements. Inorganic iodine distributions at Station ALOHA are in agreement with global patterns showing increased surface I− concentrations in tropical waters and a shift toward IO3−-dominated waters below the euphotic zone. In general, there is good agreement between concentrations reported almost thirty years ago and measurements made in this study. Salinity-predicted iodine concentrations reveal the existence of an apparent iodine deficit in surface waters, which we suspect is composed of iodine species of intermediate oxidation state, such as HOI and DOI. Indeed, we collected and analyzed large volumes of seawater and obtained a profile of DOI, a component of the dissolved iodine pool which has previously been considered negligible in the open ocean. Although total iodine abundances are dominated by the inorganic forms, our results document the existence of DOI at Station ALOHA, showing systematic decreases in abundance and complexity with increasing water depth. While additional analyses will be needed to identify the organic compounds that comprise the DOI pool, our results demonstrate its complex nature. Short-term incubation experiments further reveal a fast gain of 127I in the IO3− pool that we attribute to rapid oxidation of to 127I -bearing intermediate species, such as DOI and HOI. Net I− oxidation to IO3− occurs slower than the oxidation of intermediates to IO3−; however, assignment of reaction rate constants depends heavily on reaction order, which remains unknown. The presence of particles accelerates IO3− formation, although light levels have no effect. Overall I− oxidation must be a slow process, as confirmed by a lack of significant increase in (129I/127I)iodate over the course of 60-hour incubations.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
I-MŞ: Formal Analysis, Investigation, Writing – original draft, Writing – review & editing, Methodology. TH: Conceptualization, Formal Analysis, Funding acquisition, Supervision, Visualization, Writing – review & editing, Methodology. DR: Conceptualization, Funding acquisition, Supervision, Writing – review & editing, Methodology. JB: Formal Analysis, Investigation, Writing – review & editing, Methodology.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the Simons Foundation (Simons Collaboration on Ocean Ecology [SCOPE] Award 329108 to DR), the U.S. National Science Foundation (to TH) and by the Breene M. Kerr Early Career Scientist Endowment Fund (OCE-2023456 to TH).
Acknowledgments
We thank the crew of R/V Kilo Moana for their help in collecting the samples and Maureen Auro for assisting with the chromatographic separation of inorganic iodine. We are grateful to Lydia Babcock-Adams, Jay Li and Matthew McIlvin for their assistance with running the HPLC and ICP-MS; Gretchen Swarr for help in the WHOI Plasma Facility; Dalton Hardisty for his valuable comments on the manuscript; Alexi Schnur for sharing helpful insights about the laboratory protocols; and Michael Dotzel for expertise with exploring multiple modeling approaches.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1272968/full#supplementary-material
References
Amachi S. (2008). Microbial contribution to global iodine cycling: volatilization, accumulation, reduction, oxidation and sorption of iodine. Microbes Environ. 23, 269–276. doi: 10.1264/jsme2.ME08548
Bluhm K., Croot P., Wuttig K., Lochte K. (2010). Transformation of iodate to iodide in marine phytoplankton driven by cell senescence. Aquat. Biol. 11 (1), 1–15. doi: 10.3354/ab00284
Boiteau R. M., Fitzsimmons J. N., Repeta D. J., Boyle E. A. (2013). Detection of iron ligands in seawater and marine cyanobacteria cultures by high-performance liquid chromatography – inductively coupled plasma-mass spectroscopy. Analytical Chem. 85, 4357–4362. doi: 10.1021/ac3034568
Boiteau R. M., Repeta D. J. (2015). An extended siderophore suite from Synechococcus sp. PCC 7002 revealed by LC-ICPMS-ESIMS. Metallomics 7, 877–884. doi: 10.1039/C5MT00005J
Campos M. L. A. M. (1997). New approach to evaluating dissolved iodine speciation in natural waters using cathodic stripping voltammetry and a storage study for preserving iodine species. Mar. Chem. 57, 107–117. doi: 10.1016/S0304-4203(96)00093-X
Campos M. L. A. M., Farrenkopf A. M., Jickells T. D., Luther G. W. III (1996). A comparison of dissolved iodine cycling at the Bermuda Atlantic Time-series Station and Hawaii Ocean Time-series Station. Deep Sea Res. II 43 (2-3), 455–466. doi: 10.1016/0967-0645(95)00100-X
Campos M. L. A. M., Sanders R., Jickells T. (1999). The dissolved iodate and iodide distribution in the South Atlantic from the Weddell Sea to Brazil. Mar. Chem. 65, 167–175. doi: 10.1016/S0304-4203(98)00094-2
Carpenter L. J., Chance R. J., Sherwen T., Adams T. J., Ball S. M., Evans M. J., et al. (2021). Marine iodine emissions in a changing world. Proc. to R. Soc. A 477, 20200824. doi: 10.1098/rspa.2020.0824
Chance R., Baker A., Carpenter L., Jickells T. (2014). The distribution of iodide at the sea surface. Environ. Sciences: Processes Impacts 16 (8), 1841–1859. doi: 10.1039/C4EM00139G
Chance R., Malin G., Jickells T., Baker A. (2007). Reduction of iodate to iodide by cold water diatom cultures. Mar. Chem. 105 (1-2), 169–180. doi: 10.1016/j.marchem.2006.06.008
Chance R., Tinel L., Sarkar A., Sinha A., Mahajan A., Chacko R., et al. (2020). Surface inorganic iodine speciation in the Indian and southern oceans from 12°N to 70°S. Front. Mar. Sci. 7. doi: 10.3389/fmars.2020.00621
Dore J. E., Karl D. M. (1996). Nitrification in the euphotic zone as a source for nitrite, nitrate and nitrous oxide at Station ALOHA. Limnology Oceanography 41, 1619–1628. doi: 10.4319/lo.1996.41.8.1619
Edwards A., Truesdale V. W. (1997). Regeneration of inorganic iodine species in Loch Etive, a natural leaky incubator. Estuarine Coast. Shelf Sci. 45, 357–366. doi: 10.1006/ecss.1996.0185
Elderfield H., Truesdale V. W. (1980). On the biophilic nature of iodine in seawater. Earth Planetary Sci. Lett. 50, 105–114. doi: 10.1016/0012-821X(80)90122-3
Fuge R., Johnson C. C. (2015). Iodine and human health, the role of environmental geochemistry and diet, a review. Appl. Geochemistry 63, 282–302. doi: 10.1016/j.apgeochem.2015.09.013
Hardisty D., Horner T., Evans N., Moriyasu R., Babbin A., Wankel S., et al. (2021). Limited iodate reduction in shipboard seawater incubations from the Eastern Tropical North Pacific oxygen deficient zone. Earth Planetary Sci. Lett. 554, 116676. doi: 10.1016/j.epsl.2020.116676
Hardisty D. S., Horner T. J., Wankel S. D., Blusztajn J., Nielsen S. G. (2020). Experimental observations of marine iodide oxidation using a novel sparge-interface MC-ICP-MS technique. Chem. Geology 532, 119360. doi: 10.1016/j.chemgeo.2019.119360
He P., Hou X., Aldahan A., Possnert G., Yi P. (2013). Iodine isotopes species finger-printing environmental conditions in surface water along the northeastern Atlantic Ocean. Sci. Rep. 3, 2685. doi: 10.1038/srep02685
Hepach H., Hughes C., Hogg K., Collings S., Chance R. (2020). Senescence as the main driver of iodide release from a diverse range of marine phytoplankton. Biogeosciences 17, 2453–2471. doi: 10.5194/bg-17-2453-2020. 2020.
Herring J. R., Liss P. S. (1974). A new method for the determination of iodine species in seawater. Deep Sea Res. 21, 777–783. doi: 10.1016/0011-7471(74)90085-0
Hou X., Dahlgaard H., Rietz B., Jacobsen U., Nielsen S. P., Aarkrog A. (1999). Determination of chemical species of iodine in seawater by radiochemical neutron activation analysis combined with ion-exchange preseparation. Analytical Chem. 71 (14), 2745–2750. doi: 10.1021/ac9813639
Hughes C., Barton E., Hepach H., Chance R., Pickering M., Hogg K., et al. (2021). Oxidation of iodide to iodate by cultures of marine ammonia-oxidising bacteria. Mar. Chem. 234, 104000. doi: 10.1016/j.marchem.2021.104000
Jickells T. D., Boyd S. S., Knap A. H. (1988). Iodine cycling in the Sargasso Sea and the Bermuda inshore waters. Mar. Chem. 24, 61–82. doi: 10.1016/0304-4203(88)90006-0
Jones M. R., Chance R., Dadic R., Hannula H.-R., May R., Ward M., et al. (2023). Environmental iodine speciation quantification in seawater and snow using ion exchange chromatography and UV spectrophotometric detection. Analytica Chimica Acta 1239, 340700. doi: 10.1016/j.aca.2022.340700
Karl D. M., Church M. J. (2014). Microbial oceanography at the Hawaii Ocean Time-series programme. Nat. Reviwes 12, 699–713. doi: 10.1038/nrmicro3333
Li J., Boiteau R. M., Babcock-Adams L., Acker M., Song Z., McIlvin M. R., et al. (2021). Element-selective targeting of nutrient metabolites in environmental samples by inductively coupled plasma mass spectrometry and electrospray ionization mass spectrometry. Front. Mar. Sci. 8, 630494. doi: 10.3389/fmars.2021.630494
Liss P. S., Herring J. R., Goldberg E. D. (1973). The iodide/iodate system in seawater as a possible measure of redox potential. Nat. Phys. Sci. 242, 108–109. doi: 10.1038/physci242108a0
Lu W., Ridgwell A., Thomas E., Hardisty D., Luo G., Algeo T., et al. (2018). Late inception of a resiliently oxygenated upper ocean. Science 361, 174–177. doi: 10.1126/science.aar5372
Lu Z., Lu W., Rickaby R., Thomas E. (2020). Earth History of Oxygen and the iprOxy (Elements in Geochemical Tracers in Earth System Science) (Cambridge: Cambridge University Press). doi: 10.1017/9781108688604
Luther G. W. III (2023). Review on the physical chemistry of iodine transformations in the oceans. Front. Mar. Sci. 10. doi: 10.3389/fmars.2023.1085618
Luther G. W. III, Branson-Swartz C., Ullman W. J. (1988a). Direct determination of iodide in seawater by cathodic stripping square wave voltammetry. Analytical Chem. 60, 1721–1724. doi: 10.1021/ac00168a017
Luther G. W. III, Ferdelman T., Culberson C. H., Kostka J., Wu J. F. (1991). Iodine chemistry in the water column of the Chesapeake Bay: Evidence for organic iodine forms. Estuarine Coast. Shelf Sci. 32 (3), 267–279. doi: 10.1016/0272-7714(91)90020-C
Manley S. L., de la Cuesta J. L. (1997). Methyl iodide production from marine phytoplankton cultures. Limnology oceanography 42 (1), 142–147. doi: 10.4319/lo.1997.42.1.0142
Moffett J. W. (1994). A radiotracer study of cerium and manganese uptake onto suspended particles in Chesapeake Bay. Geochimica Cosmochimica Acta 58 (2), 695–703. doi: 10.1016/0016-7037(94)90499-5
Moriyasu R., Bolster K. M., Hardisty D. S., Kadko D. C., Stephens M. P., Moffett J. W. (2023). Meridional survey of the central Pacific reveals iodide accumulation in equatorial surface waters and benthic sources in abyssal plain. Global Biogeochemical Cycles 37, e2021GB007300. doi: 10.1029/2021GB007300
Moriyasu R., Evans N., Bolster K. M., Hardisty D. S., Moffett J. W. (2020). The distribution and redox speciation of iodine in the Eastern Tropical North Pacific Ocean. Global Biogeochemical Cycles 34, e2019GB006302. doi: 10.1029/2019GB006302
Qi Y., Matsuzaki H., Yamagata T., Nagai H. (2023). Iodine cycling in the subarctic Pacific Ocean: insights from 129I. Geochimica Cosmochimica Acta 344, 12–23. doi: 10.1016/j.gca.2023.01.006
Rue E. L., Smith G. J., Cutter G. A., Bruland K. W. (1997). The response of trace element redox couples to suboxic conditions in the water column. Deep Sea Res. Part I: Oceanographic Res. Papers 44 (1), 113–134. doi: 10.1016/S0967-0637(96)00088-X
Sherwen T., Chance R. J., Tinel L., Ellis D., Evans M. J., Carpenter L. J. (2019). A machine-learning-based global sea-surface iodide distribution. Earth System Sci. Data 11, 1239–1262. doi: 10.5194/essd-11-1239-2019
Sherwen T. M., Evans M. J., Spracklen D. V., Carpenter L. J., Chance R., Baker A. R., et al. (2016a). Global modeling of tropospheric iodine aerosol. Geophysical Res. Lett. 43, 10,012–10,019. doi: 10.1002/2016GL070062
Sherwen T. M., Schmidt J. A., Evans M. J., Carpenter L. J., Großmann K., Eastham S. D., et al. (2016b). Global impacts of tropospheric halogens (Cl, Br, I) on oxidants and composition in GEOS-Chem. Atmospheric Chem. Phys. 16, 12,239–12,271. doi: 10.5194/acp-16-12239-2016
Solomon S., Garcia R. R., Ravishankara A. R. (1994). On the role of iodine in ozone depletion. J. Geophysical Res. 99 (D10), 20,491–20,499. doi: 10.1029/94JD02028
Stracke A., Scherer E. E., Reynolds B. C. (2014). Application of isotope dilution in geochemistry. Treatise geochemistry second edition 15, 71–86. doi: 10.1016/B978-0-08-095975-7.01404-2
Sun Y.-J., Robinson L.F., Parkinson I.J., Stewart J.A., Lu W., Hardisty D. S., Liu Q., Kershaw J., LaVigne M., et al. (2023). Iodine-to-calcium ratios in deepsea scleractinian and bamboo corals. EFront. Mar. Sci. 10, 1264380. doi: 10.3389/fmars.2023.1264380
Takayanagi K., Wong G. T. F. (1986). The oxidation of iodide to iodate for the polarographic determination of total iodine in natural waters. Talanta 33, 451–454. doi: 10.1016/0039-9140(86)80115-1
Truesdale V. W., Bale A. J., Woodward E. M. S. (2000). The meridional distribution of dissolved iodine in near-surface waters of the Atlantic Ocean. Prog. Oceanography 45, 387–400. doi: 10.1016/S0079-6611(00)00009-4
Truesdale V. W., Jones K. (2000). Steady-state mixing of iodine in shelf seas off the British Isles. Continental shelf Res. 20, 1889–1905. doi: 10.1016/S0278-4343(00)00050-9
Tsunogai S., Henmi T. (1971). Iodine in the surface water of the Pacific Ocean. J. Oceanographical Soc. Japan 27, 67–72. doi: 10.1007/BF02109332
van Bergeijk S., Hernández L., Zubía E., Cañavate J. (2016). Iodine balance, growth and biochemical composition of three marine microalgae cultured under various inorganic iodine concentrations. Mar. Biol. 163 (5). doi: 10.1007/s00227-016-2884-0
Wadley M. R., Stevens D. P., Jickells T. D., Hughes C., Chance R., Hepach H., et al. (2020). A global model for iodine speciation in the upper ocean. Global Biogeochemical Cycles 34, e2019GB006467. doi: 10.1029/2019GB006467
Wong G. T. F., Brewer P. G. (1974). The determination and distribution of iodate in South Atlantic waters. J. Mar. Res. 32, 25–36.
Keywords: Station ALOHA, iodate formation, organic iodine, iodine intermediates, oxidation mechanisms
Citation: Ştreangă I-M, Repeta DJ, Blusztajn JS and Horner TJ (2024) Speciation and cycling of iodine in the subtropical North Pacific Ocean. Front. Mar. Sci. 10:1272968. doi: 10.3389/fmars.2023.1272968
Received: 04 August 2023; Accepted: 13 December 2023;
Published: 08 January 2024.
Edited by:
Gregory A. Cutter, Old Dominion University, United StatesReviewed by:
Martha Gledhill, Helmholtz Association of German Research Centres (HZ), GermanyMaija Iris Heller, University of Galway, Ireland
Copyright © 2024 Ştreangă, Repeta, Blusztajn and Horner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Iulia-Mădălina Ştreangă, aXVsaWFtc0BtaXQuZWR1