 Alexi A. Schnur
Alexi A. Schnur Kevin M. Sutherland2
Kevin M. Sutherland2 Colleen M. Hansel
Colleen M. Hansel Dalton S. Hardisty
Dalton S. Hardisty- 1Department of Earth and Environmental Sciences, Michigan State University, East Lansing, MI, United States
- 2Department of Earth and Planetary Sciences, Harvard University, Cambridge, MA, United States
- 3Department of Marine Chemistry and Geochemistry, Woods Hole Oceanographic Institution, Woods Hole, MA, United States
The distribution of iodine in the surface ocean – of which iodide-iodine is a large destructor of tropospheric ozone (O3) – can be attributed to both in situ (i.e., biological) and ex situ (i.e., mixing) drivers. Currently, uncertainty regarding the rates and mechanisms of iodide (I-) oxidation render it difficult to distinguish the importance of in situ reactions vs ex situ mixing in driving iodine’s distribution, thus leading to uncertainty in climatological ozone atmospheric models. It has been hypothesized that reactive oxygen species (ROS), such as superoxide (O2•−) or hydrogen peroxide (H2O2), may be needed for I- oxidation to occur at the sea surface, but this has yet to be demonstrated in natural marine waters. To test the role of ROS in iodine redox transformations, shipboard isotope tracer incubations were conducted as part of the Bermuda Atlantic Time Series (BATS) in the Sargasso Sea in September of 2018. Incubation trials evaluated the effects of ROS (O2•−, H2O2) on iodine redox transformations over time and at euphotic and sub-photic depths. Rates of I- oxidation were assessed using a 129I- tracer (t1/2 ~15.7 Myr) added to all incubations, and 129I/127I ratios of individual iodine species (I-, IO3-). Our results show a lack of I- oxidation to IO3- within the resolution of our tracer approach – i.e., <2.99 nM/day, or <1091.4 nM/yr. In addition, we present new ROS data from BATS and compare our iodine speciation profiles to that from two previous studies conducted at BATS, which demonstrate long-term iodine stability. These results indicate that ex situ processes, such as vertical mixing, may play an important role in broader iodine species’ distribution in this and similar regions.
1 Introduction
Iodine is a redox-sensitive element that is found ubiquitously in the surface ocean at an average concentration of about 450 nM (Elderfield and Truesdale, 1980; Chance et al., 2014; Moriyasu et al., 2023). Knowledge of the distribution of iodine’s two major redox species – iodide (I-) and iodate (IO3-) – at the sea surface is important for our understanding of iodine’s role in atmospheric cycles through the destruction of ozone (O3) by I-, a significant O3 sink (Carpenter et al., 2013; Chance et al., 2014; Luhar et al., 2017). The destruction of O3 by I• releases hypoiodous acid (HOI) and I2 to the atmosphere, which photolyzes to I atoms and continues to break down atmospheric O3 (Carpenter et al., 2013). An understanding of the rates and mechanisms that contribute to the distribution of I- and IO3- at the sea surface can aid in our understanding of tropospheric O3 destruction and its importance in the cycles of global climate change and impact on air quality.
In well-oxygenated portions of the open ocean, IO3- is found at levels exceeding 250 nM at the surface, increasing in concentration with depth in sub-photic waters (>350 nM). In much of the open ocean water column, I- concentrations are found to be inversely correlated with [IO3-]; however, I- is consistently found to be present in larger amounts (up to 250 nM) than would be expected if O2 were the direct oxidant of I- in fully oxygenated surface waters (Chance et al., 2014). Indeed, O2 is not thermodynamically favored to fully oxidize I- to IO3- and the oxidant responsible for the reaction is unknown (Luther et al., 1995; Luther, 2023). Given multiple known pathways of IO3- reduction, it is clear however that I- oxidation is occurring within marine waters but is likely sluggish (1.5-560 nM/yr) (Campos et al., 1996; Edwards and Truesdale, 1997; Truesdale et al., 2001b; Žic and Branica, 2006; He et al., 2013; Hardisty et al., 2020; Hughes et al., 2021). For example, in situ reduction of IO3- to I- by phytoplankton and bacteria is known to be a major pathway through which I- accumulates in areas of generally high primary productivity, such as upwelling zones and along coasts (Bluhm et al., 2010). Some iodine may be assimilated and later released during cell senescence (Hepach et al., 2020) and may account for “missing iodine” that is found in mass balance calculations of these areas.
The extracellular production of reactive oxygen species (ROS) by oxygenic photo- and heterotrophic bacteria promotes a variety of cell functions (Hansel and Diaz, 2021) and may also aid in the oxidation of I- in surface waters. Extracellular O2•− production by these bacteria varies as a function of species, where it ranges from 0.1-3.7 amol cell-1 h-1 (heterotrophs) to 4.3-13,400 amol cell-1 h-1 (oxygenic phototrophs) (Diaz et al., 2013; Sutherland et al., 2020). Similarly, bacteria and phytoplankton are also sources of H2O2 to the marine environment, both through the secretion of intracellular pools and extracellular production. Extracellular O2•− production by Roseobacter sp. AzwK-3b – 15-20% of coastal bacterial communities (Bond et al., 2020) – was shown in cell cultures to promote I- oxidation in the absence of Mn2+, which is preferentially oxidized (Li et al., 2014; Hansel et al., 2019) (Figure 1). Oxidation is thought to be completed extracellularly through the aid of these ROS (Li et al., 2014). This mechanism has yet to be tested under ambient marine conditions.
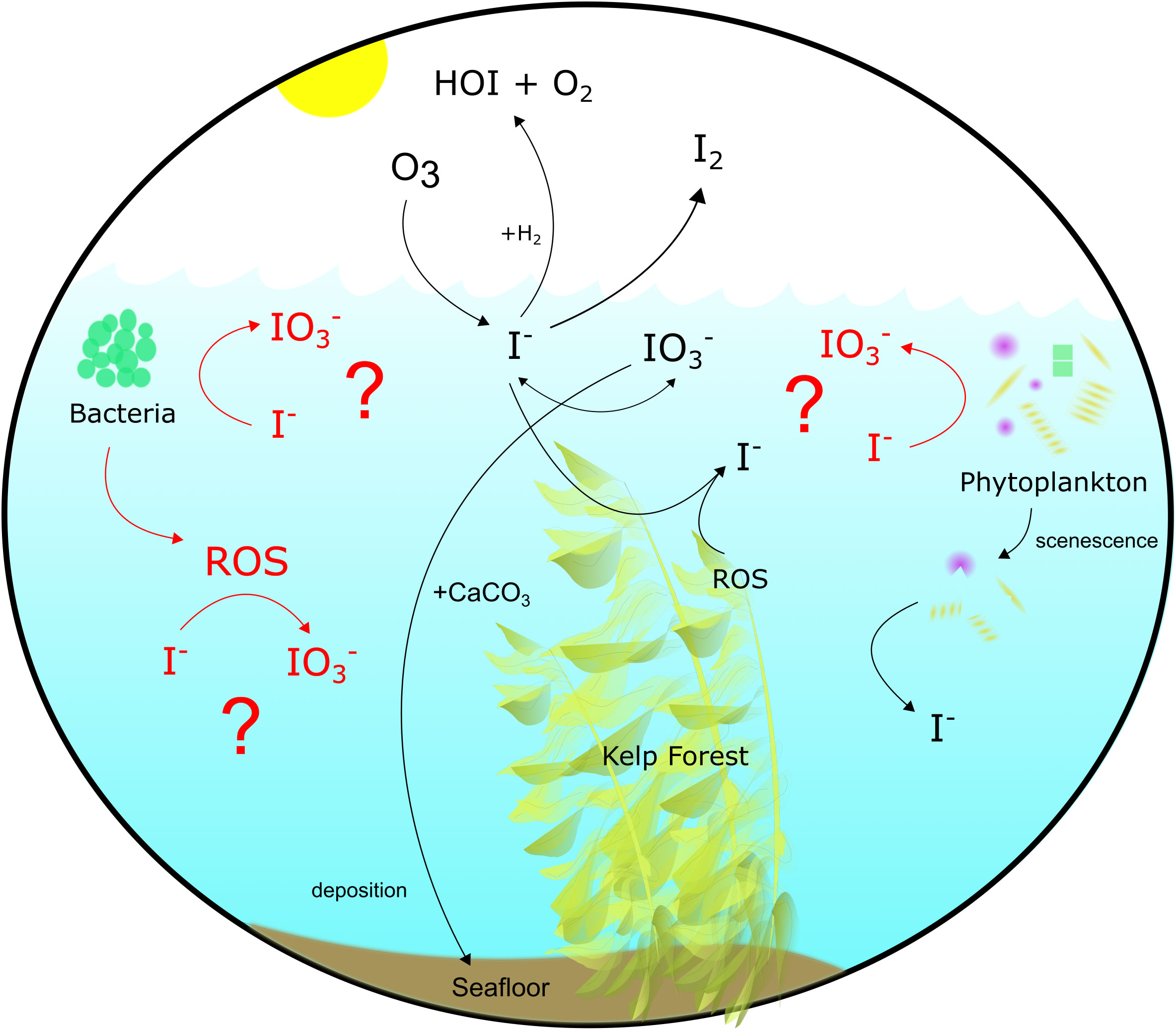
Figure 1 Iodine cycling in the modern surface ocean. Processes highlighted in red were examined in this study.
Here, we performed shipboard 129I radiotracer incubations under ambient seawater conditions to investigate the role of ROS in I- oxidation processes at the Bermuda Atlantic Time Series (BATS) in September 2018. 129I has a half-life of ~15.7 Ma and is therefore useful as a tracer on timescales of decades or less (Hardisty et al., 2020; Hardisty et al., 2021). To build on previous studies, the ROS H2O2 and O2•− were added to experiments at levels analogous to natural seawater concentrations to investigate their effect on oxidation of I- to IO3-, or vice versa. In addition, we have measured iodine speciation and O2•− in depth profiles from the BATS and the adjacent Hydrostation S. Our iodine speciation is compared to previous measurements from 1993 to 1994 and 1984 to 1985 (Jickells et al., 1988; Campos et al., 1996), thus providing the first long-term intercomparison for marine iodine.
2 Methods
2.1 Sample collection
Seawater was collected from the Bermuda Atlantic Time Series (BATS) and Hydrostation S sites in the Sargasso Sea in September 2018. Depth profile investigations at BATS were taken at 32.343°N 64.594°W at 21 separate depths between 1 m and 4500 m. Hydrostation S samples were taken at 32.165°N 64.501°W at 10 depths between 1 m and 500 m. Incubation water was taken from two depths (1 m and 240 m) and collected into four carboys (two euphotic (1 m) and two subphotic (240 m)) between 20:30 and 22:30 ADT. One carboy from each depth was filtered using a 0.2 µm filter to remove bacteria and other biology and particles while another was left unfiltered. 129I (t1/2 15.7 My) (Eckert and Ziegler Isotope Products ©) (Hardisty et al., 2020; Hardisty et al., 2021), was added directly to each of the carboys at a targeted concentration of ~70 nM 129I- for investigating iodine redox reactions in natural seawater over time. 129I- was added before aliquoting the carboy water for individual incubations to ensure homogenous 129I- concentrations at t0 for all incubations. 200 ml from each carboy were fractionated into separated incubation containers. Samples for t0 were immediately subsampled from spiked incubation containers, with this and subsequent (t1, t2, tf) subsamples being ~50 ml. All subsamples were immediately filtered at 0.2 µm to end interaction with biology after sampling. Subsamples were refrigerated and stored at 4°C until they returned to Michigan State University and were frozen for storage. Campos (1997) showed that seawater samples stored refrigerated (4°C) or frozen (-20°C) did not show signs of degradation in total iodine measurements over a one-to-three-year period – well over the eight-month timeframe in which measurement of these samples began after collection.
2.2 Incubation setup
Five incubation factors were used to create 20 incubation trials using a ship-deck light-filtering incubator to mimic at-depth light filtration, cooled with a continuous resupply of ambient surface seawater and stored in translucent and amber high-density polyethylene (HDPE) Nalgene bottles for dark incubations: each done in triplicate (Table 1). Factors included: 1) filtering of samples through a 0.2 µm syringe filter, meant as a control to screen filtered seawater of bacteria and macro-organisms and particles, kept in either the light or the dark depending on incubation, (Campos et al., 1996; Farrenkopf et al., 1997; Hardisty et al., 2020); 2) addition of O2•− dismutase (SOD) to incubations both filtered and unfiltered, but all left in the dark, intended as a control to remove ambient O2•− in seawater (Li et al., 2012; Diaz et al., 2013; Sutherland et al., 2020); 3) addition of superoxide thermal source (SOTS) or hydrogen peroxide (H2O2) to filtered samples kept in the dark in separate experiments, both suspected of being able to aid in oxidation of I- to IO3- in seawater; 4) unfiltered water in the dark to determine the role, if any, of photochemical reactions that may cause the reduction of IO3- to I- in the presence of organic matter (Spokes and Liss, 1996; Chance et al., 2014); 5) additions of MnCl2 to iterations of the above in order to consider the potential of preferential Mn2+ oxidation relative to I-. Note that controls 2 and 5 were only relevant if I- oxidation was detected in the other controls.
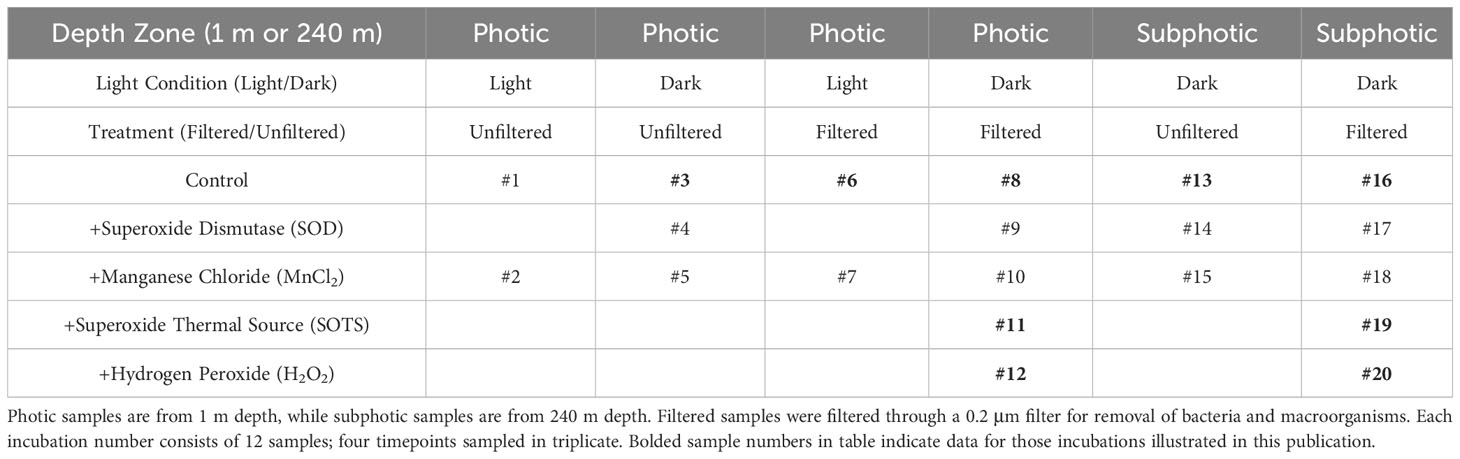
Table 1 Factor setup and inclusion for all 20 incubation trials and controls.
Superoxide thermal source was kept frozen (-80°C) until it was added daily by pipette to incubations 11 and 19 (Table 1) as a combination of 1 ml dimethyl sulfoxide (DMSO) + 1 mg SOTS (3028 µM SOTS) (Cayman Chemicals, CAS number 223507-96-8) and diluted to 15 µM SOTS within seawater samples, which provides ≥25 nM O2•− at the surface water temperatures at BATS and Hydrostation S (Heller and Croot, 2010). This was made fresh daily immediately before adding to samples and added daily to account for natural decay. The O2•− concentration of the SOTS stock was not analyzed but O2•− concentration was analyzed in one experiment a few hours post-SOTS addition – to allow to reach steady state concentrations – to confirm O2•− accumulation. Hydrogen peroxide (30%) was added at a volume targeting 50 nM H2O2 in each solution. SOD was added by pipette daily – thus accounting for decay and titration via potentially newly formed O2•− within the incubations – from a stock concentration of 4 kU/ml to incubations to produce samples with SOD concentration of 0.32 kU/ml. Given potential long oxidation timescales of I-, all incubations were performed over a 140-hour time period, with subsamples collected for iodine species measurement at t0, ~t40, ~t88, and ~t140 hours.
2.3 Analytical methods
2.3.1 Superoxide
The steady-state concentration of O2•− was determined as previously described with some minor modifications (Sutherland et al., 2020). Water samples were collected using 12 L Ocean Test Equipment bottles on a 24-position Sea-Bird CTD rosette. Samples were transferred into dark, acid washed bottles and measured between 30 minutes and six hours of the collection time. Thirty minutes was chosen as a sample delay period because it is greater than 10 half-lives of O2•− in typical marine waters, meaning that any O2•− remaining is the result of light-independent O2•− production by microbial communities in the bottles (Roe et al., 2016). Samples collected above the thermocline were incubated on deck with continuously flowing surface water (28.2°C and 29.2°C at Hydrostation S and BATS, respectively) and samples below the thermocline were incubated at 4°C.
Superoxide concentrations were measured using an FeLume Mini (Waterville Analytical) and the O2•−-specific chemiluminescent probe methyl cypridina luciferin analog (MCLA, Santa Cruz Biotechnology, Rose et al., 2008) stored at 4°C. Recent work using these methods has demonstrated that filtration of natural seawater can produce additional O2•− (Roe et al., 2016). To avoid introducing this bias into sample measurements, we used the following equation:
where represents the measured concentration of O2•− in unfiltered seawater (USW) and represents the concentration of O2•− in aged (>24 hours) filtered (0.2 µm Sterivex filter) seawater (AFSW) amended with 75 µM diethylene-triaminepentaacetic acid (DTPA) to complex any metals present in the sample. Each measurement consisted of running a 25 mL USW sample through the FeLume system (3 mL/min) for several minutes until a steady signal was recorded. After a steady signal was recorded, 2 μL superoxide dismutase (SOD; Superoxide Dismutase from bovine erythrocytes >3,000 U/mg, Sigma, stock prepared in DI water to 4,000 U/mL) was added to the sample to quench all O2•− in the sample. The same procedure was followed for the AFSW samples. The reported O2•− concentrations represent the difference between the USW and the AFSW concentrations, the latter allowing us to eliminate the portion of the measured signal due to MCLA auto-oxidation in each particular sample matrix. Calibration curves were generated daily from three or more paired observations of time-zero O2•− concentration (dependent variable) and chemiluminescence (independent variable) using linear regression. Separate calibration curves were used for each of the two storage temperatures. Because chemiluminescence values were baseline-corrected, regression lines were forced through the origin. Calibrations yielded highly linear curves (typically R2 >0.9), with a typical sensitivity of one chemiluminescence unit per pM O2•−.
2.3.2 Spectrophotometry
Iodate was measured using the spectrophotometric method outlined in Jickells et al. (1988), using 10% potassium iodide (KI) solution and 1.5 M sulfamic acid (H3NSO3). 10% KI was made fresh daily. Samples were measured using a VWR UV-Vis Scanning 3100 PC spectrophotometer and accompanying UV-Vis Analyst software using VWR® Two-Sided Disposable Plastic Cuvettes for measurements within the visible range (300-900 nm), path length 10 mm. Fisherbrand® Semi-Micro Quartz Cuvettes (Cat. No. 14-958-126) for wavelengths 200-2500 nm, were used for repeated measurements of samples 3.5 years after the initial IO3- measurements, with similar results obtained.
Jickells’ (1988) method of IO3- reaction with excess I- in acidic conditions yields triiodide (I3-) and is specific to IO3- (Jickells et al., 1988; Moriyasu et al., 2020; Moriyasu et al., 2023). Triiodide in reacted samples was measured at a ~320 nm trough, with the lowest point being found between 300 and 350 nm, peak at 350 nm, and secondary trough at 400 nm (). The concentrations of IO3- were calculated from these values using the equation:
where mstandard curve is the value of the slope of the standard curve created with potassium iodate (KIO3-), calculated between zero and 500 nM KIO3- and calibrated using standard additions of KIO3- to seawater.
2.3.3 Ion chromatography
We used a previously established ion-exchange chromatography method (Wong and Brewer, 1977; Hou et al., 1999; Hou et al., 2001; Hou et al., 2007; Hou et al., 2009; Hardisty et al., 2020; Hardisty et al., 2021; Moriyasu et al., 2023) to separate I-, IO3-, and DOI from natural seawater samples. Iodide fractions were measured via ICP-MS for I- concentrations (see section 2.3.4) since yields are known to reach ~100% (Hardisty et al., 2020), and then subsequently measured for 129I/127I ratios via MC-ICP-MS (see section 2.3.5). Previous IO3- yields have been found to commonly be between 90-95% (Hou et al., 1999; Hou et al., 2001; Hou et al., 2007; Hou et al., 2009), thus, IO3- and DOI fractions were only measured for 129I/127I ratios and not used to quantify concentrations from ICP-MS. Spot-checks on IO3- fractions were completed on ICP-MS to test reproducibility between spectrophotometric and ICP-MS measurements, with 82 to >100% agreement between methods for [IO3-] measured (Table 2). Glass columns used for iodine redox species separation were packed with PYREX glass wool and 1 ml (volumetric) AG1-X8 resin that was cleaned of any residual iodine after packing using one full ion-exchange chromatography “cleaning” procedure (substituting sample for 18.2 M-cm water) before samples were run through columns for collection of iodine redox species using the same chromatography procedure. About 10 ml of each sample – which was quantified gravimetrically before addition – were used during each procedure.

Table 2 IO3- and DOI spot-check measurements compared via spectrophotometry and ICP-MS.
The ion chromatography procedure specifically elutes I- from the seawater matrix. Iodate and DOI were independently separated prior to the I- elution step. The DOI and IO3- fractions were then reduced to I- using concentrated hydrochloric acid (HCl) and 0.3 M sodium bisulfite (NaHSO3) at pH <2 (Hou et al., 1999; Hou et al., 2009; Reifenhaüser and Heumann, 1990; Hardisty et al., 2020). Samples were left overnight and then run through ion exchange chromatography columns the next day, using the same I- elution procedure described above in an eluent of 18% TMAH/2 M HNO3. Like the samples, the eluent mass was determined gravimetrically, which together were used for determining concentrations, when relevant. The eluent was then directly diluted for measurement via ICP-MS and/or MC-ICP-MS.
For quality control, a 200 ppb I- solution diluted from a 1000 ± 4 ug/ml I- solution in 1% tetraethyl ammonium (TEA) (I-, DOI) or KIO3- (IO3-) standard was run alongside samples through columns as a monitor of iodine elution efficiency from columns to estimate yields. At least two 18.2 M-cm water blanks were also run as monitors of contamination with sample sets. At least one sample replicate was also included in each column set for assessment of reproducibility between column runs.
2.3.4 ICP-MS
[127I-] was measured by a Thermoscientific iCap triple-quad inductively coupled plasma mass spectrometer (ICP-MS-TQ) using Qtegra software version 2.10.3324.131 in both single-quad (SQ) and triple-quad (TQ) mode with O2 reaction cell gas. A Teledyne ASX 520 autosampler was used to deliver liquid solution to the ICP-MS. Samples analyzed by ICP-MS were diluted 1:20 or 1:40 in a 0.9% tetramethyl ammonium hydroxide (TMAH)/0.1 M nitric acid (HNO3) or 0.45% TMAH/0.05 M HNO3 solution, respectively. The same dilutions were used for ICP-MS rinse solutions. Data was corrected relative to the internal standards In, Rh, and Cs. Internal standards used were from Inorganic Ventures© – In was a 1001 ± 3 ug/ml solution in 2% HNO3; Rh was a 999 ± 5 ug/ml solution in 15% HCl; and Cs was a 1000 ± 4 ug/ml solution in 0.1% HNO – to create a 5 ppb internal standard solution that was spiked into each measured sample directly or using an inline mixing chamber. The 127I standard used for creating standard curves and column standard samples was a 1000 ± 4 ug/ml I- solution in 1% tetraethylammonium (TEA).
2.3.5 MC-ICP-MS ratios (129I/127I)
Iodine isotope ratios (129I/127I) were measured at Woods Hole Oceanographic Institution (WHOI) via a ThermoFinnegan Neptune MC-ICP-MS according to Hardisty et al. (2020). Specific ion beams mass/charge (m/z) were monitored for Te (126, 128, 130) and Xe (126, 128, 129, 130, 131, 132) isotopes, as well as 132Ba and 127I and 129I in faraday cups L3-L1 and H1-H3 with m/z 129 in the center position. Tuning was completed before running samples each morning to optimize beam intensity. A 500 ppb Te solution (Inorganic Ventures©) was used to account for mass bias corrections. Corrections needed for isobaric interferences were tracked via 131Xe.
We utilized a gas-based “sparge” method for iodine sample introduction and desolvation with a 300 uL/min quartz nebulizer for Ar carrier gas Te solution introduction, using a regular sample cone and x-type skimmer cone (Hardisty et al., 2020; Hardisty et al., 2021). 30 ml Teflon vials, outfitted with pre-formed “sparge caps” that allowed for Ar gas flow through the sample, held ≤6 ml sample (solution containing fractions representing I-, DOI, or IO3-). Teflon vials used for running samples were cleaned before use with each sample in 50% nitric acid for >3 hours at 90°C, then rinsed with 18.2 M-cm water and allowed to air dry in hood until next use. Teflon tubing connecting samples to the Neptune intake were changed regularly to inhibit cross-contamination between sample runs. Before connection to the torch, Ar gas flow rate was decreased to ~0.1 L min-1 and Ar was run through the connected sample for one minute to purge air out of the sample container before connecting to the torch. After the sample was connected to the torch, the gas rate was increased to ~1.2 L min-1. Te signal was monitored for stabilization, increasing to a value of 3-7 V. The sample run was started after Te signal stabilized, and then 4 ml concentrated 70% HNO3 was added upstream of the sample vial to induce volatilization of iodine samples. The sum of nitric and iodine eluent was kept <10 ml to allow for headspace to prevent bubbling over, which can inadvertently introduce liquid upstream of the sample vial, preventing sample measurement. Data were corrected as described in Hardisty et al. (2020), for a final 129I/127I ratio and standard deviation output.
3 Results
3.1 Iodine speciation and superoxide in depth profiles
Depth profiles of I- and IO3- (nM), O2•− (pM), temperature (°C), and dissolved oxygen (μM) at BATS and Hydrostation S are detailed in Figure 2 and show predictable changes throughout depth of IO3- and I-. The dark, particle-associated superoxide steady-state concentration at BATS and Hydrostation S span 4-720 pM through the water column between 1 m and 4553 m at BATS and Hydrostation S sampling sites. Superoxide concentrations were highest between the surface and 1000 meters and fall within the range of previously reported water column values (Hansard et al., 2010; Rusak et al., 2011; Roe et al., 2016). Temperature and dissolved oxygen values are available from cruise CTD data at http://batsftp.bios.edu/. Depth profiles of I- and IO3- mirror that of previous studies of iodine in an oxygenated seawater, with I- accumulation in the surface elevated at the expense of IO3- and with iodine below the euphotic zone being almost completely IO3- except for the bottom water sample at 4500 m which has relatively elevated I-. We also show I- and IO3- reported from previous studies at BATS which were limited to shallower depths (2500 m) than that studied here (4500 m).
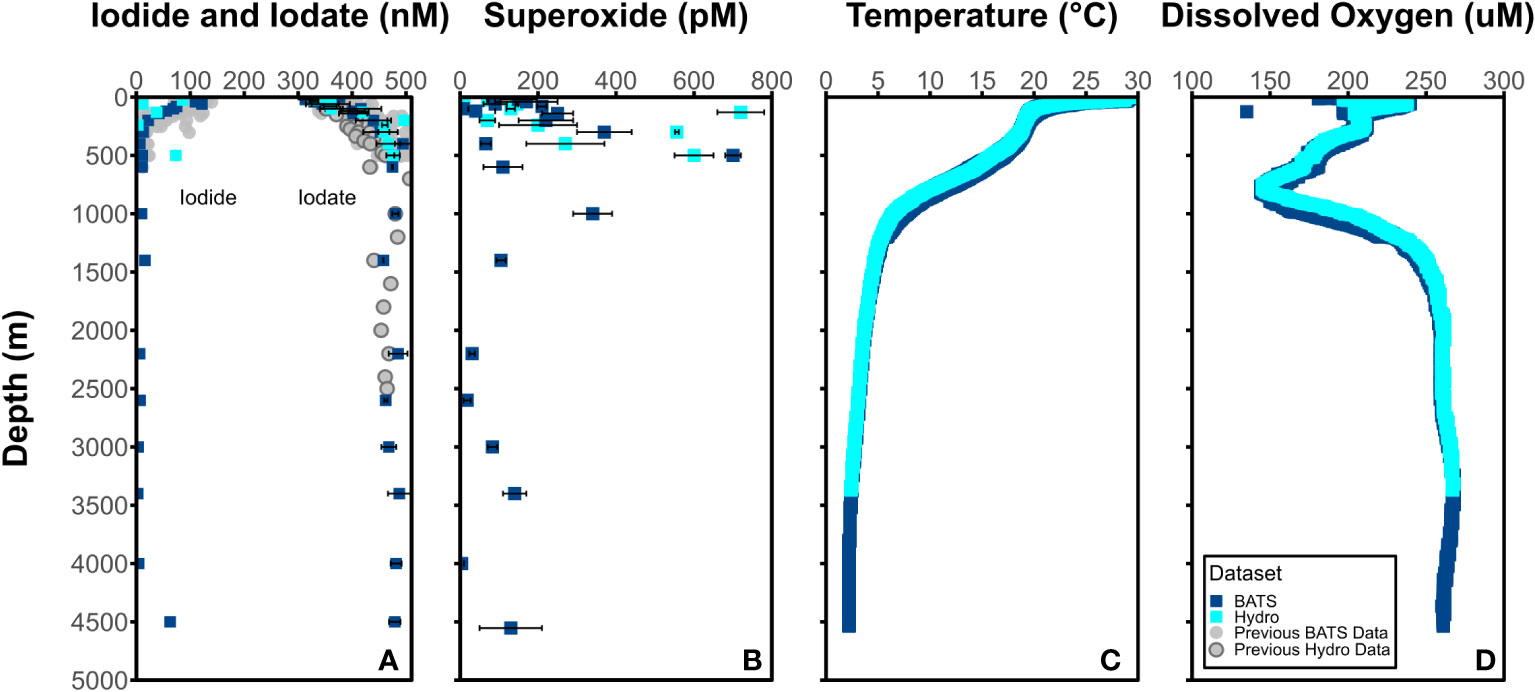
Figure 2 Concentrations of (A) I- (left) and IO3- (right) (nM) and (B) O2•− (pM) at BATS (blue square) and Hydrostation S (cyan square) stations. Average (C) temperature (°C) and (D) dissolved oxygen (μM) from BATS and Hydrostation S stations also included.
It is notable that there appears to be elevated superoxide concentrations at some intermediate water sample depths (e.g., 500 m). This is somewhat at odds with the expectation that light-independent superoxide production will scale with cell abundance and activity. While we did not measure additional biological parameters that might allow us to interpret these concentrations, it is apparent that elevated superoxide concentration do coincide with significant dissolved oxygen gradients. It is possible that elevated superoxide is either a direct or indirect result of remineralization processes occurring at these depths.
3.2 Iodine measurements from incubations
In incubation samples, initial [IO3-] values measured spectrophotometrically were found to be within the range of 209 nM to 452 nM, while I- separated from incubation samples via ion-exchange chromatography was found to be in the range of 92 nM to 235 nM from ICP-MS measurements (Table 2, Figures 3–5). Iodate values were spot-checked via exchange chromatography and ICP-MS and were found to be consistent with spectrophotometric measurements but showed a larger standard deviation in most cases, consistent with potential variability in yields (Table 2). It is interesting to note that, in some cases, filtered conditions have a lower measured [IO3-] than unfiltered conditions (Figure 3A, subphotic depth).
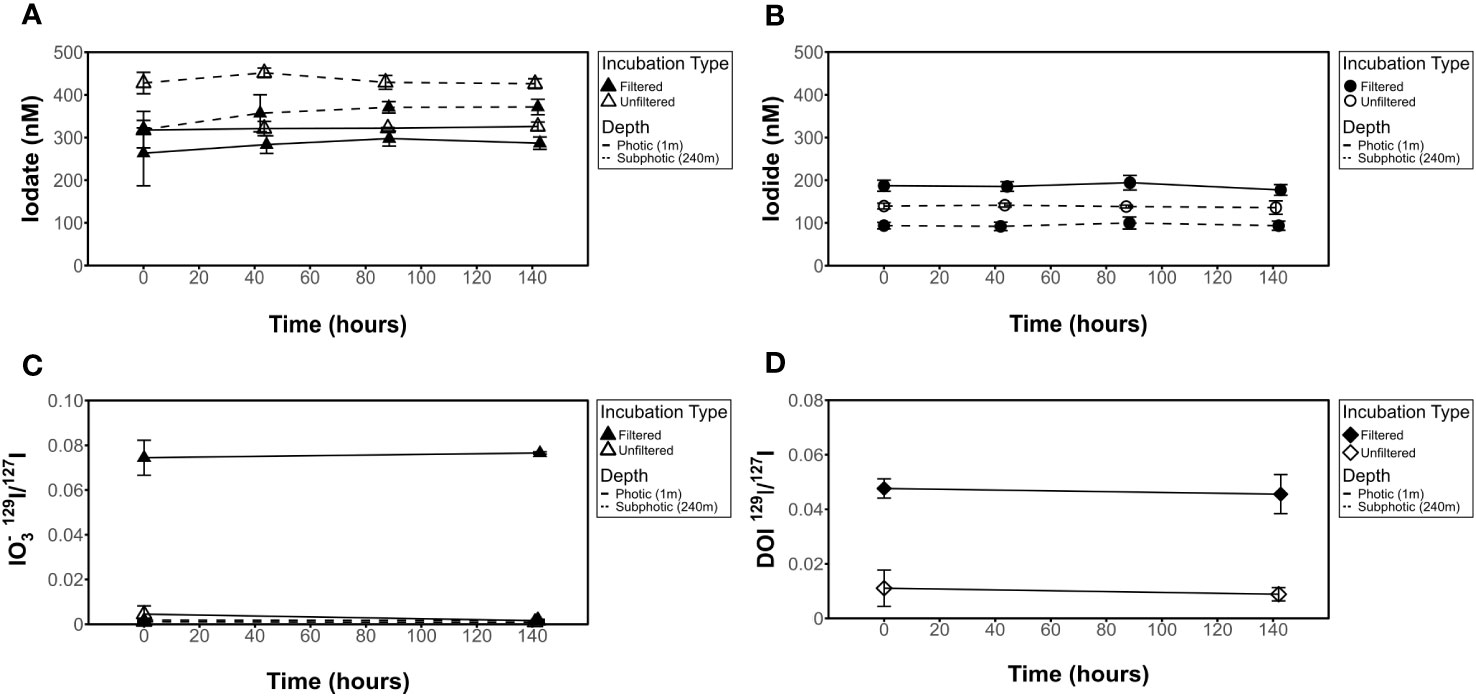
Figure 3 Influence of 0.2 μm filter on [IO3-], [I-], IO3- 129I/127I ratios, and DOI 129I/127I ratios for dark incubations. No change seen in (A) [IO3-], (B) [I-], (C) 129I/127I IO3- isotope ratio, or (D) 129I/127I DOI isotope ratio results in filtered (incubations 8 and 16) or unfiltered (incubations 3 and 13) experiments.
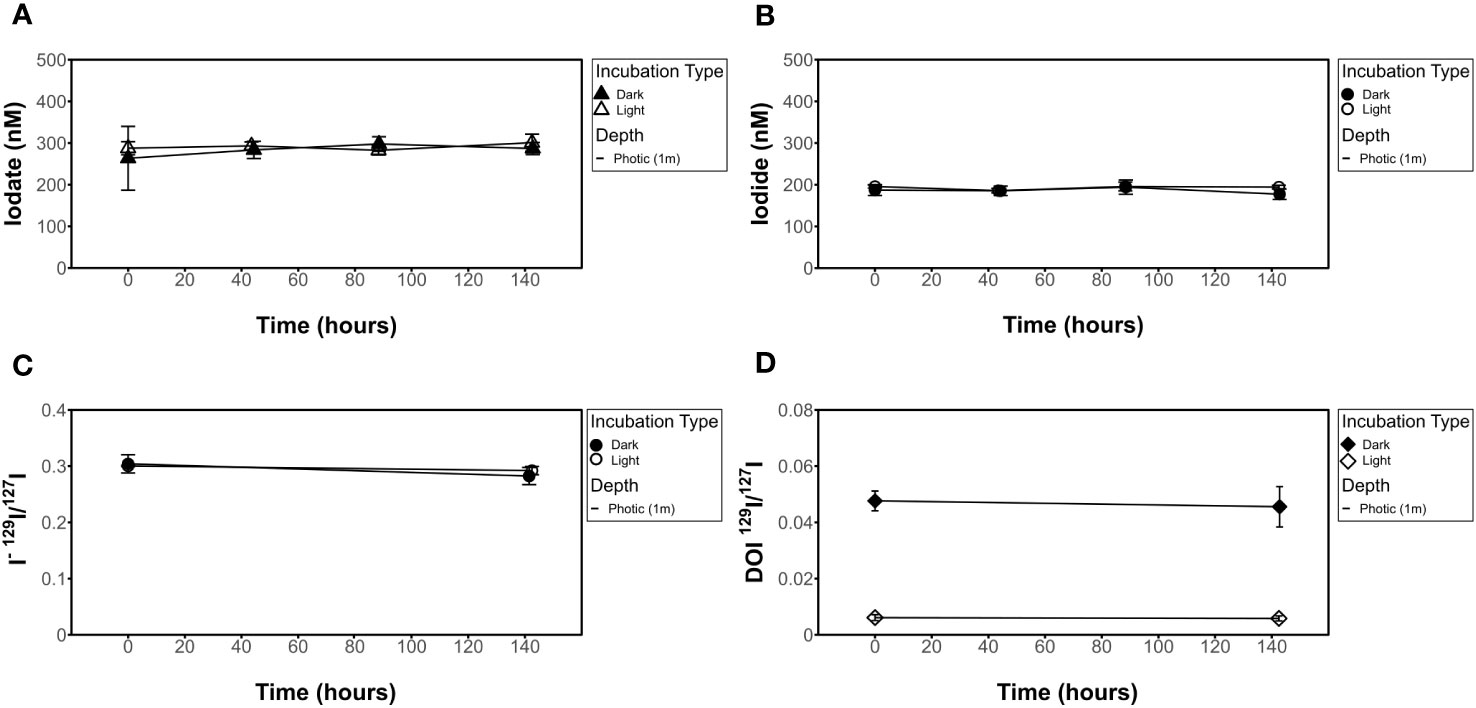
Figure 4 Influence of light on filtered seawater for [IO3-], [I-], I- 129I/127I ratios, and DOI 129I/127I ratios. No change seen in (A) [IO3-], (B) [I-], (C) 129I/127I I- isotope ratio, or (D) 129I/127I DOI isotope ratio results in light (incubation 6) vs dark (incubation 8) experiments.
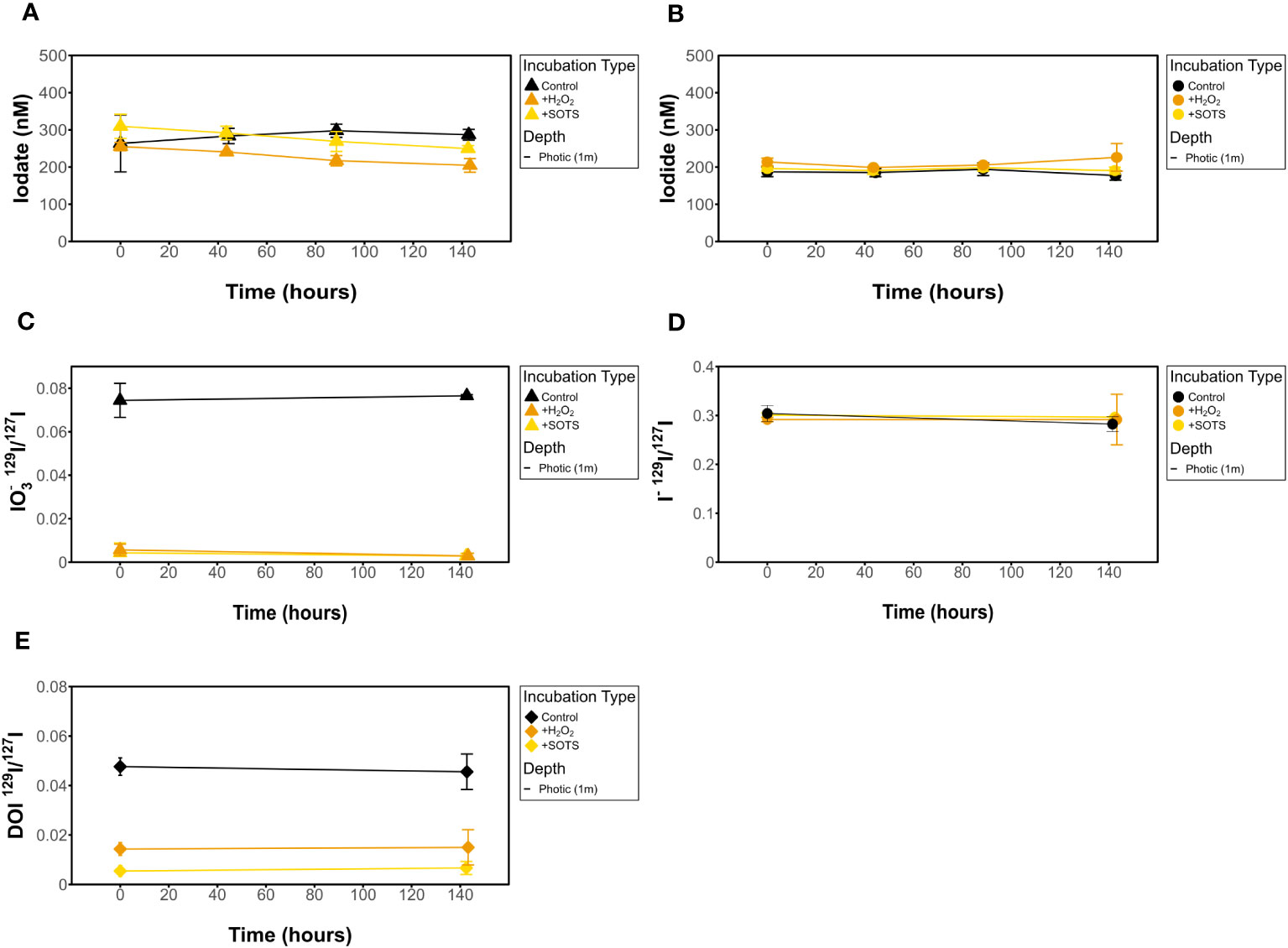
Figure 5 Influence of H2O2 and SOTS on filtered (A) [IO3-], (B) [I-], (C) IO3- 129I/127I ratios, (D) I- 129I/127I ratios, and (E) DOI 129I/127I ratios for dark incubations.
Measured [I-] shows no change over time in any of the incubations investigated (Figures 4–6). This is also true for [IO3-] for most incubation trials; however, IO3- values in incubations that included sequential additions of O2•− (10 nM/24 hrs) and H2O2 (50 nM/48 hrs) showed a decrease in [IO3-] over time, from 309 nM to 249 nM and 255 nM to 204 nM, respectively. Given an interference causing a baseline shift in these incubations (discussed in Supplementary Information), these samples were corrected for by selecting the minimum trough value between 300 nm and 350 nm for spectrophotometer concentration calculations (section 2.3.2) instead of the exact 320 nm value, which was impacted and artificially increased by the interference shift. With correction, a decrease in IO3- concentration was still observed. Since there was no corresponding increase in [I-] or decrease in 129I-/127I-, which would be anticipated for 127IO3- reduction to I-, this could indicate reduction of IO3- to an iodine intermediate not identified in this study. That said, it more likely reflects interferences from SOTS degradation products which have an overlapping absorbance range with I3 near 320 nm. Specifically, 4-Formyl Benzoic acid is a degradation product of SOTS-1 (Ingold et al., 1997; Konya et al., 2000; Heller and Croot, 2010) and ROS-induced oxidation products of CDOM have overlapping absorption peaks. Notably, the SOTS degradation products do not account for the same observations made for the same trend observed in H2O2 observations. Importantly, we acknowledge that the spectrophotometric IO3- measurement is highly prone to interferences (Truesdale, 1978; Luther et al., 1988). Seawater background measurements at 350 nm have the potential to correct these interferences (Hepach et al., 2020; Jones et al., 2023).
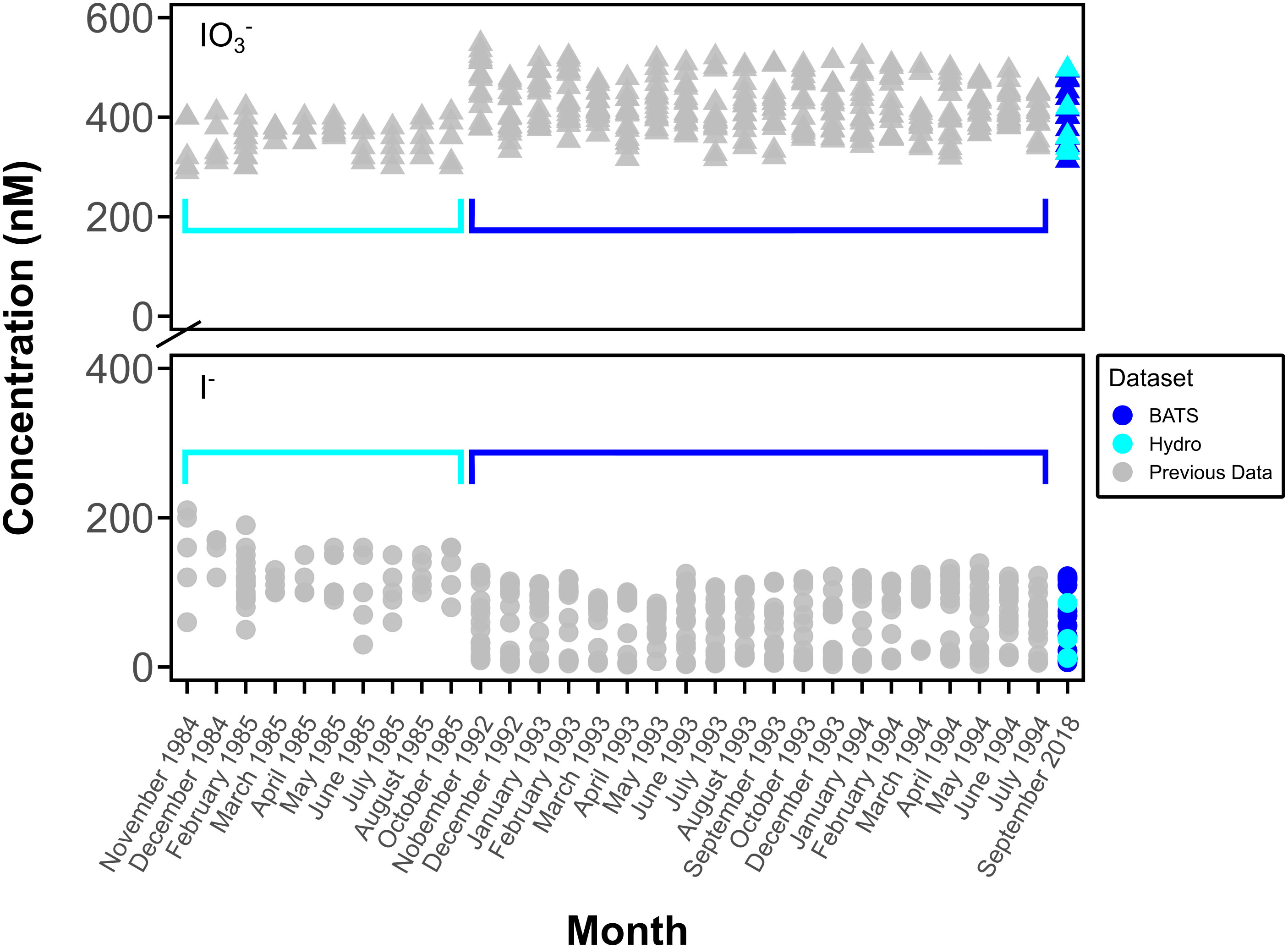
Figure 6 Comparison of previous BATS and Hydrostation S monthly concentration data of I- (●) and IO3- (▲). Data from Campos et al., 1996 (BATS) and Jickells et al., 1988 (Hydrostation S) (gray symbols, surface to 200 m (Hydrostation S) and to 600 m (BATS)) and this study (blue and cyan symbols, surface to 600 m).
Note that the controls with addition of SOD and MnCl2 (meant to test the potential to stop I- oxidation via scrubbing of superoxide or added preferred electron donor) were only relevant if I- oxidation was observed in other controls, so are not considered further. These data are available in Supplementary Table 1.
DOI was not a focus of this study but was quantified in some instances for concentration (Table 2) as well as for 129I/127I ratios in incubations (described in the next section). We note that DOI concentrations in measured incubations (~7.5-9.5% of total dissolved iodine) were larger than we anticipated for open ocean areas where DOI is commonly negligible or uncharacterized (Wong and Cheng, 1998). DOI has previously been found to account for up to 10% of the total iodine pool in coastal areas (Chance et al., 2014).
3.3 129I/127I isotope ratios
While all incubations were measured for [IO3-], we did not measure 129I/127I ratios for all incubations. This is because of the lack of variation observed in 129I/127I for the targeted ROS and other incubations. Measured 129I/127I ratios measured from chosen incubation experiments show no change over time beyond error bars in any of the incubations investigated (Figures 3–5). This includes IO3-, I-, and DOI. This also includes the 129I/127I of I- in the ROS-based incubations where a decrease in spectrophotometrically quantified [IO3-] was observed.
Initial 129I/127I ratios were consistent for measured I- samples, which was expected given that the spike was added to a larger stock volume of seawater that was then aliquoted for the incubations. Average 129I/127I ratios for I- at t0 ranged from 0.29 ± 0.004 to 0.32 ± 0.002 at the surface. Initial isotope ratios for IO3- range from 0.0016 ± 0.0006 to 0.088 ± 0.0005 and 0.0008 ± 0.0001 to 0.002 ± 0.0002 for 1 m (photic) and 240 m (subphotic) depths, respectively, with incubation 8 (photic, dark, filtered) being a slight outlier at 0.07 ± 0.00024. It is notable that 129I was observed in both the DOI and IO3- fractions, though it was added as I-. Initial IO3- and DOI values were still predictably quite low, as Hardisty et al. (2020) demonstrated the spike is mostly 129I- with only minor 129IO3-, with the same being expected for DOI. Initial DOI values ranged between 0.005 ± 0.002 and 0.01 ± 0.003, with incubation 8 again being a slight outlier at 0.05 ± 0.004 (Figure 4).
3.4 Calculating I- oxidation rates and constraining uncertainty
While oxidation was not observed, we quantified the maximum possible rates that would maintain our time series for 129I/127I of IO3- within the error (1 s.d.). Maximum daily gross rates of I- oxidation determined by incubation conditions were calculated using isotope mass balance equations outlined in Hardisty et al. (2020). Average and standard deviation of triplicate initial and final incubation timepoint spectrophotometer measurements of 127[IO3-], ICP-MS measurements of 127[I-], and MC-ICP-MS measurements of I- 129I/127I ratios and IO3- 129I/127I ratios were used to first calculate the total IO3- created from I- oxidation in incubations between t0 to t140 (tfinal). These calculations were then used to determine the rate (nM/day) of I- oxidation to IO3-.
The (initial [IO3-] pre-spike addition) and the isotope ratios of I- and IO3- at a given time t ( and , respectively) are measured (Equations 1, 2). Since I- oxidation to IO3- is the only quantifiable source of 129I to and negligibly fractionated, we can assume that changes in from 129I and 127I are from I- oxidation (i.e., in Equation 3) are contributed at an isotope ratio equivalent to (Equation 1). As such, Equation 1 can be rearranged to solve for which can be substituted into Equation 2 to solve for . Plugging into Equation 1 then allows to solve for which then can be used in Equation 3 to solve for . Based on the measured standard deviations of values and the hours of the incubation, we can then solve for the rate of I- oxidation to IO3- taking place in the incubations in nM/day (Table 3).
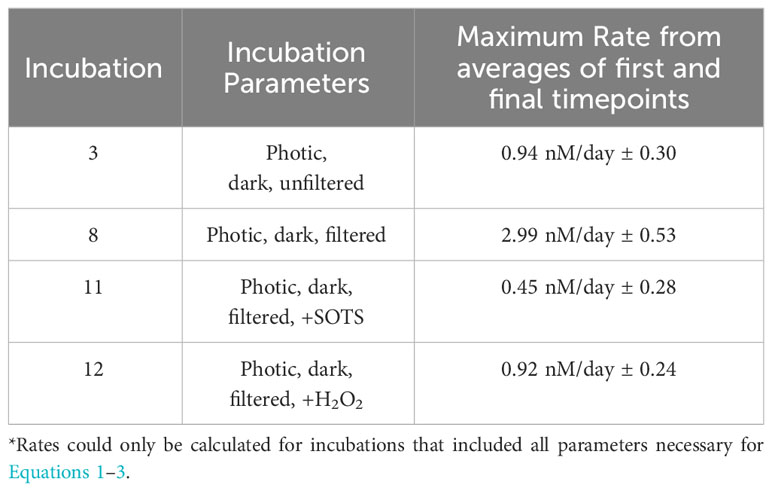
Table 3 Constrained rate measurements of I- oxidation to IO3- in control incubations (incubations 3 and 8) and those with addition of SOTS (incubation 11) and H2O2 (incubation 12).
4 Discussion
We measured the iodine speciation of shipboard incubations performed as part of the Bermuda Atlantic Time Series (BATS) in the Sargasso Sea in September of 2018, as well as depth profiles of [IO3-] and [I-] from both BATS and Hydrostation S stations. Depth profiles repeat a typical trend for iodine redox species in surface waters, including increases in I- at the sea surface and nearly complete IO3- below the euphotic zone (Figure 2). Incubation treatments described above (Table 1) tested the significance of the presence of the ROS O2•− (added as SOTS) and H2O2 on IO3- and I- concentrations in natural seawater settings.
4.1 Temporal and methodological iodine comparison
This is the first report of O2•− at BATS, although iodine speciation at BATS and Hydrostation S has been previously reported through investigations of depth profiles of [I-] and [IO3-] over time to a depth of up to 2500 m (Jickells et al., 1988; Campos et al., 1996) (Figure 2A, Figure 6). The similarities between measured iodine speciation profiles provide some constraints on the reproducibility of variable iodine speciation techniques used between labs. Our current measured data for depth profiles of IO3- and I- at BATS generally agree with that of Campos et al. (1996) and demonstrate little change in iodine speciation with time. We also extended those investigations here to the bottom waters at 4500 m. While not the focus here, we note that our new bottom water data additionally show elevated I-, consistent with other studies demonstrating likely benthic I- fluxes in abyssal plains (Francois, 1987a; Francois, 1987b; Kennedy and Elderfield 1987a; Kennedy and Elderfield 1987b; Moriyasu et al., 2023).
Our data from Hydrostation S also generally overlaps with the range observed in Jickells et al. (1988); however, we note that our I- data have a range that extends below that of these previous data. One explanation is that Jickells et al. (1988) measured I- via difference between total iodine – determined via a UV-oxidation technique – and the spectrophotometrically measured IO3-, as was done here. Note that the opposite is true for Campos et al. (1996), who reduced IO3- to I- and calculated IO3- via difference following voltametric analysis (Total I – I-). For both cases, given that total iodine includes organic iodine, this implies the potential that DOI is contributing to either the I- or IO3- pool, depending on the technique. While not thoroughly evaluated in this study, we note that DOI concentrations in measured incubations were up to 10% of the total iodine pool, so this could explain the difference between ours and earlier studies.
Alternatively, the difference is Hydrostation S data ranges between the two studies could reflect real changes in hydrography or primary production. For the seasonal study of Jickells et al. (1988), these previous seasonal IO3- lows and I- high values were generally observed in the summer and interpreted to reflect more limited water mass exchange, which allows the combination of local Bermuda inshore inputs and primary productivity to limit IO3- availability. In the absence of a seasonal iodine context, it is difficult to explain the differences from our September 2018 data, but it is likely that increased water mass exchange – increasing the supply of high IO3- and low I- from deeper waters and the Sargasso Sea generally – plays an important role. For example, the similarities in our 2018 BATS and Hydrostation S data suggest the potential for increased hydrographic exchange between these two sites. Further, the September 2018 sampling overlapped with the passage of Hurricane Florence 750 miles southeast of Bermuda on September 9th, 2018. While the mixed layer depth from September 2018 is like that of previous years, hurricanes have been documented to influence nutrient and other chemical parameters via increased mixing (Babin et al., 2004).
We note that while seasonal iodine variations may be driven by variations in primary productivity and mixed layer depth, other factors may contribute to longer-term evolution of iodine speciation. Specifically, Hughes et al. (2021) demonstrate that I- oxidation to IO3- may be linked to nitrification, which is sensitive to a range of factors – e.g., light, O2, temperature, pH, and photochemically produced ROS (Pajares and Ramos, 2019; Morris et al., 2022). Given the baseline iodine speciation conditions documented here and in previous studies, BATS may be an ideal location to track potential future changes in iodine speciation linked to predictions of evolving nitrification from global warming and ocean acidification (Bemen et al., 2012). To our knowledge, the only other locations with multiple iodine measurements are from the Black Sea (Wong and Brewer, 1977; Luther and Campbell, 1991; Truesdale et al., 2001a) and Baltic Sea (Hou et al., 2001; Truesdale et al., 2001b; Aldahan et al., 2007; Truesdale et al., 2013), but iodine speciation changes there will additionally be sensitive to the well-known presence of hypoxia and euxinia.
4.2 Limitations of experimental approach
Prior to interpretations, it is important to discuss the limitations of our experimental and analytical approaches. First, while isotope ratio variations (or lack thereof) form the basis of our interpretations, we note that variability in triplicate measurements in isotopes and concentrations measured via ICP-MS and spectrophotometry all contribute to the uncertainty of our rate calculations (Table 3). As previously mentioned, we acknowledge that the spectrophotometric method of measuring [IO3-] is prone to interferences and relatively low precision (Truesdale, 1978; Luther et al., 1988). For example, variation observed in the IO3- concentration but not in IO3- isotope ratios for the ROS-amended incubations (Figure 5) is likely attributed to interferences and not iodine species variations (see Supplementary Information). That said, beyond this example, there was general agreement between IO3- concentration and iodine isotope trends in this study and between ours and previously measured IO3- concentrations at Hydrostation S and BATS (Figures 2 and 6).
Another important limitation is the duration of the experiments, which likely induced bottle effects in unfiltered conditions. For example, a subset of our experiments was unfiltered in order to retain particles and native biological communities, but changes in community composition is well known within prolonged bottle experiments, even within 24 hours (Berg et al., 1999). Our experiments lasted ~140 hours (due to the known sluggish iodine oxidation kinetics) and the biological communities of unfiltered controls were not monitored. As there were no nutrient enrichments in our unfiltered incubations, it is likely that native cells underwent physiological changes that altered the balance between phytoplankton and bacteria. While not observed here, such a turnover could have shifted the balance between iodine oxidation and reduction, or commenced cell senescence, which favors I- release (e.g., Bluhm et al., 2010). This places a limitation on our ability to interpret biologically induced iodine oxidation and reduction in unfiltered incubations; however, biological turnover is not a factor in our filtered controls testing additional oxidation-reduction pathways.
An additional limitation on our experiments is uncertainty regarding the superoxide concentrations and decay rate in the SOTS amended incubations. Specifically, the superoxide concentration and decay rate were not quantified directly in these incubations. That said, SOTS was added at a concentration estimated to produce an excess superoxide (25-55 nM instantaneous superoxide) relative to the natural waters of BATS (Figure 2). The instantaneous superoxide concentration can be estimated from temperature-dependent decay constants – 4.3-9.9x10-5 s-1 at 28.2-29.2°C (Heller and Croot, 2010) – and the observed superoxide decay constant from analogous oligotrophic waters (0.01 s-1; (Roe et al., 2016)). While experiments were conducted in a shipdeck chamber continuously refilled with local surface water, the temperature was not directly monitored; however, we note that the biggest uncertainty in kSOTS comes from uncertainty in the values at a given temperature and not from the temperature range. Regardless, the conservative estimate of 25 nM superoxide is still in excess of that measured in water column profiles from this study (Figure 2).
4.3 Rates of I- oxidation
Understanding the rates of I- oxidation to IO3- is essential for already applied marine iodine cycling models (Lu et al., 2018; Wadley et al., 2020), but direct constraints are limited. In this study, all incubations showed no discernable change in [IO3-], [I-], or the 129I/127I of IO3- beyond uncertainties over the timeframe of the incubations (~140 hours) (Figures 3–5).
While direct observations of IO3- production from I- remains an important goal, our use of a radiotracer still allows us to place novel constraints on the maximum rates of I- oxidation to IO3- based on the limitations of our analytical uncertainties (Table 3). Specifically, a lack of change seen over time in these incubations in both concentration and 129I/127I ratios are used to constrain a rate of <2.99 nM/day ± 0.53 nM/day. Previously calculated values of I- oxidation have been reported from BATS from mass balance studies to be between 0.74 and 18 nM/day (Table 4). The value calculated for our incubation studies fits well in this range (Table 2), supporting previous claims that oxidation of I- to IO3- is slow in natural seawaters (Hardisty et al., 2020; Carpenter et al., 2021).
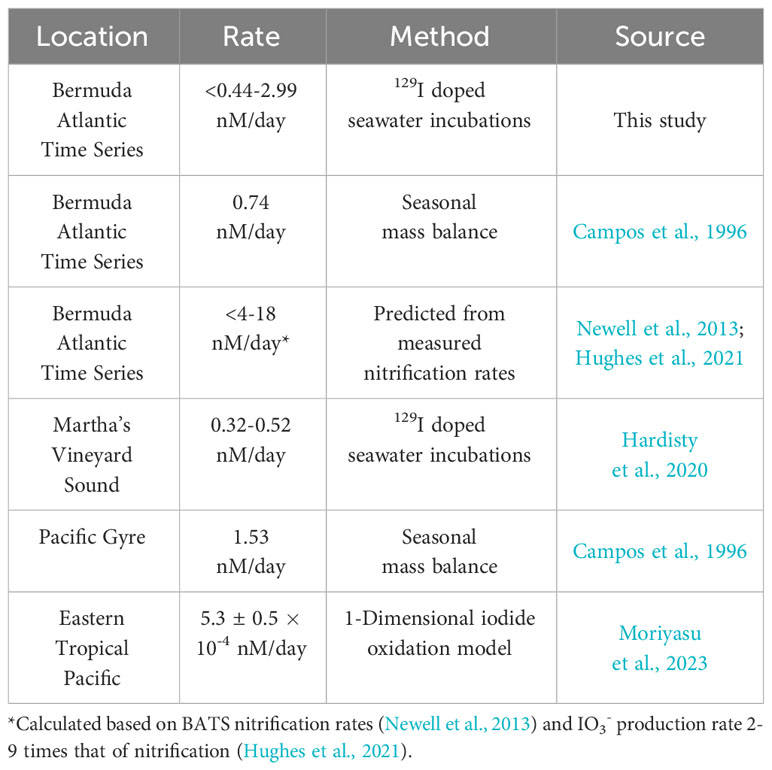
Table 4 Previously reported daily I- oxidation rates.
Further, we observe non-negligible 129I in the initial post-spike sample for both the IO3- and DOI incubation pools. Hardisty et al. (2020) made a similar observation and provided some evidence that the iodine-129 spike, while overwhelmingly I-, contains some oxidized iodine. An alternative explanation is that the IO3- or iodine intermediates rapidly form upon addition to seawater or other solutions. This observation implies the potential for near instantaneous formation of 129IO3- or DOI upon addition of spike to samples. Since the increase in 129I/127I was not ongoing, this would require rapid quenching of an existing oxidant in seawater, thus inhibiting further oxidation.
4.4 Role of reactive oxygen species in I- oxidation to IO3- in natural seawater
We provide here the most detailed and direct constraints to date of the potential for the ROS O2•− (added as the chemical source SOTS) and H2O2 to oxidize I- to IO3- in natural seawater. To the point, in our incubations with added O2•− and H2O2, no oxidation of I- to IO3- was observed in BATS seawater, as shown in 129I/127I ratios of IO3- (Figure 5). We emphasize that this does not completely rule out the role of ROS in I- oxidation broadly. Below we consider the implications of these results in coordination with previous studies evaluating the role, if any, of ROS on iodine cycling.
First, it is possible that ROS is preferentially reacting with dissolved organic matter or another component of seawater (e.g., Mn, NOx) (Wuttig et al., 2013; Li et al., 2014; Sutherland et al., 2021). We did not constrain alternative electron acceptors here but given the oligotrophic nature of BATS and known low dissolved Mn (Wu et al., 2014), this would imply that preferential extracellular O2•− scavenging relative to I- is common throughout most of the ocean, and intracellular ROS have been shown to be a significant oxygen sink (Sutherland et al., 2021), hence diminishing the likely role of O2•− in marine I- oxidation more broadly. Some evidence for preferential reaction of ROS with other redox-active species may come from the interference observed for our spectrophotometric measurements of IO3-. Second, thermodynamic calculations predict that O2•− alone forms iodine intermediates and cannot fully oxidize I- to IO3-, so it is likely that iodine intermediates such as I2, HOI, or DOI, instead of IO3-, in samples with excess H2O2 or SOTS added, are forming. This implies a potential role of ROS in iodine redox species cycling but contrasts previous findings. As discussed in detail in Luther (2023), reactions with O2•− and I- forms I2 and subsequently I2- (Bielski et al., 1985; Schwarz and Bielski, 1986). I2 reacts quickly with organic matter to form DOI, which may act as a bottleneck in some environments to titrate I2 and prevent subsequent oxidation (e.g., Hardisty et al., 2020). Since the 129I/127I ratio of DOI did not change in experiments (Figure 5E), it implies relatively little, if any, oxidation to intermediates forming DOI. Beyond this, O2•− is not favorable to subsequently oxidize I2/I2- to form IO3-. Instead, •OH or O3 are required for subsequent HOI formation, and OH, O3, and H2O2 can then oxidize to IO2-, and then subsequently to IO3-. O3 is not prevalent beyond the marine micro-layer and thus is unlikely responsible for I- oxidation elsewhere in seawater.
Ultimately, these thermodynamic considerations imply that combinations of O2•- and H2O2, and perhaps OH radicals produced during their reduction, are necessary for complete oxidation from I- to IO3- via ROS. Therefore, ROS most likely play a minor, if any, role in IO3- formation given the combination of: slow predicted kinetics (e.g., Wong and Zhang, 2008), limited ROS in seawater relative to iodine, the up to four step oxidation sequence each consuming ROS (I- ➔ I2 ➔ HOI ➔ IO2- ➔ IO3-), abundance of DOM and other preferred electron acceptors/donor – such as Br and Mn – for ROS, titration of HOI with DOM, and the likelihood of back reactions of iodine intermediates to I-.
4.5 Constraints on other redox pathways
The lack of change in [IO3-], [I-] or 129I/127I isotope ratios over the incubation period (~140 hours) (Figure 4) for the incubations exposed to dark vs light conditions additionally provide constraints on the likelihood of iodine redox reaction pathways. First, interactions between light and organic matter represent an abiotic pathway for ROS production (Morris et al., 2022). If 129IO3- formation had been observed in the light experiments, an abiotic ROS mechanism could have been inferred from a lack of 129IO3- in both the dark and light +SOD controls. Second, natural light is also suggested to aid in photo-reduction of IO3- in seawater, although it is not known how important this reduction pathway may be for I- accumulation in the surface ocean (Spokes and Liss, 1996; Chance et al., 2014). Conversion of IO3- to I- observed in both isotopes and concentrations would provide evidence for such a pathway, but this was not observed in this study.
4.6 Implications for marine iodine redox cycling and distributions
Our results support mounting evidence that iodine cycling in some ocean regions, if not more broadly, may be considered semi-conservative. From this perspective, given the relatively oligotrophic conditions at BATS (Lipschultz et al., 2002), it is perhaps not surprising that IO3- production may be slow or isolated to specific depths or even seasons. For example, laboratory cultures have identified nitrification as a possible pathway of IO3- production, which occurs specifically at the nitrite maximum at BATS and other similar localities (Newell et al., 2013; Hughes et al., 2021). In addition, Luther (2023) provides thermodynamic constraints that OH is a powerful oxidant capable of producing IO3-. Oxygenated waters supporting dissolved Fe – such as hydrothermal plumes, some low oxygen zones, and the benthic boundary layer – produce elevated •OH via Fenton chemistry, which could oxidize I- to IO3- (e.g., Shaw et al., 2021). Luther (2023) also suggests that O3 and N2O can form IO3-, indicating that marine microlayer (Carpenter et al., 2021) and the oxycline of oxygen deficient zones (Babbin et al., 2015) may be important for IO3- production, respectively. Lastly, IO3- reduction in surface waters is more clearly linked to phytoplankton, which show seasonal distributions at BATS (Michaels et al., 1994; Michaels and Knap, 1996; DuRand et al., 2001).
Ex situ mixing of source waters from “hotspots” of high primary productivity, oxygen deficient zones, and pore waters may initiate iodine speciation gradients in surface waters whose rates of change slow as water masses extend to the open ocean from coastal or productive settings. As a result, water mass mixing may have a more dramatic effect on the distribution of iodine species in the open surface ocean than previously known. For example, Chance et al. (2020) and Chance et al. (2010) provided some evidence that diffusion/advection of IO3- from below the mixed layer may be important for controlling surface ocean IO3- abundance. Even in oxygen deficient zones, Hardisty et al. (2021) provide evidence that below the oxycline that IO3- has the potential to reflect water mass mixing, as opposed to clear in situ IO3- reduction.
5 Conclusions
We performed shipboard incubation experiments of seawater at the Bermuda Atlantic Time Series in the Sargasso Sea. These included natural concentrations of iodine and the reactive oxygen species O2•− and H2O2 to better understand the mechanisms of oxidation of I- to IO3- in surface seawaters and better constrain the rates at which oxidation of I- to IO3- takes place in the open ocean. We provided evidence that rates of I- oxidation are extremely slow, if anything, on a daily timescale. We explicitly tested the potential for iodine redox reaction with ROS, which did not oxidize I- within the resolution of our analytical uncertainty.
Based on the sluggish iodine oxidation rates, it is likely that ex situ sources of transportation, such as water mass mixing and vertical diffusion, are more important in the distribution of iodine redox species from “hotspots” of formation, such as areas of very high biogeochemical activity, ODZ’s, and pore waters (Hardisty et al., 2021). This study and similar continuing work will help to inform atmospheric models of O3 destruction and paleoredox models of IO3- incorporation with carbonates for measurement of I/Ca values.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
AS: Writing – original draft, Writing – review & editing, Formal analysis. DH: Writing – review & editing, Conceptualization, Funding acquisition, Supervision, Writing – original draft. KS: Writing – review & editing, Formal analysis. CH: Writing – review & editing, Methodology, Resources.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. NSF grant (#1829406) funded the entirety of this work.
Acknowledgments
AS and DH would like to thank Jurek Blusztajn for assistance at the WHOI Plasma Facility. We thank the Chief Scientist (Rod Johnson) and Captain and Crew of the R/V Atlantic Explorer for making sampling for this dataset possible.
Conflict of interest
The reviewer, RC, is currently organizing a Research Topic with the author, DH.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1272870/full#supplementary-material
References
Aldahan A., Possnert G., Alfimov V., Cato I., Kekli A. (2007). Anthropogenic 129I in the Baltic Sea. Nucl. Instruments Methods Phys. Research Section B: Beam Interact. Materials Atoms 259 (1), 491–495. doi: 10.1016/j.nimb.2007.01.242
Babbin A. R., Bianchi D., Jayakumar A., Ward B. B. (2015). Rapid nitrous oxide cycling in the suboxic ocean. Science 348 (6239), 1127–1129. doi: 10.1126/SCIENCE.AAA8380/SUPPL_FILE/BABBIN-SM.PDF
Babin S. M., Carton J. A., Dickey T. D., Wiggert J. D. (2004). Satellite evidence of hurricane-induced phytoplankton blooms in an oceanic desert. J. Geophys. Res.-Oceans 109, Artn c03043. doi: 10.1029/2003JC001938
Beman M. J., Popp B. N., Alford S. E. (2012). Quantification of ammonia oxidation rates and ammonia-oxidizing archaea and bacteria at high resolution in the Gulf of California and eastern tropical North Pacific Ocean. Limnology Oceanography 57 (3), 711–726. doi: 10.4319/lo.2012.57.3.0711
Berg G. M., Glibert P. M., Chen C. C. (1999). Dimension effects of enclosures on ecological processes in pelagic systems. Limnol. Oceanogr. 44, 1331–1340. doi: 10.4319/lo.1999.44.5.1331
Bielski B. H. J., Cabelli D. E., Arudi R. L., Ross A. B. (1985). Reactivity of HO2/O–2 radicals in aqueous solution. J. Phys. Chem. Reference Data 14 (4), 1041. doi: 10.1063/1.555739
Bluhm K., Croot P., Wuttig K., Lochte K. (2010). Transformation of iodate to iodide in marine phytoplankton driven by cell senescence. Aquat. Biol. 11, 1–15. doi: 10.3354/ab00284
Bond R. J., Hansel C. M., Voelker B. M. (2020). Heterotrophic bacteria exhibit a wide range of rates of extracellular production and decay of hydrogen peroxide. Front. Mar. Sci. 7. doi: 10.3389/FMARS.2020.00072/BIBTEX
Campos M. L. A. M. (1997). New approach to evaluating dissolved iodine speciation in natural waters using cathodic stripping voltammetry and a storage study for preserving iodine species. Mar. Chem. 57, 107–117. doi: 10.1016/S0304-4203(96)00093-X
Campos M. L. A. M., Farrenkopf A. M., Jickells T. D., Luther G. W. (1996). A comparison of dissolved iodine cycling at the Bermuda Atlantic Time-series Station and Hawaii Ocean Time-series Station. Deep Sea Res. Part II: Topical Stud. Oceanography. 43, 455–466. doi: 10.1016/0967-0645(95)00100-x
Carpenter L. J., Chance R. J., Sherwen T., Adams T. J., Ball S. M., Evans M. J., et al. (2021). Marine iodine emissions in a changing world. Proc. R. Soc. A 477 (2247), 20200824. doi: 10.1098/RSPA.2020.0824
Carpenter L. J., MacDonald S. M., Shaw M. D., Kumar R., Saunders R. W., Parthipan R., et al. (2013). Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine. Nat. Geosci. 6, 108–111. doi: 10.1038/ngeo1687
Chance R., Baker A. R., Carpenter L., Jickells T. D. (2014). “The distribution of iodide at the sea surface,” in Environmental Sciences: Processes and Impacts. (Piccadilly, London: Royal Society of Chemistry) 118, 171–181. doi: 10.1039/c4em00139g
Chance R., Tinel L., Sarkar A., Sinha A. K., Mahajan A. S., Chacko R., et al. (2020). Surface inorganic iodine speciation in the Indian and southern oceans from 12°N to 70°S. Front. Mar. Sci. 7. doi: 10.3389/FMARS.2020.00621/BIBTEX
Chance R., Weston K., Baker A. R., Hughes C., Malin G., Carpenter L., et al. (2010). Seasonal and interannual variation of dissolved iodine speciation at a coastal Antarctic site. Mar. Chem. doi: 10.1016/j.marchem.2009.11.009
Diaz J. M., Hansel C. M., Voelker B. M., Mendes C. M., Andeer P. F., Zhang T. (2013). Widespread production of extracellular superoxide by heterotrophic bacteria. Science 340 (6137), 1223–1226. doi: 10.1126/science.1237331
DuRand M. D., Olson R. J., Chisholm S. W. (2001). Phytoplankton population dynamics at the Bermuda Atlantic Time-series station in the Sargasso Sea. Deep Sea Res. Part II: Topical Stud. Oceanography 48 (8–9), 1983–2003. doi: 10.1016/S0967-0645(00)00166-1
Edwards A., Truesdale V. W. (1997). Regeneration of inorganic iodine species in Loch Etive, a natural leaky incubator. Estuarine Coast. Shelf Sci. 45 (3), 357–366. doi: 10.1006/ECSS.1996.0185
Elderfield H., Truesdale V. W. (1980). On the biophilic nature of iodine in seawater. Earth Planetary Sci. Lett. 50, 105–114. doi: 10.1016/0012-821X(80)90122-3
Farrenkopf A. M., Dollhopf M. E., Chadhain S. N., Luther G. W., Nealson K. H. (1997). Reduction of iodate in seawater during Arabian Sea shipboard incubations and in laboratory cultures of the marine bacterium Shewanella putrefaciens strain MR-4. Mar. Chem. 57, 347–354. doi: 10.1016/S0304-4203(97)00039-X
Francois R. (1987a). The influence of humic substances on the geochemistry of iodine in nearshore and hemipelagic marine-sediments. Geochimica Et Cosmochimica Acta 51, 2417–2427. doi: 10.1016/0016-7037(87)90294-8
Francois R. (1987b). Iodine in marine sedimentary humic substances. Sci. Total Environ. 62, 341–342. doi: 10.1016/0048-9697(87)90519-5
Hansard S. P., Vermilyea A. W., Voelker B. M. (2010). Measurements of superoxide radical concentration and decay kinetics in the Gulf of Alaska. Deep-Sea Res. I 57, 1111–1119. doi: 10.1016/j.dsr.2010.05.007
Hansel C. M., Diaz J. M. (2021). Production of extracellular reactive oxygen species by marine biota. Annu. Rev. Mar. Sci. 2021 (13), 177–200. doi: 10.1146/annurev-marine-041320
Hansel C. M., Diaz J. M., Plummer S. (2019). Tight regulation of extracellular superoxide points to its vital role in the physiology of the globally relevant roseobacter clade. MBio 10 (2), e02668-18. doi: 10.1128/MBIO.02668-18/SUPPL_FILE/MBIO.02668-18-ST003.PDF
Hardisty D. S., Horner T. J., Evans N., Moriyasu R., Babbin A. R., Wankel S. D., et al. (2021). Limited iodate reduction in shipboard seawater incubations from the Eastern Tropical North Pacific oxygen deficient zone. Earth Planetary Sci. Lett. 554, 116676. doi: 10.1016/j.epsl.2020.116676
Hardisty D. S., Horner T. J., Wankel S. D., Blusztajn J., Nielsen S. G. (2020). Experimental observations of marine iodide oxidation using a novel sparge-interface MC-ICP-MS technique. Chem. Geology. 532, 119360. doi: 10.1016/j.chemgeo.2019.119360
He P., Hou X., Aldahan A., Possnert G., Yi P. (2013). Iodine isotopes species fingerprinting environmental conditions in surface water along the northeastern Atlantic Ocean. Sci. Rep. 3 (1), 1–8. doi: 10.1038/srep02685
Heller M. I., Croot P. L. (2010). Application of a superoxide (O 2 – ) thermal source (SOTS-1) for the determination and calibration of O 2 – fluxes in seawater. Analytica Chimica Acta 667, 1–13. doi: 10.1016/j.aca.2010.03.054
Hepach H., Hughes C., Hogg K., Collings S., Chance R. (2020). Senescence as the main driver of iodide release from a diverse range of marine phytoplankton. Biogeosciences. 17, 2453–2471. doi: 10.5194/bg-17-2453-2020
Hou X., Aldahan A., Nielsen S. P., Possnert G. (2009). Time series of 129I and 127I speciation in precipitation from Denmark. Environ. Sci. Technol. 43 (17), 6522–6528. doi: 10.1021/ES9012678
Hou X., Aldahan A., Nielsen S. P., Possnert G., Nies H., Hedfors J. (2007). Speciation of 129I and 127I in seawater and implications for sources and transport pathways in the North Sea. Environ. Sci. Technol. 41, 5993–5999. doi: 10.1021/es070575x
Hou X., Dahlgaard H., Nielsen S. P. (2001). Chemical speciation analysis of 129I in seawater and a preliminary investigation to use it as a tracer for geochemical cycle study of stable iodine. Mar. Chem. 74, 145–155. doi: 10.1016/S0304-4203(01)00010-X
Hou X., Dahlgaard H., Rietz B., Jacobsen U., Nielsen S. P., Aarkrog A. (1999). Determination of chemical species of iodine in seawater by radiochemical neutron activation analysis combined with ion-exchange preseparation. Analytical Chem. 71 (14), 2745–2750. doi: 10.1021/AC9813639
Hughes C., Barton E., Hepach H., Chance R., Pickering M. D., Hogg K., et al. (2021). Oxidation of iodide to iodate by cultures of marine ammonia-oxidising bacteria. Mar. Chem. 234, 104000. doi: 10.1016/J.MARCHEM.2021.104000
Ingold K. U., Paul T., Young M. J., Doiron L. (1997). Invention of the first azo compound to serve as a superoxide thermal source under physiological conditions: Concept, synthesis, and chemical properties. J. Of Am. Chem. Soc. 119, 12364–12365. doi: 10.1021/ja972886l
Jickells T. D., Boyd S. S., Knap A. H. (1988). Iodine cycling in the Sargasso Sea and the Bermuda inshore waters. Mar. Chem. 24, 61–82. doi: 10.1016/0304-4203(88)90006-0
Jones M. R., Chance R., Dadic R., Hannula H. R., May R., Ward M., et al. (2023). Environmental iodine speciation quantification in seawater and snow using ion exchange chromatography and UV spectrophotometric detection. Anal. Chim. Acta, 1239. doi: 10.1016/j.aca.2022.340700
Kennedy H. A., Elderfield H. (1987a). Iodine diagenesis in non-pelagic deep-sea sediments. Geochimica et Cosmochimica Acta 51, 2505–2514. doi: 10.1016/0016-7037(87)90301-2
Kennedy H. A., Elderfield H. (1987b). Iodine diagenesis in pelagic deep-sea sediments. Geochimica Et Cosmochimica Acta 51, 2489–2504. doi: 10.1016/0016-7037(87)90300-0
Konya K. G., Paul T., Lin S., Lusztyk J., Ingold K. U. (2000). Laser flash photolysis studies on the first superoxide thermal source. First direct measurements of the rates of solvent-assisted 1,2- hydrogen atom shifts and a proposed new mechanism for this unusual rearrangement1. J. Am. Chem. Soc. 122, 7518–7527. doi: 10.1021/ja993570b
Li H. P., Daniel B., Creeley D., Grandbois R., Zhang S., Xu C., et al. (2014). Superoxide production by a manganese-oxidizing bacterium facilitates iodide oxidation. Appl. Environ. Microbiol. 80, 2693–2699. doi: 10.1128/AEM.00400-14
Li H. P., Yeager C. M., Brinkmeyer R., Zhang S., Ho Y. F., Xu C., et al. (2012). Bacterial production of organic acids enhances H2O 2-dependent iodide oxidation. Environ. Sci. Technol. 46, 4837–4844. doi: 10.1021/es203683v
Lipschultz F., Bates N. R., Carlson C. A., Hansell D. A. (2002). New production in the Sargasso Sea: History and current status. Global Biogeochemical Cycles 16 (1), 1–1. doi: 10.1029/2000GB001319
Lu W., Ridgwell A., Thomas E., Hardisty D. S., Luo G., Algeo T. J., et al. (2018). Late inception of a resiliently oxygenated upper ocean. Science. 361, 174–177. doi: 10.1126/science.aar5372
Luhar A. K., Galbally I. E., Woodhouse M. T., Thatcher M. (2017). An improved parameterisation of ozone dry deposition to the ocean and its impact in a global climate-chemistry model. Atmospheric Chem. Phys. 17, 3749–3767. doi: 10.5194/acp-17-3749-2017
Luther G. W. I. (2023). Review on the physical chemistry of iodine transformations in the oceans. Front. Mar. Sci. 10, 1721–1724. doi: 10.3389/FMARS.2023.1085618
Luther G. W., Campbell T. (1991). Iodine speciation in the water column of the Black Sea. Deep Sea Research Part A. Oceanographic Res. Papers. 38, S875–S882. doi: 10.1016/s0198-0149(10)80014-7
Luther G. W., Swartz C. B., Ullman W. J. (1988). Direct determination of iodide in seawater by Cathodic stripping square wave voltammetry. Analytical Chem. 60 (17). doi: 10.1021/ac00168a017
Luther G. W., Wu J., Cullen J. B. (1995). Redox Chemistry of Iodine in Seawater. (Washington, D.C.: American Chemical Society), 135–155. doi: 10.1021/BA-1995-0244.CH006
Michaels A. F., Knap A. H. (1996). Overview of the U.S. JGOFS Bermuda Atlantic Time-series study and the hydrostation S program. Deep-Sea Res. Part II: Topical Stud. Oceanography 43 (2–3), 157–198. doi: 10.1016/0967-0645(96)00004-5
Michaels A. F., Knap A. H., Dow R. L., Gundersen K., Johnson R. J., Sorensen J., et al. (1994). Seasonal patterns of ocean biogeochemistry at the U.S. JGOFS Bermuda Atlantic time-series study site. Deep-Sea Res. Part I. 41, 1013–1038. doi: 10.1016/0967-0637(94)90016-7
Moriyasu R., Evans N., Bolster K. M., Hardisty D. S., Moffett J. W. (2020). The Distribution and Redox Speciation of Iodine in the Eastern Tropical North Pacific Ocean. Global Biogeochem Cycles 34 (2). doi: 10.1029/2019GB006302
Moriyasu R., Bolster K. M., Hardisty D. S., Kadko D. C., Stephens M. P., Moffett J. W. (2023). Meridional survey of the central pacific reveals iodide accumulation in equatorial surface waters and benthic sources in the abyssal plain. Global Biogeochem. Cycles 37 (3), e2021GB007300. doi: 10.1029/2021GB007300
Morris J. J., Rose A. L., Lu Z. (2022). Reactive oxygen species in the world ocean and their impacts on marine ecosystems. Redox Biol. 52, 102285. doi: 10.1016/j.redox.2022.102285
Newell S. E., Fawcett S. E., Ward B. B. (2013). Depth distribution of ammonia oxidation rates and ammonia-oxidizer community composition in the Sargasso Sea. Limnology Oceanography 58 (4), 1491–1500. doi: 10.4319/LO.2013.58.4.1491
Pajares S., Ramos R. (2019). Processes and microorganisms involved in the marine nitrogen cycle: knowledge and gaps. Front. Mar. Sci. 6. doi: 10.3389/fmars.2019.00739
Reifenhäuser C., Heumann K. G. (1990). Development of a definitive method for iodine speciation in aquatic systems. Fresenius J. Anal. Chem. 336 (7), 559–563. doi: 10.1007/BF00331416/METRICS
Roe K. L., Schneider R. J., Hansel C. M., Voelker B. M. (2016). Measurement of dark, particle-generated superoxide and hydrogen peroxide production and decay in the subtropical and temperate North Pacific Ocean. Deep-Sea Res. I 107, 59–69. doi: 10.1016/j.dsr.2015.10.012
Rose A. L., Webb E. A., Waite T. D., Moffett J. W. (2008). Measurement and implications of nonphotochemically generated superoxide in the equatorial pacific ocean. Environ. Sci. Technol. 42 (7), 2387–2393. doi: 10.1021/ES7024609/SUPPL_FILE/ES7024609-FILE003.PDF
Rusak S. A., Peake B. M., Richard E., Nodder S. D., Cooper W. J. (2011). Distributions of hydrogen peroxide and superoxide in seawater east of New Zealand. Mar. Chem. 127, 155–169. doi: 10.1016/j.marchem.2011.08.005
Schwarz H. A., Bielski B. H. J. (1986). Reactions of HO2 and O2- with iodine and bromine and the I2- and I atom reduction potentials. J. Phys. Chem. 90 (7), 1445–1448. doi: 10.1021/J100398A045/ASSET/J100398A045.FP.PNG_V03
Shaw T. J., Luther G. W., Rosas R., Oldham V. E., Coffey N. R., Ferry J. L., et al. (2021). Fe-catalyzed sulfide oxidation in hydrothermal plumes is a source of reactive oxygen species to the ocean. Proc. Natl. Acad. Sci. United States America 118 (40), e2026654118. doi: 10.1073/PNAS.2026654118/SUPPL_FILE/PNAS.2026654118.SAPP.PDF
Spokes L. J., Liss P. S. (1996). Photochemically induced redox reactions in seawater, II. Nitrogen and iodine. Mar. Chem. 54, 1–10. doi: 10.1016/0304-4203(96)00033-3
Sutherland K. M., Grabb K. C., Karolewski J. S., Plummer S., Farfan G. A., Wankel S. D., et al. (2020). Spatial heterogeneity in particle-associated, light-independent superoxide production within productive coastal waters. J. Geophys. Res. Oceans 125, e2020JC016747. doi: 10.1029/2020JC016747
Sutherland K. M., Grabb K. C., Karolewski J. S., Taenzer L., Hansel C. M., Wankel S. D. (2021). The redox fate of hydrogen peroxide in the marine water column. Limnology Oceanography 66 (10), 3828–3841. doi: 10.1002/LNO.11922
Sutherland K. M., Wankel S. D., Hansel C. M. (2020). Dark biological superoxide production as a significant flux and sink of marine dissolved oxygen. Proc. Natl. Acad. Sci. United States America. doi: 10.1073/pnas.1912313117
Truesdale V. W. (1978). The automatic determination of iodate- and total-iodine in seawater. Mar. Chem. 6 (3), 253–273. doi: 10.1016/0304-4203(78)90034-8
Truesdale V. W., Nausch G., Baker A. (2001a). The distribution of iodine in the Baltic Sea during summer. Mar. Chem. 74, 87–98. doi: 10.1016/S0304-4203(00)00115-8
Truesdale V. W., Nausch G., Waite T. J. (2013). The effects of the 2001 Barotropic intrusion of bottom-water upon the vertical distribution of inorganic iodine in the Gotland Deep. Continental Shelf Res. 55, 155–167. doi: 10.1016/j.csr.2013.01.005
Truesdale V. W., Watts S. F., Rendell A. R. (2001b). On the possibility of iodide oxidation in the near-surface of the Black Sea and its implications to iodine in the general ocean. Deep Sea Res. Part I: Oceanographic Res. Papers 48 (11), 2397–2412. doi: 10.1016/S0967-0637(01)00021-8
Wadley M. R., Stevens D. P., Jickells T. D., Hughes C., Chance R., Hepach H., et al. (2020). A global model for iodine speciation in the upper ocean. Global Biogeochemical Cycles 34 (9), e2019GB006467. doi: 10.1029/2019GB006467
Wong G. T. F., Brewer P. G. (1977). The marine chemistry of iodine in anoxic basins. Geochimica Cosmochimica Acta. 41, 151–159. doi: 10.1016/0016-7037(77)90195-8
Wong G. T. F., Cheng X. H. (1998). Dissolved organic iodine in marine waters: Determination, occurrence and analytical implications. Mar. Chem. 59, 271–281. doi: 10.1016/S0304-4203(97)00078-9
Wong G. T. F., Zhang L. S. (2008). The kinetics of the reactions between iodide and hydrogen peroxide in seawater. Mar. Chem. 111, 22–29. doi: 10.1016/j.marchem.2007.04.007
Wu J., Roshan S., Chen G. (2014). The distribution of dissolved manganese in the tropical–subtropical North Atlantic during US GEOTRACES 2010 and 2011 cruises. Mar. Chem. 166, 9–24. doi: 10.1016/J.MARCHEM.2014.08.007
Wuttig K., Heller M. I., Croot P. L., Biogeochemistry M. (2013). Pathways of Superoxide (O 2 – ) Decay in the Eastern Tropical North Atlantic. (Washington, D. C.: American Chemical Society). doi: 10.1021/es401658t
Keywords: iodine, redox transformations, superoxide & hydrogen peroxide, isotope tracer, incubations, oxidation, iodide, iodate
Citation: Schnur AA, Sutherland KM, Hansel CM and Hardisty DS (2024) Rates and pathways of iodine speciation transformations at the Bermuda Atlantic Time Series. Front. Mar. Sci. 10:1272870. doi: 10.3389/fmars.2023.1272870
Received: 04 August 2023; Accepted: 07 December 2023;
Published: 09 January 2024.
Edited by:
John Lee Ferry, University of South Carolina, United StatesReviewed by:
Rosie Chance, University of York, United KingdomPeter Leslie Croot, University of Galway, Ireland
Copyright © 2024 Schnur, Sutherland, Hansel and Hardisty. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexi A. Schnur, c2NobnVyYWxAbXN1LmVkdQ==