 Pengyuan Liu1,2
Pengyuan Liu1,2 Songbao Zou3,4,5
Songbao Zou3,4,5 Haikun Zhang1,6
Haikun Zhang1,6 Qing Liu1,2Zenglei Song1,2Yanyan Huang1,2
Qing Liu1,2Zenglei Song1,2Yanyan Huang1,2 Xiaoke Hu1,2,6*
Xiaoke Hu1,2,6*- 1Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences, Yantai, China
- 2University of Chinese Academy of Sciences, Beijing, China
- 3Key Laboratory of Healthy Freshwater Aquaculture, Ministry of Agriculture and Rural Affairs, Zhejiang Institute of Freshwater Fisheries, Huzhou, Zhejiang, China
- 4Key Laboratory of Fish Health and Nutrition of Zhejiang Province, Zhejiang Institute of Freshwater Fisheries, Huzhou, Zhejiang, China
- 5Huzhou Key Laboratory of Aquatic Product Quality Improvement and Processing Technology, Zhejiang Institute of Freshwater Fisheries, Huzhou, Zhejiang, China
- 6Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
The presence of seagrasses facilitates numerous microbial-mediated biogeochemical cycles, with sulfur- and nitrogen-cycling microorganisms playing crucial roles as regulators. Despite efforts to comprehend the diversity of microbes in seagrass ecosystems, the metabolic functions of these benthic microorganisms in seagrass sediments remain largely unknown. Using metagenomics, we provide insights into the sulfur- and nitrogen-cycling pathways and key metabolic capacities of microorganisms in both Z. japonica-colonized and unvegetated sediments over a seasonal period. Taxonomic analysis of N and S cycling genes revealed that δ- and γ- proteobacteria dominated the benthic sulfate-reducing bacteria, while α- and γ-proteobacteria played a significant role in the sulfur-oxidation processes. The proteobacterial lineages were also major contributors to the benthic nitrogen cycling. However, at a finer taxonomic resolution, microbial participants in different processes were observed to be highly diverse and mainly driven by environmental factors such as temperature and salinity. The gene pools of sulfur and nitrogen cycles in the seagrass sediments were dominated by genes involved in sulfide oxidation (fccA) and hydroxylamine oxidation (hao), respectively. Seagrass colonization elevated the relative abundance of genes responsible for sulfite production (phsC), hydroxylamine oxidation (hao), and nitrogen fixation (nifK), but suppressed sulfur oxidation (soxXYZ) and denitrification (nosZ and nirS). The prevalence of proteobacterial lineages functioned with versatile capabilities in both sulfur and nitrogen cycles in seagrass ecosystems, highlighting tight couplings between these processes, which was further supported by the recovery of 83 metagenome-assembled genomes (MAGs). These findings broaden our understanding of the biogeochemical processes that are mediated by microorganisms in seagrass ecosystems.
Introduction
Seagrass meadows, situated in the intertidal zone between land and ocean, are vital components of marine ecosystems, providing numerous ecological services (Fourqurean et al., 2012). The sediments inhabited by seagrass are characterized by high nutrient content, owing to the enrichment with organic carbon from various sources such as root exudates, seagrass litter, and the settling of phytoplankton debris (Duarte et al., 2005). These conditions foster abundant microbial activities in the seagrass sediments, which play essential roles in functioning and maintaining the health of the seagrass ecosystem by either increasing nutrient availability (Cifuentes et al., 2000; Ikenaga et al., 2010) or reducing the presence of potential phytotoxins (Holmer et al., 2005). This highlights the significance of further investigating the diversity, abundance, and functional capabilities of the microbial communities within seagrass systems, and the importance of protecting and conserving these unique ecosystems.
Sulfur is among the most abundant elements in nature and essential for all living organisms. The sulfur cycling is intricately coupled with carbon and nitrogen cycles, involving oxidation and reduction reactions that facilitate energy transfer and nutrient cycling (Muyzer and Stams, 2008). The microbial communities that inhabit the root and rhizosphere of seagrasses are mainly composed of members involved in the sulfur and nitrogen cycles (Cúcio et al., 2016; Cúcio et al., 2018). Sulfate-reducing bacteria (SRB) and sulfur-oxidizing bacteria (SOB) were identified as the most abundant groups among them (Sun et al., 2015; Cúcio et al., 2018). SRB plays a crucial role in organic matter mineralization (Holmer et al., 2001), nitrogen fixation (Nielsen et al., 2001; Bagwell et al., 2002), and phosphate regulation (Holmer et al., 2001). However, their anaerobic respirations, which are boosted in seagrass-vegetated sediments (Devereux, 2005), result in sulfide accumulation. Sulfide is a potent phytotoxin and has been identified as a primary culprit of global seagrass loss (Goodman et al., 1995; Baden et al., 2012). SOB, on the other hand, can alleviate sulfide stress by direct oxidizing it through desulfurization, or indirectly depositing to improve the seagrass living environments. A metatranscriptomic study by Cúcio et al. (2018) provided evidence for the presence of free-living forms of sulfur-oxidizing chemolithoautotrophic symbionts in the rhizosphere of Z. marina, highlighting the intimate relationship between sulfur-cycling microbes and seagrass.
Microbial-driven nitrogen cycling stands as one of the most fundamental biogeochemical processes on Earth, comprising six distinct processes: nitrogen fixation, assimilation, ammonification, nitrification, denitrification, and anaerobic ammonium oxidation (anammox) (Kuypers et al., 2018). The role of nitrogen-fixing bacteria in the seagrass sediments has been widely studied in efforts to understand the biodiversity and contribution to seagrass growth, which is often limited by nitrogen availability (Welsh, 2000; Sun et al., 2015; Garcias-Bonet et al., 2016). Studies have reported that up to 50% of nitrogen requirement of seagrass meadows can be attributed to the biological nitrogen fixation (Welsh, 2000; Smith et al., 2004; Agawin et al., 2016), with a significant involvement of SRB in this metabolic process (Smith et al., 2004; Sun et al., 2015). Despite the recognition of the importance of nitrogen-fixing bacteria, there remains a knowledge gap regarding the diversity and genetic makeup of microorganisms involved in various nitrogen cycling steps (e.g., dissimilatory nitrate reduction, ammoxidation, and denitrification). These processes not only influence seagrass growth and health but also play a pivotal role in maintaining ecosystem resilience, preventing eutrophication, and shaping the interactions between seagrasses and microbial communities (Hutchins and Capone, 2022; Suonan et al., 2022). Although the significance of microbial communities in seagrass ecosystems is increasingly acknowledged, particularly regarding nitrogen and sulfur cycles, limited research on these molecular pathways (Cúcio et al., 2018) impedes a systemic understanding of the ecological role of seagrass microorganisms.
Previously, studies investigating microbial communities from seagrass sediments have primarily concentrated on the taxonomic structure and diversity. These studies have found considerable differences in composition between rhizosphere and bulk sediments (James et al., 2006; Sun et al., 2015; Zheng et al., 2019) or between inner and outer seagrass sediments (Ettinger et al., 2017; Alsaffar et al., 2020). The high activity of sulfate-reducing diazotrophs in rhizosphere sediments, where nitrogen fixation rates are ~40 times higher than those in the bulk sediments (Nielsen et al., 2001), highlights the influence of environmental factors such as the high quality and quantity of organic matter, the steep redox gradient (Martin et al., 2019), and heavy metals accumulation (Lin et al., 2016) on the activity in these ecosystems. Additionally, the abundance and activities of bacteria in seagrass sediments exhibit strong seasonal patterns, with the highest levels observed during periods of active seagrass growth (Smith et al., 2004; James et al., 2006). These observed patterns likely reflect the seagrass growth status and seasonal shifts. Nonetheless, the influence of seagrass colonization and seasonality on the distribution of functional genes, particularly those involved in biogeochemical cycling of sulfur and nitrogen remains largely unknown.
Seagrass Zostera japonica is an ecologically important marine angiosperm found in the Northern Pacific coast, ranging from temperate to subtropical regions (Abe et al., 2009). This species has experienced a drastic decline in its population in Asia due to a range of factors, including climate change, anthropogenic disturbance, and other causes (Abe et al., 2009). Given the significance of sulfur- and nitrogen-cycling to seagrass ecosystems, a thorough understanding of these microbe-driven metabolic processes is crucial for the productivity, conservation, and recovery of seagrass systems. In the present study, seasonal patterns of diversity, abundances, and functional gene content of microbial taxa in both seagrass-vegetated and unvegetated sediments were investigated by shotgun metagenomic sequencing. The following questions were addressed: 1) Does the colonization of seagrass enhance the diversity and abundance of functional genes involved in the sulfur and nitrogen cycling? 2) Does this result in an alteration of their taxonomic and functional composition? 3) How do functional genes involved in sulfur and nitrogen metabolism respond to environmental changes?
Materials and methods
Study area, sampling and environmental characteristics
The seagrass meadow studied in the present study is situated at the estuary of the Yellow River in Dongying, Shandong Province, China (37° 51.070’ N, 119° 06.184’ W; Figure S1). The region features a temperate monsoon continental climate with pronounced seasonal variations. The meadow extends for 25-30 km, with a distribution width of 200-500 m from the shore to the sea, and encompasses an area of more than 1000 ha. It is the largest seagrass bed discovered in China to date (Yue et al., 2021). Field observation found that the eelgrass continuously distributed on the seaward side of the habitat of cordgrass (Sporobolus alterniflorus) and partially mixed with cordgrass. We collected seagrass-vegetated samples solely from an area covered by seagrass, without any cordgrass mixing, at a distance of approximately 200 m from the shore during the lowest tide period. Additionally, the unvegetated samples were collected from an unvegetated area located ~100 m further towards the seaward side, where seagrass growth was absent throughout the whole year of 2018.
Sampling was conducted during January, April, July, and September of 2018. Surface-layer (top 0-5cm) sediment samples were collected from a seagrass-covered area and an adjacent barren area, using a custom-made corer (10 cm in length and 1.5 cm in diameter). Triplicate samples, spaced roughly 5 meters apart, were pooled to form a single sample for both vegetated (V) and unvegetated (U) regions, resulting in a total of 8 samples. The samples were immediately transported in an ice box to the laboratory within an hour. Subsamples were taken from the well-mixed sediments and stored at −80°C for DNA extraction, while the remaining sediments were stored at −20°C for subsequent physicochemical determination.
The parameters of the overlying water, including temperature, salinity, dissolved oxygen (DO) concentrations, and pH, were measured using a YSI 556 (YSI, USA) (Table S1). The homogenized sediments were subjected to freeze-drying, grinding, and sieving in preparation for the determination of sediment parameters. The grain sizes of sediments were measured using a Mastersizer 2000F laser diffraction particle size analyzer (Malvern, England). The concentrations of nitrate (NO3−-N), nitrite (NO2−-N), and ammonium (NH4+-N) were analyzed using a Nutrient AutoAnalyzer (Seal, Germany). The sulfate (SO42−) concentrations were determined by an ion chromatography Dionex ICS 3000 (USA). The contents of total organic nitrogen (TON) and total organic carbon (TOC) were measured with a Vario Micro Cube elemental analyzer (Elementar, Germany). The concentrations of metals were determined using an ELAN DRC II plasma mass spectrometer (PerkinElmer, Hong Kong) (Table S1).
Metagenomic sequencing and data processing
Genomic DNA of triplicates was extracted from 0.5 to 1.0 g of sediment using a FastDNA® Spin Kit for Soil (MP Biomedical, USA) following the manufacturer’s protocol. The quantity of DNA was determined by a NanoDrop ND-3000 spectrophotometer (Thermo Scientific, NC, USA). The sequencing library was constructed with NEBNext Ultra II DNA PCR-free library prep kit (New England Biolabs, Ipswich, MA, USA). Shotgun sequencing was performed on the Illumina Hiseq 4000 platform with 2×150-bp paired-end reads at Majorbio Bio-Pharm technology company (Shanghai, China).
The raw metagenomic reads were preprocessed using KneadData v0.7.2 (Table S2), which integrates Trimmomatic (Bolger et al., 2014) and Bowtie2 (Langmead and Salzberg, 2012) to trim low-quality sequences (parameters: ‘SLIDINGWINDOW:4:20 MINLEN:50’) and remove contaminant reads originating from the seagrass Zostera marina reference genome (GCA_001185155.1 assembly) (parameters: ‘-very-sensitive-dovetail’). Each sample’s metagenomic reads were assembled individually into contigs using MEGAHIT v1.1.3 with default parameters (Li et al., 2016). To yield more genic and ecological (biomass) information of microorganisms, all clean reads were combined and co-assembled with MEGAHIT. Assembly qualities were evaluated using QUAST v5.0.2 (Gurevich et al., 2013) and are summarized in Table S3.
Protein-coding open reading frames (ORFs) were predicted from all contigs, which were at least ≥ 500 bp in length, using Prodigal v2.6.3 (Hyatt et al., 2010) with parameters: ‘-p meta -m’. The extracted ORFs, longer than 100 bp, were further clustered at a criterion of 95% identity (Sunagawa et al., 2015; Wang et al., 2022) to generate a non-redundant (NR) gene catalog (5,004,158 ORFs) using CD-HIT-EST v4.8.1 (Li and Godzik, 2006) with parameters ‘-T 86 -c 0.95 -M 0 -n 10 -G 0 -aS 0.9 -g 1 -d 0’. These NR ORFs were functionally annotated with KOfamscan v1.3.0 (Aramaki et al., 2020) and taxonomically annotated with DIAMOND-BLASTp v0.8.36.98 (parameters: ‘-evalue 1e-5’) against NCBI nr database (25 October 2019). The coverage and abundance (RPKM, reads per kilobase per million sample reads) were calculated by mapping high-quality reads to the NR gene set using BBMap v38.84 with default settings (Bushnell, 2014).
Identification and taxonomic assignment of nitrogen- and sulfur-cycling genes
The functions of genes involved in nitrogen- and sulfur-cycling were identified based on the assignment of KEGG Orthology (KO) (see Table S4). For each predicted gene, CD-HIT was utilized to further eliminate redundant sequences with a similarity of 90%. The diversity of each gene was calculated based on the number of non-redundant sequences. Nitrogen-cycling (hao, napB, narG/nxrA, narI/narV, nirS, norB, nosZ, nrfA) and sulfur-cycling genes (aprB, dsrA, fccA, phsC, sat, soeC, soxY, sqr) were selected according to following criteria: (i) sequence length ≥ 100, (ii) genes involved assimilatory reduction and mineralization processes were excluded, and (iii) the top 8 genes were with a minimum sequence count of 173 for sulfur genes and 117 for nitrogen genes (Table S4). Taxonomic annotation of these functional genes was carried out against NCBI nr database using BLASTp v.2.9.0 with an E-value cutoff of 1e-5. The full taxonomic lineages were obtained by querying the “taxids” against the NCBI taxonomy using “lineage” function in TaxonKit v0.10.1 (Shen and Ren, 2021). The composition of different phyla or classes for each functional gene was calculated as the sum of the abundance of sequences affiliated with that phylum or class.
Taxonomic composition of prokaryotes based on SSU rRNAs
The SSU rRNAs obtained from assembled contigs were screened using Metaxa2 (Bengtsson-Palme et al., 2015). These SSU rRNAs were further subjected to a denoising workflow that involved filtering, dereplication, chimera removal, and merging of paired-end reads. This workflow was carried out using the DADA2 pipeline (Callahan et al., 2017) in R version 3.6.3 (R Core Team, 2018), as previously described by (Liu et al., 2021). Taxonomic assignment was performed against the SILVA database (v138) and the community composition was visualized using the ggplot2 package in R software (Wickham, 2011).
Genome binning, refinement, de-replication, and annotation
Contigs with length ≥ 1000 bp were retained and binned into metagenome-assembled genomes (MAGs) utilizing the function modules of MetaWRAP v1.2.1 pipeline (Uritskiy et al., 2018) and three binning tools: MetaBAT2 v2.12.1 (Kang et al., 2019), MaxBin2 v2.2.6 (Wu et al., 2016), and CONCOCT v1.1.0 (Alneberg et al., 2014). The resulting MAGs were then refined using the ‘Bin_refinement’ module in MetaWRAP, and their completeness and contamination were evaluated by running CheckM v1.0.12 (Parks et al., 2015). The sets of MAGs from all samples were then combined and de-replicated using dRep v.2.6.2 (Olm et al., 2017) with default parameters, yielding 33 high-quality MAGs (completeness ≥ 90%, and contamination ≤ 5%) and 50 medium-quality MAGs (completeness ≥ 60%, and contamination ≤ 10%).
The final MAGs were re-annotated more thoroughly, with taxonomic classification performed using GTDB-Tk v1.2.0 (Chaumeil et al., 2020). Genes prediction was carried out using Prodigal, and protein functions were annotated through a combination of InterProScan v5.34 (Jones et al., 2014) and KOfamscan. Identification of the types and quantity of tRNA sequences was performed using tRNAscan-SE v2.0.5 (Meier-Kolthoff et al., 2013). The presence of subunit rRNA genes was screened using Metaxa2 v2.2.3 (Bengtsson-Palme et al., 2015). The coverage of each MAG was calculated as the average coverage of all contigs belonging to that MAG and normalized based on sequencing depth. A summary of the taxonomic classification, genome size, completeness, contamination, strain heterogeneity, N50, GC content, and coverage of recovered MAGs is presented in Table S7.
Phylogenetic analysis
The phylogeny of the final MAGs was inferred using the UBCG v3.0 pipeline (Na et al., 2018), which was based on 92 housekeeping genes. A concatenated maximum-likelihood phylogenetic tree was constructed using IQ-TREE (Nguyen et al., 2015) with 1000 bootstraps and was visualized using Interactive Tree of Life (iTOL, v5.0) platform (Letunic and Bork, 2021).
Statistical analysis
The abundance of each gene was calculated as the sum of the coverage of all ORFs assigned to that gene. Student’s t-test was performed to test the difference in relative abundance of major taxonomic groups of functional genes between seagrass-vegetated and unvegetated samples. To explore the relationships between diversity and abundance of functional genes and environmental parameters, linear regression and Spearman’s rank correlation were conducted using SPSS v.13.0 (IBM, USA).
Results
Metagenomic statistics and microbial community composition
A total of ~418 million clean metagenomic reads was retained after quality control, with an average of 52.3 ± 7.1 million reads per sample (Table S2). Following assembly, 831,728 contigs with length > 1 kb were obtained, ranging from 34,537 to 149,379. The N50, representing the length at which 50% of the assembled contigs were equal to or longer than, ranged between 699 and 895 bp, with an average G+C content of 54.5%. Co-assembly resulted in a total of 1,423,701 contigs (>1 kb), with an N50 of 1,072 bp and an average G+C content of 55.6% (Table S3).
The taxonomy of prokaryotic community in 8 samples was analyzed by screening SSU rRNAs from the assembled contigs. Our analysis identified the presence of 26 prokaryotic phyla, as well as the prominent subclasses within Proteobacteria (Figure S2). A large proportion of reads (approximately 58.1% on average) was assigned to Proteobacteria, with variations ranging from 52.8% to 64.5% in individual samples. Within Proteobacteria, γ- (14.9%-34.5%), δ- (9.4%-30.4%), and α-proteobacteria (6.6%-19.2%) were the most abundant. While ε-, and β-proteobacteria were present in relatively lower abundance, with average proportions of less than 1%. The second most abundant bacterial phylum was Bacteroidetes, accounting for 15.3% of reads, followed by Firmicutes and Actinobacteria, which accounted for 6.0% each. The rest (e.g., Planctomycetes, and Verrucomicrobia) appeared to be minor. Compared to the bacteria, archaea were less abundant, accounted for 3.5% of the total sequences, with the majority belonging to Euryarchaeota phylum (Figure S2).
Abundance of sulfur- and nitrogen-cycling genes in Z. japonica-vegetated and unvegetated sediments
After clustering protein sequences at a 90% similarity level, 4,612 ORFs were annotated as sulfur-cycling-related genes (Table S4). The gene fccA, which encodes sulfide dehydrogenase, dominated the pool of sulfur metabolism genes, particularly in the summer sediments that were seagrass-vegetated (Figure 1A). Additionally, the abundances of dissimilatory thiosulfate reduction gene phsC (average RPKM, 89.8 vs. 66.5) and assimilation gene cysC (30.1 vs. 14.9) were notably higher in vegetated sediments compared to unvegetated sediments (P < 0.05). However, genes involved in thiosulfate oxidation (soxXYZ, 73.1 vs. 110.7), sulfate reduction (aprA, 53.4 vs. 77.2), and tetrathionate reduction (ttrB, 53.8 vs. 72.6) were more abundant in unvegetated areas, indicating a potentially different mechanism of sulfur metabolism between the two sediment types. Regarding seasonal comparisons, fccA, and sseA peaked during summer in both sediment types, while sat and aprB genes were generally higher in the autumn (Figure 1A).
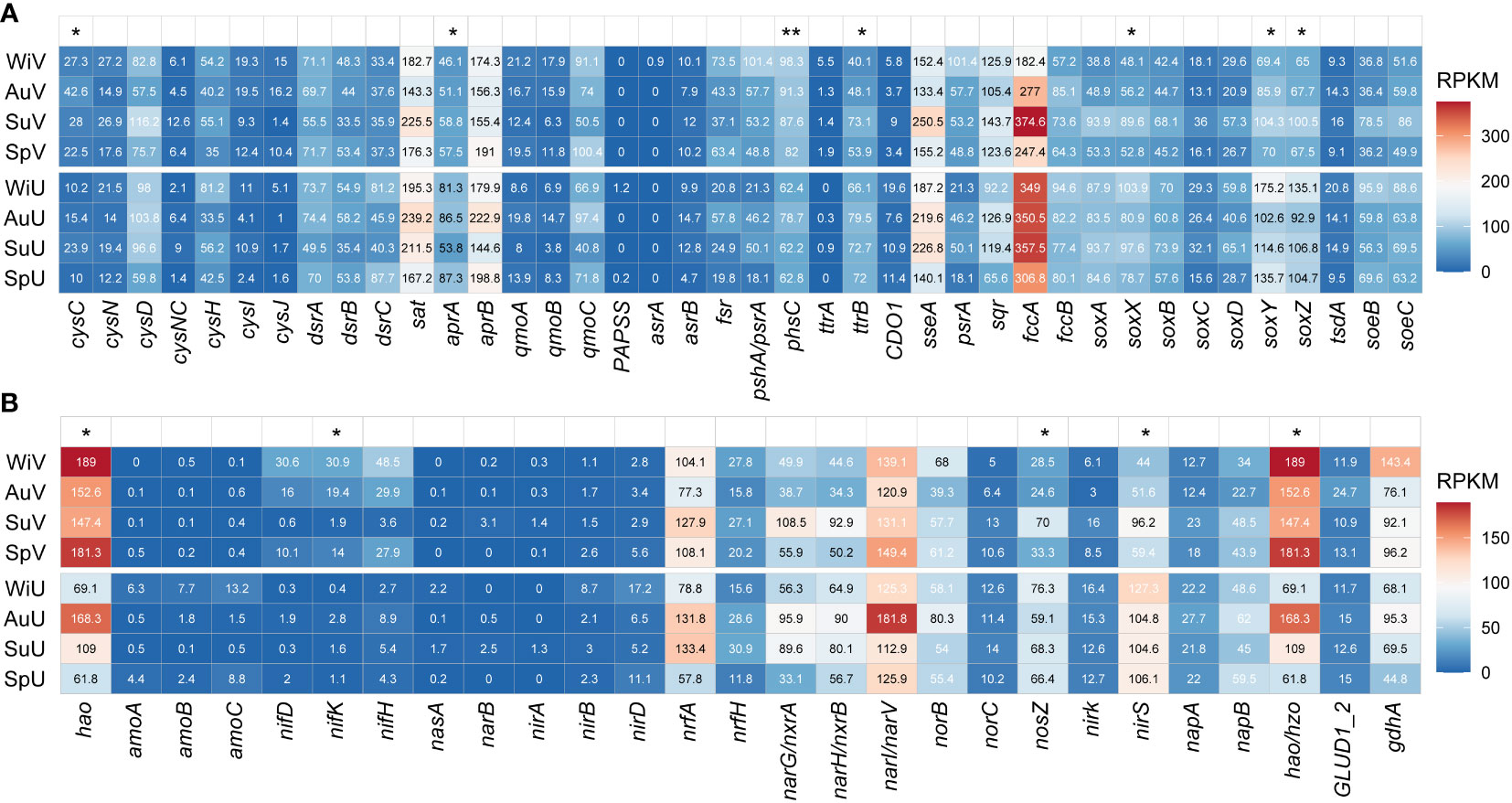
Figure 1 Heatmap showing the variation in abundance of functional genes that are involved in sulfur (A) and nitrogen (B) metabolic pathways between seagrass-vegetated (V) and unvegetated sediments (U) across four seasons of spring (Sp), summer (Su), autumn (Au), and winter (Wi). Statistical significance levels are indicated as *P < 0.05 and **P < 0.01 according to Student’s t-tests. RPKM, reads per kilobase per million sample reads.
For the nitrogen cycles, 2,343 ORFs were obtained (Table S4). The highest abundance of nitrogen-cycling-related genes was hao, encoding hydroxylamine dehydrogenase, with RPKM ranging from 61.8 to 189 (Figure 1B), followed by narI/narV encoding nitrate reductases. The proportions of the nitrogen fixation gene (nifK, 16.5 vs. 1.5) and hydroxylamine oxidation gene (hao, 167.6 vs. 102.1) were significantly higher in vegetated sediments compared to unvegetated areas. However, the relative abundances of nitrite reduction gene (nirS, 62.8 vs. 110.7) and nitrous-oxide reduction gene (nosZ, 39.1 vs. 67.5) were notably higher in unvegetated sediments. In seagrass-covered area, nitrogen metabolism genes (hao, nifH, narI/narV, and hzo) were more abundant in spring and winter compared summer and autumn. In contrast, the barren area generally had higher gene abundance for hao, narI/narV, and gdhA during summer and autumn (Figure 1B).
Taxonomic composition of functional genes involved in sulfur and nitrogen cycling
The taxonomic affiliation and niche distribution of microbial taxa involved in sulfur and nitrogen cycles were analyzed, as depicted in Figures 2 and S3. In the seagrass meadow, sulfur cycling was predominantly governed by proteobacterial lineages, constituting an average relative abundance of 80.7% (Figure 2A). Within the Proteobacteria phylum (Figure S3), δ- and γ-proteobacteria played key roles in sulfate reduction, including aprB (85%-95%), dsrA (83%-99%), phsC (19%-96%), and sat (58%-87%). The δ-proteobacteria were mainly represented by Desulfosarcina genus, while γ-proteobacteria were dominated by genera Ca. Thiodiazotropha and Sedimenticola (Table S5). Conversely, α- and γ-proteobacteria were predominantly associated with sulfide oxidation (fccA, soxY, soeC, and sqr), accounting for over 80% of the abundance. Predominant members of α-proteobacteria included Hyphomonas (fccA and soxY), Roseobacter (soeC), and Pseudoruegeria (sqr) (Table S5). Apart from Proteobacteria, other functional groups were also identified in sulfur cycle. Acidobacteria and Chloroflexi were important sources of genes related to sulfur reduction (aprB, dsrA, phsC, and sat), with an average abundance of 8.1%. Bacteroidetes contributed to both sulfide-oxidation (sqr, 20.3%) and thiosulfate-reduction (phsC, 5.2%) processes. Additionally, Planctomycetes and Gemmatimonadetes participated in sulfite oxidation (soe) and sulfide oxidation (fccA), respectively.
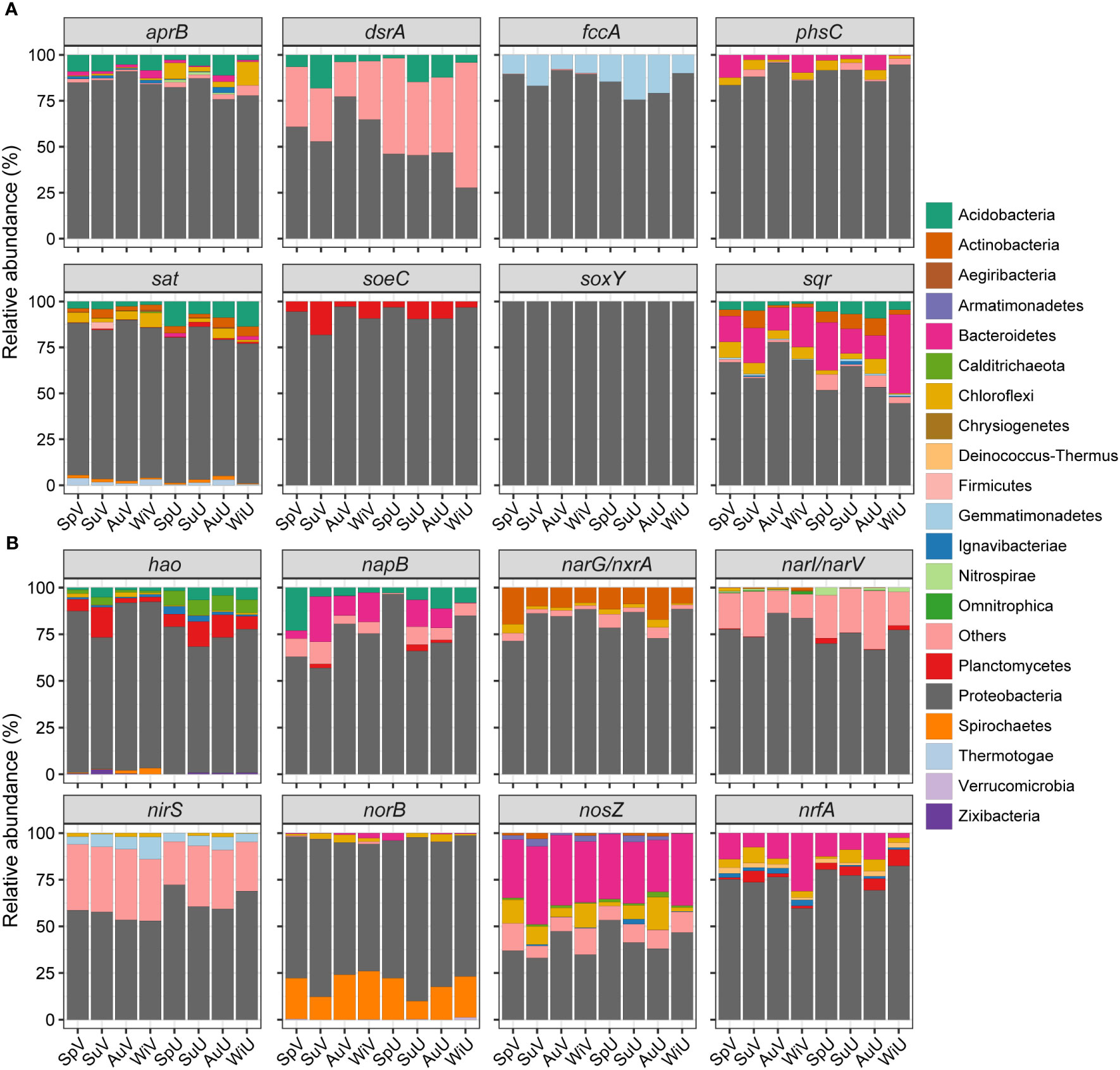
Figure 2 Taxonomic compositions of sulfur- (A) and nitrogen-cycling genes (B) at phylum level.
Regarding nitrogen cycling, Proteobacteria was also the dominant participant of nitrogen-cycling (70.6% on average) affiliated microbial taxa in the Z. japonica system (Figure 2B). The gene encoding hydroxylamine oxidoreductase (hao) was mainly assigned to δ- (62.8%, mainly Desulfosarcina) and γ -proteobacteria (16.2%, mainly Ca. Thiodiazotropha), as well as Planctomycetes (8.3%). The α-, δ-, and γ-proteobacteria collectively contributed significantly to the abundance of napB, norB, and narI/narV, with proportions ranging from 52.3% to 94.5%. Furthermore, napB could also be categorized to phylum Acidobacteria (7.9%) and Bacteroidetes (10%), while norB could partially be assigned to Spirochaetes (19.4%). Contig sequences aligning to gene narG/nxrA were mostly affiliated with α- (53.9%, mainly Aestuariivita), δ-proteobacteria (22.2%, mainly Desulfatitalea and Desulfotignum), and Actinobacteria (11.7%). nrfA sequences belonging to Proteobacteria were exclusively from δ-proteobacteria (mainly Desulfuromonas and Malonomonas), while Bacteroidetes, Planctomycetes, and Chloroflexi were also its carriers. Gene nirS was affiliated with γ- (32.5%, mainly Thiohalomonas), α-proteobacteria (12.1%, mainly Roseovarius), and, Gemmatimonadetes (6.3%). The main contributors of nosZ were α- (22.3%, mainly Roseovarius), γ-proteobacteria (18.5%, mainly Sulfurivermis), Bacteroidetes (34.7%), Chloroflexi (8.6%), and Armatimonadetes (2.1%; Table S6).
Environmental factors driving the variations of sulfur- and nitrogen-cycling genes
We utilized linear regression analysis (Figures 3, 4) and Spearman’s rank correlation analysis (Figures S4, 5) to identify key geochemical parameters influencing the diversities and abundance of sulfur- and nitrogen-cycling genes. The diversities of sulfur-cycling genes, particularly gene sat (R = 0.882, P = 0.004), increased with warming (Figure 3A). Spearman’s rank correlation further highlighted that temperature significantly promoted sat (ρ = 0.946, P < 0.001) and sqr (ρ = 0.776, P < 0.05) diversities, while salinity and specific metal elements (Mn, As, and Cd) substantially reduced the diversities of sulfate-reducing genes dsrA and aprB (ρ < -0.731, P < 0.05; Figure S4A).
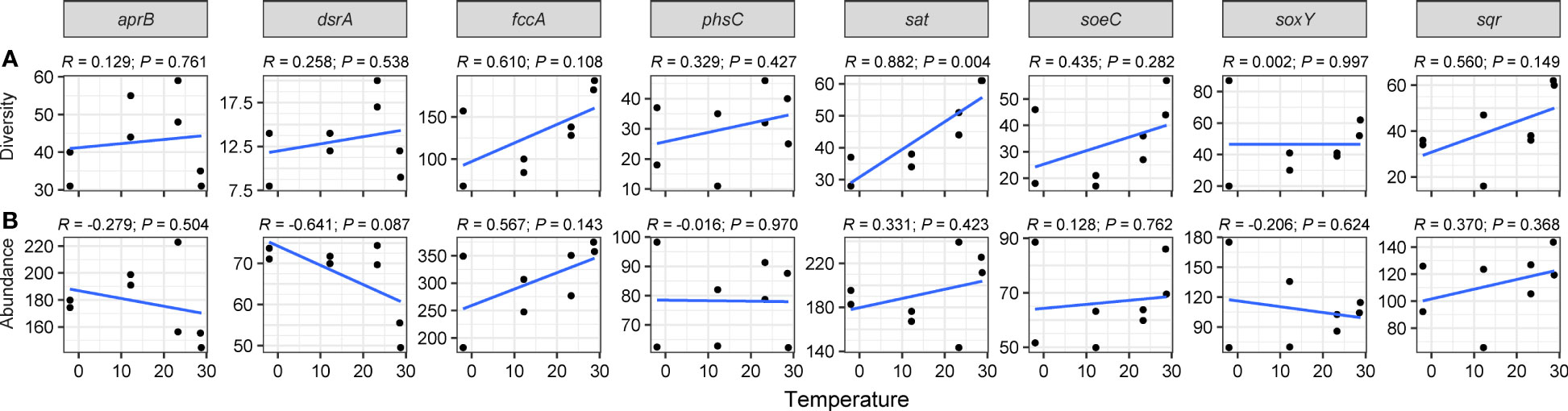
Figure 3 Linear regression modeling of the relationship between temperature and diversities (A) and abundances (B) of sulfur-cycling genes.
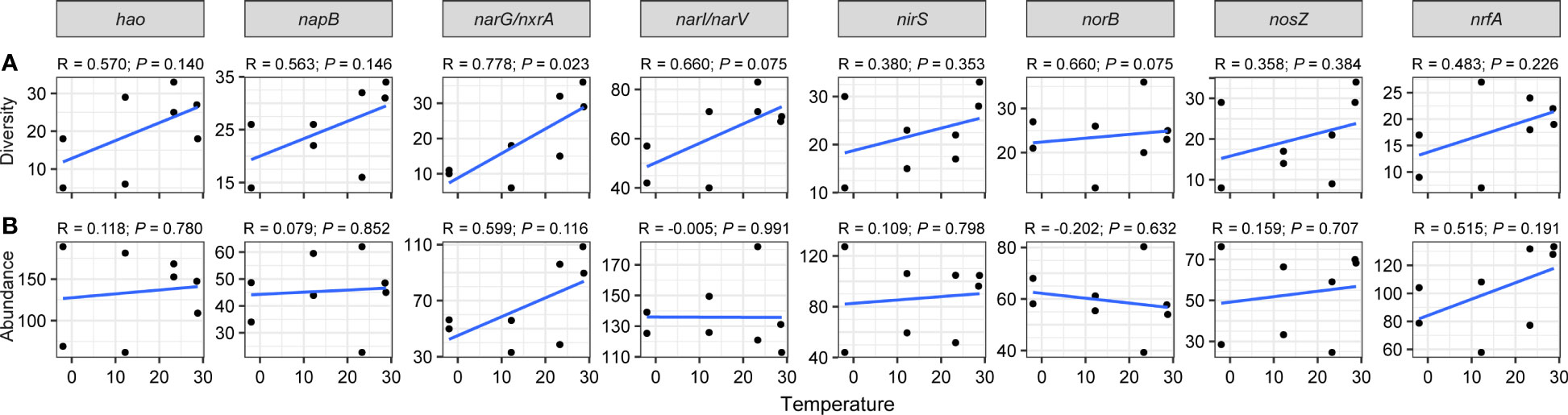
Figure 4 Linear regression modeling of the relationship between temperature and diversities (A) and abundances (B) of nitrogen-cycling genes.
In contrast, no significant linear relationship was observed between sulfur-genes abundance and temperature (Figure 3B). Spearman’s correlation revealed a positive correlation between dsrA abundance and DO (ρ = 0.714, P < 0.05) but a negative correlation with NO3- content (ρ = -0.738, P < 0.05). Additionally, gene aprB abundance was negatively correlated with Cd (ρ = -0.714, P < 0.05; Figure S4B).
As for nitrogen-cycling genes, particularly gene narG/nxrA (R = 0.778, P = 0.023), their diversities increased with rising temperature (Figure 4A). Spearman’s coefficients indicated a significant positive correlation between temperature with narG/nxrA diversity (ρ = 0.795, P < 0.05), a positive correlation between NH4+ contents and norB diversity (ρ = 0.714, P < 0.05), and a negative relationship between salinity and narI/narV diversity (ρ = -0.719, P < 0.05; Figure S5A). Although no significant linear relationship was found between nitrogen genes abundance and temperature (Figure 4B), a significantly positive correlation was observed between NH4+ and norB abundance (ρ = 0.762, P < 0.05; Figure S5B).
Metagenome-assembled genomes of prokaryotes associated with sulfur and nitrogen cycles
We employed a genome binning strategy to comprehensively explore the genomic potential for sulfur and nitrogen cycling, resulting in 83 medium- to- high quality metagenome-assembled genomes (MAGs) after removing duplicates. In accordance with recent MAGs standards (Bowers et al., 2017), all MAGs in this study exhibited >80% completeness with < 10% contamination. Detailed statistics for each MAGs are provided in the Supplementary Table S7. In summary, these 83 MAGs exhibited genome size ranging from 1.9 to 9.4 Mbp, GC content spanning 34.3 to 41.4%, and contained 2,102 to 8,478 ORFs. Using the GTDB-Tk tool, we classified the 83 MAGs in to 12 different phyla (Figure 5; Table S7), with the majority classified into Proteobacteria (30 MAGs, predominantly Halieaceae, Rhodobacteraceae, and Sedimenticolaceae), Bacteroidota (21, mainly Flavobacteriaceae), and Desulfobacterota (16, mainly Desulfocapsaceae). Smaller numbers of MAGs were assigned to various other phyla, including Actinobacteriota (4, Acidimicrobiia), Desulfuromonadota (2, Desulfuromonadales), Acidobacteriota (2, Pyrinomonadaceae), Chloroflexota (2, Anaerolineaceae) and Gemmatimonadota (2, Gemmatimonadetes), Nitrospirota (1, Thermodesulfovibrionales), Planctomycetota (1, Phycisphaerae), Cyanobacteria (1, Cyanobiaceae), and BMS3Abin14 (1).
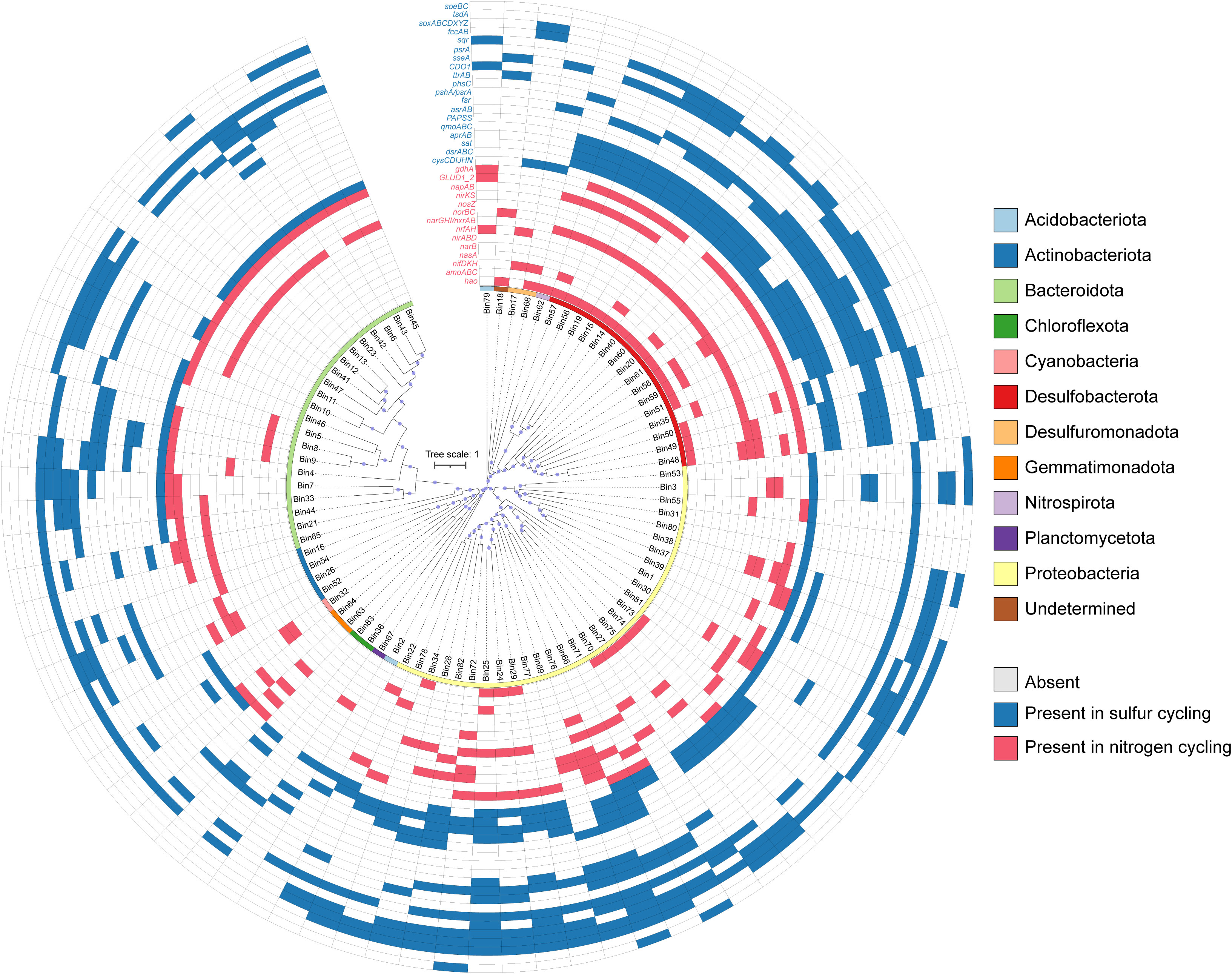
Figure 5 Presence (blue) and absence (blank) of key functional genes in metagenome-assembled genomes (MAGs) involved in sulfur and nitrogen biogeochemical cycling processes. The taxonomy (phylum level) of MAGs are indicated in different colors.
Among the 83 MAGs, each featured at least one sulfur-cycling gene (Figure 5; Table S8). Notably, a greater proportion of MAGs contained genes for sulfide oxidation (64 MAGs) and dissimilatory sulfite reduction (60 MAGs) compared to other sulfur-cycling pathways (Figure 6). Various MAGs encompassed genes encoding sulfide-oxidizing enzymes, specifically sulfide:quinone reductase (sqr, 53 MAGs) and fccAB (38 MAGs; Figure 5). Specific MAGs assigned to α- (Bin1) and γ-proteobacteria (Bin22, 24-25, 27-29, 66, 72, 75-78, and 82) exhibited the dual capability of encoding both sulfide dehydrogenase (fccA) and sulfide:quinone oxidoreductase (sqr) (Figure 5; Table S8), indicating a pronounced potential for sulfide oxidation. Moreover, three MAGs within γ- (Bin27) and α-proteobacteria (Bin37 and 38) were able to encode a complete Sox-enzyme system (Figure 5; Table S8). Additionally, 15 MAGs assigned to Desulfobacterota (Bin14-15, 19, 20, 40, 51, 56-60), Nitrospiraceae bacterium UBA6898 (Bin62), and γ-proteobacteria (Bin66, 76, and 82), were identified as hosting the full canonical pathway for sulfate reduction to sulfide (sat+qmoABC+aprAB+dsrAB) (Figure 5; Table S8). The gene encoding 3-mercaptopyruvate sulfurtransferase (TST), involved in the mineralization of organic sulfur compounds, exhibited widespread prevalence across these MAGs (58 MAGs) (Figure 5; Table S8).
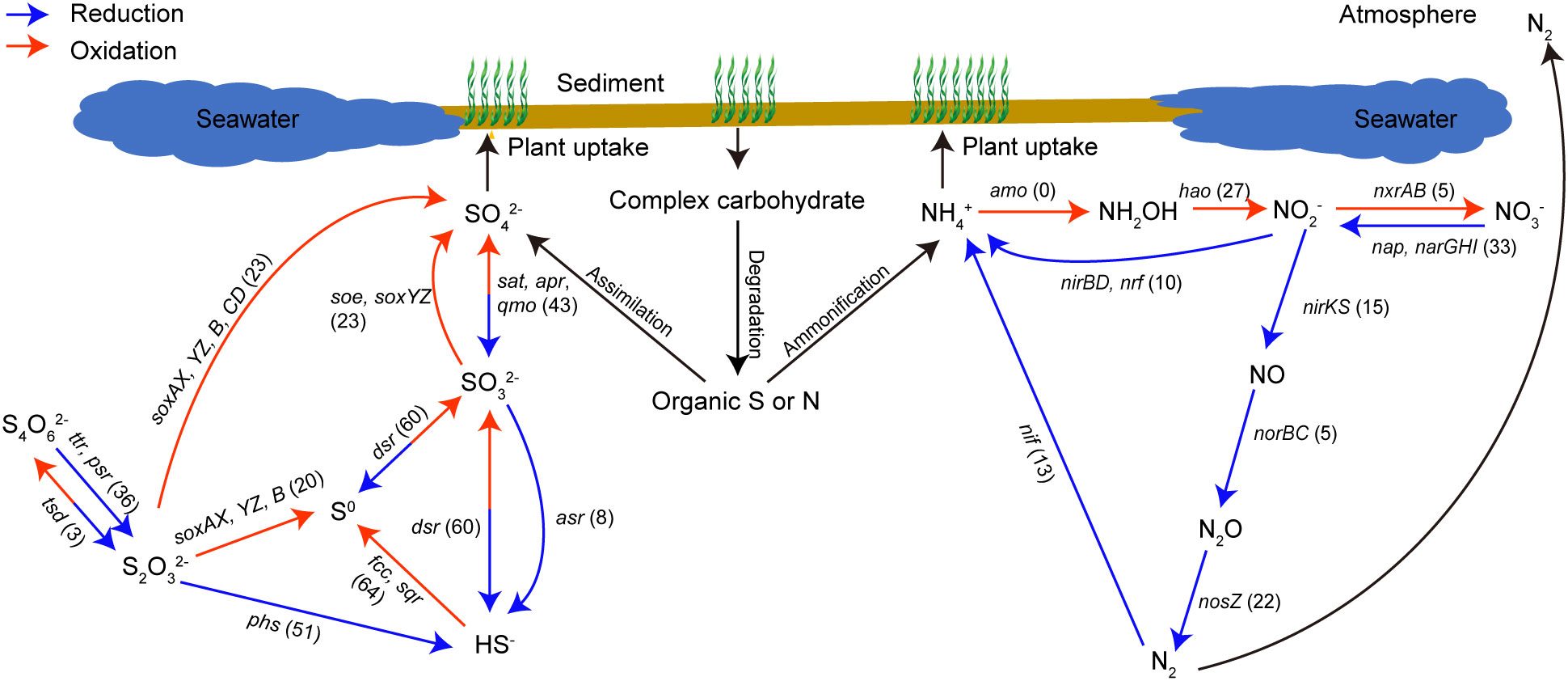
Figure 6 Number of MAGs involved in dissimilatory sulfur- and nitrogen-cycling pathways. MAGs were considered possessing the related enzyme if they contain any of the genes that form the enzyme complex, and the number of MAGs in each step was labelled in the brackets.
For the nitrogen cycle, the gene encoding glutamate dehydrogenase (E1.4.1.4), which is involved in the nitrogen-mineralization, was the most widely distributed and detected in 42 MAGs (Figure 5; Table S8), followed by narI/narV. The complete pathways of assimilatory nitrate reduction, nitrogen fixation, DNRA, and denitrification were identified in the 83 MAGs (Figure 5), and the number of MAGs containing nitrate reduction genes was more abundant (33 MAGs) than other nitrogen-cycling steps (Figure 6). Along the denitrification pathway, MAGs associated with Desulfobacterota (NO3- → NO2- and NO → N2O), γ-proteobacteria (NO2- → NO), and Bacteroidota (N2O → N2) were dominant in different steps, respectively (Figure 5; Table S8). Additionally, five MAGs from Desulfobacterota (Bin48 and 58), Gemmatimonadota (Bin63), and γ-proteobacteria (Bin72 and 76) contained both dissimilatory nitrate- and nitrite-reduction gene clusters (narGHI/napAB+nirBD/nrfAH), which have the potential to reduce nitrate to ammonium. Desulfobacterota was found to be the main nitrogen-fixing bacteria (Figure 5; Table S8).
Discussion
In this study, we investigated the functional genetic potential associated with sulfur and nitrogen cycles of seagrass (Z. japonica) benthic microbes at a seasonal scale with the consideration of seagrass colonization effects. A variety of sulfur- and nitrogen-cycling microorganisms with complex metabolic pathways involved in sulfur and nitrogen transformations were recovered. Our findings demonstrate that investigating the biogeography of microbial functional genes can give greater insight into the biogeochemical processes present in seagrass system.
Diversities and abundance of sulfur-cycling genes and their environmental drivers
In all metagenomes, we identified gene sets responsible for dissimilatory sulfate reduction and sulfide oxidation, highlighting the ecological significance of sulfate respiration and sulfide removal in the seagrass meadow ecosystem. Notably, the gene fccA was found to be the most abundant among sulfur-cycling gene pool in the seagrass sediments. The prevalence may be attributed to the provision of aerobic conditions created by seagrass, which are favorable for SOB. In contrast, sqr, the homolog of fccA, appeared relatively scarce. The gene fcc encodes flavocytochrome c, typically utilized for sulfide oxidation under low sulfide concentrations and aerobic conditions, while sqr encodes enzyme sulfide:quinone oxidoreductase, primarily employed for oxidizing sulfide under high sulfide concentrations (Cúcio et al., 2018). The observed high abundance of sulfide-oxidation genes potentially signifies a rapid recycling mechanism for reduced sulfides in sediments colonized by seagrass. This aligns with previous research by Holmer et al. (2003), which highlighted an accelerated turnover of reduced sulfides in these seagrass-covered sediments.
Our study observed that seagrass-vegetated sediments specifically promoted the abundance of thiosulfate reductase gene phsC, which is responsible for sulfide production (Table S5). This result is in line with previous reports of enrichment of sulfate-reduction bacteria in seagrass-vegetated sediments (Holmer et al., 2005; Sun et al., 2015; Cúcio et al., 2018). These findings indicate that seagrass vegetation may stimulate the microbial capacity to reduce thiosulfate, with thiosulfate serving as a major electron acceptor in sedimentary microbial communities involved in sulfur cycle. In general, SRB utilizes thiosulfate as an electron acceptor to completely oxidize various fatty acids and alcohols to CO2 (Brysch et al., 1987) or incompletely to acetate (Brenner et al., 2005). Thus, these thiosulfate-utilizing bacteria aid in the mineralization and recycling of organic matter in productive seagrass-vegetated sediments, and also provide the seagrass with an alcohol detoxification process as a compensation for the sulfide they produce (Cúcio et al., 2016). In contrast, the abundances of genes related to sulfur oxidation (soxXYZ), sulfate reduction (aprA), and tetrathionate reduction (ttrB) were significantly higher in the samples from the unvegetated areas and are involved in the formation of sulfur cycle intermediates (SCIs), including sulfite, thiosulfate, tetrathionate, elemental sulfur, polysulfides. SCIs play a significant role in ecological and biogeochemical perspective in marine environments, serving as an energy source for microbial communities (Vigneron et al., 2021) and creating shortcuts in tropical sulfur metabolism (Jørgensen et al., 2019). Hence, the enrichment of genes involved in SCIs production may provide additional energy for microbial guilds in the oligotrophic unvegetated environments.
Taxonomic annotation of sulfur-related genes revealed that Ca. Thiodiazotropha and Sedimenticola, members of γ-proteobacteria, accounted for a large portion of the genes involved in dissimilatory sulfate reduction (aprB, dsrA, phsC, and sat; Table S5). Ca. Thiodiazotropha, prominent member of γ-proteobacteria in the Z. japonica system, is typically known as a symbiotic bacteria of lucinids, which are frequently found in seagrass sediments (Osvatic et al., 2021). Notably, a comprehensive global survey of rhizosphere microbial diversity across 14 seagrass species revealed that Ca. Thiodiazotropha is abundant in seagrass environments worldwide, regardless of the presence of lucinids. This suggests that Ca. Thiodiazotropha offers a mechanism for seagrasses to alleviate sulfide stress (Martin et al., 2020). Additionally, members of δ-proteobacteria, particularly Desulfosarcina significantly contribute to dissimilatory sulfate reduction (Table S5), consistent with our metagenome annotation (Table S8). The genus Desulfosarcina predominates among δ-proteobacteria in the rhizosphere of various seagrass species, including Cymodocea nodosa, Zostera marina, Zostera noltii, Halophila ovalis, Halodule wrightii, Syringodium isoetifolium, Thalassia hemprichii, and Thalassia testudinum (Cúcio et al., 2016; Wang et al., 2020; Aires et al., 2021; Ling et al., 2021b). Previous study have demonstrated that members of genera Desulfosarcina are capable of utilizing a diverse range of electron donors, such as ethanol, benzoate, lactate, formate, H2, propionate, lactate, and 1-propanol, for sulfate reduction (Kuever et al., 2015; Cúcio et al., 2018). Consequently, the genus Desulfosarcina was conjectured to contribute significantly for nutrient availability and remineralization in seagrass sediments (Holmer et al., 2001). In seagrass-vegetated sediments, high concentrations of various carbon substrates supplied from root exudates and organic matter decomposition provide a competitive advantage for the metabolically versatile SRB (Smith et al., 2004).
Sulfide oxidation processes (fccA, soxY, soeC, and sqr) are primarily carried out by genus Hyphomonas, Roseobacter, and Pseudoruegeria (Table S5). These genera share a common ecological strategy, where Hyphomonas utilizes a cylindrical prostheca as a holdfast to attach to the surface of seagrass leaves (Halophila stipulacea) (Weidner et al., 2000). Similarly, certain Roseobacter and Pseudoruegeria strains are reported to form biofilms on surfaces of marine algae, invertebrates, and particles (Ding et al., 2023). Above-mentioned these gene-level findings aligns well with genome binning analysis, emphasizing the pivotal role of gproteobacteria in sulfur metabolism in Z. japonica meadows, participating in both sulfate reduction and sulfide oxidation.
In this study, we observed an increase in diversity of sulfur-cycling genes, specifically the gene sat, with higher temperature (Figure 3A). Previous studies have demonstrated that increased temperature can affect diversity and intensify sulfate reduction reactions of SRB (Knoblauch and Jørgensen, 1999; Robador et al., 2016). For instance, Ma et al. (2021) found that the diversity of dsrB gene was highest observed at mid-temperature in hot springs. Given the established association between seagrass loss with elevated temperatures and sulfides accumulation (Koch et al., 2007; García et al., 2012; García et al., 2013), further research on sulfur biogeochemical cycles is imperative to understand the impact of global warming on seagrass ecosystems.
Diversities and abundance of nitrogen-cycling genes and their environmental drivers
The gene hao, responsible for hydroxylamine oxidation to nitrite, was abundant in vegetated sediment compared to unvegetated sediment (Figure 1B). Hydroxylamine has been identified as a key nitrogen metabolite for microbial interactions within microbial communities and engineered systems (Soler-Jofra et al., 2021). Adding external hydroxylamine to biofilm system has been shown to disperse microcolonies into scattered cells (Kindaichi et al., 2004; Harper et al., 2009). Therefore, the prevalence of hao gene suggests a significant role in shaping the microbial communities and the biofilm dynamics in seagrass ecosystems. Additionally, we observed a higher level of the nitrogenase gene (nifK) in the vegetated sediments (Figure 1B), consistent with the findings of Sun et al. (2015). Nitrogen fixation by rhizosphere microbiomes is considered a major contributor to seagrasses’ nitrogen requirements (Welsh, 2000). The presence of diazotrophic assemblages has been characterized on seagrass surfaces through sequencing nitrogenase gene (nifH) (Sun et al., 2015) and 16S rRNA amplicons (Jensen et al., 2007). In contrast, the low abundance of nitrogen-fixing encoding genes (nifK) and the high abundance of denitrification genes (nirS and nosZ) in the unvegetated sediments imply that nitrogen was not a limiting nutrient in the unvegetated environments.
In agreement with our expectations, the majority of nitrogen-cycling genes and MAGs derived from metagenomic data exhibited affiliation with δ- and γ-Proteobacteria (Figure S3B and Figure 5). This suggests that Proteobacteria play a crucial role in the nitrogen cycling process, with various lineages of this phylum contributing differently to the abundance of different nitrogen-cycling genes. For example, genera that are potentially associated with nitrogen cycling were mainly affiliated with Desulfosarcina, Desulfuromonas, and Malonomonas (Table S6), have the capacity to use sulfate as a terminal electron acceptor (Mills et al., 2012).
In concordance with sulfur-cycling genes, we observed that the diversity of nitrogen cycle genes increased with increasing temperature (Figure 4A), especially for nitrification gene nxrA, which is responsible for nitrate (NO3–N) and nitrite-nitrogen (NO2–N) conversion. This corresponds with previous findings that temperature is positively correlated with nitrification rates in estuaries or coastal systems (Dai et al., 2008; Damashek et al., 2016). Elevated nitrification may result from increased ammonium regeneration due to enhanced microbial respiration at high temperatures (Caffrey, 2004), or it may be due to higher growth rates of nitrifying bacteria at higher temperatures. Additionally, denitrification genes narI/narV, encoding nitrate reductase responsible for the transformation of nitrate to nitrite (Sohaskey and Wayne, 2003), exhibited reduced diversity in response to salinity (Figure S5A). This observation is consistent with the widely accepted concept that salinity acts as a stressor on denitrification process by inhibiting microorganism activities (Miao et al., 2015; Zhai et al., 2020).
Tight couplings between sulfur- and nitrogen-cycling in seagrass ecosystems
Approximately 68% of the draft genomes were found to contain genes responsible for partial nitrogen and sulfur metabolisms, indicating tight couplings between sulfur and nitrogen cycles. The most abundant MAGs classified as Proteobacteria suggest that this taxonomic group may represent the most abundant active population. Among the members of Proteobacteria, γ- and α-proteobacteria were genetically equipped to perform denitrification and oxidation of reduced inorganic sulfur compounds. Different taxa were equipped with genes for different metabolic steps, implying a cooperative effort among microbial species. The dissimilatory nitrate reduction (to ammonium or denitrification) coupled with sulfur oxidation is a significant energy source for inorganic carbon fixation (Nakagawa and Takai, 2008; Li et al., 2018) and utilizes nitrate and nitrite as electron acceptors. For instance, the frequently detected SOB Sulfurovum sp. in seagrass systems (Sun et al., 2020; Zhang et al., 2020; Martin et al., 2022; Zhang et al., 2022), have been shown to use nitrate as an electron acceptor (Li et al., 2018).
It has been reported that fix nitrogen is widespread among SRB, which were previously considered the dominant diazotrophs in seagrass-colonized sediments (Welsh, 2000; Smith et al., 2004). Studies have shown that the nitrogen fixation fulfilled by the sulfate-reducing community could supply 60-95% of the nitrogen requirements of seagrasses (Welsh, 2000; Smith et al., 2004). Similarly, the dominant members of the nifDHK host guild in the Z. japonica system were found to be sulfate-reducing Desulfobacterota, including the families Desulfococcaceae, Desulfocapsaceae, and Desulfosarcinaceae (Figure 5; Table S6). This finding provides molecular evidence for the tight coupling of nitrogen fixation and sulfate reduction in seagrass meadows (Welsh, 2000; Smith et al., 2004; Ling et al., 2021a). Additionally, we also identified the potential of the SRB genomes from Desulfobacterota and γ-proteobacteria participate in the reductive sulfur cycle processes by oxidizing hydroxylamine or nitrite (Figure 5). This suggests that they may facilitate the connection between sulfate reduction and nitrification processes. Alternatively, the presence of diverse functional profiles in these microorganisms allows for metabolic switch between available compounds in seagrass ecosystems (Cúcio et al., 2018). Our results suggest a tight interdependence between sulfur- and nitrogen-metabolite exchange, with cooperation among various microbial species being crucial for the completion of benthic nitrogen and sulfur cycles in seagrass meadows.
Concluding remarks
This study sheds light on the unknown seasonal variations in sulfur- and nitrogen-cycling genes between seagrass-vegetated and unvegetated sediments, thus supporting the hypothesis that seagrass colonization could affect microbial-mediated biogeochemical cycles. We found that genes involved in sulfide oxidation (fccA) and hydroxylamine oxidation (hao) dominated the gene pools of sulfur- and nitrogen cycles. Microorganisms residing in vegetated sediments exhibited higher proficiency in sulfite reduction (via phsC) and nitrogen fixation (nifK), whereas those in unvegetated sediments displayed enhanced activity in sulfur oxidation (soxXYZ) and denitrification (nosZ and nirS). We provided a finer taxonomic resolution, revealing that microbial participants in different processes are highly diverse and their relationship with environmental drivers such as temperature and salinity. The dominance of proteobacterial lineages with versatile functions in both sulfur and nitrogen cycles, indicating tight coupling between these processes in seagrass ecosystems. This was reinforced by annotation of 83 MAGs recovered from our metagenomic dataset. Taken together, our work provides valuable insights into the complex and interconnected biogeochemical processes within seagrass ecosystems, emphasizing the importance of microbial communities in mediating biogeochemical cycling in seagrass Z. japonica habitats.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov, BioProject: PRJNA882256.
Author contributions
PL conceived and designed the experiments, performed the sampling, analyzed the data, drafted the manuscript, prepared the figures and/or tables. SZ and HZ assisted on the drafting of the manuscript and substantially contributed to its intellectual content. SZ, QL, ZS and YH edited and reviewed the drafts of the manuscript. XH supervised the study. All authors contributed to the article and approved the submitted version.
Funding
This work was supported provided by the National Key Research and Development Program (2020YFD0901003), National Natural Science Foundation of China (No. 92051119, 42077305, and 42070112), Open Research Fund of National Key Laboratory of Coastal Ecological Environment (202107), Jilin Province and Chinese Academy of Sciences Technology Cooperation High-Tech Industrialization Special Project (2021SYHZ0006).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author’s note
This material is the authors’ own original work, which has not been previously published elsewhere. The paper is not currently being considered for publication elsewhere. The paper reflects the authors’ own research and analysis in a truthful and complete manner.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1245288/full#supplementary-material
References
Abe M., Yokota K., Kurashima A., Maegawa M. (2009). Temperature characteristics in seed germination and growth of Zostera japonica Ascherson & Graebner from Ago Bay, Mie Prefecture, central Japan. Fish Sci. 75 (4), 921–927. doi: 10.1007/s12562-009-0123-z
Agawin N. S., Ferriol P., Cryer C., Alcon E., Busquets A., Sintes E., et al. (2016). Significant nitrogen fixation activity associated with the phyllosphere of Mediterranean seagrass Posidonia oceanica: first report. Mar. Ecol. Prog. Ser. 551, 53–62. doi: 10.3354/meps11755
Aires T., Stuij T. M., Muyzer G., Serrão E. A., Engelen A. H. (2021). Characterization and comparison of bacterial communities of an invasive and two native caribbean seagrass species sheds light on the possible influence of the microbiome on invasive mechanisms. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.653998
Alneberg J., Bjarnason B. S., De Bruijn I., Schirmer M., Quick J., Ijaz U. Z., et al. (2014). Binning metagenomic contigs by coverage and composition. Nat. Methods 11 (11), 1144–1146. doi: 10.1038/nmeth.3103
Alsaffar Z., Pearman J. K., Curdia J., Ellis J., Calleja M. L., Ruiz-Compean P., et al. (2020). The role of seagrass vegetation and local environmental conditions in shaping benthic bacterial and macroinvertebrate communities in a tropical coastal lagoon. Sci. Rep. 10 (1), 1–17. doi: 10.1038/s41598-020-70318-1
Aramaki T., Blanc-Mathieu R., Endo H., Ohkubo K., Kanehisa M., Goto S., et al. (2020). KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36 (7), 2251–2252. doi: 10.1093/bioinformatics/btz859
Baden S., Emanuelsson A., Pihl L., Svensson C.-J., berg P. (2012). Shift in seagrass food web structure over decades is linked to overfishing. Mar. Ecol. Prog. Ser. 451, 61–73. doi: 10.3354/meps09585
Bagwell C. E., La Rocque J. R., Smith G. W., Polson S. W., Friez M. J., Longshore J. W., et al. (2002). Molecular diversity of diazotrophs in oligotrophic tropical seagrass bed communities. FEMS Microbiol. Ecol. 39 (2), 113–119. doi: 10.1111/j.1574-6941.2002.tb00912.x
Bengtsson-Palme J., Hartmann M., Eriksson K. M., Pal C., Thorell K., Larsson D. G. J., et al. (2015). metaxa2: improved identification and taxonomic classification of small and large subunit rRNA in metagenomic data. Mol. Ecol. Resour. 15 (6), 1403–1414. doi: 10.1111/1755-0998.12399
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Bowers R. M., Kyrpides N. C., Stepanauskas R., Harmon-Smith M., Doud D., Reddy T., et al. (2017). Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35 (8), 725–731. doi: 10.1038/nbt.3893
Brenner D. J., Boone D. R., Garrity G. M., Goodfellow M., Krieg N. R., Rainey F. A., et al. (2005). The Proteobacteria, part C: the Alpha-, Beta-, Delta-, and Epsilonproteobacteria. (Springer). Athens, GA, USA: Bergey's Manual Trust.
Brysch K., Schneider C., Fuchs G., Widdel F. (1987). Lithoautotrophic growth of sulfate-reducing bacteria, and description of Desulfobacterium autotrophicum gen. nov., sp. nov. Arch. Microbiol. 148 (4), 264–274. doi: 10.1007/BF00456703
Bushnell B. (2014). BBMap: a fast, accurate, splice-aware aligner (Berkeley, CA (United States: Lawrence Berkeley National Lab. (LBNL).
Caffrey J. M. (2004). Factors controlling net ecosystem metabolism in U.S. estuaries. Estuaries 27 (1), 90–101. doi: 10.1007/BF02803563
Callahan B. J., Mcmurdie P. J., Holmes S. P. (2017). Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11 (12), 2639–2643. doi: 10.1038/ismej.2017.119
Chaumeil P.-A., Mussig A. J., Hugenholtz P., Parks D. H. (2020). GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36 (6), 1925–1927. doi: 10.1093/bioinformatics/btz848
Cifuentes A., Antón J., Benlloch S., Donnelly A., Herbert R. A., Rodríguez-Valera F. (2000). Prokaryotic diversity in Zostera noltii-colonized marine sediments. Appl. Environ. Microbiol. 66 (4), 1715–1719. doi: 10.1128/AEM.66.4.1715-1719.2000
Cúcio C., Engelen A. H., Costa R., Muyzer G. (2016). Rhizosphere microbiomes of European seagrasses are selected by the plant, but are not species specific. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00440
Cúcio C., Overmars L., Engelen A. H., Muyzer G. (2018). Metagenomic analysis shows the presence of bacteria related to free-living forms of sulfur-oxidizing chemolithoautotrophic symbionts in the rhizosphere of the seagrass Zostera marina. Front. Mar. Sci. 5. doi: 10.3389/fmars.2018.00171
Dai M., Wang L., Guo X., Zhai W., Li Q., He B., et al. (2008). Nitrification and inorganic nitrogen distribution in a large perturbed river/estuarine system: the Pearl River Estuary, China. Biogeosciences 5 (5), 1227–1244. doi: 10.5194/bg-5-1227-2008
Damashek J., Casciotti K. L., Francis C. A. (2016). Variable nitrification rates across environmental gradients in turbid, nutrient-rich estuary waters of San Francisco Bay. Estuaries Coasts 39 (4), 1050–1071. doi: 10.1007/s12237-016-0071-7
Devereux R. (2005). “Seagrass rhizosphere microbial communities,” in Interactions between macro-and microorganisms in marine sediments,. Eds. Kristensen E., Haese R. R., Kostka J. E. (Washington, DC: American Geophysical Union), 199.
Ding W., Wang S., Qin P., Fan S., Su X., Cai P., et al. (2023). Anaerobic thiosulfate oxidation by the Roseobacter group is prevalent in marine biofilms. Nat. Commun. 14 (1), 2033. doi: 10.1038/s41467-023-37759-4
Duarte C. M., Holmer M., Marbà N. (2005). “Plant-microbe interactions in seagrass meadows,” in Interactions between macro-and microorganisms in marine sediments,. Eds. Kristensen E., Haese R. R., Kostka J. E. (Washington, DC: American Geophysical Union), 31.
Ettinger C. L., Voerman S. E., Lang J. M., Stachowicz J. J., Eisen J. A. (2017). Microbial communities in sediment from Zostera marina patches, but not the Z. marina leaf or root microbiomes, vary in relation to distance from patch edge. PeerJ 5, e3246. doi: 10.7717/peerj.3246
Fourqurean J. W., Duarte C. M., Kennedy H., Marbà N., Holmer M., Mateo M. A., et al. (2012). Seagrass ecosystems as a globally significant carbon stock. Nat. Geosci. 5 (7), 505–509. doi: 10.1038/NGEO1477
García R., Holmer M., Duarte C. M., Marbà N. (2013). Global warming enhances sulphide stress in a key seagrass species (NW Mediterranean). Glob. Change Biol. 19 (12), 3629–3639. doi: 10.1111/gcb.12377
García R., Sánchez-Camacho M., Duarte C. M., Marbà N. (2012). Warming enhances sulphide stress of Mediterranean seagrass (Posidonia oceanica). Estuar. Coast. Shelf Sci. 113, 240–247. doi: 10.1016/j.ecss.2012.08.010
Garcias-Bonet N., Arrieta J. M., Duarte C. M., Marbà N. (2016). Nitrogen-fixing bacteria in Mediterranean seagrass (Posidonia oceanica) roots. Aquat. Bot. 131, 57–60. doi: 10.1016/j.aquabot.2016.03.002
Goodman J. L., Moore K. A., Dennison W. C. (1995). Photosynthetic responses of eelgrass (Zostera marina L.) to light and sediment sulfide in a shallow barrier island lagoon. Aquat. Bot. 50 (1), 37–47. doi: 10.1016/0304-3770(94)00444-Q
Gurevich A., Saveliev V., Vyahhi N., Tesler G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29 (8), 1072–1075. doi: 10.1093/bioinformatics/btt086
Harper W., Terada A., Poly F., Le Roux X., Kristensen K., Mazher M., et al. (2009). The effect of hydroxylamine on the activity and aggregate structure of autotrophic nitrifying bioreactor cultures. Biotechnol. Bioeng. 102 (3), 714–724. doi: 10.1002/bit.22121
Holmer M., Andersen F., Nielsen S. L., Boschker H. T. (2001). The importance of mineralization based on sulfate reduction for nutrient regeneration in tropical seagrass sediments. Aquat. Bot. 71 (1), 1–17. doi: 10.1016/S0304-3770(01)00170-X
Holmer M., Duarte C. M., Marbá N. (2003). Sulfur cycling and seagrass (Posidonia oceanica) status in carbonate sediments. Biogeochemistry 66 (3), 223–239. doi: 10.1023/B:BIOG.0000005326.35071.51
Holmer M., Frederiksen M. S., Møllegaard H. (2005). Sulfur accumulation in eelgrass (Zostera marina) and effect of sulfur on eelgrass growth. Aquat. Bot. 81 (4), 367–379. doi: 10.1016/j.aquabot.2004.12.006
Hutchins D. A., Capone D. G. (2022). The marine nitrogen cycle: new developments and global change. Nat. Rev. Microbiol. 20 (7), 401–414. doi: 10.1038/s41579-022-00687-z
Hyatt D., Chen G.-L., Locascio P. F., Land M. L., Larimer F. W., Hauser L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11 (1), 119. doi: 10.1186/1471-2105-11-119
Ikenaga M., Guevara R., Dean A. L., Pisani C., Boyer J. N. (2010). Changes in community structure of sediment bacteria along the Florida coastal everglades marsh-mangrove-seagrass salinity gradient. Microb. Ecol. 59 (2), 284–295. doi: 10.1007/s00248-009-9572-2
James J., Sherman T., Devereux R. (2006). Analysis of bacterial communities in seagrass bed sediments by double-gradient denaturing gradient gel electrophoresis of PCR-amplified 16S rRNA genes. Microb. Ecol. 52 (4), 655–661. doi: 10.1007/s00248-006-9075-3
Jensen S. I., Kühl M., Priemé A. (2007). Different bacterial communities associated with the roots and bulk sediment of the seagrass Zostera marina. FEMS Microbiol. Ecol. 62 (1), 108–117. doi: 10.1111/j.1574-6941.2007.00373.x
Jones P., Binns D., Chang H.-Y., Fraser M., Li W., Mcanulla C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30 (9), 1236–1240. doi: 10.1093/bioinformatics/btu031
Jørgensen B. B., Findlay A. J., Pellerin A. (2019). The biogeochemical sulfur cycle of marine sediments. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.00849
Kang D. D., Li F., Kirton E., Thomas A., Egan R., An H., et al. (2019). MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359. doi: 10.7717/peerj.7359
Kindaichi T., Okabe S., Satoh H., Watanabe Y. (2004). Effects of hydroxylamine on microbial community structure and function of autotrophic nitrifying biofilms determined by in situ hybridization and the use of microelectrodes. Water Sci. Technol. 49 (11-12), 61–68. doi: 10.2166/wst.2004.0805
Knoblauch C., Jørgensen B. B. (1999). Effect of temperature on sulphate reduction, growth rate and growth yield in five psychrophilic sulphate-reducing bacteria from Arctic sediments. Environ. Microbiol. 1 (5), 457–467. doi: 10.1046/j.1462-2920.1999.00061.x
Koch M. S., Schopmeyer S. A., Holmer M., Madden C. J., Kyhn-Hansen C. (2007). Thalassia testudinum response to the interactive stressors hypersalinity, sulfide and hypoxia. Aquat. Bot. 87 (2), 104–110. doi: 10.1016/j.aquabot.2007.03.004
Kuever J., Rainey F. A., Widdel F. (2015). “Desulfobulbus,” in Bergey's manual of systematics of archaea and bacteria,. Eds. Brenner D., Krieg N. R., Staley J. T. (New York: Springer), 1–6.
Kuypers M. M. M., Marchant H. K., Kartal B. (2018). The microbial nitrogen-cycling network. Nat. Rev. Microbiol. 16 (5), 263–276. doi: 10.1038/nrmicro.2018.9
Langmead B., Salzberg S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9 (4), 357–359. doi: 10.1038/nmeth.1923
Letunic I., Bork P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49 (W1), W293–W296. doi: 10.1093/nar/gkab301
Li W., Godzik A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22 (13), 1658–1659. doi: 10.1093/bioinformatics/btl158
Li D., Luo R., Liu C.-M., Leung C.-M., Ting H.-F., Sadakane K., et al. (2016). MEGAHIT v1. 0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11. doi: 10.1016/j.ymeth.2016.02.020
Li Y., Tang K., Zhang L., Zhao Z., Xie X., Chen C.-T. A., et al. (2018). Coupled carbon, sulfur, and nitrogen cycles mediated by microorganisms in the water column of a shallow-water hydrothermal ecosystem. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.02718
Lin H., Sun T., Xue S., Jiang X. (2016). Heavy metal spatial variation, bioaccumulation, and risk assessment of Zostera japonica habitat in the Yellow River Estuary, China. Sci. Total Environ. 541, 435–443. doi: 10.1016/j.scitotenv.2015.09.050
Ling J., Zhou W., Yang Q., Lin X., Zhang Y., Ahmad M., et al. (2021a). Effect of PAHs on nitrogen-fixing and sulfate-reducing microbial communities in seagrass Enhalus acoroides sediment. Arch. Microbiol. 203 (6), 3443–3456. doi: 10.1007/s00203-021-02321-7
Ling J., Zhou W., Yang Q., Yin J., Zhang J., Peng Q., et al. (2021b). Spatial and species variations of bacterial community structure and putative function in seagrass rhizosphere sediment. Life 11 (8), 852. doi: 10.3390/life11080852
Liu P., Zhang H., Song Z., Huang Y., Hu X. (2021). Seasonal dynamics of bathyarchaeota-dominated benthic archaeal communities associated with seagrass (Zostera japonica) meadows. J. Mar. Sci. Eng. 9 (11), 1304. doi: 10.3390/jmse9111304
Ma L., She W., Wu G., Yang J., Phurbu D., Jiang H. (2021). Influence of temperature and sulfate concentration on the sulfate/sulfite reduction prokaryotic communities in the Tibetan hot springs. Microorganisms 9 (3), 583. doi: 10.3390/microorganisms9030583
Martin B. C., Bougoure J., Ryan M. H., Bennett W. W., Colmer T. D., Joyce N. K., et al. (2019). Oxygen loss from seagrass roots coincides with colonisation of sulphide-oxidising cable bacteria and reduces sulphide stress. ISME J. 13 (3), 707–719. doi: 10.1038/s41396-018-0308-5
Martin B. C., Middleton J. A., Fraser M. W., Marshall I. P. G., Scholz V. V., Hausl B., et al. (2020). Cutting out the middle clam: lucinid endosymbiotic bacteria are also associated with seagrass roots worldwide. ISME J. 14 (11), 2901–2905. doi: 10.1038/s41396-020-00771-3
Martin B. C., Middleton J. A., Skrzypek G., Kendrick G. A., Cosgrove J., Fraser M. W. (2022). Composition of seagrass root associated bacterial communities are linked to nutrients and heavy metal concentrations in an anthropogenically influenced estuary. Front. Mar. Sci. 2145. doi: 10.3389/fmars.2021.768864
Meier-Kolthoff J. P., Auch A. F., Klenk H.-P., Göker M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 14 (1), 1–14. doi: 10.1186/1471-2105-14-60
Miao Y., Liao R., Zhang X.-X., Liu B., Li Y., Wu B., et al. (2015). Metagenomic insights into salinity effect on diversity and abundance of denitrifying bacteria and genes in an expanded granular sludge bed reactor treating high-nitrate wastewater. J. Chem. Eng. 277, 116–123. doi: 10.1016/j.cej.2015.04.125
Mills H., Kiel Reese B., Shepard A., Dowd S., Riedinger N., Morono Y., et al. (2012). Characterization of metabolically active bacterial populations in subseafloor Nankai Trough sediments above, within, and below the sulfate–methane transition zone. Front. Microbiol. 3. doi: 10.3389/fmicb.2012.00113
Muyzer G., Stams A. J. M. (2008). The ecology and biotechnology of sulphate-reducing bacteria. Nat. Rev. Microbiol. 6 (6), 441–454. doi: 10.1038/nrmicro1892
Na S.-I., Kim Y. O., Yoon S.-H., Ha S.-M., Baek I., Chun J. (2018). UBCG: up-to-date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J. Microbiol. 56 (4), 280–285. doi: 10.1007/s12275-018-8014-6
Nakagawa S., Takai K. (2008). Deep-sea vent chemoautotrophs: diversity, biochemistry and ecological significance. FEMS Microbiol. Ecol. 65 (1), 1–14. doi: 10.1111/j.1574-6941.2008.00502.x
Nguyen L.-T., Schmidt H. A., Von Haeseler A., Minh B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi: 10.1093/molbev/msu300
Nielsen L. B., Finster K., Welsh D. T., Donelly A., Herbert R. A., De Wit R., et al. (2001). Sulphate reduction and nitrogen fixation rates associated with roots, rhizomes and sediments from Zostera noltii and Spartina maritima meadows. Environ. Microbiol. 3 (1), 63–71. doi: 10.1046/j.1462-2920.2001.00160.x
Olm M. R., Brown C. T., Brooks B., Banfield J. F. (2017). dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11 (12), 2864–2868. doi: 10.1038/ismej.2017.126
Osvatic J. T., Wilkins L. G., Leibrecht L., Leray M., Zauner S., Polzin J., et al. (2021). Global biogeography of chemosynthetic symbionts reveals both localized and globally distributed symbiont groups. Proc. Natl. Acad. Sci. U.S.A. 118 (29), e2104378118. doi: 10.1073/pnas.2104378118
Parks D. H., Imelfort M., Skennerton C. T., Hugenholtz P., Tyson G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25 (7), 1043–1055. doi: 10.7287/peerj.preprints.554
R Core Team. (2018). “R: a language and environment for statistical computing,” in R Foundation for Statistical Computing. Vienna. Available at: https://www.R-project.org/.
Robador A., Müller A. L., Sawicka J. E., Berry D., Hubert C. R., Loy A., et al. (2016). Activity and community structures of sulfate-reducing microorganisms in polar, temperate and tropical marine sediments. ISME J. 10 (4), 796–809. doi: 10.1038/ismej.2015.157
Shen W., Ren H. (2021). TaxonKit: a practical and efficient NCBI taxonomy toolkit. J. Genet. Genomics 48 (9), 844–850. doi: 10.1016/j.jgg.2021.03.006
Smith A. C., Kostka J. E., Devereux R., Yates D. F. (2004). Seasonal composition and activity of sulfate-reducing prokaryotic communities in seagrass bed sediments. Aquat. Microb. Ecol. 37 (2), 183–195. doi: 10.3354/ame037183
Sohaskey C. D., Wayne L. G. (2003). Role of narK2X and narGHJI in Hypoxic Upregulation of Nitrate Reduction by Mycobacterium tuberculosis. J. Bacteriol. 185 (24), 7247–7256. doi: 10.1128/jb.185.24.7247-7256.2003
Soler-Jofra A., Pérez J., Van Loosdrecht M. C. (2021). Hydroxylamine and the nitrogen cycle: a review. Water Res. 190, 116723. doi: 10.1016/j.watres.2020.116723
Sun Y., Song Z., Zhang H., Liu P., Hu X. (2020). Seagrass vegetation affect the vertical organization of microbial communities in sediment. Mar. Environ. Res. 162, 105174. doi: 10.1016/j.marenvres.2020.105174
Sun F., Zhang X., Zhang Q., Liu F., Zhang J., Gong J. (2015). Seagrass (Zostera marina) colonization promotes the accumulation of diazotrophic bacteria and alters the relative abundances of specific bacterial lineages involved in benthic carbon and sulfur cycling. Appl. Environ. Microbiol. 81 (19), 6901–6914. doi: 10.1128/AEM.01382-15
Sunagawa S., Coelho L. P., Chaffron S., Kultima J. R., Labadie K., Salazar G., et al. (2015). Structure and function of the global ocean microbiome. Science 348 (6237), 1261359. doi: 10.1126/science.1261359
Suonan Z., Kim S. H., Qin L.-Z., Kim H., Zhang F., Lee K.-S. (2022). Increased coastal nutrient loading enhances reproductive intensity of Zostera marina: implications for seagrass meadow resilience. Front. Mar. Sci. 9. doi: 10.3389/fmars.2022.832035
Uritskiy G. V., Diruggiero J., Taylor J. (2018). MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6 (1), 1–13. doi: 10.1186/s40168-018-0541-1
Vigneron A., Cruaud P., Culley A. I., Couture R.-M., Lovejoy C., Vincent W. F. (2021). Genomic evidence for sulfur intermediates as new biogeochemical hubs in a model aquatic microbial ecosystem. Microbiome 9 (1), 1–14. doi: 10.1186/s40168-021-00999-x
Wang L., English M. K., Tomas F., Mueller R. S. (2020). Recovery and community succession of the Zostera marina Rhizobiome after transplantation. Appl. Environ. Microbiol. 87 (3), e02326–e02320. doi: 10.1128/AEM.02326-20
Wang P., Li J.-L., Luo X.-Q., Ahmad M., Duan L., Yin L.-Z., et al. (2022). Biogeographical distributions of nitrogen-cycling functional genes in a subtropical estuary. Funct. Ecol. 36 (1), 187–201. doi: 10.1111/1365-2435.13949
Weidner S., Arnold W., Stackebrandt E., Pühler A. (2000). Phylogenetic analysis of bacterial communities associated with leaves of the seagrass Halophila stipulacea by a culture-independent small-subunit rRNA gene approach. Microb. Ecol. 39 (1), 22–31. doi: 10.1007/s002489900194
Welsh D. T. (2000). Nitrogen fixation in seagrass meadows: regulation, plant–bacteria interactions and significance to primary productivity. Ecol. Lett. 3 (1), 58–71. doi: 10.1046/j.1461-0248.2000.00111.x
Wu Y.-W., Simmons B. A., Singer S. W. (2016). MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32 (4), 605–607. doi: 10.1093/bioinformatics/btv638
Yue S., Zhang X., Xu S., Liu M., Qiao Y., Zhang Y., et al. (2021). The super typhoon Lekima, (2019) resulted in massive losses in large seagrass (Zostera japonica) meadows, soil organic carbon and nitrogen pools in the intertidal Yellow River Delta, China. Sci. Total Environ. 793, 148398. doi: 10.1016/j.scitotenv.2021.148398
Zhai S., Ji M., Zhao Y., Su X. (2020). Shift of bacterial community and denitrification functional genes in biofilm electrode reactor in response to high salinity. Environ. Res. 184, 109007. doi: 10.1016/j.envres.2019.109007
Zhang X., Liu S., Jiang Z., Wu Y., Huang X. (2022). Gradient of microbial communities around seagrass roots was mediated by sediment grain size. Ecosphere 13 (2), e3942. doi: 10.1002/ecs2.3942
Zhang X., Zhao C., Yu S., Jiang Z., Liu S., Wu Y., et al. (2020). Rhizosphere microbial community structure is selected by habitat but not plant species in two tropical seagrass beds. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.00161
Keywords: metagenomics, biogeochemical cycling, environmental drivers, seagrass sediments, plant-microbe interactions
Citation: Liu P, Zou S, Zhang H, Liu Q, Song Z, Huang Y and Hu X (2023) Genome-resolved metagenomics provides insights into the microbial-mediated sulfur and nitrogen cycling in temperate seagrass meadows. Front. Mar. Sci. 10:1245288. doi: 10.3389/fmars.2023.1245288
Received: 25 June 2023; Accepted: 08 November 2023;
Published: 28 November 2023.
Edited by:
Danny Ionescu, Leibniz-Institute of Freshwater Ecology and Inland Fisheries (IGB), GermanyReviewed by:
Melissa R. Kardish, Naval Research Laboratory, United StatesJiwen Liu, Ocean University of China, China
Copyright © 2023 Liu, Zou, Zhang, Liu, Song, Huang and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoke Hu, eGtodUB5aWMuYWMuY24=