
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 20 October 2023
Sec. Aquatic Microbiology
Volume 10 - 2023 | https://doi.org/10.3389/fmars.2023.1244849
This article is part of the Research TopicMicrobial Diversity and Resources in Tidal FlatsView all 17 articles
 Lei Gao1,2†
Lei Gao1,2† Bao-Zhu Fang1*†
Bao-Zhu Fang1*† Chun-Yan Lu3†Kun-Hui Hong4
Chun-Yan Lu3†Kun-Hui Hong4 Xin-Yu Huang4Ting-Ting She4Min Xiao3
Xin-Yu Huang4Ting-Ting She4Min Xiao3 Wen-Jun Li1,3*
Wen-Jun Li1,3*During an investigation of the culturable microbial diversity of sediments with salinity from tidal flats and saline lake, seven strains of the genus Demequina were harvested. The genomic analysis and physiological characteristics of strains of this genus have unveiled their significant potential in degrading complex carbon source such as lignin, hemicellulose, chitin, and oligosaccharides. In addition, these strains show potential abilities in nitrite ammonification and sulfide oxidation. These findings not only improved our understanding of their metabolic model, but also provided valuable insights into their ecological roles. Four new species of the genus Demequina are described: Demequina litoralis sp. nov., with SYSU T00192T designated as the type species; Demequina zhanjiangensis sp. nov., with SYSU T00b26T as the type species; Demequina lignilytica sp. nov., with SYSU T00068T as the type species; and Demequina muriae sp. nov., with EGI L300058T as the type species. Additionally, strains SYSU T0a273, SYSU T00039-1, and SYSU T00039 are identified as different strains of Demequina lignilytica. Our study thus sheds light on the diversity, biological significance, and ecological contribution of the Demequina genus in different habitats.
Demequina was a genus in the family Demequinaceae of the phylum Actinomycetota (Ue et al., 2011; Oren and Garrity, 2021). The genus Demequina, a group of Gram-stain positive bacteria, was first discovered from tidal flats sediment by Yi et al., 2007 (Yi et al., 2007) and currently comprises 21 known species (https://lpsn.dsmz.de/genus/demequina, accessed on 15 May 2023). Then, the description of the genus Demequina is amended by Ue et al., 2011 and Park et al., 2016 Species of the genus Demequina exhibited diverse morphologies, including short rod-shaped, oval, coccoid forms and were widely distributed in tidal flats sediment (Yi et al., 2007), mangrove (Matsumoto et al., 2010; Hamada et al., 2015b), soil (Finster et al., 2009), and marine related niches (Hamada et al., 2013). The species within the genus Demequina displayed diverse morphologies and biochemical characteristics and were found to be widely distributed in natural environments. These traits provided interesting objects for researchers to explore their genomes and biological properties (Chao et al., 2021).
With technological advances, more researches had been conducted on the genomics (Hamada et al., 2015a) and biological properties of strains of the genus Demequina, revealing their ability to perform ecological functions in various niches (Meng et al., 2009; Al-Naamani et al., 2015; Peruzzi et al., 2017; Wei et al., 2018; Duan et al., 2019). Comparative genome analysis was an effective approach to decipher the genetic diversity and evolutionary relationships of microorganisms (Wei et al., 2002). However, the lack of understanding genomic data on Demequina strains hindered our comprehension of their ecological roles and potential applications. Comparative genome analysis of species in the genus Demequina would therefore make an important contribution to the field of microbial genomics and could have practical implications for biotechnology and human health.
In our study, we obtained seven strains of the genus Demequina. Our research has three main objectives: first, to determine the taxonomic status of these new strains; second, to compare the metabolic potential of the genus Demequina; and finally, to evaluate the potential ecological and physiological functions of the genus Demequina. Overall, our efforts have resulted in a successful expansion of diversity within the genus Demequina, which will be critical for future implementation of industrial applications.
During our investigation into the culturable microbial diversity of saline aquatic environments, we successfully isolated a strain (EGI L300058) from sediment samples of Dabancheng saline lake in Xinjiang, PR China. In the meantime, tidal flats as other saline niche, we also obtained six strains (SYSU T0a273, SYSU T00068, SYSU T00039-1, SYSU T00039, SYSU T00192, and SYSU T00b26) from sediment samples of tidal flats from Zhanjiang city in Guangdong. All strains were routinely maintained on marine agar 2216 (28°C, 1 week) and biomass of these strains for the later experiment was collected under the same conditions.
DNA extraction from all strains was carried out using the TIANamp Bacteria DNA kit, following the kit instructions. The DNA library was prepared according to the instructions of the NGS Fast DNA Library Prep Set for Illumina. The genomes of these strains were sequenced using paired-end sequencing on the Illumina Hiseq X platform, which was performed at the Guangdong Institute of Microbiology. The reads obtained from each dataset were trimmed using Sickle (Joshi and Fass, 2011) and subsequently, the high-quality paired-end reads were assembled using the SPAdes program (Bankevich et al., 2012) with options “-k 21, 33, 55, 77, 99, 121 -careful” in Unicycler software (Wick et al., 2017). The identification of protein-coding sequences within the assembled sequences was carried out using Prodigal (Hyatt et al., 2010). Meanwhile, tRNA-scan (Chan and Lowe, 2019) and RNAmmer (Lagesen et al., 2007) were utilized to identify tRNA and rRNA, respectively. To identify the gene functions, we applied an E-value cut-off of 1e-5 and filtered for the best hit using the Clusters of Orthologous Groups (COG) (Galperin et al., 2019), Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2016), and carbohydrate-active enzymes (CAZy) databases (Drula et al., 2021). We determined the G + C content from the genome sequences and constructed genome maps with the GenoVi: Genome Visualizer (Cumsille et al., 2023).
16S rRNA gene sequences were amplified using genomic DNA as the template with universal bacterial primers 27F and 1492R following the protocol described by Fang et al., 2020. Almost full-length 16S rRNA gene sequences were compared to related sequences in the EzBioCloud (Yoon et al., 2017) and NCBI (Sayers et al., 2021) databases, respectively (Altschul et al., 1990). To conduct phylogenetic analysis, 16S rRNA gene sequences of related species within the genus Demequina were downloaded from the above-mentioned databases and aligned using the CLustalW MEGAX (Thompson et al., 1994; Kumar et al., 2018). The Kimura 2-parameter model (Casanellas et al., 2020) was used to calculate genetic distances and clustering, and phylogenetic trees were constructed using the neighbor-joining (Saitou and Nei, 1987) and maximum likelihood (Felsenstein, 1981) methods. The phylogenomic tree of the genus Demequina was constructed according to the method described before (Jiao et al., 2021) and shown in brief below. Multiple sequence alignments (MSAs) of 120 bacterial marker genes were generated using GTDB-Tk (Chaumeil et al., 2019), and the maximum-likelihood phylogeny of the MSAs was calculated using IQ-Tree (Nguyen et al., 2015) with the parameters -alrt 1000, -bb 1000, and -nt AUTO.
Nineteen Demequina genomes were retrieved from NCBI databases for comparative analysis. Average nucleotide identity (ANI) and amino acid identity (AAI) were computed using IPGA (Liu et al., 2022) and CompareM software (https://github.com/dparks1134/CompareM, ver. 3), respectively. To perform comparative analysis of orthologous and unique genes among species of the genus Demequina, we filtered out low-quality protein sequences based on length (10 AA) and percent stop codons (20%) in FASTA format and then used the OrthoFinder program (Emms and Kelly, 2019) within the PGCGAP pipeline (Liu et al., 2020) to cluster the proteomes and generate gene families based on reciprocal DIAMOND best hits of predicted amino acid sequences. We also leveraged the IPGA pipeline to perform pan- and core-genome analyses of species of the genus Demequina with a minimum sequence identity threshold of 70% (https://nmdc.cn/ipga/, accessed on 8 April 2023). Metabolic and biogeochemical functional trait profiles for the genome datasets were further predicted using the METABOLIC software (Zhou et al., 2022). The antiSMASH (ver. 6.0) was used for the analysis of biosynthetic gene clusters (BGCs) in all genomes (Blin et al., 2021). All plots were generated using the ggplot2 package in R (https://www.r-project.org/, ver. 4.1.2).
The physiological characteristics of these strains were determined by the methods previously described (Xu et al., 2005; Gao et al., 2022). We also analyzed the morphology of our strains by negative staining with ammonium molybdate and transmission electron microscopy. After seven days of incubation on marine agar 2216, we recorded the size, shape, and color of the colonies, and conducted Gram staining to examine the bacterial cell wall. We assessed the utilization of substrates by the target strain at 30°C using GEN III Microplates (BIOLOG Inc., Hayward, California, United States) and measured oxidase activity using the Oxidase Reagent kit (bioMerieux SA). We also evaluated the ability of strains to peptonize and coagulate milk, as well as their urease activity, starch and Tween degradation, and liquefaction of gelatin, following Smibert and Krieg’s instructions (Smibert and Krieg, 1994). We measured protease activity by spot inoculating strains onto a 5% (v/v) skim milk agar medium (Rohban et al., 2009). Additionally, we analyzed nitrate and nitrite reduction and other biochemical properties using API 20NE and API ZYM kits (manufactured by bioMérieux in France). The production for ligninase, cellulase, chitinase, IAA (indole-3-acetic acid), ACC (1-Aminocyclopropane-1-carboxylic Acid) deaminase of Demequina strains were tested by following protocols of manufacturer for the Microbial Ligninase Detection Kit (JINMEI, China), Microorganism Cellulase (CE) ELISA Kit (JINMEI, China), Microbial Chitinase ELISA Kit (JINMEI, China), Microbial Indoleacetic Acid (IAA) ELISA Kit (JINMEI, China), and Microbial ACC Deaminase (ACCD) ELISA Kit (JINMEI, China), respectively.
Biomasses for the chemotaxonomic analysis of seven strains were obtained from cultures grown on the marine agar 2216 at 30 °C for 7 days. The cellular fatty acids of both strains were extracted from the collected biomasses, methylated, and analyzed by the Microbial Identification System (Sherlock Version 6.1; MIDI database: TSBA6). The respiratory quinones were extracted and purified from lyophilized cells (Collins et al., 1977; Minnikin et al., 1984) and the extracts were analyzed by using HPLC (High Performance Liquid Chromatography) (Kroppenstedt, 1982; Groth et al., 1997). After extraction, the polar lipids were analyzed by the two-dimensional TLC (thin-layer chromatography) method on silica gel G 60 plates (Merck; Germany) (Hasegawa et al., 1983; Yin et al., 2021).
The genome size of species in the genus Demequina displayed a range of 2,359,865 bp to 3,228,371 bp, with a corresponding GC content range of 62.73% to 72.39%, and numbers of predicted genes varied from 2172 to 3013 (Dataset S1). For a strain form saline lake, genome size of strain EGI L300058T was 2,359,865 bp with a GC content of 69.41% and contained 8 contigs, 2172 genes, 3 rRNAs, and 39 tRNAs. For six strains from tidal flats, strain SYSU T00192T had a genome size of 3,024,536 bp and a GC content of 72.26%, consisting of 21 contigs, 2804 genes, 3 rRNAs, and 52 tRNAs. Strain SYSU T00b26T had a genome size of 3,125,962 bp and a GC content of 69.58%, consisting of 17 contigs, 2882 genes, 3 rRNAs, and 49 tRNAs. Strain SYSU T00068T had a genome size of 2,927,318 bp and a GC content of 71.88%, consisting of 23 contigs, 2705 genes, 4 rRNAs, and 52 tRNAs (Supplementary Figures 1A–D). Strain SYSU T0a273 had a genome size of 2,838,212 bp and a GC content of 72.08%, consisting of 16 contigs, 2626 genes, 3 rRNAs, and 53 tRNAs. Strain SYSU T00039-1 had a genome size of 2,896,852 bp and a GC content of 71.95%, consisting of 15 contigs, 2677 genes, 3 rRNAs, and 45 tRNAs. Strain SYSU T00039 had a genome size of 2,482,171 bp and a GC content of 71.93%, consisting of 17 contigs, 2309 genes, 3 rRNAs, and 52 tRNAs (Supplementary Figure 1E).
The results of KEGG annotation revealed that the species in this genus were classified into 22 KEGG level two pathways. In particular, the metabolic pathways, including carbohydrate metabolism, amino acid metabolism, and overview, exhibited high abundance (Figure 1A). The results of COG annotation revealed that species were classified into 22 functional categories, including A, C-V, and Z (Figure 1B). The most abundant COGs of the genomes in this genus were exclusively assigned to general function prediction, closely followed by CDSs dedicated to carbohydrate transport and metabolism, amino acid transport and metabolism, function unknown, and transcription. Notably, the COG annotation results were also enriched for genes associated with metabolism, which was largely consistent with the KEGG annotation results. Functional annotation of the Demequina strains revealed a high degree of similarity in their functional repertoire, as indicated by the relatively concentrated distribution of similar functions across different strains. These new strains contained 6 families of carbohydrate-active enzymes involved in glycosidic bond degradation, modification, and generation. These enzyme families include Carbohydrate Esterases (CEs), Glycoside Hydrolases (GHs), Auxiliary Activities (AAs), Polysaccharide Lyases (PLs), Glycoside Transferases (GTs), and Carbohydrate Binding Modules (CBMs) (Figure 1C). Glycoside Hydrolases, which were present in the highest amounts, were a ubiquitous group of enzymes that catalyze the hydrolysis of glycosidic bonds between two or more carbohydrates or between a carbohydrate and a non-carbohydrate moiety (Li et al., 2021). CBMs in CAZymes, which ranked third in quantity, bind specifically to carbohydrates and played critical roles in enzyme targeting, activity enhancement, facilitation of recycling, assembly of multi-enzyme complex, and modulation of substrate specificity and catalytic activity of associated enzymes (Attia and Brumer, 2021). GTs, which ranked third in terms of quantity, had also been identified and were involved in the biosynthesis of disaccharides, oligosaccharides, and polysaccharides (Sánchez-Rodríguez et al., 2014). In addition, our analysis revealed there were few PLs in the genomes of this genus that removed uronic acid from polysaccharide chains via a β-elimination mechanism, resulting in the formation of an unsaturated hexenuronic acid residue and a new reducing end (MacDonald and Berger, 2014). This suggested that species of the genus Demequina might have a versatile substrate utilization ability.
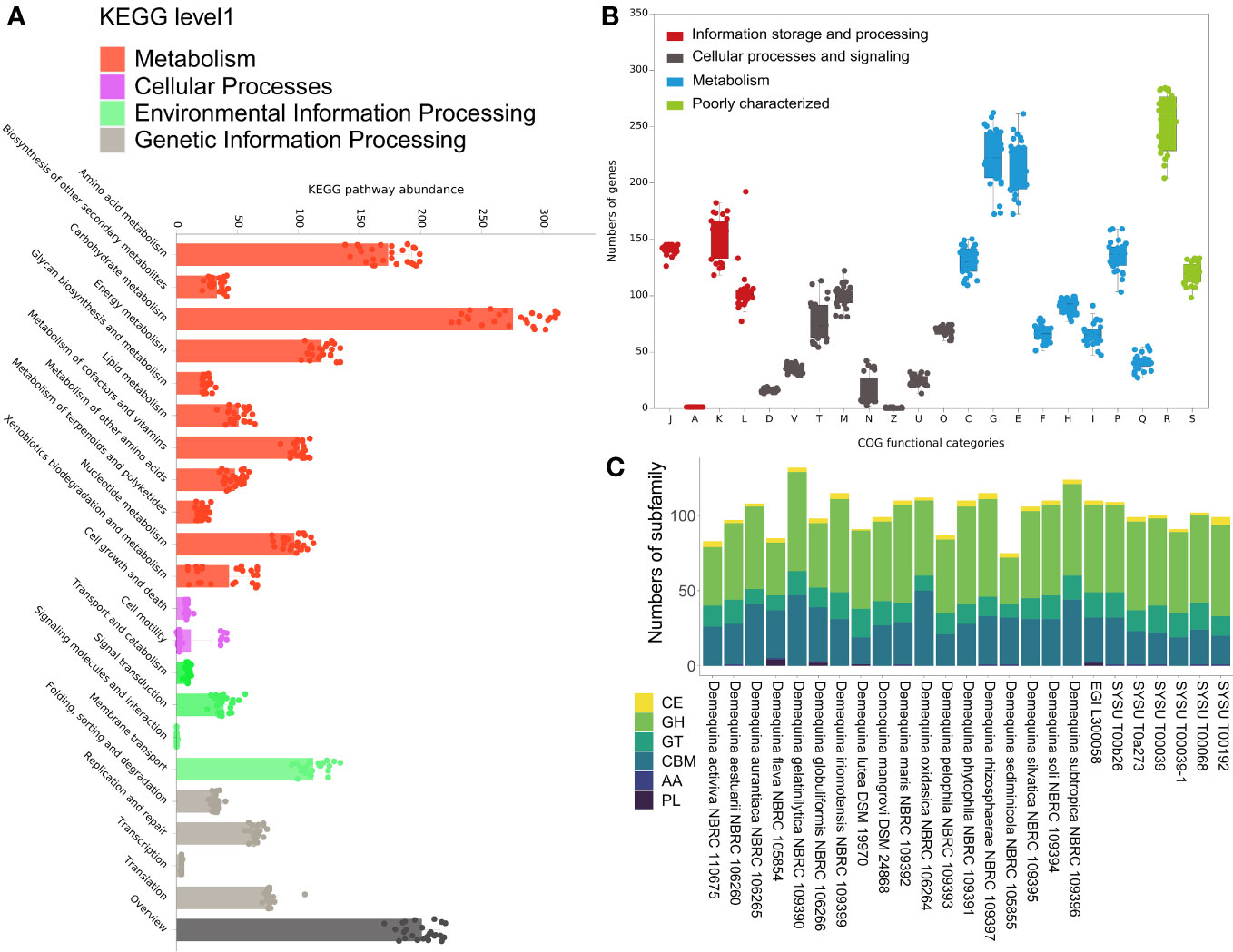
Figure 1 Metabolic potentials of Demequina strains. (A) KEGG pathway abundance of species of the genus Demequina. (B) COG functional categories of Demequina strains. (C) Carbohydrate-active enzymes (CAZymes) family associated with the CAZy database. (A), RNA processing and modification; (B), Chromatin structure and dynamics; (C), Energy production and conversion; (D), Cell cycle control, cell division, chromosome partitioning; (E), Amino acid transport and metabolism; (F), Nucleotide transport and metabolism; (G), Carbohydrate transport and metabolism; (H), Coenzyme transport and metabolism; (I), Lipid transport and metabolism; (J), Translation, ribosomal structure and biogenesis; (K), Transcription; (L), Replication, recombination and repair; (M), Cell wall/membrane/envelope biogenesis; (N), Cell motility; (O), Posttranslational modification, protein turnover; (P), Inorganic ion transport and metabolism; (Q), Secondary metabolites biosynthesis, transport and catabolism; (R), General function prediction only; (S), Function unknown; (T), Signal transduction mechanisms; (U), Intracellular trafficking, secretion, and vesicular transport; (V), Defense mechanisms; (W), Extracellular structures; (X), Mobilome: prophages, transposons; (Z), Cytoskeleton.
In addition, the nearly-complete 16S rRNA gene sequences of related species of this genus were submitted to the IMNGS platform and 1513 OTUs with a high sequence similarity cutoff value (99%) were discovered (Lagkouvardos et al., 2016). Our analysis revealed the global distribution of strains of the genus Demequina in various sample types worldwide, including soil, rhizosphere, and plant samples, and suggested that related strains of this genus might be more abundant in aquatic environments (Supplementary Figure 2).
The EZBioCloud comparison results based on almost complete16S rRNA gene sequences indicated that these sequences shared significant similarity with bacteria of the genus Demequina. The highest sequence similarity was found between strain EGI L300058T and D. aestuarii NBRC 106260T (97.74%). Strain SYSU T00b26T had the highest homology of 99.22% with D. salsinemoris NBRC 105323T, while strain SYSU T00192T had a homology of 98.45% with D. maris NBRC 109392T. D. iriomotensis NBRC 109399T had the highest homology with strains SYSU T00068T, SYSU T00039, SYSU T00039-1 and SYSU T00a273 with homology of 97.82%, 98.16%, 97.87%, and 97.96%, respectively. The phylogenetic trees constructed by the neighbor-joining and maximum likelihood methods using the 16S rRNA gene also confirmed the above relationship and similarity results for the seven novel strains (Supplementary Figures 3, 4). The phylogenomic tree (Figure 2) based on the genomes of seven strains and their related species of the genus Demequina also confirmed the above results of the phylogenetic analysis.
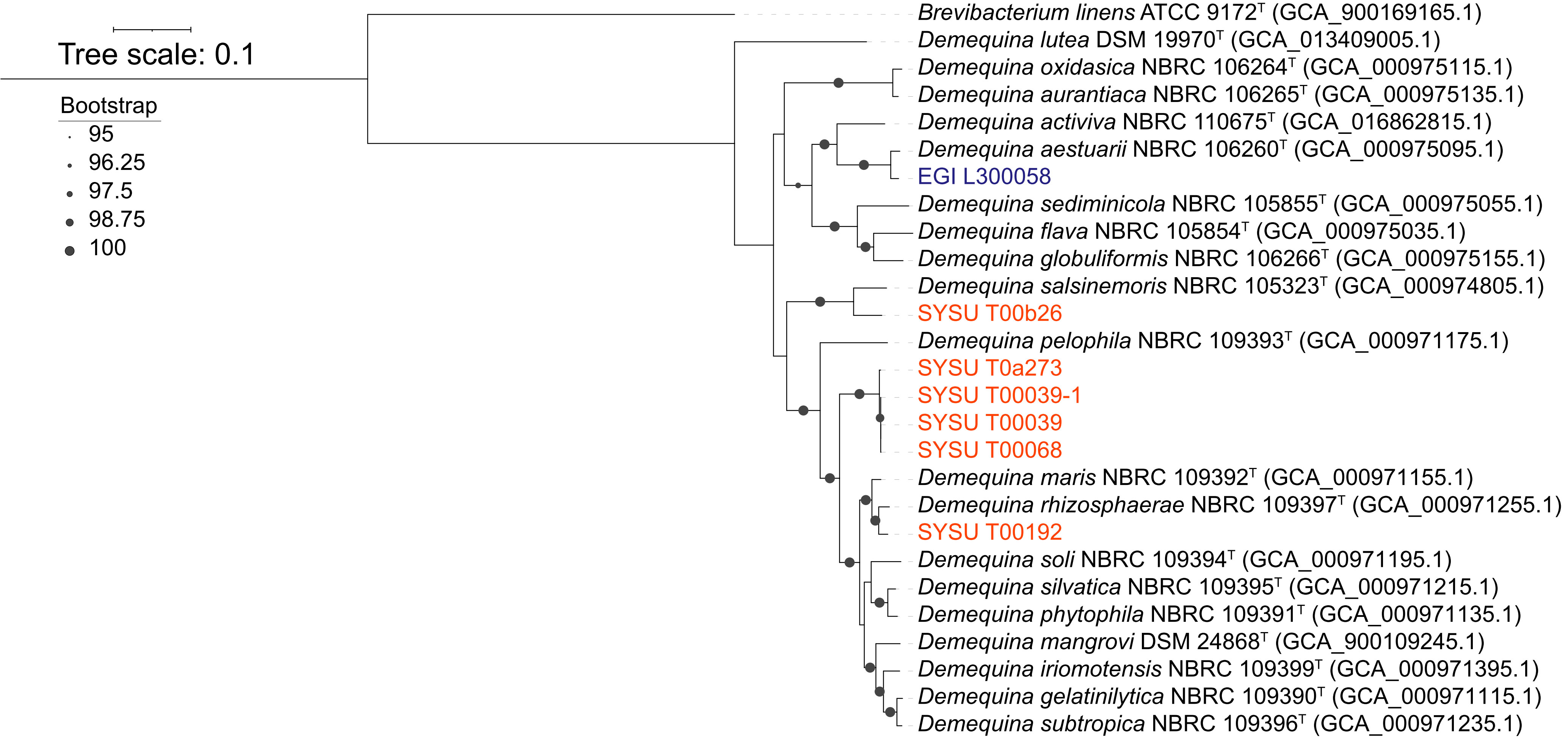
Figure 2 Phylogenomic tree of species of the genus Demequina. Bootstrap values (>95) based on 1000 resamplings are marked gray points at the nodes. Brevibacterium linens ATCC 9172T (GCA_900169165.1) was used as an outgroup. ModelFinder used the Akaike Information Criterion (AIC), Corrected Akaike Information Criterion (Correct AIC), and Bayesian Information Criterion (BIC) to select the best-fit model (LG+F+R3).
Based on average nucleotide identity (ANI), average amino acid identity (AAI), and digital DNA-DNA hybridization (dDDH), we found that strains EGI L300058T, SYSU T00b26T, SYSU T00192T and SYSU T00068T were genetically distinct from related type species of this genus, indicating that they belonged to different species. Furthermore, values of ANI, AAI, and dDDH showed that strains SYSU T00039, SYSU T00039-1, SYSU T00a273 and SYSU T00068T were closely related, indicating that they were different strains of the same species (Figures 3A, B and Dataset S2).
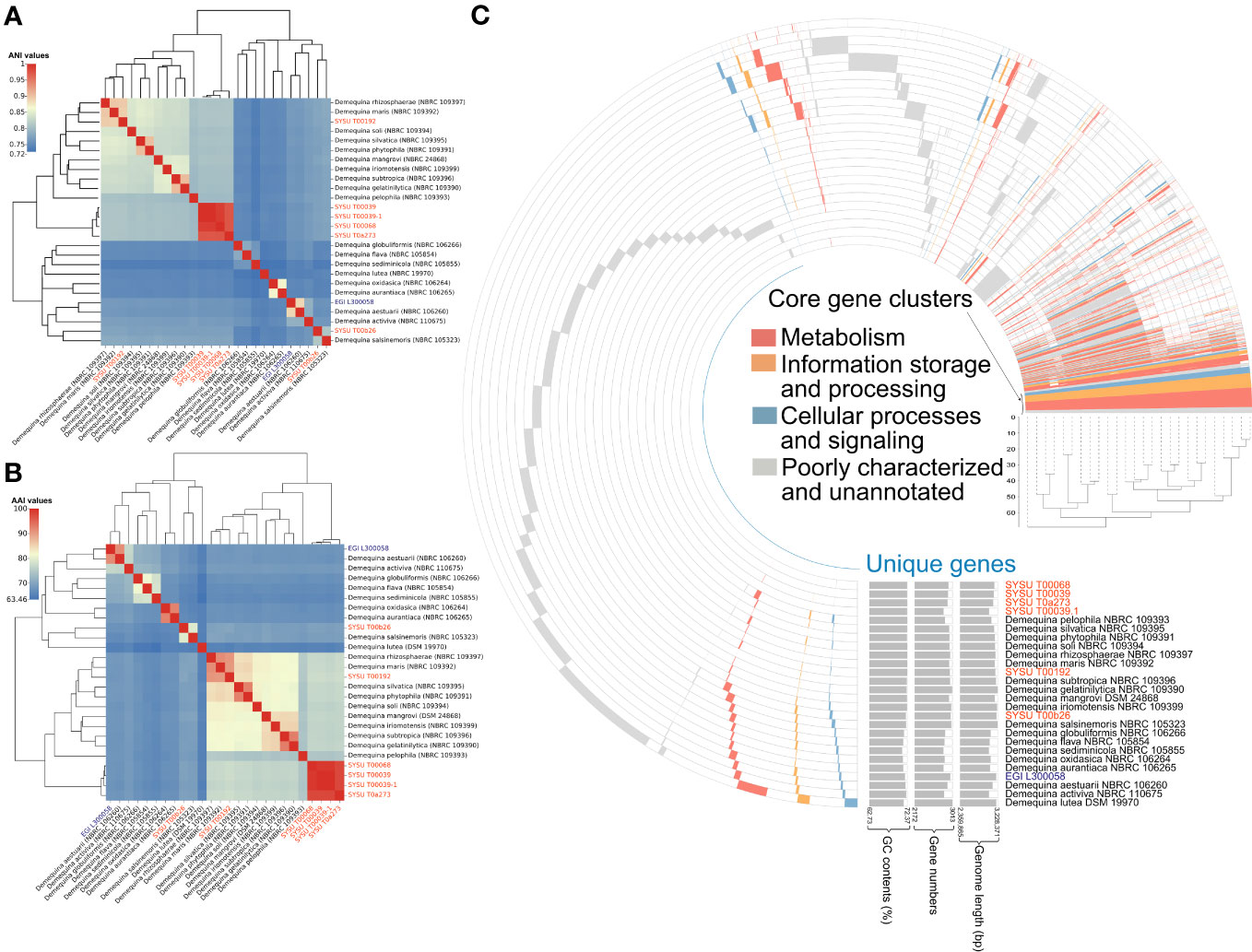
Figure 3 Comparative genomic of species of the genus Demequina. (A), Average nucleotide identity (ANI) values shared among the related type species; (B), Average amino acid identity (AAI) values shared among the related type species; (C), The pan-genome profile of all Demequina strains. “GC,” “Gene number,” and “Genome length” represent GC content, gene number, genome length, respectively.
The extent of genetic variation among species of the genus Demequina, consisting of 19 type species and 7 novel strains genomes, was investigated by analyzing the distribution of conserved (core) and species-specific (unique) genes using a pan-genome analysis. A limited number of core gene clusters, involved in various basic biological processes such as metabolism, information storage and processing, cellular processes and signaling, were identified in these genomes. Nevertheless, numerous strain-specific genes involved in metabolism, information storage and processing, cellular processes, and signal transduction, as well as uncharacterized and insufficiently annotated genes contributing to the unique metabolic functions of each strain in this genus (Figure 3C). Comparative genomic analysis based on the orthologs of the recovered genomes revealed that all strains of the genus Demequina possessed 1002 core proteins, whereas each strain contained more than 1000 unique proteins (Supplementary Figure 5A). Besides, core/pan genome analysis also showed that there were significant genomic differences among the species of the genus Demequina (Supplementary Figure 5B).
Genome annotation of genus Demequina revealed that they might play a vital role in the biogeochemical cycles of various crucial elements such as nitrogen, sulfur, and carbon, which was crucial for maintaining the stability of aquatic ecosystem.
Members of the genus Demequina were able to convert nitrite, the intermediate compound of dissimilatory nitrate reduction, to ammonia via the nrfADH and nirBD pathways under different oxygen concentrations (Figure 4). In particular, many species of this genus had been isolated from different marine niches that could express nitrite reductase encoded by nrfADH genes to adapt to low-oxygen environments. At the same time, members of this genus played important roles in various steps of sulfur cycling in aquatic ecosystems. In our study, we predicted the presence of some functional genes (fccB, sqr) responsible for sulfide oxidation from the genomes of this genus (Figure 4). The process of sulfide oxidation, in which the key genes fccB and sqr were involved, was of critical ecological importance for maintaining the sulfur cycle in aquatic ecosystems. The fccB gene, a subunit of the flavocytochrome c complex, facilitated the transfer of electrons from reduced sulfur compounds to oxygen. Similarly, the sqr gene, encoding a sulfide: quinone oxidoreductase, catalyzed the oxidation of sulfide to elemental sulfur, transferring electrons to quinones. In summary, the presence of these two genes was critical for detoxification of sulfide in the oxic-anoxic interface of aquatic habitats, thereby preventing potential damage to other organisms and generating elemental sulfur as an energy source for sulfur-oxidizing bacteria. Furthermore, genome annotation results revealed that strains of the genus Demequina had the ability to reduce iron. The iron reduction process encompasses various genes and proteins, such as CytC3 and CytC4 genes, Fe (III) reductase proteins, electron transfer chain genes, thiol-containing proteins, and electron carrier protein genes, working collaboratively to convert iron oxides into soluble iron. Iron-reducing bacteria (FeRB) not only contributed to the reduction of minerals and humus, but also participated in the oxidation of sulfur compounds and organic matter. FeRB played a crucial role in many engineering applications, such as wastewater treatment, bioremediation, and bioelectrochemical systems (Garber et al., 2020).
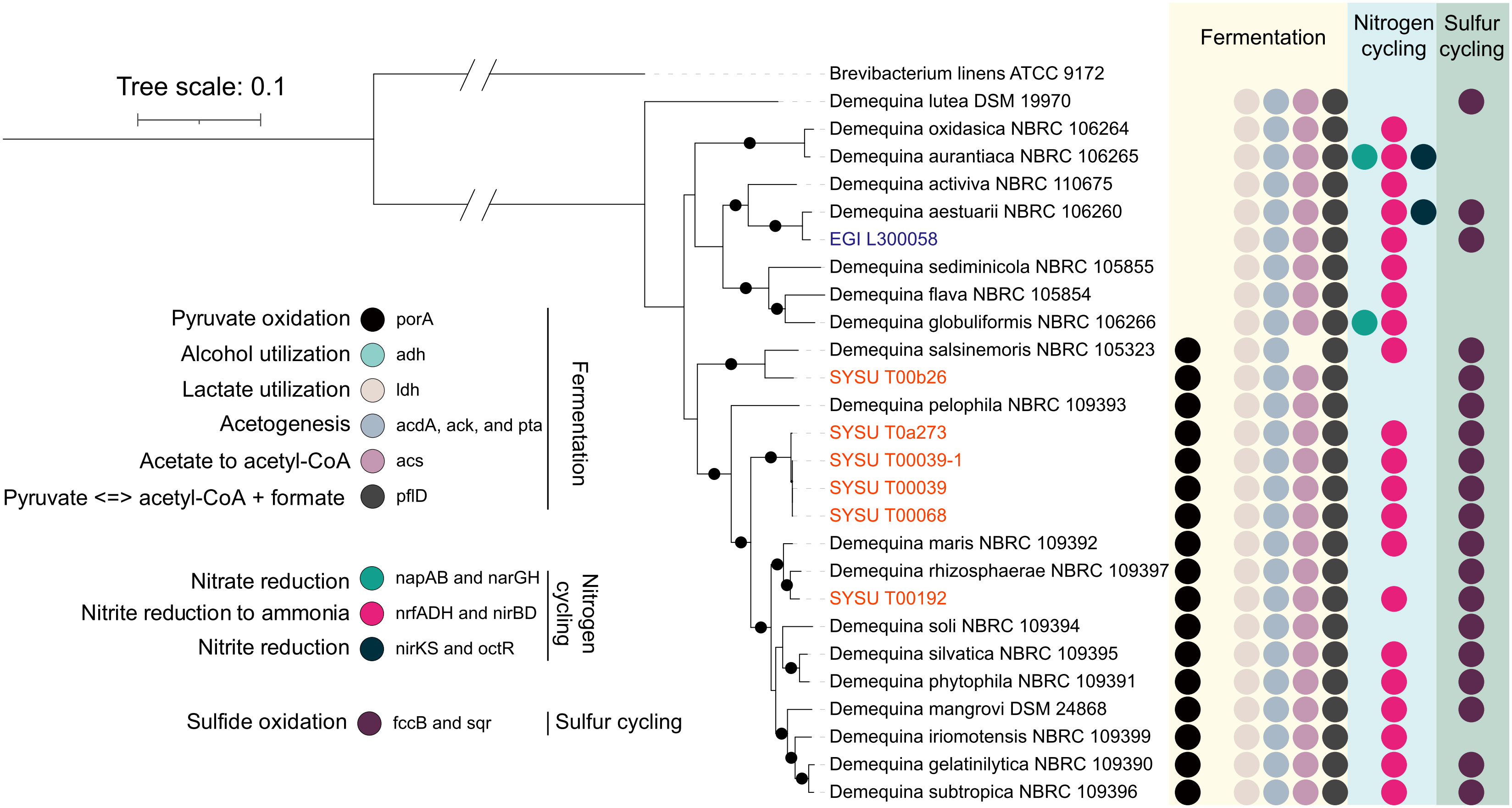
Figure 4 Complex carbon degradation potentials of species of the genus Demequina. Bootstrap values (>95) based on 1000 resamplings are marked gray points at the nodes. Brevibacterium linens ATCC 9172T (GCA_900169165.1) was used as an outgroup. ModelFinder used the Akaike Information Criterion (AIC), Corrected Akaike Information Criterion (Correct AIC), and Bayesian Information Criterion (BIC) to select the best-fit model (LG+F+R3). Different colored stripes show different metabolisms. Various colored circles represent different metabolic genes.
Species of the genus Demequina from aquatic environments also played critical roles in metabolic and energy cycles and showed the ability to oxidize organic carbon or acetate as a substrate for fermentation. Annotation of these genomes revealed a diverse repertoire of complex carbon degradation enzymes, including cellobiosidase, cellulase, and β-glucosidase for cellulose degradation, and arabinosidase, β-glucuronidase, α-L-rhamnosidase for hemicellulose debranching, and mannan endo-1,4-β-mannosidase, α-D-xyloside xylohydrolase for endohemicellulase degradation; β-xylosidase, β-mannosidase, and β-galactosidase for degradation of other oligosaccharides; α-amylase, glucoamylase, pullulanase, and isoamylase for amylolytic enzymes; and chitinase and hexosaminidase for chitin degradation (Figures 4, 5, and Dataset S3). In addition, species of this genus harbored the Embden-Meyerhof pathway for glycolysis, as well as the pentose phosphate pathway and genes responsible for phosphoribosyl diphosphate (PRPP) biosynthesis and facilitating the metabolism of purine, pyrimidine, and histidine. For energy metabolism, the genome had a complete oxidative phosphorylation pathway, including an NADH dehydrogenase (nuoABCDEFGHIJKLMN) for proton shift, a ubiquinol-cytochrome c reductase (qerABC), a cytochrome c oxidase (ctaCDE), and an F-type ATPase (atpABCDEFGH) (Dataset S3). Finally, basic organic substrate utilization processes such as pyruvate metabolism and the tricarboxylic acid cycle were also found in their genomes, representing the metabolic pathways of the three major nutrients (sugars, lipids, and amino acids) and serving as metabolic hubs for their cross-linking. In summary, the species of the genus Demequina were involved in the main carbon cycle processes and played an important role in the carbon cycle of the aquatic ecosystem (Dataset S4).
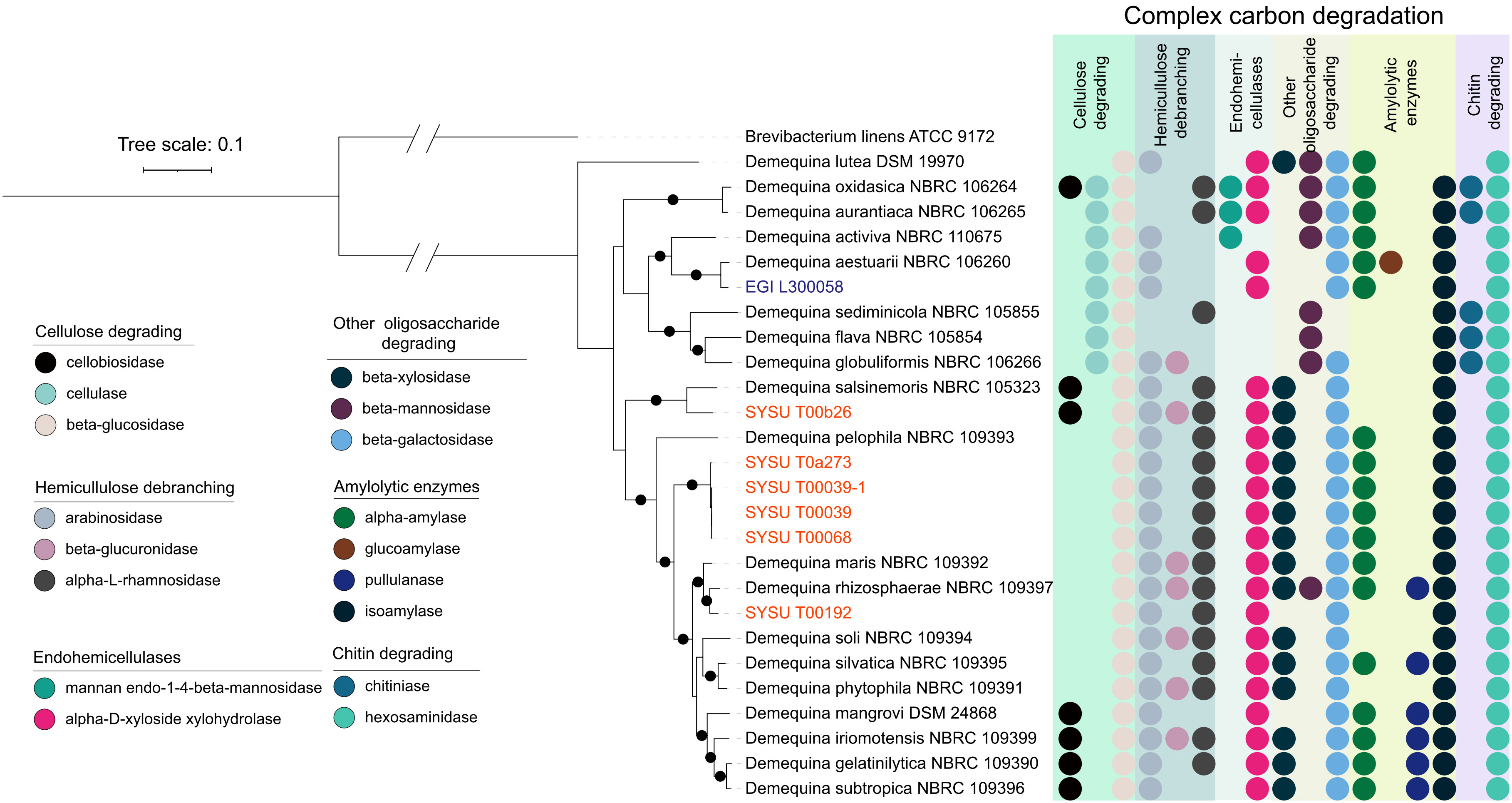
Figure 5 Fermentation, nitrogen, sulfur metabolism of species of the genus Demequina. Bootstrap values (>95) based on 1000 resamplings are marked gray points at the nodes. Brevibacterium linens ATCC 9172T (GCA_900169165.1) was used as an outgroup. ModelFinder used the Akaike Information Criterion (AIC), Corrected Akaike Information Criterion (Correct AIC), and Bayesian Information Criterion (BIC) to select the best-fit model (LG+F+R3). Different colored stripes show different metabolisms. Various colored circles represent different metabolic genes.
Comparative analysis of genes related to salt tolerance revealed that species of the genus Demequina had the ability to regulate extracellular osmotic balance through outer membrane pore proteins (OmpR), maintained optimal potassium ion concentrations (Hasan et al., 2023), and excreted excessive sodium ions via ion transport proteins (NhaA) (Tan et al., 2019; Quick et al., 2021). It also synthesized the osmoregulatory substance proline via ProA, ProB, glnA (Wu et al., 2018), and relied on chaperones and proteases (DnaK, GroEL, ClpP) (Susin et al., 2006; Nouri et al., 2020) to ensure proper protein folding and repair in response to osmotic stress (Supplementary Figure 6). These functional systems allowed species in this genus to adapt to different osmotic niches, maintained cellular homeostasis, and ensured optimal physiological function and survival.
Furthermore, secondary metabolites of species in this genus were categorized into eight types of biosynthetic gene clusters (BGCs), including terpenes, RRE-containing, betalactones, NAP-AA, T3PKS, NRPS-like, resorcinol, and lanthipeptide-class-v. Differences in the type and abundance of BGCs were observed among these species, but some BGCs were present in most strains (Supplementary Figure 7). This diversity of BGCs suggests that Demequina strains might have the potential to be resistant to pathogens, which could have various functions in defending against pathogens in different niches.
Seven novel strains were characterized as Gram-stain positive, non-motile, and aerobic. The colony morphology of these strains was characterized by a large, circular shape with a smooth and moist texture, well-defined edges, and yellow. The electron micrograph of seven strains showed the short-rod structure with a length of roughly 0.71-1.37 μm and a width of approximately 0.34-0.45 μm (Supplementary Figure 8). The seven strains exhibited a wide range of growth temperatures, from 4-37 °C, and could tolerate different pH values between 6-9 and salt concentrations of 0-8% (w/v). According to results of Biolog Gen III, strain SYSU T0a273 could utilize D-fructose, D-fructose-6-PO4, D-glucuronic acid, glucuronamide, nalidixic acid, lithium chloride, and potassium tellurite. Strain SYSU T00039-1 was able to utilize 33 different substrates, while strain SYSU T00039 was capable of utilizing 29 substrates, and SYSU T00068T was capable of utilizing 31 substrates. SYSU T00039-1, SYSU T00039, and SYSU T00068T shared the ability to utilize 16 substrates, including dextrin, D-maltose, D-trehalose, D-cellobiose, gentiobiose, sucrose, D-turanose, stachyose, α-D-lactose, D-melibiose, β-methyl-D-glucoside, D-fructose, D-galactose, inosine, lithium chloride, and sodium butyrate. Significant differences in substrate utilization were observed between the above four strains of the same species. Strains SYSU T00192T, SYSU T00b26T, and EGI L300058T showed the ability to utilize different substrates by using 27, 28, and 50 substrates, respectively. These results demonstrated the metabolic versatility of these strains and indicated their potential applications in biotechnology and environmental fields. The API 20NE experiments showed that SYSU T0a273, SYSU T00039-1, SYSU T00039, and SYSU T00068T produced only a single enzyme, β-galactosidase. Although SYSU T0a273, SYSU T00039, and SYSU T00068T produced β-glucosidase, SYSU T00039-1 was found to have other abilities, including hydrolysis of gelatin, production of urease and indole, and the ability to assimilate glucose, arabinose, mannose, and N-acetyl-β-glucosamine. SYSU T00192T produced both β-glucosidase and β-galactosidase and was able to assimilate glucose, arabinose, and maltose. SYSU T00b26T was found to produce β-galactosidase and assimilate glucose, arabinose, and maltose. EGI L300058T produced both β-glucosidase and β-galactosidase. According to the results of experiments conducted by API ZYM, these strains shared the following enzyme activities: (C4) esterase, (C8) esterase lipase, leucine arylamidase, valine arylamidase, naphthol-AS-BI-phosphohydrolase, β-galactosidase, and α-glucosidase. Substantial variations in enzyme activities were observed within the Demequina strains. However, the results of the H2S production, starch degradation, Tween (60 and 80) degradation, and gelatin liquefaction assays showed that these novel strains could not perform these activities.
Upon analysis with Microbial Ligninase Detection Kit analysis, all seven strains showed the ability to produce ligninase, with EGI L300058T producing the highest amount of up to 800 ng/mL, while SYSU T00b26T had the lowest production at 500 ng/mL (Figure 6A). When analyzed with the Microbial Cellulase Detection Kit, all seven strains showed the ability to produce cellulase, with EGI L300058T and SYSU T00068T producing the highest amount of up to 1200 pg/mL, while SYSU T00b26T and SYSU T00039-1 exhibited the lowest production at 600 pg/mL (Figure 6B). According to Microbial Chitinase Detection Kit analysis, all seven strains showed the ability to produce chitinase, with SYSU T00068T producing the highest amount of up to 60 pg/mL, while SYSU T00b26T exhibited the lowest production 3 to 30 pg/mL (Figure 6C). Analysis of the microbial indoleacetic acid (IAA) with ELISA kit showed that all novel strains were able to produce IAA (SYSU T00068T with the highest IAA production of 28.5 μg/L on average) (Figure 6D), suggesting that these strains had the potential to promote plant growth. ACC is one of the precursors involved in the synthesis of ethylene in plants. The enzyme ACC deaminase degrades ACC into ammonia and α-keto butyric acid, thereby lowering the ethylene levels in plants and promoting plant growth (Glick et al., 2007). In addition, the Demequina strains exhibited high-yielding ACC deaminase properties (SYSU T00039 with the highest ACCD production on average: 17.9 ng/L), as shown by the result of the physiological experiment (Figure 6E).
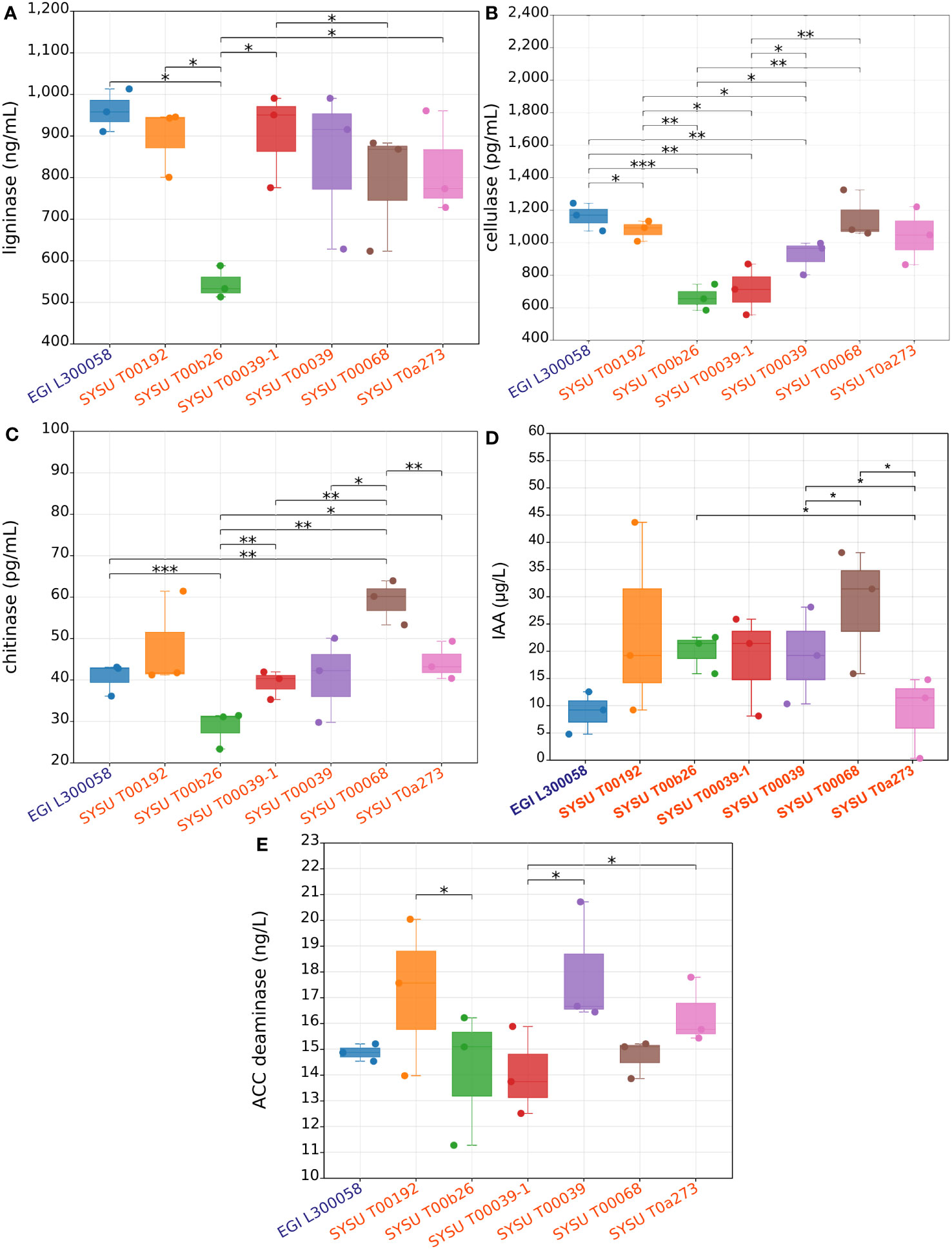
Figure 6 Ligninase, cellulase, chitinase, IAA, and ACCD production species of the genus Demequina. (A), comparison of ligninase production capacity among tested Demequina strains; (B), comparison of cellulase production capacity among tested Demequina strains; (C), comparison of chitinase production capacity among tested Demequina strains; (D), comparison of indole-3-acetic acid (IAA) production capacity among tested Demequina strains; (E), comparison of ACC (1-aminocyclopropane-1-carboxylate) deaminase production capacity among tested Demequina strains. The method employed for the differential comparison was the t-test. * refers to p < 0.05; ** refers to p < 0.01; *** refers to p < 0.001.
The major fatty acids (>5%) of Demequina strains were anteiso-C15:0, iso-C19:0, anteiso-C17:1 ω9c, iso-C16:0, iso-C14:0, and iso-C15:0 (Supplementary Table 1). MK-9 (H4) was found to be the respiratory quinone of these strains. The polar lipids of these novel strains were found to consist of diphosphatidylglycerol (DPG), phosphoglyceride (PG), phosphatidylinositol (PI), phosphatidylinositol mannosides (PIMs), and unidentified lipid (L) (Supplementary Figure 9). Detailed phenotypic and physiological characteristics of novel strains from the genus of Demequina were listed in Dataset S5.
As previous studies description, numerous species of the genus Demequina were isolated from marine related niches such as marine, tidal flat, arctic permafrost soil, rhizosphere of a mangrove, etc. In our study, six strains of the genus Demequina (SYSU T00068T, SYSU T0a273, SYSU T00039, SYSU T00039-1, SYSU T00192T and SYSU T00b26T) were isolated from tidal flats and one strain (EGI L300058T) was isolated from saline lake. Strains EGI L300058T and SYSU T00192T displayed the low 16S rRNA gene homologies with their most related type species, which were satisfied the species delineation (98.65%) (Kim et al., 2014). The phylogenetic analysis of the 16S rRNA genes and genomes, along with comparative genomics (specifically ANI and AAI values below species delimitation thresholds), confirmed that EGI L300058T and SYSU T00192T represented two novel species of this genus. Although the 16S rRNA gene sequence of strain SYSU T00b26T showed a high homology of 99.22% with D. salsinemoris NBRC 105323T, phylogenetic analysis of the 16S rRNA gene and genome, as well as comparative genomics (especially ANI and AAI values below the species delimitation thresholds) confirmed that SYSU T00b26T represented a novel species of this genus (Richter and Rosselló-Móra, 2009). The 16S rRNA gene homologies between D. iriomotensis NBRC 109399T and strains SYSU T00068T, SYSU T00039, SYSU T00039-1 and SYSU T00a273 were lower than the species delineation threshold. Furthermore, based on the phylogenetic analysis of the 16S rRNA genes and genomes, as well as comparative genomics using ANI, AAI, dDDH values below the species delimitation thresholds (Richter and Rosselló-Móra, 2009), it was confirmed that SYSU T00068T, SYSU T00039, SYSU T00039-1and SYSU T00a273 were seen as one novel species of this genus. Additionally, the comparative genomic results demonstrated that these four strains belong to different strains within the same species. In summary, we acquired seven strains from different salinity niches that could be classified into 4 novel species, and expended our understanding of microbial biodiversity in the genus Demequina.
The genus Demequina from the order Micrococcales played a vital role in the nitrogen cycle of aquatic ecosystems, particularly in the reduction of nitrite to ammonia via the nrfADH and nirBD pathways (as shown in Figure 4). The nrfADH pathway utilized the nrfA and nrfH genes, which encoded key enzymes for the reduction of nitrite to ammonia, particularly in oxygen deficient aquatic niches (Rütting et al., 2011). The nirBD pathway, on the other hand, used the nirB and nirD genes to reduce nitrite to ammonia, which was critical in high-oxygen environments (Akhtar et al., 2013). This implied that strains of this genus could express different functional genes for the conversion of nitrite to ammonia to expand habitat with different oxygen concentrations. Our study also shed light on the possible involvement of strains of this genus in the sulfur cycle in aquatic ecosystems. The sulfur cycle was an important biogeochemical process that involved the conversion of sulfur compounds between different oxidation states. Sulfide, a reduced form of the sulfur element, was toxic to most organisms and could cause severe ecological damage if not properly detoxified (Lamers et al., 2013). Our research revealed that most strains of the genus Demequina possessed key genes, namely fccB and sqr, which were involved in sulfide oxidation. fccB, a subunit of the flavocytochrome c complex, could transfer electrons from reduced sulfur compounds to oxygen (Friedrich et al., 2001), while sqr, a sulfide: quinone oxidoreductase, catalyzed the oxidation of sulfide to elemental sulfur and transferred electrons to quinones (Cherney et al., 2012). Taken together, these genes were essential for detoxification of sulfide in the aquatic environment and prevented other organisms from being harmed. Moreover, sulfide oxidation contributes to the production of elemental sulfur, which serves as an energy source for sulfur-oxidizing bacteria, supporting the diversity of microbial communities in aquatic ecosystems. The presence of these key genes in Demequina strains suggested that they were actively involved in sulfur cycling of aquatic ecosystems, which was a significant finding because the role of Demequina strains in sulfur cycling had not been previously reported. Overall, our study highlights the ecological importance of Demequina strains in maintaining the balance of nitrogen and sulfur cycling in aquatic ecosystems, thereby maintaining the diversity of microbial communities in these environments. Demequina strains had also been shown to have the ability to reduce Fe3+ to Fe2+. This process, known as iron reduction, was an important biogeochemical process that occurs in both soil and aquatic environments. The reduction of Fe3+ to Fe2+ can be carried out by a variety of microorganisms, including Demequina strains, and can play a significant role in the cycling of iron in ecosystems. This process was important because Fe2+ was more soluble than Fe3+ and more easily taken up by plants and other organisms (Morrissey and Guerinot, 2009). In addition, Fe2+ could react with other compounds in the environment and lead to the formation of minerals such as magnetite and siderite, which could affect the composition and stability of soil and sediments environments (Huang et al., 2021; Shen et al., 2021). Therefore, the ability of Demequina strains to reduce Fe3+ to Fe2+ was of ecological importance to the biogeochemical cycling of iron and the maintenance of ecosystem functions.
Our study also revealed that strains of the genus Demequina had the potential for antagonistic activity against pathogens, as evidenced by genome annotations. In particular, the strains were found to contain a number of biosynthetic gene clusters (BGCs) involved in antifungal activities, including terpenes, RRE-containing, betalactones, NAP-AA, T3PKS, NRPS-like, resorcinol or lanthipeptide-class-V. Previous researches had revealed that these types of secondary metabolites might exhibit various biological activities such as antibacterial, antioxidant, and anti-inflammatory properties (Modjinou et al., 2017; Shimizu et al., 2017; Lima et al., 2020; Xu et al., 2020; Shi et al., 2021). Overall, these results suggested that strains of this genus had a role in biocontrol and bioremediation in various environments and might be suitable for the pharmaceutical industry.
Our results showed that genomes of the genus Demequina had diverse complex carbon degradation enzymes, including cellulose and hemicellulose degradation, oligosaccharide degradation, amylolytic enzymes, and chitin degradation. The presence of these enzymes suggested that strains of this genus, as heterotrophic microorganisms, were capable of degrading a variety of complex organic compounds, which was essential for maintaining the balance of carbon and nutrient cycling to sustain life and stabilize microbial communities in different aquatic niches. Hence wood or herbs as a carbon source for aquatic niches consisted of lignocellulose, lignin, chitin, or other compounds that made them resistant to the degradation of these complex carbohydrates (Janusz et al., 2017; Zoghlami and Paës, 2019; Börcsök and Pásztory, 2021). Wood-degrading enzymes, especially lignin-degrading enzymes, played a critical role in the utilization of wood or herbs as a renewable energy and carbon source (Ayuso-Fernández et al., 2018). Our research results had shown that strains of the genus Demequina possessed a significant amount of ligninase, cellulase, chitinase, IAA, and ACCD related genes and were capable of utilizing a wide range of substrates based on genomic and physiological evidences. These results might shed a light on the reason for the distribution of most species of this genus from mangrove or tidal flat niches, where lignin or cellulose had normally been enriched. Further studies of the ligninase produced by strains of the genus Demequina could lead to a better understanding of its function and potential applications in the biorefinery and bioenergy industries.
These research findings have greatly improved our understanding of the diversity and potential functions of Demequina, which are abundant in aquatic environments, and facilitate study of microorganisms in more complex ecosystems.
Our research results showed that seven novel strains of the genus Demequina were isolated from different salinity aquatic niches (tidal flats and saline lake). Phylogenetic relationships, based on 16S rRNA gene sequences and genomes, comparative genomics (ANI, AAI, and dDDH values), and physiological characteristics, showed that the seven isolated strains could be classified into 4 novel species. In particular, SYSU T00039, SYSU T00039-1, SYSU T00a273, and SYSU T00068T were different strains of the same novel species. However, EGI L300058T, SYSU T00192T, and SYSU T00b26T belonged to three different species in the genus Demequina. Genomic analysis and physiological characteristics of these strains had led to the development of a metabolic model that showed their ability to degrade various polysaccharides such as lignin, hemicellulose, and chitin, as well as oligosaccharides, and to participate in nitrite ammonification and sulfide oxidation. These results shed light on the potential ecological role of strains from the genus Demequina. Therefore, we propose four novel strains of the genus Demequina, namely Demequina muriae sp. nov. with strain EGI L300058T (=GDMCC 1.3270T=KCTC 59052T) as the type species, Demequina litoralis sp. nov. with strain SYSU T00192T (=GDMCC 1.3840T=KCTC 49956T) as the type species, Demequina zhanjiangensis sp. nov. with strain SYSU T00b26T (=GDMCC 1.3841T =KCTC 49950T) as the type species, and Demequina lignilytica sp. nov. with strain SYSU T00068T (=GDMCC 1.3838T= KCTC 49954T) as the type species, while SYSU T0a273, SYSU T00039-1, and SYSU T00039 were different strains of Demequina lignilytica.
Description of Demequina muriae sp. nov.
Demequina muriae (mu’ri.ae. L. gen. n. muriae, of a brine).
Each cell is about 0.80 μm in length and 0.39 μm in width, giving them a short rod-like shape. Cells are not motile and have no flagella. Cells are Gram-stain positive and aerobic. Cells grow at pH values of 8 to 9, temperatures between 25 and 37°C, as well as NaCl concentration at 0-5%. The organism exhibits negative activity for urease, milk peptonization and coagulation, gelatin liquefaction and coagulation, and degradation of Tweens (40, 60, and 80) and starch, while positive activity is observed for indole-3-acetic acid production, 1-aminocyclopropane-1-carboxylic acid (ACC) deaminase production, catalase, esterase, β-glucosidase, ligninase, cellulase, chitinase, and other enzymes. The major fatty acids (>5%) were anteiso-C15:0, iso-C19:0, anteiso-C17:1 ω9c, iso-C16:0, iso-C14:0, and iso-C15:0. The predominant menaquinone is MK-9 (H4). The polar lipid profile consists of diphosphatidylglycerol, phosphoglyceride, phosphatidylinositol, phosphatidylinositol mannosides, and unidentified lipid. The G+C content of the genomic DNA is 69.41%.
The type strain, EGI L300058T (=GDMCC 1.3270T=KCTC 59052T), was isolated from the Dabancheng saline lake in Xinjiang, PR China. The G+C content is 69.41%. The GenBank accession numbers for the 16S rRNA gene and genome sequences of strain EGI L300058T are OR098447 and NMDC60064244 respectively.
Description of Demequina litoralis sp. nov.
Demequina litoralis (lit.to.ra.lis. L. fem. adj. litoralis, coastal).
Each cell is about 0.92 μm in length and 0.42 μm in width, giving them a short rod-like shape. Cells are not motile and have no flagella. Cells are Gram-stain positive and aerobic. Cells grow at pH values of 6 to 7, temperatures between 25 and 37°C, as well as NaCl concentration at 0-4%. The organism exhibits negative activity for urease, milk peptonization and coagulation, gelatin liquefaction and coagulation, and degradation of Tweens (40, 60, and 80) and starch, while positive activity is observed for indole-3-acetic acid production, 1-aminocyclopropane-1-carboxylic acid (ACC) deaminase production, esterase, lipase, β-glucosidase, ligninase, cellulase, chitinase, and other enzymes. The major fatty acids (>5%) were anteiso-C15:0, iso-C19:0, anteiso-C17:1 ω9c, iso-C16:0, iso-C14:0, and iso-C15:0. The predominant menaquinone is MK-9 (H4). The polar lipid profile consists of diphosphatidylglycerol, phosphoglyceride, phosphatidylinositol, phosphatidylinositol mannosides, and unidentified lipid. The G+C content of the genomic DNA is 72.26%.
The type strain, SYSU T00192T (=GDMCC 1.3840T=KCTC 49956T), was isolated from tidal flats sediments of Zhanjiang city in Guangdong, PR China. The G+C content is 72.26%. The GenBank accession numbers for the 16S rRNA gene and genome sequences of strain SYSU T00192T are OR098449 and NMDC60064242 respectively.
Description of Demequina zhanjiangensis sp. nov.
Demequina zhanjiangensis (zhan.jiang.en’sis. N.L. fem. adj. zhanjiangensis, from Zhanjiang, China).
Each cell is about 0.73 μm in length and 0.34 μm in width, giving them a short rod-like shape. Cells are not motile and have no flagella. Cells are Gram-stain positive and aerobic. Cells grow at pH values of 6 to 8, temperatures between 25 and 37°C, as well as NaCl concentration at 0-8%. The organism exhibits negative activity for urease, milk peptonization and coagulation, gelatin liquefaction and coagulation, and degradation of Tweens (40, 60, and 80) and starch, while positive activity is observed for indole-3-acetic acid production, 1-aminocyclopropane-1-carboxylic acid (ACC) deaminase production, esterase, lipase, β-glucosidase, β-mannosidase, ligninase, cellulase, chitinase, and other enzymes. The major fatty acids (>5%) were anteiso-C15:0, iso-C19:0, anteiso-C17:1 ω9c, iso-C16:0, iso-C14:0, and iso-C15:0. The predominant menaquinone is MK-9 (H4). The polar lipid profile consists of diphosphatidylglycerol, phosphoglyceride, phosphatidylinositol, phosphatidylinositol mannosides, and unidentified lipid. The G+C content of the genomic DNA is 69.58%.
The type strain, SYSU T00b26T (=GDMCC 1.3841T =KCTC 49950T), was isolated from tidal flats sediments of Zhanjiang city in Guangdong, PR China. The G+C content is 69.58%. The GenBank accession numbers for the 16S rRNA gene and genome sequences of strain SYSU T00b26T are OR098448 and NMDC60064243 respectively.
Description of Demequina lignilytica sp. nov.
Demequina lignilytica (lig.ni.ly’ti.ca. L. neut. n. lignum, wood; Gr. masc. adj. lytikos, loosening, dissolving; N.L. fem. adj. lignilytica, splitting lignin).
Each cell is about 1.37 μm in length and 0.49 μm in width, giving them a short rod-like shape. Cells are not motile and have no flagella. Cells are Gram-stain positive and aerobic. Cells grow at pH values of 6 to 7, temperatures between 25 and 37°C, as well as NaCl concentration at 0-8%. The organism exhibits negative activity for urease, milk peptonization and coagulation, gelatin liquefaction and coagulation, and degradation of Tweens (40, 60, and 80) and starch, while positive activity is observed for indole-3-acetic acid production, 1-aminocyclopropane-1-carboxylic acid (ACC) deaminase production, esterase, lipase, β-glucosidase, ligninase, cellulase, chitinase, and other enzymes. The major fatty acids (>5%) were anteiso-C15:0, iso-C19:0, anteiso-C17:1 ω9c, iso-C16:0, iso-C14:0, and iso-C15:0. The predominant menaquinone is MK-9 (H4). The polar lipid profile consists of diphosphatidylglycerol, phosphoglyceride, phosphatidylinositol, phosphatidylinositol mannosides, and unidentified lipid. The G+C content of the genomic DNA is 71.88%.
The type strain, SYSU T00068T (=GDMCC 1.3838T= KCTC 49954T), was isolated from tidal flats sediments of Zhanjiang city in Guangdong, PR China. The G+C content is 71.88%. The GenBank accession numbers for the 16S rRNA gene and genome sequences of strain SYSU T00068T are OR098453 and NMDC60064239 respectively.
The datasets presented in this study can be found in online repositories. The names of the repository or repositories and accession number(s) can be found in the article or Supplementary Material.
W-JL and B-ZF designed research and project outline. B-ZF and C-YL obtained samples. LG, K-HH, and X-YH performed isolation, deposition, and other experiments. LG and T-TS performed genome analysis. LG, B-ZF, MX, and W-JL drafted the manuscript. All authors contributed to the article and approved the submitted version.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
This research was supported by National Science and Technology Fundamental Resources Investigation Program of China (Nos 2019FY100701 and 2021FY100900), The Third Xinjiang Scientific Expedition Program (No.2022xjkk1204), Natural Science Foundation of Xinjiang Uygur Autonomous Region (2022D01A154), Xinjiang Uygur Autonomous Region regional coordinated innovation project (Shanghai cooperation organization science and technology partnership program) (No. 2021E01018). We thank Prof. Aharon Oren for his recommendations on the nomenclature of these novel strains in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1244849/full#supplementary-material
Akhtar S., Khan A., Sohaskey C. D., Jagannath C., Sarkar D. (2013). Nitrite reductase NirBD is induced and plays an important role during in vitro dormancy of Mycobacterium tuberculosis. J. Bacteriol 195, 4592–4599. doi: 10.1128/JB.00698-13
Al-Naamani L., Dobretsov S., Al-Sabahi J. N., Soussi B. (2015). Identification and characterization of two amylase producing bacteria Cellulosimicrobium sp. and Demequina sp. isolated from marine organisms. J. Agric. Mar. Sci. 20, 8–15. doi: 10.24200/jams.vol20iss0pp8-15
Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Attia M. A., Brumer H. (2021). New family of carbohydrate-binding modules defined by a galactosyl-binding protein module from a cellvibrio japonicus endo-xyloglucanase. Appl. Environ. Microbiol. 87, e0263420. doi: 10.1128/AEM.02634-20
Ayuso-Fernández I., Ruiz-Dueñas F. J., Martínez A. T. (2018). Evolutionary convergence in lignin-degrading enzymes. Proc. Natl. Acad. Sci. U.S.A. 115, 6428–6433. doi: 10.1073/pnas.1802555115
Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Blin K., Shaw S., Kloosterman A. M., Charlop-Powers Z., Van Wezel G. P., Medema M. H., et al. (2021). antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 49, W29–w35. doi: 10.1093/nar/gkab335
Börcsök Z., Pásztory Z. (2021). The role of lignin in wood working processes using elevated temperatures: an abbreviated literature survey. Eur. J. Wood Wood Products 79, 511–526. doi: 10.1007/s00107-020-01637-3
Casanellas M., Fernández-Sánchez J., Roca-Lacostena J. (2020). Embeddability and rate identifiability of Kimura 2-parameter matrices. J. Math Biol. 80, 995–1019. doi: 10.1007/s00285-019-01446-0
Chan P. P., Lowe T. M. (2019). tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol. Biol. 1962, 1–14. doi: 10.1007/978-1-4939-9173-0_1
Chao S., Xiang Z., Guangyu L., Yaping D., Zhaoshou W., Zongze S. (2021). Diversity of anaerobic degrading bacteria for natural organic polymers in mangrove sediments and isolation of novel groups of bacteria. Acta Microbiologica Sin. 61, 987–1001. doi: 10.13343/j.cnki.wsxb.20200715
Chaumeil P. A., Mussig A. J., Hugenholtz P., Parks D. H. (2019). GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927. doi: 10.1093/bioinformatics/btz848
Cherney M. M., Zhang Y., James M. N., Weiner J. H. (2012). Structure-activity characterization of sulfide:quinone oxidoreductase variants. J. Struct. Biol. 178, 319–328. doi: 10.1016/j.jsb.2012.04.007
Collins M. D., Pirouz T., Goodfellow M., Minnikin D. E. (1977). Distribution of menaquinones in actinomycetes and corynebacteria. J. Gen. Microbiol. 100, 221–230. doi: 10.1099/00221287-100-2-221
Cumsille A., Durán R. E., Rodríguez-Delherbe A., Saona-Urmeneta V., Cámara B., Seeger M., et al. (2023). GenoVi, an open-source automated circular genome visualizer for bacteria and archaea. PloS Comput. Biol. 19, e1010998. doi: 10.1371/journal.pcbi.1010998
Drula E., Garron M.-L., Dogan S., Lombard V., Henrissat B., Terrapon N. (2021). The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 50, D571–D577. doi: 10.1093/nar/gkab1045
Duan Y., Wang Y., Liu Q., Xiong D., Zhang J. (2019). Transcriptomic and microbiota response on Litopenaeus vannamei intestine subjected to acute sulfide exposure. Fish Shellfish Immunol. 88, 335–343. doi: 10.1016/j.fsi.2019.02.021
Emms D. M., Kelly S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238. doi: 10.1186/s13059-019-1832-y
Fang B. Z., Han M. X., Jiao J. Y., Xie Y. G., Zhang X. T., Liu L., et al. (2020). Streptomyces cavernae sp. nov., a novel actinobacterium isolated from a karst cave sediment sample. Int. J. Syst Evol. Microbiol. 70, 120–125. doi: 10.1099/ijsem.0.003724
Felsenstein J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376. doi: 10.1007/BF01734359
Finster K. W., Herbert R. A., Kjeldsen K. U., Schumann P., Lomstein B. A. (2009). Demequina lutea sp. nov., isolated from a high Arctic permafrost soil. Int. J. Syst. Evol. Microbiol. 59, 649–653. doi: 10.1099/ijs.0.004929-0
Friedrich C. G., Rother D., Bardischewsky F., Quentmeier A., Fischer J. (2001). Oxidation of reduced inorganic sulfur compounds by bacteria: emergence of a common mechanism? Appl. Environ. Microbiol. 67, 2873–2882. doi: 10.1128/AEM.67.7.2873-2882.2001
Galperin M. Y., Kristensen D. M., Makarova K. S., Wolf Y. I., Koonin E. V. (2019). Microbial genome analysis: the COG approach. Brief Bioinform. 20, 1063–1070. doi: 10.1093/bib/bbx117
Gao L., Fang B. Z., Liu Y. H., Jiao J. Y., Li M. M., Antunes A., et al. (2022). Rhabdothermincola salaria sp. nov., a novel actinobacterium isolated from a saline lake sediment. Int. J. Syst. Evol. Microbiol. 72, 005361. doi: 10.1099/ijsem.0.005361
Garber A. I., Nealson K. H., Okamoto A., Mcallister S. M., Chan C. S., Barco R. A., et al. (2020). FeGenie: A comprehensive tool for the identification of iron genes and iron gene neighborhoods in genome and metagenome assemblies. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.00037
Glick B. R., Cheng Z., Czarny J., Duan J. (2007). Promotion of plant growth by ACC deaminase-producing soil bacteria. Eur. J. Plant pathology. 119, 329–339. doi: 10.1007/s10658-007-9162-4
Groth I., Schumann P., Rainey F. A., Martin K., Schuetze B., Augsten K. (1997). Demetria terragena gen. nov., sp. nov., a New Genus of Actinomycetes Isolated from Compost Soil. Int. J. Systematic Evolutionary Microbiol. 47, 1129–1133. doi: 10.1099/00207713-47-4-1129
Hamada M., Ichikawa N., Oguchi A., Komaki H., Tamura T., Fujita N. (2015a). Draft genome sequences of eight type strains of the genus demequina. Genome Announcements 3, e00281–15. doi: 10.1128/genomeA.00281-15
Hamada M., Shibata C., Saitou S., Tamura T., Komaki H., Ichikawa N., et al. (2015b). Proposal of nine novel species of the genus Lysinimicrobium and emended description of the genus Lysinimicrobium. Int. J. Syst. Evol. Microbiol. 65, 4394–4402. doi: 10.1099/ijsem.0.000587
Hamada M., Tamura T., Yamamura H., Suzuki K. I., Hayakawa M. (2013). Demequina flava sp. nov. and Demequina sediminicola sp. nov., isolated from sea sediment. Int. J. Syst. Evol. Microbiol. 63, 249–253. doi: 10.1099/ijs.0.039297-0
Hasan M. K., Scott N. E., Hays M. P., Hardwidge P. R., El Qaidi S. (2023). Salmonella T3SS effector SseK1 arginine-glycosylates the two-component response regulator OmpR to alter bile salt resistance. Sci. Rep. 13, 9018. doi: 10.1038/s41598-023-36057-9
Hasegawa T., Takizawa M., Tanida S. (1983). A rapid analysis for chemical grouping of aerobic actinomycetes. J. Gen. Appl. Microbiol. 29, 319–322. doi: 10.2323/jgam.29.319
Huang J., Jones A., Waite T. D., Chen Y., Huang X., Rosso K. M., et al. (2021). Fe(II) redox chemistry in the environment. Chem. Rev. 121, 8161–8233. doi: 10.1021/acs.chemrev.0c01286
Hyatt D., Chen G. L., Locascio P. F., Land M. L., Larimer F. W., Hauser L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11, 119. doi: 10.1186/1471-2105-11-119
Janusz G., Pawlik A., Sulej J., Swiderska-Burek U., Jarosz-Wilkolazka A., Paszczynski A. (2017). Lignin degradation: microorganisms, enzymes involved, genomes analysis and evolution. FEMS Microbiol. Rev. 41, 941–962. doi: 10.1093/femsre/fux049
Jiao J. Y., Fu L., Hua Z. S., Liu L., Salam N., Liu P. F., et al. (2021). Insight into the function and evolution of the Wood-Ljungdahl pathway in Actinobacteria. ISME J. 15, 3005–3018. doi: 10.1038/s41396-021-00935-9
Joshi N. A., Fass J. N. (2011). Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33) [Software]. Available at: https://github.com/najoshi/sickle.
Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/nar/gkv1070
Kim M., Oh H. S., Park S. C., Chun J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. doi: 10.1099/ijs.0.059774-0
Kroppenstedt R. M. (1982). Separation of bacterial menaquinones by HPLC using reverse phase (RP18) and a silver loaded ion exchanger as stationary phases. J. Liquid Chromatogr. 5, 2359–2367. doi: 10.1080/01483918208067640
Kumar S., Stecher G., Li M., Knyaz C., Tamura K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lagesen K., Hallin P., Rødland E. A., Staerfeldt H. H., Rognes T., Ussery D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lagkouvardos I., Joseph D., Kapfhammer M., Giritli S., Horn M., Haller D., et al. (2016). IMNGS: A comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci. Rep. 6, 33721. doi: 10.1038/srep33721
Lamers L. P., Govers L. L., Janssen I. C., Geurts J. J., van der Welle M. E., Van Katwijk M. M., et al. (2013). Sulfide as a soil phytotoxin-a review. Front. Plant Sci. 4, 268. doi: 10.3389/fpls.2013.00268
Li D. D., Wang J. L., Liu Y., Li Y. Z., Zhang Z. (2021). Expanded analyses of the functional correlations within structural classifications of glycoside hydrolases. Comput. Struct. Biotechnol. J. 19, 5931–5942. doi: 10.1016/j.csbj.2021.10.039
Lima L. M., Silva B., Barbosa G., Barreiro E. J. (2020). β-lactam antibiotics: An overview from a medicinal chemistry perspective. Eur. J. Med. Chem. 208, 112829. doi: 10.1016/j.ejmech.2020.112829
Liu H., Xin B., Zheng J., Zhong H., Yu Y., Peng D., et al. (2020). Build a bioinformatics analysis platform and apply it to routine analysis of microbial genomics and comparative genomics. Protoc Exch. doi: 10.21203/rs.2.21224/v3
Liu D., Zhang Y., Fan G., Sun D., Zhang X., Yu Z., et al. (2022). IPGA: A handy integrated prokaryotes genome and pan-genome analysis web service. iMeta 1, e55. doi: 10.1002/imt2.55
MacDonald L. C., Berger B. W. (2014). A polysaccharide lyase from Stenotrophomonas maltophilia with a unique, pH-regulated substrate specificity. J. Biol. Chem. 289, 312–325. doi: 10.1074/jbc.M113.489195
Matsumoto A., Nakai K., Morisaki K., Ōmura S., Takahashi Y. (2010). Demequina salsinemoris sp. nov., isolated on agar media supplemented with ascorbic acid or rutin. Int. J. Syst. Evol. Microbiol. 60, 1206–1209. doi: 10.1099/ijs.0.012617-0
Meng X., Shao Z., Hong Y., Lin L., Li C., Liu Z. (2009). A novel pH-stable, bifunctional xylanase isolated from a deep-sea microorganism, Demequina sp. JK4. J. Microbiol. Biotechnol. 19, 1077–1084. doi: 10.4014/JMB.0901.0017
Minnikin D. E., O’donnell A. G., Goodfellow M., Alderson G., Athalye M., Schaal A., et al. (1984). An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J. Microbiological Methods 2, 233–241. doi: 10.1016/0167-7012(84)90018-6
Modjinou T., Versace D.-L., Abbad-Andaloussi S., Langlois V., Renard E. (2017). Antibacterial and antioxidant photoinitiated epoxy co-networks of resorcinol and eugenol derivatives. Materials Today Commun. 12, 19–28. doi: 10.1016/j.mtcomm.2017.03.005
Morrissey J., Guerinot M. L. (2009). Iron uptake and transport in plants: the good, the bad, and the ionome. Chem. Rev. 109, 4553–4567. doi: 10.1021/cr900112r
Nguyen L. T., Schmidt H. A., Von Haeseler A., Minh B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nouri K., Feng Y., Schimmer A. D. (2020). Mitochondrial ClpP serine protease-biological function and emerging target for cancer therapy. Cell Death Dis. 11, 841. doi: 10.1038/s41419-020-03062-z
Oren A., Garrity G. M. (2021). Valid publication of the names of forty-two phyla of prokaryotes. Int. J. Systematic Evolutionary Microbiol. 71, 005056. doi: 10.1099/ijsem.0.005056
Park S., Jung Y.-T., Won S.-M., Yoon J.-H. (2016). Demequina litorisediminis sp. nov., isolated from a tidal flat, and emended description of the genus Demequina. Int. J. Systematic Evolutionary Microbiol. 66, 4197–4203. doi: 10.1099/ijsem.0.001335
Peruzzi E., Franke-Whittle I. H., Kelderer M., Ciavatta C., Insam H. (2017). Microbial indication of soil health in apple orchards affected by replant disease. Appl. Soil Ecol. 119, 115–127. doi: 10.1016/j.apsoil.2017.06.003
Quick M., Dwivedi M., Padan E. (2021). Insight into the direct interaction of Na+ with NhaA and mechanistic implications. Sci. Rep. 11, 7045. doi: 10.1038/s41598-021-86318-8
Richter M., Rosselló-Móra R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rohban R., Amoozegar M. A., Ventosa A. (2009). Screening and isolation of halophilic bacteria producing extracellular hydrolyses from Howz Soltan Lake, Iran. J. Ind. Microbiol. Biotechnol. 36, 333–340. doi: 10.1007/s10295-008-0500-0
Rütting T., Boeckx P., Müller C., Klemedtsson L. (2011). Assessment of the importance of dissimilatory nitrate reduction to ammonium for the terrestrial nitrogen cycle. Biogeosciences 8, 1779–1791. doi: 10.5194/bg-8-1779-2011
Saitou N., Nei M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Sánchez-Rodríguez A., Tytgat H. L. P., Winderickx J., Vanderleyden J., Lebeer S., Marchal K. (2014). A network-based approach to identify substrate classes of bacterial glycosyltransferases. BMC Genomics 15, 349. doi: 10.1186/1471-2164-15-349
Sayers E. W., Beck J., Bolton E. E., Bourexis D., Brister J. R., Canese K., et al. (2021). Database resources of the national center for biotechnology information. Nucleic Acids Res. 49, D10–d17. doi: 10.1093/nar/gkaa892
Shen X., Dong W., Wan Y., Feng K., Liu Y., Wei Y. (2021). Influencing mechanisms of siderite and magnetite, on naphthalene biodegradation: Insights from degradability and mineral surface structure. J. Environ. Manage 299, 113648. doi: 10.1016/j.jenvman.2021.113648
Shi J., Xu X., Liu P. Y., Hu Y. L., Zhang B., Jiao R. H., et al. (2021). Discovery and biosynthesis of guanipiperazine from a NRPS-like pathway. Chem. Sci. 12, 2925–2930. doi: 10.1039/D0SC06135B
Shimizu Y., Ogata H., Goto S. (2017). Type III polyketide synthases: functional classification and phylogenomics. ChemBioChem 18, 50–65. doi: 10.1002/cbic.201600522
Smibert R., Krieg N. (1994). “Phenotypic characterization. In: Methods for General and Molecular Bacteriology Gerhardt P., Murray R. G. E., Wood W. A., Krieg N. R. Eds. American Society for Microbiology, Washington, DC, 607–654.
Susin M. F., Baldini R. L., Gueiros-Filho F., Gomes S. L. (2006). GroES/GroEL and DnaK/DnaJ have distinct roles in stress responses and during cell cycle progression in Caulobacter crescentus. J. Bacteriol 188, 8044–8053. doi: 10.1128/JB.00824-06
Tan S., Liu J., Fang Y., Hedlund B. P., Lian Z.-H., Huang L.-Y., et al. (2019). Insights into ecological role of a new deltaproteobacterial order Candidatus Acidulodesulfobacterales by metagenomics and metatranscriptomics. ISME J. 13, 2044–2057. doi: 10.1038/s41396-019-0415-y
Thompson J. D., Higgins D. G., Gibson T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Ue H., Matsuo Y., Kasai H., Yokota A. (2011). Demequina globuliformis sp. nov., Demequina oxidasica sp. nov. and Demequina aurantiaca sp. nov., actinobacteria isolated from marine environments, and proposal of Demequinaceae fam. nov. Int. J. Systematic Evolutionary Microbiol. 61, 1322–1329. doi: 10.1099/ijs.0.024299-0
Wei L., Liu Y., Dubchak I., Shon J., Park J. (2002). Comparative genomics approaches to study organism similarities and differences. J. BioMed. Inform 35, 142–150. doi: 10.1016/S1532-0464(02)00506-3
Wei H., Wang L., Hassan M., Xie B. (2018). Succession of the functional microbial communities and the metabolic functions in maize straw composting process. Bioresource Technol. 256, 333–341. doi: 10.1016/j.biortech.2018.02.050
Wick R. R., Judd L. M., Gorrie C. L., Holt K. E. (2017). Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PloS Comput. Biol. 13, e1005595. doi: 10.1371/journal.pcbi.1005595
Wu Z., Li Y., Gu Z., Ding Z., Zhang L., Shi. G. (2018). Function of glnA, proB and proA genes in L-proline anabolic pathway of Bacillus subtilis. Acta Microbiologica Sin. 58, 39–50. doi: 10.13343/j.cnki.wsxb.20170011
Xu P., Li W. J., Tang S. K., Zhang Y. Q., Chen G. Z., Chen H. H., et al. (2005). Naxibacter alkalitolerans gen. nov., sp. nov., a novel member of the family ‘Oxalobacteraceae’ isolated from China. Int. J. Syst. Evol. Microbiol. 55, 1149–1153. doi: 10.1099/ijs.0.63407-0
Xu M., Zhang F., Cheng Z., Bashiri G., Wang J., Hong J., et al. (2020). Functional genome mining reveals a class V lanthipeptide containing a d-amino acid introduced by an F(420) H(2) -dependent reductase. Angew Chem. Int. Ed Engl. 59, 18029–18035. doi: 10.1002/anie.202008035
Yi H., Schumann P., Chun J. (2007). Demequina aestuarii gen. nov., sp. nov., a novel actinomycete of the suborder Micrococcineae, and reclassification of Cellulomonas fermentans Bagnara et al. 1985 as Actinotalea fermentans gen. nov., comb. nov. Int. J. Systematic Evolutionary Microbiol. 57, 151–156. doi: 10.1099/ijs.0.64525-0
Yin L. Z., Liu Z. T., Li J. L., Wang P. D., Dong L., Duan L., et al. (2021). Agilicoccus flavus gen. nov., sp. nov., a novel member of the family Dermatophilaceae isolated from the Pearl River. Int. J. Syst. Evol. Microbiol. 71, 005076. doi: 10.1099/ijsem.0.005076
Yoon S. H., Ha S. M., Kwon S., Lim J., Kim Y., Seo H., et al. (2017). Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617. doi: 10.1099/ijsem.0.001755
Zhou Z., Tran P. Q., Breister A. M., Liu Y., Kieft K., Cowley E. S., et al. (2022). METABOLIC: high-throughput profiling of microbial genomes for functional traits, metabolism, biogeochemistry, and community-scale functional networks. Microbiome 10, 33. doi: 10.1186/s40168-021-01213-8
Keywords: Demequina, comparative genomic analysis, diversity and identification, tidal flat, saline lake
Citation: Gao L, Fang B-Z, Lu C-Y, Hong K-H, Huang X-Y, She T-T, Xiao M and Li W-J (2023) Unraveling the genomic diversity and ecological potential of the genus Demequina: insights from comparative analysis of different saline niche strains. Front. Mar. Sci. 10:1244849. doi: 10.3389/fmars.2023.1244849
Received: 23 June 2023; Accepted: 10 October 2023;
Published: 20 October 2023.
Edited by:
Xue-Wei Xu, Ministry of Natural Resources, ChinaReviewed by:
Dao-Feng Zhang, Hohai University, ChinaCopyright © 2023 Gao, Fang, Lu, Hong, Huang, She, Xiao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bao-Zhu Fang, ZmFuZ2Jhb3podTIwMDlAMTI2LmNvbQ==; Wen-Jun Li, bGl3ZW5qdW4zQG1haWwuc3lzdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.