 Yan Wang
Yan Wang Lin Gong
Lin Gong Zhaoming Gao
Zhaoming Gao Yong Wang
Yong Wang Feng Zhao
Feng Zhao Lulu Fu
Lulu Fu Xinzheng Li
Xinzheng Li- 1Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China
- 2Laboratory of Marine Organism Taxonomy and Phylogeny, Qingdao Key Laboratory of Marine Biodiversity and Conservation, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China
- 3Graduate University, University of Chinese Academy of Sciences, Beijing, China
- 4Institute of Deep-Sea Science and Engineering, Chinese Academy of Sciences, Sanya, Hainan, China
- 5Institute for Ocean Engineering, Shenzhen International Graduate School, Tsinghua University, Shenzhen, China
- 6Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
The cold-seep sponge holobionts are attracting growing attention in recent years. In this study, we utilized 16S rRNA amplicons to characterize the bacterial communities of six deep-sea sponge species found in sponge grounds at the Formosa Ridge cold seep in the South China Sea. Bacterial communities in these geographically proximal sponge species are dominated by Proteobacteria (mainly Gammaproteobacteria and Alphaproteobacteria) but exhibit distinct diversity and compositions among communities. Further analysis revealed that the SUP05 clade (Thioglobaceae) dominated most of the sponge samples. Meanwhile, phylogenetic analysis showed that the six sponge species harbored diverse SUP05 OTU phylotypes, indicating significant divergence within this clade. Additionally, operational taxonomic units (OTUs) of the family Methylomonadaceae, another abundant group in these sponges, displayed a significant genetic distance both from each other and from known species. Our findings support the hypothesis of the host-species specificity of sponge-associated bacterial communities, a widely accepted concept in shallow-water and other deep-sea sponges. The presence of dominant functional microbes, such as sulfur- and methanol-oxidizing bacteria, suggests their crucial role as chemosynthetic symbionts in facilitating the niche adaption of sponge hosts to the cold seep ecosystem. In conclusion, our study reveals the diverse and novel bacterial communities in deep-sea sponges from cold seep environments, contributing new knowledge to the host-species specificity of bacterial communities within sponges and highlighting the potential significance of functional microbes in cold seep ecosystems with dynamic energy supplies.
1 Introduction
Sponges (phylum Porifera) are evolutionarily ancient animals (Li, 1998), originating from the early Cambrian period (Reitner and Wörheide, 2002). They are found in all oceans and at all depths, even living depths greater than 8,000 m. Most sponges are highly effective filter feeders, while some species exhibit a surprising adaptation by reducing their filtering capacity in favor of a carnivorous lifestyle (Van Soest et al., 2012). In some areas, the abundance of sponge resources leads to the formation of sponge grounds—aggregations of large sponges that develop under specific geological, hydrological, and biological conditions, creating unique structural habitats (Sogin et al., 2021). Demosponges aggregations always have a mixture of different sponges, while hexactinellids aggregations are always typically composed of monospecific species (Maldonado et al., 2017). Due to their unique loose and porous body structure, sponges can harbor a large number of microorganisms that coexist and evolve together in the ocean, forming inseparable symbiotic entities known as holobionts (Webster and Thomas, 2016). The microbial communities coexisting with sponges exhibit a high degree of diversity and complexity, with 63 phyla having been discovered in sponges to date, making a major contributor to the overall prokaryotic diversity of the world’s oceans (Webster et al., 2010; Schmitt et al., 2012; Thomas et al., 2016; Moitinho-Silva et al., 2017). Within these holobionts, microbial symbionts perform critical functions for the health and survival of sponges. They provide essential functions such as nutrient provisioning (particularly, for the nitrogen and carbon cycle, vitamin production, photosynthesis, and the synthesis of secondary metabolites) and chemical defense of sponges by aiding in cellular stress responses, motility, and adhesion (Taylor et al., 2007; Fan et al., 2012; De Goeij et al., 2013; Webster and Thomas, 2016; Botté et al., 2019). Simultaneously, the host sponge also provides a habitat for these microorganisms (Osinga et al., 2001). As integral elements of the ocean, inseparable holobionts exert significant influence on community structure and ecosystem functioning through cascading effects, including nutrient cycling, primary productivity, community structure, and food webs (Pita et al., 2018).
Based on the abundance of associated microbial communities, sponges can be broadly classified into two groups: “high microbial abundance (HMA)” and “low microbial abundance (LMA)” (Hentschel et al., 2003). HMA sponges and LMA sponges exhibit distinct characteristics in terms of indicator taxa, diversity and the abundance of microbial communities, pumping rates, higher frequency of hosting photosynthetic symbionts, and similarity to the environmental community (Hentschel et al., 2003; Weisz et al., 2007; Schläppy et al., 2010; Erwin et al., 2011; Freeman and Thacker, 2011; Giles et al., 2013). Numerous studies consistently demonstrated that sponge microbial communities exhibit significant host specificity across different seasons, environments, and geographical regions (Hardoim et al., 2012; Cleary et al., 2013; Reveillaud et al., 2014). This suggests that the composition of microbial communities within sponges is influenced by their hosts, potentially indicating a co-evolutionary relationship between them (Fan et al., 2012; Pita et al., 2013; Easson and Thacker, 2014). Furthermore, extensive research on symbiotic microbial communities in sponges has revealed additional features, such as the existence of both specialists and generalists, the reliably stable core microbiota, and functional modularity (Thomas et al., 2016). In terms of interaction with the environment, it appears that non-biological factors predominantly influence the overall sponge microbiota, whereas biological factors primarily impact the prokaryotic core (Lurgi et al., 2019).
Seeps are special ecosystems formed by continuous or intermittent emission or eruption of reduced components such as methane and hydrogen sulfide on the seafloor (Paull et al., 1984). Seeps and hydrothermal systems, with their unique characteristics providing ideal conditions for diverse and specialized biological communities, particularly distinct microbiomes, have revolutionized our understanding of life activities under extreme environmental conditions in the deep sea (Jørgensen and Boetius, 2007; Smedile et al., 2012). To date, less than 5% of the deep sea has been explored (Ramirez-Llodra et al., 2010), leading to a lack of research on the community structure and metabolic spectrum of deep-sea sponge organisms, in contrast to the extensive research conducted on shallow-water sponges (Meyer and Kuever, 2008; Radax et al., 2012). Deep-sea chemoautotrophic sponge holobionts serve as a key component of deep-sea ecosystems, constitute the complex habitats hosting many other species, and contribute to ecosystem primary productivity (Nishijima et al., 2010; Arellano et al., 2012; Li et al., 2015), which are essential for supporting life in these extreme environments (Dubilier et al., 2008).
Over 40 additional cold seeps have been identified along the continental margin of the South China Sea (SCS) (Chen et al., 2005; Feng et al., 2018). The chemosynthetic community we investigated is known as the Formosa Ridge (also called Site F). The Formosa Ridge is located on the top of the northern continental slope of the SCS and is considered one of the most active cold seeps in the SCS (Tong et al., 2011; Wang et al., 2021). Spanning approximately 30 km in length and 5 km in width, the Formosa Ridge consists of two summits. The southern summit, where the active cold seep is located, has a depth of approximately 1,125 m (Cai et al., 2022). Previous studies (Chen et al., 2005) have suggested that a major source of cold seep fluids is methane released from the dissolution of the gas hydrate under the seabed. Furthermore, the Formosa Ridge is known to exhibit a novel sulfur oxidation pathway, which plays an important role in the biogeochemical sulfur cycle (Cai et al., 2022).
In this study, we collected 16 samples of six sponge species at the Formosa Ridge cold seep. Based on 16S rRNA gene sequencing, we investigated the symbiotic microbial communities of these sponges in cold-seep sponge ground to explore three aspects: 1) The composition, structure, and diversity of symbiotic microorganisms in different sponge species found in the cold-seep sponge ground; 2) The relationship between sponge symbiotic microorganisms and the phylogenetic relationship and the evolutionary status of the host; and 3) The special characteristics of dominant bacterial species.
2 Materials and methods
2.1 Sample collection
Sponges were collected from the Formosa Ridge cold seep (Figure 1A) during the scientific cruise of Research Vessel (R/V) KEXUE organized by the Institute of Oceanology, Chinese Academy of Sciences in 2021. The cold seep covers a long circular area (W-E: ~200 m; N-S: ~150 m) (Cao et al., 2021) and a sponge ground was found on the rocks surrounding this area (Figure 1B). We brought back rocks from the seafloor and collected the samples on board (Figure 1C). Upon arrival at the deck, the samples were rinsed several times with 0.22-µm membrane-filtered seawater to remove loosely attached microbes and debris. Small sections of clean tissues were sub-sampled and placed into separated sterile plastic tubes and instantly frozen at -80 °C for molecular analyses, while the majority of tissues were stored in ethanol (80%) as vouchers for morphological identification. Our sponge samples were collected from three stations: station 267 (119.285636°E, 22.115354°N; 1128 m depth; 13 June 2021), station ZD-10 (119.285152°E, 22.115256°N; 1164 m depth; 18 June 2021), and station ZD-7 (119.284662°E, 22.114870°N; 1208 m depth; 14 June 2021). Among these, station 267 was located closest to the seepage and belongs to the gas flares area. Station ZD-7 was the farthest from the active seepage and had shell debris covering the seabed. Station ZD-10 was a biologically rich area with flourishing benthic communities. Additional information about the Formosa Ridge cold seep, including the methane and hydrogen sulfide concentrations, can be found in the study by Cao et al. (2021).

Figure 1 The sponges colonize cold seeps in the South China Sea (SCS) drawn by an ocean data view. (A) Geographic location of the sponge sampling site, the Site F cold seep. The map was drawn by the Ocean Data View. (B) An ROV image of the sponge ground. (C) A photograph of the fresh sponge samples on a rock after being taken back to the deck.
2.2 Sponge taxonomy
A total of six sponge species were identified according to both morphological and molecular characteristics, with two species having two biological replicates and four species having three biological replicates. To perform morphological identification, the sponge spicules were isolated by digesting a small piece of sponge tissue using concentrated nitric acid. Subsequently, the spicules were examined using both a light microscope (LM) and a scanning electron microscope (SEM). For molecular data, DNA libraries were prepared from individual genomic DNA samples and subsequently, sequencing was performed using the Illumina PE150 strategy. The sequencing reads were quality-filtered using Trimmomatic and assembled using Megahit. The assembly contigs were then used to extract the 18S ribosomal RNA gene sequences of the sponge host. The 18S rRNA gene sequences of 16 individual samples are presented in Supplementary Table S1.
2.3 16S rRNA gene amplicon sequencing and processing
Microbial DNA was extracted from sponge samples using the E.Z.N.A.® Soil DNA Kit (Omega Bio-tek, Norcross, GA, U.S.) following the manufacturer’s protocols. The V1-V9 regions of the bacterial 16S rRNA gene were amplified by PCR (95°C for 2 min, followed by 27 cycles at 95°C for 30 s, 55°C for 30 s, and 72°C for 60 s, and a final extension at 72°C for 5 min) using primers 27F 5’-AGRGTTYGATYMTGGCTCAG-3’ and 1492R 5’-RGYTACCTTGTTACGACTT-3’ ligated with an eight-base barcode unique to each sample. PCR reactions were performed in triplicate 20 μL mixture containing 4 μL of 5 × FastPfu Buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL of FastPfu Polymerase, and 10 ng of template DNA. Amplicons were extracted from 2% agarose gels and purified using the Axy Prep DNA Gel Extraction Kit (Axygen Biosciences, Union City). SMRT bell libraries were prepared from the purified amplicons by blunt ligation according to the manufacturer’s instructions (Pacific Biosciences). Purified SMRT bell libraries from the pooled and barcoded samples were sequenced on a single PacBio Sequel II cell by Shanghai Biozeron Biotechnology Co. Ltd (Shanghai, China).
PacBio raw reads were processed using the SMRT Link Analysis software version 9.0 to obtain demultiplexed circular consensus sequence (CCS) reads with the following settings: minimum number of passes = 3 and minimum predicted accuracy = 0.99. Raw fastq files were first demultiplexed using in-house perl scripts according to the barcode sequences information for each sample with the following criteria: (i) The 250bp reads were truncated at any site receiving an average quality score <20 over a 10 bp sliding window, discarding the truncated reads that were shorter than 50bp; (ii) exact barcode matching, 2 nucleotide mismatch in primer matching, and reads containing ambiguous characters were removed; and (iii) only sequences that overlapped longer than 10 bp were assembled according to their overlap sequence. Reads which could not be assembled were discarded. Operational taxonomic units (OTUs) were clustered with a 97% similarity cutoff using UPARSE (version 7.1 http://drive5.com/uparse/) and chimeric sequences were identified and removed using UCHIME. Pick representative sequences for each OTU using Qiime2 (https://qiime2.org) Taxonomic assignment was performed using UCLUST (http://www.drive5.com/usearch/manual/uclust_algo.html) against the Silva 138.1 (https://www.arb-silva.de/) 16S rRNA database at a similarity threshold of 0.8. OTUs that did not belong to bacteria and were annotated as mitochondria and chloroplast were removed. The number of Pacbio reads assigned to each OTU and the OTU taxonomy details are stored in Supplementary Table 2. To account for different sequencing depths, the OTU abundance matrix was rarefied to 4592 reads per sample.
2.4 Data analyses
The rarefaction analysis was performed using Mothur v.1.21.1 (Schloss et al., 2009) to reveal the Chao1, Simpson, and Shannon diversity indices. The principal coordinate analysis (PCoA) with a weighted-unifrac matrix (Lozupone and Knight, 2006) was performed using the community ecology R-forge package (Vegan 2.0 package was used to generate a PCoA figure). Mantel tests were carried out to examine the analysis of similarities (ANOSIM) correlation between the sponge taxonomy and the bacterial community similarity using weighted-unifrac distance matrices with 999 permutations, using the vegan package in R. Non-parametric multivariate analysis of variance (ADONIS) was conducted to further confirm the observed differences.
The PhyloSuite workflow desktop platform (Zhang et al., 2020) was used to construct phylogenetic trees of bacteria. The most abundant OTUs belonging to the clade SUP05 and the family Methylomonadaceae in each sponge species were used as queries to search against the NCBI NR database and close relatives were selected. The symbionts and their relatives were aligned together using MAFFT with the default parameters, the SUP05 dataset consisted of 1475 bp, while the Methylomonadaceae dataset comprised 1465 bp. Phylogenetic relationships were using both maximum likelihood (ML) and Bayesian inference (BI) methods. The most suitable nucleotide substitution model was determined by ModelFinder 2 using the Akaike Information Criterion (AIC) and Bayesian Information Criterion (BIC). Maximum likelihood (ML) analysis was performed in PhyloSuite batch mode, employing the IQ-TREE plug-in with the “Auto” mode and 100,000 ultrafast bootstrap replicates. Bayesian analysis was conducted using MrBayes. Two runs were carried out with four Markov Chains for 2,000,000 generations (SUP05 dataset) and 10,000,000 generations (Methylomonadaceae dataset) started from a random tree, with sampling every 1,000 generations. The first 25% of trees were discarded as burn-in, and a 50% majority-rule consensus tree was obtained from the remaining trees. The average standard deviation of split frequencies fell below 0.01. The phylogenetic trees and node labels were visualized using FigTree.
In order to identify biomarkers for highly dimensional bacteria, linear discriminant analysis effect size (LEfSe) analysis was performed (Segata et al., 2011). Kruskal-Wallis sum-rank test was performed to examine the changes and dissimilarities among classes followed by LDA analysis to determine the size effect of each distinctively abundant taxa (Li et al., 2018).
3 Result
3.1 The sponge taxonomy
Morphological identification and molecular analysis revealed that our sponge samples belong to six sponge species affiliated with six different families in two classes (Demospongiae and Homoscleromorpha) (Figure 2). Most of the species were classified at the genus level, except for Petrosiidae sp., which could only be classified at the family level. Among the six species, only Plakina sp. belongs to Homoscleromorpha. As for the five demosponges, Euchelipluma sp. and Coelosphaera sp. belong to the same order (Poecilosclerida), while Janulum sp. and Petrosiidae sp. belong to the same order (Haplosclerida). Notably, Euchelipluma sp. is the only carnivorous sponge. Several sponges are new species, and their detailed identification will be reported in a separate article. In this study, we mainly provide general taxonomic information about the sponges (Table 1).

Figure 2 The photographs of six cold-seep sponge species with their taxa indicated.

Table 1 Sponge taxonomy and sampling locations.
3.2 Bacterial richness and diversity
In all, 16 sponge samples yielded a total of 179,026 qualified reads, with an average length of 1456.52 bp (the length of 99.98% reads >1350 bp). The reads were clustered into 1,783 unique operational taxonomic units (OTUs) using a similarity threshold of 97%. Out of these OTUs, 1,123 were classified at the species level, accounting for 63.02% of the total, and 532 were exclusively classified at the genus level, representing 29.83% of the total, whereas the remaining 128 can be classified at other higher taxonomic levels, comprising 7.18% of the total.
The coverage rates ranged from 95.34% (Euchelipluma sp.-2) to 99.87% (Stylocordyla sp.-2) (Figure 3), suggesting most of the bacteria have been covered by our sequencing efforts. The number of OTUs (reads can be seen in Table 2) and the Chao1 index indicated that Stylocordyla sp.-1 has the lowest species richness and diversity, while Euchelipluma sp.-2 has the highest species richness and diversity (Table 2). Significant differences were found among sponge species in the Shannon index (ANOVA: df = 5, P < 0.001) and Simpson index (ANOVA: df = 5, P < 0.001), whereas no significant difference was found in the Chao1 index (ANOVA: df = 5, P > 0.05) (Figure 3).
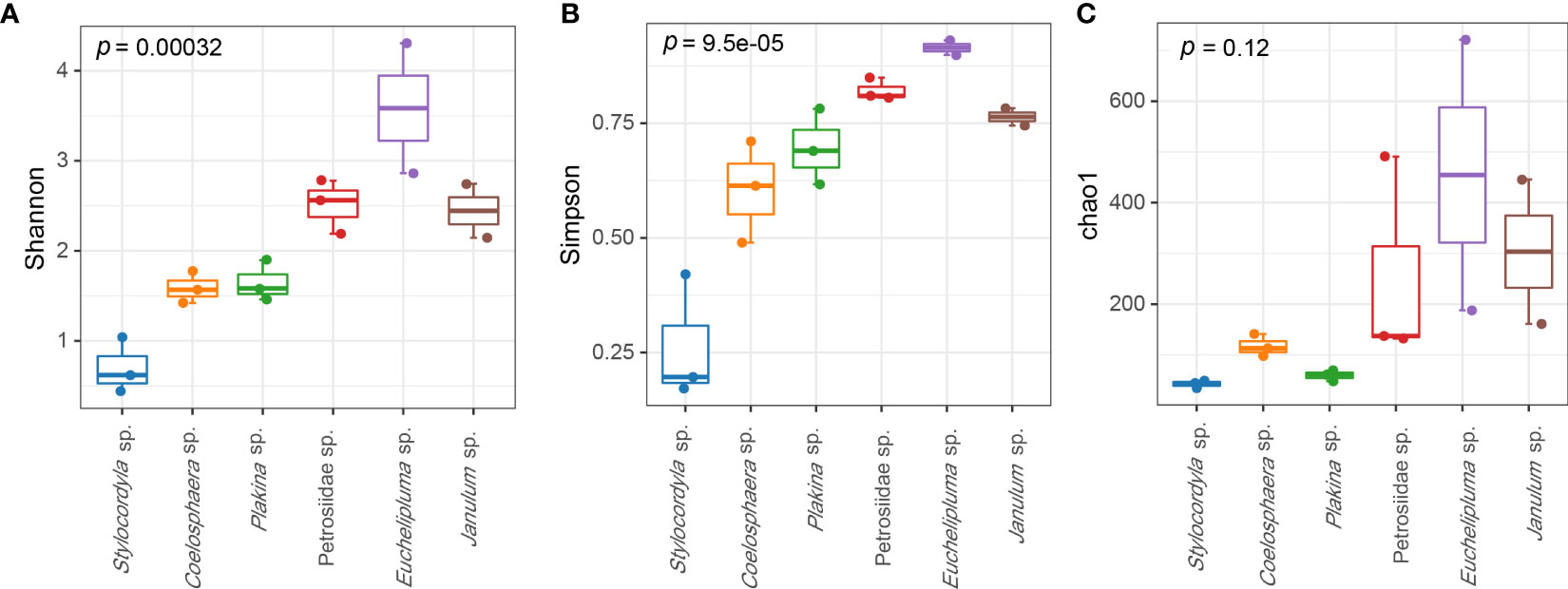
Figure 3 Alpha diversity of bacterial communities in the six cold-seep sponge species. The alpha diversity indices including Shannon diversity (A), Chao1 (B), and Simpson (C) are shown, respectively. The p values were calculated by ANOVA (and indicate the significance of inter-group differences).
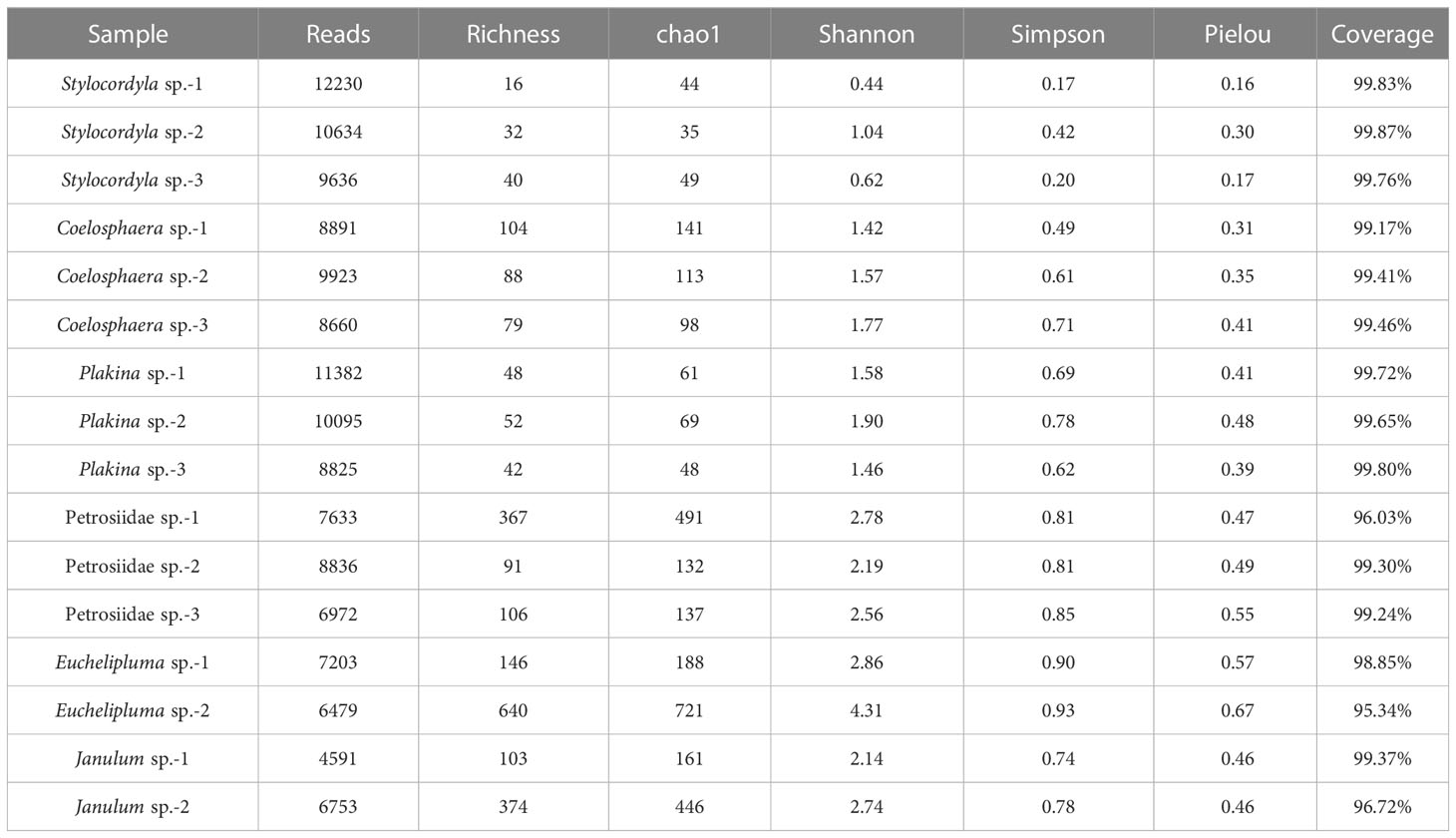
Table 2 Diversity summary of bacterial communities in the six cold-seep sponge species.
3.3 Abundance of the sponge-associated bacterial communities
The taxonomic abundance of the sponge-associated microbial communities was summarized at both the phylum level and genus level (Figure 4). In total, 25 bacterial phyla were detectable among all sponge samples (based on SILVA classification). The dominant bacterial phyla are Proteobacteria (0.907 ± 0.107), Bacteroidota (0.025 ± 0.001), Actinobacteriota (0.0165 ± 0.0028), Bdellovibrionota (0.0118 ± 0.0005), Planctomycetota, and Acidobacteriota. The predominant phylum Proteobacteria is mainly composed of the classes Gammaproteobacteria and Alphaproteobacteria, of which Gammaproteobacteria are found in all the sponge samples and represent the most abundant group in most of the samples. Especially, the relative abundance of Gammaproteobacteria in the Stylocordyla sp. samples was up to 99.78% (Stylocordyla sp.-1), 99.23% (Stylocordyla sp.-2), and 97.80% (Stylocordyla sp.-3), respectively, and that in the Coelosphaera sp. and Janulum sp. samples exceeded 73.43%. Alphaproteobacteria is relatively abundant in Petrosiidae sp. and Plakina sp. sponges, with an average relative abundance of 36.50% and 51.10%, respectively. Bacteroidota were mainly found in Euchelipluma sp., with an average relative abundance of 10.51%. Actinobacteriota accounts for 3.40% and 21.00% of the carnivorous sponge Euchelipluma sp. and 1.61% in Petrosiidae sp.-3. Bdellovibrionota is mainly found in Janulum sp. and Euchelipluma sp., with an average relative abundance of 10.51% and 6.15%, respectively. Planctomycetota were found in all samples, with up to 6.12% in Petrosiidae sp.-3.
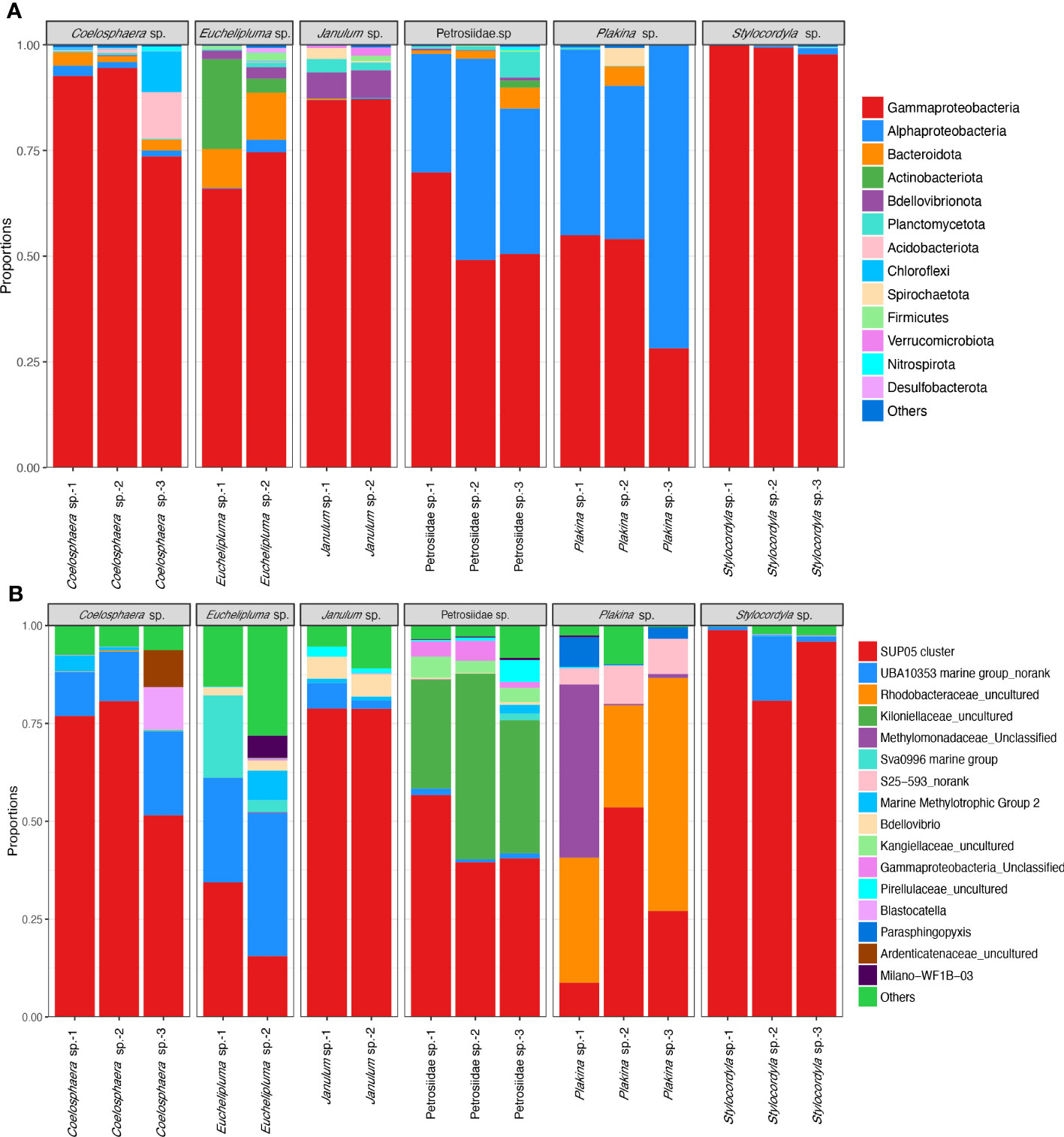
Figure 4 Taxon abundance of bacterial reads in the six cold-seep sponge species. (A) Relative abundance of microbes at the phylum level. The phylum Proteobacteria was shown with class-level taxa including Gammaproteobacteria and Alphaproteobacteria. The phylum-level taxa with a relative abundance of less than 0.1% in all samples were assigned to the category of “Others”. (B) Relative abundance of microbes at the genus level. The species-level taxa of less than 0.5% in all samples were assigned to the category of “Others”.
At the genus level, the bacterial symbionts associated with sponges are characterized as specialists and generalists rather than opportunists. The SUP05 clade at the genus level is present in all sponges and appears to be a generalist in the sponge ground. It makes up more than half of the total bacterial reads and is the predominant bacterial species in the Stylocordyla sp., Coelosphaera sp., and Janulum sp. samples. An unclassified UBA10353 marine group in the class Gammaproteobacteria is more closely related to samples from the carnivorous sponge Euchelipluma sp. samples. An uncultured group in the family Rhodobacteraceae belonging to order Rhodobacterales is abundantly present in Plakina sp. but is nearly absent or found in trace amounts in the remaining five Demospongiae sponges. This suggests that the group potentially specializes in Homoscleromorpha sponges. Similarly, an unclassified group in the family Methylomonadaceae is exclusively found in the three Homoscleromorpha samples, suggesting another specialist associates with Homoscleromorpha sponges. Alternatively, an unclassified group in the family Kiloniellaceae, belonging to the class Alphaproteobacteria, is exclusively found in the Petrosiidae sp. and accounts for 27.05% to 47.43% of the total bacterial reads. In addition, bacterial reads assigned to genus-level taxa in the Sva0996 marine group, the S25−593 group, and the Marine Methylotrophic Group 2 also occupy a relatively high proportion in these cold seep sponges.
Given a large number of OTUs cannot be accurately classified at the species or genus level, we conducted a detailed analysis of the convergence and divergence of OTUs among sponge species (Figure 5B). The results indicate that around 85.73% of all OTUs were exclusive to a single sponge species, 4.05% of all OTUs were shared by two sponge species, and 10.23% of all OTUs were shared among three or more sponge species. Notably, only OTU55 was detected in all samples, making it the most widely distributed taxon. OTU55 was classified as unclassified Gammaproteobacteria and may have a correlation with environmental microbiota. Interestingly, most highly abundant OTUs are not shared by sponge species.
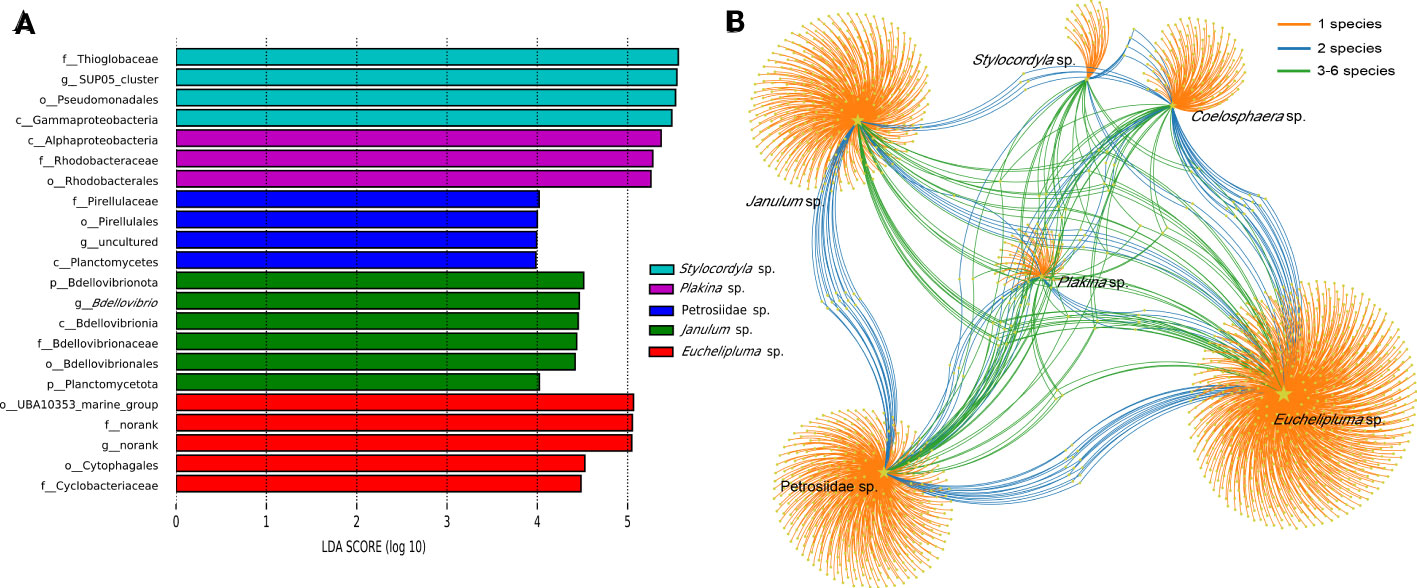
Figure 5 Linear discriminant analysis effect size (LEfSe) analyses of bacterial communities in the six cold-seep sponge species (A). Microbial taxa belonging to different sponge species were labeled with different colors, while the length of the bars represents the LDA scores, which were log10-transformed using the default threshold value of 2.0. “The labels c_, o_, f_, and g_ “ in the figure represent the taxonomic ranks of class level, order level, family level, and genus level, respectively. OTU distribution across sponge species by a bipartite network (B). Total number of OTUs detected in each sampled species is indicated by yellow stars with different sizes. Colored links are referred to the prevalence of OTUs in one (orange), two (blue), and more than two (green) sponge species.
3.4 Beta diversity
The Principal Coordinates Analysis (PCoA) clearly demonstrated the distinct separation of microbial communities among the six sponge species (Figure 6A). The analysis based on weighted-unifrac revealed that the host species diversity accounted for a substantial proportion of variation in community compositions (ANOSIM: test statistic=1.0, p<0.001; ADONIS: F=20.47, R2 = 0.91099, p<0.001). However, ANOSIM analysis revealed no significant variation in community compositions at the host sub-order level (from order level to Plakina sp.) (test statistic = 0.2995, p = 0.011). The PCoA plots (Figure 6B) indicated that the differences among samples from different sub-orders were partially attributed to variations in dispersion between groups. Specifically, the sub-order groups exhibited greater dispersion compared to the species groups. On the host class level (Figure 6C), a highly significant difference in microbial community structure was observed based on ANOSIM (test statistic=0.3625, p =0.018).
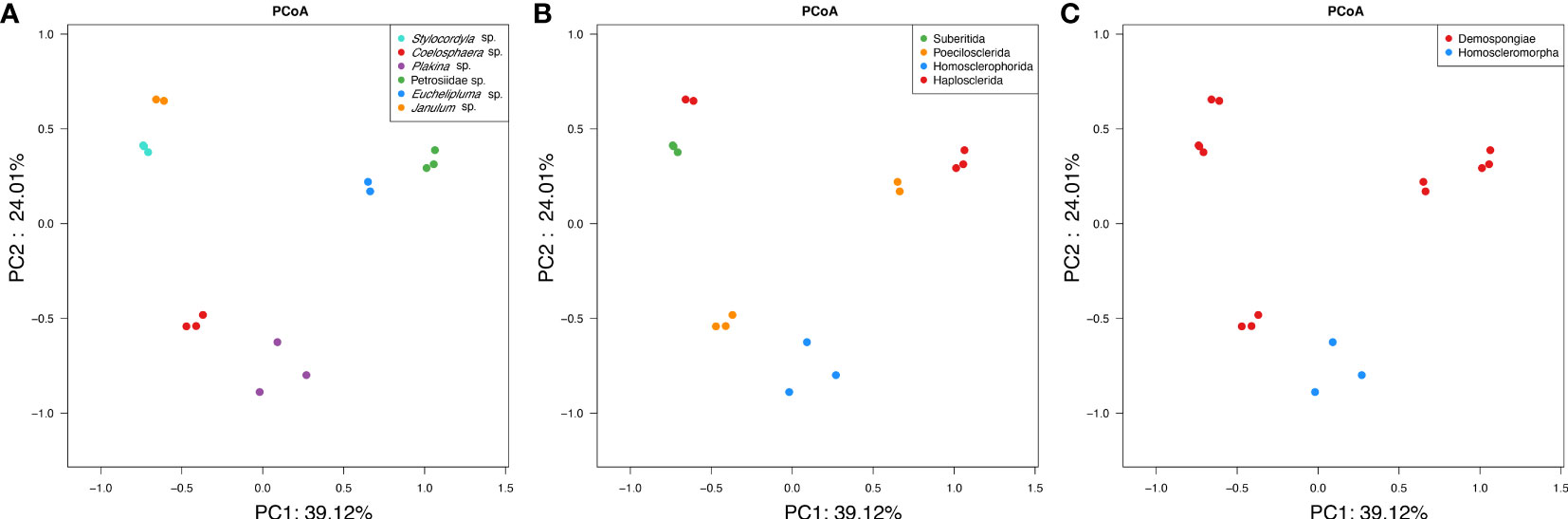
Figure 6 PCoA clustering of bacterial communities in the six cold-seep sponge species. The clustering was conducted based on weighted-unifrac dissimilarities of OTUs among samples. The three plots represent the same analysis, whereas samples were colored based on the sponge taxonomy at the species (A), family (B), and class level (C).
3.5 Difference in microbes among sponge species
LEfSe analyses were conducted to identify significantly different microbial abundance across sponge species and a bar plot was used to display the distribution of LDA scores above the default value of 2.0 for taxa deemed significant, identified as biomarkers (Figure 5A). The result indicated that all sponge species except for Coelosphaera sp. have unique biomarkers. Four species of sponge have their unique bacterial class, including Alphaproteobacteria in Plakina sp., Planctomycetia in Petrosiidae sp., Gammaproteobacteria in Stylocordyla sp., and Bdellovibrionales in Janulum sp. Biomarkers at lower taxonomic levels were also identified (Figure 5A).
3.6 The dominant sulfur-oxidizing bacteria
The microbes in the SUP05 clade, particularly those symbiotic with macrofauna such as clams and sponges, are characteristic sulfur-oxidizing bacteria (SOB). The SUP05 clade is highly abundant in our cold-seep sponges. A Venn diagram illustrates that each sponge species has a specific SUP05 OTU (Figure 7A). The phylogenetic tree based on 16S rDNA of ML and BI analyses were highly congruent (Figure 7B). Furthermore, it also revealed that the predominant SUP05 OTUs in each sponge species do not cluster together or with any known species. The six predominant SUP05 OTUs in each sponge species were searched further against the 16S rRNA gene amplicons of sediment and seawater samples adjacent to them obtained during the same research expedition, and no closely related OTUs were found (the data has not been published yet). Additionally, we searched these OTUs against a large size of metagenomic reads produced from seawater and sediment samples in the Site F cold seep (NCBI accession numbers: SRX10272306, SRX10272305, SRX10272304, SRX10272316, and SRX10272320). Only a limited number of reads matched, which likely resulted from matches of conservative regions in 16S rRNA genes. Consequently, we hypothesize the uniqueness of our SUP05 OTUs to each sponge species.
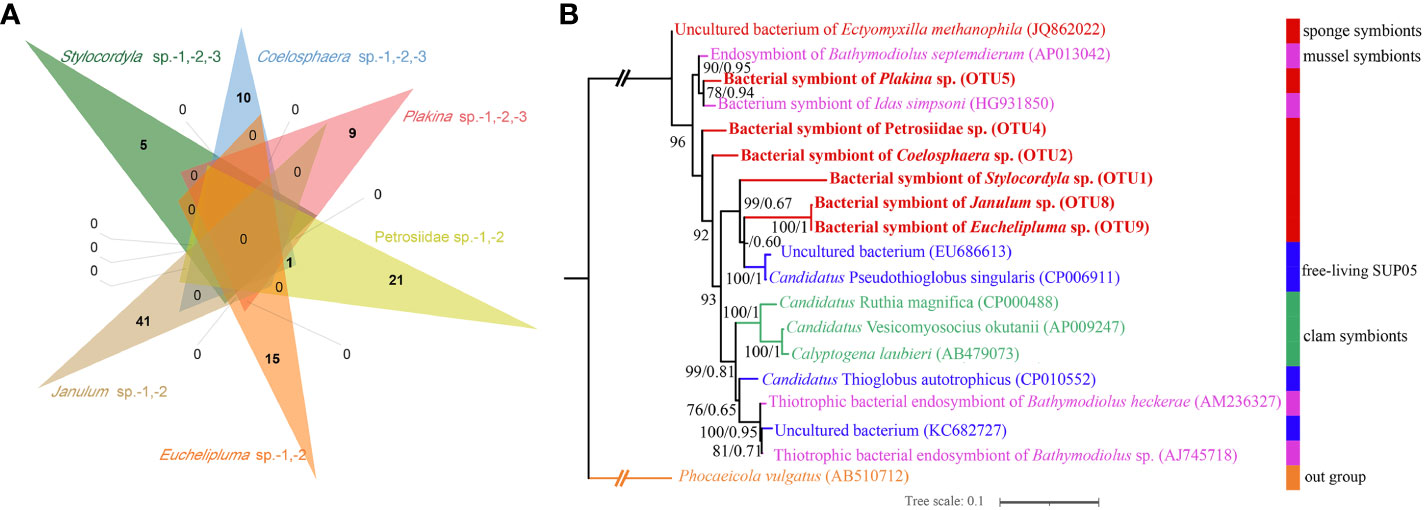
Figure 7 Venn diagram depicting the shared and unique OTUs about SUP05 clade for each sponge species (A), Maximum-likelihood phylogenetic tree based on 16S rRNA sequence data of the dominant sponge symbionts and selected symbiotic and free-living SUP05 bacteria (B). ML analysis bootstrap values (left) and Bayesian posterior probabilities (right) are shown on each branch.
3.7 The methane-oxidizing bacteria in cold-seep sponges
Porifera in chemosynthetic environments, such as hydrothermal vents and cold seeps, have been reported to harbor substantial populations of bacteria capable of sulfide and methane oxidation (Rubin-Blum et al., 2019; Zhou, 2019). OTUs belonging to methane-oxidizing bacteria (MOX) in the family Methylomonadaceae were also present in our cold-seep sponges. Each sponge species possesses specific MOX OTUs in the family Methylomonadaceae, similar to the phylogenetically diverse SUP05 OTUs found in these sponges. Carnivorous sponges of Euchelipluma sp. exhibited the highest diversity of MOXs, while Plakina sp. had the highest abundance of MOXs. Phylogenetic trees based on the predominant MOX OTUs in each sponge species of ML and BI analyses were generally congruent (Figure 8), revealing that these OTUs do not form a single clade or are clustered with any known species. The process of sponge animals acquiring MOX from the environment is hypothesized to have occurred multiple times throughout their evolutionary history.
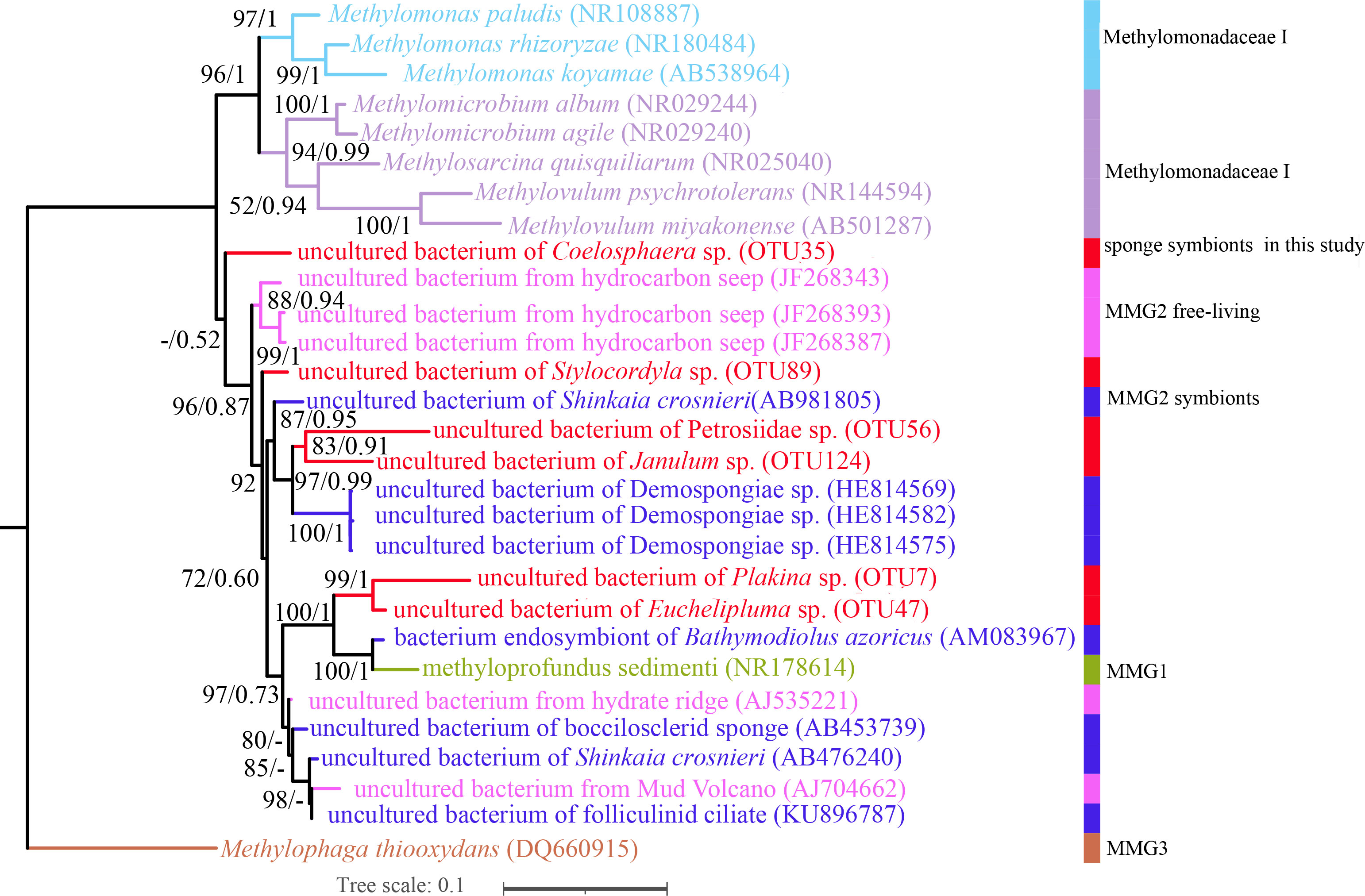
Figure 8 Maximum-likelihood phylogenetic tree based on 16S rRNA sequence data of the dominant sponge symbionts and selected symbiotic and free-living Methylococcaceae. ML analysis bootstrap values (left) and Bayesian posterior probabilities (right) are shown on each branch.
4 Discussion
4.1 Novelty and divergence of bacterial communities in cold-seep sponges
Early studies were mainly focused on sponge-associated microbial communities in shallow-water marine habitats. These studies demonstrated that sponges maintain highly diverse and specific microbial communities in the face of a constant influx of seawater microorganisms due to their filter-feeding activities (Margalef, 1963). The Sponge Microbiome Project (SMP) employed high-throughput sequencing technologies to analyze 81 sponge species comprising 804 samples, primarily collected from shallow-water habitats (Moitinho-Silva et al., 2017; Busch et al., 2022). Limited sampling challenges research on sponges from cold-water and deep-sea environments. The Deep-sea Sponge Microbiome Project (D-SMP), conducted in 2022, aimed to characterize the microbial communities associated with 1,077 sponge samples, predominantly collected from the deep sea, representing 169 sponge species (Busch et al., 2022). Both studies utilized extensive sequencing data to demonstrate the microbial diversity and composition of global sponge microbiomes. The chemoautotrophic environment of the cold seeps in the SCS represents unexplored sponge habitats, with sponge grounds consisting of up to six sponge species rarely reported in deep-sea environments. In this study, we investigated the microbial communities of six cold-seep sponge species in the SCS using nearly full-length sequencing, thereby expanding the datasets of sponge-associated microbial communities.
At high taxonomic levels, such as the phylum, the bacterial compositions of the six sponge species showed similarities to shallow-water demosponges. The most abundant phyla included Proteobacteria (Gamma- and Alpha-), Bacteroidota, and Acidobacteriota. Generally, Gammaproteobacteria were the dominant bacterial communities associated with sponges, irrespective of geographical region or sampling depth (Thomas et al., 2016; Cárdenas et al., 2018; Steinert et al., 2019). Additionally, Plakina sp. and Petrosiidae sp. exhibit a high abundance of Alphaproteobacteria, which are commonly found in shallow-water and other deep-sea sponges. Notably, Cyanobacteria are generally absent or occur in low abundance in deep-sea sponges, attributed to the absence of sunlight in deep-sea environments (Thomas et al., 2016). In contrast, the phylum Poribacteria, which is highly specific to sponges, appears to be less prevalent in deep-sea and cold habitat sponges (Cárdenas et al., 2018; Steinert et al., 2020). Nevertheless, the district chemoautotrophic environment of the cold seep in the SCS could potentially influence the composition of sponge-associated microbial communities. Our analysis revealed that 63.02% of OTUs found in the six cold-seep sponges could be classified at the species level, while the remaining OTUs represent novel taxa. These findings highlight the novelty of sponge-associated microbes in deep-sea sponges that reside in unique environments.
The host-species specificity is a major characteristic of the global sponge microbiomes. The six sponge species in our study exhibit substantial variations in both bacterial diversity and compositions (Figures 3, 4, 6). These results align with previous studies on shallow-water and deep-sea sponges (Moitinho-Silva et al., 2017; Busch et al., 2022), underscoring the existence of distinct microbial communities within each sponge species. This rule also applies to cold-seep sponges in narrow geographical sponge grounds. Analyses of the global sponge microbiome have revealed that the majority of OTUs are specialists (that is, found in only one or a few sponge species), while only a few are truly core (that is, found across many sponge species) (Busch et al., 2022). In our result, 85.73% of all OTUs were found only in one sponge species, indicating their specialization. This divergence is evident in the varying species composition biomarker quantities among the five sponge species (Figure 5). These findings indicate significant differences in bacterial communities among sponges inhabiting the same restricted geographic area. However, due to the inherent randomness and limitations of collecting samples from deep-sea sponge grounds, only a single sponge species from class Homoscleromorpha was collected. Further analyses will be conducted upon supplementing the sample with additional sponge samples from sponge grounds.
Sponges can be broadly classified into two groups: “high microbial abundance (HMA)” and “low microbial abundance (LMA)” (Hentschel et al., 2003). Generally, LMA sponges have fewer microbial species and lower alpha diversity. In our study, the microbial diversity of Plakina sp., Stylocordyla sp., and Coelosphaera sp. was significantly lower than the other three sponges, referring to both diversity indices (Figure 3) and the number of unique OTUs (Figure 5B). However, other direct evidence, such as observations using transmission electron microscopy to detect the presence of microorganisms in the mesohyl matrix, quantitative comparative analyses between fresh samples, and in situ seawater assessments, still need to be comprehensively considered to further determine the HMA and LMA types of sponges.
4.2 The functional adaption of symbionts to cold seep
The SUP05 clade (class: Gammaproteobacteria) is mainly composed of unculturable chemoautotrophic SOBs in marine environments (Anantharaman et al., 2013; Anantharaman et al., 2014). Additionally, these SOBs establish symbiotic relationships with invertebrate animals in deep-sea environments (Kuwahara et al., 2007; Zhou, 2019). As an example, Candidatus Ruthia magnifica, bacteria affiliated with the SUP05 clade, is an obligate chemolithoautotrophic bacterium with a fully sequenced genome (Nelson and Fisher, 1995). Throughout the extended process of symbiosis, SUP05 symbionts have undergone significant genome reduction (Kuwahara et al., 2007), while retaining the complete set of genes necessary for sulfur oxidation, which allows them to obtain energy from environments abundant in reduced sulfur compounds (Ikuta et al., 2015). Existing evidence of strict maternal co-transmission suggests that certain endosymbiotic SUP05 bacteria may have lost their capability to live independently in the marine environment and re-infect invertebrates (Hurtado et al., 2003). Previous studies have reported the presence of sulfur-oxidizing SUP05 strains with high abundance in deep-sea sponges inhabiting asphalt seeps and hydrothermal vents (Tong et al., 2011; Rubin-Blum et al., 2019), but the association of SUP05 symbionts with sponges remains unclear (Kuwahara et al., 2007; Zhou, 2019; Georgieva et al., 2020). The genome of sponge-associated SUP05 was found to contain a variety of genes related to sulfur oxidization, including sat, aprAB, dsrAB, soxA, soxB, soxX, soxY, and soxZ (Zhou, 2019). The genomes possess a complete set of genes necessary for sulfur oxidation, allowing the bacteria to use surrounding reduced sulfur compounds as an energy source (Kuwahara et al., 2007; Newton et al., 2007; Ikuta et al., 2015). The generated energy benefits various symbiotic processes, such as the synthesis of essential amino acids and cofactors, which drives carbon dioxide fixation through the Calvin cycle to fulfill the sponges’ nutritional needs, as well as the uptake and utilization of nitrate produced by the sponge during nitrate respiration(Zhou, 2019).
Our study demonstrates that the microbial communities of cold-seep sponges in the SCS are dominated by sulfur-oxidizing bacteria belonging to the SUP05 group. The predominant SUP05 16S rRNA genes in six sponges exhibited genetic divergence from each other and from other symbiotic and free-living SUP05 members of the SUP05 group (Figure 7), indicating their novelty. In the cold seep environment that we investigated, dark sulfide oxidation appears to serve as the primary source for the sponge microbiome, thus highlighting the potentially significant roles of the SUP05 symbionts.
In cold seep environments, it has been observed that nutritionally symbiotic MOXs constitute a significant proportion (50% to 70%) of the microbiota associated with sponges, consisting of nutritionally symbiotic MOXs (Rubin-Blum et al., 2019). These MOXs utilize methane from seeps to provide the chemosynthetic supply of organic matter to support the host, thereby sustaining the symbiotic system. In our sponge samples, we also found the presence of MOX. Phylogenetic analysis of the 16S rRNA genes indicated that the MOXs associated with the sponges largely belong to the family Methylomonadaceae in the order Methylococcales with some being classified as members of the Marine Methylotrophic Group (MMG) 2 clade (also referred as deep-sea clade 2) (Parks et al., 2018). Pure cultures from the MMG2 clade have not yet been obtained, although three MMG2 genomes have been reported previously (Vekeman et al., 2016; Rubin-Blum et al., 2019). The MMG2 clade is not exclusively associated with sponges, as its 16S rRNA gene clones have also been found in ciliates and the squat lobster Shinkaia crosnieri (Rubin-Blum et al., 2019). MMG2 sequences were also found in certain sponge species. For instance, they were discovered in an unidentified poecilosclerida sponge from seeps in the Gulf of Mexico (Nishijima et al., 2010) and in Cladorhiza methanophila from mud volcanoes off Barbados (Hestetun et al., 2016). These findings suggest that the presence of MMG2 clade bacteria is not restricted to a specific symbiotic relationship but might be more widely distributed in various deep-sea environments and associated with different host organisms. The symbiotic MOXs in sponges assimilate methane through a limited number of essential enzymes, and they exclusively employ the highly efficient Embden-Meyerhof-Parnas (EMP) variant of the ribulose monophosphate (RuMP) cycle to assimilate single carbon compounds (Kalyuzhnaya et al., 2013; Rubin-Blum et al., 2019). Importantly, the symbiotic MOXs in sponges can provide nutrition to their hosts even at lower concentrations of methane, unlike the symbionts found in mussels (Rubin-Blum et al., 2019). Free-living MMG2 bacteria are also commonly found in methane-rich environments, including the sediment and water column of vents, hydrothermal fields, and cold seeps, in addition to their symbiotic relationships with animals (Ruff et al., 2013).
In our study, we observed lower OTU-sharing ratios of MOXs among the six different sponge species. This phenomenon may be attributed to the vertical transmission of symbiotic MOXs from one generation to the next (Rubin-Blum et al., 2019). The six dominant OTUs do not cluster in a single clade and are not clustered together with other known symbionts, indicating their genetic diversity. In future work, more attention should be given to understanding how these symbiotic MOXs adapt to different sponge hosts. In Figure 4B, the analysis of microbial community composition in the 16 samples collected from the six sponge species revealed that Plakina sp. has a higher predominance of MOXs from Methylomonadaceae compared to other sponge species. Plakina sp. stands out among the host sponges as it belongs to the class Homoscleromorpha, distinguishing it from other species. The distribution of MOXs among these sponge species appears to reflect the host-specificity of sponge symbiotic microorganisms. Furthermore, within the three samples of Plakina sp., Plakina sp.-1 exhibits significantly higher MOX enrichment compared to the other two samples. Plakina sp.-1 was collected from the area of gas flares in this cold-seep site, where abundant methane bubbles emerge, while the other two samples were collected from areas farther away from active seepage. Therefore, the distribution of MOXs in Plakina sp., a specific sponge species, is presumed to be influenced by the surrounding methane concentrations. However, in order to understand the impact of class Homoscleromorpha and methane concentration on the content of MOXs in symbiotic microorganisms within sponges, further comparisons should be conducted with more biological samples and environmental parameters collected from the cold-seep site. Furthermore, more attention is needed to investigate this potential pattern.
5 Conclusion
In the present study, we collected six sponge species belonging to two classes from the cold seep sponge ground of the South China Sea (SCS), and we analyzed their bacterial communities using full-length 16S rRNA amplicons. The results revealed significant differences in both microbial diversity and composition among sponge species, highlighting the host-species specificity of the sponge-associated bacterial communities. Potential SOBs within the SUP05 (Thioglobaceae) clade and MOXs in the family Methylomonadaceae were found to occupy important proportions in these sponge microbiomes. These microorganisms likely play important chemosynthetic roles in supporting the thriving of symbionts in the cold seep environment. Phylogenetic analyses indicated that the predominant SOB and MOX OTUs belong to divergent phylotypes, suggesting the novelty of bacterial symbionts associated with each sponge species. Our study provides a preliminary insight into the bacterial communities associated with cold-seep sponges in deep-sea sponge grounds and expands our understanding of the sponge microbiome across diverse environments. Further metagenomic studies could shed light on the functions of these divergent phylotypes, their adaption to the sponge hosts and cold seep environments, and the interaction between sponges and their associated microorganisms in these unique environments.
Data availability statement
Sequences were deposited at NCBI as BioProject with accession ID PRJNA987203.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
YaW, LG, and X-ZL designed and conducted the experiments. YaW, LG, Z-MG, FZ, and L-LF analyzed the data. YaW and LG wrote the manuscript. Z-MG and X-ZL reviewed and edited the manuscript. LG provided initial identification and curation of taxonomic vouchers of sponges. All authors contributed to the article and approved the submitted version.
Funding
Supported by the National Natural Science Foundation of China (No. 42176114) and the Senior User Project of R/V Kexue (No. KEXUE2020GZ01).
Acknowledgments
We would like to express our gratitude to Dr. Xue-Feng Fang, a Ph.D. candidate from the Institute of Oceanology, Chinese Academy of Sciences, for his assistance with the sample collection. We also thank Yong Xu from the Institute of Oceanology, Chinese Academy of Sciences for his guidance in data analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1243952/full#supplementary-material
Supplementary Table 1 | The 18S rRNA gene sequences of six cold-seep sponge species.
Supplementary Table 2 | Number of Pacbio reads assigned to each OTU and the OTU taxonomy details.
References
Anantharaman K., Breier J. A., Sheik C. S., Dick G. J. (2013). Evidence for hydrogen oxidation and metabolic plasticity in widespread deep-sea sulfur-oxidizing bacteria. Proc. Natl. Acad. Sci. 110, 330–335. doi: 10.1073/pnas.1215340110
Anantharaman K., Duhaime M., Breier J., Wendt K., Toner B., Dick G. (2014). Sulfur oxidation genes in diverse deep-sea viruses. Sci. (New. York. N.Y.) 344, 757–760. doi: 10.1126/science.1252229
Arellano S., Lee O., Lafi F., Yang J., Wang Y., Young C., et al. (2012). Deep sequencing of myxilla (Ectyomyxilla) methanophila, an epibiotic sponge on cold-seep tubeworms, reveals methylotrophic, thiotrophic, and putative hydrocarbon-degrading microbial associations. Microbial. Ecol. 65, 450–461. doi: 10.1007/s00248-012-0130-y
Botté E., Nielsen S., Abdul Wahab M. A., Webster J., Robbins S., Thomas T., et al. (2019). Changes in the metabolic potential of the sponge microbiome under ocean acidification. Nat. Commun. 10. doi: 10.1038/s41467-019-12156-y
Busch K., Slaby B., Bach W., Boetius A., Clefsen I., Colaco A., et al. (2022). Biodiversity, environmental drivers, and sustainability of the global deep-sea sponge microbiome. Nat. Commun. 13, 5160. doi: 10.1038/s41467-022-32684-4
Cai R., He W., Liu R., Zhang J., Zhang X., Sun C. (2022). Deep-sea in situ insights into the formation of zero-valent sulfur driven by a bacterial thiosulfate oxidation pathway. mBio 13, e0014322. doi: 10.1128/mbio.00143-22
Cao L., Lian C., Zhang X., Zhang H., Wang H., Zhou L., et al (2021). In situ detection of the fine scale heterogeneity of active cold seep environment of the Formosa ridge, the south China Sea. J. Mar. Syst. 218, 103530. doi: 10.1016/j.jmarsys.2021.103530
Cárdenas C., Gonzalez M., Font A., Hestetun J., Hajdu E., Trefault N., et al. (2018). High similarity in the microbiota of cold-water sponges of the Genus Mycale from two different geographical areas. PeerJ. 6, e4935. doi: 10.7717/peerj.4935
Chen D., Huang Y., Yuan X., Cathles L. (2005). Seep carbonates and preserved methane oxidizing archaea and sulfate reducing bacteria fossils suggest recent gas venting on the seafloor in the Northeastern South China Sea. Mar. Petroleum. Geol. 22, 613–621. doi: 10.1016/j.marpetgeo.2005.05.002
Cleary D., Becking L., De Voogd N., Pires A., Polónia A., Egas C., et al. (2013). Habitat- and host-related variation in sponge bacterial symbiont communities in Indonesian waters. FEMS Microbiol. Ecol. 85 (3), 465–482. doi: 10.1111/1574-6941.12135
De Goeij J. M., Van Oevelen D., Vermeij M. J. A., Osinga R., Middelburg J. J., De Goeij A. F. P. M., et al. (2013). Surviving in a marine desert: the sponge loop retains resources within coral reefs. Science 342, 108–110. doi: 10.1126/science.1241981
Dubilier N., Bergin C., Lott C. (2008). Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat. Rev. Microbiol. 6, 725–740. doi: 10.1038/nrmicro1992
Easson C., Thacker R. (2014). Phylogenetic signal in the community structure of host-specific microbiomes of tropical marine sponges. Front. Microbiol. 5, 1–11. doi: 10.3389/fmicb.2014.00532
Erwin P., Olson J., Thacker R. (2011). Phylogenetic diversity, host-specificity and community profiling of sponge-associated bacteria in the northern gulf of Mexico. PloS One 6, e26806. doi: 10.1371/journal.pone.0026806
Fan L., Reynolds D., Liu M., Stark M., Kjelleberg S., Webster N., et al. (2012). Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc. Natl. Acad. Sci. U.S.A 109, E1878–E1887. doi: 10.1073/pnas.1203287109
Feng D., Qiu J.-W., Hu Y., Peckmann J., Guan H., Tong H., et al. (2018). Cold seep systems in the South China Sea: An overview. J. Asian Earth Sci. 168, 3–16. doi: 10.1016/j.jseaes.2018.09.021
Freeman C., Thacker R. (2011). Complex interactions between marine sponges and their symbiotic microbial communities. Limnol. Oceanogr. 56, 1577–1586. doi: 10.4319/lo.2011.56.5.1577
Georgieva M., Taboada S., Riesgo A., Diez-Vives C., De Leo F., Jeffreys R., et al. (2020). Evidence of vent-adaptation in sponges living at the periphery of hydrothermal vent environments: ecological and evolutionary implications. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.01636
Giles E., Kamke J., Moitinho-Silva L., Taylor M., Hentschel U., Ravasi T., et al. (2013). Bacterial community profiles in low microbial abundance sponges. FEMS Microbiol. Ecol. 83 (1), 232–241. doi: 10.1111/j.1574-6941.2012.01467.x
Hardoim C., Esteves A., Pires F., Gonçalves J., Cox C., Xavier J., et al. (2012). Phylogenetically and spatially close marine sponges harbour divergent bacterial communities. PloS One 7, e53029. doi: 10.1371/journal.pone.0053029
Hentschel U., Fieseler L., Wehrl A., Gernert C., Steinert M., Hacker J., et al. (2003). Microbial diversity of marine sponges. Sponges.: (Porifera). 37, 59–88. doi: 10.1007/978-3-642-55519-0_3
Hestetun J. T., Dahle H., Jørgensen S. L., Olsen B. R., Rapp H. T. (2016). The microbiome and occurrence of methanotrophy in carnivorous sponges. Front. Microbiol. 7, 1781. doi: 10.3389/fmicb.2016.01781
Hurtado L., Mateos M., Lutz R., Vrijenhoek R. (2003). Coupling of bacterial endosymbiont and host mitochondrial genomes in the hydrothermal vent clam calyptogena magnifica. Appl. Environ. Microbiol. 69, 2058–2064. doi: 10.1128/AEM.69.4.2058-2064.2003
Ikuta T., Takaki Y., Nagai Y., Shimamura S., Tsuda M., Kawagucci S., et al. (2015). Heterogeneous composition of key metabolic gene clusters in a vent mussel symbiont population. ISME. J. 10, 990–1001. doi: 10.1038/ismej.2015.176
Jørgensen B., Boetius A. (2007). Feast and famine-microbial life in the deep-sea bed. Nat. Rev. Microbiol. 5, 770–781. doi: 10.1038/nrmicro1745
Kalyuzhnaya M., Yang S., Rozova O., Smalley N., Clubb J., Lamb A., et al. (2013). Highly efficient methane biocatalysis revealed in methanotrophic bacterium. Nat. Commun. 4, 2785. doi: 10.1038/ncomms3785
Kuwahara H., Yoshida T., Takaki Y., Shimamura S., Nishi S., Harada M., et al. (2007). Reduced genome of the thioautotrophic intracellular symbiont in a deep-sea clam, calyptogena okutanii. Curr. Biol.: CB. 17, 881–886. doi: 10.1016/j.cub.2007.04.039
Li C. W. (1998). Precambrian sponges with cellular structures (vol 280, pg 882, 1998). Science 285, 838–838. doi: 10.1126/science.279.5352.879
Li C., Ijaz M., Ahmad M., Zou X., Hussain M., Zhang M., et al. (2018). Citation: beef, casein, and soy proteins differentially affect lipid metabolism, triglycerides accumulation and gut microbiota of high-fat diet-fed C57BL/6J mice. Front. Microbiol. 9, 2200. doi: 10.3389/fmicb.2018.02200
Li Z.-Y., Wang Y., He L.-M., Zheng H.-J. (2015). CORRIGENDUM: Metabolic profiles of prokaryotic and eukaryotic communities in deep-sea sponge Neamphius huxleyi indicated by metagenomics. Sci. Rep. 5, 8176. doi: 10.1038/srep08176
Lozupone C., Knight R. (2006). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005
Lurgi M., Thomas T., Wemheuer B., Webster N., Montoya J. (2019). Modularity and predicted functions of the global sponge-microbiome network. Nat. Commun. 10. doi: 10.1038/s41467-019-08925-4
Maldonado M., Aguilar R., Bannister R. J., James J., Conway K. W., Dayton P. K., et al. (2017). “Sponge grounds as key marine habitats: A synthetic review of types, structure, functional roles, and conservation concerns” in Marine animal forests: the ecology of benthic biodiversity hotspots. Ed. Rossi S. (Switzerland: Springer International Publishing), 145–183. doi: 10.1007/978-3-319-21012-4
Meyer B., Kuever J. (2008). Phylogenetic Diversity and Spatial Distribution of the Microbial Community Associated with the Caribbean Deep-water Sponge Polymastia cf. corticata by 16S rRNA, aprA, and amoA Gene Analysis. Microbial. Ecol. 56, 306–321. doi: 10.1007/s00248-007-9348-5
Moitinho-Silva L., Nielsen S., Amir A., Gonzalez A., Ackermann G., Cerrano C., et al. (2017). The sponge microbiome project. GigaScience 6, 1–7. doi: 10.1093/gigascience/gix077
Margalef R. (1963). On certain unifying principles in ecology. Am. Nat. 97, 357–374. doi: 10.1086/282286
Nelson D., Fisher C. R. (1995). Chemoautotrophic and methanotrophic endosymbiotic bacteria at deep-sea vents and seeps. Microbiol. Deep-sea. Hydrothermal. Vents. (CRC Press), 125–167.
Newton I., Woyke T., Auchtung T. A., Dilly G. F., Dutton R., Fisher M. C., et al. (2007). The calyptogena magnifica chemoautotrophic symbiont genome. Sci. (New. York. N.Y.) 315, 998–1000. doi: 10.1126/science.1138438
Nishijima M., Lindsay D., Hata J., Nakamura A., Kasai H., Ise Y., et al. (2010). Association of thioautotrophic bacteria with deep-sea sponges. Mar. Biotechnol. (New. York. N.Y.) 12, 253–260. doi: 10.1007/s10126-009-9253-7
Osinga R., Armstrong E., Burgess J. G., Hoffmann F., Reitner J., Schumann-Kindel G. (2001). Sponge-microbe associations and their importance for sponge bioprocess engineering. Hydrobiologia 461, 55–62. doi: 10.1023/A:1012717200362
Parks D., ChuvoChina M., Waite D., Rinke C., Skarshewski A., Chaumeil P.-A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Paull C., Hecker B., Commeau R., Freeman-Lynde R., Neumann C., Corso W., et al. (1984). Biological communities at the florida escarpment resemble hydrothermal vent taxa. Sci. (New. York. N.Y.) 226, 965–967. doi: 10.1126/science.226.4677.965
Pita L., Rix L., Slaby B. M., Franke A., Hentschel U. (2018). The sponge holobiont in a changing ocean: from microbes to ecosystems. Microbiome 6, 46. doi: 10.1186/s40168-018-0428-1
Pita L., Turon X., Lopez-Legentil S., Erwin P. (2013). Host rules: spatial stability of bacterial communities associated with marine sponges (Ircinia spp.) in the Western Mediterranean Sea. FEMS Microbiol. Ecol. 86 (2), 268–276. doi: 10.1111/1574-6941.12159
Radax R., Rattei T., Lanzén A., Bayer C., Rapp H., Urich T., et al. (2012). Metatranscriptomics of the marine sponge Geodia barretti: Tackling phylogeny and function of its microbial community. Environ. Microbiol. 14, 1308–1324. doi: 10.1111/j.1462-2920.2012.02714.x
Ramirez-Llodra E., Brandt A., Danovaro R., E, E., German C., Levin L., Martinez Arbizu P., et al. (2010). Deep, diverse and definitely different: Unique attributes of the world’s largest ecosystem. Biogeosci. Discussions. 7, 2851–2899. doi: 10.5194/bg-7-2851-2010
Reitner J., Wörheide G. (2002). Non-lithistid fossil demospongiae? Origins of theirPalaeobiodiversity and highlights in history of preservation. Eds. Hooper J. N. A., Van Soest R. W. M., Willenz P. (Systema Porifera. Springer US), 52–68.
Reveillaud J., Maignien L., Eren A., Huber J., Apprill A., Sogin M., et al. (2014). Host-specificity among abundant and rare taxa in the sponge microbiome. ISME. J. 8 (6), 1198–1209. doi: 10.1038/ismej.2013.227
Rubin-Blum M., Chakkiath A. C., Sayavedra L., Martínez-Pérez C., Birgel P. J., Wu Y. -C, et al. (2019). Fueled by methane: deep-sea sponges from asphalt seeps gain their nutrition from methane-oxidizing symbionts. ISME. J. 13, 1209–1225. doi: 10.1038/s41396-019-0346-7
Ruff S. E., Arnds J., Knittel K., Amann R., Wegener G., Ramette A., et al. (2013). Microbial communities of deep-sea methane seeps at Hikurangi continental margin (New Zealand). PloS One 8, e72627. doi: 10.1371/journal.pone.0072627
Schläppy M.-L., Schöttner S., Lavik G., Kuypers M., De Beer D., Hoffmann F. (2010). Evidence of nitrification and denitrification in high and low microbial abundance sponges. Mar. Biol. 157, 593–602. doi: 10.1007/s00227-009-1344-5
Schloss P., Westcott S., Ryabin T., Hall J., Hartmann M., Hollister E., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schmitt S., Tsai P., Bell J., Fromont J., Ilan M., Lindquist N., et al. (2012). Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. Isme. J. 6, 564–576. doi: 10.1038/ismej.2011.116
Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. doi: 10.1186/gb-2011-12-6-r60
Smedile F., Messina E., Cono V., Tsoy O., Monticelli L., Borghini M., et al. (2012). Metagenomic analysis of hadopelagic microbial assemblages thriving at the deepest part of Mediterranean Sea, Matapan-Vavilov Deep. Environ. Microbiol. 15, 167–182. doi: 10.1111/j.1462-2920.2012.02827.x
Sogin E., Kleiner M., Borowski C., Gruber-Vodicka H., Dubilier N. (2021). Life in the dark: phylogenetic and physiological diversity of chemosynthetic symbioses. Annu. Rev. Microbiol. 75, 695–718. doi: 10.1146/annurev-micro-051021-123130
Steinert G., Busch K., Bayer K., Khodami S., Martinez Arbizu P., Kelly M., et al. (2020). Compositional and quantitative insights into bacterial and archaeal communities of south pacific deep-sea sponges (Demospongiae and hexactinellida). Front. Microbiol. 11, 716. doi: 10.3389/fmicb.2020.00716
Steinert G., Wemheuer B., Janussen D., Erpenbeck D., Daniel R., Simon M., et al. (2019). Prokaryotic diversity and community patterns in antarctic continental shelf sponges. Front. Mar. Sci. 6, 297. doi: 10.3389/fmars.2019.00297
Taylor M. W., Radax R., Steger D., Wagner M. (2007). Sponge-associated microorganisms: Evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 71, 295–29+. doi: 10.1128/MMBR.00040-06
Thomas T., Moitinho-Silva L., Lurgi M., Björk J., Easson C., Astudillo-García C., et al. (2016). Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 7, 11870. doi: 10.1038/ncomms11870
Tong H., Feng D., Cheng H., Yang S., Wang H., Min A., et al. (2011). Authigenic carbonates from cold seeps of the South China sea: new insights into fluid sources, formation condition and geochronology. Mar. Pet. Geol. 43, 260–271. doi: 10.1371/journal.pone.0035105
Van Soest R., Boury-Esnault N., Vacelet J., Dohrmann M., Erpenbeck D., De Voogd N., et al. (2012). Global diversity of sponges (Porifera). PloS One 7, e35105. doi: 10.1371/journal.pone.0035105
Vekeman B., Speth D., Wille J., Cremers G., De Vos P., Op Den Camp H., et al. (2016). Genome characteristics of two novel type I methanotrophs enriched from north sea sediments containing exclusively a lanthanide-dependent XoxF5-type methanol dehydrogenase. Microbial. Ecol. 72, 503–509. doi: 10.1007/s00248-016-0808-7
Wang B., Du Z., Luan Z., Zhang X., Wang M., Wang X., et al. (2021). Seabed features associated with cold seep activity at the Formosa Ridge, South China Sea: Integrated application of high-resolution acoustic data and photomosaic images. Deep. Sea. Res. Part I.: Oceanogr. Res. Papers. 177, 103622. doi: 10.1016/j.dsr.2021.103622
Webster N. S., Taylor M. W., Behnam F., Luecker S., Rattei T., Whalan S., et al. (2010). Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ. Microbiol. 12, 2070–2082. doi: 10.1111/j.1462-2920.2009.02065.x
Webster N. S., Thomas T. (2016). The sponge hologenome. Mbio 7, e00135–16. doi: 10.1128/mBio.00135-16
Weisz J. B., Hentschel U., Lindquist N., Martens C. S. (2007). Linking abundance and diversity of sponge-associated microbial communities to metabolic differences in host sponges. Mar. Biol. 152, 475–483. doi: 10.1007/s00227-007-0708-y
Zhang D., Gao F., Jakovlić I., Zou H., Zhang J., Li W., et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Keywords: symbiont, host-species specificifity, sponge ground, SUP05 clade, Methylomonadaceae, 16S rRNA amplicons
Citation: Wang Y, Gong L, Gao Z, Wang Y, Zhao F, Fu L and Li X (2023) Host-specific bacterial communities associated with six cold-seep sponge species in the South China Sea. Front. Mar. Sci. 10:1243952. doi: 10.3389/fmars.2023.1243952
Received: 21 June 2023; Accepted: 26 July 2023;
Published: 21 August 2023.
Edited by:
Conghui Liu, Chinese Academy of Agricultural Sciences, ChinaCopyright © 2023 Wang, Gong, Gao, Wang, Zhao, Fu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinzheng Li, bGl4emhAcWRpby5hYy5jbg==; Zhaoming Gao, Z2Fvem1AaWRzc2UuYWMuY24=
†These authors have contributed equally to this work and share first authorship