 Ro Allen
Ro Allen Kimberley E. Bird
Kimberley E. Bird J. Colin Murrell
J. Colin Murrell Michael Cunliffe
Michael Cunliffe
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 15 August 2023
Sec. Ocean Observation
Volume 10 - 2023 | https://doi.org/10.3389/fmars.2023.1241333
This article is part of the Research TopicThe Atlantic Meridional Transect Programme (1995-2023)View all 12 articles
The activities of bacterioplankton sustain open ocean biogeochemical and ecological processes, however, little is known about the activity of specific bacterioplankton, especially related to their biogeography across oceanic scales. The Atlantic is the second largest of the world’s oceans and has an essential role in the global carbon cycle. Here, we show congruence in the structure of 16S rRNA and 16S rRNA gene derived bacterioplankton communities throughout the Atlantic Ocean from temperate to tropical regions. We used 16S rRNA:16S rRNA gene ratios as a phylogenetically resolved proxy for potential activity, demonstrating ocean-scale patterns of putative oligotrophy and copiotrophy in major bacterioplankton groups, with spatial niche partitioning being evident at single-nucleotide resolution within some groups, including the Flavobacteria and SAR86. This study examines the potential structure of the active microbiome of the Atlantic Ocean, providing novel insights into the ecology and life history strategies of both well-known and currently understudied bacterioplankton taxa.
Bacterioplankton regulate biogeochemical and ecological processes in the oceans at local and global scales (Falkowski et al., 2008). For example, approximately 50% of global primary production occurs in the marine environment (Field et al., 1998), a large fraction of which is converted to dissolved organic matter (DOM) that is processed by heterotrophic bacterioplankton through the microbial loop (Azam and Malfatti, 2007). Important ecological interactions in planktonic ecosystems depend on the activity of bacterioplankton, including the production and exchange of metabolites (Kazamia et al., 2016), growth inhibiting compounds (Van Tol et al., 2017), and the provision of ‘public-good’ functions (Reintjes et al., 2019). Despite their established functional importance, the diversity and distribution of active open ocean bacterioplankton remain poorly understood.
High-throughput sequencing of 16S rRNA genes amplified from genomic DNA (gDNA) is a standard approach to survey the diversity and distribution of bacterioplankton in the marine environment (Sunagawa et al., 2015). However, in addition to metabolically-active cells, 16S rRNA gene surveys capture dormant cells (i.e. metabolically-inactive), dead cells, and cell-free DNA. A large proportion of bacterioplankton are dormant in oceanic ecosystems (Lennon and Jones, 2011), ranging from ~50–95% of cells in oligotrophic regions (Del Giorgio and Scarborough, 1995; Longnecker et al., 2010; Alonso-Sáez et al., 2012), and cell-free DNA can accumulate in substantial concentrations in seawater, despite rapid turnover rates (Taylor et al., 2018). Consequently, the recovery of dormant cells, dead cells, and cell-free DNA from 16S rRNA gene surveys can mask important patterns in the diversity and distribution of the active bacterioplankton that underpin biogeochemical and ecological processes.
High-throughput sequencing of complementary DNA (cDNA) synthesised from 16S rRNA is an approach used to characterize the active components of bacterial communities, whilst minimizing the influence of dormant cells, dead cells, and cell-free DNA as outlined above (Bowsher et al., 2019). While dormant cells still contain some ribosomes (Chambon et al., 1968; Sukenik et al., 2012), 16S rRNA concentrations generally correlate with growth rates in cultured marine bacteria (Kemp et al., 1993; Kerkhof and Ward, 1993; Worden and Binder, 2003; Lin et al., 2013; Salter et al., 2015). However, the relationship between 16S rRNA and growth rate is often non-linear and can differ substantially between taxa (Kerkhof and Kemp, 1999; Blazewicz et al., 2013; Lankiewicz et al., 2016). Despite these caveats, 16S rRNA surveys can provide a more refined view of active bacterial communities than DNA-based 16S rRNA gene surveys. Furthermore, the ratio of 16S rRNA to 16S rRNA genes (hereafter 16S rRNA:16S rRNA gene ratios) can act as an in situ proxy for the protein synthesis potential (potential activity) of specific bacterial populations (Jones and Lennon, 2010; Campbell et al., 2011).
In the South China Sea, cDNA derived bacterioplankton communities display stronger coupling to prevailing environmental conditions than gDNA derived communities (Zhang et al., 2014). This suggests that 16S rRNA surveys may reduce stochasticity introduced by dormant cells, dead cells, and cell-free DNA, to more accurately resolve the relationships between bacterioplankton communities and their environment. In coastal waters, 16S rRNA:16S rRNA gene ratios have been used to demonstrate that large proportions of bacterioplankton operational taxonomic units (OTUs) cycle between states of high and low potential activity (Campbell et al., 2011). Whilst at the Hawaii Ocean Time-Series station ALOHA, 16S rRNA:16S rRNA gene ratios indicated that some rare populations display high potential activity, suggesting that they contribute disproportionately to biogeochemical processes relative to their abundance (Hunt et al., 2013). Importantly, these studies were focused on local or regional environments, and employed 97% similarity clustering of OTUs which is does not resolve ecologically relevant microdiversity within bacterioplankton communities (Needham et al., 2017). The biogeographic relationship between active and total bacterioplankton communities, and the potential activity of specific bacterioplankton populations at high phylogenetic resolution, remain poorly understood across oceanic basin.
The Atlantic is the second largest oceanic basin on Earth, and plays a major role in the global carbon cycle (Hoppe et al., 2002; Tilstone et al., 2017). The Atlantic is a net sink for atmospheric CO2 and is responsible for the drawdown of approximately 0.5 Pg C yr−1 (Landschützer et al., 2014). The biogeochemical function of the Atlantic Ocean is primarily modulated by the balance between phytoplankton primary production and the metabolic activity of heterotrophic bacterioplankton (Hoppe et al., 2002). Current understanding of the structure and function of Atlantic Ocean bacterioplankton communities is primarily founded on DNA-based 16S rRNA gene surveys (Milici et al., 2016a; Milici et al., 2016b; Logares et al., 2020; Bolaños et al., 2021) and in situ estimates of secondary production and respiration (Hoppe et al., 2002; García-Martín et al., 2017). The biogeography of potentially active bacterioplankton is poorly understood across the Atlantic, limiting efforts to comprehensively link bacterioplankton community structure and function across this regionally and globally important ocean.
Here, we present an ocean-scale survey of cDNA and gDNA derived bacterioplankton communities, from samples collected across three variable depths dependent on light levels (97%, 55% and 1% photosynthetically active radiation; PAR) throughout the Atlantic Ocean during the Austral spring. We show congruence in the structure of cDNA and gDNA derived communities across oceanic scales, which was greatest in the nutrient-rich waters of the Southwest Atlantic Shelves where cDNA and gDNA derived bacterioplankton diversity also converge. We then explore the use 16S rRNA:16S rRNA gene ratios to examine patterns in the protein synthesis potential (potential activity) of bacterioplankton groups and populations through space. Based on 16S rRNA:16S rRNA gene ratios, we speculate on possible signatures of putative oligotrophy and copiotrophy in biogeochemically important bacterioplankton groups, and propose how spatial niche partitioning underlies broad-scale patterns in potential activity and distribution.
Seawater sampling was conducted between 18th September and 4th November 2015 aboard the RRS James Clark Ross during the Atlantic Meridional Transect (AMT) research voyage from the UK to the Falkland Islands (AMT25/JR15001). Forty eight seawater samples were collected from 16 stations spanning 8 oceanographic provinces, ranging from the North Atlantic Drift to the South Subtropical Convergence and Southwest Atlantic Shelves (Reygondeau et al., 2013) (Figure 1A; Supplementary Figure 1). At each station, 9 litre seawater samples were collected from three light-dependent depths (97%, 55%, and 1% PAR) using Niskin bottles attached to a CTD rosette system. Seawater samples were immediately filtered through 0.2 µm cellulose nitrate filters and transferred to sterile 2 ml microcentrifuge tubes containing 1 ml of RNAlater (Merck, Germany) before storage at −80°C. Filtration took on average 2hr 22 min (range 1hr11 to 3hr11).
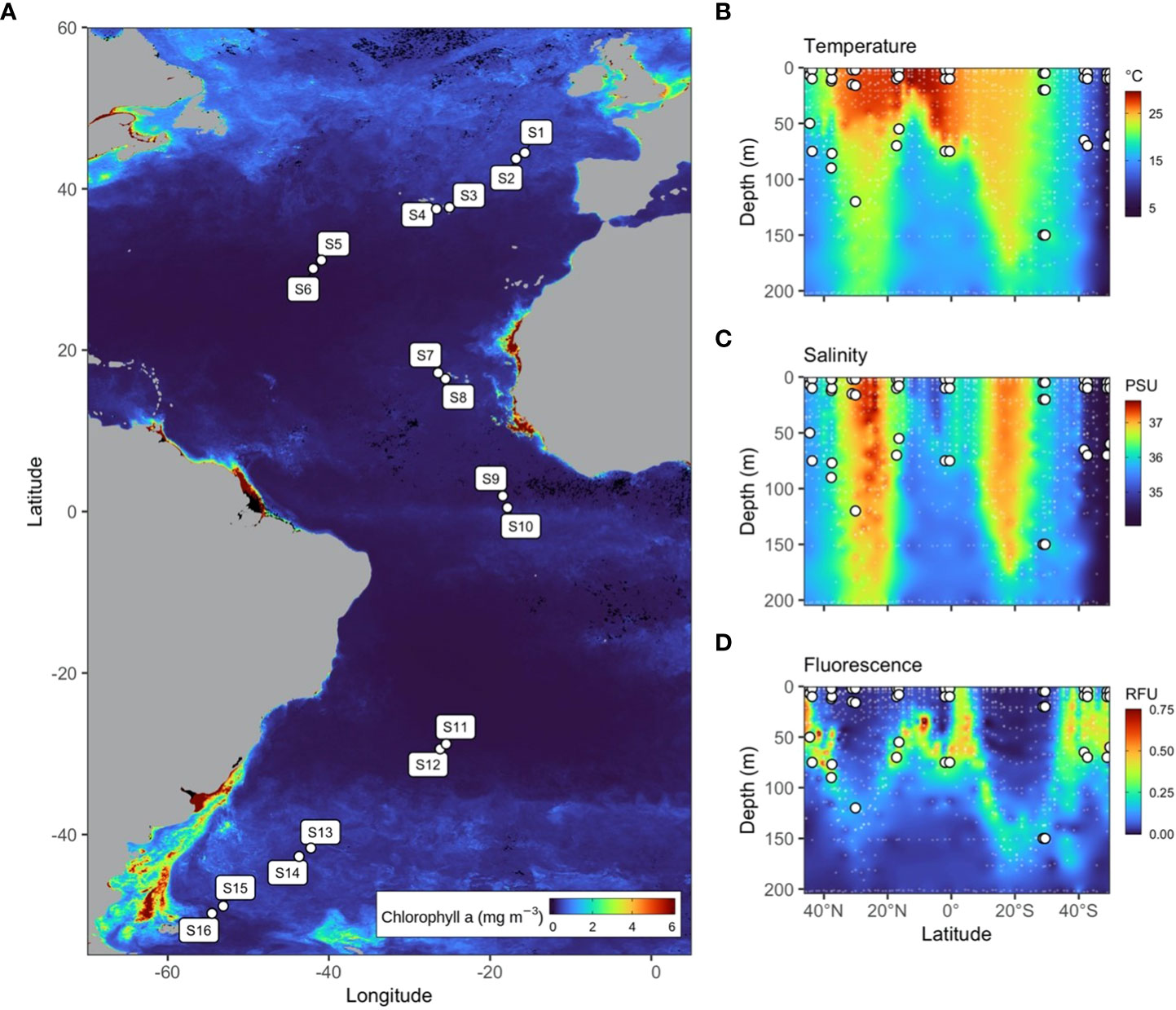
Figure 1 (A) Map of 16 stations sampled during the AMT25 latitudinal transect of the Atlantic Ocean during the Austral spring of 2015. Colour represents MODIS-Aqua satellite-derived chlorophyll-a concentration averaged from 21/09/2015 to 21/12/2015 at 4 km resolution. (B–D) Temperature, salinity, and fluorescence depth profiles throughout the Atlantic Ocean measured using the Sea-Bird SBE 911plus CTD and Aquatracka III Fluorometer, respectively.
Depth profiles for temperature (Figure 1B) and salinity (Figure 1C) were measured using a Sea-Bird SBE 911 plus CTD (Sea-Bird Scientific, USA), and dissolved oxygen was measured using a Sea-Bird SBE 43 dissolved oxygen sensor. Fluorescence (Figure 1D) was measured using an Aquatracka III Fluorometer (Chelsea Technologies, United Kingdom) and PAR was measured using a QCD-905L sensor (Biospherical Instruments, USA). Inorganic nutrient (nitrate NO3−, nitrite NO2−, phosphate PO43− and silicate SiO2) concentrations were measured onboard from discrete water samples using an AutoAnalyzer 3 (Bran+Luebbe, Germany) according to GO-SHIP protocols and determined according to standard analytical techniques (Hydes et al., 2010).
Filters were thawed on ice and transferred to sterile 2 ml FastPrep Lysing Matrix B tubes (MP Biomedicals, USA) before being snap frozen in liquid nitrogen and fragmented using a sterile pellet pestle. GTC lysis buffer (700 µl) (Omega Bio-Tek, USA) was added to the filters followed by further mechanical disruption using a Mini-BeadBeater-8 (BioSpec, USA) for 3 minutes at maximum speed. RNA and DNA were co-extracted from the lysate using the E.Z.N.A. DNA/RNA isolation kit (Omega Bio-Tek, USA) according to the manufacturer’s instructions, including an on-column DNase digestion with RNase-Free DNase (Omega Bio-Tek, USA). cDNA was synthesised from 200 ng of extracted RNA using the Omniscript Reverse Transcription kit (Qiagen, Germany) according to the manufacturer’s instructions, with the primer PROK1492R (GGWTACCTTGTTACGACTT) (Suzuki et al., 2000).
Sequencing libraries were constructed from gDNA and the raised cDNA according to protocols described in Comeau et al. (2017). In brief, the V4–V5 regions of the 16S rRNA gene were PCR amplified in duplicate using the primers 515F-Y (GTGYCAGCMGCCGCGGTAA) and 926R (CCGYCAATTYMTTTRAGTTT) (Parada et al., 2016) modified to include Illumina Nextera adaptors and indices. Pooled duplicate PCR products were purified and normalised in one-step using the SequalPrep 96-well plate kit (Invitrogen, USA) according to manufacturer’s instructions and then pooled to generate a single library, which was quantified using the Qubit dsDNA High Sensitivity Assay (ThermoFisher Scientific, USA). Sequencing was performed on the Illumina MiSeq platform using the V3 reagent kit, yielding 2 × 300 bp paired-end reads (Illumina, USA).
A total of 7,997,448 paired-end reads were recovered from 96 samples (48 cDNA, 48 gDNA) and were processed according to the Bioconductor workflow (Callahan et al., 2016; Bioconductor version 3.11). Primer sequences were trimmed (forward reads: position 19, reverse reads: position 20), low-quality bases were truncated (forward reads: position 270, reverse reads: position 210), and reads with greater than 2 expected errors were removed, using the ‘filterandtrim’ function in the R package DADA2 (Callahan et al., 2016; DADA2 version 1.18). The DADA2 algorithm was used to resolve amplicon sequence variants (ASVs) at single-nucleotide resolution. Paired-end reads were merged using the ‘mergePairs’ function, and chimeras were removed using the ‘removeBimeraDenovo’ function in DADA2. Taxonomy was assigned according to the SILVA version 132 database (Quast et al., 2013) using the RDP naïve Bayseian classifier (Wang et al., 2007). Read count, taxonomic and sample data, were assembled as a single object in the R package phyloseq (McMurdie and Holmes, 2013). ASVs not classified as bacteria were excluded. Samples were rarefied to a depth of 15,973 reads, resulting in the exclusion of two samples (Station 5, 1% PAR, cDNA and gDNA). ASVs which did not account for a minimum of 0.01% of reads in at least one sample were excluded from the total dataset prior to downstream analysis.
The effect of latitude (continuous), light-dependent depth (categorical: 1%, 55%, or 97% PAR), and nucleic acid type (categorical: cDNA or gDNA) on bacterioplankton community composition was tested using a permutational multivariate analysis of variance (PERMANOVA) (Anderson, 2001) based on Bray-Curtis dissimilarity, implemented through the R package vegan (Oksanen et al., 2014). Principal coordinates analysis (PCoA) based on Bray-Curtis dissimilarity was then conducted for cDNA and gDNA communities independently, to visualize the structuring of these communities across latitudes and light-dependent depths. To examine congruence in potentially active and total community structure throughout the Atlantic Ocean, the first two principal coordinates were extracted from each PCoA and subjected to Procrustes analysis (Peres-Neto and Jackson, 2001). Procrustes analysis is a form of multivariate correlation analysis that aims to minimize the sum of squared deviations between samples in two multivariate matrices, through translation, rotation, and dilation of one matrix to match the other. Significance testing was performed using a procrustean randomization test (PROTEST) (Peres-Neto and Jackson, 2001). Procrustes residuals were then extracted and used to examine the effect of latitude and light-dependent depth on the congruence between communities using a generalised additive model (GAM) implemented using the R package mgcv (Wood, 2012) and ANOVA with Tukey’s HSD, respectively. Chao1 richness (Chao, 1984) and Pielou’s evenness (Pielou, 1966) were calculated as components of alpha diversity. The effect of light-dependent depth and nucleic acid type on richness and evenness was compared using a two-way ANOVA with Tukey’s HSD. The relationship between cDNA and gDNA richness and evenness was further examined using a linear model.
As a proxy for in situ potential activity (that is, protein synthesis potential), 16S rRNA:16S rRNA gene ratios were calculated by dividing the cDNA read count by corresponding gDNA read count for each ASV in each sample (Bowsher et al., 2019). For a given ASV in each sample, if the cDNA read count was equal to or greater than 1, but the gDNA read count was 0, 1 was added to gDNA read count prior to ratio calculation. Based on 16S rRNA:16S rRNA gene ratios, trends in the potential activity of biogeochemically and ecologically significant bacterioplankton groups, selected to represent a range from known copiotrophs (e.g. Flavobacteriaceae) to known oligotrophs (e.g. SAR11) and taxa with understudied life history strategies (e.g. SAR86), are described in detail. All statistical analyses were performed in R (version 3.3.6) (R-Core-Team, 2017). Maximum likelihood trees were constructed in RAxML using the GTRGAMMA model (Stamatakis, 2014), based on 16S ASV sequences.
cDNA and gDNA derived bacterioplankton community composition was surveyed at sixteen stations across three light-dependent depths throughout the Atlantic Ocean during the Austral spring 2015 (Figure 1A) by sequencing cDNA synthesised from 16S rRNA, and the 16S rRNA gene, respectively. Bacterioplankton community composition significantly differed between nucleic acid types (i.e. cDNA versus gDNA; PERMANOVA; R2 = 0.05, p < 0.001), latitudes (R2 = 0.13, p < 0.001), and light-dependent depths (R2 = 0.09, p < 0.001) throughout the Atlantic Ocean, with no significant interaction between these variables (p > 0.05). Independent principal coordinates analyses for cDNA and gDNA communities revealed distinct clustering according to latitude and light-dependent depth (Figures 2A, B). Northern temperate regions (S1–S2), southern temperate regions (S13–S16), the fringe of the North Atlantic Gyre and South Atlantic Gyre (S3–S4 and S11–S12), and the centre of the North Atlantic Gyre and tropical North Atlantic (S5–S10) each formed distinct clusters (Figures 2A, B), reflecting the different environmental regimes of these provinces (Figure 1A). In stratified waters, bacterioplankton communities at 1% PAR were distinct from those at 55% PAR and 97% PAR, and displayed less variation across the transect (S3–S12). In temperate regions where 1% PAR fell within the mixed layer, this trend was less pronounced (S1–S2, S13–S16; Figures 2A, B).
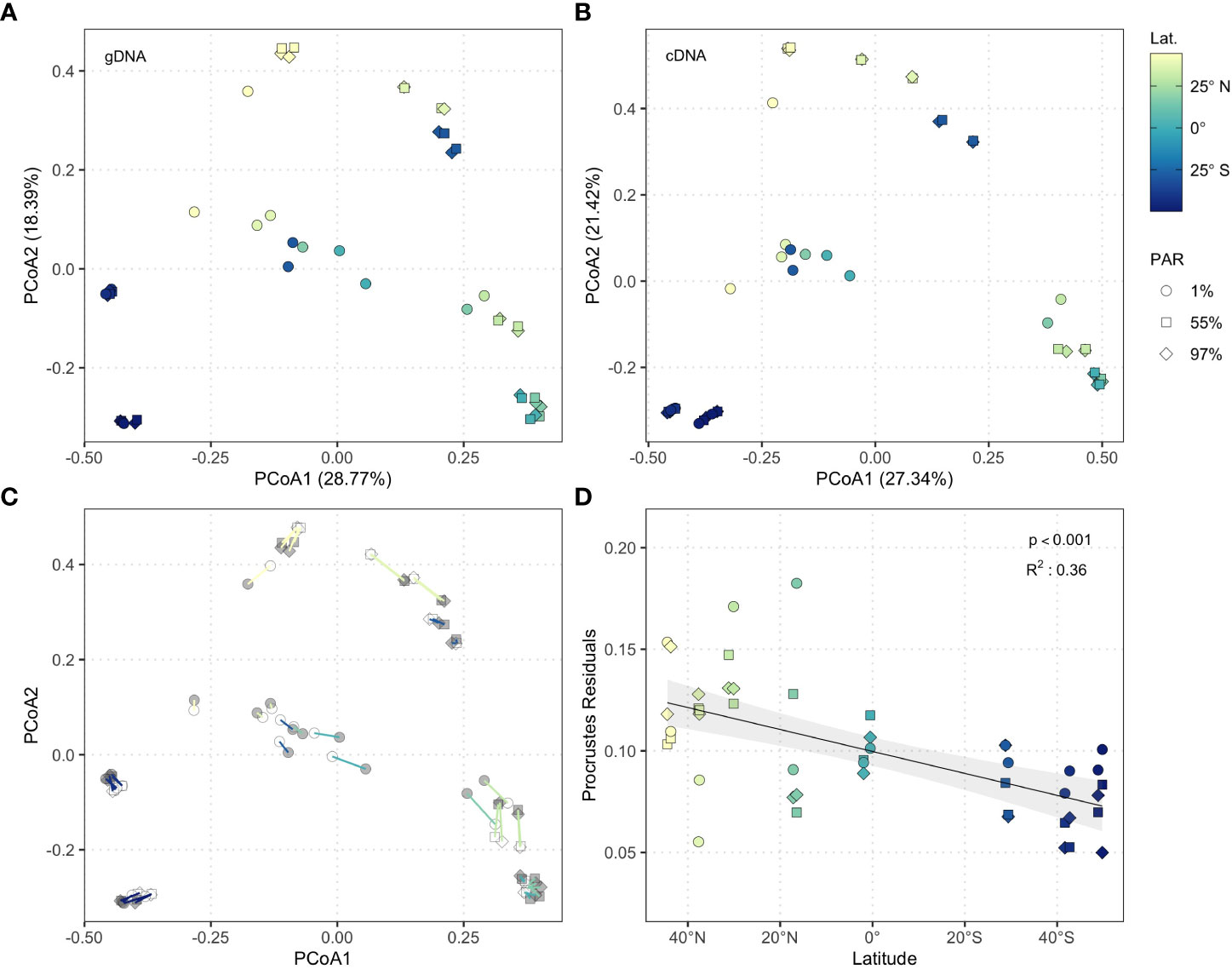
Figure 2 Principle coordinates analysis of (A) gDNA (n = 47) and (B) cDNA (n = 47) community structure based on Bray-Curtis dissimilarity. Shapes represent light-dependent depth (squares = 97% PAR, diamonds = 55% PAR, circles = 1% PAR), colours indicate latitude. (C) Procrustes superimposition of cDNA data onto gDNA data. Segments connect pairs of cDNA and gDNA samples and represent Procrustes residual values. Point colours indicate nucleic acid (grey: gDNA, white: cDNA), segment colours correspond to latitude. (D) Generalised additive model of Procrustes residuals against latitude (n = 47). Shading represents 95% confidence interval.
The structure of cDNA and gDNA communities was highly congruent throughout the Atlantic Ocean (PROTEST; r = 0.98, p < 0.001; Figure 2C). Procrustes residuals were extracted to examine the effect of latitude and light-dependent depth on congruence between cDNA and gDNA community structure, revealing a significant linear decrease from temperate Northern hemisphere latitudes to temperate Southern hemisphere latitudes (GAM; adjusted R2 = 0.36, p < 0.001; Figure 2D). Congruence between cDNA and gDNA derived communities was greatest in the productive South Subtropical Convergence and Southwest Atlantic Shelves (S13–S16; Figure 1A). These data align with a survey of the Indian sector of the Southern Ocean, which suggested that cDNA and gDNA bacterioplankton communities display similar structuring in highly productive waters (Liu et al., 2019).
Comparisons of cDNA and gDNA bacterioplankton biogeography across basin scales are scarce. However, distinct biogeographic patterns in cDNA and gDNA bacterioplankton communities have been shown on regional scales in the South China Sea, where cDNA bacterioplankton communities were structured predominantly by prevailing environmental conditions, whilst gDNA communities were comparatively more influenced by water mass identity (Zhang et al., 2014). Thus, differences in the relative importance of environmental and spatial processes at a regional scale underpinned the distinct biogeography of cDNA and gDNA bacterioplankton communities. The congruence between cDNA and gDNA bacterioplankton communities demonstrated in this study across the Atlantic basin indicates that similar processes structure cDNA and gDNA bacterioplankton communities across oceanic scales (Supplementary Figure 2), suggesting that the relationship between these communities may be scale-dependent in the marine environment. Our data show that the structure of both cDNA and gDNA bacterioplankton communities reflects the oceanographic province from which samples were collected (Reygondeau et al., 2013).
The overall richness of gDNA communities was greater than of cDNA communities (ANOVA; p < 0.005; Supplementary Figure 3), and pairwise comparisons revealed that gDNA community richness and evenness were greater than cDNA community richness evenness at each light-dependent depth (Tukey’s HSD; p < 0.05; Supplementary Figure3). cDNA communities consisted of a subset of the gDNA community, dominated by a small cohort of ASVs with high potential activity that are apparently able to capitalize on prevailing environmental conditions. Both richness and evenness were greater at 1% PAR, which closely tracked the position of the deep chlorophyll maximum over the transect (Figure 1D), than at 55% or 97% PAR (Tukey’s HSD; p < 0.001).
cDNA and gDNA community richness were significantly related (linear model; slope = 0.50, R2 = 0.56, p < 0.001; Figure 3A) but displayed a shallow slope indicating that as gDNA community richness increased, the disparity between cDNA and gDNA community richness also increased. cDNA and gDNA community evenness displayed a stronger relationship (linear model; slope = 1.11, R2 = 0.52, p < 0.001; Figure 3B) including a slope of ~1. cDNA and gDNA community richness and evenness converged in nutrient-rich waters of the Southwest Atlantic Shelves where large phytoplankton dominate primary production (Marañón et al., 2000; Marañón et al., 2001; Tilstone et al., 2017). This convergence was driven by the high relative abundance and potential activity of copiotrophic bacterioplankton (see below), which likely capitalize on phytoplankton-derived organic carbon and nutrient availability.
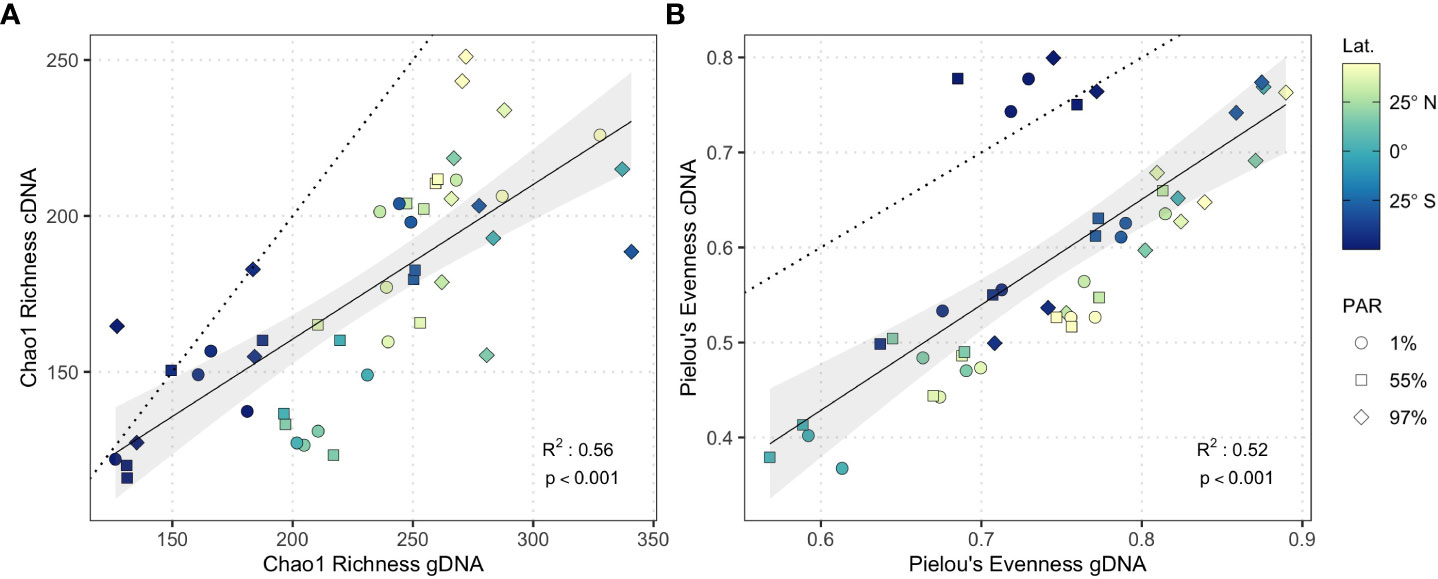
Figure 3 Linear model of (A) Chao1 Richness and (B) Pielou’s evenness from gDNA (n = 47) and cDNA (n = 47) samples. Solid line represents linear model fit, shading represents 95% confidence interval. Dotted guideline represents 1:1 cDNA:gDNA ratio. Shapes represent light-dependent depth.
The potential activity and relative abundance of selected biogeochemically and ecologically important bacterioplankton groups was structured by both latitude and light-dependent depth (Figure 4). An inverse relationship between the potential activity of the cyanobacteria Prochlorococcus and Synechococcus was evident throughout the transect. Prochlorococcus displayed high potential activity north of the South Subtropical Convergence, whilst Synechococcus displayed high potential activity at the South Subtropical Convergence and Southwest Atlantic Shelves (S13–S16), aligning with their characteristically distinct thermal niches (Flombaum et al., 2013). Interestingly, the potential activity of Synechococcus was highest at the Southwest Atlantic Shelves despite low relative abundance, which may suggest that local environmental conditions are optimal for the growth of Synechococcus, but that top-down controls, such as viral infection (Suttle, 2007) and grazing (Apple et al., 2011), constrain relative abundance. However, these data must be interpreted carefully as the relationship between growth rate and cellular 16S rRNA content is non-linear in Synechococcus (Binder and Liu, 1998; Worden and Binder, 2003).
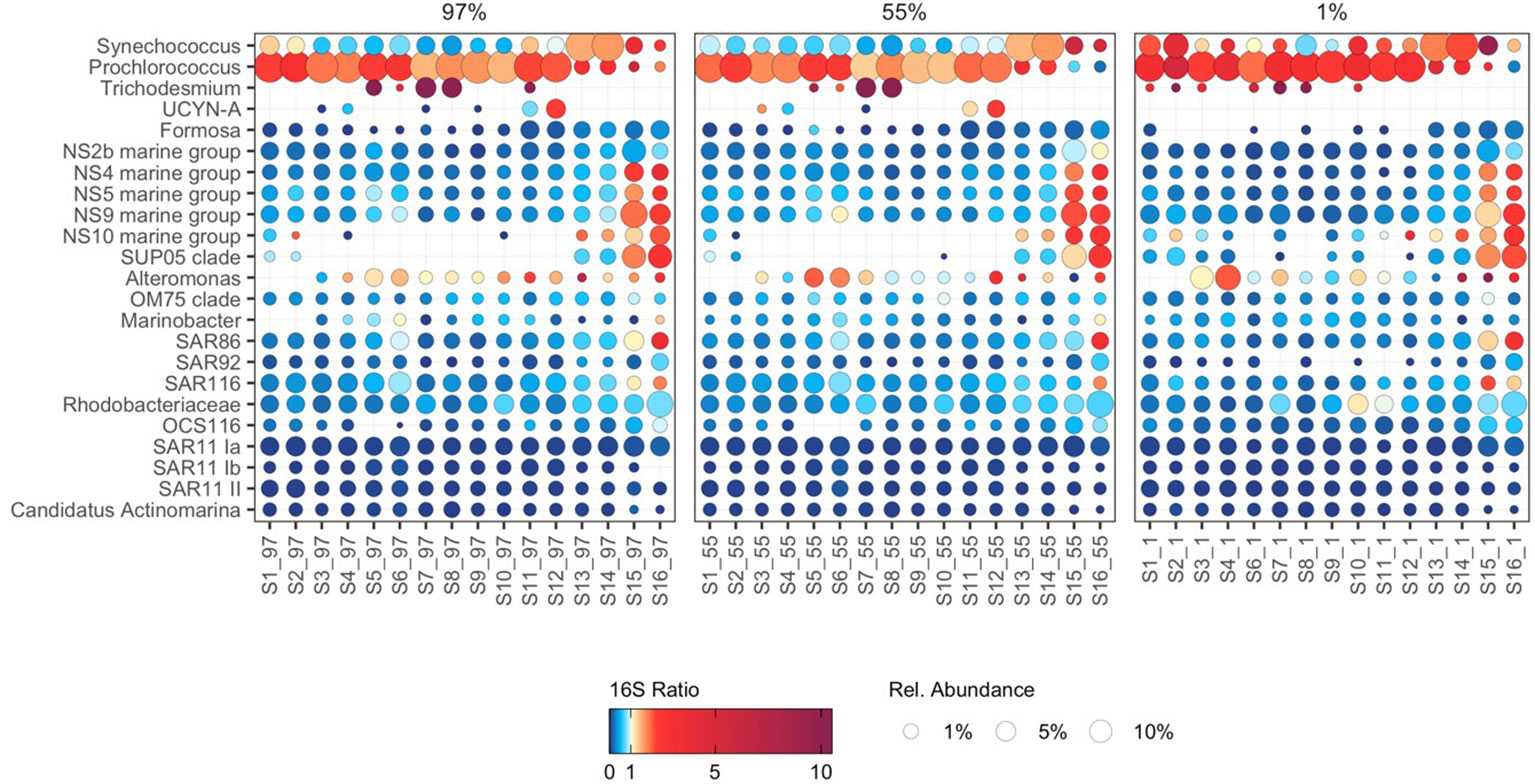
Figure 4 The potential activity and abundance of major bacterioplankton groups. Point size represents the average relative abundance in cDNA and gDNA derived bacterioplankton communities. Colours represent 16S rRNA:16S rRNA gene ratios, a proxy for protein synthesis potential (potential activity). 16S rRNA:16S rRNA gene ratios greater than 10 are represented by a dark plum colour (e.g. in Trichodesmium).
The filamentous diazotrophic cyanobacteria Trichodesmium displayed high potential activity in the tropical North Atlantic (S7–S8; Figure 4), corresponding to areas of known iron supply from atmospheric dust deposition (Fernández et al., 2010). These data underscore the crucial role that Trichodesmium play in nitrogen fixation in this region and suggest that they may contribute disproportionately to biogeochemical function relative to their abundance. Conversely, the unicellular diazotroph UCYN-A (Candidatus Atelocyanobacterium thalassa) were restricted to surface water masses, and displayed high potential activity in the South Atlantic Subtropical Gyre (S11–S12) (Figure 4), illustrating latitudinal switching in the potential activity of the two major nitrogen fixing cyanobacteria in the Atlantic Ocean.
The potential activity of NS4, NS5, NS9, and NS10 marine group Flavobacteria was enhanced at the Southwest Atlantic Shelves (S15–S16; Figure 4) and was related to nutrient availability (Supplementary Figure 4). The observed pattern of enhanced potential activity in nutrient-rich waters where large phytoplankton dominate primary production likely reflects the copiotrophic life history strategy of these taxa, which possess physiological adaptations to capitalize on the availability of phytoplankton-derived organic carbon (Buchan et al., 2014) and are frequently associated with diatom blooms (Teeling et al., 2012; Teeling et al., 2016). Possible niche partitioning was indicated between phylogenetically coherent clusters of active NS4 and NS5 marine group Flavobacteria ASVs, aligning with evidence of ecologically distinct units within these groups (Díez-Vives et al., 2019). ASVs displaying the highest potential activity at the Southwest Atlantic Shelves were largely absent from subtropical and tropical latitudes. In contrast, ASVs distributed throughout the subtropical and tropical Atlantic were absent at the Southwest Atlantic Shelves (Figure 5A). These findings indicate that the cosmopolitan distribution of NS4 and NS5 Marine Groups throughout the Atlantic Ocean (Figure 4) is underpinned by ecological niche partitioning between specific ASVs (Figure 5A) and highlights the value of detailed phylogenetic resolution when investigating bacterioplankton ecology.

Figure 5 The potential activity and abundance of bacterioplankton ASVs at single-nucleotide resolution within (A) Flavobacteriaceae, (B) SAR86, and (C) SAR11 Ia. Point size represents the average relative abundance in cDNA and gDNA derived bacterioplankton communities. Colours represent 16S rRNA:16S rRNA gene ratios, a proxy for protein synthesis potential (potential activity). 16S rRNA:16S rRNA gene ratios greater than 10 are represented by a dark plum colour.
The ubiquitous SAR86 are proteorhodopsin-containing Gammaproteobacteria with streamlined genomes that lack biosynthesis pathways for key vitamins, cofactors, and amino acids (Dupont et al., 2012). As SAR86 have only relatively recently been cultivated, significant questions remain about their ecology and life history strategies. We show that SAR86 are cosmopolitan in their distribution throughout the Atlantic Ocean yet display enhanced potential activity at the Southwest Atlantic Shelves (Figure 4). Group-level patterns in the potential activity of SAR86 resemble those of typical copiotrophic bacterioplankton, suggesting that SAR86 are capable of substantially upregulating potential activity in response to resource availability. Recent evidence indicates that SAR86 can be subdivided into ecotypes based on functional gene content, and that these ecotypes possess distinct biogeographic distributions (Hoarfrost et al., 2020). In this study, niche partitioning was indicated between SAR86 ASVs, which were typically constrained to either tropical and subtropical latitudes, or temperate latitudes (Figure 5B). A cohort of SAR86 ASVs displayed enhanced potential activity in the Southwest Atlantic Shelves, suggesting that they may play an important role in processing phytoplankton-derived organic carbon. Many SAR86 ASVs were restricted to surface water masses (97% and 55% PAR) in oligotrophic tropical and subtropical regions (Figure 5B). We speculate that this trend may relate to the role of proteorhodopsins, a light-driven proton pump, which contributes to cellular energy budget of SAR86 in these regions (Beja et al., 2000; Sabehi et al., 2004). Single-cell and metagenomic evidence indicates substantial variation in organic carbon utilization capabilities and proteorhodopsin content within SAR86 (Hoarfrost et al., 2020), suggesting that a wide array of life history strategies may coexist within the group. Here, we provide ocean scale insights into the potential activity of SAR86, revealing distinct clusters putatively adapted to oligotrophic surface waters and nutrient-rich waters, respectively (Figure 5B).
The Gammaproteobacteria clade SUP05 play an important role in open ocean sulfur metabolism (Moran and Durham, 2019), degrading algal-derived dimethylsulphoniopropionate (DMSP), and utilising energy generated from sulfur oxidation to drive autotrophic carbon fixation (Walsh et al., 2009; Spietz et al., 2019). Heterotrophic carbon metabolism has also been demonstrated in the SUP05 clade member Thioglobus singularis strain PS1, indicative of their ability to utilise diatom cell lysate to support heterotrophic growth (Spietz et al., 2019). To date, most studies of SUP05 are related to the deep ocean and oxygen minimum zones (Walsh et al., 2009; Shah et al., 2019), though viable SUP05 cells have been cultivated from surface waters (Marshall and Morris, 2013). Here we show that SUP05 display high potential activity at the Southwest Atlantic Shelves (Figure 4) where large phytoplankton dominate primary production (Marañón et al., 2000; Marañón et al., 2001; Tilstone et al., 2017). The high potential activity of SUP05 may be underpinned by the production of DMSP and associated organic sulfur compounds by diatoms and other large phytoplankton. Alternatively, the high potential activity of SUP05 may be resultant of heterotrophic growth fuelled by phytoplankton primary production. Regardless of mechanism, these data suggest that SUP05 play an important role in biogeochemical cycling during phytoplankton blooms in these regions, highlighting the need for further investigation to elucidate the role of this enigmatic group.
SAR11 clades Ia, Ib, and II generally displayed uniform relative abundance and low potential activity throughout the Atlantic Ocean (Figure 4). Similar uniformity in activity and abundance of SAR11 has been demonstrated across the Atlantic Ocean using flow-cytometry and 35S-methionine uptake (Mary et al., 2006). These trends may emerge from the generally limited ability of SAR11 to respond to transient increases in substrate availability, and their survival through scavenging widespread labile organic compounds and nutrients present at low background concentrations (Cottrell and Kirchman, 2016; Giovannoni, 2017). SAR11 Ia ASVs displayed distinct distribution patterns, despite minimal variability in potential activity (Figure 5C). Interestingly, we identified a minor increase in the potential activity of a small cluster of SAR11 Ia ASVs at the Southwest Atlantic Shelves (Figure 5C), suggesting that these populations possess some capacity for metabolic upregulation. We speculate that this may be the result of differences between carbon-source utilization pathways in SAR11 genomes (Schwalbach et al., 2010), which may elicit subtle responses to phytoplankton-derived organic carbon for some specialised ASVs. Some SAR11 populations can be particle-associated and therefore may occupy alternative niches other than the generalized archetypical free-living oligotroph (Yeh and Fuhrman, 2022). Our findings provide additional evidence supporting the paradigm that SAR11 generally maintain a consistent state of low metabolic activity across oceanic scales, but some SAR11 have the ability to respond to changes in environmental conditions or substrate availability (Brown et al., 2012; Bolaños et al., 2021; Bolaños et al., 2022). Given that the 16S rRNA gene has limited taxonomic resolution for SAR 11 (Eren et al., 2013) we should be cautious in interpretation of these data and complementary approaches to assess SAR11 diversity should also considered.
This study provides an investigation of the relationship between cDNA and gDNA derived bacterioplankton communities on an oceanic scale. We showed congruence in cDNA and gDNA community structure throughout the Atlantic Ocean, indicating that similar processes structure these communities across oceanic scales. We used 16S rRNA:16S rRNA gene ratios to explore patterns in the potential activity of bacterioplankton groups, from which cyanobacteria, copiotrophic, and oligotrophic bacterioplankton could be broadly distinguished. Furthermore, we provide ASV-level investigation of potential activity in the marine environment, demonstrating possible niche partitioning of Flavobacteria and SAR86 ASVs that underlie the cosmopolitan distribution of these taxa. While potential activity derived through 16S rRNA:16S rRNA gene ratios are indirect indicators of metabolic activity, they are possible proxies of protein synthesis potential providing important ecological insights at detailed phylogenetic resolution which can be used to guide the development of hypotheses for further investigation (Blazewicz et al., 2013). Integrating 16S rRNA:16S rRNA gene ratios with other molecular tools such as metatranscriptomics, which offers insights into the transcription of specific genes within a community but does not directly indicate translation, and metaproteomics, which offers insights into the realised expression of proteins but do not directly indicate the activity of these proteins, are clear avenues for developing a more holistic understanding of bacterioplankton metabolic activity on the community level. Combined, the findings of this study shed light on the active microbiome of the Atlantic Ocean and provide insights towards the ecology and life history strategies of biogeochemically and ecologically significant bacterioplankton.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/ena/browser/view/PRJEB38834.
KB and MC conceived and designed the study. KB conducted the fieldwork, seawater sample processing, DNA/RNA extraction and initial data analysis. RA performed further data and statistical analysis. All authors contributed to the interpretation of the results. RA wrote the first draft of the manuscript working with MC and KB. All authors contributed to manuscript revision, read, and approved the submitted version. All authors contributed to the article and approved the submitted version.
KB was supported by the UKRI Natural Environment Research Council (NERC) Environment East (EnvEast) Doctoral Training Partnership (grant number NE/L002582/1). Sampling was supported by NERC National Capability funding for Atlantic Meridional Transect (AMT) Programme to Plymouth Marine Laboratory. AMT is funded by the NERC through its National Capability Long-term Single Centre Science Programme, Climate Linked Atlantic Sector Science (grant number NE/R015953/1).
We are indebted to the all the scientists, the captain and crew of the RRS James Clark Ross AMT25 who helped make this work happen, especially those who contributed metadata (nutrients, etc.) used in this study and helped with sampling.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1241333/full#supplementary-material
Supplementary Figure 1 | Map of 16 stations sampled during the AMT25 latitudinal transect of the Atlantic Ocean during the Austral spring of 2015 with Longhurst oceanographical spatial codes (NADR, North Atlantic Drift; NASTE, Northeast Atlantic subtropical gyral; NASTW, Northwest Atlantic subtropical gyral; NATR, North Atlantic tropical gyral; ETRA, Eastern Tropical Atlantic; SATL, South Atlantic gyral; SSTC, South subtropical convergence; FKLD, Southwest Atlantic shelves).
Supplementary Figure 2 | Canonical correspondence analysis (CCA) of (A) gDNA (n = 47) and (B) cDNA (n = 47) derived bacterioplankton community composition. Vectors represent statistically significant environmental variables (p < 0.01).
Supplementary Figure 3 | (A) Chao1 Richness and (B) Pielou’s evenness at each light-dependent depth from gDNA (n = 47; white) and cDNA (n = 47; grey) samples. Crossbar represents median value.
Supplementary Figure 4 | The relative activity (16S ratio) of copiotrophic (top row), oligotrophic (middle row) and understudied (bottom row) ASVs against nitrate/nitrite concentration (n = 38). Black lines indicate significant linear model fits (p < 0.05), shaded areas represent 95% confidence intervals. Red guidelines indicate threshold above which ASVs are considered active (16S ratio ≥ 1).
Alonso-Sáez L., Sánchez O., Gasol J. M. (2012). Bacterial uptake of low molecular weight organics in the subtropical Atlantic: Are major phylogenetic groups functionally different? Limnology Oceanography 57, 798–808. doi: 10.4319/lo.2012.57.3.0798
Anderson M. J. (2001). A new method for non-parametric multivariate analysis of variance. Austral Ecology 26 (1), 32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x
Apple J. K., Strom S. L., Palenik B., Brahamsha B. (2011). Variability in protist grazing and growth on different marine Synechococcus isolates. Appl. Environ. Microbiol. 77, 3074–3084. doi: 10.1128/AEM.02241-10
Azam F., Malfatti F. (2007). Microbial structuring of marine ecosystems. Nat. Rev. Micro 5, 782–791. doi: 10.1038/nrmicro1747
Beja O., Aravind L., Koonin E. V., Suzuki M. T., Hadd A., Nguyen L. P., et al. (2000). Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science 289, 1902–1906. doi: 10.1126/science.289.5486.1902
Binder B. J., Liu Y. C. (1998). Growth rate regulation of rRNA content of a marine Synechococcus (Cyanobacterium) strain. Appl. Environ. Microbiol. 64, 3346–3351. doi: 10.1128/AEM.64.9.3346-3351.1998
Blazewicz S. J., Barnard R. L., Daly R. A., Firestone M. K. (2013). Evaluating rRNA as an indicator of microbial activity in environmental communities: limitations and uses. ISME J. 7, 2061–2068. doi: 10.1038/ismej.2013.102
Bolaños L. M., Choi C. J., Worden A. Z., Baetge N., Carlson C. A., Giovannoni S. (2021). Seasonality of the microbial community composition in the north atlantic. Front. Mar. Sci. 8. doi: 10.3389/fmars.2021.624164
Bolaños L. M., Tait K., Somerfield P. J., Parsons R. J., Giovannoni S. J., Smyth T., et al. (2022). Influence of short and long term processes on SAR11 communities in open ocean and coastal systems. ISME Commun. 2, 116. doi: 10.1038/s43705-022-00198-1
Bowsher A. W., Kearns P. J., Shade A. (2019). 16S rRNA/rRNA gene ratios and cell activity staining reveal consistent patterns of microbial activity in plant-associated soil. mSystems 4, e00003–19. doi: 10.1128/msystems.00003-19
Brown M. V., Lauro F. M., Demaere M. Z., Muir L., Wilkins D., Thomas T., et al. (2012). Global biogeography of SAR11 marine bacteria. Mol. Syst. Biol. 8, 595–595. doi: 10.1038/msb.2012.28
Buchan A., Lecleir G. R., Gulvik C. A., González J. M. (2014). Master recyclers: features and functions of bacteria associated with phytoplankton blooms. Nat. Rev. Microbiol. 12, 686–698. doi: 10.1038/nrmicro3326
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., Holmes S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods 13, 581–583. doi: 10.1038/nmeth.3869
Campbell B. J., Yu L., Heidelberg J. F., Kirchman D. L. (2011). Activity of abundant and rare bacteria in a coastal ocean. Proc. Natl. Acad. Sci. U.S.A. 108, 12776–12781. doi: 10.1073/pnas.1101405108
Chambon P., Deutscher M. P., Kornberg A. (1968). Biochemical studies of bacterial sporulation and germination. X. Ribosomes and nucleic acids of vegetative cells and spores of Bacillus megaterium. J. Biol. Chem. 243, 5110–5116. doi: 10.1016/S0021-9258(18)91998-8
Chao A. (1984). Nonparametric estimation of the number of classes in a population. Scandinavian J. Stat 11, 265–270. Available at: https://www.jstor.org/stable/4615964.
Comeau A. M., Douglas G. M., Langille M. G. (2017). Microbiome helper: a custom and streamlined workflow for microbiome research. mSystems 2, e00127–16. doi: 10.1128/mSystems.00127-16
Cottrell M. T., Kirchman D. L. (2016). Transcriptional control in marine copiotrophic and oligotrophic bacteria with streamlined genomes. Appl. Environ. Microbiol. 82, 6010–6018. doi: 10.1128/AEM.01299-16
Del Giorgio P. A., Scarborough G. (1995). Increase in the proportion of metabolically active bacteria along gradients of enrichment in freshwater and marine plankton: implications for estimates of bacterial growth and production rates. J. Plankton Res. 17, 1905–1924. doi: 10.1093/plankt/17.10.1905
Díez-Vives C., Nielsen S., Sánchez P., Palenzuela O., Ferrera I., Sebastián M., et al. (2019). Delineation of ecologically distinct units of marine Bacteroidetes in the Northwestern Mediterranean Sea. Mol. Ecol. 28, 2846–2859. doi: 10.1111/mec.15068
Dupont C. L., Rusch D. B., Yooseph S., Lombardo M. J., Richter R. A., Valas R., et al. (2012). Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J. 6, 1186–1199. doi: 10.1038/ismej.2011.189
Eren A. M., Maignien L., Sul W. J., Murphy L. G., Grim S. L., Morrison H. G., et al. (2013). Oligotyping: differentiating between closely related microbial taxa using 16S rRNA gene data. Methods Ecol. Evol. 4, 1111–1119. doi: 10.1111/2041-210X.12114
Falkowski P. G., Fenchel T., Delong E. F. (2008). The microbial engines that drive Earth's biogeochemical cycles. Science 320, 1034–1039. doi: 10.1126/science.1153213
Fernández A., Mouriño-Carballido B., Bode A., Varela M., Marañón E. (2010). Latitudinal distribution of Trichodesmium spp. and N2 fixation in the Atlantic Ocean. Biogeosciences 7, 3167–3176. doi: 10.5194/bg-7-3167-2010
Field C. B., Behrenfeld M. J., Randerson J. T., Falkowski P. (1998). Primary production of the biosphere: integrating terrestrial and oceanic components. Science 281, 237–240. doi: 10.1126/science.281.5374.237
Flombaum P., Gallegos J. L., Gordillo R. A., Rincon J., Zabala L. L., Jiao N., et al. (2013). Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. Proc. Natl. Acad. Sci. U.S.A. 110, 9824–9829. doi: 10.1073/pnas.1307701110
García-Martín E. E., Aranguren-Gassis M., Hartmann M., Zubkov M. V., Serret P. (2017). Contribution of bacterial respiration to plankton respiration from 50°N to 44°S in the Atlantic Ocean. Prog. Oceanography 158, 99–108. doi: 10.1016/j.pocean.2016.11.006
Giovannoni S. J. (2017). SAR11 bacteria: the most abundant plankton in the oceans. Annu. Rev. Mar. Sci. 9, 231–255. doi: 10.1146/annurev-marine-010814-015934
Hoarfrost A., Nayfach S., Ladau J., Yooseph S., Arnosti C., Dupont C. L., et al. (2020). Global ecotypes in the ubiquitous marine clade SAR86. ISME J. 14, 178–188. doi: 10.1038/s41396-019-0516-7
Hoppe H. G., Gocke K., Koppe R., Begler C. (2002). Bacterial growth and primary production along a north-south transect of the Atlantic Ocean. Nature 416, 168–171. doi: 10.1038/416168a
Hunt D. E., Lin Y., Church M. J., Karl D. M., Tringe S. G., Izzo L. K., et al. (2013). Relationship between abundance and specific activity of bacterioplankton in open ocean surface waters. Appl. Environ. Microbiol. 79, 177–184. doi: 10.1128/AEM.02155-12
Hydes D., Aoyama M., Aminot A., Bakker K., Becker S., Coverly S., et al. (2010). Determination of dissolved nutrients (N, P, Si) in seawater with high precision and inter-comparability using gas-segmented continuous flow analysers. In The GO-SHIP repeat hydrography manual : a collection of expert reports and guidelines. IOCCP Report No 14, ICPO Publication Series No. 134, version 1, 2010 (UNESCO/IOC). Available at: https://archimer.ifremer.fr/doc/00020/13141/.
Jones S. E., Lennon J. T. (2010). Dormancy contributes to the maintenance of microbial diversity. Proc. Natl. Acad. Sci. U.S.A. 107, 5881–5886. doi: 10.1073/pnas.0912765107
Kazamia E., Helliwell K. E., Purton S., Smith A. G. (2016). How mutualisms arise in phytoplankton communities: building eco-evolutionary principles for aquatic microbes. Ecol. Lett. 19, 810–822. doi: 10.1111/ele.12615
Kemp P. F., Lee S., Laroche J. (1993). Estimating the growth rate of slowly growing marine bacteria from RNA content. Appl. Environ. Microbiol. 59, 2594–2601. doi: 10.1128/aem.59.8.2594-2601.1993
Kerkhof L., Kemp P. (1999). Small ribosomal RNA content in marine Proteobacteria during non-steady-state growth. FEMS Microbiol. Ecol. 30, 253–260. doi: 10.1111/j.1574-6941.1999.tb00653.x
Kerkhof L., Ward B. B. (1993). Comparison of nucleic acid hybridization and fluorometry for measurement of the relationship between RNA/DNA ratio and growth rate in a marine bacterium. Appl. Environ. Microbiol. 59, 1303–1309. doi: 10.1128/aem.59.5.1303-1309.1993
Landschützer P., Gruber N., Bakker D. C. E., Schuster U. (2014). Recent variability of the global ocean carbon sink. Global Biogeochemical Cycles 28, 927–949. doi: 10.1002/2014GB004853
Lankiewicz T. S., Cottrell M. T., Kirchman D. L. (2016). Growth rates and rRNA content of four marine bacteria in pure cultures and in the Delaware estuary. ISME J. 10, 823–832. doi: 10.1038/ismej.2015.156
Lennon J. T., Jones S. E. (2011). Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat. Rev. Microbiol. 9, 119–130. doi: 10.1038/nrmicro2504
Lin Y., Gazsi K., Lance V. P., Larkin A. A., Chandler J. W., Zinser E. R., et al. (2013). In situ activity of a dominant Prochlorococcus ecotype (eHL-II) from rRNA content and cell size. Environ. Microbiol. 15, 2736–2747. doi: 10.1111/1462-2920.12135
Liu Y., Debeljak P., Rembauville M., Blain S., Obernosterer I. (2019). Diatoms shape the biogeography of heterotrophic prokaryotes in early spring in the Southern Ocean. Environ. Microbiol. 21, 1452–1465. doi: 10.1111/1462-2920.14579
Logares R., Deutschmann I. M., Junger P. C., Giner C. R., Krabberød A. K., Schmidt T. S. B., et al. (2020). Disentangling the mechanisms shaping the surface ocean microbiota. Microbiome 8, 55. doi: 10.1186/s40168-020-00827-8
Longnecker K., Lomas M. W., Van Mooy B. A. (2010). Abundance and diversity of heterotrophic bacterial cells assimilating phosphate in the subtropical North Atlantic Ocean. Environ. Microbiol. 12, 2773–2782. doi: 10.1111/j.1462-2920.2010.02247.x
Marañón E., Holligan P. M., Barciela R., González N., Mouriño B., Pazó M. J., et al. (2001). Patterns of phytoplankton size structure and productivity in contrasting open-ocean environments. Mar. Ecol. Prog. Ser. 216, 43–56. doi: 10.3354/meps216043
Marañón E., Holligan P. M., Varela M., Mouriño B., Bale A. J. (2000). Basin-scale variability of phytoplankton biomass, production and growth in the Atlantic Ocean. Deep Sea Res. Part I: Oceanographic Res. Papers 47, 825–857. doi: 10.1016/S0967-0637(99)00087-4
Marshall K. T., Morris R. M. (2013). Isolation of an aerobic sulfur oxidizer from the SUP05/Arctic96BD-19 clade. ISME J. 7, 452–455. doi: 10.1038/ismej.2012.78
Mary I., Heywood J., Fuchs B., Amann R., Tarran G., Burkill P., et al. (2006). SAR11 dominance among metabolically active low nucleic acid bacterioplankton in surface waters along an Atlantic meridional transect. Aquat. Microbial Ecol. 45, 107–113. doi: 10.3354/ame045107
McMurdie P. J., Holmes S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8, e61217. doi: 10.1371/journal.pone.0061217
Milici M., Tomasch J., Wos-Oxley M. L., Decelle J., Jáuregui R., Wang H., et al. (2016a). Bacterioplankton biogeography of the Atlantic Ocean: a case study of the distance-decay relationship. Front. Microbiol. 7, 590. doi: 10.3389/fmicb.2016.00590
Milici M., Tomasch J., Wos-Oxley M. L., Wang H., Jáuregui R., Camarinha-Silva A., et al. (2016b). Low diversity of planktonic bacteria in the tropical ocean. Sci. Rep. 6, 19054. doi: 10.1038/srep19054
Moran M. A., Durham B. P. (2019). Sulfur metabolites in the pelagic ocean. Nat. Rev. Microbiol. 17, 665–678. doi: 10.1038/s41579-019-0250-1
Needham D. M., Sachdeva R., Fuhrman J. A. (2017). Ecological dynamics and co-occurrence among marine phytoplankton, bacteria and myoviruses shows microdiversity matters. ISME J. 11, 1614–1629. doi: 10.1038/ismej.2017.29
Oksanen J., Blanchet F. G., Kindt R., Legendre P., Minchin P. R., O’Hara R. B., et al. (2014). Vegan: Community Ecology Package. R Package Version 2.2-0. Available at: http://CRAN.Rproject.org/package=vegan.
Parada A. E., Needham D. M., Fuhrman J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414. doi: 10.1111/1462-2920.13023
Peres-Neto P. R., Jackson D. A. (2001). How well do multivariate data sets match? The advantages of a Procrustean superimposition approach over the Mantel test. Oecologia 129, 169–178. doi: 10.1007/s004420100720
Pielou E. C. (1966). The measurement of diversity in different types of biological collections. J. Theor. Biol. 13, 131–144. doi: 10.1016/0022-5193(66)90013-0
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R-Core-Team (2017). R: A language and environment for statistical computing (Vienna, Austria: R Foundation for Statistical Computing). Available at: https://www.R-project.org/.
Reintjes G., Arnosti C., Fuchs B., Amann R. (2019). Selfish, sharing and scavenging bacteria in the Atlantic Ocean: a biogeographical study of bacterial substrate utilisation. ISME J. 13, 1119–1132. doi: 10.1038/s41396-018-0326-3
Reygondeau G., Longhurst A., Martinez E., Beaugrand G., Antoine D., Maury O. (2013). Dynamic biogeochemical provinces in the global ocean. Global Biogeochemical Cycles 27, 1046–1058. doi: 10.1002/gbc.20089
Sabehi G., Béjà O., Suzuki M. T., Preston C. M., Delong E. F. (2004). Different SAR86 subgroups harbour divergent proteorhodopsins. Environ. Microbiol. 6, 903–910. doi: 10.1111/j.1462-2920.2004.00676.x
Salter I., Galand P. E., Fagervold S. K., Lebaron P., Obernosterer I., Oliver M. J., et al. (2015). Seasonal dynamics of active SAR11 ecotypes in the oligotrophic Northwest Mediterranean Sea. ISME J. 9, 347–360. doi: 10.1038/ismej.2014.129
Schwalbach M. S., Tripp H. J., Steindler L., Smith D. P., Giovannoni S. J. (2010). The presence of the glycolysis operon in SAR11 genomes is positively correlated with ocean productivity. Environ. Microbiol. 12, 490–500. doi: 10.1111/j.1462-2920.2009.02092.x
Shah V., Zhao X., Lundeen R. A., Ingalls A. E., Nicastro D., Morris R. M. (2019). Morphological plasticity in a sulfur-oxidizing marine bacterium from the SUP05 clade enhances dark carbon fixation. mBio 10, e00216–19. doi: 10.1128/mbio.00216-19
Spietz R. L., Lundeen R. A., Zhao X., Nicastro D., Ingalls A. E., Morris R. M. (2019). Heterotrophic carbon metabolism and energy acquisition in Candidatus Thioglobus singularis strain PS1, a member of the SUP05 clade of marine Gammaproteobacteria. Environ. Microbiol. 21, 2391–2401. doi: 10.1111/1462-2920.14623
Stamatakis A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sukenik A., Kaplan-Levy R. N., Welch J. M., Post A. F. (2012). Massive multiplication of genome and ribosomes in dormant cells (akinetes) of Aphanizomenon ovalisporum (Cyanobacteria). ISME J. 6, 670–679. doi: 10.1038/ismej.2011.128
Sunagawa S., Coelho L. P., Chaffron S., Kultima J. R., Labadie K., Salazar G., et al. (2015). Structure and function of the global ocean microbiome. Science 348 (6237). doi: 10.1126/science.1261359
Suttle C. A. (2007). Marine viruses—major players in the global ecosystem. Nat. Rev. Microbiol. 5, 801–812. doi: 10.1038/nrmicro1750
Suzuki M. T., Taylor L. T., Delong E. F. (2000). Quantitative Analysis of Small-Subunit rRNA Genes in Mixed Microbial Populations via 5′-Nuclease Assays. Appl. Environ. Microbiol. 66, 4605–4614. doi: 10.1128/AEM.66.11.4605-4614.2000
Taylor J. D., Bird K. E., Widdicome C. E., Cunliffe M. (2018). Active bacterioplankton community response to dissolved ‘free’ deoxyribonucleic acid (dDNA) in surface coastal marine waters. FEMS Microbiol. Ecol. 94, fiy132–fiy132. doi: 10.1093/femsec/fiy132
Teeling H., Fuchs B. M., Becher D., Klockow C., Gardebrecht A., Bennke C. M., et al. (2012). Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336, 608–611. doi: 10.1126/science.1218344
Teeling H., Fuchs B. M., Bennke C. M., Kruger K., Chafee M., Kappelmann L., et al. (2016). Recurring patterns in bacterioplankton dynamics during coastal spring algae blooms. Elife 5, e11888. doi: 10.7554/eLife.11888
Tilstone G. H., Lange P. K., Misra A., Brewin R. J. W., Cain T. (2017). Micro-phytoplankton photosynthesis, primary production and potential export production in the Atlantic Ocean. Prog. Oceanography 158, 109–129. doi: 10.1016/j.pocean.2017.01.006
Van Tol H. M., Amin S. A., Armbrust E. V. (2017). Ubiquitous marine bacterium inhibits diatom cell division. ISME J. 11, 31–42. doi: 10.1038/ismej.2016.112
Walsh D. A., Zaikova E., Howes C. G., Song Y. C., Wright J. J., Tringe S. G., et al. (2009). Metagenome of a versatile chemolithoautotroph from expanding oceanic dead zones. Science 326, 578–582. doi: 10.1126/science.1175309
Wang Q., Garrity G. M., Tiedje J. M., Cole J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wood S. N. (2012). mgcv:Mixed GAM Computation Vehicle with GCV/AIC/REML smoothness estimation. Available at: https://researchportal.bath.ac.uk/en/publications/mgcv-mixed-gam-computation-vehicle-with-gcvaicreml-smoothness-est.
Worden A. Z., Binder B. J. (2003). Growth regulation of rRNA content in Prochlorococcus and Synechococcus (marine cyanobacteria) measured by whole-cell hybridization of rRNA-targeted peptide nucleic acids. J. Phycology 39, 527–534. doi: 10.1046/j.1529-8817.2003.01248.x
Yeh Y.-C., Fuhrman J. A. (2022). Contrasting diversity patterns of prokaryotes and protists over time and depth at the San-Pedro Ocean Time series. ISME Commun. 2, 36. doi: 10.1038/s43705-022-00121-8
Keywords: 16S rRNA, bacterioplankton, biogeography, microbial activity, Atlantic Ocean
Citation: Allen R, Bird KE, Murrell JC and Cunliffe M (2023) Latitudinal variation in the potential activity of Atlantic Ocean bacterioplankton revealed through 16S rRNA and 16S rRNA gene metabarcoding. Front. Mar. Sci. 10:1241333. doi: 10.3389/fmars.2023.1241333
Received: 16 June 2023; Accepted: 28 July 2023;
Published: 15 August 2023.
Edited by:
Andrew Paul Rees, Plymouth Marine Laboratory, United KingdomReviewed by:
Luis Manuel Bolaños, University of Exeter, United KingdomCopyright © 2023 Allen, Bird, Murrell and Cunliffe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Cunliffe, bWljbmxpQG1iYS5hYy51aw==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.