 Yongjie Wu1
Yongjie Wu1 Yang Zhang1
Yang Zhang1 Huaiyang Fang1
Huaiyang Fang1 Cheng Wang2Zengrui Wang3Wucai Zhang1Bixian Mai4,5
Cheng Wang2Zengrui Wang3Wucai Zhang1Bixian Mai4,5 Zhili He6Renren Wu1*Kaiming Li1*
Zhili He6Renren Wu1*Kaiming Li1*- 1State Environmental Protection Key Laboratory of Water Environmental Simulation and Pollution Control, South China Institute of Environmental Sciences, Ministry of Ecology and Environment of the People’s Republic of China, Guangzhou, China
- 2Environmental Microbiomics Research Center, School of Environmental Science and Engineering, Sun Yat-Sen University, Guangzhou, China
- 3School of Resources and Environmental Engineering, Wuhan University of Technology, Wuhan, Hubei, China
- 4State Key Laboratory of Organic Geochemistry and Guangdong Key Laboratory of Environmental Resources Utilization and Protection, Guangzhou Institute of Geochemistry, Chinese Academy of Sciences, Guangzhou, China
- 5Guangdong-Hong Kong-Macao Joint Laboratory for Environmental Pollution and Control, Guangzhou Institute of Geochemistry, Chinese Academy of Sciences, Guangzhou, China
- 6Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai), Zhuhai, China
Karst rivers are highly susceptible to environmental disturbance due to their robust hydraulic connectivity. However, current knowledge of the mechanisms that regulate the assembly of bacterial and protistan subcommunities in river ecosystems, particularly in karst regions, is limited. By employing 16S and 18S rRNA gene amplicon sequencing, we explored how the diversities and assembly of abundant and rare bacterial and protistan subcommunities adapt to the local environmental variables in a karst river. Both bacterial and protistan rare subcommunities in karst river environments showed a similar biogeography to their abundant subcommunities. Also, a significant distance-decay pattern was observed in all components of the bacterial and protistan subcommunities along the Chishui River, with the rare subcommunities showing a more pronounced distance-decay pattern compared to the abundant subcommunities. Except protist rare subcommunity, the abundant and rare bacterial and abundant protistan subcommunities were strongly structured by the dispersal limitation processes rather than heterogeneous selection. Either bacteria or temperature, elevation and conductivity were the primary drivers for both abundant and rare subcommunities. Additionally, our results suggested that the rare subcommunities contribute significantly to the persistence and stability of microbial networks in the Chishui River, as they exhibited a higher number of keystones compared to the abundant subcommunities. Overall, our study revealed that in the karst river ecosystem, abundant bacterial subcommunities had a higher potential for environmental adaptation than rare bacterial and protistan subcommunities and identified the factors that moderate their assembly processes.
1 Introduction
The microbial determinants can shape the biogeography and structure of the microbial communities, which are crucial research areas in the ecology. These communities significantly influence the global biogeochemical cycles, bioremediation, and food webs (Hosen et al., 2017). A typical microbial community is a mixed culture of an abundant subcommunity (with low diversity) and a rare subcommunity (with high diversity). Among them, the abundant subcommunities are essentially important for biomass production and element cycling (Nemergut et al., 2011). However, studies showed that the rare subcommunity could remarkably affect the function and stability of the microbial community (Jia et al., 2018). In either abundant or rare subcommunities, a comprehensive understanding of the biogeography and assembly mechanisms is of outstanding subjects to advance our knowledge about microbial-driven ecosystem processes and functions. Therefore, it is vital to completely study the biogeography and assembly mechanisms in these subcommunities. Despite previous works that mainly emphasized the abundant microbial subcommunity due to their role in carbon cycling and biomass production, it is essential to understand the assembly mechanisms in the rare subcommunity (Jia et al., 2018). A methodical analysis of both subcommunities can cause a better understanding of microbial ecosystem dynamics and functions (Nemergut et al., 2011; Pikitch, 2018). However, recent studies highlighted the importance of the rare microbial subcommunities, which exhibited higher metabolic activity in comparison to the abundant subcommunities and is critical to ecosystems (Jia et al., 2018). Furthermore, the rare subcommunity indicates distinctive functional properties and dissimilar biogeographical patterns compared to their abundant counterparts (Jiao and Lu, 2020). However, the current research on the abundant and rare subcommunities has been around the comparison among the structure and dynamics in soil (Gao et al., 2020; Jiao and Lu, 2020), ocean (Mo et al., 2018; Li et al., 2021), and lakes (Zhang et al., 2022). Therefore, despite the importance of the assembly mechanisms of abundant and rare microbes in rivers for ecosystem response prediction during environmental changes, the current findings have not been sufficient in this domain.
A thorough understanding of the composition of microbial communities is essential to characterize the typical function of individual species and their links to ecosystem functions, and to influence biogeochemical processes (Lu et al., 2020; Pan et al., 2022). Although some resemblances between abundant and rare subcommunities have been observed, diverse microbial types have showed unique biogeographical patterns, even for identical environments (Liu et al., 2015; Hou et al., 2020; Li et al., 2021; Bai et al., 2023). In our paper, the objective was to investigate bacterial and protistan subcommunities as essential constituents of the microbial food chain in river ecosystems (Cruaud et al., 2019; Hirakata et al., 2020; Husnik et al., 2021). Despite previous studies about the metacommunity dynamics of river microbiomes (Carraro et al., 2018; Chen et al., 2019), there is still limited research on the main factors of organism types’ patterns, especially analyzing abundant and rare subcommunities separately (Cruaud et al., 2020).
Deterministic factors (e.g., heterogeneous or homogeneous selection), stochastic factors (e.g., dispersal limitation and homogenizing dispersal), and drift processes show a significant effect on shaping the microbiome of a community containing individual organisms (Stegen et al., 2013). Recent works proposed that the configuration of microbial communities in aquatic ecosystems is affected by deterministic and stochastic mechanisms. The interplay between these procedures is vulnerable to the influence of complicated factors (Gweon et al., 2021; Zhang et al., 2022). In flowing water ecosystems (like rivers and streams), the variables such as pH, temperature, dissolved oxygen, and levels of nitrogen and phosphorus nutrients have caused a substantial influence on species sorting (Chen et al., 2020; Wang et al., 2021). Additionally, the hydrological characteristics of these ecosystems (such as dams, flow direction, and water retention time) are likely critical to regulating the balance between deterministic and stochastic mechanisms (Cruaud et al., 2019; Shen et al., 2020). Regardless of the numerous studies about the bacterial, protistan, and fungal communities in flowing water ecosystems, a significant portion of them has concentrated on the overall community. Also, trivial research has been performed on the biogeographical distribution of abundant and rare subcommunities and the mechanisms of the communities’ diversity in river ecosystems. The lack of this knowledge limits our ability to anticipate the response of microbiomes to environmental changes in rivers.
Despite numerous studies in marine, soil, and lake ecosystems (Jiao et al., 2020; Wang Z, et al., 2020; Cheng et al., 2021; Li et al., 2021), the studies on the microbial community assembly in river ecosystems (especially karst rivers) are still relatively limited. It is a challenge to make broad generalizations about freshwater systems. The Chishui River is located at the intersection of the Yunnan-Guizhou Plateau in Southwest China as a karst river, which is the ultimate unblocked primary tributary of the upper Yangtze River (Buckerfield et al., 2019; Xiang et al., 2020; Wu T, et al., 2022). With a length of 436.5 km and an elevation gradient from 2200 m to 200 m at the outlet, the Chishui River ecosystem offers unique opportunity to study the spatial variability of microbial community structures and their assembly mechanisms. Our knowledge has already been limited about the microbial community assembly in the natural rivers of the karst regions (Xiang et al., 2020). The main emphasis of earlier research on microbiomes in karst rivers was on the analysis of diversity, composition, migration, pathogenic bacteria, and the characterization of specified functional microbiomes (Menning et al., 2017; Ender et al., 2018; Buckerfield et al., 2019). However, the assembly mechanisms of the microbial communities in karst river ecosystems and their adaption to the environment are not still fully understood.
This work aimed to explore the bacterial and protistan communities in the Chishui River through 16S rRNA and 18S rRNA gene amplicon sequencing. The main purpose was to find the biogeographical patterns and assembly mechanisms in microbial subcommunities. A particular emphasis was on investigating two scientific subjects: 1) the diversity and co-occurrence relationships of the abundant and rare bacterial subcommunities and the abundant and rare protistan subcommunities, 2) the factors for the assembly of these subcommunities in a karst river. The mentioned subjects were investigated for a deeper comprehension of the ecological role of microbial communities in karst rivers, along with their adaptation possibility to the environment.
2 Materials and methods
2.1 Sampling and data collection
In this study, the methodology and data published by Wu Y, et al. (2022) were used. It involved the evaluation of water physicochemical parameters and samples. The samples were collected from eleven sites along the Chishui River on 14 September 2021 (Supplementary Figure 1). At each site, three repetitive samples were prepared. The dissolved oxygen (DO), pH, conductivity, and temperature (T) were measured in situ by using a multi-parameter water quality probe YSIEXO2 (Yellow Springs Instruments, USA). The chemical oxygen demand (COD), turbidity, total phosphate (TP), total nitrogen (TN), and heavy metal concentrations were also analyzed by standardized testing methods. The concentration of heavy metals (e.g., V, Cr, Mn, Co) were simultaneously determined with an inductively coupled plasma mass spectrometry (ICP-MS) (Agilent 7500 series, USA). Furthermore, the elevation of each sampling location was documented by a GPS device.
2.2 Sequencing of 16S and 18S rRNA gene amplicons and data analysis
The analysis and detailed data of bacterial communities for the Chishui River were presented by Wu Y, et al. (2022). A dual-index sequencing strategy was used to amplify the V3-V4 regions with universal primers 338F (50-ACTCCTACGGGAGGCAGCA-30) and 806R (50GGACTACHVGGGTWTCTAAT-3). The amplification conditions were as follows: initial denaturation at 98°C for 2 min, followed by 25 cycles of 98°C for 30 s, 50°C for 30 s and 72°C for 1 min, ending with a final extension at 72°C for 5 min. Also, protistan communities data were obtained using the primer sequences TAReukREV3 (5′-ACTTTCGTTCTTGAT(C/T)(A/G)A-3′) and TAReuk454FWD1 (5′-CCAGCA(G/C)C(C/T)GCGGTAATTCC-3′) for 18S rRNA sequencing (Stoeck et al., 2010). According to our used protocol, a PCR test was performed that started with an initial denaturation phase at 95°C for 5 mins, followed by 30 cycles of 94°C for 30 s, 47°C for 45 s, and 72°C for 1 min, and culminating with a final extension phase at 72°C for 5 mins. Following the PCR amplification, the QIAquick PCR purification kit (QIAGEN, Germany) was utilized to collect, purify and concentrate all PCR products. Subsequently, paired-end sequencing (2 × 250 bp) was performed on the Illumina HiSeq platform at Beijing Nuohe Biotechnology (Beijing, China) to obtain the sequencing data.
In this work, a processing technique similar to that in16S rRNA gene sequencing was adopted to preserve the integrity of 18S rRNA gene sequencing. First, the raw reads were mapped to sample barcodes, and primer sequences were trimmed with one nucleotide mismatch. Then, Btrim was then used to remove those reads with an average quality thread of 20 at a window size of five. Second, the FLASH program was used to merge the paired-end sequences (> 200 bases) with at least 10 bp overlap (Magoc and Salzberg, 2011). Only the sequences without any ambiguous bases and > 250 bp were kept for forwarding analysis. Next, chimeras were removed against the SILVA database (Quast et al., 2013) and Protist Ribosomal Reference databases (PR2, version 4.11.1) (Guillou et al., 2013) for 16S and 18S rRNA gene sequences, respectively. Finally, a clustering threshold of 97% was applied to classify the sequences into operational taxonomic units (OTUs) by UPARSE, while singletons were discarded (Edgar, 2013). All analyses were performed on a publicly available Galaxy pipeline (http://mem.rcees.ac.cn:8080/) (Feng et al., 2017). The microbial data has been stored in the NCBI SRA and assigned the accession number PRJNA938286, making the data is made publicly available for future research and analysis.
2.3 Ecological process and molecular ecological network of the metacommunities
The ecological dynamics of microbial subcommunities in Chishui River water were quantified using the same methods as described before (Stegen et al., 2013; Wu Y, et al., 2022). The β-Nearest Taxon Index (β-NTI) and Raup-Crick (RCBray) were used to indicate the phylogenetic and taxonomic diversity variation or inversion based on null model-based β-diversity. Then, β-NTI was combined with RCBray to evaluate the contribution of deterministic and stochastic drivers in structuring the bacterial and protistan subcommunities in the Chishui River. Furthermore, a molecular ecological network analysis pipeline (MENA, available at http://ieg2.ou.edu/MENA/) (Deng et al., 2012) was used to examine microbial interactions in the Chishui River ecosystem. Specifically, four phylogenetic molecular ecological networks were generated for the abundant and rare subcommunities. MENA utilizes an interface method that is based on random matrix theory to detect and visualize microbial interactions among subcommunities. Only the OTUs appeared in over 25 samples (a total of 33 for each group) were utilized to construct the network. Then, the MENA pipeline was employed to calculate topological properties and modularity and visualize via Gephi 0.9. Another objective of our study was to identify keystone nodes for each subcommunity by assessing their within-module connectivity (Zi) and among-module connectivity (Pi) (Guimerà and Amaral, 2005). Four types of nodes were identified as potential keystones: (i) module hubs (highly connected nodes within modules with Zi > 2.5 and Pi ≤ 0.62), (ii) network hubs (highly connected nodes within the entire network with Zi > 2.5 and Pi > 0.62), (iii) connectors (nodes that connect modules with Zi ≤ 2.5 and Pi > 0.62), and (iv) peripherals (nodes with Zi ≤ 2.5 and Pi ≤ 0.62) (Deng et al., 2012).
2.4 Statistical analysis
For taxa classification, a relative abundance threshold of 0.1% in total sequences was defined as “abundant” taxa, while those with relative abundances below 0.01% were classified as “rare” (Liu et al., 2015; Jiao and Lu, 2020). One-way ANOVA was used to examine the variation in the environmental parameters, including α-diversity, relative abundance, and intra-cluster and inter-cluster dissimilarity in the Chishui River water. For the sequence normalization, the minimum number of reads was used to resample the differences in sequencing depth and was determined to be 38,026 (for bacteria) and 68,668 (for protists) per sample. The β-diversity was evaluated by means of the Bray-Curtis distance calculation. Different statistical approaches (including correlation analysis, Adonis test, and Mantel test) were utilized to determine the correlations or differences between variables and/or samples.
3 Results
3.1 Geochemical characteristics
A comparison of 20 environmental variables measured along the Chishui River was performed, as presented in Supplementary Figure 2. It was observed that elevation and turbidity displayed a decreasing trend along the river, while conductivity and temperature showed an opposite pattern. The P3 site represented the maximum TN, TP, and COD. The variation in heavy metal concentrations differed among sampling sites (Supplementary Figure 3). For example, the concentration of Fe, Co, and Ba showed an increasing trend alongside the river, while Mn was significantly (p < 0.01) higher in P1 compared with in other sites. Additionally, the Ca in the P11 site was remarkably (p < 0.05) lower than in other places.
3.2 Distribution and diversity of microbial subcommunities in the Chishui River
A total number of 33 water samples was analyzed, 14,142 bacterial and 11,112 protistan OTUs were characterized. Rarefaction curves showed a trend towards saturation, indicating that the sequencing depth was sufficient (Supplementary Figure 4). A small fraction of highly abundant OTUs accounted fora significantly higher proportion of the total relative abundance compared to a larger fraction of rare OTUs. Specifically, on average, only 1.01% of bacterial OTUs and 0.26% of protistan OTUs were classified as abundant, while they yet accounted for a much higher portion of the relative abundance (57.68% and 65.37%, respectively) (Supplementary Figure 5A). Comparatively, 16.13% of bacterial OTUs and 11.53% of protistan OTUs were identified as rare, but only 10.64% and 2.22% were attributed to the relative abundance, respectively, for bacteria and protistans (Supplementary Figure 5B). There were 13,103 rare bacterial OTUs and only 157 abundant OTUs, while 10,083 rare protistan OTUs and only 144 abundant OTUs were characterized for protists (Supplementary Figures 5C, D). Generally, significant abundance-occupancy correlations were observed between both bacterial and protistan OTUs, showing a stronger correlation between rare subcommunities than the abundant subcommunities.
Substantial dissimilarities were observed in biodiversity and community composition between the rare and abundant subcommunities of bacterial and protistan subcommunities. Compared to the rare subcommunity, the abundant subcommunity showed significantly (p < 0.05) higher diversity values of observed richness, Inv_simpson index, and Chao1 and Shannon diversity (Supplementary Figure 6). Also, the rare subcommunity showed a significantly (p < 0.05) higher community dissimilarity than the abundant subcommunity for both bacteria and protists (Supplementary Figure 7). For 33 samples, the population of bacteria was higher than the protists’ number (Supplementary Figures 8, 9). Both rare and abundant subcommunities represented great phylogenetic diversity, with most species belonging to Proteobacteria, Actinobacteria, Firmicutes, and Bacteroidetes for bacteria (Supplementary Figure 10A), and Chlorophyta, Diatomea, and Streptophyta for protists (Supplementary Figure 10B). From Figure 1, an analysis was performed to determine the phylogenetic pattern of the rare and abundant subcommunities and their correlation with environmental factors (whether positive or negative). The results showed that the abundant bacterial subcommunities were mainly comprised of Proteobacteria and Acidobacteria. However, those found in rare subcommunities were predominantly composed of Bacteroidetes. Similarly, abundant protistan subcommunities were mostly included Chlorophyta, c_Chrysophyceae, and c_Cryptophyceae, but rare protistan subcommunities were composed of Chlorophyta, c_Spirotrichea, and unclassified organisms.
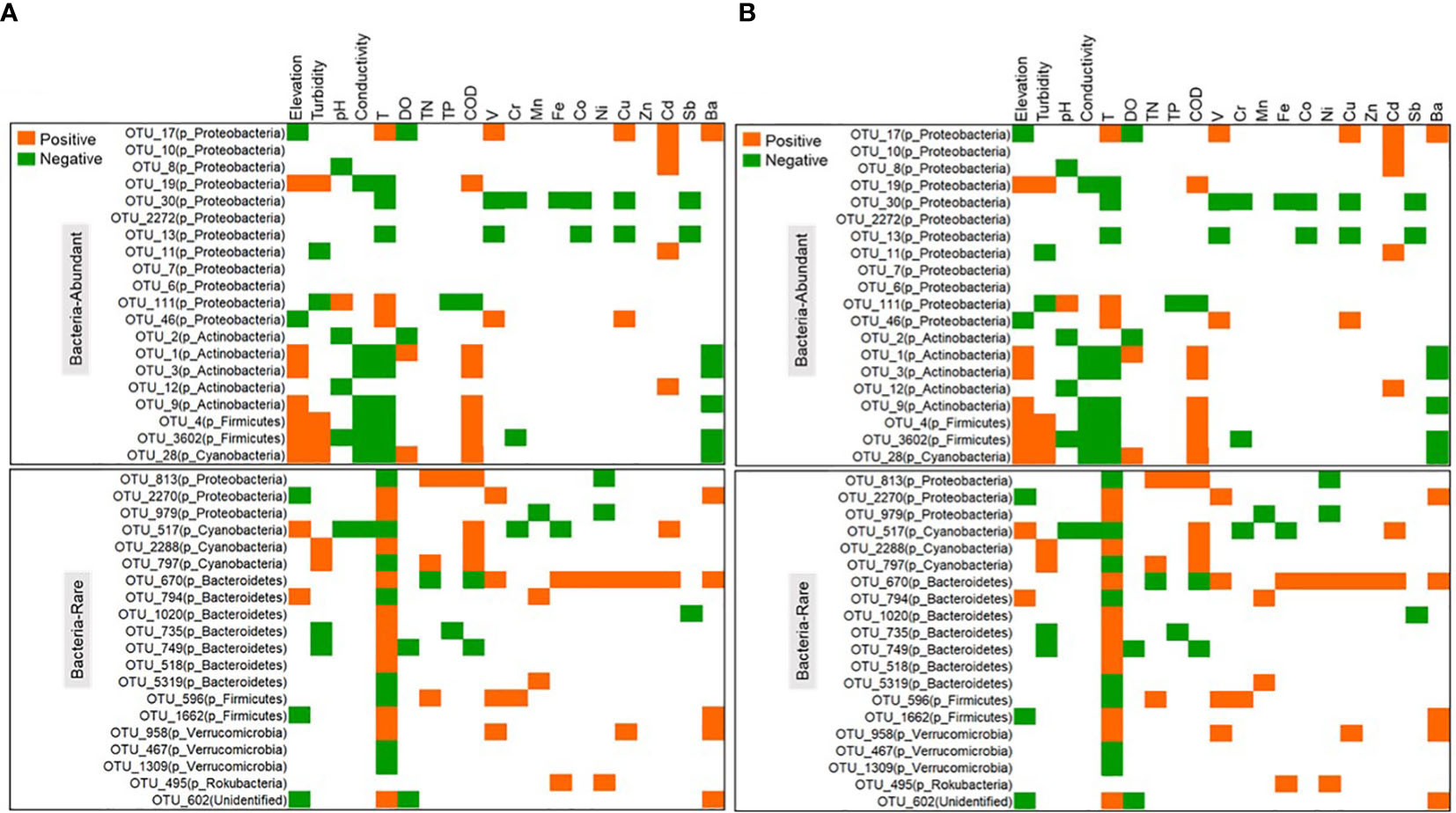
Figure 1 The correlation between the environment and the taxonomy of the abundant and rare subcommunities of bacteria (A) and protists (B) in the water of the Chishui River.
3.3 Assembly processes for microbial subcommunities in the Chishui River
A consistent pattern of community differentiation was achieved concerning geographical distance for both bacterial and protistan subcommunities in the Chishui River ecosystem (Figure 2). Spearman’s correlation coefficients for β-diversity versus geographic distance were significant (p <0.001) for both abundant and rare subcommunities (Figure 2). In either abundant or rare subcommunities, the slopes of the distance-dissimilarity curves were remarkably steeper in protists (0.001, abundant; 0.0006, rare) compared with in bacteria (0.0009, abundant; 0.0003, rare). Also, they were steeper in the abundant subcommunities compared to the rare ones. According to these findings, both bacterial and protistan subcommunities represented a consistent decrease in community similarity alongside the Chishui River. Additionally, higher community differentiation was observed in the protistan subcommunities compared to their bacterial counterparts.
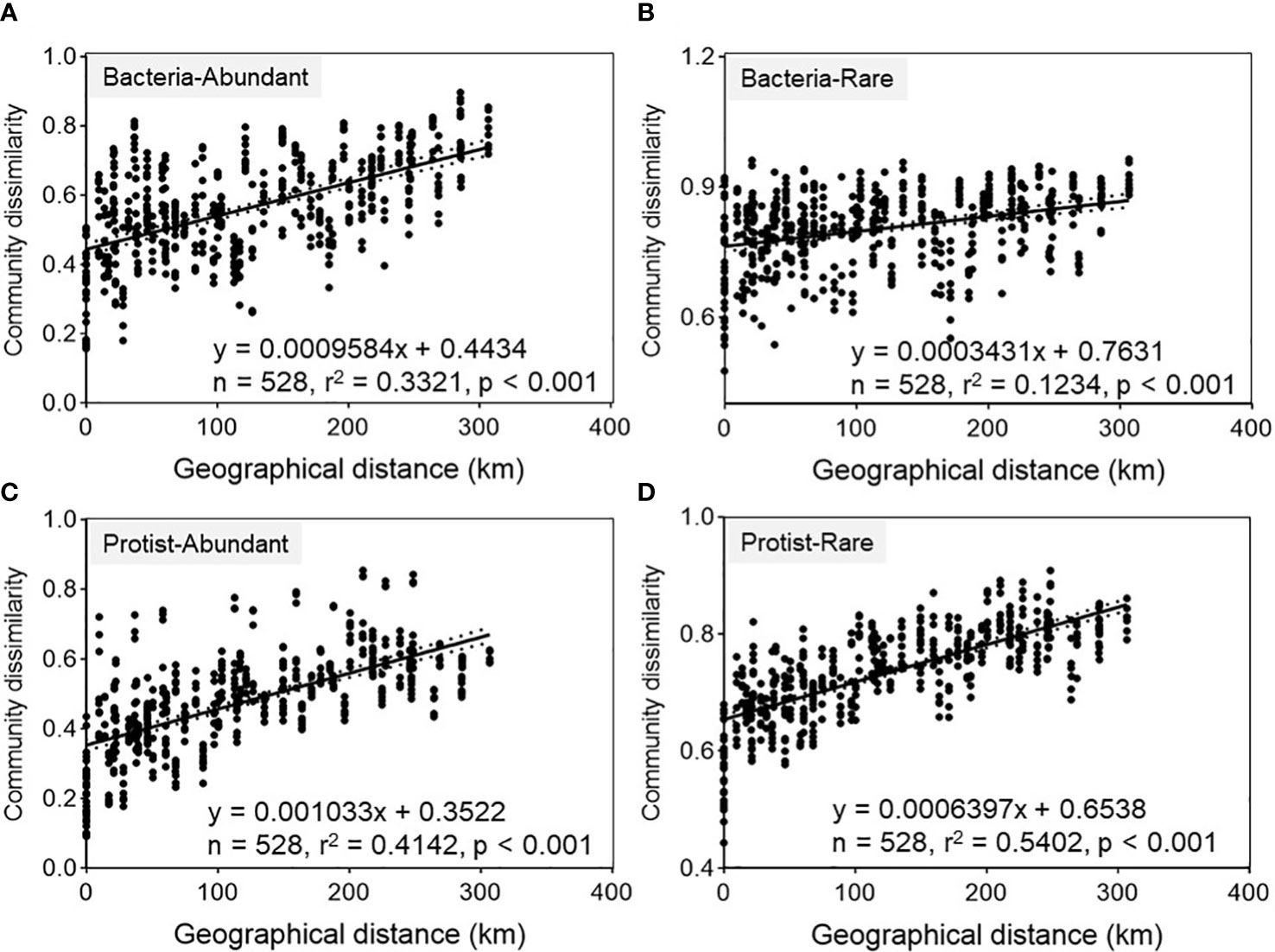
Figure 2 Spearman’s correlations between geographical distance and Bray-Curtis dissimilarity of bacteria-abundant (A), bacteria-rare (B), protist-abundant (C) and protist-rare (D) subcommunity (with n representing the number of comparisons).
To gain a comprehension of the assembly mechanisms that drive the biogeographical pattern of the abundant and rare subcommunities, the extent to which ecological processes contribute to the assembly of bacteria and protists was assessed for the Chishui River ecosystem. In either abundant or rare subcommunities, significant (p <0.001) phylogenetic signals were shown for both bacteria and protists (Supplementary Figure 11). The rare protist subcommunity was shaped by both deterministic (44.51%) and stochastic (55.49%) processes, while the abundant bacteria, abundant protists, and rare bacteria were mainly driven by stochastic processes (78.87%, 82.2%, and 80.87%, respectively). Additionally, dispersal limitation was identified as the primary factor governing the assembly of bacterial subcommunities (abundant vs rare: 77.08% vs 69.89%), and the abundant protistan subcommunity (82.87%). Comparatively, the rare protistan subcommunity was remarkably affected by a heterogeneous selection process (44.32%) (Figure 3A).
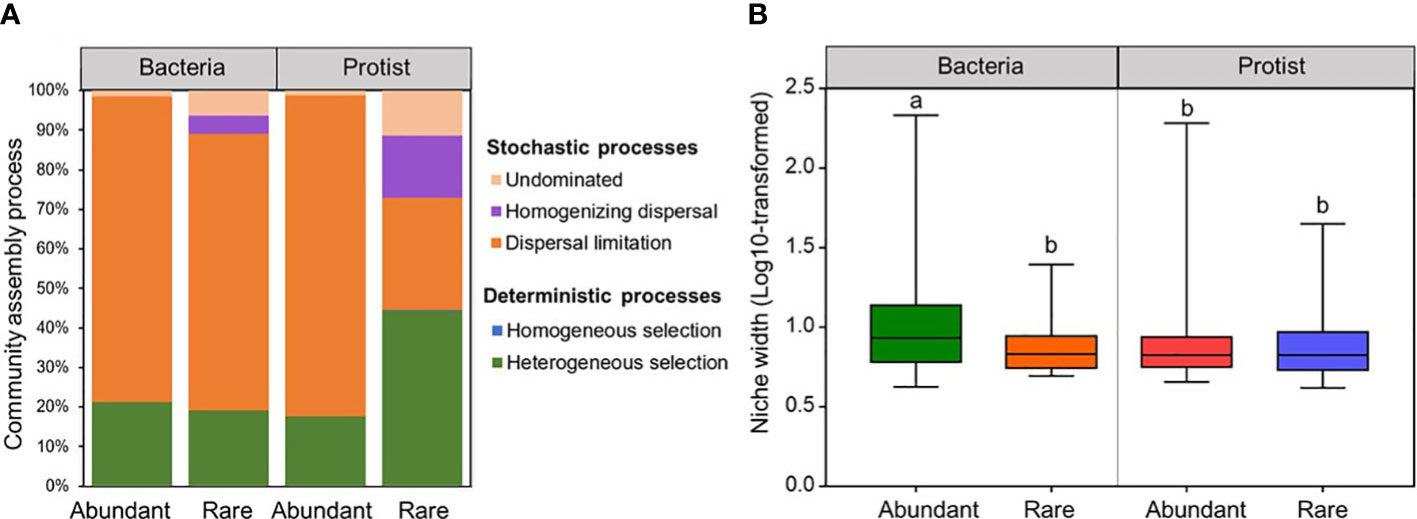
Figure 3 The ecological process (A) and niche width (B) (log10 transformed) of the bacterial and protistan subcommunities in the Chishui River water.
3.4 Driving factors of assembly processes for the microbial subcommunities in the Chishui River
Niche widths were investigated to study the abundant and rare subcommunities which adapted to environmental changes. The water habitat niche widths of the abundant bacterial subcommunity in the Chishui River were significantly (p < 0.05) higher than those of the rare bacterial and protistan subcommunities. However, the protist showed insignificant (p > 0.05) differences between its abundant and rare subcommunities (Figure 3B). These findings showed that the abundant bacterial subcommunity likely possesses higher metabolic flexibility in comparison to other subcommunities at the community level. A correlation analysis was performed between the abundant and rare subcommunities and environmental factors for further exploring the environmental drivers in our dataset. Multiple factors, as determined by Spearman’s correlation analysis, played a significant role in structuring the diversity of bacterial and protistan subcommunities in the Chishui River ecosystem. Specifically, significant factors included elevation, temperature, conductivity, dissolved oxygen, and barium (Supplementary Table 1). For example, elevation showed a significant (p < 0.05) positive correlation with Shannon diversity of all microbial subcommunities but an inverse correlation with temperature (Supplementary Table 1). These findings were further confirmed by the Mantel test, which showed a significant (p < 0.05) relationship between the β-diversity of both the abundant and rare subcommunities of bacteria and protists with elevation, temperature, and conductivity (Figures 4A, B). Heavy metals represented a weak correlation with the microbial subcommunities in the Chishui River ecosystem. The Adonis analysis showed that elevation caused the most significant effect on the diversity of microbial subcommunities. Protists were more affected by elevation than bacteria (Figure 4C). Turbidity, pH, conductivity, and temperature followed elevation in their contribution to the observed variation among subcommunities, while heavy metals showed a trivial effect (Figure 4C). These results suggested that the subcommunities showed distinct preferences for different environmental conditions. Additionally, the abundant bacterial and protistan subcommunities showed a more significant relationship with environmental factors than the rare subcommunity.
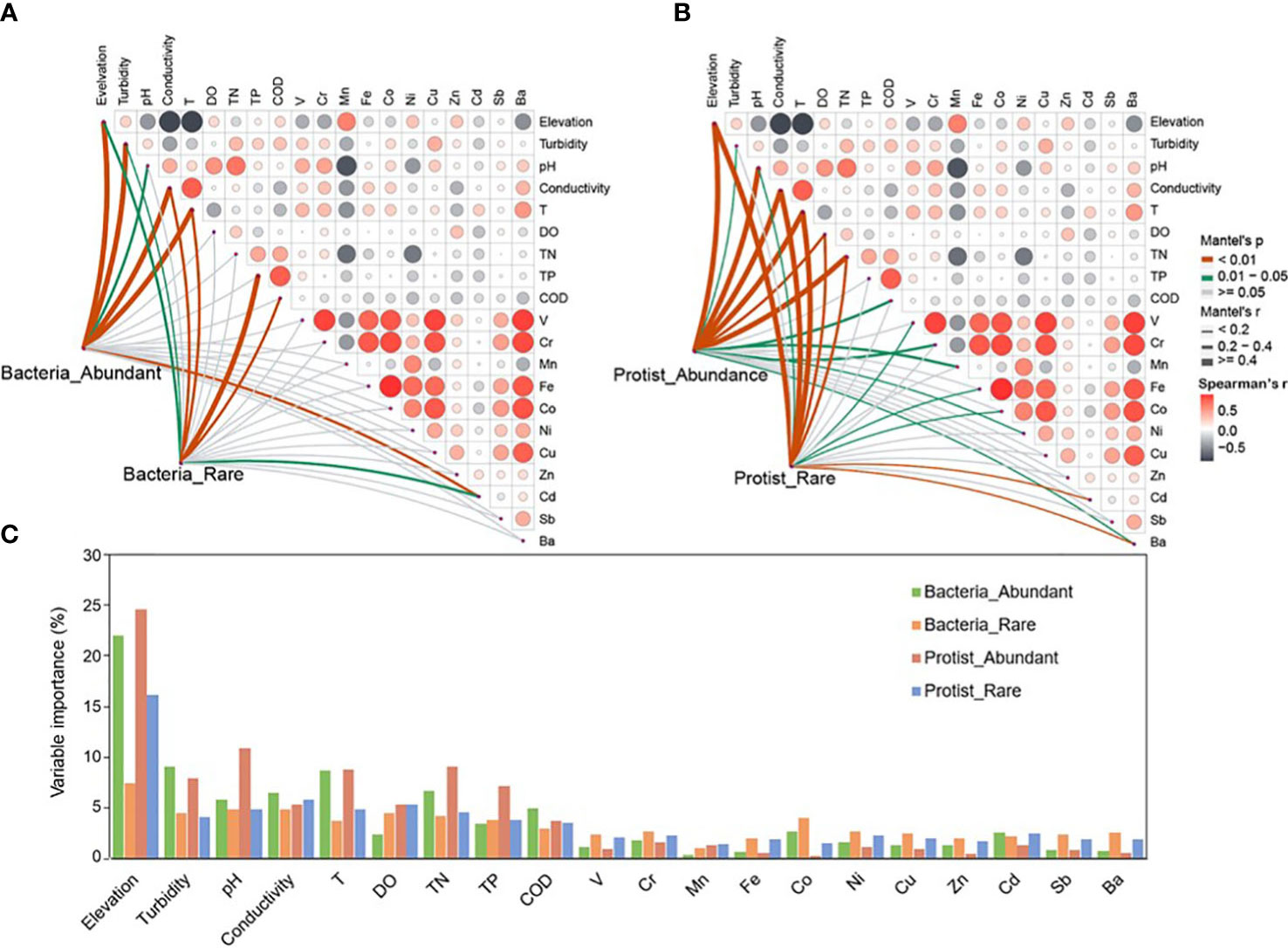
Figure 4 Environmental drivers of bacterial (A) and protistan (B) subcommunities composition in the Chishui River water. The Spearman’s correlation coefficients between environmental factors are displayed using a color gradient in the pairwise comparisons. Partial mantel tests were employed to examine the relationship between the composition and each environmental factor, and to evaluate the potential drivers’ contribution to the variation of abundant and rare taxa and different subcommunities in the water of the Chishui River (C).
A correlation analysis was conducted using main variables such as elevation, temperature, pH, turbidity, and conductivity (Figure 4; Supplementary Table 1) to investigate the correlation between environmental variables and the assembly process of these microbial subcommunities. Our results represented a remarkable (p < 0.05) positive relationship between temperature and βNTI. It suggested that higher temperatures affected the assembly of the protistan subcommunities as a deterministic process (Figures 5A, B). A similar correlation was observed between βNTI of abundant bacteria and temperature (Figure 5C). Additionally, both elevation and conductivity for rare protists showed a highly positive correlation with βNTI (Figures 5D, E). With the increase in Δtemperature, Δelevation, and Δconductivity, the ecological process of rare protistan subcommunity changed from stochastic to deterministic processes.
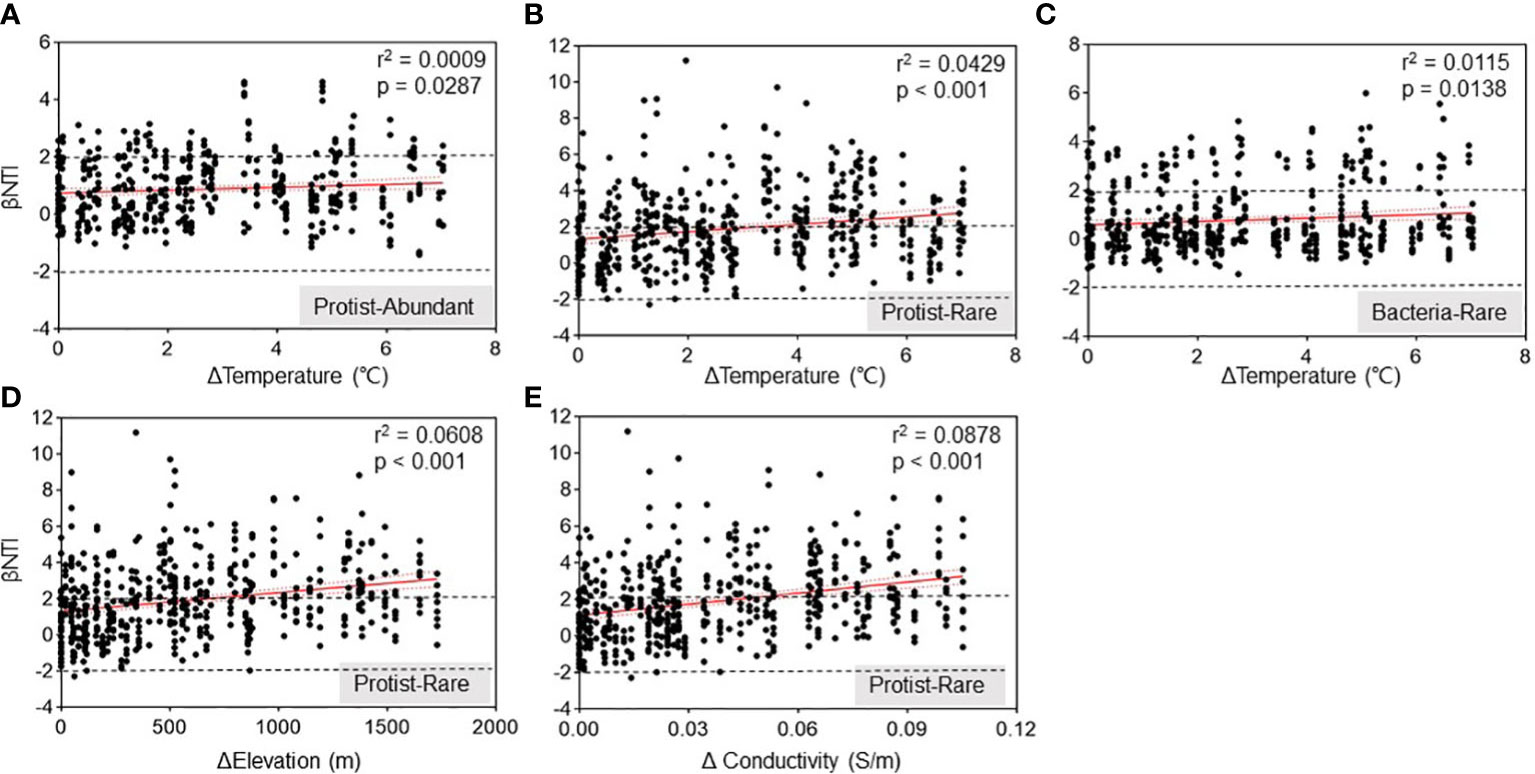
Figure 5 Spearman’s correlations between βNTI of the subcommunities and variations in Chishui River water. Relationships between βNTI and variations in water temperature for protistan abundant (A), protistan rare (B) and bacterial abundant (C) subcommunity, respectively. Relationships between βNTI and variations in elevation (D) and water conductivity (E) for protistan rare subcommunity, respectively. Δ: changes of water conditions among different samples.
3.5 Co-occurrence network analysis of microbial subcommunities in the Chishui River
The metacommunity co-occurrence networks were constructed to evaluate the co-occurrence among abundant and rare bacteria and protists in the Chishui River ecosystem. The network of the microbial subcommunities was established using Spearman’s correlation coefficients (Figure 6; Supplementary Table 2). The empirical networks for bacterial and protistan subcommunities showed higher average clustering coefficient, harmonic geodesic distance, and modularity compared with their random networks (Supplementary Table 2). According to the results, the co-occurrence network of microbial communities in the river showed a non-random modular structure. This structure was primarily influenced by the taxonomic similarity of the coexisting species. Network complexity was found to be higher for the abundant subcommunity compared to the rare subcommunity, with the average degree for the Chishui River bacterial and protistan community networks being 11.712, 2.46, and 5.178, 2.38 (Supplementary Table 2), respectively. The bacterial subcommunity networks were higher and more complicated than those of the protistan subcommunities, with statistical significance (p < 0.05) indicated by several network topological metrics. Further examination of the networks of these subcommunities (Figure 6) showed that the abundant subcommunity for bacteria and protists was characterized by positive links, and the network modularity was >0.451 in all networks (Supplementary Table 2). However, the rare subcommunity was identified by negative links. Additionally, potential keystones were characterized in the microbial subcommunities of the Chishui River by using the values of within-module connectivity (Zi) and among-module connectivity (Pi). A total of 3, 5, 2, and 17 connectors were observed, respectively, for the abundant bacteria, rare bacteria, abundant protist and rare protist. (Figure 7; Supplementary Table 3). The potential keystone taxa in the rare protistan subcommunity were mostly Eukaryota (e.g., the OTU_9879, OTU_5441, OTU_653, OTU_6981, OTU_792, OTU_1043, OTU_2290, OTU_1090, OTU_1438, OTU_1180, OTU_1217, OTU_881, and OTU_3362). The results showed that these bacterial and protistan subcommunities possessed unique network configurations and topological roles for each OTU and potential keystone taxa.
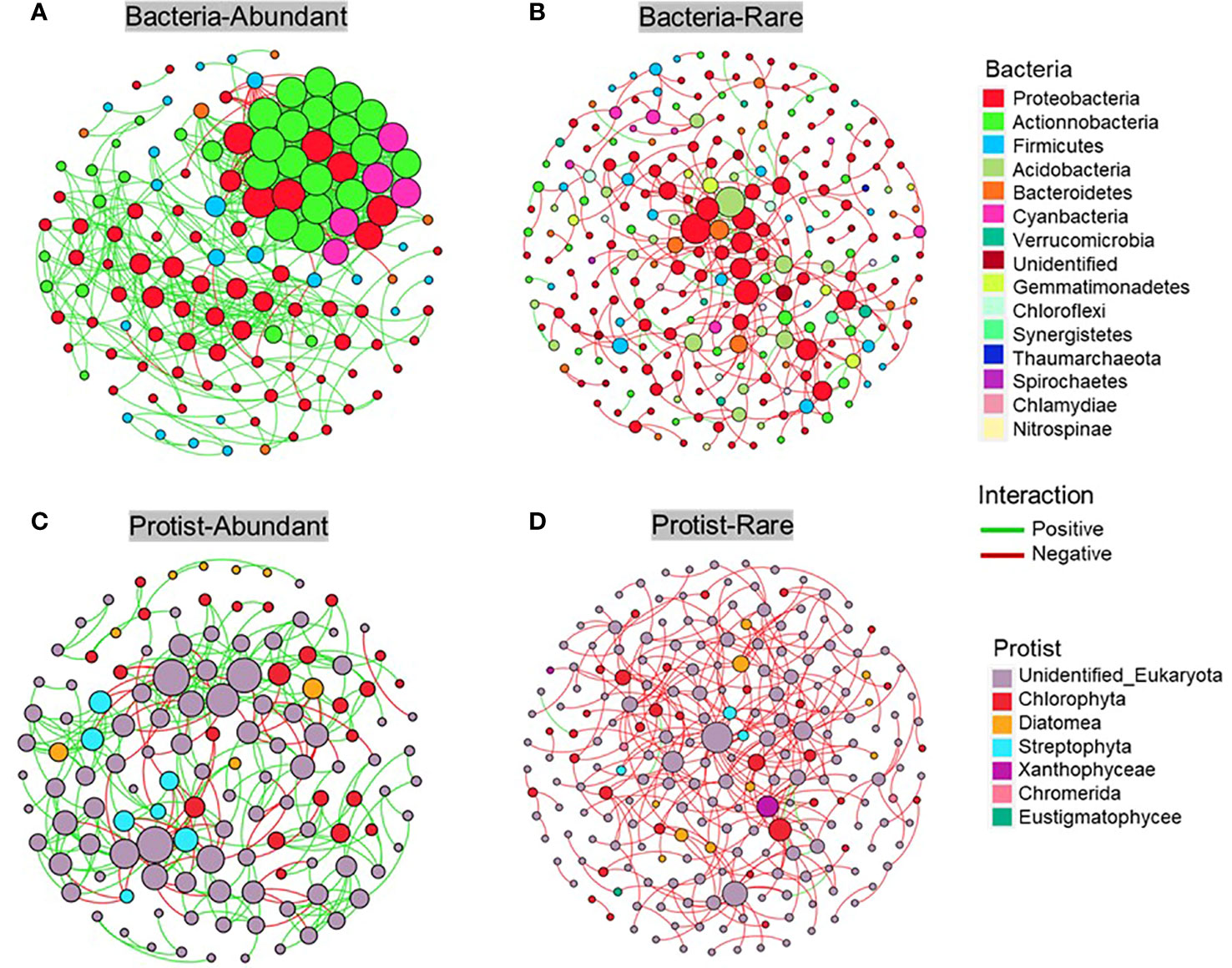
Figure 6 The correlation-based network displays the associations within subcommunities, nodes represent individual Operational Taxonomic Units (OTUs) and their respective phyla. Co-occurrence network of bacterial abundant (A), bacterial rare (B), protistan abundant (C) and protistan rare (D) subcommunity, respectively.
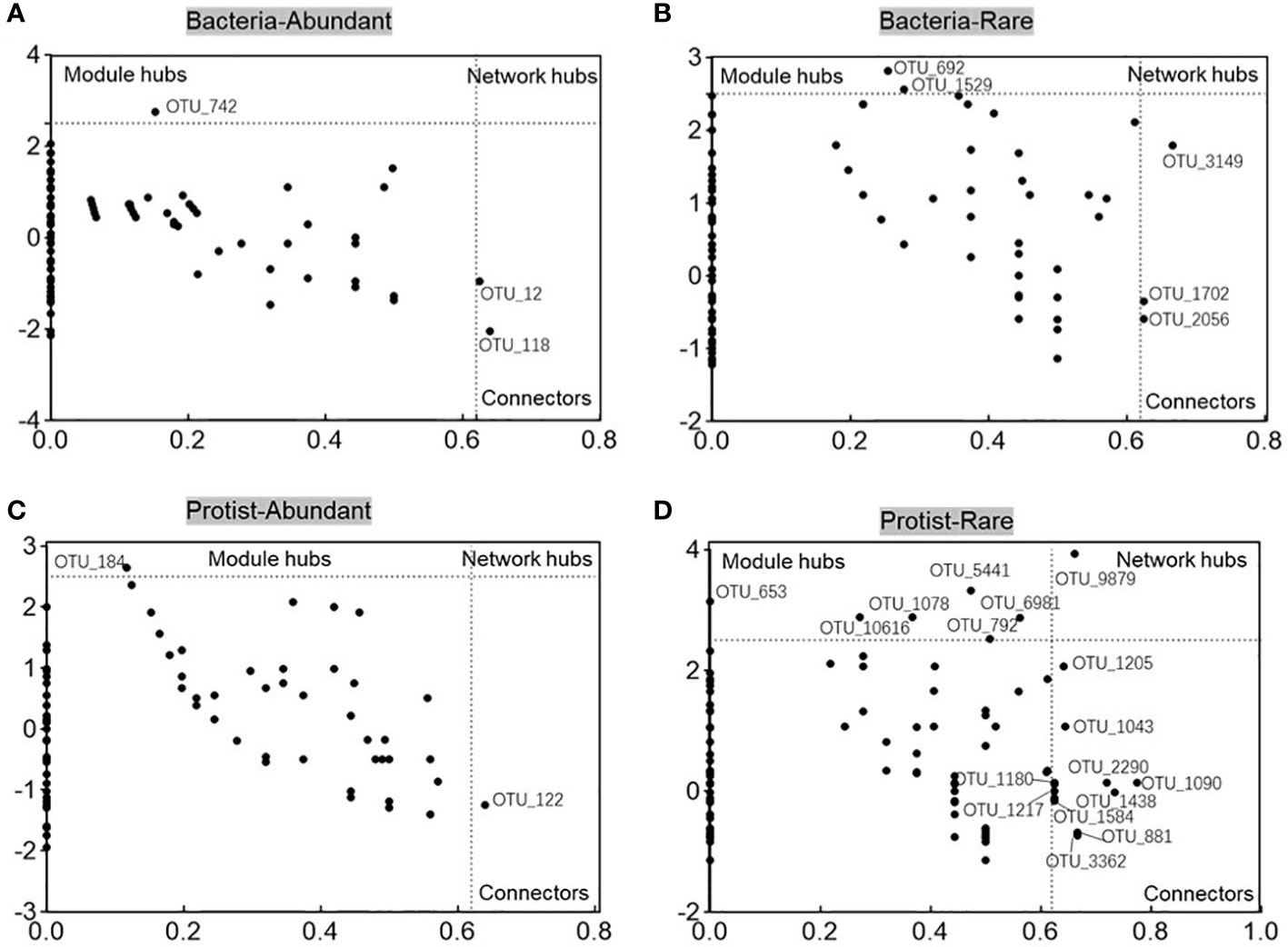
Figure 7 The Zi-Pi plots depict the dispersion of OTUs based on their topological positions as potential keystone species within bacterial abundant (A), bacterial rare (B), protistan abundant (C), and protistan rare (D) subcommunities.
4 Discussion
Acquiring knowledge about the dissimilarities in assembly procedures between abundant and rare subcommunities is of utmost importance due to the modifications in river ecosystems. In this work, the ecological mechanisms and the biogeographical distribution of bacterial and protistan subcommunities was investigated, regulating their assembly in the Chishui River. Our findings showed that the biogeography of abundant and rare subcommunities of bacteria and protists in the Chishui River was comparable. Additionally, our results showed that dispersal limitation caused a considerable impact on generating the assembly of the abundant bacterial and protistan subcommunities. Also, selective pressures were the primary driving force behind the rare protistan subcommunity. These findings were essential to comprehend the mechanisms in establishing and sustaining the microbial diversity of karst rivers, particularly under global environmental changes.
A thorough understanding of the microbial biogeography and assembly mechanisms of abundant and rare subcommunities is vital to predicting the health of river ecosystems and water quality. Despite being lower prevalent, rare subcommunities showed a considerably higher population higher α-diversity compared to abundant subcommunities. These results emphasized the significance of taking into account the rare microbial taxa for investigating the general diversity and health of the river ecosystems (Liang et al., 2020). Also, earlier studies demonstrated that rare species could function as a protection for the entire community in the event of external disturbances (Chen et al., 2020). Additionally, evidence showed that the rare subcommunity displayed a lower level competition for nutrients compared to the abundant subcommunity, which likely led to a decrease in the α-diversity of the overall composition (Xue et al., 2018). According to our results, the abundant and rare subcommunities of bacteria and protists showed similar spatial patterns alongside the Chishui River. Similar observations were obtained in previous studies for the large river in the Cascade Reservoirs (Chen et al., 2020), the Yangtze River (Wang Y. et al., 2020), and the river networks of the Taihu Basin (Zhang et al., 2022). The abundant bacterial and protistan subcommunities were likely the dominant taxa in the water of the Chishui River, and these subcommunities showed lower community dissimilarity (Xue et al., 2018). Furthermore, these findings implied that the rare subcommunities of either bacteria or protists were not collected randomly. Also, it showed that these subcommunities responded similarly to environmental changes in the river, just like the abundant subcommunities (Liao et al., 2016; Zhang et al., 2022).
The study of the mechanisms that govern assembly is a critical area of microbial ecology and is essential for assessing the adaptation of microbial communities to local environmental changes (Stegen et al., 2013; Zhou et al., 2013). The composition of the abundant and rare subcommunities for bacteria and protists was primarily determined by stochastic processes, which showed greater resilience compared to deterministic processes. It was consistent with studies for the Yarlung Tsangpo River (Wang et al., 2021), lakes and reservoirs (Liu et al., 2015), in which the microbial subcommunities were predominantly affected by the dispersion. There were several interpretations that could be associated with this observation. First, the abundant species were likely to have greater dispersal abilities and were more likely to experience significant immigration compared to rare species (Chen et al., 2020; Jiao and Lu, 2020). Additionally, the abundant species had a wider range of nutrient resources available to them than the rare species, which were often restricted to specific substrates (Jia et al., 2018). It could make the dispersal of abundant species easier to colonize in new environments (Vergin et al., 2013; Jia et al., 2018). However, previous studies have suggested that the dispersion of rare subcommunities was limited in subtropical bays (Mo et al., 2018), salt marsh ecosystems (Du et al., 2020), and oil- contaminated soils (Jiao et al., 2017), and Plitvice Lakes (Čačković et al., 2023). The variation in habitats and spatial scales likely accounts for the difference in results (Shi et al., 2018). It is imperative to conduct further research that investigates the effect of stochastic factors such as extinctions or mutations to develop our insight into the distribution of microbial communities in karst rivers. The balance between stochastic and deterministic ecological processes has been remarkably influenced by environmental factors (Jiao et al., 2020). Previous studies have showed that various factors (including pH (Cao et al., 2017), elevation (Liu et al., 2018), temperature and nutrients (Chen et al., 2021; Geisen et al., 2021), the surrounding landscape (Kolmakova et al., 2014), and the remaining time of water in a particular location [Read et al., 2015) or additional stress factors (Hosen et al., 2014)], could directly or indirectly affect the makeup of the entire bacterial or microbial communities. However, there is still no consensus on the specific drivers that influence microbial communities in different river ecosystems. The current insufficient empirical evidence highlights the need to evaluate the effect of drivers on the structures of both abundant and rare subcommunities in river ecosystems. However, the elevation, temperature, and conductivity in either abundant or rare subcommunities were the primary drivers for bacteria and protists, while elevation had the most significant effect on their structures. Furthermore, these findings were consistent with those of a major river bacterioplankton community (Chen et al., 2020), where the environmental factors showed a higher effect on the structure of abundant subcommunity compared to rare subcommunity for both bacteria and protists. This observation likely explained the broader niche breadth of abundant bacterial subcommunities compared to rare bacterial subcommunities. The significance of function and composition could not be neglected for considering the stability of the microbial community (Wang Y, et al., 2020; Wang et al., 2021). However, there were limited studies on microbial function compared with taxonomic composition. It was mainly due to the challenges in discerning between abundant and rare subcommunities (Wang et al., 2021). In the future, it is essential to conduct research that simultaneously reveals the biogeographical pattern of both taxonomy and function. It will help distinguish between abundant and rare subcommunities to maintain ecological stability in river ecosystems.
Within the network of co-occurring microbial communities, positive interactions between different species were commonly observed as cooperative relationships. However, our results showed that positive connections were prevalent in the abundant subcommunities of bacterial and protistan communities. The negative connections were more common in the rare subcommunity. Therefore, the frequent cooperation in either the abundant or rare subcommunities likely bolstered their resilience to environmental disturbances by serving as a safeguard against potential disruptions (Konopka et al., 2015). These results likely provided insight into the wider niche breadth of the abundant bacterial subcommunity. A higher number of keystone taxa in bacterial and protistan rare subcommunities compared with abundant subcommunities showed that the rare subcommunity likely had a crucial effect on the overall health and stability of the community. It was consistent with a prior investigation of bacterial communities in the Yangtze River (Wang Y, et al., 2020). Differing from abundant taxa, studies have shown that metabolically active microbes are more commonly found among rare ones. These rare taxa could serve as keystones with a significant effect on regulating the aquatic ecosystem functions (Lynch and Neufeld, 2015). Although the population of rare species was not as much as that of abundant taxa, evidence suggested that they could serve as an inexhaustible source of genetic diversity and perform crucial functions in ecological systems (Galand et al., 2009; Jia et al., 2018). Furthermore, rare taxa possessed the ability to survive and become prevalent in favorable environments (Shade et al., 2014), which could help the durability and stability of a community (Shade and Gilbert, 2015). These results suggested that rare subcommunities could play a critical role in the community composition (Ma et al., 2016). Therefore, it could be inferred that the rare subcommunities was likely more crucial compared with abundant subcommunities in preserving the overall composition of the community.
5 Conclusions
In this study, the assemblage of bacteria and protists in the Chishui River was systematically investigated by analyzing abundant and rare subcommunities. We confirmed that abundant and rare subcommunities along the Chishui River showed comparable biogeographic trends, regardless of whether they were bacteria or protists. Stochastic processes were the primary driving force behind the structures of abundant subcommunities for both bacteria and protists. A more pronounced dispersal limitation was observed in the structuring of the abundant subcommunity compared to the rare subcommunity. Also, rare bacteria were more susceptible to this process than rare protists. According to our results, changes in elevation, temperature, and conductivity of either abundant or rare subcommunities were the main drivers of the structure of bacteria and protists. Additionally, the co-occurring networks revealed that the occurrence patterns of bacteria and protists were not random. The rare subcommunity had a higher number of keystones, indicating a important role in maintaining the stability and durability of the microbial communities. Our study provided important insights into how abundant and rare microbial species adjusted to the variations in the surroundings, and it may help predict the potential loss of biodiversity by anthropogenic activities and climate change in karst riverine ecosystems.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
RW, KL, and BM conceptualized and supervised the study. RW and KL contributed to the funding acquisition and project administration. YW, YZ, WZ and ZW investigated the study and performed the resources. YW, HF, and CW contributed to the formal analysis. YW, ZH, and BM visualized the manuscript. YW wrote the original draft of the manuscript. All authors contributed to the data analysis and interpretation and commented on the manuscripts. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Basic and Applied Basic Research Foundation of Guangdong Province (2020A1515011109), and Special Basic Research Fund for Central Public Research Institutes of China (PM-zx703-202104-103, PM-zx703-202104-128).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1228813/full#supplementary-material
References
Bai S., Zhang J., Qi X., Zeng J., Wu S., Peng X. (2023). Changes of in situ prokaryotic and eukaryotic communities in the upper Sanya river to the sea over a nine-hour period. Microorganisms 11 (536), 1–19. doi: 10.3390/genes1030413
Buckerfield S. J., Waldron S., Quilliam R. S., Naylor L. A., Li S., Oliver D. M. (2019). How can we improve understanding of faecal indicator dynamics in karst systems under changing climatic, population, and land use stressors? - Research opportunities in SW China. Sci. Total Environ. 646, 438–447. doi: 10.1016/j.scitotenv.2018.07.292
Čačković A., Kajan K., Selak L., Marković T., Brozičević A., Pjevac P., et al. (2023). Hydrochemical and seasonally conditioned changes of microbial communities in the tufa-forming freshwater network ecosystem. mSphere 8 (3), 1–17. doi: 10.1128/msphere.00602-22
Cao X., Wang J., Liao J., Gao Z., Jiang D., Sun J., et al. (2017). Bacterioplankton community responses to key environmental variables in plateau freshwater lake ecosystems: A structural equation modeling and change point analysis. Sci. Total Environ. 580, 457–467. doi: 10.1016/j.scitotenv.2016.11.143
Carraro L., Hartikainen H., Jokela J., Bertuzzo E., Rinaldo A. (2018). Estimating species distribution and abundance in river networks using environmental DNA. PNAS 115 (46), 11724–11729. doi: 10.1073/pnas.1813843115
Chen Q., Chen Y., Yang J., Maberly S. C., Zhang J., Ni J., et al. (2021). Bacterial communities in cascade reservoirs along a large river. Limnol. Oceanogr. 66 (12), 4363–4374. doi: 10.1002/lno.11967
Chen W., Ren K., Isabwe A., Chen H., Liu M., Yang J. (2019). Stochastic processes shape microeukaryotic community assembly in a subtropical river across wet and dry seasons. Microbiome 7 (1), 138. doi: 10.1186/s40168-019-0749-8
Chen J., Wang P., Wang C., Wang X., Miao L., Liu S., et al. (2020). Distinct assembly mechanisms underlie similar biogeographic patterns of rare and abundant bacterioplankton in cascade reservoirs of a large river. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.00158
Cheng X., Yun Y., Wang H., Ma L., Tian W., Man B., et al. (2021). Contrasting bacterial communities and their assembly processes in karst soils under different land use. Sci. Total Environ. 751, 142263. doi: 10.1016/j.scitotenv.2020.142263
Cruaud P., Vigneron A., Dorea C. C., Rodriguez M. J., Charette S. J. (2020). Rapid changes in microbial community structures along a meandering river. Microorganisms 8 (11), 1–21. doi: 10.3390/microorganisms8111631
Cruaud P., Vigneron A., Fradette M. S., Dorea C. C., Culley A. I., Rodriguez M. J., et al. (2019). Annual protist community dynamics in a freshwater ecosystem undergoing contrasted climatic conditions: the Saint-Charles river (Canada). Front. Microbiol. 10. doi: 10.3389/fmicb.2019.02359
Deng Y., Jiang Y.-H., Yang Y., He Z., Luo F., Zhou J. (2012). Molecular ecological network analyses. BMC Bioinf. 13 (113), 1–20. doi: 10.1186/1471-2105-13-113
Du S., Dini-Andreote F., Zhang N., Liang C., Yao Z., Zhang H., et al. (2020). Divergent co-occurrence patterns and assembly processes structure the abundant and rare bacterial communities in a salt marsh ecosystem. Appl. Environ. Microbiol. 86 (13), e00322-20. doi: 10.1128/AEM.00322-20
Edgar R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10 (10), 996–998. doi: 10.1038/nmeth.2604
Ender A., Goeppert N., Goldscheider N. (2018). Hydrogeological controls of variable microbial water quality in a complex subtropical karst system in Northern Vietnam. Hydrogeol. J. 26 (7), 2297–2314. doi: 10.1007/s10040-018-1783-5
Feng K., Zhang Z., Cai W., Liu W., Xu M., Yin H., et al. (2017). Biodiversity and species competition regulate the resilience of microbial biofilm community. Mol. Ecol. 26 (21), 6170–6182. doi: 10.1111/mec.14356
Galand P. E., Casamayor E. O., Kirchman D. L., Lovejoy C. (2009). Ecology of the rare microbial biosphere of the Arctic Ocean. PNAS 106 (52), 22427–22432. doi: 10.1073/pnas.0908284106
Gao G.-F., Peng D., Tripathi B., Zhang and Chu Y. (2020). Distinct community assembly processes of abundant and rare soil bacteria in coastal wetlands along an inundation gradient. mSystems 5 (6), e01150–e01120. doi: 10.1128/mSystems.01150-20
Geisen S., Hu S., Dela Cruz T. E. E., Veen G. F. C. (2021). Protists as catalyzers of microbial litter breakdown and carbon cycling at different temperature regimes. ISME J. 15 (2), 618–621. doi: 10.1038/s41396-020-00792-y
Guillou L., Bachar D., Audic S., Bass D., Berney C., Bittner L., et al. (2013). The Protist Ribosomal Reference database (PR2): a catalog of unicellular eukaryote small sub-unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 41 (Database issue), D597–D604. doi: 10.1093/nar/gks1160
Guimerà R., Amaral L. A. N. (2005). Functional cartography of complex metabolic networks. Nature 25 (433(7028), 895–900. doi: 10.1038/nature03288
Gweon H. S., Bowes M. J., Moorhouse H. L., Oliver A. E., Bailey M. J., Acreman M. C., et al. (2021). Contrasting community assembly processes structure lotic bacteria metacommunities along the river continuum. Environ. Microbiol. 23 (1), 484–498. doi: 10.1111/1462-2920.15337
Hirakata Y., Hatamoto M., Oshiki M., Watari T., Araki N., Yamaguchi T. (2020). Food selectivity of anaerobic protists and direct evidence for methane production using carbon from prey bacteria by endosymbiotic methanogen. ISME J. 14 (7), 1873–1885. doi: 10.1038/s41396-020-0660-0
Hosen J. D., Febria C. M., Crump B. C., Palmer M. A. (2017). Watershed urbanization linked to differences in stream bacterial community composition. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.01452
Hosen J. D., McDonough O. T., Febria C. M., Palmer M. A. (2014). Dissolved organic matter quality and bioavailability changes across an urbanization gradient in headwater streams. Environ. Sci. Technol. 48 (14), 7817–7824. doi: 10.1021/es501422z
Hou J., Wu L., Liu W., Ge Y., Mu T., Zhou T., et al. (2020). Biogeography and diversity patterns of abundant and rare bacterial communities in rice paddy soils across China. Sci. Total Environ. 730, 139116. doi: 10.1016/j.scitotenv.2020.139116
Husnik F., Tashyreva D., Boscaro V., George E. E., Lukes J., Keeling P. J. (2021). Bacterial and archaeal symbioses with protists. Curr. Biol. 31 (13), R862–R877. doi: 10.1016/j.cub.2021.05.049
Jia X., Dini-Andreote F., Falcao Salles J. (2018). Community assembly processes of the microbial rare biosphere. Trends Microbiol. 26 (9), 738–747. doi: 10.1016/j.tim.2018.02.011
Jiao S., Chen W., Wei G. (2017). Biogeography and ecological diversity patterns of rare and abundant bacteria in oil-contaminated soils. Mol. Ecol. 26 (19), 5305–5317. doi: 10.1111/mec.14218
Jiao S., Lu Y. (2020). Abundant fungi adapt to broader environmental gradients than rare fungi in agricultural fields. Glob Chang. Biol. 26 (8), 4506–4520. doi: 10.1111/gcb.15130
Jiao S., Yang Y., Xu Y., Zhang J., Lu Y. (2020). Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across eastern China. ISME J. 14 (1), 202–216. doi: 10.1038/s41396-019-0522-9
Kolmakova O. V., Gladyshev M. I., Rozanov A. S., Peltek S. E., Trusova M. Y. (2014). Spatial biodiversity of bacteria along the largest Arctic river determined by next-generation sequencing. FEMS Microbiol. Ecol. 89 (2), 442–450. doi: 10.1111/1574-6941.12355
Konopka A., Lindemann S., Fredrickson J. (2015). Dynamics in microbial communities: unraveling mechanisms to identify principles. ISME J. 9 (7), 1488–1495. doi: 10.1038/ismej.2014.251
Li L., Pujari L., Wu C., Huang D., Wei Y., Guo C., et al. (2021). Assembly processes and co-occurrence patterns of abundant and rare bacterial community in the Eastern Indian Ocean. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.616956
Liang Y., Xiao X., Nuccio E. E., Yuan M., Zhang N., Xue K., et al. (2020). Differentiation strategies of soil rare and abundant microbial taxa in response to changing climatic regimes. Environ. Microbiol. 22 (4), 1327–1340. doi: 10.1111/1462-2920.14945
Liao J., Cao X., Wang J., Zhao L., Sun J., Jiang D., et al. (2016). Similar community assembly mechanisms underlie similar biogeography of rare and abundant bacteria in lakes on Yungui Plateau, China. Limnol. Oceanogr. 62 (2), 723–735. doi: 10.1002/lno.10455
Liu L., Yang J., Yu Z., Wilkinson D. M. (2015). The biogeography of abundant and rare bacterioplankton in the lakes and reservoirs of China. ISME J. 9 (9), 2068–2077. doi: 10.1038/ismej.2015.29
Liu T., Zhang A. N., Wang J., Liu S., Jiang X., Dang C., et al. (2018). Integrated biogeography of planktonic and sedimentary bacterial communities in the Yangtze River. Microbiome 6 (1), 16. doi: 10.1186/s40168-017-0388-x
Lu L., Zou X., Yang J., Xiao Y., Wang Y., Guo J., et al. (2020). Biogeography of eukaryotic plankton communities along the upper Yangtze River: The potential impact of cascade dams and reservoirs. J. Hydrol. 590, 125495. doi: 10.1016/j.jhydrol.2020.125495
Lynch M. D., Neufeld J. D. (2015). Ecology and exploration of the rare biosphere. Nat. Rev. Microbiol. 13 (4), 217–229. doi: 10.1038/nrmicro3400
Ma B., Wang H., Dsouza M., Lou J., He Y., Dai Z., et al. (2016). Geographic patterns of co-occurrence network topological features for soil microbiota at continental scale in eastern China. ISME J. 10 (8), 1891–1901. doi: 10.1038/ismej.2015.261
Magoc T., Salzberg S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27 (21), 2957–2963. doi: 10.1093/bioinformatics/btr507
Menning D. M., Carraher-Stross W. A., Graham E. D., Thomas D. N., Phillips A. R., Scharping R. J., et al. (2017). Aquifer discharge drives microbial community change in karst estuaries. Estuar. Coasts. 41 (2), 430–443. doi: 10.1007/s12237-017-0281-7
Mo Y., Zhang W., Yang J., Lin Y., Yu Z., Lin S. (2018). Biogeographic patterns of abundant and rare bacterioplankton in three subtropical bays resulting from selective and neutral processes. ISME J. 12 (9), 2198–2210. doi: 10.1038/s41396-018-0153-6
Nemergut D. R., Costello E. K., Hamady M., Lozupone C., Jiang L., Schmidt S. K., et al. (2011). Global patterns in the biogeography of bacterial taxa. Environ. Microbiol. 13 (1), 135–144. doi: 10.1111/j.1462-2920.2010.02315.x
Pan H., Gao H., Peng Z., Chen B., Chen S., Liu Y., et al. (2022). Aridity threshold induces abrupt change of soil abundant and rare bacterial biogeography in dryland ecosystems. mSystems 7 (1), e0130921. doi: 10.1128/msystems.01309-21
Pikitch E. K. (2018). A tool for finding rare marine species. Science 360 (6394), 1180–1182. doi: 10.1126/science.aao3787
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41 (Database issue), D590–D596. doi: 10.1093/nar/gks1219
Read D. S., Gweon H. S., Bowes M. J., Newbold L. K., Field D., Bailey M. J., et al. (2015). Catchment-scale biogeography of riverine bacterioplankton. ISME J. 9 (2), 516–526. doi: 10.1038/ismej.2014.166
Shade A., Gilbert J. A. (2015). Temporal patterns of rarity provide a more complete view of microbial diversity. Trends Microbiol. 23 (6), 335–340. doi: 10.1016/j.tim.2015.01.007
Shade A., Jones S. E., Caporaso J. G., Handelsman J., Knight R., Fierer N., et al. (2014). Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio 5 (4), e01371–e01314. doi: 10.1128/mBio.01371-14
Shen C., Gunina A., Luo Y., Wang J., He J. Z., Kuzyakov Y., et al. (2020). Contrasting patterns and drivers of soil bacterial and fungal diversity across a mountain gradient. Environ. Microbiol. 22 (8), 3287–3301. doi: 10.1111/1462-2920.15090
Shi Y., Li Y., Xiang X., Sun R., Yang T., He D., et al. (2018). Spatial scale affects the relative role of stochasticity versus determinism in soil bacterial communities in wheat fields across the North China Plain. Microbiome 6 (1), 27. doi: 10.1186/s40168-018-0409-4
Stegen J. C., Lin X., Fredrickson J. K., Chen X., Kennedy D. W., Murray C. J., et al. (2013). Quantifying community assembly processes and identifying features that impose them. ISME J. 7 (11), 2069–2079. doi: 10.1038/ismej.2013.93
Stoeck T., Bass D., Nebel M., Christen R., Jones M. D., Breiner H. W., et al. (2010). Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 19 (Suppl 1), 21–31. doi: 10.1111/j.1365-294X.2009.04480.x
Vergin K. L., Done B., Carlson C. A., Giovannoni S. J. (2013). Spatiotemporal distributions of rare bacterioplankton populations indicate adaptive strategies in the oligotrophic ocean. Aquat. Microbial Ecol. 71 (1), 1–13. doi: 10.3354/ame01661
Wang Z., Han M., Li E., Liu X., Wei H., Yang C., et al. (2020). Distribution of antibiotic resistance genes in an agriculturally disturbed lake in China: Their links with microbial communities, antibiotics, and water quality. J. Hazard Mater. 393, 122426. doi: 10.1016/j.jhazmat.2020.122426
Wang X., Wang P., Wang C., Chen J., Hu B., Liu S., et al. (2021). Distinct strategies of abundant and rare bacterioplankton in river-reservoir system: Evidence from a 2800 km plateau river. Environ. Res. 199, 111418. doi: 10.1016/j.envres.2021.111418
Wang Y., Ye F., Wu S., Wu J., Yan J., Xu K., et al. (2020). Biogeographic pattern of bacterioplanktonic community and potential function in the Yangtze River: Roles of abundant and rare taxa. Sci. Total Environ. 747, 141335. doi: 10.1016/j.scitotenv.2020.141335
Wu T., Zhang Y., Wang B., Chen C., Cheng Z., Li Y., et al. (2022). Antibiotic resistance genes in Chishui River, a tributary of the Yangtze River, China: Occurrence, seasonal variation and its relationships with antibiotics, heavy metals and microbial communities. Sci. Total Environ. 846, 157472. doi: 10.1016/j.scitotenv.2022.157472
Wu Y., Zhang Y., Yang X., Li K., Mai B., He Z., et al. (2022). Deterministic processes shape bacterial community assembly in a karst river across dry and wet seasons. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.938490
Xiang S., Wang X., Ma W., Liu X., Zhang B., Huang F., et al. (2020). Response of microbial communities of karst river water to antibiotics and microbial source tracking for antibiotics. Sci. Total Environ. 706, 135730. doi: 10.1016/j.scitotenv.2019.135730
Xue Y., Chen H., Yang J. R., Liu M., Huang B., Yang J. (2018). Distinct patterns and processes of abundant and rare eukaryotic plankton communities following a reservoir cyanobacterial bloom. ISME J. 12 (9), 2263–2277. doi: 10.1038/s41396-018-0159-0
Zhang T., Xu S., Yan R., Wang R., Gao Y., Kong M., et al. (2022). Similar geographic patterns but distinct assembly processes of abundant and rare bacterioplankton communities in river networks of the Taihu Basin. Water Res. 211, 118057. doi: 10.1016/j.watres.2022.118057
Keywords: karst river, biogeography, community assembly, rare bacteria, rare protist, stochastic processes
Citation: Wu Y, Zhang Y, Fang H, Wang C, Wang Z, Zhang W, Mai B, He Z, Wu R and Li K (2023) The assembly, biogeography and co-occurrence of abundant and rare microbial communities in a karst river. Front. Mar. Sci. 10:1228813. doi: 10.3389/fmars.2023.1228813
Received: 25 May 2023; Accepted: 10 July 2023;
Published: 01 August 2023.
Edited by:
Cristina García-Aljaro, University of Barcelona, SpainReviewed by:
Sandi Orlic, Rudjer Boskovic Institute, CroatiaShijie Bai, Institute of Deep-Sea Science and Engineering, Chinese Academy of Sciences (CAS), China
Copyright © 2023 Wu, Zhang, Fang, Wang, Wang, Zhang, Mai, He, Wu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kaiming Li, bGlrYWltaW5nQHNjaWVzLm9yZw==; Renren Wu, d3VyZW5yZW5Ac2NpZXMub3Jn