 Kuo-Jian Ma
Kuo-Jian Ma Yong-Lian Ye
Yong-Lian Ye Yun-Han Fu
Yun-Han Fu Ge-Yi Fu
Ge-Yi Fu Cong Sun
Cong Sun Xue-Wei Xu
Xue-Wei Xu- 1Key Laboratory of Marine Ecosystem Dynamics, Ministry of Natural Resources and Second Institute of Oceanography, Ministry of Natural Resources, Hangzhou, China
- 2Ocean College, Zhejiang University, Zhoushan, China
- 3College of Life Sciences and Medicine, Zhejiang Sci-Tech University, Hangzhou, China
- 4Center of Marine Microbial Resource, Zhejiang Sci-Tech University Shaoxing Academy of Biomedicine Co., Ltd., Shaoxing, China
Special geographical location and abundant organic matter profiles in tidal flats have resulted in great microbial diversity, in which Bacteroidota strains are considered as one of the primary degraders of polysaccharides, playing a crucial role in the carbon cycle. In this study, we collected sediment or sand samples from 34 bare tidal flats in China and investigated the profile of culturable bacteria, selected three Bacteroidota for polyphasic taxonomic analysis and revealed their polysaccharide metabolic potential. Totally, we isolated 352 pure cultured bacteria and they mainly distributed in Bacteroidota, Pseudomonadota, Bacillota, and Actinomycetota. It is shown that the bare tidal flats contained a large number of potential novel species, mainly distributed in Flavobacteriales and Cytophagales within Bacteroidota. Three Bacteroidota strains, M17T, M82T, and M415T, isolated from mudflat were selected for polyphasic taxonomic analysis. The 16S rRNA gene sequence similarity between strain M17T and Mangrovivirga cuniculi KCTC 72349T was 99.28%, and less than 90.09% with other species; strain M82T shared the highest 16S rRNA gene sequence similarity of 97.85% with Pontibacter litorisediminis KCTC 52252T, and less than 97.43% with other species; strain M415T had higher 16S rRNA gene sequence similarities with type species of genera Eudoraea (92.62-93.68%), Zeaxanthinibacter (92.02-92.91%), and Muriicola (92.21-92.83%). Phylogenetic analysis based on 16S rRNA gene sequences and single-copy orthologous clusters showed that strains M17T and M82T represent novel species within the genus Mangrovivirga and Pontibacter, respectively, and strain M415T represents a novel species of a novel genus within the family Flavobacteriaceae. The potential in polysaccharide metabolism of all these three strains was analyzed by genomes. The analysis revealed that glycoside hydrolases and glycosyltransferases account for more than 70% of the total CAZymes. Additionally, the numbers of polysaccharide utilization loci (PULs) and annotated CAZymes in Cytophagales spp. M17T and M82T were found to be higher than those in Flavobacteriales sp. M415T. Highly specialized saccharolytic systems and the presence of numerous diversified CAZymes for obtaining energy through polysaccharide metabolism were speculated to help the three novel strains adapt to the utilization of both terrestrial and marine polysaccharides.
1 Introduction
Tidal flats are located in the intertidal zone between the high- and low-tide levels (Murray et al., 2019; Wang et al., 2020b). Recent research showed that tidal flats occupy at least 127,921 km2 globally, mainly distributed along the coast of Asia, especially China (Murray et al., 2019). Due to their unique geographical location and periodic changes in environmental factors such as salinity, temperature, dissolved oxygen, light intensity, tides, ocean currents, and human disturbance, tidal flats have become one of the most productive and vulnerable environments in the world (Underwood and Kromkamp, 1999; Mayor et al., 2018; Chang et al., 2022). At the same time, tidal flats play important roles in carbon sequestration (Howard et al., 2014; Sasmito et al., 2020), aquaculture (Ni et al., 2020), microbial diversity, and function research (Mayor et al., 2018; Perillo et al., 2018). Lots of studies mainly focused on the special vegetated tidal flats such as mangrove and salt marsh; however, the studies on bare tidal flats (also referred to unvegetated tidal flats) were relatively limited.
With the rapid development of high-throughput sequencing technologies, our understanding of microbial diversity, structure, and function in bare tidal flats has tremendously expanded in recent years (Gong et al., 2019; Zhang et al., 2021; Rinke et al., 2022). Microbial diversity in mudflats is extremely high and significantly varies with different substrates and depths, playing an essential role in organic matter catabolism and even carbon fixation (Molari et al., 2012; Choi et al., 2018; Gaubert-Boussarie et al., 2020; Mohapatra et al., 2021). The degradation and metabolism process involved by bacteria and archaea in bare tidal flats is an important part of the global cycle of carbon, nitrogen, phosphorus, sulfur, and other elements (Ettwig et al., 2010; Bauer et al., 2013), which has a profound impact on the content of atmospheric greenhouse gases and the rate of element cycling.
Although the well-known hypothesis said “only 1% of microbes are culturable” (Amann et al., 1995; Torsvik and Ovreas, 2002), more and more scientists believe that more than 1% of microbes can be culturable (Martiny, 2019; Steen et al., 2019). High-throughput sequencing technologies provide a comprehensive understanding of microbial diversity; pure culture of microorganisms is also of great importance, which can help us to elucidate the physiological mechanism and ecological function of microorganisms, as well as find new metabolic pathways and metabolites (Guo et al., 2006), such as Bacteroidota strains which play important roles in polysaccharide degradation, which account for roughly 75% of the annually renewable biomass (Lichtenthaler and Peters, 2004; Lapebie et al., 2019; Gavriilidou et al., 2020; McKee et al., 2021).
Marine representatives of the phylum Bacteroidota possess diverse enzyme repertoires and flexible polysaccharide metabolism, actively participating in numerous biogeochemical processes (Fernández-Gómez et al., 2013). Highly specialized bacterial strains of the phylum Bacteroidota exhibit prolific proliferation during phytoplankton blooms and serve as primary degraders of microalgal polysaccharides (Unfried et al., 2018). In phylum Bacteroidota, members of the family Flavobacteriaceae exhibit a high proportion and diversity of carbohydrate-active enzymes (CAZymes) within their polysaccharide utilization loci (PULs), which supports their ability to utilize a wide range of polysaccharides (Kappelmann et al., 2019). For instance, Zobellia galactanivorans DsijT has emerged as a model organism for studying polysaccharide degradation in marine flavobacteria (Barbeyron et al., 2016), Z. amurskyensis KMM 3526T and Z. laminariae KMM 3676T possess a relatively high proportion of CAZymes (accounting for 6.49% and 5.93% of all predicted coding sequences, respectively) and are specialized in the degradation of algal polysaccharides (Chernysheva et al., 2019). In this study, we investigated the culturable bacterial proportion in the sediments from 34 sampling stations of bare tidal flats, selected three novel Bacteroidota strains for polyphasic taxonomy, and further analyzed their potential in polysaccharide metabolism.
2 Materials and methods
2.1 Sample collection and strain isolation
All samples were collected by a self-made cylindrical plexiglass tube sampler, transferred into sterile sample tubes after being fully homogenized, stored in a 4°C–6°C incubator, and transported to the laboratory as soon as possible. The distribution of samples is presented in Figure 1. The particle size of the sediments was determined using a laser particle size analyzer, and the sediment types were classified according to the modification of Folk’s classification of sediments: sand (greater than 10% of particles with a diameter less than 63 µm), muddy sand (10%–90% of particles with a diameter less than 63 µm), and mud (less than 10% of particles with a diameter less than 63 µm) (Folk et al., 1970). Strain M17T was isolated from an intertidal mudflat (0–5 cm) collected from Qingdao, Shandong Provence (36°10′ N, 120°07′ E); strains M82T and M415T were isolated from two intertidal mudflats (0–5 cm) collected from Taizhou, Zhejiang Provence (28°27′ N, 121°37′ E and 29°4′ N, 121°37′ E, respectively).
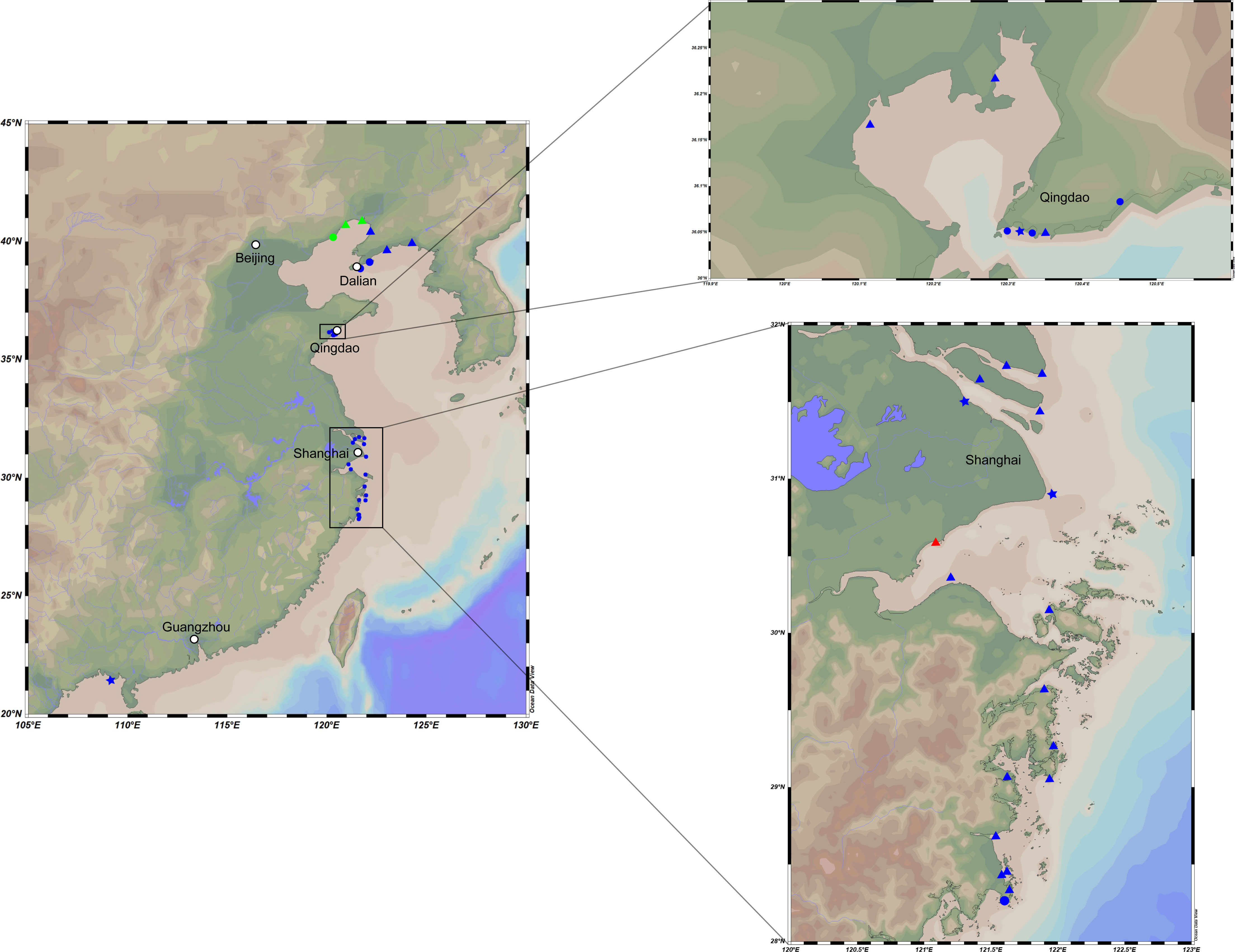
Figure 1 Map representing locations of the intertidal sediment sampling stations. Dots in these plots represent different substrate types and sample depths. Triangle, mud flat; circle, sand flat; star, muddy sand flat. Blue, 0–5-cm depth; red, 5–15-cm depth; green, 15–25-cm depth.
Samples were serially diluted to 10−3 with sterile seawater using the standard dilution-plating method (Williams and Davies, 1965). Generally, a 100-μL aliquot of each dilution was spread on modified marine agar (per liter of distilled water: Bacto yeast extract 0.1 g, Bacto peptone 0.5 g, ferric citrate 0.1 g, NaCl 19.45 g, MgCl2·6H2O 12.6 g, MgSO4 3.24 g, CaCl2 1.8 g, KCl 0.55 g, NaHCO3 0.16 g, KBr 0.08 g, SrCl2 34.0 mg, NaSiO3 4.0 mg, NH4NO3 1.6 mg, H3BO4 22.0 mg, NaF 2.4 mg, Na2HPO4 8.0 mg, agar 20 g) and incubated at 30°C for 3–7 days to a simulated oligotrophic environment. Afterward, the strains were isolated from different plates and purified by repeating streaking. All bacterial cultures were stored in Marine Broth 2216 (MB) medium containing 25% glycerol at −80°C. Six type strains, namely, Mangrovivirga cuniculi KCTC 72349T, Pontibacter actiniarum KCTC 12367T, Pontibacter litorisediminis KCTC 52252T, Poritiphilus flavus MCCC 1K03853T, Eudoraea chungangensis KCTC 42048T, and Zeaxanthinibacter enoshimensis NBRC 101990T, were purchased from the Korean Collection for Type Cultures (KCTC), the Marine Culture Collection of China (MCCC), and the NITE Biological Resource Center (NBRC), and used as reference strains in this study.
2.2 Morphological, physiological, and chemotaxonomic characteristics
Strains M17T, M82T, and M415T were cultured on Marine Agar (MA) medium for 3 days to observe their morphological characteristics, including colonial size, shape, edge, bulge, transparency, and color characteristics. Cell morphology, size, and special structure were examined using a transmission electron microscope (JEM-1230; JEOL). Gram staining reaction was performed according to the method described by Dong and Cai (Dong and Cai, 2001). Motility of the strains was assessed in semisolid MB medium containing 0.5% agar. The growth range and optimum temperature of the strains were determined in MB medium at 4°C, 10°C, 20°C, 25°C, 28°C, 30°C, 37°C, 40°C, 45°C, and 50°C, whereas their growth and optimal pH range were measured by adding appropriate buffer (40 mM) to MB medium (0.5 pH unit intervals), which include MES (pH 5.0–5.5), MOPS (pH 6.0–7.5), Tricine buffers (pH 8.0–8.5), and CAPSO (pH 9.0–10.0). By adding 0%, 0.5%, 1%, 1.5%, 2%, 2.5%, 3%, 3.5%, 4%, 6%, 8%, 10%, and 12% (w/v) NaCl to saltness-MB medium, the growth and optimal salinity range were determined. The optimal growth conditions and growth range were determined after 3 days and 1 month of culture, respectively. The growth condition of the strains was measured using a UV/visible spectrophotometer (Ultrospec 6300 Pro, Amersham Biosciences) at OD600.
Sodium nitrate (20 mM) was used as a potential electron acceptor to assess the anaerobic growth of strains in the anaerobic system (AnaeroPack-MicroAero, 2.5 L, MGC, Japan) (Shi et al., 2017). Catalase activity was measured by dripping 3% (v/v) hydrogen peroxide solution to the colonies placed on sterile slides. Oxidase activity was determined by observing whether the cell color turned red within 1 min after dripping 1% p-amino dimethylaniline oxalate solution. Carotenoid was extracted by acetone/methanol (7:2, v/v) solution, and their absorption spectra were determined using a scanning UV/visible spectrophotometer (Bowman and Nichols, 2005). Strains were cultured in suitable medium containing sodium thiosulfate (5 g/L), and their hydrogen sulfide production capacity was determined using sterile filter strips soaked in the solution of lead acetate. Amylase, cellulase, and hydrolysis of Tweens 20, 40, 60, and 80 were carried out according to the previously described methods (Liu et al., 2019). Carbon source oxidation was tested using Biolog GEN III MicroPlates, and activity of other common bacterial enzymes was examined using the API ZYM kit. Other physiological characteristics of the strains were analyzed using the API 20NE kit. All of the BIOLOG and API tests were carried out according to the manufacturer’s instructions except for adjusting salinity to 2%.
Strains M17T, M82T, and M415T and the six type strains were cultured on MB medium under optimal conditions, and the cells at the end of exponential growth stage were collected for chemotaxonomic analysis. Bacteria were collected and freeze-dried, then saponified, methylated, extracted, and washed to obtain the bacterial fatty acids (Sasser, 1990). Identification and quantification of the extracted cellular fatty acids of these strains were performed using a gas chromatograph (Agilent G6890N) and the Sherlock Microbial Identification System (MIDI database: Version 6.0). Polar lipids were extracted according to the procedure described by Minnikin et al. (1984), and composition analysis was performed on silica gel 60 F254 plates (10 × 10 cm, Merck) (Komagata and Suzuki, 1988). Isoprenoid quinones were extracted by a mixture of chloroform:methanol (2:1 v/v), and the further identification was performed by the HPLC-MS system.
2.3 16S rRNA gene sequence similarities and phylogenetic analysis
A total of 352 strains were isolated from 34 sampling stations. The 16S rRNA gene was amplified by PCR using the universal primers 27F/1492R and sequenced by Guangdong Magigene Biotechnology Co., Ltd. (Guangzhou, China). The sequences were submitted to NCBI under the accession numbers OQ617539–OQ617890. The complete 16S rRNA gene sequences of strains M17T, M82T, and M415T were extracted from their draft genomes. The similarities of all 16S rRNA gene sequences were identified by aligning these sequences against the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/) and the EzBioCloud database (https://www.ezbiocloud.net/).
The 16S rRNA gene sequence alignment of strains M17T, M82T, and M415T and their phylogenetically related taxa were performed by the ClustalW algorithm within MEGA11 v11.0.13 (Tamura et al., 2021), and the phylogenetic trees were reconstructed by the neighbor-joining (NJ), maximum-parsimony (MP), and maximum-likelihood (ML) algorithms within the MEGA11 software (Felsenstein, 1981; Saitou and Nei, 1987). The robustness of phylogenetic trees was assessed through bootstrap analysis based on 1,000 replications.
2.4 Genome sequencing and analysis
The genomes of strains M17T, M82T, and M415T were extracted using a bacterial genomic DNA kit (Takara), and draft genomes were sequenced using the Illumina NovaSeq 6000 platform (PE150) in Guangdong Magigene Biotechnology Co., Ltd. (Guangzhou, China). The sequences were assembled using SPAdes v3.10.1 (Zou et al., 2020), and the completeness and contamination of the assembled draft genomes were accessed using CheckM v1.1.3 (Parks et al., 2015).
Phylogenomic analysis based on single-copy orthologous clusters (OCs) of strains M17T, M82T, and M415T and the related type strains were performed as described (Xu et al., 2018). Briefly, the orthologous clusters were filtered based on the blastp+ program and 50% sequence identity using Proteinortho v5.16 (Lechner et al., 2011), and their formats were converted into OrthoMCL for subsequent analysis. The single-copy orthologous clusters were aligned through MAFFT v7.310 (Katoh and Standley, 2013), and the aligned sequences were concatenated after further refining by trimAL v1.4.1 (Capella-Gutierrez et al., 2009). The IQ-TREE v1.6.2 software was used to predict the best-fit models. The phylogenetic trees were constructed by maximum-likelihood algorithms based on the concatenated aligned single-copy orthologous clusters (Lam-Tung et al., 2015), and the best-fit models of strains M17T, M82T, and M415T were LG+F+R6, LG+F+R5, and LG+F+R8, respectively. Finally, MEGA11 software was employed to visualize the phylogenetic trees.
Functional and metabolic pathway predictions were realized by Kyoto Encyclopedia of Genes and Genomes (KEGG) and Evolutionary Genealogy of Genes with enhanced Non-supervised Orthologous Groups (EggNOG) (Kanehisa et al., 2017; Hernández-Plaza et al., 2023). The Average Nucleotide Identity (ANI) was calculated using the online ANI calculator based on the OrthoANIu algorithm, which is an improved iteration of the original OrthoANI algorithm (Yoon et al., 2017). Digital DNA–DNA hybridization (dDDH) and Two-way Average Amino Acid Identity (AAI) were calculated through Genome-to-Genome Distance Calculator 3.0 and the AAI calculator, respectively (Rodriguez-R and Konstantinidis, 2014; Meier-Kolthoff et al., 2022).
As for the potential in polysaccharide metabolism of strains M17T, M82T, and M415T, carbohydrate-active enzymes (CAZymes) were predicted by the HMMER tools within dbCAN2 software based on Carbohydrate-Active enZYmes Database V11 (CAZy database), and SusC/D-like proteins (SusC, outer membrane TonB-dependent transporter; SusD, surface glycan-binding protein) and other auxiliary proteins and genes were predicted and annotated using PROKKA v1.12 and Rapid Annotation using Subsystem Technology (RAST) version 2.0 (Aziz et al., 2008; Seemann, 2014; Zhang et al., 2018; Drula et al., 2022). The PUL prediction relies on the identification in each genome of the PUL markers: the presence of adjacent genes encoding SusC/D-like proteins, and according to the position of CAZymes and SusC/D-like proteins in the genome, combined with the position of corresponding auxiliary proteins and genes (Terrapon et al., 2015). In addition, the substrates of these PULs were predicted by searching through the PUL database (PULDB) and BLAST the CAZymes through UniProt (Terrapon et al., 2018; Consortium, 2019).
3 Results and discussion
3.1 Diversity of culturable bacteria in bare tidal flats
The sampling stations are widely distributed in the bare tidal flats of China. The samples from 34 sampling stations were used for the isolation of strains (Figure 1). A total of 352 bacterial strains were isolated. Compared with the 16S rRNA gene sequences with validly published species, the isolates were assigned to 180 species, belonging to 4 phyla, 7 classes, 22 orders, 37 families, and 94 genera; among them, 126 species and 52 genera had only one isolate (Table 1 and Figure 2). In our study, a higher number of species were discovered among the culturable strains compared with mangrove sediments (116 pure culture strains distributed in 13 species, 1 sample; Sefrji et al., 2022), plant rhizosphere (59 pure culture strains distributed in 22 species, 4 samples; Brígido et al., 2019), and Tabernas Desert (236 strains distributed in 37 genera, 3 samples; Molina-Menor et al., 2021); our results showed that bare tidal flats had a greater diversity of culturable bacteria. Flavobacteriales and Bacillales represented two most abundant orders, accounting for approx. 50% of the total isolates (Figure 3). A total of 17 strains were identified as Fictibacillus phosphorivorans, which was widely distributed in 12 sampling stations located in the different areas such as Jiangsu Province, Zhejiang Province, Guangxi Zhuang Autonomous Region, and Shanghai City. It became the most abundant and widely distributed culturable species in this study. Recent studies showed that Fictibacillus phosphorivorans was able to produce biosurfactant and displayed high nematicidal capability against root-knot nematodes (RKNs), which could infect almost all crops and lead to huge economic losses in agriculture around the world (Zheng et al., 2016; Pandey et al., 2021).
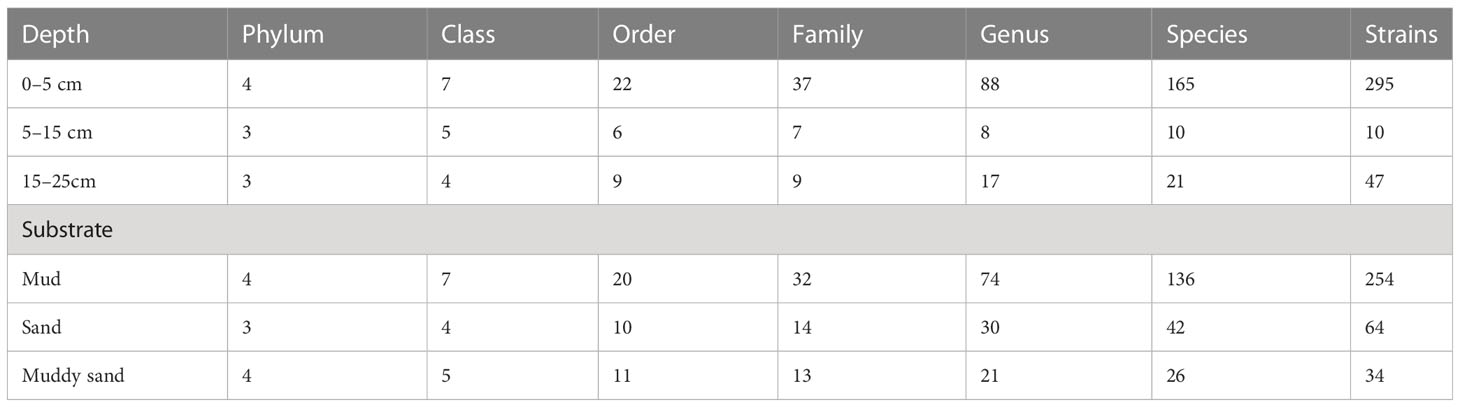
Table 1 Taxonomic profile of isolates in different depths and substrates.
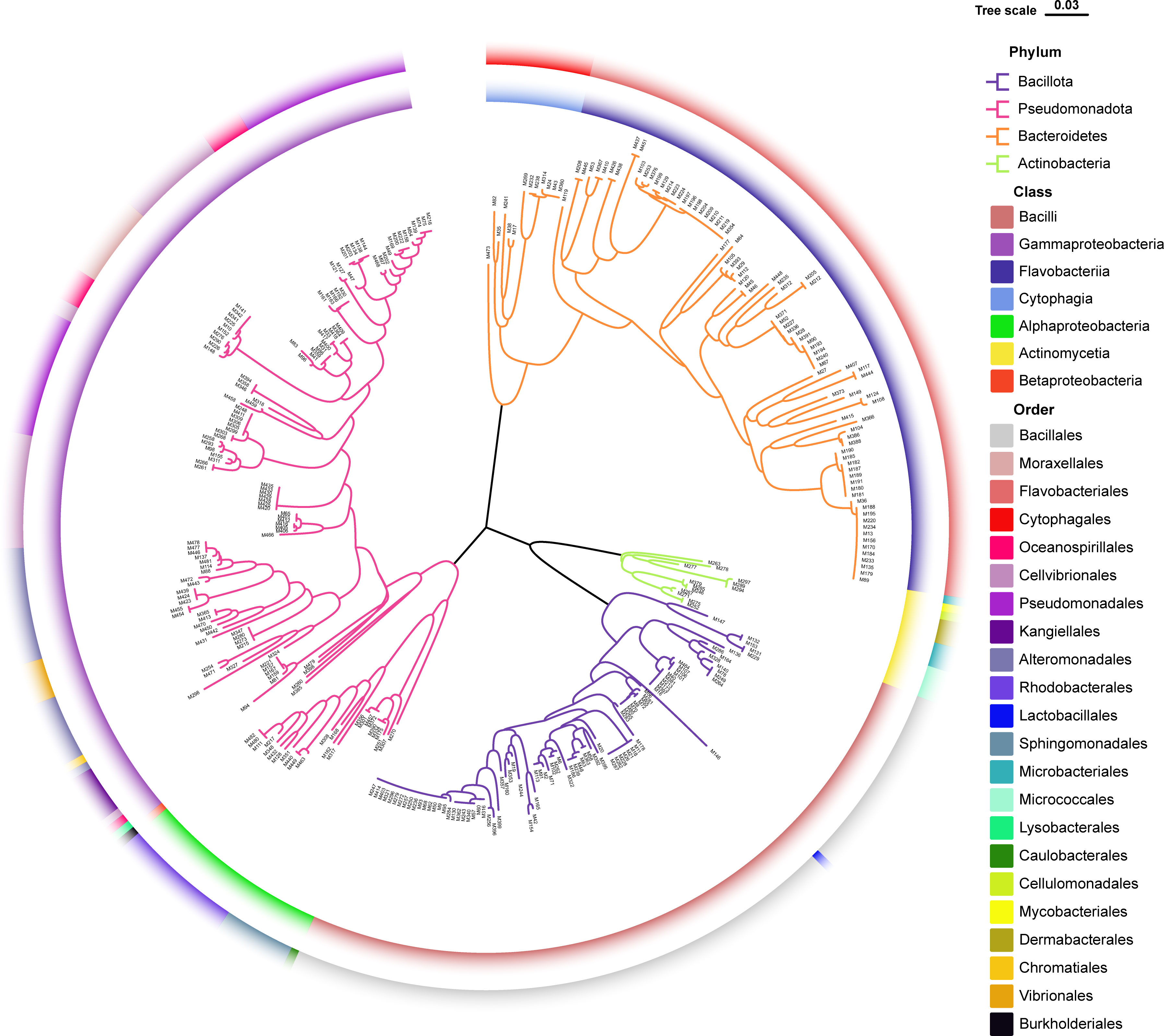
Figure 2 Diversity of culturable bacteria from unvegetated tidal flats of China. Neighbor-joining phylogenetic tree reconstructed from 16S rRNA gene sequences of 352 pure culture isolates. Branches in different colors represent different bacterial phyla; colors on the inner ring refer to bacterial classes; and colors on the outer ring refer to bacterial orders.
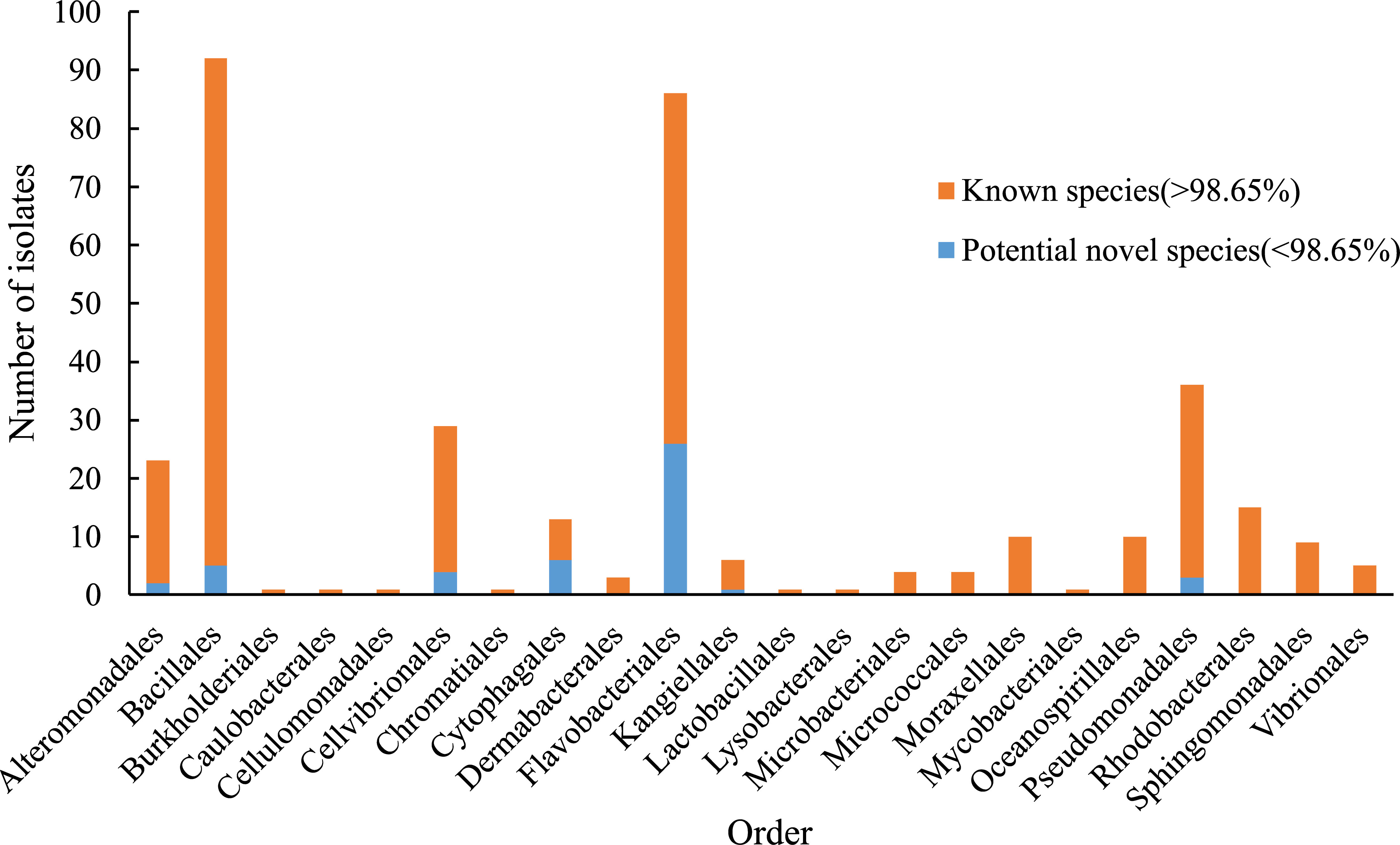
Figure 3 Number of isolates in different orders. Blue bar, the number of potential novel species in each order (16S rRNA gene similarity < 98.65%). Orange bar, the number of identified isolates in each order (16S rRNA gene similarity > 98.65%).
In addition, the effects of different depths and substrates on culturable bacteria were compared (Table 1). Actinomycetota strains, a total of 13 species in 5 orders, were isolated only in 0–5-cm non-sandy sediment, some of which were reported to have great ecological functions and economic values; e.g., strains of Rhodococcus qingshengii and Brachybacterium paraconglomeratum have the ability to repair heavy metal pollution (Du et al., 2022; Harboul et al., 2022) and pesticide contamination (Chuang et al., 2021; Wang et al., 2021), strains of Arthrobacter pascens, known as indole-3-acetic acid (IAA)-producing bacteria, could regulate plant growth and development (Li et al., 2021), and strains of Cellulosimicrobium cellulans are able to produce ginsenoside Rg3, a known anticancer agent (Hu et al., 2019). Different from Actinomycetota species, Pseudomonadota species Psychrobacter nivimaris was the only species distributed in all three types of sediment (Supplementary Figure 1), and strains in it may have the ecological function of repairing heavy metal pollution (Staloch et al., 2022).
In addition, a large number of novel species were discovered in bare tidal flats; a total of 47 strains showed less than 98.65% sequence similarities of the 16S rRNA gene with validly published species and may represent novel species (Kim et al., 2014). Most of them were assigned to Cytophagales and Flavobacteriales, both of which belong to the phylum Bacteroidota, with ratios of 46.2% and 30.2%, respectively. Three Bacteroidota strains (M17T, M82T, and M415T) were chosen for further phylogenetic and functional characterization.
3.2 Morphological, physiological, and chemotaxonomic characteristics
The morphological observations by transmission electron microscopy showed that the cells of strains M17T, M82T, and M415T were slender and long (2.0–10.0 µm × 0.3–0.5 µm), ellipsoidal to ovoid (0.9–3.2 µm × 0.6–1.1 µm), and slender and long (1.8–8.0 µm × 0.3–0.5 µm), respectively (Supplementary Figure 2 - Figure 4). For strain M17T, after 3 days of cultivation, the colony was round, 1–3 mm in diameter, and orange in color; mobility was not observed in the semisolid MB medium; and anaerobic growth and carotenoid production were not detected. The colony of M82T was round, 1–2 mm in diameter, and red in color after 3 days of incubation; mobility was not observed in the semisolid MB medium; and anaerobic growth and carotenoid production were both observed. The colony of M415T was round, 0.5 mm in diameter, and orange in color; mobility was not observed; and anaerobic growth and carotenoid production were detected. All the three strains were positive in the oxidations of D-fucose, L-galactonic acid lactone, D-glucuronic acid, glucuronamide, and tetrazolium violet. Detailed differences between strains M17T, M82T, and M415T and reference strains are summarized in Table 2 and Supplementary Table 1.
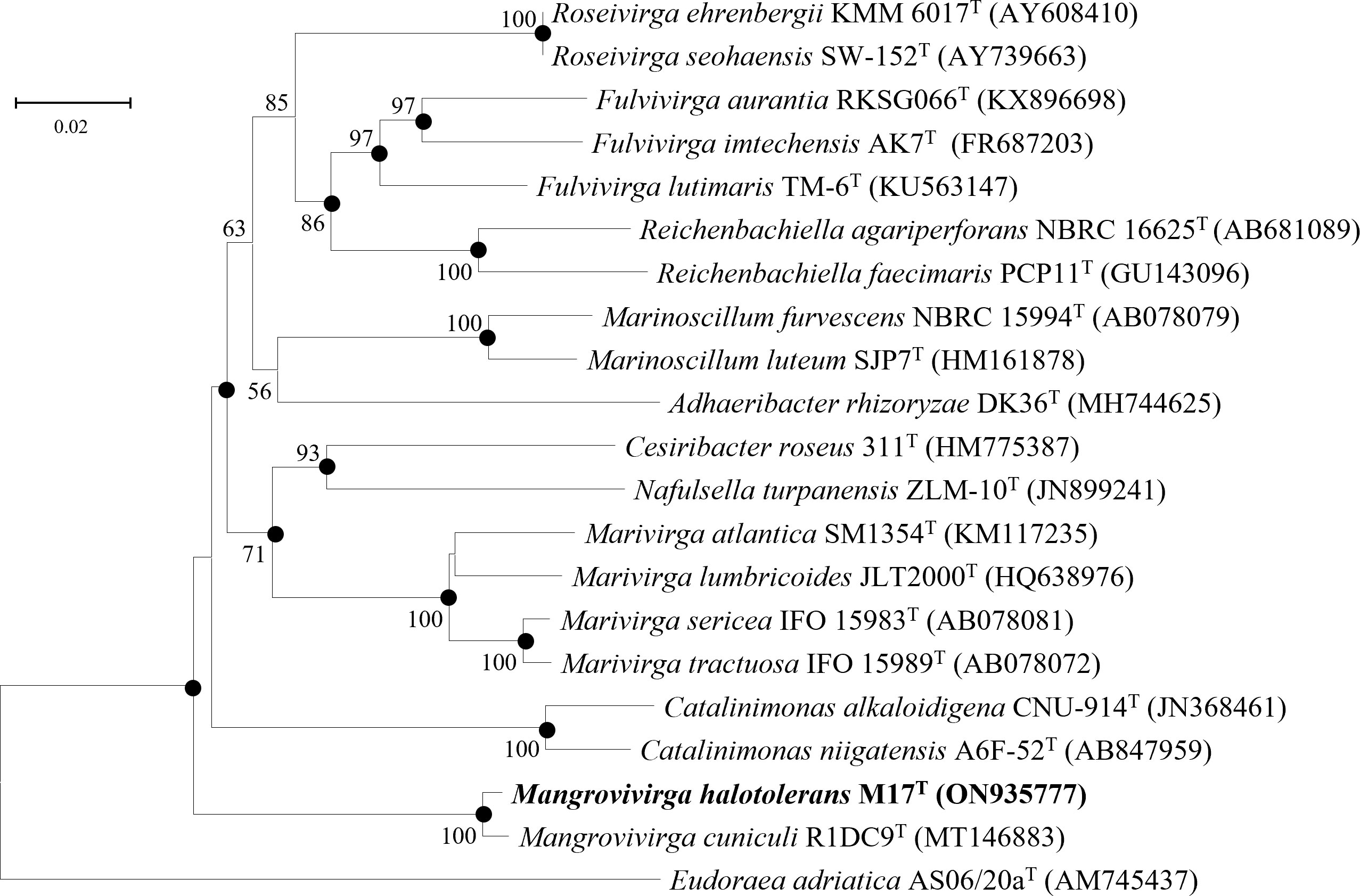
Figure 4 Neighbor-joining phylogenetic tree reconstructed from 16S rRNA gene sequences of strains M17T and related species. Bootstrap values <50% (based on 1,000 replications) are not shown. Filled circles indicate branches that were also recovered using maximum-likelihood and maximum-parsimony methods. Eudoraea adriatica AS06/20aT (AM745437) was used as the outgroup; bar, 0.02 nt substitutions per nucleotide position.

Table 2 Differential phenotypic characteristics between strains M17T, M82T, and M415T and their reference strains.
The major respiratory quinone of strains M17T and M82T was MK-7, which is consistent with their reference strains Mangrovivirga cuniculi KCTC 72349T, Pontibacter actiniarum KCTC 12367T, and Pontibacter litorisediminis KCTC 52252T. The major respiratory quinone of strain M415T was MK-6, identical with that of Poritiphilus flavus MCCC 1K03853T, Eudoraea chungangensis KCTC 42048T, and Zeaxanthinibacter enoshimensis NBRC 101990T.
The main polar lipids of strain M17T were phosphatidylethanolamine (PE), aminoglycolipid (AGL), one unidentified phospholipid (PL), three unidentified aminolipids (ALs), three unidentified glycolipids (GLs), and six unidentified lipids (L1–6) (Supplementary Figure 5). Compared with its reference strain M. cuniculi KCTC 72349T, they both contained PE as main polar lipids, but strain M17T comprised more polar lipids such as AGL, ALs, and GLs. The major polar lipids of strain M82T were PE, two ALs, and nine unidentified lipids (L1–9), in which PE and plenty of unidentified lipids were also detected in other species of the genus Pontibacter (Nedashkovskaya et al., 2005; Zhang et al., 2008; Subhash et al., 2013; Park et al., 2016). The major polar lipids detected in M415T were PE, phosphoglycolipid (PGL), aminophospholipid (APL), one GL, two ALs, and five unidentified lipids (L1–5).
Similar to M. cuniculi KCTC 72349T, iso-C15:0 and iso-C17:0 3-OH were the main cellular fatty acids (>10%) in strain M17T. Meanwhile, strain M82T contained iso-C15:0 and summed feature 4 (SF4) as the major cellular fatty acids (>10%), which was also found in its reference strains. However, some differences in the ratio of main fatty acids (such as iso-C15:0) existed between the two novel isolates and their reference strains (Table 3). Similar to its reference strains, strain M415T contained iso-C15:0 and iso-C17:0 3-OH as the main cellular fatty acids, but the relatively higher ratio of iso-C15:1 -G and the lower ratio of summed feature 3 (SF3) differed strain M415T from its reference strains (Table 3).
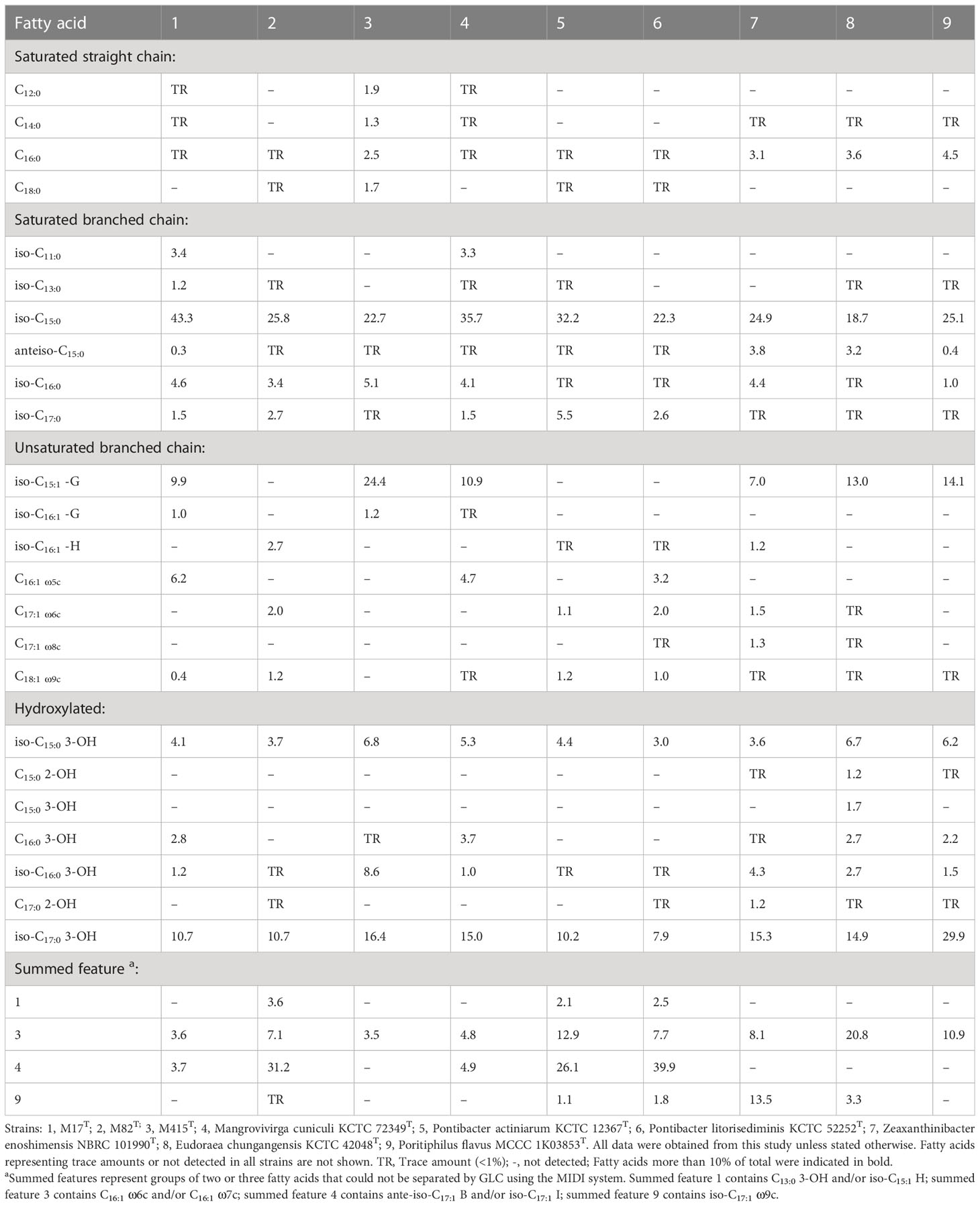
Table 3 Cellular fatty acid composition of strains M17T, M82T, and M415T and their reference strains.
3.3 Phylogenetic analysis and genomic properties
The 16S rRNA gene sequences between strain M17T and M. cuniculi KCTC 72349T shared the highest similarity of 99.28%, and less than 90.09% with other species (Table 4). In both of the phylogenetic trees based on the 16S rRNA gene and single-copy orthologous clusters (concatenated protein sequences), strain M17T formed a closest and robust cluster with M. cuniculi KCTC 72349T (Figure 4 and Supplementary Figure 6), indicating that strain M17T was affiliated with the genus Mangrovivirga. Although sequence similarity of the 16S rRNA gene reached 99.28% between strains M17T and M. cuniculi KCTC 72349T, the dDDH, ANI, and AAI values between strain M17T and M. cuniculi KCTC 72349T were 57.9%, 84.0%, and 88.8%, respectively (Table 4), all lower than the thresholds for species delimitation (Konstantinidis and Tiedje, 2005; Tindall et al., 2010; Kim et al., 2014), indicating that strain M17T represents a new species within the genus Mangrovivirga. In addition, the cell morphology of the strain M. cuniculi KCTC 72349T was short rod-shaped (1.0–1.2 µm × 0.3–0.5 µm; Sefrji et al., 2021), which was different from that of strain M17T, showing a long rod shape (2.0–10.0 µm × 0.3–0.5 µm; Supplementary Figure 2). In addition, there were also a great number of differences between them in phenotypic and genomic properties (Tables 2-5). Therefore, strain M17T represents a novel species of the genus Mangrovivirga.
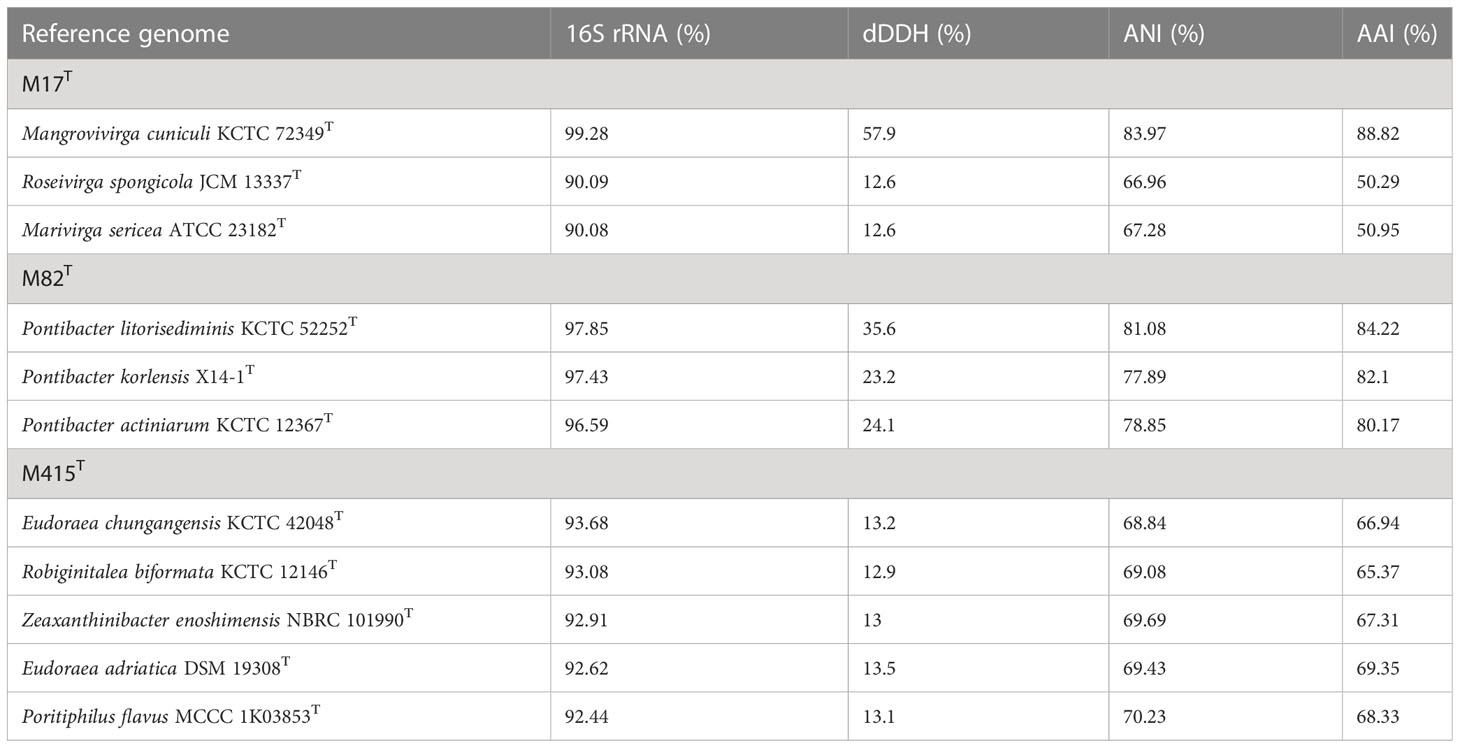
Table 4 16S rRNA gene sequence similarities, digital DNA-DNA hybridization (dDDH), Average Nucleotide Identity (ANI), and Average Amino acid Identity (AAI) of strains M17T, M82T, and M415T and their related type strains.
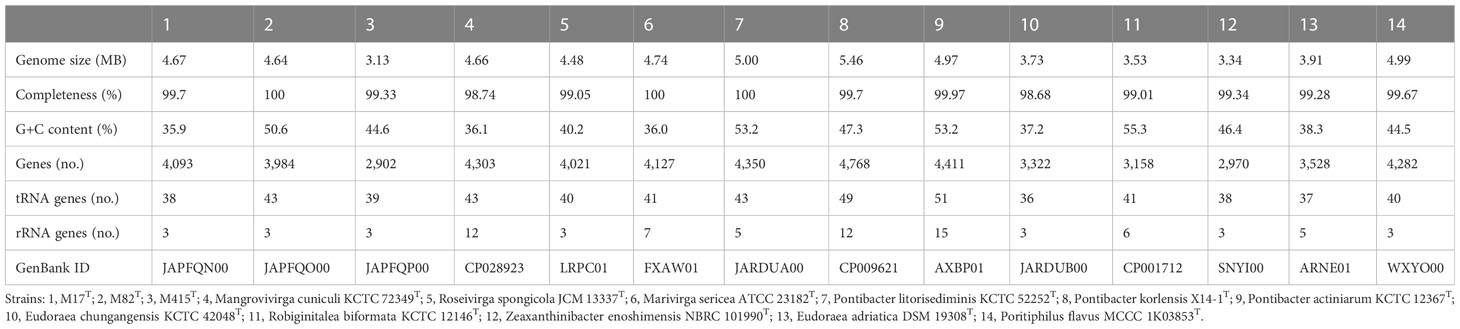
Table 5 Genomic statistics of strains M17T, M82T, and M415T and their related type strains.
Strain M82T shares the highest 16S rRNA gene sequence similarity of 97.85%, 97.43%, and 96.59%, respectively, with Pontibacter litorisediminis KCTC 52252T, Pontibacter korlensis X14-1T, and Pontibacter actiniarum KCTC 12367T, lower than the threshold for species delimitation (Kim et al., 2014). Based on the phylogenetic analysis of 16S rRNA gene sequences and single-copy orthologous clusters, strain M82T was closely clustered within Pontibacter strains (Figure 5 and Supplementary Figure 7). The highest values of dDDH, ANI, and AAI between strain M82T and the type strains within the genus Pontibacter were 35.6%, 81.1%, and 84.2%, respectively, which are lower than the thresholds of species delimitation (Table 4).

Figure 5 Neighbor-joining phylogenetic tree reconstructed from 16S rRNA gene sequences of strains M82T and related species. Bootstrap values < 50% (based on 1,000 replications) are not shown. Filled circles indicate branches that were also recovered using maximum-likelihood and maximum-parsimony methods. Eudoraea adriatica AS06/20aT (AM745437) was used as the outgroup; bar, 0.02 nt substitutions per nucleotide position.
Low 16S rRNA gene sequence similarities were found between strain M415T and its type species of the genera Eudoraea (92.62%–93.68%), Zeaxanthinibacter (92.02%–92.91%), Muriicola (92.21%–92.83%), Robiginitalea (91.48%–92.74%), and Poritiphilus (92.44%). Phylogenetic analysis based on 16S rRNA gene sequences and single-copy orthologous clusters showed that strain M415T was clearly separated from the related genera (Figure 6 and Supplementary Figure 8), representing a novel species of a new genus within the family Flavobacteriaceae. The highest values of dDDH, ANI, and AAI between strain M415T and its related species were 13.5%, 70.2%, and 69.4%, respectively (Table 4).
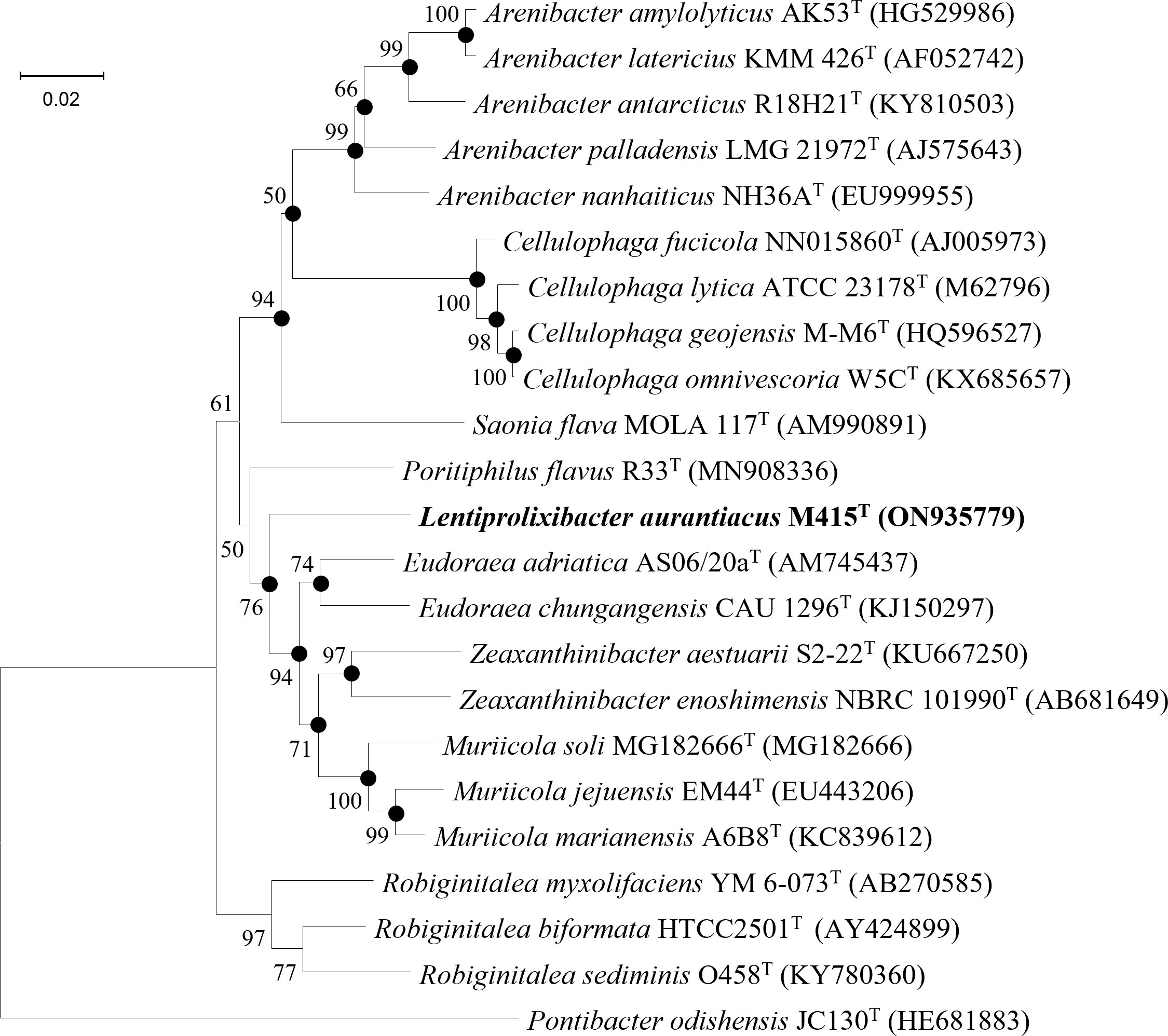
Figure 6 Neighbor-joining phylogenetic tree reconstructed from 16S rRNA gene sequences of strains M415T and related species. Bootstrap values < 50% (based on 1,000 replications) are not shown. Filled circles indicate branches that were also recovered using maximum-likelihood and maximum-parsimony methods. Pontibacter odishensis JC130T (HE681883) was used as the outgroup; bar, 0.02 nt substitutions per nucleotide position.
The genomic features of strains M17T, M82T, and M415T and their related strains were analyzed to further confirm the taxonomic status of these three novel strains (Table 5). Strain M17T showed a similar genome size and GC content with M. cuniculi KCTC 72349T, but the latter has more protein-coding genes and obvious multicopy of the rRNA gene than strain M17T. The genomic composition of strain M82T was different from the related species, showing a higher GC content and a lower number of rRNA. Strain M415T has the smallest genome size and the lowest protein-coding gene number, compared with its related species. According to the annotation result against the COG database, the most abundant category in strains M17T and M82T was cell wall/membrane/envelope biogenesis; however, in strain M415T, it changed to amino acid transport and metabolism (Supplementary Figure 9). The genome sequences of strains M17T, M82T, and M415T were annotated against the KEGG database and 1,614 (39.43%), 1,775 (44.55%), and 1,368 (47.14%) genes were assigned to putative functions, respectively. These functions were mainly composed of carbohydrate metabolism, genetic information processing, signaling and cellular processing, and amino acid metabolism, and notably, strain M82T contained much more function genes associated with energy metabolism and environmental information processing (Supplementary Figure 10).
As indicated by KEGG pathway annotation, these three strains have many differences in metabolic pathways. As for sulfur metabolism, the assimilatory sulfate reduction (M00176) was devoid only in strain M415T. In the lipid metabolism, phosphatidylcholine (PC) biosynthesis (M00091) was only found in strain M82T; nevertheless, strain M82T and strain M415T both contained the threonine biosynthesis (M00018) which was not found in strain M17T. In the metabolism of major nutrients, the complete β-oxidation pathway only existed in strain M17T; the phosphate acetyltransferase-acetate kinase pathway was complete in strains M17T and M415T but incomplete in strain M82T (Figure 7). In addition, we also found that the glyoxylate cycle was presented in both strains M17T and M82T but not in strain M415T (Figure 7); the activation of the glyoxylate cycle could provide malic acid and NADH, which serve as precursors and energy sources for metabolic reactions to sustain cell survival under stress conditions (Schroeter et al., 2011; Yuan et al., 2019). Therefore, the existence of the glyoxylate cycle for resisting the stress condition in tidal flats might indicate the adaptation mechanism of strains M17T and M82T to such environment.
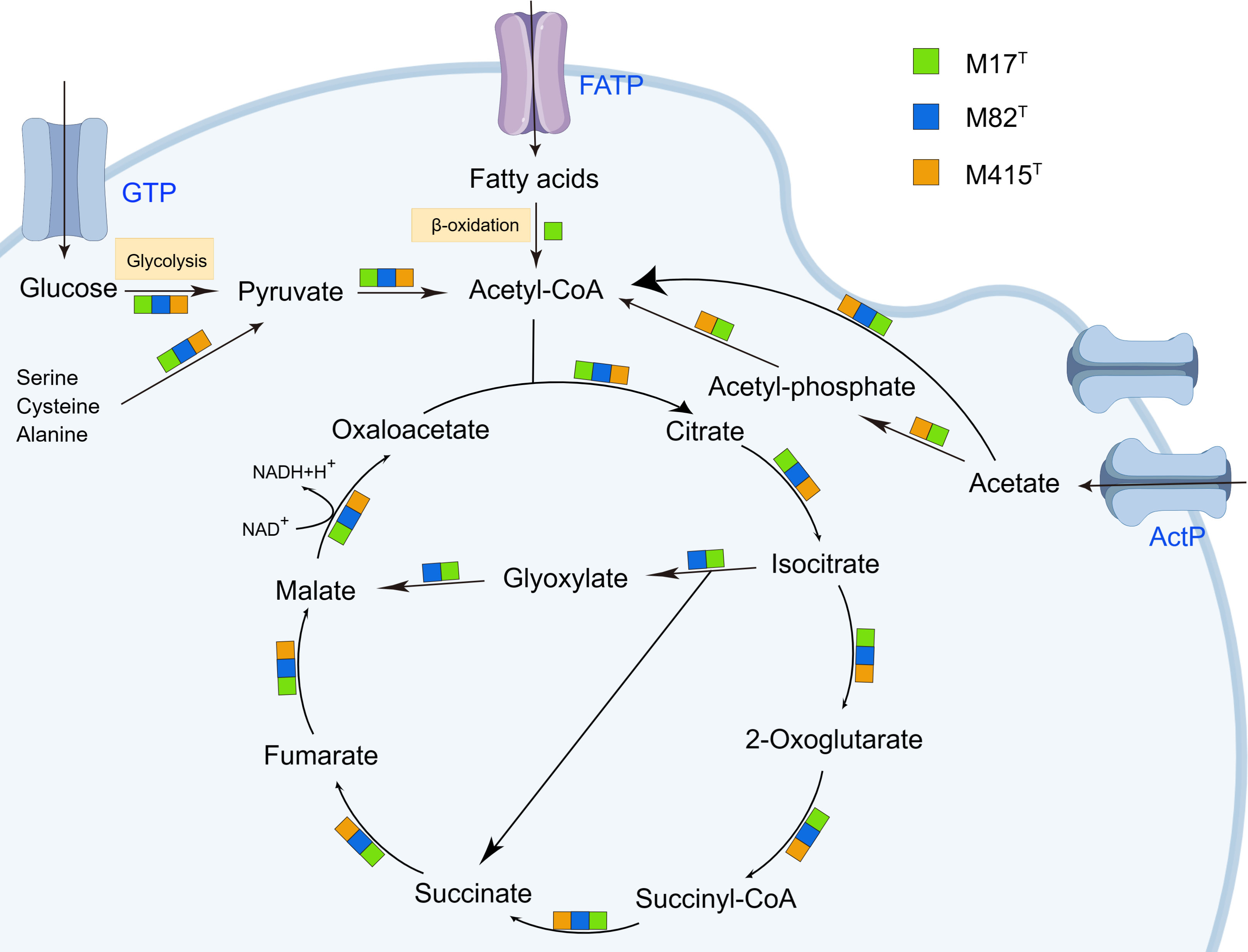
Figure 7 The citrate cycle (TCA cycle) and glyoxylate cycle in strains M17T, M82T, and M415T. Color blocks indicated the reaction occurred in specific strains. GTP, glucose transport protein. FATP, fatty acid transport proteins. ActP, acetate permease. Other products have been omitted.
3.4 The potential in polysaccharide metabolism
Considering the outstanding polysaccharide metabolic abilities of marine Bacteroidota (Krüger et al., 2019), especially Flavobacteriaceae strains (Kappelmann et al., 2019; Gavriilidou et al., 2020), and special geographical location of mudflats—the source of strains M17T, M82T, and M415T, we further analyzed their CAZyme profiles and potential polysaccharide substrates. The result showed that strains M17T, M82T, and M415T had 114, 155, and 103 CAZymes, respectively (Figure 8A), which were similar as the KEGG results, where 157, 175, and 153 genes for the carbohydrate metabolism pathway were detected, respectively. The detailed numbers of each CAZyme family presented in three strains are summarized in Supplementary Table 2. GH and GT were the most abundant CAZyme classes, accounting for more than 70% of the total CAZymes; this finding was consistent with studies on carbohydrate-active enzymes in Flavobacteriaceae species by Gavriilidou et al., which also indicated that GH and GT were the most abundant CAZyme classes (Gavriilidou et al., 2020). Strain M82T comprised the highest number of CAZymes assigned to different classes and SusC/D-like proteins, indicating that it may have great potential in polysaccharide metabolism. However, the CAZyme count per Mb genome in strain M415T was similar with strain M82T and much higher than that in strain M17T, even though the total number of CAZymes of strain M415T was smaller than strains M17T and M82T due to the smallest genome size of strain M415T (Figure 8B). A great number of GT2 and GT4 were present in strain M415T, and they were involved in critical glycan synthesis, such as cellulose, chitin, and mannan; in addition, a higher number of GH2, GH3, GH16, and GH30 revealed that strain M415T can metabolize plant polysaccharides and oligosaccharides (Coutinho et al., 2003; Gómez-Silva et al., 2019).
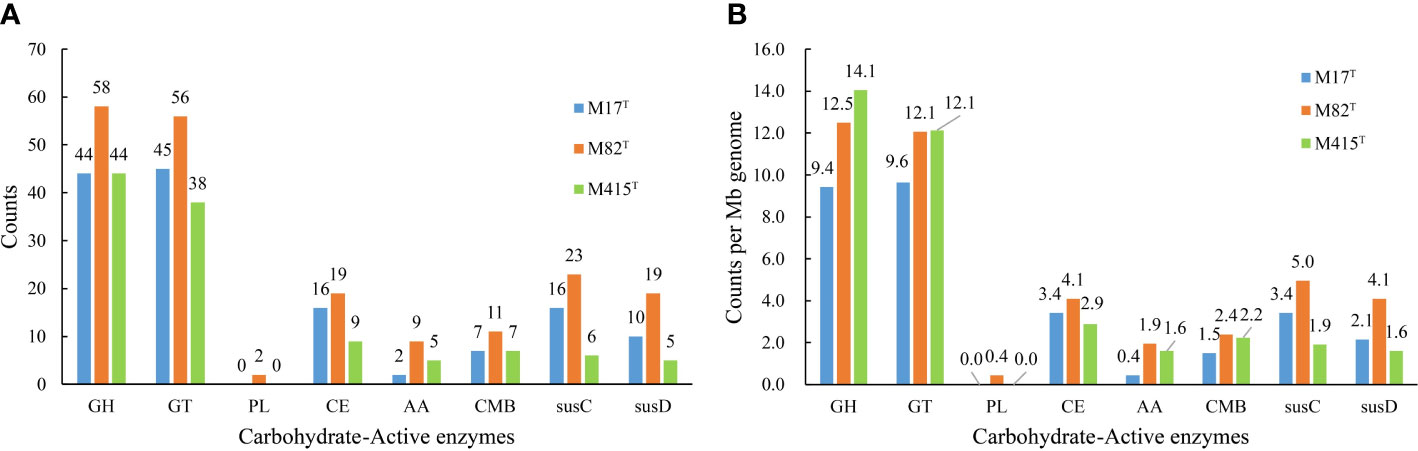
Figure 8 The number of predicted carbohydrate-active enzymes in strains M17T, M82T, and M415T. (A) number of predicted CAZymes per genome and (B) average number of predicted CAZymes per Mb genome. GH, glycoside hydrolases; GT, glycosyltransferases; PL, polysaccharide lyases; CE, carbohydrate esterases; AA, auxiliary activities; CBM, carbohydrate-binding modules.
Polysaccharide utilization loci (PULs) are specialized saccharolytic systems that exhibit functional homology to the paradigmatic starch utilization system; the number and type of PULs determine the potential of polysaccharide utilization (Xu et al., 2003; McKee et al., 2021). According to the annotation results of carbohydrate-active enzymes and the SusC/D-like protein complex, we manually sorted the PULs of three novel strains (Figure 9); strains M17T, M82T, and M415T have five, six, and two putative PULs, respectively. The PULs in strains M17T and M82T were specific for marine polysaccharide metabolism; for strain M17T, PUL2 may be associated with the degradation of ulvan as the existence of GH2 (β-xylosidase), PUL3 may be associated with the degradation of laminarin as the existence of GH3 and GH16_3 (Tang et al., 2017; Chen et al., 2018), and PUL4 may be associated with the degradation of chitin as the existence of GH18 (Kappelmann et al., 2019). For strain M82T, PUL1 may be associated with the degradation of fucoidan as the existence of GH29 (fucosidase) and PUL4 may be associated with the degradation of laminarin as the existence of GH16_3 (Tang et al., 2017; Chen et al., 2018). However, with the ability to utilize marine polysaccharides as strains M17T and M82T, strains M415T can also utilize polysaccharides that are mainly present in land; for instance, it contained more glycogen and starch degrading CAZymes compared to another two strains (e.g., GH13, GH13_8; Berlemont and Martiny, 2015), and the PUL1 of strain M415T may be related to the degradation of glycogen or starch.
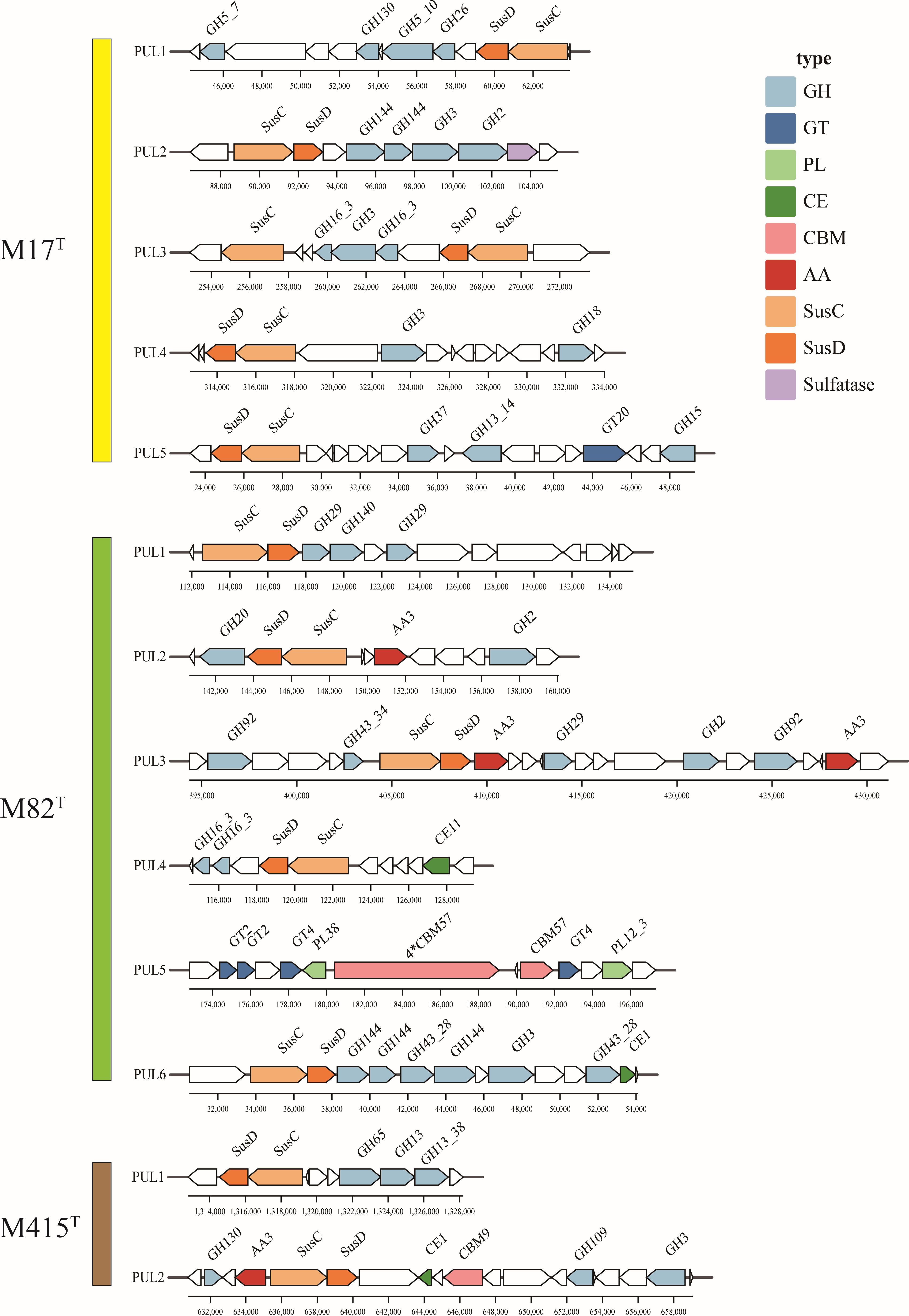
Figure 9 The putative polysaccharide utilization loci and genes in strains M17T, M82T, and M415T. GH, glycoside hydrolases; GT, glycosyltransferases; PL, polysaccharide lyases; CE, carbohydrate esterases; AA, auxiliary activities; CBM, carbohydrate-binding modules.
3.5 Description of Lentiprolixibacter gen. nov.
Lentiprolixibacter (L. masc. adj. lentus, slow, delayed; L. masc. adj. prolixus, long, extended; N.L. masc. n. bacter, a rod; N.L. masc. n. Lentiprolixibacter, slowly growing long rod)
Cells are Gram-stain-negative, non-spore-forming, and non-motile aerobic rods. Catalase- and oxidase-positive. Predominant menaquinone is menaquinone 6 (MK-6). Major polar lipids are phosphatidylethanolamine, phosphoglycolipid, aminophospholipid, unidentified glycolipids, aminolipids, and lipids. The type species is Lentiprolixibacter aurantiacus.
3.6 Description of Lentiprolixibacter aurantiacus sp. nov.
Lentiprolixibacter aurantiacus (au.ran.ti.a’cus. N.L. masc. adj. aurantiacus, orange-colored)
Displays the following characteristics in addition to those given in the genus description. Cells are rod shaped and usually 0.3–0.5 µm wide and 1.8–8.0 µm long. Colonies (0.5 mm in diameter) are circular, convex, smooth, shiny, and orange pigmented after 3 days of incubation. Cells grow at 25°C–37°C (optimum, 25°C–30°C) in a medium of pH 6–8 (optimum, pH 6.5–7) and contain 1%–4% NaCl (optimum, 2%–3%). Catalase and oxidase are positive; nitrate reduction, carotenoid production, and H2S production are negative. Starch is weakly hydrolyzed, but Tweens 20, 40, 60, and 80 are not. Dextrin, D-maltose, D-trehalose, D-cellobiose, gentiobiose, sucrose, D-turanose, β-methyl-D-glucoside, D-salicin, N-acetyl-D-glucosamine, α-D-glucose, fusidic acid, D-serine, D-glucose-6-PO4, D-fructose-6-PO4, pectin, glucuronamide, L-malic acid, bromo-succinic acid, nalidixic acid, and sodium butyrate are oxidized. In assays with the API ZYM system, alkaline phosphatase, esterase (C4), esterase lipase (C8), leucine arylamidase, valine arylamidase, cystine arylamidase, trypsin, chymotrypsin, acid phosphatase, naphthol-AS-BI-phosphohydrolase, β-glucosidase, and N-acetylglucosaminidase are positive and α-glucosidase is weakly positive. The major cellular fatty acids (>10%) are iso-C15:0, iso-C15:1 -G, and iso-C17:0 3-OH. The polar lipids are phosphatidylethanolamine, phosphoglycolipid, aminophospholipid, one unidentified glycolipids, two unidentified aminolipids, and five unidentified lipids. The DNA G+C content of the type strain is 44.6%.
The type strain, M415T (MCCC 1K08058T = KCTC 92534T), was isolated from an intertidal mudflat (0–5 cm) collected from Taizhou, Zhejiang Provence, PR China. The GenBank accession numbers for the 16S rRNA gene sequence and the draft genome sequence of strain M415T are ON935779 and JAPFQP000000000, respectively.
3.7 Description of Mangrovivirga halotolerans sp. nov.
Mangrovivirga halotolerans (ha.lo.to’le.rans. Gr. masc. n. hals, salt; L. pres. part. tolerans, tolerating, enduring; N.L. part. adj. halotolerans, salt-tolerating)
Cells are Gram-stain negative, non-motile, strictly aerobic rod-shaped, usually 0.3–0.5 µm wide, and 2–10 µm long, some more than 10 µm in length. Colonies (1–3 mm in diameter) are circular, convex, smooth, shiny, and orange pigmented after 3 days of incubation. Cells grow at 20°C–45°C (optimum, 20°C–28°C) in a medium of pH 6.5–9 (optimum, pH 6.5-7.5) and contain 0.5%–10% NaCl (optimum, 3.5%–6%). Catalase and oxidase activities are positive; nitrate reduction, carotenoid production, and H2S production are negative. Starch is hydrolyzed, but Tweens 20, 40, 60, and 80 are not. Sucrose, stachyose, D-fucose, fusidic acid, L-alanine, glucuronamide, tetrazolium violet, α-keto-glutaric acid, L-malic acid, nalidixic acid, acetoacetic acid, propionic acid, acetic acid, formic acid, aztreonam, and sodium butyrate are oxidized. In assays with the API ZYM system, alkaline phosphatase, esterase (C4), esterase lipase (C8), leucine arylamidase, valine arylamidase, cystine arylamidase, trypsin, chymotrypsin, acid phosphatase, naphthol-AS-BI-phosphohydrolase, β-glucuronidase, β-glucosidase, and N-acetylglucosaminidase are positive. The predominant menaquinone is MK-7. The major cellular fatty acids (>10%) are iso-C15:0 and iso-C17:0 3-OH. The polar lipids are phosphatidylethanolamine, aminoglycolipid, one unidentified phospholipid, three unidentified aminolipids, three unidentified glycolipids, and six unidentified lipids. The DNA G+C content of the type strain is 35.9%.
The type strain, M17T (MCCC 1K08105T = KCTC 92592T), was isolated from an intertidal mudflat (0–5 cm) collected from Qingdao, Shandong Provence, PR China. The GenBank accession numbers for the 16S rRNA gene sequence and the draft genome sequence of strain M17T are ON935777 and JAPFQN000000000, respectively.
3.8 Description of Pontibacter anaerobius sp. nov.
Pontibacter anaerobius (an.ae.ro’bi.us. Gr. pref. an-, not; Gr. masc. n. aêr, air; Gr. masc. n. bios, life; N.L. masc. adj. anaerobius, able to live in the absence of oxygen)
Cells are Gram-stain negative, non-motile, facultative aerobic rod-shaped, and usually 0.6–1.1 µm wide and 0.9–3.2 µm long. Colonies (1–2 mm in diameter) are circular, convex, smooth, shiny, and red pigmented after 3 days of incubation. Cells grow at 20°C–40°C (optimum, 28°C–37°C) in a medium of pH 6–9 (optimum, pH 6.5–7.5) and contain 0%–8% NaCl (optimum, 2%–3%). Carotenoid production and catalase and oxidase activity are positive. Nitrate reduction and H2S production are negative. Starch and Tweens 20, 40, 60, and 80 are not hydrolyzed. Dextrin, D-maltose, D-trehalose, D-cellobiose, gentiobiose, sucrose, D-turanose, stachyose, D-raffinose, α-D-lactose, D-melibiose, β-methyl-D-glucoside, D-salicin, N-acetyl-D-glucosamine, α-D-glucose, D-mannose, D-galactose, D-fucose, L-fucose, fusidic acid, D-arabitol, myo-inositol, D-glucose-6-PO4, D-fructose-6-PO4, gelatin, glycyl-L-proline, L-aspartic acid, L-glutamic acid, L-serine, D-galacturonic acid, glucuronamide, acetoacetic acid, acetic acid, formic acid, and sodium butyrate are oxidized. In assays with the API ZYM system, alkaline phosphatase, esterase (C4), esterase lipase (C8), leucine arylamidase, valine arylamidase, cystine arylamidase, trypsin, chymotrypsin, acid phosphatase, α-galactosidase, α-glucosidase, β-glucosidase, and N-acetylglucosaminidase are positive; naphthol-AS-BI-phosphohydrolase and β-galactosidase are weakly positive. The predominant menaquinone is MK-7. The major cellular fatty acids (>10%) are iso-C15:0, iso-C17:0 3-OH, and summed feature 4 (ante-iso-C17:1 B and/or iso-C17:1 I). The polar lipids are phosphatidylethanolamine, two unidentified aminolipids, and nine unidentified lipids. The DNA G+C content of the type strain is 50.6%.
The type strain, M82T (MCCC 1K08048T = KCTC 92537T), was isolated from an intertidal mudflat (0–5 cm) collected from Taizhou, Zhejiang Provence, PR China. The GenBank accession numbers for the 16S rRNA gene sequence and the draft genome sequence of strain M82T are ON935778 and JAPFQO000000000, respectively.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
K-JM, Y-LY, and Y-HF collected the samples and isolated these strains. K-JM performed data collection and analysis. G-YF performed project guidance. K-JM and CS wrote the manuscript. X-WX and CS performed project guidance and critical revision of manuscripts. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Science and Technology Fundamental Resources Investigation Program of China (2019FY100700), the National Natural Science Foundation of China (No. 31900003), and the Key R&D Program of Zhejiang (#2023C03011).
Acknowledgments
We appreciate the helpful suggestion of Prof. Aharon Oren on the nomenclature and the help of Dr. Zhi-Cheng Wu and Dr. Maripat Xamxidin in sample collection and detection of polar lipids, respectively.
Conflict of interest
Author CS was employed by company Zhejiang Sci-Tech University Shaoxing Academy of Biomedicine Co., Ltd..
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1222157/full#supplementary-material
References
Amann R. I., Ludwig W., Schleifer K. H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiological Rev. 59 (1), 143–169. doi: 10.1128/mmbr.59.1.143-169.1995
Asker D., Beppu T., Ueda K. (2007). Zeaxanthinibacter enoshimensis gen. nov., sp nov., a novel zeaxanthin-producing marine bacterium of the family Flavobacteriaceae, isolated from seawater off Enoshima Island, Japan. Int. J. Systematic Evolutionary Microbiol. 57 (4), 837–843. doi: 10.1099/ijs.0.64682-0
Aziz R. K., Bartels D., Best A. A., DeJongh M., Disz T., Edwards R. A., et al. (2008). The RAST server: Rapid annotations using subsystems technology. BMC Genomics 9 (1), 75. doi: 10.1186/1471-2164-9-75
Barbeyron T., Thomas F., Barbe V., Teeling H., Schenowitz C., Dossat C., et al. (2016). Habitat and taxon as driving forces of carbohydrate catabolism in marine heterotrophic bacteria: example of the model algae-associated bacterium Zobellia galactanivorans DsijT. Environ. Microbiol. 18 (12), 4610–4627. doi: 10.1111/1462-2920.13584
Bauer J. E., Cai W. J., Raymond P. A., Bianchi T. S., Hopkinson C. S., Regnier P. A. (2013). The changing carbon cycle of the coastal ocean. Nature 504 (7478), 61–70. doi: 10.1038/nature12857
Berlemont R., Martiny A. C. (2015). Genomic potential for polysaccharide deconstruction in bacteria. Appl. Environ. Microbiol. 81 (4), 1513–1519. doi: 10.1128/aem.03718-14
Bowman J. P., Nichols D. S. (2005). Novel members of the family Flavobacteriaceae from Antarctic maritime habitats including Subsaximicrobium wynnwilliamsii gen. nov., sp. nov., Subsaximicrobium saxinquilinus sp. nov., Subsaxibacter broadyi gen. nov., sp. nov., Lacinutrix copepodicola gen. nov., sp. nov., and novel species of the genera Bizionia, Gelidibacter and Gillisia. Int. J. Systematic Evolutionary Microbiol. 55 (4), 1471–1486. doi: 10.1099/ijs.0.63527-0
Brígido C., Singh S., Menéndez E., Tavares M. J., Glick B. R., Félix M. D. R., et al. (2019). Diversity and functionality of culturable endophytic bacterial communities in chickpea plants. Plants 8 (2), 42. doi: 10.3390/plants8020042
Capella-Gutierrez S., Silla-Martinez J. M., Gabaldon T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25 (15), 1972–1973. doi: 10.1093/bioinformatics/btp348
Chang M. X., Li P., Li Z. H., Wang H. J. (2022). Mapping tidal flats of the bohai and yellow seas using time series sentinel-2 images and google earth engine. Remote Sens. 14 (8), 1789. doi: 10.3390/rs14081789
Chen J., Robb C. S., Unfried F., Kappelmann L., Markert S., Song T., et al. (2018). Alpha- and beta-mannan utilization by marine Bacteroidetes. Environ. Microbiol. 20 (11), 4127–4140. doi: 10.1111/1462-2920.14414
Chernysheva N., Bystritskaya E., Stenkova A., Golovkin I., Nedashkovskaya O., Isaeva M. (2019). Comparative Genomics and CAZyme Genome Repertoires of Marine Zobellia amurskyensis KMM 3526T and Zobellia laminariae KMM 3676T. Mar. Drugs 17 (12), 661. doi: 10.3390/md17120661
Choi H. J., Jeong T. Y., Yoon H., Oh B. Y., Han Y. S., Hur M. J., et al. (2018). Comparative microbial communities in tidal flats sediment on Incheon, South Korea. J. Gen. Appl. Microbiol. 64 (5), 232–239. doi: 10.2323/jgam.2017.12.007
Chuang S. C., Yang H. X., Wang X., Xue C., Jiang J. D., Hong Q. (2021). Potential effects of Rhodococcus qingshengii strain djl-6 on the bioremediation of carbendazim-contaminated soil and the assembly of its microbiome. J. Hazardous Materials 414, 125496. doi: 10.1016/j.jhazmat.2021.125496
Consortium U. (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47 (D1), D506–D515. doi: 10.1093/nar/gky1049
Coutinho P. M., Deleury E., Davies G. J., Henrissat B. (2003). An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 328 (2), 307–317. doi: 10.1016/s0022-2836(03)00307-3
Dong X. Z., Cai M. Y. (2001). “Determination of Biochemical Characteristics,” in Manual for the Systematic Identification of General Bacteria. Eds. Dong X. Z., Cai M. Y. (Beijing: Science Press), 370–398.
Drula E., Garron M. L., Dogan S., Lombard V., Henrissat B., Terrapon N. (2022). The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 50 (D1), D571–D577. doi: 10.1093/nar/gkab1045
Du S. T., Lu Q., Liu L. J., Wang Y., Li J. X. (2022). Rhodococcus qingshengii facilitates the phytoextraction of Zn, Cd, Ni, and Pb from soils by Sedum alfredii Hance. J. Hazardous Materials 424, 127638. doi: 10.1016/j.jhazmat.2021.127638
Ettwig K. F., Butler M. K., Le Paslier D., Pelletier E., Mangenot S., Kuypers M. M., et al. (2010). Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464 (7288), 543–548. doi: 10.1038/nature08883
Felsenstein J. (1981). Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 17 (6), 368–376. doi: 10.1007/bf01734359
Fernández-Gómez B., Richter M., Schüler M., Pinhassi J., Acinas S. G., González J. M., et al. (2013). Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 7 (5), 1026–1037. doi: 10.1038/ismej.2012.169
Folk R. L., Andrews P. B., Lewis D. W. (1970). Detrital sedimentary rock classification and nomenclature for use in New Zealand. N. Z. J. Geology Geophysics 13 (4), 937–968. doi: 10.1080/00288306.1970.10418211
Gaubert-Boussarie J., Prado S., Hubas C. (2020). An untargeted metabolomic approach for microphytobenthic biofilms in intertidal mudflats. Front. Mar. Sci. 7. doi: 10.3389/fmars.2020.00250
Gavriilidou A., Gutleben J., Versluis D., Forgiarini F., van Passel M. W., Ingham C. J., et al. (2020). Comparative genomic analysis of Flavobacteriaceae: insights into carbohydrate metabolism, gliding motility and secondary metabolite biosynthesis. BMC Genomics 21 (1), 1–21. doi: 10.1186/s12864-020-06971-7
Gómez-Silva B., Vilo-Muñoz C., Galetović A., Dong Q., Castelán-Sánchez H. G., Pérez-Llano Y., et al. (2019). Metagenomics of Atacama lithobiontic extremophile life unveils highlights on fungal communities, biogeochemical cycles and carbohydrate-active enzymes. Microorganisms 7 (12), 619. doi: 10.3390/microorganisms7120619
Gong B., Cao H. M., Peng C. Y., Perculija V., Tong G. X., Fang H. Y., et al. (2019). High-throughput sequencing and analysis of microbial communities in the mangrove swamps along the coast of Beibu Gulf in Guangxi, China. Sci. Rep. 9, 9377. doi: 10.1038/s41598-019-45804-w
Guo B., Wu X. L., Qian Y. (2006). Approaches for increasing the culturability of microorganisms. Weishengwu Xuebao 46 (3), 504–507. doi: 10.3321/j.issn:0001-6209.2006.03.036
Harboul K., Alouiz I., Hammani K., El-Karkouri A. (2022). Isotherm and kinetics modeling of biosorption and bioreduction of the Cr (VI) by Brachybacterium paraconglomeratum ER41. Extremophiles 26 (3), 30. doi: 10.1007/s00792-022-01278-9
Hernández-Plaza A., Szklarczyk D., Botas J., Cantalapiedra C. P., Giner-Lamia J., Mende D. R., et al. (2023). eggNOG 6.0: enabling comparative genomics across 12 535 organisms. Nucleic Acids Res. 51 (D1), D389–D394. doi: 10.1093/nar/gkac1022
Howard J., Hoyt S., Isensee K., Telszewski M., Pidgeon E. (2014). Coastal blue carbon: methods for assessing carbon stocks and emissions factors in mangroves, tidal salt marshes, and seagrasses (Arlington, VA: Conservation International, Intergovernmental Oceanographic Commission of UNESCO, International Union for Conservation of Nature).
Hu Y. B., Wang N., Yan X. C., Yuan Y., Luo F., Jiang Z. Y., et al. (2019). Ginsenoside Re impacts on biotransformation products of ginsenoside Rb1 by Cellulosimicrobium cellulans sp. 21 and its mechanisms. Process Biochem. 77, 57–62. doi: 10.1016/j.procbio.2018.11.019
Kanehisa M., Furumichi M., Tanabe M., Sato Y., Morishima K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45 (D1), D353–D361. doi: 10.1093/nar/gkw1092
Kappelmann L., Krüger K., Hehemann J. H., Harder J., Markert S., Unfried F., et al. (2019). Polysaccharide utilization loci of North Sea Flavobacteriia as basis for using SusC/D-protein expression for predicting major phytoplankton glycans. ISME J. 13 (1), 76–91. doi: 10.1038/s41396-018-0242-6
Katoh K., Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780. doi: 10.1093/molbev/mst010
Kim M., Oh H. S., Park S. C., Chun J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Systematic Evolutionary Microbiol. 64 (Pt_2), 346–351. doi: 10.1099/ijs.0.059774-0
Komagata K., Suzuki K. I. (1988). 4 Lipid and cell-wall analysis in bacterial systematics. Methods Microbiol. 19, 161–207. doi: 10.1016/S0580-9517(08)70410-0
Konstantinidis K. T., Tiedje J. M. (2005). Towards a genome-based taxonomy for prokaryotes. J. Bacteriology 187 (18), 6258–6264. doi: 10.1128/jb.187.18.6258-6264.2005
Krüger K., Chafee M., Ben Francis T., Glavina del Rio T., Becher D., Schweder T., et al. (2019). In marine Bacteroidetes the bulk of glycan degradation during algae blooms is mediated by few clades using a restricted set of genes. ISME J. 13 (11), 2800–2816. doi: 10.1038/s41396-019-0476-y
Lam-Tung N., Schmidt H. A., von Haeseler A., Bui Quang M. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi: 10.1093/molbev/msu300
Lapebie P., Lombard V., Drula E., Terrapon N., Henrissat B. (2019). Bacteroidetes use thousands of enzyme combinations to break down glycans. Nat. Commun. 10, 2043. doi: 10.1038/s41467-019-10068-5
Lechner M., Findeiss S., Steiner L., Marz M., Stadler P. F., Prohaska S. J. (2011). Proteinortho: Detection of (Co-)orthologs in large-scale analysis. BMC Bioinf. 12, 124. doi: 10.1186/1471-2105-12-124
Li M. S., Li T., Zhou M., Li M. D., Zhao Y. X., Xu J. J., et al. (2021). Caenorhabditis elegans extracts stimulate IAA biosynthesis in Arthrobacter pascens ZZ21 via the indole-3-pyruvic acid pathway. Microorganisms 9 (5), 970. doi: 10.3390/microorganisms9050970
Lichtenthaler F. W., Peters S. (2004). Carbohydrates as green raw materials for the chemical industry. Comptes Rendus Chimie 7 (2), 65–90. doi: 10.1016/j.crci.2004.02.002
Liu Y. L., Meng D., Li R. R., Gu P. F., Fan X. Y., Huang Z. S., et al. (2019). Rhodoligotrophos defluvii sp. nov., isolated from activated sludge. Int. J. Systematic Evolutionary Microbiol. 69 (12), 3830–3836. doi: 10.1099/ijsem.0.003691
Martiny A. C. (2019). High proportions of bacteria are culturable across major biomes. ISME J. 13 (8), 2125–2128. doi: 10.1038/s41396-019-0410-3
Mayor D. J., Thornton B., Jenkins H., Felgate S. L. (2018). “Chapter 3 microbiota: the living foundation,” in Mudflat ecology. Ed. Beninger P. G. (Amsterdam, Netherlands: Springer), 43–61.
McKee L. S., La Rosa S. L., Westereng B., Eijsink V. G., Pope P. B., Larsbrink J. (2021). Polysaccharide degradation by the Bacteroidetes: mechanisms and nomenclature. Environ. Microbiol. Rep. 13 (5), 559–581. doi: 10.1111/1758-2229.12980
Meier-Kolthoff J. P., Carbasse J. S., Peinado-Olarte R. L., Goeker M. (2022). TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 50 (D1), D801–D807. doi: 10.1093/nar/gkab902
Minnikin D., O'donnell A., Goodfellow M., Alderson G., Athalye M., Schaal A., et al. (1984). An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J. Microbiological Methods 2 (5), 233–241. doi: 10.1016/0167-7012(84)90018-6
Mohapatra M., Yadav R., Rajput V., Dharne M. S., Rastogi G. (2021). Metagenomic analysis reveals genetic insights on biogeochemical cycling, xenobiotic degradation, and stress resistance in mudflat microbiome. J. Environ. Manage. 292, 112738. doi: 10.1016/j.jenvman.2021.112738
Molari M., Giovannelli D., d’Errico G., Manini E. (2012). Factors influencing prokaryotic community structure composition in sub-surface coastal sediments. Estuarine Coast. Shelf Sci. 97, 141–148. doi: 10.1016/j.ecss.2011.11.036
Molina-Menor E., Gimeno-Valero H., Pascual J., Peretó J., Porcar M. (2021). High culturable bacterial diversity from a European desert: The Tabernas desert. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.583120
Murray N. J., Phinn S. R., DeWitt M., Ferrari R., Johnston R., Lyons M. B., et al. (2019). The global distribution and trajectory of tidal flats. Nature 565 (7738), 222–225. doi: 10.1038/s41586-018-0805-8
Nedashkovskaya O. I., Kim S. B., Suzuki M., Shevchenko L. S., Lee M. S., Lee K. H., et al. (2005). Pontibacter actiniarum gen. nov., sp nov., a novel member of the phylum 'Bacteroidetes', and proposal of Reichenbachiella gen. nov as a replacement for the illegitimate prokaryotic generic name Reichenbachia Nedashkovskaya et al. 2003. Int. J. Systematic Evolutionary Microbiol. 55, 2583–2588. doi: 10.1099/ijs.0.63819-0
Ni M., Yuan J. L., Hua J. Q., Lian Q. P., Guo A. H., Liu M., et al. (2020). Shrimp–vegetable rotational farming system: An innovation of shrimp aquaculture in the tidal flat ponds of Hangzhou Bay, China. Aquaculture 518, 734864. doi: 10.1016/j.aquaculture.2019.734864
Pandey R., Sharma P., Rathee S., Singh H. P., Batish D. R., Krishnamurthy B., et al. (2021). Isolation and characterization of a novel hydrocarbonoclastic and biosurfactant producing bacterial strain: Fictibacillus Phosphorivorans RP3. 3 Biotech. 11 (2), 105. doi: 10.1007/s13205-021-02655-5
Park S., Park J. M., Lee K. H., Yoon J. H. (2016). Pontibacter litorisediminis sp. nov., isolated from a tidal flat. Int. J. Systematic Evolutionary Microbiol. 66 (10), 4172–4178. doi: 10.1099/ijsem.0.001331
Parks D. H., Imelfort M., Skennerton C. T., Hugenholtz P., Tyson G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25 (7), 1043–1055. doi: 10.1101/gr.186072.114
Perillo G., Wolanski E., Cahoon D. R., Hopkinson C. S. (2018). Coastal Wetlands: An Integrated Ecosystem Approach (Amsterdam: Elsevier).
Rinke M., Maraun M., Scheu S. (2022). Spatial and temporal variations in salt marsh microorganisms of the Wadden Sea. Ecol. Evol. 12 (3), e8767. doi: 10.1002/ece3.8767
Rodriguez-R L. M., Konstantinidis K. T. (2014). Bypassing cultivation to identify bacterial species. Microbe 9 (3), 111–118. doi: 10.1128/microbe.9.111.1
Saitou N., Nei M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4 (4), 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Sasmito S. D., Kuzyakov Y., Lubis A. A., Murdiyarso D., Hutley L. B., Bachri S., et al. (2020). Organic carbon burial and sources in soils of coastal mudflat and mangrove ecosystems. Catena 187, 104414. doi: 10.1016/j.catena.2019.104414
Sasser M. (1990). Identification of bacteria by gas chromatography of cellular fatty acids (Newark, DE: Microbial ID Inc). Available at: http://www.microbialid.com/PDF/TechNote_101.pdf.
Schroeter R., Voigt B., Jürgen B., Methling K., Pöther D. C., Schäfer H., et al. (2011). The peroxide stress response of Bacillus licheniformis. Proteomics 11 (14), 2851–2866. doi: 10.1002/pmic.201000461
Seemann T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30 (14), 2068–2069. doi: 10.1093/bioinformatics/btu153
Sefrji F. O., Marasco R., Michoud G., Seferji K. A., Merlino G., Daffonchio D. (2022). Insights Into the Cultivable Bacterial Fraction of Sediments From the Red Sea Mangroves and Physiological, Chemotaxonomic, and Genomic Characterization of Mangrovibacillus cuniculi gen. nov., sp. nov., a Novel Member of the Bacillaceae Family. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.777986
Sefrji F. O., Michoud G., Marasco R., Merlino G., Daffonchio D. (2021). Mangrovivirga cuniculi gen. nov., sp. nov., a moderately halophilic bacterium isolated from bioturbated Red Sea mangrove sediment, and proposal of the novel family Mangrovivirgaceae fam. nov. Int. J. Systematic Evolutionary Microbiol. 71 (7), 4866. doi: 10.1099/ijsem.0.004866
Shi X. L., Wu Y. H., Jin X. B., Wang C. S., Xu X. W. (2017). Alteromonas lipolytica sp. nov., a poly-beta-hydroxybutyrate-producing bacterium isolated from surface seawater. Int. J. Systematic Evolutionary Microbiol. 67 (2), 237–242. doi: 10.1099/ijsem.0.001604
Siamphan C., Chang Y. H., Kim W. (2015). Eudoraea chungangensis sp nov., isolated from an aquafarm waste water sludge. Antonie Van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 107 (4), 1009–1015. doi: 10.1007/s10482-015-0393-7
Staloch B. E. K., Niero H., de Freitas R. C., Ballone P., Rodrigues-Costa F., Trivella D. B. B., et al. (2022). Draft genome sequence of Psychrobacter nivimaris LAMA 639 and its biotechnological potential. Data Brief 41, 107927. doi: 10.1016/j.dib.2022.107927
Steen A. D., Crits-Christoph A., Carini P., DeAngelis K. M., Fierer N., Lloyd K. G., et al. (2019). High proportions of bacteria and archaea across most biomes remain uncultured. ISME J. 13 (12), 3126–3130. doi: 10.1038/s41396-019-0484-y
Subhash Y., Tushar L., Sasikala C., Ramana C. V. (2013). Erythrobacter odishensis sp nov and Pontibacter odishensis sp nov isolated from dry soil of a solar saltern. Int. J. Systematic Evolutionary Microbiol. 63, 4524–4532. doi: 10.1099/ijs.0.052183-0
Tamura K., Stecher G., Kumar S. (2021). MEGA11 molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38 (7), 3022–3027. doi: 10.1093/molbev/msab120
Tang K., Lin Y., Han Y., Jiao N. (2017). Characterization of potential polysaccharide utilization systems in the marine bacteroidetes gramella flava JLT2011 using a multi-omics approach. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00220
Terrapon N., Lombard V., Drula E., Lapebie P., Al-Masaudi S., Gilbert H. J., et al. (2018). PULDB: the expanded database of Polysaccharide Utilization Loci. Nucleic Acids Res. 46 (D1), D677–D683. doi: 10.1093/nar/gkx1022
Terrapon N., Lombard V., Gilbert H. J., Henrissat B. (2015). Automatic prediction of polysaccharide utilization loci in Bacteroidetes species. Bioinformatics 31 (5), 647–655. doi: 10.1093/bioinformatics/btu716
Tindall B. J., Rosselló-Móra R., Busse H. J., Ludwig W., Kämpfer P. (2010). Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Systematic Evolutionary Microbiol. 60 (1), 249–266. doi: 10.1099/ijs.0.016949-0
Torsvik V., Ovreas L. (2002). Microbial diversity and function in soil: from genes to ecosystems. Curr. Opin. Microbiol. 5 (3), 240–245. doi: 10.1016/s1369-5274(02)00324-7
Underwood G. J. C., Kromkamp J. (1999). Primary production by phytoplankton and microphytobenthos in estuaries. Adv. Ecol. Res. 29, 93–153. doi: 10.1016/s0065-2504(08)60192-0
Unfried F., Becker S., Robb C. S., Hehemann J.-H., Markert S., Heiden S. E., et al. (2018). Adaptive mechanisms that provide competitive advantages to marine bacteroidetes during microalgal blooms. ISME J. 12 (12), 2894–2906. doi: 10.1038/s41396-018-0243-5
Wang Y., Nie M. Q., Diwu Z. J., Chang F., Nie H. Y., Zhang B., et al. (2021). Toxicity evaluation of the metabolites derived from the degradation of phenanthrene by one of a soil ubiquitous PAHs-degrading strain Rhodococcus qingshengii FF. J. Hazardous Materials 415, 125657. doi: 10.1016/j.jhazmat.2021.125657
Wang X. X., Xiao X. M., Zou Z. H., Chen B. Q., Ma J., Dong J. W., et al. (2020b). Tracking annual changes of coastal tidal flats in China during 1986–2016 through analyses of Landsat images with Google Earth Engine. Remote Sens. Environ. 238, 110987. doi: 10.1016/j.rse.2018.11.030
Wang G. H., Xu S. L., Dang G., Liu J. F., Su H. F., Chen B. A., et al. (2020a). Poritiphilus flavus gen. nov., sp. nov., a member of the family Flavobacteriaceae isolated from coral Porites lutea. Int. J. Systematic Evolutionary Microbiol. 70 (11), 5620–5626. doi: 10.1099/ijsem.0.004452
Williams S., Davies F. (1965). Use of antibiotics for selective isolation and enumeration of actinomycetes in soil. Microbiology 38 (2), 251–261. doi: 10.1099/00221287-38-2-251
Xu J., Bjursell M. K., Himrod J., Deng S., Carmichael L. K., Chiang H. C., et al. (2003). A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science 299 (5615), 2074–2076. doi: 10.1126/science.1080029
Xu L., Wu Y. H., Zhou P., Cheng H., Liu Q., Xu X. W. (2018). Investigation of the thermophilic mechanism in the genus Porphyrobacter by comparative genomic analysis. BMC Genomics 19, 385. doi: 10.1186/s12864-018-4789-4
Yoon S. H., Ha S. M., Lim J., Kwon S., Chun J. (2017). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 110 (10), 1281–1286. doi: 10.1007/s10482-017-0844-4
Yuan H. L., Xu Y., Chen Y. Z., Zhan Y. Y., Wei X. T., Li L., et al. (2019). Metabolomics analysis reveals global acetoin stress response of Bacillus licheniformis. Metabolomics 15, 1–12. doi: 10.1007/s11306-019-1492-7
Zhang R., Sun M. R., Zhang H. L., Zhao Z. H. (2021). Spatial separation of microbial communities reflects gradients of salinity and temperature in offshore sediments from Shenzhen, south China. Ocean Coast. Manage. 214, 105904. doi: 10.1016/j.ocecoaman.2021.105904
Zhang H., Yohe T., Huang L., Entwistle S., Wu P. Z., Yang Z. L., et al. (2018). dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46 (W1), W95–W101. doi: 10.1093/nar/gky418
Zhang L., Zhang Q. J., Luo X. S., Tang Y. L., Dai J., Li Y. W., et al. (2008). Pontibacter korlensis sp nov., isolated from the desert of Xinjiang, China. Int. J. Systematic Evolutionary Microbiol. 58, 1210–1214. doi: 10.1099/ijs.0.65667-0
Zheng Z. G., Zheng J. S., Liu H. L., Peng D. H., Sun M. (2016). Complete genome sequence of Fictibacillus phosphorivorans G25-29, a strain toxic to nematodes. J. Biotechnol. 239, 20–22. doi: 10.1016/j.jbiotec.2016.09.014
Zou H. Y., Berglund B., Xu H., Chi X. H., Zhao Q., Zhou Z. Y., et al. (2020). Genetic characterization and virulence of a carbapenem-resistant Raoultella ornithinolytica isolated from well water carrying a novel megaplasmid containing bla(NDM-1). Environ. pollut. 260, 114041. doi: 10.1016/j.envpol.2020.114041
Keywords: bare tidal flats, culturable bacteria, CAZymes, polysaccharide utilization loci, bacteroidota
Citation: Ma K-J, Ye Y-L, Fu Y-H, Fu G-Y, Sun C and Xu X-W (2023) Genomic and phylotypic properties of three novel marine Bacteroidota from bare tidal flats reveal insights into their potential of polysaccharide metabolism. Front. Mar. Sci. 10:1222157. doi: 10.3389/fmars.2023.1222157
Received: 13 May 2023; Accepted: 03 July 2023;
Published: 20 July 2023.
Edited by:
Danny Ionescu, Leibniz-Institute of Freshwater Ecology and Inland Fisheries (IGB), GermanyCopyright © 2023 Ma, Ye, Fu, Fu, Sun and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xue-Wei Xu, eHV4d0BzaW8ub3JnLmNu; Cong Sun, bWljaGFlbF9zY0BzaW5hLmNvbQ==