 Jennifer M. Polinski
Jennifer M. Polinski Mattie Rodrigue
Mattie Rodrigue Jason D. Meyer
Jason D. Meyer Matthew J. Harke
Matthew J. Harke- 1Gloucester Marine Genomics Institute, Gloucester, MA, United States
- 2OceanX, New York, NY, United States
The deep sea is the largest habitat on our planet, supporting a vast diversity of organisms which have yet to be fully described. This habitat is punctuated by hydrothermal vents in which energy derived from chemosynthesis drives carbon fixation, supporting a complex and rich food web. Connectivity between vent systems remains an active area of research, with questions as to how vent-influenced microbial function and diversity persists over space and time. In particular, the role hydrothermal vent plumes play as potential highways for connectivity and biogeography is not well understood. To add to the growing body of research, this study sampled plume waters above the Moytirra hydrothermal vent field, located just north of the Azores. We examined how hydrothermal vent plume community biodiversity and metabolic activities change with distance from the vent using a combination of metabarcoding and metatranscriptomic sequencing. We detected a rich diversity of both prokaryotic and eukaryotic organisms inhabiting the plume, which remained metabolically active for kilometers from the vent source. Enriched sulfur metabolism functional signals and relative abundance of sulfur oxidizing bacteria suggest reduced sulfur compounds are a fundamental energy source within plume waters. Additionally, we observed evidence of top-down controls on primary production through both known grazers and putative viral activity. Although community-level functional signals suggest active metabolic functions for over a kilometer north or south of the vent field, these functions grew increasingly dissimilar to those observed directly above the vent site, and bacterial communities displayed indications of entering quiescent stages, likely due to decreasing resources and reduced temperatures. These data provide a first glimpse of Moytirra’s microbial biodiversity, in addition to providing a high-resolution understanding of life on the drift within a hydrothermal plume, its persistence with distance, and implications for connectivity.
Introduction
Deep-sea hydrothermal vents are found in all ocean basins and host a wide diversity of organisms fueled by chemosynthetic processes (Jannasch and Mottl, 1985; Dick, 2019; Beaulieu and Szafrański, 2020). Geological setting, host rock, rate of seafloor spreading, temperature gradients, and degree of mixing along the flow path, among other factors, all influence the chemical and physical aspects which shape community structure and metabolism at any given vent ecosystem (Orcutt et al., 2011; Sievert and Vetriani, 2012; Dick, 2019). As hydrothermal fluids interact with source rock, the concentration and composition of reduced chemical species (e.g. H2, H2S, Fe, Mn, methane) emerging from vents support a wide variety of metabolically versatile chemolithoautotrophic microbial organisms (Jannasch, 1995). These microbial organisms in turn support a complex ecosystem structured by bottom-up (e.g. chemosynthetic primary production) and top-down (e.g. grazing, viral infection) processes (Govenar, 2012), with generated biomass comparable to photosynthetically-based marine ecosystems (Sarrazin and Juniper, 1999). Not only do these plumes drive local vent biology, but also significantly contribute to the deep-sea carbon cycle (Le Bris et al., 2019) as well as global metal inventories (Tagliabue et al., 2010; Gartman and Findlay, 2020).
As vent-derived chemically rich waters mix with cold, oxidized seawater, they rise hundreds of meters above the vent and disperse laterally for hundreds of kilometers, potentially acting as highways between vent ecosystems (Dick et al., 2013). The absolute height of a plume is determined by the density contrast between it and the surrounding seawater, with plume density controlled primarily by the heat output of the vent and rate of ambient water entrainment (Turner and Campbell, 1987). Lateral flow of the resulting neutrally buoyant plume is in turn influenced by mixing and ocean currents which are steered by bathymetry (Speer et al., 2003; Tyler and Young, 2003). Tidal or inertial currents can also be superimposed on the background flow of water in the region, potentially altering trajectories and creating complex flow patterns (Walter et al., 2010). As such, plume dispersal can be impacted by both small and large scale oceanic processes, influencing both chemistry and biology within the plume, as well as the distance from a vent these processes may travel.
The hydrothermal vents of the Mid-Atlantic Ridge (MAR) occur across a range of depths (between 840 and >3500 m) and geologic settings (e.g. massive sulfide mounds vs clusters of small sulfide chimneys), differing widely in chemical composition and subsequent community composition (Desbruyères et al., 2000). Unlike vents of the fast-spreading East Pacific Rise, where endemic Riftia tubeworms are found (Tunnicliffe et al., 1998), MAR vent systems tend to be dominated by Rimicaris and Bathymodiolus assemblages (Van Dover, 1995) and many of the currently characterized and well-studied vent systems lie south of the Azores (e.g. Lucky Strike, TAG). Only recently were vents observed between the Azores and Iceland, with tow-yo data from a cruise in 2008 identifying several neutrally buoyant plumes in the region, suggesting the presence of multiple vent sources (Wheeler et al., 2013). In 2011, further surveying with CTD tow-yos and ROV missions led to the discovery of the Moytirra hydrothermal vent system (Wheeler et al., 2013). Two published expeditions have visited the site, providing high-resolution mapping and characterizing the geology and animal fauna (Wheeler et al., 2013; Somoza et al., 2020). Located at 45°N, this high-temperature vent field is basalt-hosted, consisting of multiple black smokers and sitting midway up the 300 m high fault scarp of the eastern axial wall, 3.5 km from the axial volcanic ridge crust at ~2900 m depth (Wheeler et al., 2013). Numerous fauna have been observed inhabiting vent chimneys including gastropods, alvinocaridid shrimp, mussels, and zoarcid fish with some species thought to be similar to those south of the Azores, potentially extending the known latitudinal range of these organisms (Wheeler et al., 2013). However, to date, no studies have investigated the microbial communities associated with the Moytirra vent field or their potential connectivity with other vents in the region.
Although hydrothermal vent systems have been explored for decades, understanding the biogeography of microbial organisms and the role dispersal and isolation have in shaping these communities, remains a challenge (Dick, 2019). Variations in local geology and subsequent chemistry may impart potential barriers to exchange within vent fields, effectively isolating distinct bacterial communities (Reveillaud et al., 2016; Fortunato et al., 2018; Galambos et al., 2019). Similarly, the observation of potential endemic or novel vent-associated protists occurring across geographically and geochemically distinct vent ecosystems suggests environmental constraints may select for unique species in these extreme environments (Murdock and Juniper, 2019; Hu et al., 2022). However, there is also evidence that most, if not all, microbial communities inhabiting vents can be found in surrounding deep seawater, suggesting frequent dispersal between vent sites (Gonnella et al., 2016), potentially discounting the idea of endemism in microorganisms. As microbial communities within vent plumes are thought to persist for tens to hundreds of kilometers (Reed et al., 2015), plumes potentially act as seed banks for colonization of new vent sites as they arise (Gibbons et al., 2013). However, biodiversity appears to decline with distance from the vent site, suggesting a temporal succession driven by changing resources as plumes age and drift from the vent source (Haalboom et al., 2020). As such, there is still much to learn about whether vents host endemic microbes not found elsewhere and if so, how these may be transported to colonize new vent sites. Further, few studies have examined how the metabolisms within hydrothermal plumes change over time and space and how this translates to our understanding of connectivity and biogeography. In this study, we combine prokaryote-specific 16S SSU ribosomal RNA (rRNA) gene sequencing, eukaryote-specific 18S SSU rRNA gene sequencing, and metatranscriptomics to examine how hydrothermal vent plume community biodiversity and metabolic activities change with distance from the Moytirra hydrothermal vent.
Materials and methods
Bathymetric data
High-resolution bathymetric data were acquired aboard the R/V OceanXplorer (OXR20210703) on July 4th, 2021, as part of the OceanX Young Explorers Program, over the course of two days throughout the region surrounding the Moytirra vent site prior to CTD and water sampling operations. The R/V OceanXplorer is equipped with a Kongsberg Maritime EM304 multibeam echosounder (MBES), supported by a Seapath 380/MRU 5+ for precise positioning and inertial motion sensor data. The multibeam data acquisition software used during the survey was Kongsberg SIS v5. The EM304 MBES operates within a frequency range of 26.5 and 34.5 kHz and can receive 1600 soundings per ping using dual swath mode. This, along with the narrow beamwidth of each ping (0.5° TX/1.0° RX), slow survey speed (~6 knots) during acquisition, and >200% survey coverage, produced high-quality bathymetric data with maximum sounding density. Updated sound velocity profiles (SVP) were routinely applied to the acquisition software throughout the survey to calibrate the MBES for changing temperature/salinity variables. To generate SVP information, a Lockheed Martin expendable bathythermograph (XBT) probe was used to collect temperature/depth profiles. These profiles were processed using HydrOffice Sound Speed Manager software wherein location specific salinity values from the World Ocean Atlas database (WOA18) were utilized to generate extended SV profiles. Acquired bathymetric data proceeded through quality control and processing stages using QPS Qimera software and the resultant processed data was then used to create a gridded digital terrain model (DTM) and used for analysis and ship positioning during CTD operations using QPS Fledermaus and Qinsy software, respectfully. Due to the extreme data density of soundings collected, we were able to generate a spatial resolution of 20 m throughout the study region which extended to over 3,600 m ocean depth (Figure 1).
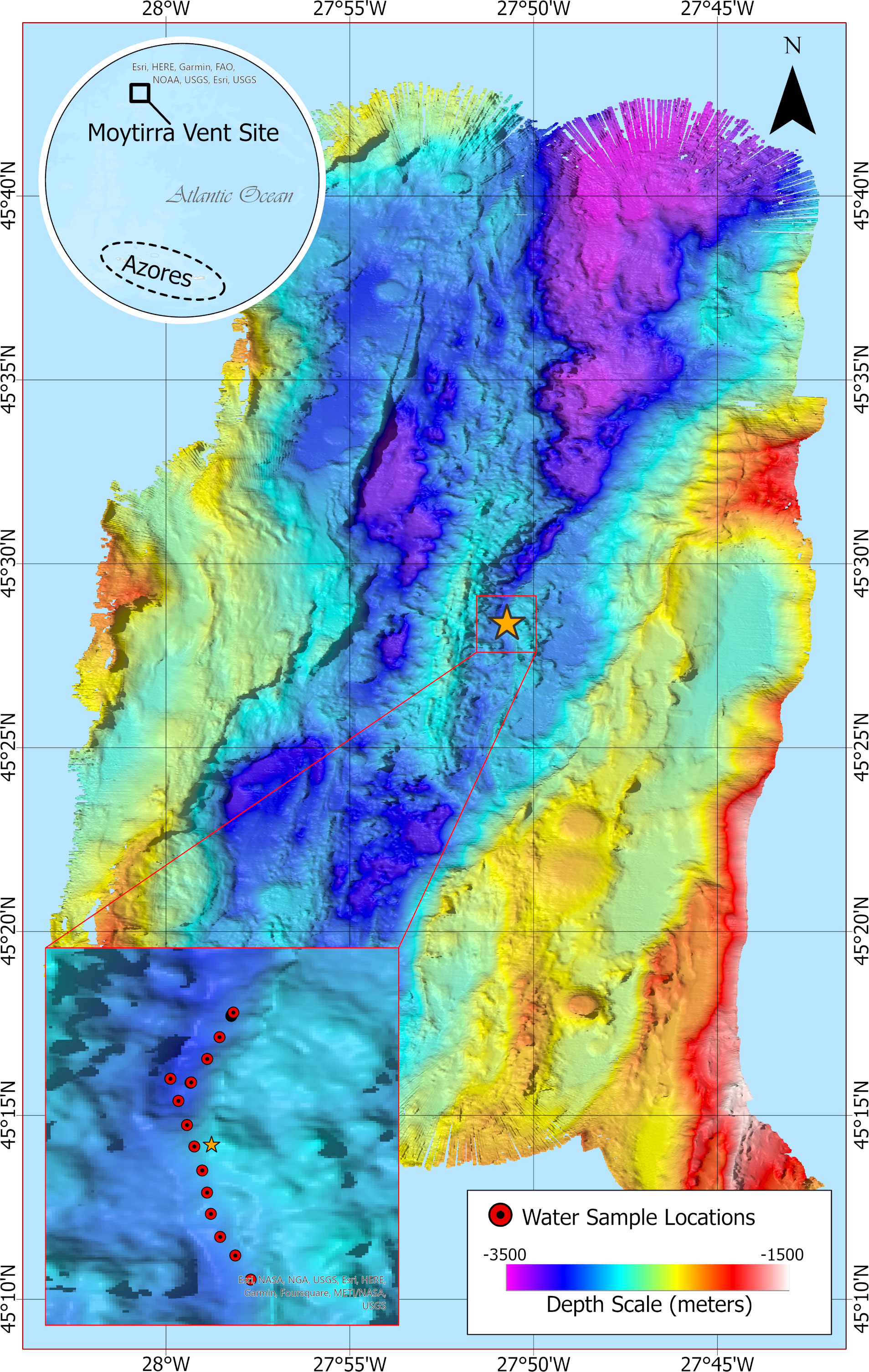
Figure 1 Bathymetric map at 20 m resolution showing sampling locations (inset, red dots) following a transect across the hydrothermal vent Moytirra (orange star). Benthic depth is indicated by color scale.
Sample collection
After survey operations, CTD rosette sampling was conducted on July 6th, 2021, within the hydrothermal plume near the Moytirra vent site (Lat 45.4833 N, Lon -27.8500 W). Vent plume seawater sampling was performed using a Seabird 911+ CTD rosette including a Wet Labs C-Star transmissometer. A primary goal of this research was to characterize vent plume biology and metabolic activity across a spatial gradient, and as such, sample collection was conducted along a 2.5 km transect following an axial ridge along the Mid-Atlantic Ridge so that sampling was conducted south of, above, and north of the Moytirra vent (Figures 1, 2). Two CTD casts were conducted in total. An initial tow-yo survey was planned to locate the plume but in the initial CTD deployment, hydrothermal plumes were detected at ~2450 m based upon a change in beam transmission (turbidity) at which point a transect was begun following the axial ridge (Figure 2, Supplementary Figure 1). Niskin bottle sampling began at this time, firing bottles approximately every 200 m. Once all bottles were filled, the CTD was retrieved, emptied, and redeployed for a total of two casts. The first bottle of cast 1 was triggered at 10:08 UTC with the final bottle triggered at 12:24 UTC. The first bottle of cast 2 was triggered at 15:41UTC with the last being triggered at 16:52 UTC (Supplementary Table 1). Samples were taken both within (n=15), under (n=1), and over (n=1) turbidity peaks to characterize both within and out of plume biology along the transect.
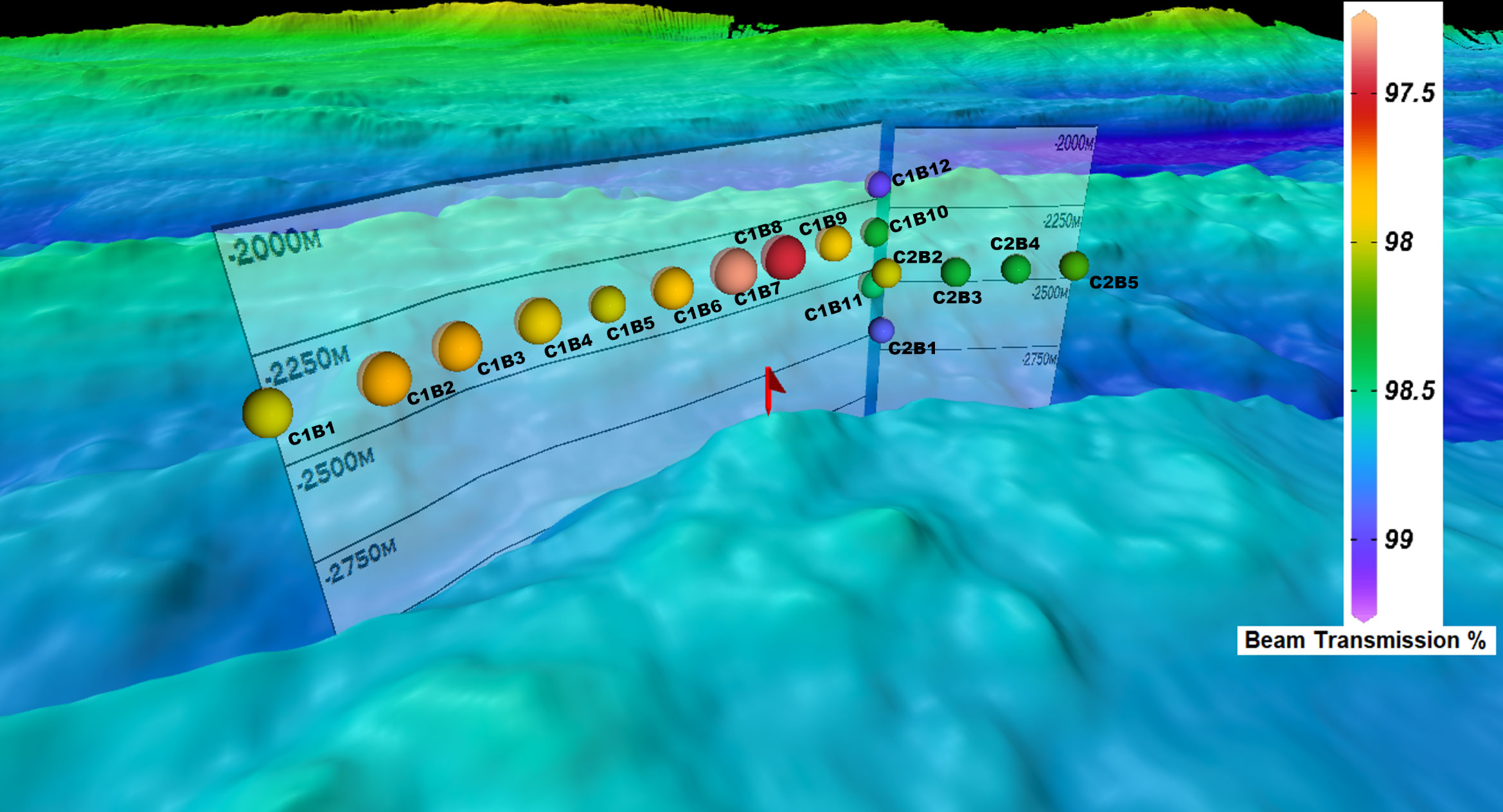
Figure 2 Sampling stations plotted in reference to bathymetry displaying beam transmission (%) at the time bottles were fired. Each station is labeled to guide the reader with the first sample taken at C1B1 and the last being C2B5. The letter “C” denotes cast number while “B” denotes bottle number on the CTD rosette and corresponds to headings in Supplementary Table 1.
For each CTD cast, 12 L Niskin bottles were drained into sterile carboys by attaching sterile tubing to the drain plug to allow for laminar flow, reducing bubbling. Once filled, carboys were immediately placed in a walk-in cooler (~4°C) in the dark to maintain temperature similar to that at time of collection. Each sample was then filtered across three 47 mm 0.2 µm Whatman ME filters using Sentino® Microbiology Peristaltic Pumps equipped with 500 mL Sentino® Magnetic Filter Funnels and disposable sterile fluid paths. Total volume ranged between 1-2 L per filter per sample. After the initial 500 mL was filtered, additional filtered volume was collected in a 250 mL LDPE bottle from the fluid path exiting the Sentino pump for dissolved water chemistry characterization and stored at -20°C until analysis. Fluid paths were changed between samples and the magnetic filter was rinsed with UltraPure water and 0.2 µm filtered bottom water between samples. After all volume was filtered, each filter was placed in a 2 mL cryovial and frozen at -80°C. Samples were stored at this temperature or colder until extraction and sequencing. Samples were labeled according to cast number, Niskin bottle number, and filter number (e.g., C1B1F1 is cast 1, bottle 1, and filter 1). Since only a single Niskin bottle was collected from each location, replicate filters are considered “technical” replicates (n=3) and we acknowledge significant time occurred (~4-6 h) between capturing water samples, completion of CTD cast, retrieval, and filtering which may have altered metatranscriptomic and chemical signals due to not only temporal but alterations in pressure and temperature. Pressure changes may have altered detected community compositions as lysis of obligate barophilic bacteria has been observed within 7h of retrieval from depth (Chastain and Yayanos, 1991). However, as the water temperature was maintained in the dark at ~4°C, variability in transcriptional signals may have been lessened to just those signals responding with time, pressure, and bottle effect. For instance, a recent study found that CTD retrieval of deep-sea water to the surface has little impact on transcriptional profiles with exception of thermal stress response (e.g., heat shock proteins) as well as softening of the overall metabolic responses due to potential loss of oxygen due to depressurization (Fortunato et al., 2021).
For all analyses, samples are grouped as within plume (C1B1-B11, C2B2-B5) and out of plume (C1B12 and C2B1). Within plume samples are subdivided relative to the vent location: south of vent or “South Plume” (C1B-C1B6), “Above Vent” (C1B7-C1B8), and north of vent or “North Plume” (C1B9-C1B11, C2B2-C2B5) (Supplemental Table 1).
Water chemistry
Filtered seawater samples were processed at the University of Maryland Center for Environmental Science Nutrient Analytical Services Laboratory (https://www.umces.edu/nutrient-analytical-services-laboratory) according to center practices. For each sample, 0.2 µm filtered seawater was analyzed for ammonium (NH4), nitrate/nitrite (NO3/NO2), phosphate (PO4), silicate (Si), total dissolved nitrogen and phosphorus (TDN/TDP), dissolved organic carbon (DOC), dissolved inorganic carbon (DIC), and sulfate (SO4) according to center protocols (https://www.umces.edu/nasl/methods). Dissolved organic phosphate (DOP) and dissolved organic nitrogen (DON) were calculated by subtracting PO4 and NO3/NO2 from TDP and TDN respectively. As NH4 was below detection limits, this was not included in the DOP/DON calculation.
DNA/RNA extraction
DNA and RNA were simultaneously extracted from filters using the Macherey-Nagel NucleoMag® DNA/RNA Water kit. Lysis steps were performed in 5 mL MN Bead Tubes with 900 µL lysate volume to accommodate the 47 mm filters. The resulting lysate was split into two equal volumes, and manufacturer’s instructions were followed for the remainder of the extraction. After extraction, DNA was removed from one extraction replicate using the Qiagen DNase Max® Kit for total RNA isolation. Resulting quantities of DNA and RNA were measured using the Qubit HS dsDNA assay and HS RNA assay, respectively, and were stored at -80°C until library prep and sequencing.
Metabarcoding library prep, sequencing, & analysis
To assess captured diversity within filtered samples, DNA extracts were used in reactions targeting the V4 region of the prokaryotic 16S SSU rRNA gene and the V2 region of the eukaryotic 18S SSU rRNA gene. 16S reactions used primers V4_515F (Parada et al., 2016) and V4_806R (Apprill et al., 2015) while 18S reactions used SSU_FO4 and SSU_R22 (Blaxter et al., 1998) with linking sequences for addition of Illumina sequencing adapters and indices at their 5′ end (forward: 5′–TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG–3′; reverse: 5′–GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG–3′). Reactions of 50 µL contained 1X OneTaq® Master Mix with standard buffer (New England Biolabs, Ipswich, MA), 0.2 µM forward primer, and 0.2 µM reverse primer. PCR conditions were as follows: 94°C initial denaturation for 3 minutes, followed by 30 cycles of 94°C for 30 seconds, 62°C for 16S or 60°C for 18S for 30 seconds, and 68°C for 30 seconds, and a final extension at 68°C for 5 minutes. Amplicon products were cleaned with PCRclean DX beads (Aline Biosciences, Woburn, MA) using a 1:1 sample-to-bead ratio. A second 25 µL PCR reaction was performed to incorporate Nextera sequencing adapters following the same conditions as the first reaction but using only seven cycles. Excess sequencing adapter was removed with an additional 1:1 bead cleanup with PCRclean DX beads. Adapter-ligated library concentrations were measured using the NEBNext Library Quant for Illumina kit (New England Biolabs), adjusting for the 435 bp and 565 bp fragment sizes for 16S and 18S, respectively. Negative controls were prepped alongside samples and qPCR results indicated no contamination was present. Libraries were pooled in equimolar concentrations and sequenced on a PE300 Illumina MiSeq run using v3 reagents. A total of 3,917,135 16S and 4,921,787 18S read pairs were generated.
The first fifteen bases of resulting sequencing reads were trimmed to remove the primer sequence, and then reads were quality trimmed with Trimmomatic v0.38 (Bolger et al., 2014) (TRAILING:15 MINLEN:150 for 16S; TRAILING:15 MINLEN 200 for 18S). This resulted in a total of 3,916,612 (99.9%) and 4,914,883 (99.8%) high-quality read pairs for 16S and 18S ASV analysis, respectively. Read pairs were merged, filtered, dereplicated, and denoised using the UNOISE3 pipeline in USEARCH v11.0.667 (Edgar, 2010). Merged reads were mapped to the resulting amplicon sequence variants (ASVs) to generate a count table. The “classify.seqs” command from Mothur (Schloss et al., 2009) was used with the SILVA v138.1 (Quast et al., 2013) database to classify 16S and 18S ASVs. For 16S ASVs not assigned taxonomy with SILVA, a phylogenetic tree was generated to determine a phylum-level classification. Briefly, 16S ASVs were subset by phyla and clustered at 90% similarity to generate a set of representative sequences. The representative sequences and unclassified sequences were then aligned using MUSCLE v5.1 (Edgar, 2004) and used to generate a neighbor-joining tree with MEGA X (https://www.megasoftware.net/). Phylum-level classifications were assigned to any previously unknown ASV that grouped in the same branch as representative sequences from a single phylum. For 18S classified ASVs, manual curations were imposed similarly to Hu et al. (2021) for direct comparison.
Metatranscriptomic library prep, sequencing, & assembly
Quantity and quality of RNA extracts were determined on a Fragment Analyzer with an HS RNA kit (Agilent, Santa Clara, CA). Metatranscriptome libraries were prepared using a TruSeq Stranded Total RNA Prep kit with Ribo-Zero Plus (Illumina, San Diego, CA). Sequencing was conducted at the University of Connecticut Center for Genome Innovation (https://cgi.uconn.edu/) on an Illumina NovaSeq 6000 using an SP200 cycle v1.5 flow cell targeting 40M 100bp paired end reads per sample. Resulting sequences were assessed for quality using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and trimmed using Trimmomatic (SLIDINGWINDOW:5:20 HEADCROP:12 MINLEN:50 and removing Truseq adapters).
Reads from each set of triplicate libraries were combined and metatranscriptomes were assembled for each bottle using Trinity v2.13.2 (Grabherr et al., 2011) and IDBA-UD (Peng et al., 2012) using default parameters for stranded libraries. The resulting metatranscriptome assemblies were combined and clustered at 90% similarity with CD-HIT (Li and Godzik, 2006). The final transcript set was generated by using Prokka v1.14.6 (Seemann, 2014) and TransDecoder v5.5.0 (https://github.com/TransDecoder/TransDecoder) to identify bacterial and eukaryotic genes, respectively. The Trinity “align_and_estimate_abundance.pl” script was used with RSEM (Li and Dewey, 2011) as the estimation method and Bowtie2 (Langmead and Salzberg, 2012) as the alignment method to generate read counts. Differential expression analysis was conducted with DESeq2 v1.32.0 (Love et al., 2014). Within plume bottles were contrasted against those obtained over (C1B12) and under (C2B1) the plume, representing background water or that outside of the influence of the hydrothermal plume, here referred to as “out of plume”. Resulting expression analysis was filtered for significance using a Padj value of <0.05. To aggregate results across all comparisons, the DEVis (Price et al., 2019) package was used. Kraken2 v2.1.3 with the Kraken Standard PlusPF database was used to classify taxonomic origin of the quality-trimmed metatranscriptomic reads, and abundance estimates were generated with Bracken v2.8 (Lu et al., 2017; Wood et al., 2019).
The Kyoto Encyclopedia of Genes and Genomes (KEGG) database was used to annotate amino acid sequences generated during assembly using the GhostKOALA mapping service against the “genus_prokaryotes + family_eukaryotes + viruses” database files. To look at differential expression at a functional level, normalized read counts for all transcripts identified as the same KO term were summed for each sample. Summed KO term counts were analyzed for differential expression as previously described. Transcripts not assigned a KO term were excluded from this analysis.
Viral signals were identified from the combined assembly using VirSorter2 v2.2.3 (Guo et al., 2021) on contigs >1 kb. Identified viral signatures were further filtered for non-viral sequences or regions with CheckV (Nayfach et al., 2021) and resultant hits were annotated with ViroBLAST against the Viral Genbank database (Deng et al., 2007). Additionally, following Thomas et al. (2021), VirFinder v1.1 (Ren et al., 2017) was used to employ a k-mer frequency-based method to search for potential novel viral signals. As with VirSorter, searches were limited to contigs >1 kb as both tools tend to have better true prediction rates with larger contigs. Results for VirFinder were filtered for p-value <0.05.
Statistical analysis
Spearman rank correlations between diversity and biogeochemical data were calculated and visualized with the R package “corrplot’ and resulting p-values controlled for false discovery using the ‘p.adjust’ function in R, Benjamini-Hochberg (BH) method (Benjamini and Hochberg, 1995). Shannon diversity index was calculated within the ‘microbiome’ R package and the difference between environmental groupings was tested with the non-parametric Kruskal-Wallis test with pairwise comparisons determined with Wilcoxon test corrected for multiple testing using the Benjamini-Hochberg (BH) method. For assessment of community-level diversity and functional dissimilarity metrics, Bray-Curtis dissimilarity was calculated using ‘vegdist’ function within the R package “vegan” (version 2.6-2) on DESeq2-normalized read count data derived from the metatranscriptomic analysis for functional signals and square-root transformed diversity count data derived from the metabarcoding analysis and Kraken2 for diversity signals. Here we define functional diversity like Carmona et al. (2016) whereby functional diversity is the variation of traits (here transcripts) between organisms in the functional space occupied by an ecological unit (here community). All transcripts were included in the dissimilarity analysis regardless of annotation. A linear regression model was then fit to the dissimilarity data and compared against distance using the ‘lm’ function in R to test for significant differences.
Results
Hydrothermal vent plumes were identified in the water column by an increase in turbidity as visualized by the reduction in beam transmission between 2250 and 2750 m with the largest reduction in beam transmission centered around 2450 m (Supplementary Figure 1). Beam transmission was lowest (highest turbidity) above the vent location, averaging ~97.4% (cast 1, Niskin 7 & 8; Supplementary Table 1) but did not correlate with any measured environmental parameter beyond sampling depth (p < 0.01; Figure 3). Neutrally buoyant plumes detected both north and south of the vent site along the transect suggests other vent systems farther south of Moytirra and/or a complex flow pattern in and around the vent (Walter et al., 2010). Water flow at bathypelagic and deeper (where hydrothermal vents typically occur) likely follows thermohaline circulation patterns, flowing north along the MAR valley with nuanced flow and turbulence due to benthic topography and tidal influence (Thurnherr et al., 2002; Lahaye et al., 2019; Haalboom et al., 2020). In the current study, it was unclear which overall direction water was flowing, suggesting more turbulent fluid dynamics characteristic of slow spreading ridges (German et al., 1998; Walter et al., 2010). Alternatively, we may have detected plume signatures of vents south of Moytirra if indeed mean water flow was in a northward direction.
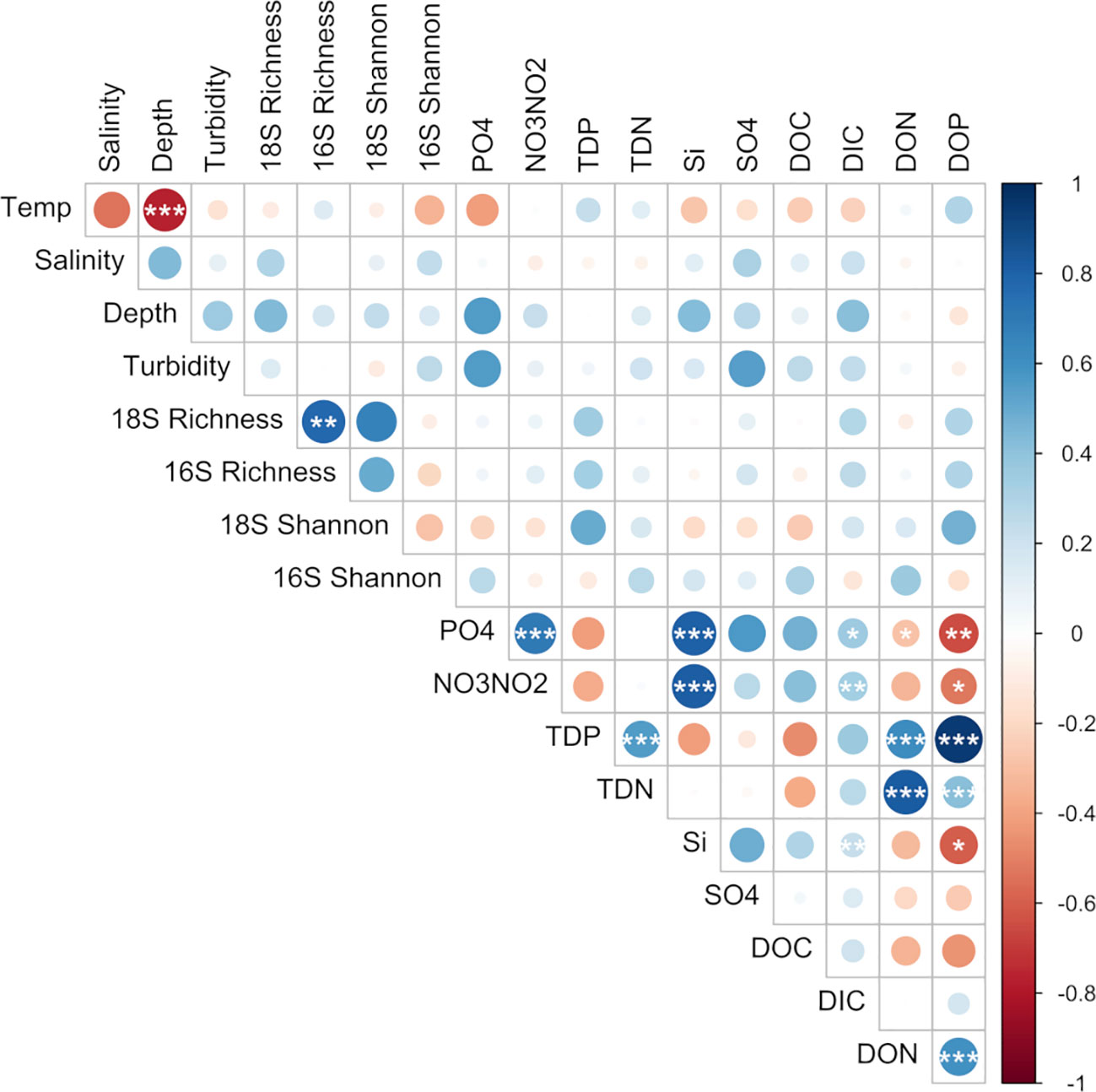
Figure 3 Pairwise comparisons of environmental factors and species richness with a color gradient denoting Spearman’s correlation coefficient. Stars indicate significant correlations after multiple testing (BH-method; *p < 0.05, **p < 0.01, ***p < 0.001).
Measures of dissolved water chemistry were variable across the transect, however water collected just above and near the vent site (cast 1, bottles 8 & 9) displayed lower levels of DIC, Si, NO3/NO2, PO4, and SO4 (Supplementary Table 1). Ammonium levels were below detection limits for all samples (Supplementary Table 1). To determine if transect velocity was the same as bottom current velocity, an estimate was made based on the sampling time and distance of the first cast. For the first cast, the total distance traveled was ~1800 m for ~2 h 16 m for a velocity of ~22 cm/s. This was 2-3-fold faster than documented bottom water currents in the region (Lahaye et al., 2019; Haalboom et al., 2020) suggesting sampling was not Lagrangian in nature.
Biodiversity
Amplicon sequencing of eukaryotic (18S rRNA) and prokaryotic (16S rRNA) communities resulted in 3,405 and 3,017 amplicon sequence variants (ASVs), respectively (Supplementary Tables 2, 3). No discernible patterns of beta diversity (Bray-Curtis dissimilarity) were observed across the sample set for eukaryotes; however prokaryotic communities directly above the vent and outside of the plume were more similar than those found within the plume (Supplementary Figure 2). With regards to overall species richness, as the diversity of prokaryotes shifted, so too did the diversity of eukaryotic organisms where species richness was significantly positively correlated (p <0.01, Figure 3). In addition, when grouped by location (sites over and under the plume or "out", above the vent, south and north of the vent), prokaryote diversity at sites south of the vent were significantly different from sites out of the plume as well as north of the vent, but not above the vent site itself (Figure 4A).
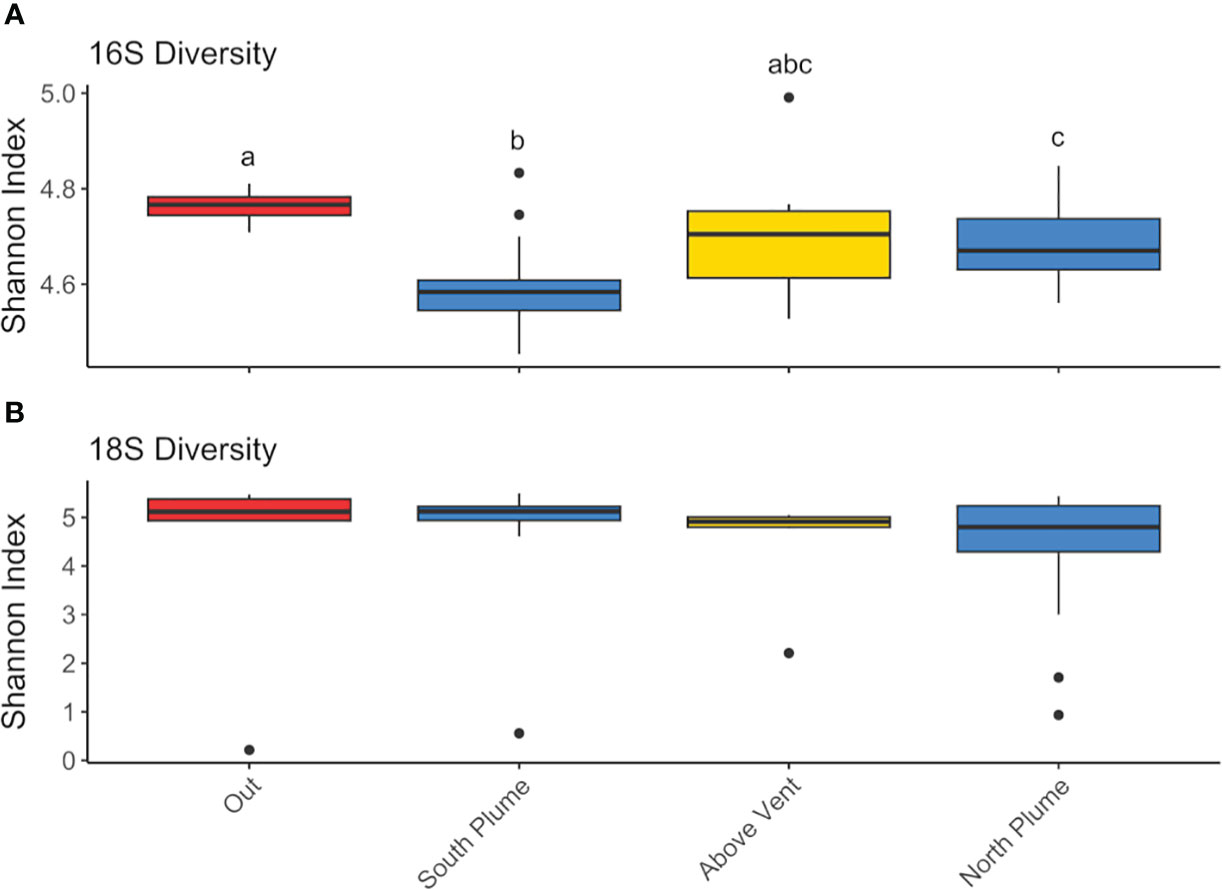
Figure 4 Box plots of Shannon diversity index for (A) 16S and (B) 18S diversity at each grouping. Groupings include “Out” of the plume, which includes all technical replicates for C1B12 and C2B1 (n=6), “South Plume” includes all technical replicates from C1B1-B6 (n=18), “Above Vent” includes technical replicates from C1B7 and C1B8 (n=6), and “North Plume” includes technical replicates C1B9-11 and C2B2-B5 (n=21). Significant differences by groupings are designated with lowercase letters. Significance was tested with the non-parametric Kruskal-Wallis test with pairwise comparisons determined with Wilcoxon test corrected for multiple testing using the Benjamini-Hochberg (BH) method.
For archaea, where more than half the reads were recruited (Supplementary Figure 3), diversity was similar across the sampled range represented by four phyla and largely composed of Crenarchaeota of the class Nitrososphaeria (87 to 89.52%), followed by Marine Group II and III Thermoplasmatota (5.92 to 8.45%, 1.89 to 2.87%, respectively; Figure 5A). Rarer were reads attributed to Halobacteria (0.06 to 0.15%), Nanoarchaeota (including Woesearchaeales, 0.01 to 0.60%), Thermoplamata (including an unclassified ASV, 0.06 to 0.14%), and Crenarchaeota of Marine Benthic Group A (1.57 to 2.06%, Figure 5A). Bacteria were more diverse, represented by over 30 phyla, with the majority of reads attributed to Alphaproteobacteria (17.63 to 25.73%) and the sulfur-oxidizing SUP05 cluster (2.49 to 28.69%) in the Gammaproteobacteria (Dede et al., 2022), with SUP05 significantly enriched within plume waters relative to out (Padj < 0.01, DESeq2; Figure 5B). In addition, bacterial communities were also represented by Campylobacterota (8.51 to 11.81%), Chloroflexi (10.26 to 16.10%), Acidobacteriota (2.77 to 4.22%), Planctomycetota (6.97 to 9.84%), SAR324 (4.61 to 10.58%), SAR406 (2.69 to 4.46%), Nitrospinota (2.58 to 4.65%), and Verrucomicrobiota (2.68 to 4.11%, Figure 5B).
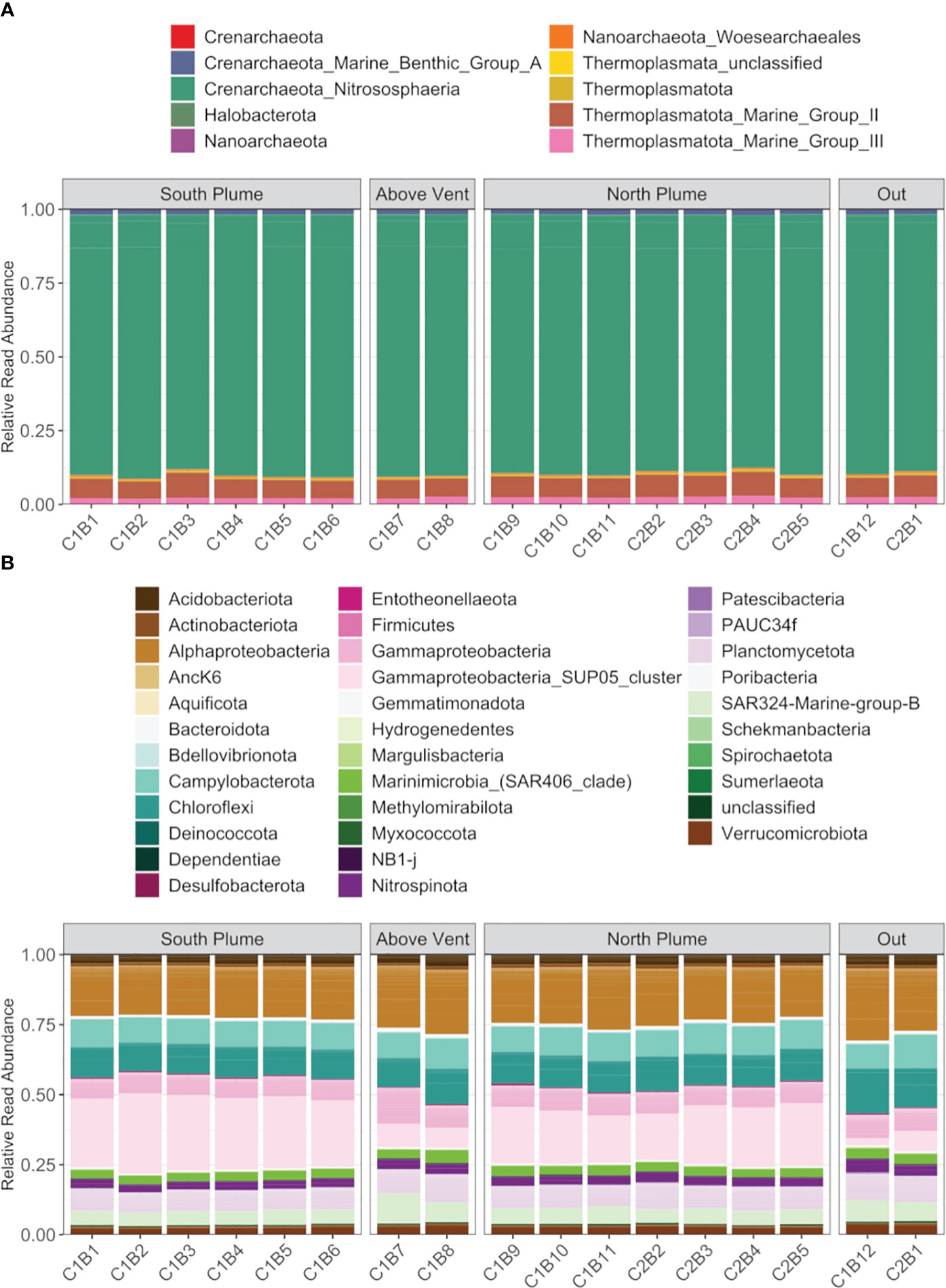
Figure 5 Summary of (A) archaeal diversity and (B) bacteria diversity along the hydrothermal plume transect. Each column represents the average read abundance of technical replicates (n=3) retrieved from the same Niskin bottle. Samples are grouped by location. Colors designate manually curated taxonomic groups.
Eukaryotic (18S) diversity was similar regardless of sample location but in general, displayed higher diversity than the prokaryotic (16S) community (Figure 4B). Of the eukaryotes detected, most reads were associated with taxa represented by Alveolates, Opisthokonts, and Rhizaria (Figure 6). Of these, a large proportion of reads were assigned to ASVs classified as the parasitic dinoflagellate order Syndiniales (10.87 to 35.43%), Metazoa (9.22 to 67.71%), and Radiolaria (4.86 to 17.25%, Figure 6). Metazoan diversity was dominated by a few classes, including Arthropoda (0.33 to 84.47%), Cnidaria (8.44 to 81.39%), and Ctenophora (3.06 to 41.83%, Supplementary Figure 4). Within the arthropods, the largest relative abundance of reads was attributed to an unclassified copepod (0.34 to 99.97%) followed by the genera Paracalanus (0.1 to 89.06%) and Clausocalanus (0 to 87.73%), both of which have been observed at black smokers (Skebo, 1994). For Cnidaria, almost all reads were attributed to an unclassified hydrozoan while Ctenophores were largely represented by an unclassified Ctenophore and the genus Lampocteis, a lobate ctenophore common in deeper waters (Supplementary Table 2; Lindsay et al., 2015). Radiolarians were represented mostly by the Acantharea (49%) and Polycystinea (29%) classes and RAD-B/Taxopodida to a lesser extent (14%) with a portion of reads attributed to unclassified Radiolarians (6%; Supplementary Figure 5). Lastly, a large proportion of reads attributed to Mollusca of the family Heterobranchia (18%) was also observed in a sample obtained under the plume (C2B1, Supplementary Figure 4), while just after north of the vent there was a high percentage of reads attributed to the class Craniata (26%). However, most of these reads (Craniata) were documented in just one of the three technical replicates, classified as a member of the Auxis genus (Supplementary Table 2) and may represent signals from pelagic waters (e.g. fecal matter or other) as Auxis are not known to live below 200 m (Graham and Dickson, 2004). Although representing a relatively small proportion of detected reads (<2%) in this study, ciliates form an important component of deep-sea communities (Schoenle et al., 2017; Hu et al., 2021). Of the ciliates detected, many reads were attributed to the Oligohymenophorea and Spirotrichea classes. Most could not be classified beyond family apart from Protocruzia and Pseudocollinia (Supplementary Figure 6). The genus Protocruzia was recently detected in sediments near the Mariana Trench (Liang et al., 2021) while the genus Pseudocollinia is a parasitoid ciliate, often infecting euphausiids (Gómez-Gutiérrez et al., 2012).
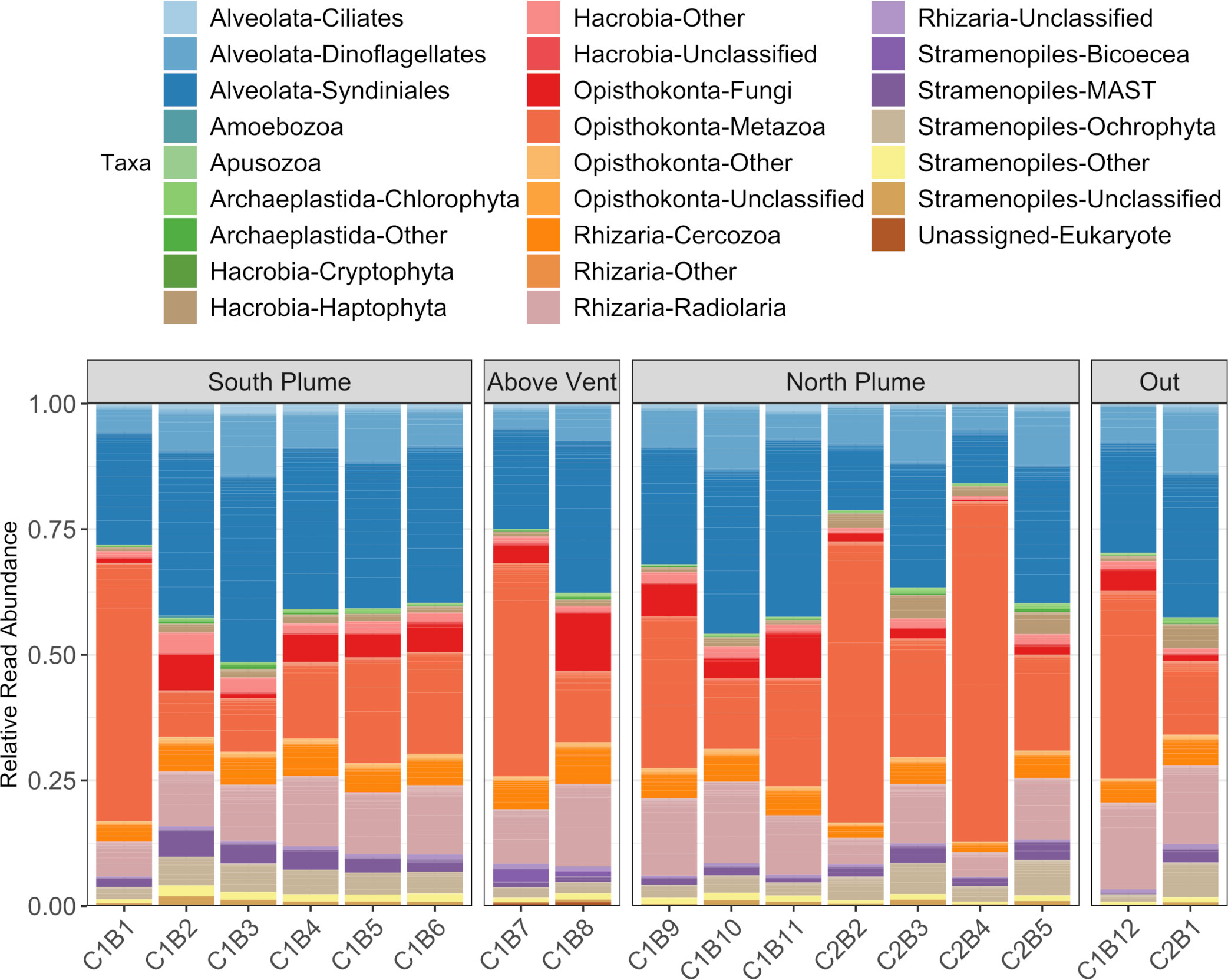
Figure 6 Summary of eukaryotic diversity along the hydrothermal plume transect. Each column represents the average read abundance of technical replicates (n=3) retrieved from the same Niskin bottle. Samples are grouped by location. Colors designate manually curated taxonomic groups.
In addition, while metabarcoding data can infer the diversity of organisms present within an ecosystem by their DNA, we also used the taxonomic classification system Kraken2 to infer ‘active’ organisms from metatranscriptomic data (RNA). Of the 1.7 billion sequenced reads, 29.2% were classified by Kraken2 to the genus level. Based upon Bracken abundance estimates, the 5 genera receiving the most reads included ammonia oxidizing archaea (Candidatus Nitrosopelagicus, Nitrosopumilus, Nitrosarchaeum, Candidatus Nitrosotenuis) as well as the sulfur oxidizing SUP05 cluster (Candidatus Thioglobus) mirroring metabarcoding data (Supplementary Table 4).
Functional responses
Trinity and IDBA-UD were used to assemble metatranscriptomes for each bottle, and resulting transcript sets were combined and clustered at 90% similarity, resulting in a total of 611,500 contigs (Supplementary Table 5). As these data cannot distinguish individual organismal responses, a community-level perspective was employed to explore microbial metabolic responses. After annotation with KEGG, transcript counts were aggregated across KO terms (Supplementary Table 6), henceforth referred to as “functional clusters”, and analyzed for differential expression by comparing samples collected within the plume versus out of the plume (i.e.C1B1 vs Out, C1B2 vs. Out, etc. where Out is C1B12 and C2B1). For this community-level integrated approach, of the 9,755 functional clusters analyzed for differential expression, 9,178 functional clusters showed significant differences in at least one within-plume sample compared to outside the plume (DESeq2, Padj < 0.05; Supplementary Table 7). Of these, 913 were significantly different directly above the vent only, while 79 showed a significant difference in all within-plume samples relative to out of plume (Supplementary Table 7). In general, samples collected within the plume exhibited transcriptional patterns distinct from those collected outside of the plume (Supplementary Figure 7), and a large proportion of reads (between 2 and 21%) recruited to pathways involved in energy metabolism (nitrogen and sulfur metabolism, oxidative phosphorylation, carbon fixation) and carbohydrate metabolism (glyoxylate/dicarboxylate metabolism, glycolysis/glucogenesis, TCA cycle) among others, at any given location within the plume (Supplementary Figure 8).
One of the key chemical energy sources at hydrothermal vents for microbial organisms is the oxidation of sulfur (Dick, 2019). Within the plume, genes involved in dissimilatory sulfate oxidation displayed significant upregulation at almost all plume samples relative to out of the plume with all genes in this pathway upregulated in samples collected above the vent (Figure 7). The sqr gene (encoding sulfide-quinone reductase), involved in conversion of hydrogen sulfide to elemental sulfur, was also significantly upregulated within most plume samples. In addition, genes within the Sox pathway, a multi-enzyme system involved in the oxidation of thiosulfate and widely distributed in chemo-lithotrophic sulfur-oxidizing prokaryotes (Ghosh and Dam, 2009), were significantly upregulated in the majority of plume samples relative to out of the plume (Figure 7). With exception were soxX and soxZ, which showed significantly lower expression directly above the vent (C1B8; Padj < 0.05). Both the Sox pathway and genes involved in dissimilatory sulfur oxidation were some of the highest transcribed genes within the community, representing over 1.2% of all reads (including unclassified reads) for some samples (Supplementary Figure 9). Conversely, many of the genes responsible for assimilatory sulfate reduction displayed decreased expression above the vent and in adjacent plume samples (Figure 7).
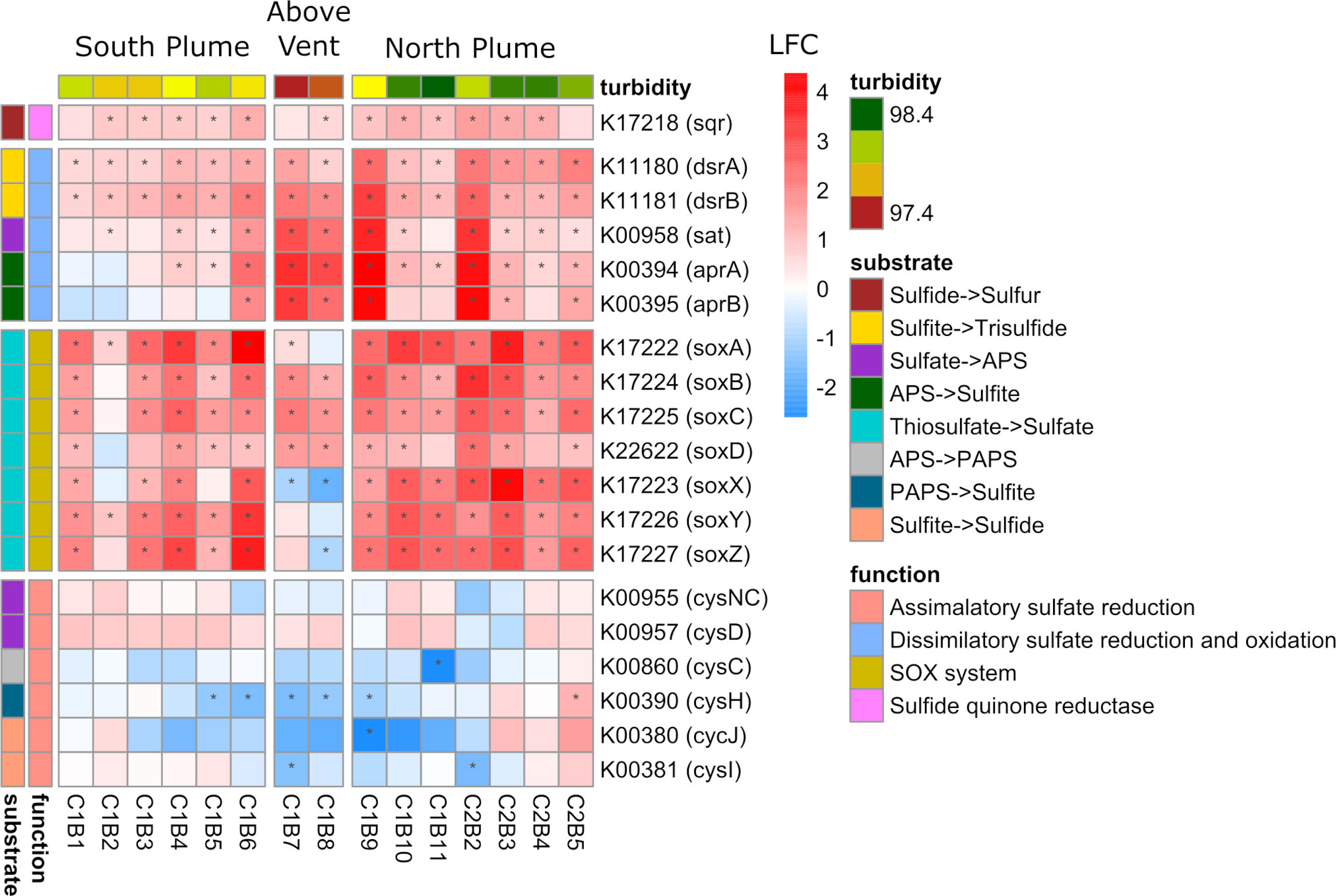
Figure 7 Community-level transcription of functional clusters involved in sulfur metabolism showing the log2 fold change (LFC) in gene expression of samples collected within the plume vs. out of the plume. Stars indicate significant expression (Padj <0.05). Heatmap is grouped and colored by substrate transformations and pathway functions as well as by location defined by turbidity and position on the transect. KEGG identifiers and gene names are labeled for each row.
One of the most abundantly transcribed pathways, in addition to sulfur metabolism, was that of nitrogen metabolism (Supplementary Figure 8). Significantly upregulated across the sample set were genes involved in transporting and converting nitrogen for biological assimilation into organic matter (Figure 8). All parts of the nitrogen metabolism pathway were represented, including the transport of nitrate/nitrite, reduction of nitrate to nitrite, reduction of nitrate to ammonia, and conversion of ammonia to glutamate (Figure 8). In addition, genes involved in nitrification (amoAB and hao) displayed significant upregulation in many of the samples south and north of the vent, but downregulation above the vent (Figure 8). Contrary to these patterns was the ammonium transporter (amt) gene which was significantly downregulated across the sample set, mirroring below detection limits of ammonium in the water column (Supplementary Table 1) as well as a gene involved in denitrification (nirK) which also showed significant downregulation across all samples (Figure 8). In addition, two genes involved in dissimilatory nitrate reduction (narG and narH) were significantly downregulated within the plume relative to out of the plume, except for the narI gene which displayed the opposite pattern (Figure 8).
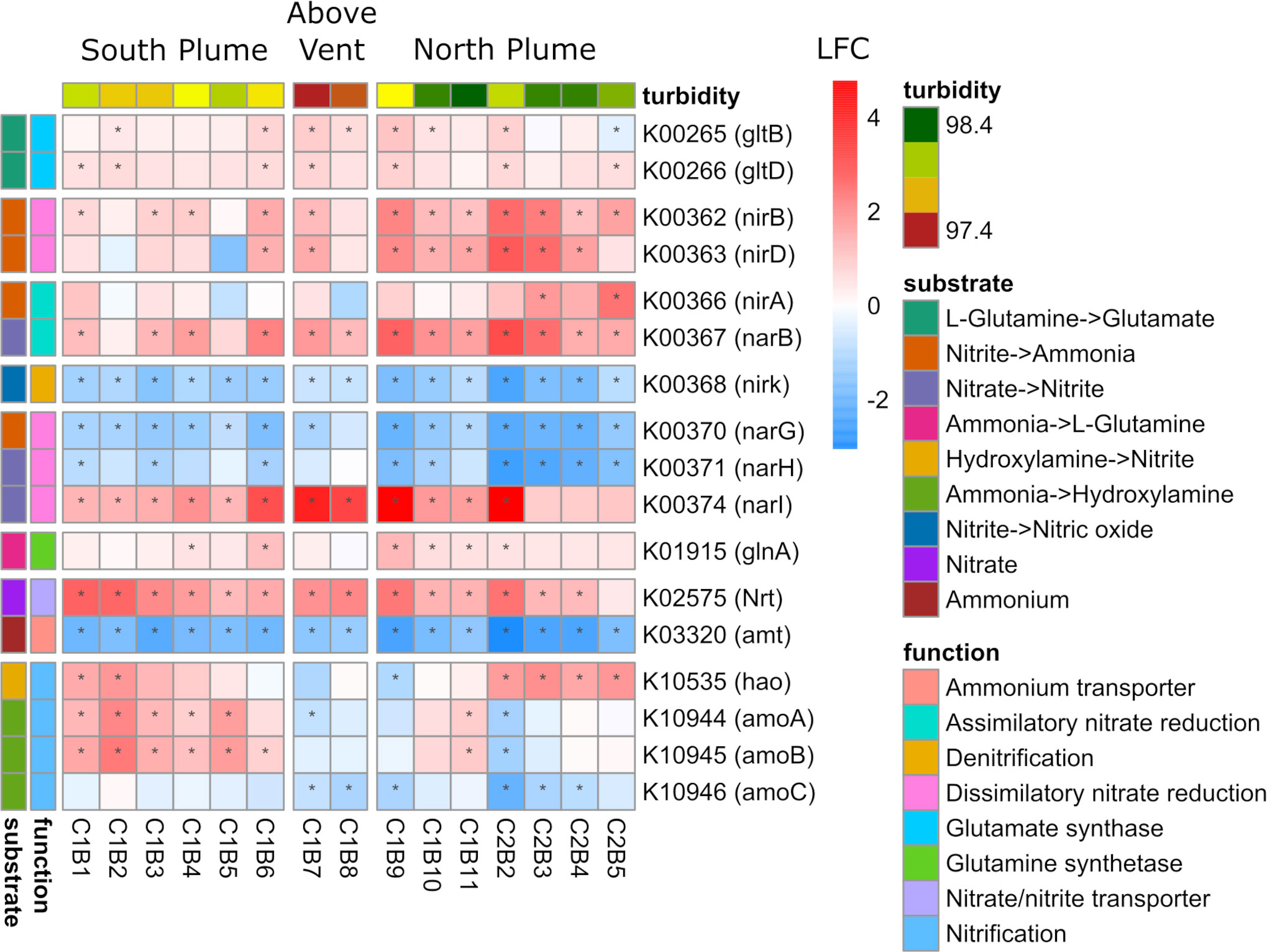
Figure 8 Community-level transcription of functional clusters involved in nitrogen metabolism showing the log2 fold change (LFC) in gene expression of samples collected within the plume vs. out of the plume. Stars indicate significant expression (Padj <0.05). Heatmap is grouped and colored by substrate transformations and pathway functions as well as by location defined by turbidity and position on the transect. KEGG identifiers and gene names are labeled for each row.
Enriched within the plume vs out of the plume were also several pathways related to energy generation, carbon fixation, or in some cases, primarily represented by gene sets involved in these or other pathways. For instance, the pathway nitrotoluene degradation had a large proportion of reads mapping to KEGG orthologs involved in dissimilatory sulfite reduction (dsrA/B genes), processing of hydrogen (hydrogenases hyaA/B and uptake hydrogenases hupU/V), as well as energy generation through pyruvate oxidation and/or carbon fixation (porABCD; Supplementary Figure 10). Other pathways related to energy generation include the oxidative phosphorylation pathway which is composed of genes involved in harnessing the reduction of oxygen to generate ATP. Within this pathway were 223 KEGG orthologs, of which the entire NADH: quinone oxidoreductase gene set (nuoA-N genes) was recovered with almost all showing significant upregulation within the plume relative to out of the plume (Supplementary Figure 11). In addition, there were several genes encoding f-type ATPases (associated with prokaryotes) with significant upregulation while significant downregulation was observed for eukaryote-associated f-type ATPases. Lastly, a cbb3-type cytochrome c oxidase, found almost exclusively in proteobacteria (Pitcher and Watmough, 2004) was significantly upregulated in many of the plume samples both north and south of the vent (Supplementary Figure 11).
With regards to carbon fixation, in addition to partially recovering active transcription of the Wood-Ljungdahl pathway (Supplementary Figure 12), a common carbon fixation pathway in anaerobic conditions in which hydrogen is used as an electron donor (Wood, 1991), the entire reductive TCA cycle was recovered. Individual gene responses varied within this pathway including significantly increased transcription in almost all samples within the plume versus out of the plume for ATP-citrate lyase subunits (K15230 and K15231; aclA/B), a gene encoding aconitate hydratase (K01682; acnB), phosphoenolpyruvate carboxylase (K01595; ppc), fumarate hydratase (K01676; fumA), a malate dehydrogenase (K00025, mdh), and a pyruvate carboxylase subunit (K01960, pycB). Significantly downregulated across most of the samples within this pathway were genes involved in 2-oxoglutarate/2-oxoacid ferredoxin oxidoreductase (K00174 and K00175), a pyruvate-ferredoxin/flavodoxin oxidoreductase (K03737) as well as another gene encoding aconitate hydratase (K01681; acnA) displaying the opposite transcription pattern of acnB (Supplementary Figure 13).
Lastly, at the community level, four genes involved in chemotaxis were significantly upregulated in the plume, including the chemotaxis protein A (cheA) which interacts with transmembrane receptors (mcp), and the motor-binding protein cheY which controls flagellar motor switching. However, genes involved in motility were largely downregulated (motA/B) as well as the flagellar motor switch proteins fliG, fliM, and fliN (Figure 9) suggesting that although chemotactic movement was not initiated, chemosensory processes remained active for over a kilometer from the vent.
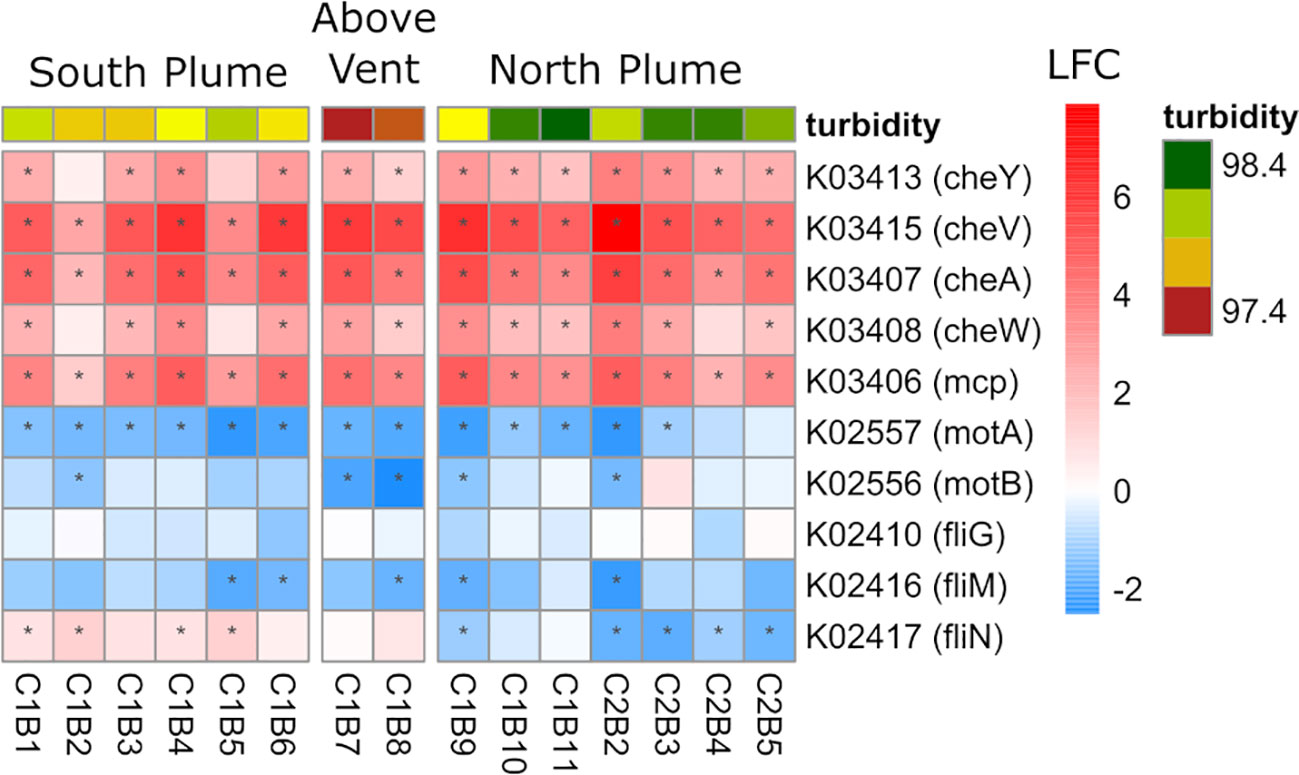
Figure 9 Community-level transcription of functional clusters involved in bacterial chemotaxis showing the log2 fold change (LFC) in gene expression of samples collected within the plume vs. out of the plume. Stars indicate significant expression (Padj <0.05). Heatmap is grouped by location defined by turbidity and position on the transect. KEGG identifiers and gene names are labeled for each row.
Viral responses
In addition to the metabolic responses of prokaryotic and eukaryotic organisms, metatranscriptomes were also searched for evidence of viral activity. In the sequenced metatranscriptomes, VirSorter2 detected 135 contigs with putative viral origin that were significantly expressed at one or more stations along the transect, with the majority showing activity north and south of the vent site (C1B7, C1B8) relative to out of the plume (Figure 10). These contigs were also identified using a kmer-based approach (VirFinder) further confirming viral identification. Annotations revealed a mixed community of Caudovirales including Podoviridae and Siphoviridae suggesting active bacterial and archaeal infections occurring within plume waters (Supplementary Tables 8, 9), including several sequences matching targets in SUP05-infecting viral genomes (Anantharaman et al., 2014).
Figure 10 Community-level transcription of transcripts with putative viral origin showing the log2 fold change (LFC) in gene expression of samples collected within the plume vs. out of the plume. Stars indicate significant expression (Padj <0.05). Heatmap is grouped by location defined by turbidity and position on the transect.
Survival on the drift
As neutrally buoyant plumes are advected away from hydrothermal vents, microbial communities and plankton swept up in these resource rich plumes must adapt to these rapidly changing conditions as the plume ages and drifts farther away. To explore these changes, we compared both community-level diversity (metabarcoding) and function (metatranscriptome) and how these shifted with distance by calculating Bray-Curtis dissimilarity metrics for each sample relative to sites above the vent (C1B7 and C1B8). There was greater dissimilarity in function observed than diversity and this dissimilarity significantly increased with distance from the vent (p < 0.01; Figures 11A, B), suggesting that vent-influenced dynamics of microbial community function decreased with distance while diversity remained the same. When this analysis was applied to functionally active diversity (Kraken2 analysis of metatranscriptomes), similar dissimilarity patterns were observed downstream of the vent. However, upstream of the vent, dissimilarity significantly increased with distance from the vent (Figure 11C).
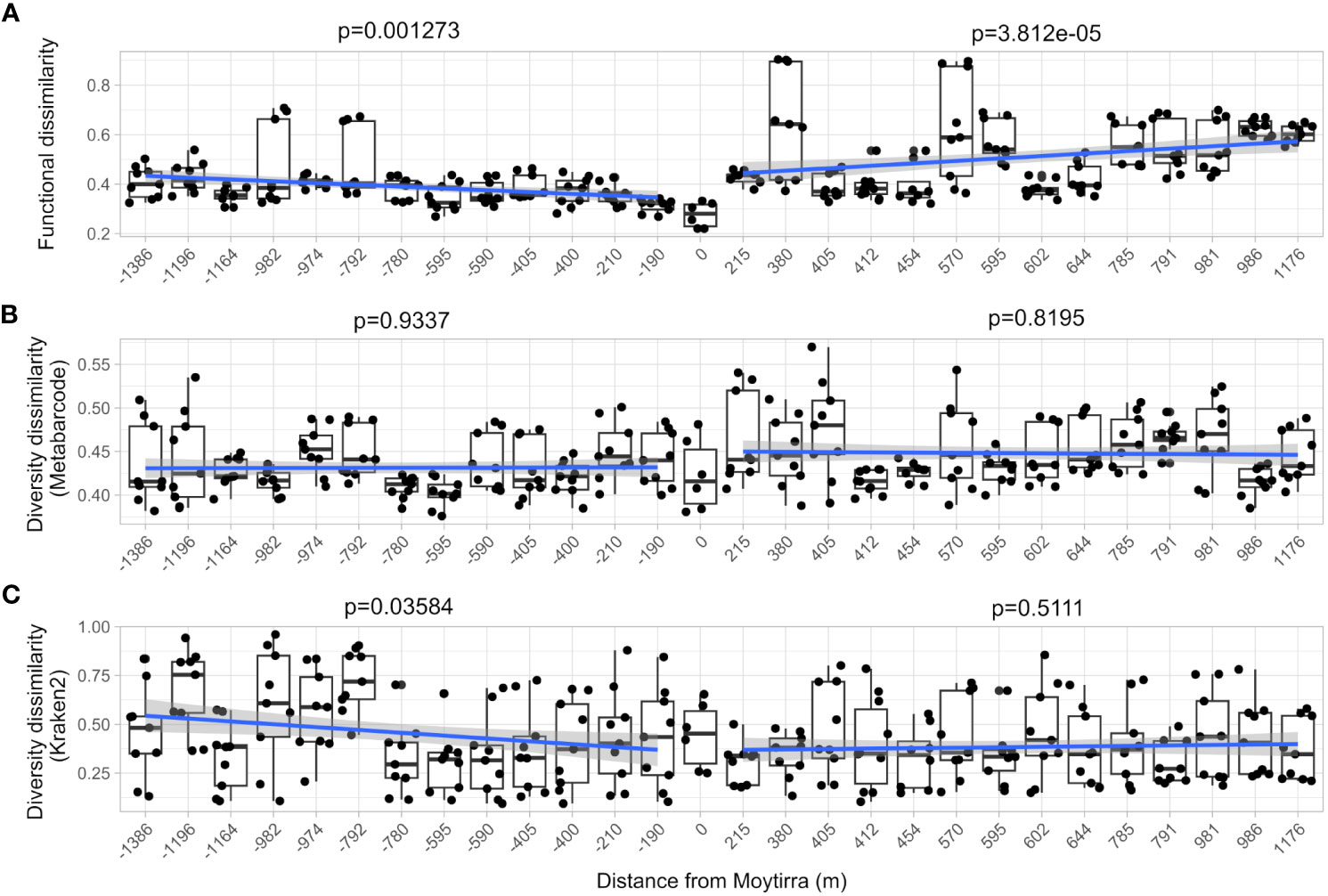
Figure 11 Community-level comparison of functional (A; metatranscriptome) and diversity (B; metabarcode), and functional diversity (C; Kraken2) dissimilarity of samples with distance from the vent site (zero on the plot). Each point represents the Bray-Curtis dissimilarity of function and diversity for each technical replicate comparison between stations within the vent plume in relation to vent site (samples C1B7 & C1B8). A linear model trend line is drawn between stations sampled south of and north of the vent. The F-statistic p-value of the linear model is displayed.
In addition, we also specifically looked for evidence of growth, quiescence, or senescence within plume communities. To assess if microbial communities were undergoing cell division or moving towards stationary states, we interrogated prokaryotic marker genes for exponential growth (rpoD), quiescence (rpoS), and cell division (ftsH) (Ishihama, 1997; Adams and Errington, 2009). Within the plume vs out of the plume, significant upregulation of the rpoS gene was observed while significant downregulation of the rpoD gene (Figure 12). In addition, significant upregulation of the cell division gene ftsH was observed directly above the vent and at one station north of the vent (Figure 12). For the eukaryotic communities, a suite of genes commonly associated with cell cycle and senescence including cyclins A and D (Yam et al., 2002; Yang et al., 2006), a proliferating cell nuclear antigen (PCNA; Strzalka and Ziemienowicz, 2011), the KI-67 gene (Uxa et al., 2021), and the p27 cyclin-dependent kinase inhibitor (Toyoshima and Hunter, 1994) were looked at. Of these, only the PCNA gene displayed any significant response at 5 stations (C1B6, C1B7, C1B9, C2B2, C2B4) with an overall pattern of downregulation (K04802, Supplementary Table 7). Further, in support of these observations, RNA polymerase subunits were also probed, finding significant upregulation of bacterial RNA polymerase subunits rpoA, rpoB, and rpoB’ above and in close proximity to the vent while archaeal and eukaryotic subunits were largely downregulated (Supplementary Figure 14).
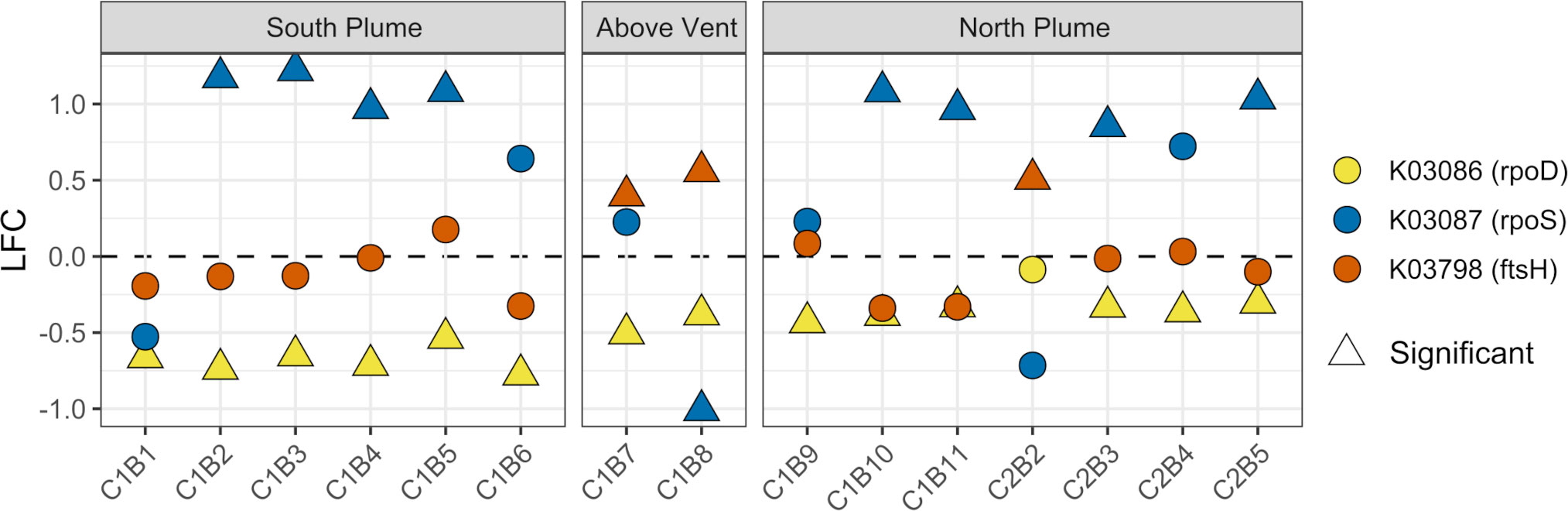
Figure 12 Community-level transcription of functional clusters involved in bacterial cell division (ftsH), exponential growth (rpoD), and quiescence (rpoS) showing the log2 fold change in gene expression (LFC) of samples collected within the plume vs. out of the plume. Triangles indicate significant expression (Padj <0.05). Plot is grouped by location on the transect. KEGG identifiers and gene names are labeled for each row.
Discussion
It has been suggested that hydrothermal vent plumes provide ‘highways’ for dispersal of endemic organisms, but how long this highway persists, its role in dispersal, and how organisms persist in such extreme environments as they are advected away from the hydrothermal vent and into the cold dark seas have yet to be fully elucidated. The current study aimed to shed light on how community diversity and metabolic function within a hydrothermal plume shift with distance from the vent site. When plume waters were compared to those outside of the vent plume, similar community composition was observed while function changed with distance from the vent source, potentially representing survival mechanisms that may contribute to dispersal and connectivity.
Plumes were inhabited by a rich diversity of microorganisms and plankton with prokaryotic communities displaying distinct biodiversity patterns from surrounding waters. Unlike vent plumes south of the Azores which are dominated by Epsilonproteobacteria (Haalboom et al., 2020), the most relatively abundant bacteria within the Moytirra plume were Gammaproteobacteria SUP05 cluster, which are known to form sulfur globules and oxidize sulfur via the reverse dissimilatory sulfate reduction pathway (rDSR) and thiosulfate through the SOX pathway (Anantharaman et al., 2013; Dede et al., 2022). In addition, the ammonia oxidizing Crenarchaeota Nitrososphaeria (Thaumarchaeota) represented the majority of reads within archaea. Both taxa can also be found in oxygen minimum zones as well as other vent environments (Dick et al., 2013). Within eukaryotic communities, a number of microbial taxa were detected having known roles in grazing and parasitism (e.g., Syndiniales, ciliates, rhizaria, and copepods) (Hu et al., 2021). Both prokaryotic and eukaryotic diversity observations point to numerous trophic modes involving top-down controls on chemosynthetic primary productivity within the vent plume, suggesting that these communities remain active even at over 1 km from the vent. Additionally, signals from vent animals such as gastropods from the Heterobranchia clade were present in eukaryotic community data, which may indicate transport of larvae within the plume (Mullineaux et al., 2010) and have implications for connectivity between vent systems (Supplementary Table 2).
When considering diversity from the perspective of Shannon Index, we observed shifts in prokaryotic but not eukaryotic diversity along the transect, suggesting no relationship between prokaryotic and eukaryotic organisms. However, Shannon Index accounts for relative abundances of ASVs, or “evenness”, which may be skewed by the multicopy nature of the 16S and 18S SSU rRNA genes. From the perspective of richness, we observed that as the number of detected ASVs for prokaryotes shifted, so did the number of detected ASVs for eukaryotic organisms, suggesting linkages between these two taxonomic groups (Figure 3). In surface waters, there is evidence of connectivity between bacteria and microeukaryote diversity driven, in part, by changes in photosynthetically derived dissolved organic matter (Töpper et al., 2013; Harke et al., 2021). When the diversity of phytoplankton shifts, so too does the diversity of dissolved organic matter substrates, in turn shifting the bacterial diversity that feeds on those substrates. The opposite pattern may be occurring within vent plume waters which are instead supported by chemosynthetic prokaryotes. As the richness of bacteria changes due to changing resources as the plume drifts away from the vent source, so too does the richness of microeukaryotes grazing on these organisms. Analogs of this relationship have been observed in surface waters between nanoflagellates and bacteria through predator/prey relationships (Yang et al., 2018) and microeukaryote grazing has been shown to play a significant role in deep-sea carbon cycling (Pasulka et al., 2019; Hu et al., 2021).
In addition to microeukaryote grazing, viruses are known to play important roles in microbial mortality, shaping community structure and, as such, contributing to biogeochemical cycling (Suttle, 2005; Weitz and Wilhelm, 2012; Breitbart et al., 2018). Since the early documentation of deep-sea viruses (Juniper et al., 1998), hydrothermal vents have been shown to host a high diversity and abundance of viruses (Ortmann and Suttle, 2005), being found in all vent environments except chimney interiors (Yoshida-Takashima et al., 2012). There appear to be population differences between vent environment types where deep-sea sediments tend to harbor Circoviridae and Microviridae while plumes tend to harbor Caudovirales viruses (Castelán-Sánchez et al., 2019). In the current study, active viral signals within the vent plume were primarily composed of Caudovirales, mirroring prior observations. In addition to having important roles in the carbon cycle through the viral shunt, viruses may also influence sulfur cycles within vent ecosystems, as recent genomic sequencing has highlighted the presence of phage-encoded sulfur-oxidizing enzymes (Anantharaman et al., 2014) which may be acting upon stored sulfur globules in the SUP05 clade (Shah et al., 2019). There is also evidence that viruses are largely endemic to localized vent ecosystems with potentially restricted dispersal based on viral-host network analysis (Thomas et al., 2021). Of the viral genomes with deep-sea origin, most have originated from hydrothermal vents (Mizuno et al., 2016), providing resources for further characterization and potential metatranscriptomic insights to be leveraged.
Here we identified and showed active expression of genes with putative viral origin suggesting active infection occurring more than 1 km from the vent site (both south and north). Increased activity, as inferred by changes in transcript abundance within plume versus out of the plume, began primarily within waters 2-300 m away from the vent site. Observed viral activity did not appear to diminish relative abundance of detected taxa along the transect, even though recent evidence suggests grazing may contribute >60% daily losses (Hu et al., 2021). However, grazing rates in that study were measured relatively close to the active vent and may diminish in more dilute plume waters where contact distances increase. The lower viral activity above the hydrothermal vent suggests these waters are more similar (from a viral activity perspective) to waters over or under the neutrally buoyant plume. This may highlight fluid dynamics in the region as cold seawater is entrained and mixed with hydrothermal fluids rising above the vent but also highlights the impact mixing plays in viral-host interactions (Ortmann and Suttle, 2005). As hydrothermal fluids rapidly cool, rise, and mix with surrounding seawater to become neutrally buoyant, this turbulent mixing may increase encounter rates causing the observed increase in activity. Alternatively, higher particulate rates closer to the vent site (as indicated by increased turbidity) may play a role in removing viral particles through adsorption (Juniper et al., 1998), which is suggested as a major source of viral loss (Suttle and Chen, 1992), potentially influencing observed reductions in activity above the vent. Taken together, it appears that hydrothermal vents, through alterations in seawater chemistry which in turn drives changes in microbial biodiversity, are also influencing subsets of deep-sea viral populations by enhancing activity. This activity extends for over a kilometer from the vent source, suggesting active transport of viral particles and shedding new light on the extent of dispersal by these important deep-sea ecosystems.
Despite observing a rich diversity of both prokaryotic and eukaryotic organisms within the Moytirra vent plume, community diversity was not shown to change with distance from the vent as observed in a recent transect near the Rainbow vent field (Haalboom et al., 2020). The distances traveled in that study were an order of magnitude greater (>20 km) than the current study (2.5 km) indicating diversity likely persists for 10’s of kilometers within hydrothermal plumes. However, despite no change in diversity, functional signals were increasingly dissimilar to those observed at the vent, suggesting changes in overall community-level function with distance, potentially driven by changing element ratios as plumes aged (Haalboom et al., 2020). Additionally, functionally active taxa determined by Kraken2 became increasingly dissimilar upstream of the vent but not downstream, possibly suggesting that while the overall community stayed similar across the transect, metabolically active organisms shift south of the vent. To the extent the sampling scheme herein revealed, seawater chemistry patterns, although variable with distance from the vent, did not correlate with any obvious diversity or functional expression patterns, at least when considered from a community-level. With exception to these global trends, waters within the Moytirra plume were found to be enriched in both sulfur oxidizing bacteria (e.g., SUP05) as well as expression of many genes involved in sulfur metabolism, representing one of the most highly expressed KEGG pathways cumulatively. Sulfur oxidizing microorganisms oxidize hydrogen sulfide or other reduced sulfur compounds for incorporation of CO2 into organic matter using either oxygen or nitrate as electron acceptors (Sievert et al., 2008). The elevated percentage of reads attributed to both nitrogen and sulfur metabolism suggests sulfur oxidation is a major source of energy within the plume emanating from Moytirra. Sulfur oxidation is thought to be the major energy-yielding reaction in vent plumes, over methane or hydrogen, supporting recent observations of its importance within the global plume microbiome (Zhou et al., 2022). The SUP05 clade was only recently discovered through 16S rRNA sequencing of hydrothermal plume waters (Sunamura et al., 2004). Since then, two main genera have been proposed, Thiomultimodus (Ansorge et al., 2020) and Thioglobus (Marshall and Morris, 2013), which have been found in numerous habitats ranging from oxic to anoxic seawater and vent systems to free living and endosymbiotic associations (Morris and Spietz, 2022). In vent ecosystems, they are known to play important roles in biogeochemical cycling of carbon, nitrogen, sulfur, and iron, connecting vent-derived nutrients to the wider ocean through storage and transport via plumes (Dick et al., 2013; Li et al., 2014; Shah et al., 2019; Spietz et al., 2019). The relative abundance of reads recruiting to SUP05 ASVs was lower both outside of the plume and directly above the vent, supporting prior observations where this clade is absent in the early stages of plume development (Lesniewski et al., 2012; Sheik et al., 2015). As such, vent plumes may act as ‘growth chambers’ for SUP05, entraining and releasing these microorganisms into the surrounding environment and supporting their geographical spread and recruitment to other vent ecosystems (Dede et al., 2022).
Sulfur metabolism within the plume was captured primarily in pathways involved in dissimilatory sulfate reduction and oxidation through the reverse-acting dissimilatory sulfite reductase (Dsr) system and the sulfate oxidation (SOX) system. Genes within both systems were found to be upregulated in all stations within the plume relative to out of the plume. With exception to this were parts of the SOX pathway (soxX and soxZ) which were significantly downregulated in samples directly above the vent. Located in the periplasm, soxX forms a complex with soxA, being the first step in the SOX pathway (Ghosh and Dam, 2009), a multienzyme complex capable of oxidizing sulfide, sulfite, sulfur, and thiosulfate to sulfate (Friedrich et al., 2001). Products from the soxAX complex are then carried through the SOX pathway on a swinging arm structure formed by the soxYZ complex, which participates in all reactions in the SOX pathway, carrying intermediates through each step (Sauve et al., 2007). Downregulation of these complexes above the vent may be related to oxygen availability directly above the vent, as there is evidence of reduced protein expression for parts of the sox pathway in SUP05, including soxY under anoxic conditions (Shah et al., 2019). However, anoxic fluids emerging from hydrothermal vents are rapidly oxygenated through mixing with surrounding seawater (Johnson et al., 1986), discounting this possibility. Nitrate availability in sulfur oxidation has also been shown to influence expression of genes within the SOX pathway in certain sulfur-oxidizing denitrifiers (Ghosh and Dam, 2009; Russ et al., 2019). Indeed, at a station directly above the vent, NO3 was reduced relative to other stations but not at levels considered limiting. Lastly, several genes were identified in Paracoccus pantotrophus which have been shown to regulate expression of the sox genes (soxR and soxS) and appear to be substrate responsive, repressing expression in the absence of thiosulfate (Rother et al., 2005). However, we did not identify KEGG orthologs of these genes within the assembled metatranscriptome suggesting either low expression or absence within organisms sequenced here (as seen in some purple sulfur bacteria; Grimm et al., 2011). In summary it is unclear from the current dataset what is driving reduced transcription of soxX and soxZ directly above the vent. Future more targeted studies may reveal further controls of these critical components of the SOX system and their role in emerging hydrothermal plumes and implications for connectivity.
A large proportion of reads were expressed in genes involved in nitrogen metabolism. All parts of the nitrogen metabolism pathway were represented in the functional data, suggesting these genes likely play additional roles in vent plume energy production outside of the role of nitrate in sulfur oxidation and SOX pathways. Ammonia oxidizing Crenarchaeota in the class Nitrososphaeria (Thaumarchaeota) represented the most abundant archaeal component of the prokaryotic diversity. Ammonia oxidizing archaea are known to drive nitrification in hydrothermal plume ecosystems (Baker et al., 2012), and Nitrososphaeria have been identified in hydrothermal vent and other deep sea ecosystems, including within plumes and surrounding waters, globally (Haalboom et al., 2020; Seyler et al., 2021; Kuppa Baskaran et al., 2023). The nitrification genes amoAB and hao were significantly upregulated upstream and downstream of the vent, but not directly above the vent. This downregulation of amo genes may be related to substrate limitations. Evidence has shown that ammonium is rapidly consumed by chemoautotrophs once it is discharged from the vent (Lam et al., 2004). This is further supported by our observations of below detection limits of ammonium throughout the transect. However, as with genes involved in sulfur metabolism, future studies investigating these transcriptional patterns would benefit from higher resolution sampling coupled to expanded nutrient profiling and measures of turbulent flow. As mixing of hydrothermal vent fluids with surrounding seawater can have important impacts on both diversity and function, getting a better understanding of the scale of this mixing would help decipher the observed patterns.
In order to sense the environment and move towards more favorable conditions, microbial organisms have evolved numerous sensory systems allowing them to respond to physical or chemical changes in the environment (Alex and Simon, 1994). For prokaryotes, these systems allow for guided movement towards more optimal growth conditions through well-defined chemotactic systems (Armitage and Schmitt, 1997). These include transmembrane methyl-accepting chemotaxis proteins (mcp) which bind to extracellular ligands and are coupled to the cheA kinase via the scaffold protein cheW (Alexander et al., 2010) and/or cheV (Huang et al., 2019). The response regulator cheY, which is phosphorylated by cheA, then interacts with the flagellar motor, stimulating movement of the cell (Sarkar et al., 2010). After being swept up in rising currents near the vent, these sensory networks likely become more important for microorganisms adapting to a rapidly changing chemical environment (Takaki et al., 2010). Within the Moytirra plume, we detected upregulation of genes involved in sensing (mcp, cheW,cheV, cheA, and cheY) but this did not appear to be translated to active movement as flagellar proteins encoded by motA and motB where downregulated, as well as the rotational switch complex encoded by fliG, fliM, fliN genes. Hydrothermal vents are known to have steep geochemical gradients which create niche environments for microbial organisms in close proximity to the vent (Reysenbach et al., 2000). Once outside the immediate environs of the vent, these chemical gradients become more homogeneous as plume water mixes with deep seawater. However, these chemical signals are likely still enriched compared to outside the plume, invoking activation of chemosensory networks within prokaryotic communities (upregulation of mcp, cheA/W/V). Due to this well mixed nutrient rich environment, movement may not be required resulting in downregulated motility genes (motA/B, FliG/M/N).
In addition to these activated chemosensory systems and aforementioned sulfur and nitrogen metabolism pathways, numerous genes involved in energy generation (oxidative phosphorylation, particularly in prokaryotes) and carbon fixation (including portions of the Wood-Ljungdahl and reductive TCA cycle pathways, pathways often enriched in vent ecosystems (Hügler et al., 2010; Hou et al., 2020)), displayed increased transcript abundance both north and south of the plume. Taken together, it appears microbial communities remain metabolically active despite increased functional dissimilarity with distance. This suggests that as hydrothermal plumes drift and mix with surrounding seawater, undergoing changes to chemical and physical properties, microorganisms are responding to these environmental changes resulting in shifts in global metabolic signatures (as illuminated through KEGG). What drives these alterations was not apparent with the current analysis. Further, as the current study focused on community-level KEGG reconstruction of metabolic functions, the uncharacterized portion of gene expression patterns (those not receiving KEGG annotation) may hold evidence for the observed dissimilarities in function with distance. As reference databases grow and allow for more comprehensive characterizations of all genes within the environment, future work may uncover what, if any, specific changes were occurring.
Despite continued metabolic activity within the plume, bacterial communities also displayed evidence for quiescence through increased transcription of the marker gene rpoS while the exponential growth marker gene rpoD was downregulated (Zambrano et al., 1993; Ishihama, 1997). Further, the cell division marker gene ftsH was only found to be significantly upregulated directly above the vent (as well as one station north of the vent). Overall, these patterns suggest bacterial cells were not only slowing down growth and cell division processes but may be entering quiescent stages as they drift farther away from the vent. At very low nutrient levels, bacteria can become dormant or, in some cases, form spores to survive these conditions, often for months (Gray et al., 2019), allowing them to survive until conditions are favorable again. Entering a state of dormancy as nutrients become depleted along the hydrothermal plume may describe survival strategies while drifting in the plume, explaining how these organisms survive and connect with hydrothermal vent systems downstream to eventually colonize. Transcriptional signals suggest that Moytirra plume communities may be entering into this state. In addition, the bacterial DNA-dependent multi-subunit RNA polymerase (RNAP), a key enzyme in gene expression which is responsible for synthesis of all RNAs within a cell (Lee and Borukhov, 2016), showed patterns of transcription suggesting a slowdown in overall gene expression within bacteria. RNA polymerase genes rpoA, rpoB, and rpoB’ only displayed significant upregulation above or near the vent while rpoZ was, in general, downregulated. The rpoZ RNA polymerase subunit omega is a non-essential subunit of the bacterial RNAP core (Kurkela et al., 2021) and thought to be involved in directing transcription efficiency of highly expressed genes, as observed in some cyanobacteria (Gunnelius et al., 2014). Downregulation here may indicate promotion of lowly expressed genes over highly expressed genes and perhaps additional evidence of quiescence within bacteria. Unlike bacteria, within archaea and eukaryote communities, RNA polymerase genes were largely downregulated. Overall, although plume communities displayed transcript patterns indicative of increased metabolic activity relative to outside the plume, there is also evidence of a slowdown of these processes resulting in slowing growth, cell division, and increasing quiescence.
Conclusions
Within the neutrally buoyant hydrothermal plume emanating from the Moytirra vent field persists a high level of microbial diversity which remains metabolically active for kilometers from the vent source. Reduced sulfur compounds appear to be a fundamental energy source within plume waters as evidenced by enriched sulfur metabolism functions and sulfur oxidizing bacteria. Although community-level functional signals suggest active metabolic functions for over a kilometer north or south of the vent field, these functions grew increasingly dissimilar to those observed at the vent site and bacterial communities displayed indications of beginning to enter quiescent stages, likely due to decreasing resources and reduced temperatures. These changes were more rapid than modelled communities suggest, where growth continues for 10’s of kilometers (Reed et al., 2015). Despite observable changes in function, the diversity of organisms within the plume remained similar both upstream and downstream of the vent. If the observed patterns here are characteristic of all vent plumes or just Moytirra remains to be studied and may have implications on our understanding of vent plume contributions to biogeochemical cycles, how long these persist, and their roles in vent ecosystem connectivity. How the observed patterns of diversity and function herein change beyond the sampled distance remains an open question, but plume transects south of the vent (Rainbow, Dog’s Head, and Longqi vent fields) suggest community changes occur farther downstream (Djurhuus et al., 2017; Haalboom et al., 2020) and may take >2 km to observe.
The deep sea is increasingly threatened by anthropogenic disturbance due to increased interest and technological advances in mining, which may have large impacts on the biodiversity of these unique and important habitats (Paulus, 2021). Vent systems have a disproportionate ecological significance in relation to their size and are vulnerable to natural and anthropogenic disturbances which can impact community composition and, as such, functional connectivity to global ocean processes (Van Dover et al., 2018). There is still much to learn regarding the importance of hydrothermal plumes in connecting vent ecosystems, providing highways for colonization, and how mining or other disturbances to vent ecosystems might disrupt this connectivity and its biogeochemical importance to deep sea carbon cycles, a major contributor to organic carbon pools (Le Bris et al., 2019). As such, there is need for future studies that cover greater spatial distances, such as the current study, to fully capture the biogeography of vent-derived microorganisms, their contribution to global biogeochemical cycles, and the risks future technological advancement may place on these unique and vulnerable habitats.
Data availability statement
The sequencing datasets (metatranscriptomic and metabarcode) generated for this study can be found in the at the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) https://www.ncbi.nlm.nih.gov/sra under accession number PRJNA950338. All bathymetric data have been deposited at the Global Multi-Resolution Topography Data Synthesis (GMRT) repository as part of the Seabed 2030 project. CTD data files can be found at https://doi.org/10.5281/zenodo.7908795. All code and data files used in analysis and to generate data products can be found at https://github.com/jmpolinski/Moytirra-Hydrothermal-Vent-Plume.
Author contributions
JP generated, analyzed, and interpreted sequencing data. MR assisted research and coordinated ship operations. JM conducted bathymetric mapping and interpretation of resulting data. MH designed and performed research, analyzed and interpreted data, and led the writing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by a grant from OceanX - Dalio Philanthropies (award ID 21-08532).
Acknowledgments
We thank the captain and crew of the R/V OceanXplorer and the operational staff of OceanX for their tireless efforts in making this work possible. We thank the participants of the Young Explorers Program who assisted with sampling, including Chelsea Gardiner, Alannah Vellacott, Shellby Johnson, Adam Wolffbrandt, Adam Lees, Anna Racanelli, Jade Asiu, Justin Lessing, Kayla Archer, Lilah McCormick, Madeleine Stewart, Madison Quarles, Miyah Brooks, and Tyler Newman. We would also like to thank Atle Foss of Akvaplan-Niva for sheltering our samples while we arranged for cryoshipping from Norway and Luis Somoza of Geological Survey of Spain IGME for mapping data that assisted in mission planning.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1219784/full#supplementary-material
References
Adams D. W., Errington J. (2009). Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nat. Rev. Microbiol. 7 (9), 642–653. doi: 10.1038/nrmicro2198
Alex L. A., Simon M. I. (1994). Protein histidine kinases and signal transduction in prokaryotes and eukaryotes. Trends Genet. 10 (4), 133–138. doi: 10.1016/0168-9525(94)90215-1
Alexander R. P., Lowenthal A. C., Harshey R. M., Ottemann K. M. (2010). CheV: CheW-like coupling proteins at the core of the chemotaxis signaling network. Trends Microbiol. 18 (11), 494–503. doi: 10.1016/j.tim.2010.07.004
Anantharaman K., Breier J. A., Sheik C. S., Dick G. J. (2013). Evidence for hydrogen oxidation and metabolic plasticity in widespread deep-sea sulfur-oxidizing bacteria. Proc. Natl. Acad. Sci. U.S.A. 110 (1), 330–335. doi: 10.1073/pnas.1215340110
Anantharaman K., Duhaime M. B., Breier J. A., Wendt K. A., Toner B. M., Dick G. J. (2014). Sulfur oxidation genes in diverse deep-sea viruses. Science 344 (6185), 757–760. doi: 10.1126/science.1252229
Ansorge R., Romano S., Sayavedra L., Rubin-Blum M., Gruber-Vodicka H., Scilipoti S., et al. (2020). The hidden pangenome: comparative genomics reveals pervasive diversity in symbiotic and free-living sulfur-oxidizing bacteria. bioRxiv. doi: 10.1101/2020.12.11.421487
Apprill A., McNally S., Parsons R., Weber L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microbial. Ecol. 75 (2), 129–137. doi: 10.3354/ame01753
Armitage J. P., Schmitt R. (1997). Bacterial chemotaxis: Rhodobacter sphaeroides and Sinorhizobium meliloti–variations on a theme? Microbiol. (Reading). 143 (Pt 12), 3671–3682. doi: 10.1099/00221287-143-12-3671
Baker B. J., Lesniewski R. A., Dick G. J. (2012). Genome-enabled transcriptomics reveals archaeal populations that drive nitrification in a deep-sea hydrothermal plume. ISME. J. 6 (12), 2269–2279. doi: 10.1038/ismej.2012.64
Beaulieu S. E., Szafrański K. M. (2020). Interridge global database of active submarine hydrothermal vent fields version 3.4. (PANGAEA). doi: 10.1594/PANGAEA.917894
Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat Soc Ser. B. 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Blaxter M. L., De Ley P., Garey J. R., Liu L. X., Scheldeman P., Vierstraete A., et al. (1998). A molecular evolutionary framework for the phylum Nematoda. Nature 392 (6671), 71–75. doi: 10.1038/32160
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Breitbart M., Bonnain C., Malki K., Sawaya N. A. (2018). Phage puppet masters of the marine microbial realm. Nat. Microbiol. 3 (7), 754–766. doi: 10.1038/s41564-018-0166-y
Carmona C. P., de Bello F., Mason N. W. H., Leps J. (2016). Traits without borders: integrating functional diversity across scales. Trends Ecol. Evol. 31 (5), 382–394. doi: 10.1016/j.tree.2016.02.003
Castelán-Sánchez H. G., Lopéz-Rosas I., García-Suastegui W. A., Peralta R., Dobson A. D. W., Batista-García R. A., et al. (2019). Extremophile deep-sea viral communities from hydrothermal vents: Structural and functional analysis. Mar. Genomics 46, 16–28. doi: 10.1016/j.margen.2019.03.001
Chastain R. A., Yayanos A. A. (1991). Ultrastructural changes in an obligately barophilic marine bacterium after decompression. Appl. Environ. Microbiol. 57 (5), 1489–1497. doi: 10.1128/aem.57.5.1489-1497.1991
Dede B., Hansen C. T., Neuholz R., Schnetger B., Kleint C., Walker S., et al. (2022). Niche differentiation of sulfur-oxidizing bacteria (SUP05) in submarine hydrothermal plumes. ISME. J. 16 (6), 1479–1490. doi: 10.1038/s41396-022-01195-x
Deng W., Nickle D. C., Learn G. H., Maust B., Mullins J. I. (2007). ViroBLAST: a stand-alone BLAST web server for flexible queries of multiple databases and user's datasets. Bioinformatics 23 (17), 2334–2336. doi: 10.1093/bioinformatics/btm331
Desbruyères D., Almeida A., Biscoito M., Comtet T., Khripounoff A., Le Bris N., et al. (2000). A review of the distribution of hydrothermal vent communities along the northern Mid-Atlantic Ridge: dispersal vs. environmental controls. Hydrobiologia 440 (1/3), 201–216. doi: 10.1023/a:1004175211848
Dick G. J. (2019). The microbiomes of deep-sea hydrothermal vents: distributed globally, shaped locally. Nat. Rev. Microbiol. 17 (5), 271–283. doi: 10.1038/s41579-019-0160-2
Dick G. J., Anantharaman K., Baker B. J., Li M., Reed D. C., Sheik C. S. (2013). The microbiology of deep-sea hydrothermal vent plumes: ecological and biogeographic linkages to seafloor and water column habitats. Front. Microbiol. 4. doi: 10.3389/fmicb.2013.00124
Djurhuus A., Mikalsen S. O., Giebel H. A., Rogers A. D. (2017). Cutting through the smoke: the diversity of microorganisms in deep-sea hydrothermal plumes. R. Soc. Open Sci. 4 (4), 160829. doi: 10.1098/rsos.160829
Edgar R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32 (5), 1792–1797. doi: 10.1093/nar/gkh340
Edgar R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26 (19), 2460–2461. doi: 10.1093/bioinformatics/btq461
Fortunato C. S., Butterfield D. A., Larson B., Lawrence-Slavas N., Algar C. K., Zeigler Allen L., et al. (2021). Seafloor incubation experiment with deep-sea hydrothermal vent fluid reveals effect of pressure and lag time on autotrophic microbial communities. Appl. Environ. Microbiol. 87 (9), e00078-21. doi: 10.1128/AEM.00078-21
Fortunato C. S., Larson B., Butterfield D. A., Huber J. A. (2018). Spatially distinct, temporally stable microbial populations mediate biogeochemical cycling at and below the seafloor in hydrothermal vent fluids. Environ. Microbiol. 20 (2), 769–784. doi: 10.1111/1462-2920.14011
Friedrich C. G., Rother D., Bardischewsky F., Quentmeier A., Fischer J. (2001). Oxidation of reduced inorganic sulfur compounds by bacteria: emergence of a common mechanism? Appl. Environ. Microbiol. 67 (7), 2873–2882. doi: 10.1128/AEM.67.7.2873-2882.2001
Galambos D., Anderson R. E., Reveillaud J., Huber J. A. (2019). Genome-resolved metagenomics and metatranscriptomics reveal niche differentiation in functionally redundant microbial communities at deep-sea hydrothermal vents. Environ. Microbiol. 21 (11), 4395–4410. doi: 10.1111/1462-2920.14806
Gartman A., Findlay A. J. (2020). Impacts of hydrothermal plume processes on oceanic metal cycles and transport. Nat. Geosci. 13 (6), 396–402. doi: 10.1038/s41561-020-0579-0
German C., Richards K., Rudnicki M., Lam M., Charlou J., Party F. S. (1998). Topographic control of a dispersing hydrothermal plume. Earth Planetary. Sci. Lett. 156 (3-4), 267–273. doi: 10.1016/S0012-821X(98)00020-X
Ghosh W., Dam B. (2009). Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol. Rev. 33 (6), 999–1043. doi: 10.1111/j.1574-6976.2009.00187.x
Gibbons S. M., Caporaso J. G., Pirrung M., Field D., Knight R., Gilbert J. A. (2013). Evidence for a persistent microbial seed bank throughout the global ocean. Proc. Natl. Acad. Sci. U.S.A. 110 (12), 4651–4655. doi: 10.1073/pnas.1217767110
Gómez-Gutiérrez J., Struder-Kypke M. C., Lynn D. H., Shaw T. C., Aguilar-Mendez M. J., Lopez-Cortes A., et al. (2012). Pseudocollinia brintoni gen. nov., sp. nov. (Apostomatida: Colliniidae), a parasitoid ciliate infecting the euphausiid Nyctiphanes simplex. Dis. Aquat. Organ 99 (1), 57–78. doi: 10.3354/dao02450
Gonnella G., Bohnke S., Indenbirken D., Garbe-Schonberg D., Seifert R., Mertens C., et al. (2016). Endemic hydrothermal vent species identified in the open ocean seed bank. Nat. Microbiol. 1 (8), 16086. doi: 10.1038/nmicrobiol.2016.86
Govenar B. (2012). Energy transfer through food webs at hydrothermal vents: linking the lithosphere to the biosphere. Oceanography 25 (1), 246–255. doi: 10.5670/oceanog.2012.23
Grabherr M. G., Haas B. J., Yassour M., Levin J. Z., Thompson D. A., Amit I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29 (7), 644–652. doi: 10.1038/nbt.1883
Graham J. B., Dickson K. A. (2004). Tuna comparative physiology. J. Exp. Biol. 207 (23), 4015–4024. doi: 10.1242/jeb.01267
Gray D. A., Dugar G., Gamba P., Strahl H., Jonker M. J., Hamoen L. W. (2019). Extreme slow growth as alternative strategy to survive deep starvation in bacteria. Nat. Commun. 10 (1), 890. doi: 10.1038/s41467-019-08719-8
Grimm F., Franz B., Dahl C. (2011). Regulation of dissimilatory sulfur oxidation in the purple sulfur bacterium Allochromatium vinosum. Front. Microbiol. 2. doi: 10.3389/fmicb.2011.00051
Gunnelius L., Hakkila K., Kurkela J., Wada H., Tyystjarvi E., Tyystjarvi T. (2014). The omega subunit of the RNA polymerase core directs transcription efficiency in cyanobacteria. Nucleic Acids Res. 42 (7), 4606–4614. doi: 10.1093/nar/gku084
Guo J., Bolduc B., Zayed A. A., Varsani A., Dominguez-Huerta G., Delmont T. O., et al. (2021). VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 9 (1), 37. doi: 10.1186/s40168-020-00990-y
Haalboom S., Price D. M., Mienis F., van Bleijswijk J. D. L., de Stigter H. C., Witte H. J., et al. (2020). Patterns of (trace) metals and microorganisms in the Rainbow hydrothermal vent plume at the Mid-Atlantic Ridge. Biogeosciences 17 (9), 2499–2519. doi: 10.5194/bg-17-2499-2020
Harke M. J., Frischkorn K. R., Hennon G. M. M., Haley S. T., Barone B., Karl D. M., et al. (2021). Microbial community transcriptional patterns vary in response to mesoscale forcing in the North Pacific Subtropical Gyre. Environ. Microbiol. 23 (8), 4807–4822. doi: 10.1111/1462-2920.15677
Hou J., Sievert S. M., Wang Y., Seewald J. S., Natarajan V. P., Wang F., et al. (2020). Microbial succession during the transition from active to inactive stages of deep-sea hydrothermal vent sulfide chimneys. Microbiome 8 (1), 102. doi: 10.1186/s40168-020-00851-8
Hu S. K., Herrera E. L., Smith A. R., Pachiadaki M. G., Edgcomb V. P., Sylva S. P., et al. (2021). Protistan grazing impacts microbial communities and carbon cycling at deep-sea hydrothermal vents. Proc. Natl. Acad. Sci. U. S. A. 118 (29), e2102674118. doi: 10.1073/pnas.2102674118
Hu S. K., Smith A. R., Anderson R. E., Sylva S. P., Setzer M., Steadmon M., et al. (2022). Globally-distributed microbial eukaryotes exhibit endemism at deep-sea hydrothermal vents. Mol. Ecol 00, 1–19. doi: 10.1111/mec.16745
Huang Z., Pan X., Xu N., Guo M. (2019). Bacterial chemotaxis coupling protein: Structure, function and diversity. Microbiol. Res. 219, 40–48. doi: 10.1016/j.micres.2018.11.001
Hügler M., Gärtner A., Imhoff J. F. (2010). Functional genes as markers for sulfur cycling and CO2 fixation in microbial communities of hydrothermal vents of the Logatchev field. FEMS Microbiol. Ecol. 73 (3), 526–537. doi: 10.1111/j.1574-6941.2010.00919.x
Ishihama A. (1997). Adaptation of gene expression in stationary phase bacteria. Curr. Opin. Genet. Dev. 7 (5), 582–588. doi: 10.1016/s0959-437x(97)80003-2
Jannasch H. W. (1995). “Microbial interactions with hydrothermal fluids,",” in Seafloor hydrothermal systems: physical, chemical, BIological, and geological interactions,. Eds. Humphris S. E., Zierenberg R. A., Mullineaux L. S., Thomson. R. E. (Washington, D.C: American Geophysical Union), 273–296.
Jannasch H. W., Mottl M. J. (1985). Geomicrobiology of deep-sea hydrothermal vents. Science 229 (4715), 717–725. doi: 10.1126/science.229.4715.717
Johnson K. S., Beehler C. L., Sakamoto-Arnold C. M., Childress J. J. (1986). In situ measurements of chemical distributions in a deep-sea hydrothermal vent field. Science 231 (4742), 1139–1141. doi: 10.1126/science.231.4742.1139
Juniper S. K., Bird D. F., Summit M., Vong M. P., Baker E. T. (1998). Bacterial and viral abundances in hydrothermal event plumes over northern Gorda Ridge. Deep. Sea. Res. Part II.: Topical. Stud. Oceanogr. 45 (12), 2739–2749. doi: 10.1016/s0967-0645(98)00091-5
Kuppa Baskaran D. K., Umale S., Zhou Z., Raman K., Anantharaman K. (2023). Metagenome-based metabolic modelling predicts unique microbial interactions in deep-sea hydrothermal plume microbiomes. ISME. Commun. 3 (1), 42. doi: 10.1038/s43705-023-00242-8
Kurkela J., Fredman J., Salminen T. A., Tyystjarvi T. (2021). Revealing secrets of the enigmatic omega subunit of bacterial RNA polymerase. Mol. Microbiol. 115 (1), 1–11. doi: 10.1111/mmi.14603
Lahaye N., Gula J., Thurnherr A. M., Reverdin G., Bouruet-Aubertot P., Roullet G. (2019). Deep currents in the rift valley of the north mid-atlantic ridge. Front. Mar. Sci. 6. doi: 10.3389/fmars.2019.00597
Lam P., Cowen J. P., Jones R. D. (2004). Autotrophic ammonia oxidation in a deep-sea hydrothermal plume. FEMS Microbiol. Ecol. 47 (2), 191–206. doi: 10.1016/S0168-6496(03)00256-3
Langmead B., Salzberg S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9 (4), 357–359. doi: 10.1038/nmeth.1923
Le Bris N., Yücel M., Das A., Sievert S. M., LokaBharathi P., Girguis P. R. (2019). Hydrothermal energy transfer and organic carbon production at the deep seafloor. Front. Mar. Sci. 5. doi: 10.3389/fmars.2018.00531
Lee J., Borukhov S. (2016). Bacterial RNA polymerase-DNA interaction-the driving force of gene expression and the target for drug action. Front. Mol. Biosci. 3. doi: 10.3389/fmolb.2016.00073
Lesniewski R. A., Jain S., Anantharaman K., Schloss P. D., Dick G. J. (2012). The metatranscriptome of a deep-sea hydrothermal plume is dominated by water column methanotrophs and lithotrophs. ISME. J. 6 (12), 2257–2268. doi: 10.1038/ismej.2012.63
Li B., Dewey C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 12 (232), 232. doi: 10.1186/1471-2105-12-323
Li W., Godzik A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22 (13), 1658–1659. doi: 10.1093/bioinformatics/btl158
Li M., Toner B. M., Baker B. J., Breier J. A., Sheik C. S., Dick G. J. (2014). Microbial iron uptake as a mechanism for dispersing iron from deep-sea hydrothermal vents. Nat. Commun. 5, 3192. doi: 10.1038/ncomms4192
Liang C., Wang W., Dong L., Mukhtar I., Wang F., Chen J. (2021). A new protocruzia species (Ciliophora: protocruziida) isolated from the mariana trench area. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.743920
Lindsay D., Umetsu M., Grossmann M., Miyake H., Yamamoto H. (2015). “The gelatinous macroplankton community at the hatoma knoll hydrothermal vent,” in Subseafloor biosphere linked to hydrothermal systems,. Eds. Ishibashi J., Okino K., Sunamura M. (Tokyo: Springer).
Love M. I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550. doi: 10.1186/s13059-014-0550-8
Lu J., Breitwieser F. P., Thielen P., Salzberg S. L. (2017). Bracken: estimating species abundance in metagenomics data. PeerJ. Comput. Sci. 3, e104. doi: 10.7717/peerj-cs.104
Marshall K. T., Morris R. M. (2013). Isolation of an aerobic sulfur oxidizer from the SUP05/Arctic96BD-19 clade. ISME. J. 7 (2), 452–455. doi: 10.1038/ismej.2012.78
Mizuno C. M., Ghai R., Saghai A., Lopez-Garcia P., Rodriguez-Valera F. (2016). Genomes of abundant and widespread viruses from the deep ocean. mBio 7 (4), e00805-16. doi: 10.1128/mBio.00805-16
Morris R. M., Spietz R. L. (2022). The physiology and biogeochemistry of SUP05. Ann. Rev. Mar. Sci. 14, 261–275. doi: 10.1146/annurev-marine-010419-010814
Mullineaux L. S., Adams D. K., Mills S. W., Beaulieu S. E. (2010). Larvae from afar colonize deep-sea hydrothermal vents after a catastrophic eruption. Proc. Natl. Acad. Sci. U.S.A. 107 (17), 7829–7834. doi: 10.1073/pnas.0913187107
Murdock S. A., Juniper S. K. (2019). Hydrothermal vent protistan distribution along the Mariana arc suggests vent endemics may be rare and novel. Environ. Microbiol. 21 (10), 3796–3815. doi: 10.1111/1462-2920.14729
Nayfach S., Camargo A. P., Schulz F., Eloe-Fadrosh E., Roux S., Kyrpides N. C. (2021). CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 39 (5), 578–585. doi: 10.1038/s41587-020-00774-7
Orcutt B. N., Sylvan J. B., Knab N. J., Edwards K. J. (2011). Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol. Mol. Biol. Rev. 75 (2), 361–422. doi: 10.1128/MMBR.00039-10
Ortmann A. C., Suttle C. A. (2005). High abundances of viruses in a deep-sea hydrothermal vent system indicates viral mediated microbial mortality. Deep. Sea. Res. Part I.: Oceanogr. Res. Papers. 52 (8), 1515–1527. doi: 10.1016/j.dsr.2005.04.002
Parada A. E., Needham D. M., Fuhrman J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18 (5), 1403–1414. doi: 10.1111/1462-2920.13023
Pasulka A., Hu S. K., Countway P. D., Coyne K. J., Cary S. C., Heidelberg K. B., et al. (2019). SSU-rRNA gene sequencing survey of benthic microbial eukaryotes from guaymas basin hydrothermal vent. J. Eukaryot. Microbiol. 66 (4), 637–653. doi: 10.1111/jeu.12711
Paulus E. (2021). Shedding light on deep-sea biodiversity—A highly vulnerable habitat in the face of anthropogenic change. Front. Mar. Sci. 8. doi: 10.3389/fmars.2021.667048
Peng Y., Leung H. C., Yiu S. M., Chin F. Y. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28 (11), 1420–1428. doi: 10.1093/bioinformatics/bts174
Pitcher R. S., Watmough N. J. (2004). The bacterial cytochrome cbb3 oxidases. Biochim. Biophys. Acta 1655 (1-3), 388–399. doi: 10.1016/j.bbabio.2003.09.017
Price A., Caciula A., Guo C., Lee B., Morrison J., Rasmussen A., et al. (2019). DEvis: an R package for aggregation and visualization of differential expression data. BMC Bioinf. 20 (1), 110. doi: 10.1186/s12859-019-2702-z
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41 (Database issue), D590–D596. doi: 10.1093/nar/gks1219
Reed D. C., Breier J. A., Jiang H., Anantharaman K., Klausmeier C. A., Toner B. M., et al. (2015). Predicting the response of the deep-ocean microbiome to geochemical perturbations by hydrothermal vents. ISME. J. 9 (8), 1857–1869. doi: 10.1038/ismej.2015.4
Ren J., Ahlgren N. A., Lu Y. Y., Fuhrman J. A., Sun F. (2017). VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 5 (1), 69. doi: 10.1186/s40168-017-0283-5
Reveillaud J., Reddington E., McDermott J., Algar C., Meyer J. L., Sylva S., et al. (2016). Subseafloor microbial communities in hydrogen-rich vent fluids from hydrothermal systems along the Mid-Cayman Rise. Environ. Microbiol. 18 (6), 1970–1987. doi: 10.1111/1462-2920.13173
Reysenbach A. L., Banta A. B., Boone D. R., Cary S. C., Luther G. W. (2000). Microbial essentials at hydrothermal vents. Nature 404 (6780), 835. doi: 10.1038/35009029
Rother D., Orawski G., Bardischewsky F., Friedrich C. G. (2005). SoxRS-mediated regulation of chemotrophic sulfur oxidation in Paracoccus pantotrophus. Microbiol. (Reading). 151 (Pt 5), 1707–1716. doi: 10.1099/mic.0.27724-0
Russ L., van Alen T. A., Jetten M. S. M., Op den Camp H. J. M., Kartal B. (2019). Interactions of anaerobic ammonium oxidizers and sulfide-oxidizing bacteria in a substrate-limited model system mimicking the marine environment. FEMS Microbiol. Ecol. 95 (9), fiz137. doi: 10.1093/femsec/fiz137
Sarkar M. K., Paul K., Blair D. (2010). Chemotaxis signaling protein CheY binds to the rotor protein FliN to control the direction of flagellar rotation in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 107 (20), 9370–9375. doi: 10.1073/pnas.1000935107
Sarrazin J., Juniper S. K. (1999). Biological characteristics of a hydrothermal edifice mosaic community. Mar. Ecol. Prog. Ser. 185, 1–19. doi: 10.3354/meps185001
Sauve V., Bruno S., Berks B. C., Hemmings A. M. (2007). The SoxYZ complex carries sulfur cycle intermediates on a peptide swinging arm. J. Biol. Chem. 282 (32), 23194–23204. doi: 10.1074/jbc.M701602200
Schloss P. D., Westcott S. L., Ryabin T., Hall J. R., Hartmann M., Hollister E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75 (23), 7537–7541. doi: 10.1128/AEM.01541-09
Schoenle A., Nitsche F., Werner J., Arndt H. (2017). Deep-sea ciliates: Recorded diversity and experimental studies on pressure tolerance. Deep. Sea. Res. Part I.: Oceanogr. Res. Papers. 128, 55–66. doi: 10.1016/j.dsr.2017.08.015
Seemann T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30 (14), 2068–2069. doi: 10.1093/bioinformatics/btu153
Seyler L. M., Trembath-Reichert E., Tully B. J., Huber J. A. (2021). Time-series transcriptomics from cold, oxic subseafloor crustal fluids reveals a motile, mixotrophic microbial community. ISME. J. 15 (4), 1192–1206. doi: 10.1038/s41396-020-00843-4
Shah V., Zhao X., Lundeen R. A., Ingalls A. E., Nicastro D., Morris R. M. (2019). Morphological plasticity in a sulfur-oxidizing marine bacterium from the SUP05 clade enhances dark carbon fixation. mBio 10 (3), e00216-19. doi: 10.1128/mBio.00216-19
Sheik C. S., Anantharaman K., Breier J. A., Sylvan J. B., Edwards K. J., Dick G. J. (2015). Spatially resolved sampling reveals dynamic microbial communities in rising hydrothermal plumes across a back-arc basin. ISME. J. 9 (6), 1434–1445. doi: 10.1038/ismej.2014.228
Sievert S. M., Hügler M., Taylor C. D., Wirsen C. O. (2008). “"Sulfur oxidation at deep-sea hydrothermal vents",” in Microbial sulfur metabolism. Eds. Dahl C., Friedrich C. G. (Berlin, Heidelberg: Springer Berlin Heidelberg).
Sievert S., Vetriani C. (2012). Chemoautotrophy at deep-sea vents: past, present, and future. Oceanography 25 (1), 218–233. doi: 10.5670/oceanog.2012.21
Skebo K. M. (1994). Distribution of Zooplankton and Nekton above Hydrothermal Vents on the Juan de Fuca and Explorer Ridges (University of Victoria: MS).
Somoza L., Medialdea T., González F. J., Calado A., Afonso A., Albuquerque M., et al. (2020). Multidisciplinary scientific cruise to the northern mid-atlantic ridge and azores archipelago. Front. Mar. Sci. 7. doi: 10.3389/fmars.2020.568035
Speer K., Maltrud M., Thurnherr A. (2003). “A global view of dispersion on the mid-oceanic ridge,” Energy and Mass Transfer in Marine Hydrothermal Systems eds. Halbach P., Tunnicliffe V., Hein J.. (Berlin: DUP), 287–302.
Spietz R. L., Lundeen R. A., Zhao X., Nicastro D., Ingalls A. E., Morris R. M. (2019). Heterotrophic carbon metabolism and energy acquisition in Candidatus Thioglobus singularis strain PS1, a member of the SUP05 clade of marine Gammaproteobacteria. Environ. Microbiol. 21 (7), 2391–2401. doi: 10.1111/1462-2920.14623
Strzalka W., Ziemienowicz A. (2011). Proliferating cell nuclear antigen (PCNA): a key factor in DNA replication and cell cycle regulation. Ann. Bot. 107 (7), 1127–1140. doi: 10.1093/aob/mcq243
Sunamura M., Higashi Y., Miyako C., Ishibashi J., Maruyama A. (2004). Two bacteria phylotypes are predominant in the Suiyo seamount hydrothermal plume. Appl. Environ. Microbiol. 70 (2), 1190–1198. doi: 10.1128/AEM.70.2.1190-1198.2004
Suttle C. A., Chen F. (1992). Mechanisms and rates of decay of marine viruses in seawater. Appl. Environ. Microbiol. 58 (11), 3721–3729. doi: 10.1128/aem.58.11.3721-3729.1992
Tagliabue A., Bopp L., Dutay J.-C., Bowie A. R., Chever F., Jean-Baptiste P., et al. (2010). Hydrothermal contribution to the oceanic dissolved iron inventory. Nat. Geosci. 3 (4), 252–256. doi: 10.1038/ngeo818
Takaki Y., Shimamura S., Nakagawa S., Fukuhara Y., Horikawa H., Ankai A., et al. (2010). Bacterial lifestyle in a deep-sea hydrothermal vent chimney revealed by the genome sequence of the thermophilic bacterium Deferribacter desulfuricans SSM1. DNA Res. 17 (3), 123–137. doi: 10.1093/dnares/dsq005
Thomas E., Anderson R. E., Li V., Rogan L. J., Huber J. A. (2021). Diverse viruses in deep-sea hydrothermal vent fluids have restricted dispersal across ocean basins. mSystems 6 (3), e0006821. doi: 10.1128/mSystems.00068-21
Thurnherr A. M., Richards K. J., German C. R., Lane-Serff G. F., Speer K. G. (2002). Flow and mixing in the rift valley of the mid-atlantic ridge. J. Phys. Oceanogr. 32 (6), 1763–1778. doi: 10.1175/1520-0485(2002)032<1763:Famitr>2.0.Co;2
Töpper B., Thingstad T. F., Sandaa R. A. (2013). Effects of differences in organic supply on bacterial diversity subject to viral lysis. FEMS Microbiol. Ecol. 83 (1), 202–213. doi: 10.1111/j.1574-6941.2012.01463.x
Toyoshima H., Hunter T. (1994). p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 78 (1), 67–74. doi: 10.1016/0092-8674(94)90573-8
Tunnicliffe V., McArthur A. G., McHugh D. (1998). A biogeographical perspective of the deep-sea hydrothermal vent fauna. Adv. Mar. Biol. 34, 353–442. doi: 10.1016/S0065-2881(08)60213-8
Turner J. S., Campbell I. H. (1987). Temperature, density and buoyancy fluxes in “black smoker” plumes, and the criterion for buoyancy reversal. Earth Planetary. Sci. Lett. 86 (1), 85–92. doi: 10.1016/0012-821x(87)90191-9
Tyler P. A., Young C. M. (2003). Dispersal at hydrothermal vents: a summary of recent progress. Hydrobiologia 503 (1-3), 9–19. doi: 10.1023/B:HYDR.0000008492.53394.6b
Uxa S., Castillo-Binder P., Kohler R., Stangner K., Muller G. A., Engeland K. (2021). Ki-67 gene expression. Cell Death Differ. 28 (12), 3357–3370. doi: 10.1038/s41418-021-00823-x
Van Dover C. L. (1995). Ecology of Mid-Atlantic Ridge hydrothermal vents. Geological. Soc. London. Special. Publications. 87 (1), 257–294. doi: 10.1144/gsl.Sp.1995.087.01.21
Van Dover C. L., Arnaud-Haond S., Gianni M., Helmreich S., Huber J. A., Jaeckel A. L., et al. (2018). Scientific rationale and international obligations for protection of active hydrothermal vent ecosystems from deep-sea mining. Mar. Policy 90, 20–28. doi: 10.1016/j.marpol.2018.01.020
Walter M., Mertens C., Stöber U., German C. R., Yoerger D. R., Sültenfuß J., et al. (2010). Rapid dispersal of a hydrothermal plume by turbulent mixing. Deep. Sea. Res. Part I.: Oceanogr. Res. Papers. 57 (8), 931–945. doi: 10.1016/j.dsr.2010.04.010
Weitz J. S., Wilhelm S. W. (2012). Ocean viruses and their effects on microbial communities and biogeochemical cycles. F1000. Biol. Rep. 4, 17. doi: 10.3410/B4-17
Wheeler A. J., Murton B., Copley J., Lim A., Carlsson J., Collins P., et al. (2013). Moytirra: Discovery of the first known deep-sea hydrothermal vent field on the slow-spreading Mid-Atlantic Ridge north of the Azores. Geochem. Geophys. Geosyst. 14 (10), 4170–4184. doi: 10.1002/ggge.20243
Wood H. G. (1991). Life with CO or CO2 and H2 as a source of carbon and energy. FASEB J. 5 (2), 156–163. doi: 10.1096/fasebj.5.2.1900793
Wood D. E., Lu J., Langmead B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20 (1), 257. doi: 10.1186/s13059-019-1891-0
Yam C. H., Fung T. K., Poon R. Y. (2002). Cyclin A in cell cycle control and cancer. Cell Mol. Life Sci. 59 (8), 1317–1326. doi: 10.1007/s00018-002-8510-y
Yang K., Hitomi M., Stacey D. W. (2006). Variations in cyclin D1 levels through the cell cycle determine the proliferative fate of a cell. Cell Div. 1, 32. doi: 10.1186/1747-1028-1-32
Yang J. W., Wu W., Chung C. C., Chiang K. P., Gong G. C., Hsieh C. H. (2018). Predator and prey biodiversity relationship and its consequences on marine ecosystem functioning-interplay between nanoflagellates and bacterioplankton. ISME. J. 12 (6), 1532–1542. doi: 10.1038/s41396-018-0111-3
Yoshida-Takashima Y., Nunoura T., Kazama H., Noguchi T., Inoue K., Akashi H., et al. (2012). Spatial distribution of viruses associated with planktonic and attached microbial communities in hydrothermal environments. Appl. Environ. Microbiol. 78 (5), 1311–1320. doi: 10.1128/AEM.06491-11
Zambrano M. M., Siegele D. A., Almiron M., Tormo A., Kolter R. (1993). Microbial competition: Escherichia coli mutants that take over stationary phase cultures. Science 259 (5102), 1757–1760. doi: 10.1126/science.7681219
Keywords: hydrothermal, microbe, biogeography, metatranscriptome, diversity
Citation: Polinski JM, Rodrigue M, Meyer JD and Harke MJ (2023) Drifting in the deep: Metatranscriptomics and metabarcoding reveal sustained metabolic activity and community composition in hydrothermal vent plume microbial communities. Front. Mar. Sci. 10:1219784. doi: 10.3389/fmars.2023.1219784
Received: 09 May 2023; Accepted: 25 September 2023;
Published: 17 October 2023.
Edited by:
Paraskevi Polymenakou, Hellenic Centre for Marine Research (HCMR), GreeceReviewed by:
Sean M. McAllister, University of Washington, United StatesSe-Jong Ju, Korea Institute of Ocean Science and Technology (KIOST), Republic of Korea
Copyright © 2023 Polinski, Rodrigue, Meyer and Harke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew J. Harke, bWF0dGhldy5oYXJrZUBnbWdpLm9yZw==