
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 18 July 2023
Sec. Microbial Symbioses
Volume 10 - 2023 | https://doi.org/10.3389/fmars.2023.1201434
This article is part of the Research Topic Diversity, Dynamics and Function of Host-Associated Microbiomes in Marine Organisms View all 11 articles
 Jianhua Sun1†Xinran Zhang1†
Jianhua Sun1†Xinran Zhang1† Yantao Liang1,2*Kaiyang Zheng1
Yantao Liang1,2*Kaiyang Zheng1 Fraser Kennedy3Meiaoxue Han1Gang Liu1Yundan Liu1Ziyue Wang1
Fraser Kennedy3Meiaoxue Han1Gang Liu1Yundan Liu1Ziyue Wang1 Xuechao Chen1
Xuechao Chen1 Yeong Yik Sung2,4
Yeong Yik Sung2,4 Wen Jye Mok2,4
Wen Jye Mok2,4 Li Lian Wong2,4
Li Lian Wong2,4 Andrew McMinn1,3*
Andrew McMinn1,3* Min Wang1,2,5,6*
Min Wang1,2,5,6*Pseudoalteromonas is a ubiquitous and abundant genus of marine bacteria commonly associated with algae. In this study, a novel siphoviral-morphological bacteriophage, vB_PhoS_XC, was isolated from the coastal seawaters of Qingdao (China) during a bloom of the Ulva prolifera (U. prolifera) green tide. The morphology of this phage (icosahedron head 51 ± 1 nm in diameter; a tail length of 86 ± 1 nm) was characterized through transmission electron microscope. The biological properties of this virus showed a short latent period (45 minutes), a large burst size (241 virions per cell) and a relatively wide range of temperatures/pH level tolerance (-20°C to 45°C and pH 4 to pH 10, respectively). The vB_PhoS_XC has a 46,490-bp double-stranded DNA genome with a G+C content of 40.0%, and encodes 72 open reading frames (ORFs). Thirty-five of these ORFs were assigned into known functions based on BLAST-based algorithm against NR database of GenBank. In addition, eco-genomic analysis provides the evidence of vB_PhoS_XC accompanied by bloom of U. prolifera, and confirmed the high expression of two phosphatase-metabolism-related auxiliary metabolic genes (AMGs). This study provides new insights into the functional and ecological roles of the Pseudoalteromonas phage vB_PhoS_XC, shedding light on the virological study approach combined with traditional isolation and meta-omics data.
In coastal marine ecosystems, algae are a naturally abundant food source for marine organisms and contribute to the production of polysaccharide-rich detritus (Jain et al., 2020; Akter et al., 2022). However, macroalgae blooms create great pressure on these ecosystems, resulting in hypoxia and acidification (Zhang et al., 2019). The largest causal agent of green tides is the macroalgae Ulva prolifera (U. prolifera), which has been affecting Qingdao since 2007. Following a bloom, huge amounts of dissolved organic matter (DOM) are produced due to the degradation of U. prolifera (Zhang and Wang, 2017). This leads to serious negative economic impacts and complex ecological consequences, most notably on the structure of the microbial community (Xiao et al., 2021; Zhang et al., 2021). Nevertheless, the majority of studies on U. prolifera control and microenvironment typically concentrated on the relationships between U. prolifera and bacteria, for instance, changes in bacterial communities and responses to U. prolifera without taking into consideration the potential contribution of viruses (Qu et al., 2020; Liang et al., 2021; Qu et al., 2021; Zhao et al., 2022).
Viruses are the most abundant and diverse biological entities in the ocean (Culley et al., 2006), playing an important role in global biogeochemical cycles (Fuhrman, 1999). They are also the most abundant and genetically diverse “life forms” on Earth (Fuhrman, 1999). Viruses mediate horizontal gene transfer by infecting host cells, and they also play a key role in the formation of bacterial community structure. Moreover, they can reduce the abundance of the major host by infecting and inducing the community structure into homeostasis (Zhang et al., 2007). The largest community of viruses in marine ecosystems are bacteriophages, which are bacterial specific. It is estimated that abundance of bacteriophages is around 10-15 times higher than bacteria (Culley et al., 2006), and the expression of phage-encoded auxiliary metabolic genes (AMGs) can influence bacterial host with reprogramming metabolic progress and improves their adaptability to environmental changes (Gregory et al., 2019; Zimmerman et al., 2020).
Pseudoalteromonas is the most abundant bacteria genus found in coastal seawater and is dominant during U. prolifera blooms (Wang et al., 2020; Qu et al., 2021). Metabolically, this species exhibits high productivity and can effectively use a variety of carbon sources, providing a competitive advantage over other microorganisms. Further, it has been explored to secrete a range of extracellular substances, including enzymes, toxins, antibiotics and polysaccharides (Ivanova et al., 2003), and found to make significant contributions to dissolved algae through the action of alginate lyases. This organism’s strong capacity to produce extracellular degrading enzymes, as well as a variety of bioactive substances, leads to its important role in marine biogeochemical cycles (Bowman, 2007). Although Pseudoalteromonas is a significant alginate-degrading group in the ocean, with its presence being particularly prominent in coastal seawater (Rong et al., 2018; Ren et al., 2022), few Pseudoalteromonas phages have been isolated. Thus, we sought to explore the possible regulatory role of viruses in the U. prolifera blooms in the coastal marine ecosystems of Qingdao in this study. To do this, the Pseudoalteromonas phage vB_PhoS_XC was isolated from these waters, its biological characteristics were investigated, and its genome was analyzed. Additionally, the metagenome and metatranscriptome outcomes of the phage-host interaction were used as models to suggest that the virus could influence the dynamic population of U. prolifera. This work thus adds to our understanding of U. prolifera blooms and provides a framework for further studies on the complex interactions between U. prolifera, Pseudoalteromonas, and their phages.
Seven samples were collected from the coastal seawaters of Qingdao between July to December in 2020 year. Eighty-liters of surface seawater were sampled from the coast of Qingdao (36.06° N, 120.31° E), and then pre-filtered on nylon sieves with a mesh of 200 μm and 20 μm to remove large zooplankton. Seawater samples were pre-filtered 3 μm filters and then followed by filtration through 0.22 μm Polycarbonate Membranes filters (Millipore, MA, USA) using a peristaltic pump (Stewart et al., 2012; Pesant et al., 2015). Filters were transferred immediately to DNase/RNase-Free tubes with RNAlater and stored at -80°C for RNA extraction. To further concentrate the filtered seawater containing viruses, tangential flow filtration (laboratory scale, 50 kDa; Millipore) was used to reduce samples to 100 mL for DNA extraction. The samples and the original seawater were stored at 4°C in the dark until experimentation (Wang et al., 2015).
Host bacterial strain was isolated from the original seawater at the coast seawaters of Qingdao during the U. prolifera blooms in 2020 year, and then incubated in liquid Zobell medium at 28°C for next experiments (Liu et al., 2018). The 16S rRNA gene of host was amplified by PCR. The phylogenetic tree was calculated on the bias of the 16S rRNA sequences using IQ-TREE with MFP model as the best-fitted substitution model undergoing 1,000 bootstraps (Supplementary Figure S1).
The phage vB_PhoS_XC was prepared using the standard virus enrichment approach and doubled-layer agar methods (Jamalludeen et al., 2007), followed by using the soft-agar overlay method for plaque analysis (Hyman and Abedon, 2010). Briefly, 1 mL host cell culture was incubated with 0.2 mL seawater, and then was filtered through 0.22 μm pore-size membranes (Millipore) for 20min. The mixture was placed into 5 mL of soft and warm agar (0.6%), and poured onto petri dishes to form plaques. Phages were purified by picking a single plaque, suspending it in SM buffer (100 mM NaCl, 81.2 mM MgSO4, 50 mM Tris HCl [pH 7.5], 0.01% gelatin) for 1 h at 37°C. The suspension was filtered through 0.22 μm pore-size membranes (Millipore), and was used to obtain the plaque through double-layer plate method. The purification step was repeated for three times. Then the purified phages were amplified and stored at 4°C.
20 µL viral suspension was negatively stained with 2% uranyl acetate, and was examined by energy-filtering transmission electron microscopy (TEM, JEOL Model JEM-1200EX) at 100 kV at NICEM to obtain the viral morphological data (Li Y et al., 2016). The average size of virion was determined by measuring three phage particles within the same field of vision.
The one-step growth curve assay was used to identify the burst size of the phage vB_PhoS_XC following the previous method with some modifications (Middelboe et al., 2010; Ma et al., 2021). The burst size was calculated by dividing the final number of virions by the initial number of infected host cells at the beginning of assay (Wang et al., 2017). A bacterial culture in the exponential growth phase (2 x 108 CFU/mL) was mixed with 1 mL of the phage suspension (MOI=0.01) at 25°C for 20 min. After centrifugation at 12,800 g for 30 seconds to remove unabsorbed phage, samples were re-suspended to 50 mL liquid Zobell medium. The re-suspended virions were used to infect bacteria with the 5-min/10-min/30-min interval in first/second/third hour, respectively (Gong et al., 2017). This assay was re-performed by three times (Li et al., 2016). The virion/bacteria co-culture was plated on a double-layer plate to incubate overnight. The number of phage plaques were counted to determine the titers of the phage at different time points to assess its growth states.
We tested pH/thermal stability to gain the information on the biological properties of the phage vB_PhoS_XC. Briefly, to test capsid tolerance to pH, 100 µL phage suspension mixed with 900 μL SM buffer with a range of pH from 3 to 12 at 30 °C for 2 h. To test the capsid tolerance to temperature, the phage suspension was incubated at -20°C, 4°C, 25°C, 35°C, 45°C, 55°C, 65°C, and 75°C for 2 h, then incubated with host at 25°C for 20min. The double-layer plate method was used to calculate the loss of virus titer of each sample. Each sample was performed in triplicate.
Bacteriophage DNA was extracted by Virus DNA Kit (OMEGA) and sequenced using the Illumina HiSeq PE150 paired-end sequence method by Novogene (Tianjin, China) company (Ma et al., 2021). The gaps between the remaining contigs were closed using GapCloser and GapFiller, with purified genomic DNA as the template (Nadalin et al., 2012; Xu et al., 2020). Open reading frames (ORFs) were predicted by GeneMarkS (http://topaz.gatech.edu/GeneMark/) and RAST (http://rast.nmpdr.org/). The protein function and homology of ORFs were predicted through the BLASTP-algorithm and Hidden-Markov-Module (HMM) based algorithm against NR (2023/02) and Pfam (v.35) database with 1e-5 as the E-value threshold (Jacob et al., 2008; Katoh and Standley, 2013; Mistry et al., 2013; Mistry et al., 2021). The genomic organization diagram was visualized by the CLC Main Workbench 20. The tRNAscan-SE (http://lowelab.ucsc.edu/tRNAscan-SE/) was used to detect tRNA sequences is in the viral genome (Chan et al., 2021). The complete viral genome sequence of vB_PhoS_XC is now available to access under the accession number MT002874.2 in GenBank. The auxiliary metabolic genes (AMGs) were blast through the Integrated Microbial Genomes/Virus 4 database with the E-value 1e-5, high search quality genes were chosen for homologous genes, and details were given in Supplementary Table 1.
Total viral DNA was extracted using QIAamp DNA MiniKit (QIAGEN) according to the manufacturer’s instructions. Library construction and high-throughput sequencing were carried out by Novogene (Tianjin, China) using Illumina NovaSeq 6000 (pair-end sequencing, 2 × 150 bp). Raw reads were quality-controlled by removing the adapters using Cutadapt (Kechin et al., 2017). The high-quality paired-end reads were filtered by Perl scripts following these criteria: 1) without N; 2) no more than 20% bases with a quality score less than 20 (Q20); 3) no more than 30% bases with a quality score less than 30 (Q30) (Yang et al., 2019). To calculate the relative abundances of the vB_PhoS_XC in all meta-omics samples, clean reads were mapped to the vB_PhoS_XC contig using the contig section of CoverM v0.6.1 (https://github.com/wwood/CoverM) with default parameter. The relative abundance of the vB_PhoS_XC was quantified as reads per kilobase million mapped reads (RPKM) (Zhang et al., 2023). We also calculated the relative abundance of other representative phages abundant in the ocean including those infecting Pelagibacter (Zhao et al., 2013; Chen et al., 2019), Cyanobacteria (Labrie et al., 2013), and all Pseudoalteromonas which belong to the same subfamily as vB_PhoS_XC (Table 1).
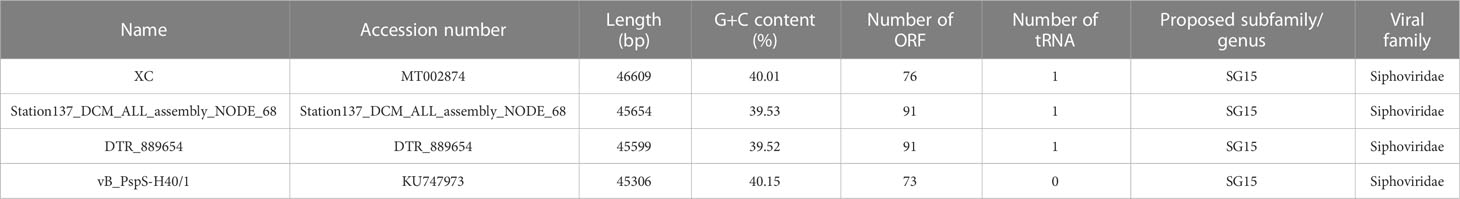
Table 1 The genome information of Pseudoalteromonas phages belonging to the same subfamily as Pseudoalteromonas phage vB_PhoS_XC.
Total RNA extraction, library construction and next-generation sequencing of viral RNA was carried out by Novogene (Tianjin, China) using Illumina NovaSeq 6000 (pair-end sequencing, 2 × 150 bp). Clean reads were picked from the raw reads by Novagene. Clean reads were then quality trimmed using Trimmomatic v0.39 (Bolger et al., 2014), and then were dropped below SLIDINGWINDOW:4:30, MINLEN:70. The potential rRNA-associated reads were removed using the SortMeRNA v4.3.2 against all databases (Kopylova et al., 2012). Then we got the high-quality reads. The relative abundances of the genes encoded by vB_PhoS_XC were calculated on the basis of all meta-omics samples through CoverM v0.6.1 with default parameter (Salazar et al., 2019). The relative abundances of these genes were then determined as reads per kilobase million mapped reads (RPKM).
Temperature, salinity, Dissolved Oxygen (DO) and pH were measured at the sampling site using a YSI proplus multi-parameter water quality analyzer (YSI, Yellow Springs, OH, USA). Seawater filtered through membranes, The concentrations of nitrate (NO3-), nitrite (NO2-), ammonium (NH4+) and phosphate (PO43-) were measured with a QuAAtro nutrient auto analyzer (Seal Analytical Ltd., King’s Lynn, UK). Picoplankton (Pico), Synechococcus (Syn), Bacterioplankton (Bac) counting using flow cytometry (Beckman Coulter Inc, USA). All environmental parameters described in Supplementary Table S2. Pearson correlation coefficient (R-value) was used to describe the correlation between vB_PhoS_XC and environmental parameters, including biological parameters (such as Pico, Syn, Bac) and non-biological parameters (such as NH4: Ammonium (NH3-N), Nitrite (NO2-N), Phoshate (PO43-), pH value, Nitrate (NO3-N), Salinity and Dissolved Oxygen (DO)). The analysis was performed by psych and vegan which are both powered by R (Dixon, 2003).
A novel marine phage, vB_PhoS_XC, was isolated from the coastal waters of Qingdao during an U. prolifera bloom (Figure 1A). By the 16srRNA analysis, we determined that the host of the phage vB_PhoS_XC is Pseudoalteromonas hodoensis strain H7 (Supplementary Figure S1). Characterization of vB_PhoS_XC revealed an icosahedral head (~51 ± 1 nm in diameter) with a long non-contractile tail (~86 ± 1 nm) (Figures 1B, C), combined with previous research, indicating that it could be classified into the Sipovirus subfamily (Zheng et al., 2023).
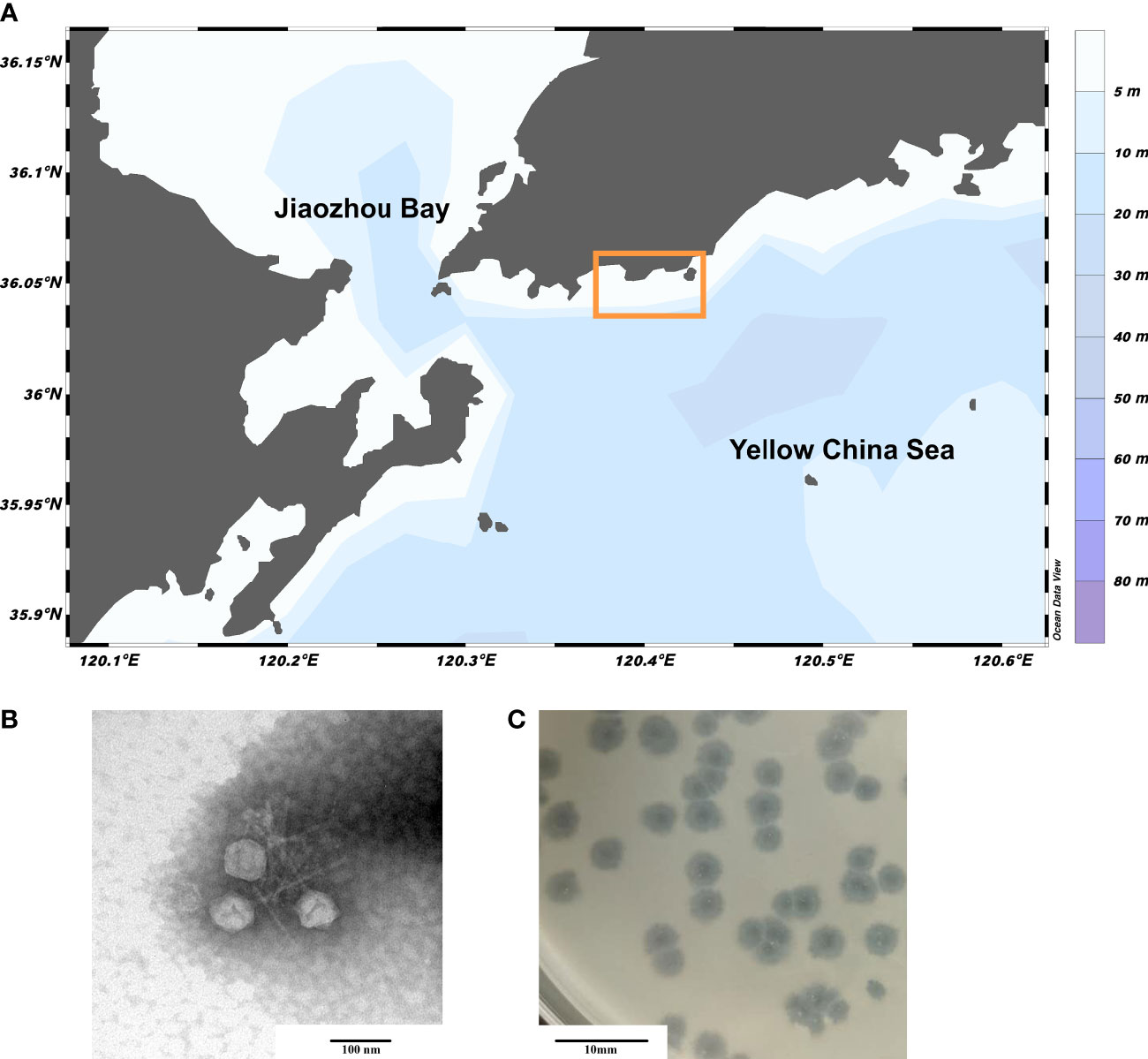
Figure 1 Sampling area of Qingdao and Morphology of phage vB_PhoS_XC. (A) Sampling region bounded by the yellow box. (B) Transmission electron micrograph (TEM) of Pseudoalteromonas phage vB_PhoS_XC. The scale bar is 100 nm. (C) The phage plaques of Pseudoalteromonas phage vB_PhoS_XC. The scale bar is 10 mm.
The phage vB_PhoS_XC demonstrated a stable titer across a range of pH levels, from 4 to 10. While it could withstand environments of slightly lower (pH3) and higher (pH10-12) acidity, damage did occur in both cases (Figure 2A). Moreover, the phage displayed a more resistance to acidic environments over alkaline. In addition, thermal stability was observed from -20°C to 45°C, although a rapid decrease in phage titer occurred at temperatures of 55°C or higher, with a maximum tolerance of 65°C (Figure 2B). Overall, the phage vB_PhoS_XC displayed a wide range of tolerance to pH and temperature.
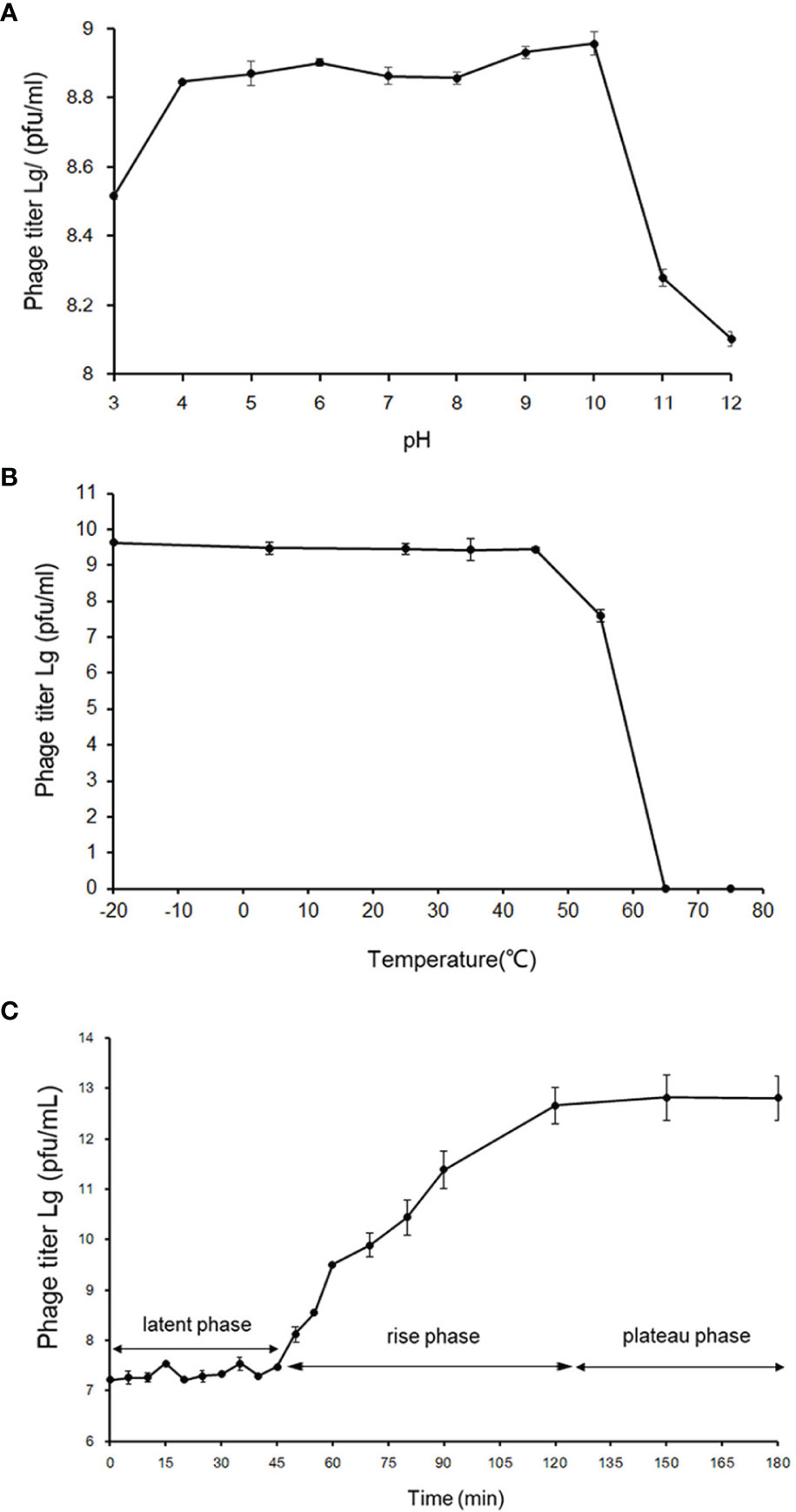
Figure 2 Biological properties of phage vB_PhoS_XC. (A) pH stability of Pseudoalteromonas phage vB_PhoS_XC. Y-axis shows the log of plaque-forming units per milliliter (PFU/mL). (B) Temperature stability of Pseudoalteromonas phage vB_PhoS_XC. Y-axis shows the log of plaque-forming units per milliliter (PFU/mL). (C) One-step growth curve of Pseudoalteromonas phage vB_PhoS_XC. Y-axis shows the log of plaque-forming units per milliliter (PFU/mL).
The one-step growth curve of phage vB_Phos_XC demonstrated a latent period of approximately 45 minutes, reaching a plateau stage after 120 minutes (Figure 2C). The final count of released phage particles was 5.0×1012 PFU/mL, indicating a burst size of approximately 241 virions per cell. This suggests that VB_PHOS_XC has a lytic activity and a relatively short replication cycle (Yuan et al., 2022).
The genome of the phage vB_PhoS_XC consisted a double-stranded DNA (dsDNA) molecule with a GC content of 40.0% and a total length of 46,490 bp (Figure 3). The genome contained 72 open reading frames (ORFs) and one tRNA. Of the ORFs, 37 had no homologous proteins in current public databases, and 35 had known functions, which were grouped into three functional modules: DNA replication and nucleotide metabolism, phage packing and lysis, and phage structure (Table 2).In these 35 known genes, two auxiliary metabolic genes (AMGs) were also predicted to be associated with its host (ORF 11 and ORF 51).
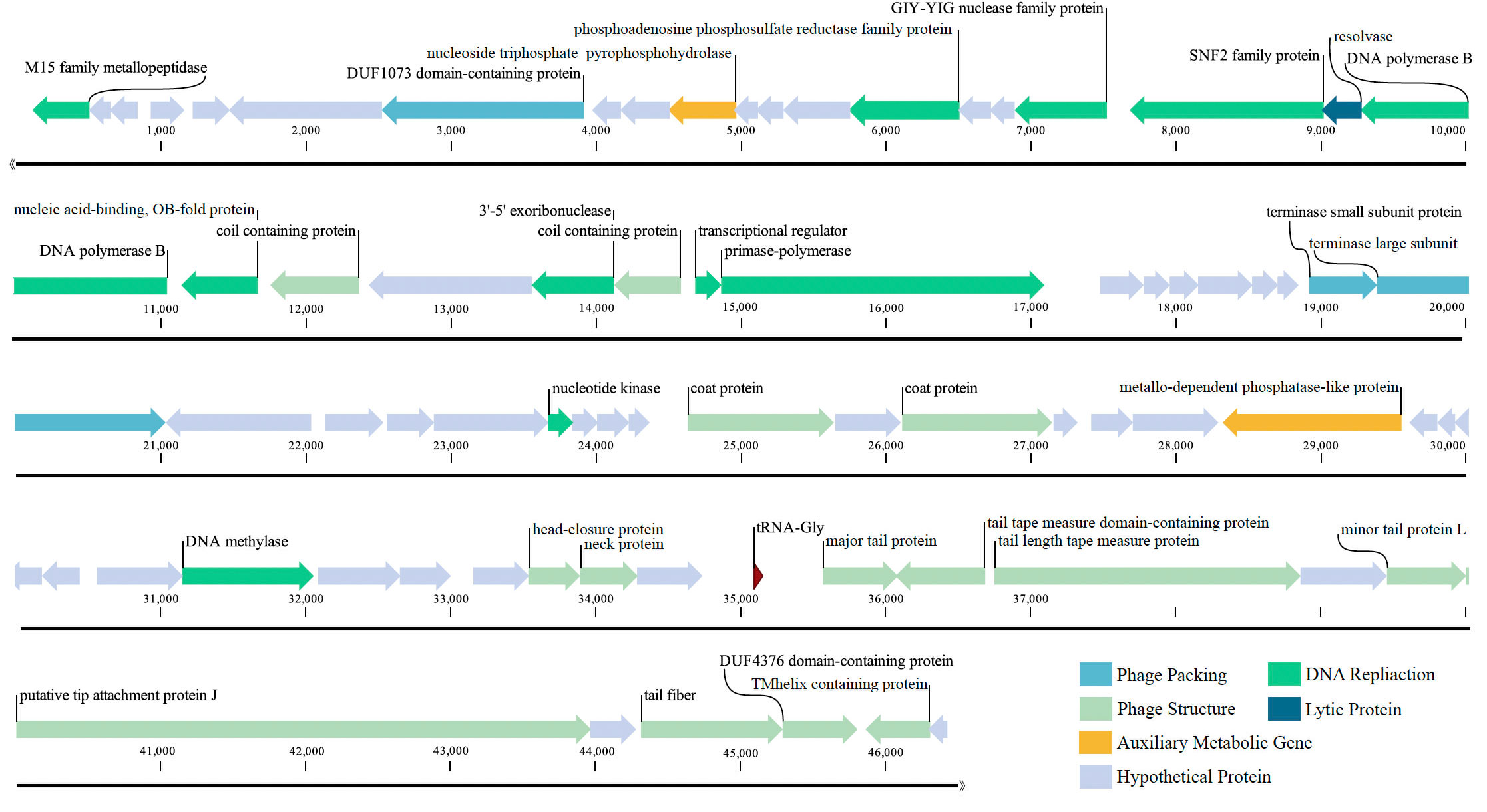
Figure 3 Genome map of Pseudoalteromonas phage vB_PhoS_XC. Putative functional categories were defined according to annotation and are represented by different colors. The length of each arrow represents the length of each gene.
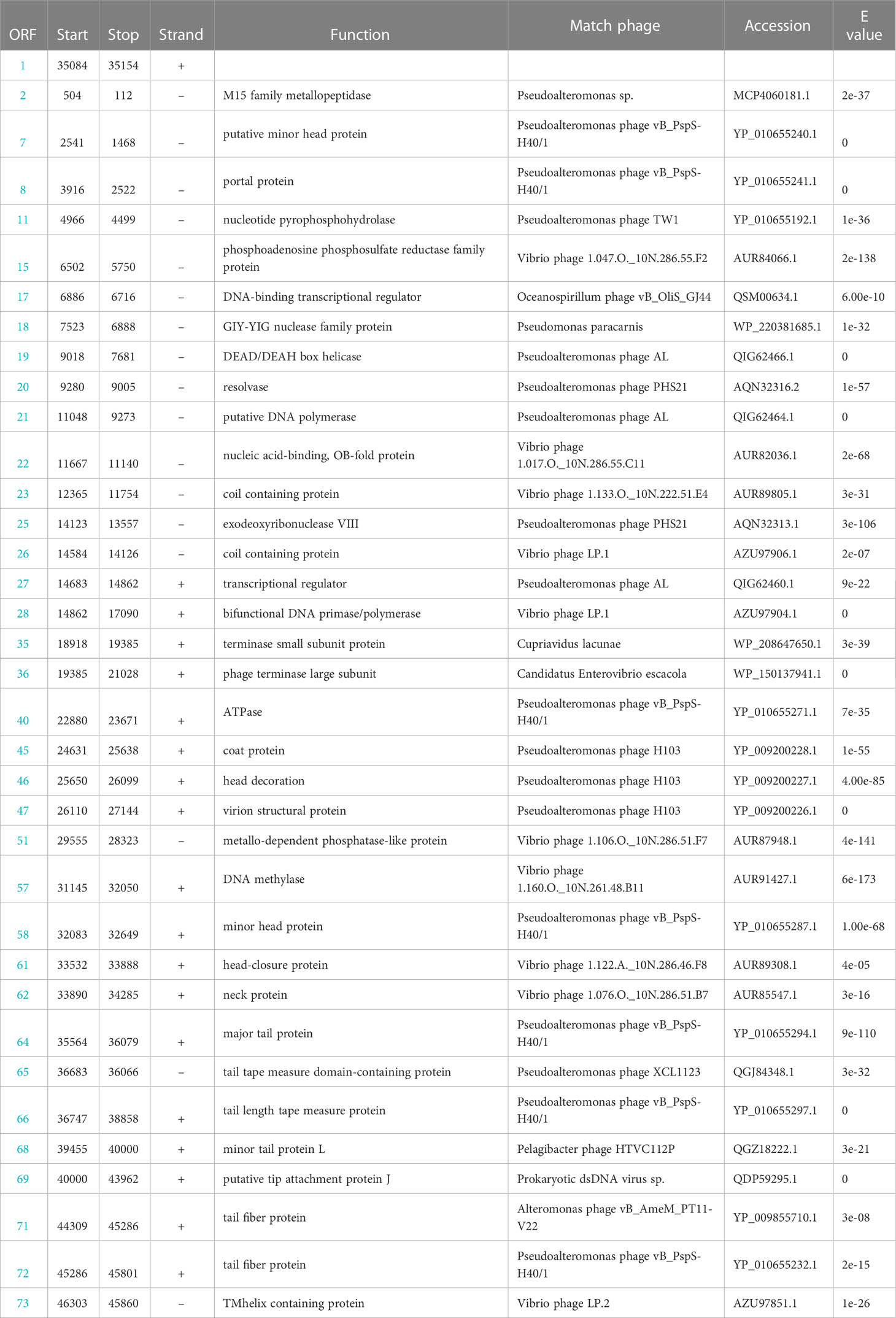
Table 2 The conserved domains information of Pseudoalteromonas phage vB_PhoS_XC genome.
Most of the ORFs associated with the phage structure in the VB_PHOS_XC genome. In addition to common structure proteins of Pseudoalteromonas phage, the genome contained two unique ORFs associated with Siphovirus structure proteins, namely Tail Tape Measure Proteins (TMPs; ORF 65 and ORF 66). This family of proteins is characterized by short repeats and its function is related to the assembly of the phage, as it participates in determining tail length (Pedersen et al., 2000). TMPs are also known to assist in phage genome delivery into the host cell cytoplasm by interacting with a cognate receptor on the cells surface (Mahony et al., 2016). Studies on the mechanisms of host infection by the Myoviridae phage T4 and Siphoviridae phage T5 have revealed the role of TMP in forming a transmembrane channel that allows the passage of phage DNA into the cell cytoplasm (Boulanger et al., 2008; Hu et al., 2015). Moreover, it has been found that the TMP is a multifunction protein responsible for both tail length determination and viral genome delivery. Consequently, the bacteriophage vB_PhoS_XC containing a TMPs may be used as a model to further investigate the mechanism of Pseudoalternomonas infection. Genes related to the packing of the genome in the vB_PhoS_XC were identified. The terminase, composed of two unique subunits (TerS and TerL), was found to be responsible for this process (Esterman et al., 2021). TerS (ORF 35) was found to be responsible for recognizing and binding to the packaging initiation site, as well as regulating the ATPase activity of the TerL (Dixit et al., 2019; Lokareddy et al., 2022b). Additionally, TerL (ORF 36) acted as an ATP-driven molecular motor that transferred viral DNA into the capsid regions, and as an endonuclease that cut the viral genome from the concatemer (Esterman et al., 2021; Lokareddy et al., 2022a). Finally, these two genes were docked on the portal protein (ORF 8) to facilitate the generation of circular phage heads (Al-Zahrani et al., 2009).
Analysis of the vB_PhoS_XC genome revealed the presence of genes related to some important metabolic processes. ORF 2 was predicted to encode a member of the M15 family metallopeptidase, which is involved in bacterial cell wall biosynthesis and metabolism (Rawlings and Barrett, 2004). Additionally, ORF 15 was observed to encode a phosphoadenosine phosphosulfate (PAPS) reductase enzyme, a member of sulfonucleotide reductases (SRs) that catalyzes the reduction of adenylated sulfate to sulfite and an essential part of the cysteine biosynthesis pathway (Chartron et al., 2007). PAPS reductase was an important enzyme found in many organisms and played a crucial role in bacteria as it is involved in the metabolism of sulfur-containing compounds such as cysteine and methionine (Kredich and Tomkins, 1966, Chartron et al., 2007). PAPS reductase regulates these compounds by reducing PAPS to 3’-phosphoadenosine-5’-phosphate (PAP). This process involves NADPH (nicotinamide adenine dinucleotide phosphate) as a cofactor and helps maintain proper levels of sulfur-containing compounds within bacterial cells (Murray-Rust et al., 2001).
ORF 17 encodes a DNA-binding transcriptional regulator, which regulates gene transcription by recognizing promoters and enhancers near the transcription initiation site (Wang et al., 2013). ORF 18 is a protein with the catalytic structural domain of GIY-YIG, used to catalyze phosphodiester bond breaking, DNA repair, recombination and genetic element transfer (Truglio et al., 2005; Dunin-Horkawicz et al., 2006). ORF 19 is a DEAD/DEAH box helicase belonging to the family of SvB_ValS_NF2, containing sequence motifs similar to those of DNA helicase proteins; involved in various aspects of transcriptional regulation, recombination and DNA repair (Xu et al., 2021). Additionally, Exodeoxyribonuclease VIII (ORF 25) acts to break dsDNA on both 5’ ends, allowing kinked and abnormal portions of the genome to be straightened and repaired through homologous recombination. Interestingly, Exodeoxyribonuclease VIII facilitates genomic repair even in low energy environments and remains stable (Sharma et al., 2019; Kumar et al., 2021).
ORF 28 encodes a bifunctional DNA primase-polymerase, which is widespread in diverse bacteria, archaea, and viruses (Lipps et al., 2003; Geibel et al., 2009; Halgasova et al., 2012). This enzyme has the capacity to act as a primase and a polymerase, and can synthesize long DNA from dNTPs without a preexisting primer bound to the DNA template (Zhu et al., 2017). ORF 57 is predicted to encode a DNA methylase that modifies the DNA of the phage, ultimately avoiding degradation of enzymes from the host cell, as well as protecting phage marked with methylation from its own nucleases which degrade host DNA during the early period of infection (Aravind et al., 2013). Therefore, it may play an important role in the lytic life cycle of the phage (Aravind et al., 2013). The phage vB_PhoS_XC has a relatively well-developed functional composition of the genome. In addition to structural, packing and metabolic genes, some other genes also contribute to its independent survival strategy, such as DNA polymerase (ORF 21), Nucleic acid-binding (ORF22), transcriptional regulator (ORF 27) and ATPase (ORF 40) and most nucleotide metabolism genes.
Two host-derived auxiliary metabolic genes (AMGs) were identified in the genome of phage vB_PhoS_XC: a nucleotide pyrophosphohydrolase gene (ORF 11) and a metallo-dependent phosphatase-like protein gene (ORF 51). These genes were found to contribute to enhanced viral replication through regulation of the host metabolism (Hurwitz and U'Ren, 2016).
ORF 11 encodes nucleotide pyrophosphohydrolase (NTPPHase), a protein found in many marine bacteriophages, suggesting it has an important role in phage proliferation (Bryan et al., 2008; Duhaime et al., 2011). Studies on bacteria have shown NTPPHase is involved in stress responses and removal of noncanonical nucleotides (Lu et al., 2010; Gonçalves et al., 2011), potentially improving the survival of its host in nutrient-depleted environments (Duhaime et al., 2011; Kang et al., 2013; Wang et al., 2021). In addition, ORF 51 encodes a metallo-dependent phosphatase-like protein (PPM). This protein was highly conserved in its sequence and structure with homologous genes identified from prokaryotes, animals, and plants (Kamada et al., 2020). It also regulates the activity of other proteins involved in glycolysis and amino acid biosynthesis (Kamada et al., 2020). Evidence also indicates that PPM has an important role in signaling processes, such as proliferation, apoptosis and metabolism (Tamura et al., 2006; Lammers and Lavi, 2007, Lu and Wang, 2008). As the viral AMG, it is particularly crucial for bacterial survival and growth under certain conditions (Aravind and Koonin, 1998). Furthermore, its homologs were also found to facilitate viral replication by manipulating host cell metabolism (Wang et al., 2021).
The temporal abundance of the phage vB_PhoS_XC was characterized in several viral metagenome data from the coastal seawaters of Qingdao in 2020 year. After analysis of viral community structure in each viral metagenome and together with other common ocean bacteriophages. The results revealed that the relative abundance of Pelagibacter phages was consistently high because it was the most abundance phages in the ocean (Zhao et al., 2019), while the relative abundance of Pseudoalternomonas vB_PhoS_XC phage increased in July (Figure 4). This finding correlated with an observed outbreak and extinction process of U. prolifera green tide in the seawater region of Qingdao in the same time period (Figure 5A). This suggests that the increase in availability of Pseudoalternomonas species likely resulted in the corresponding increase in abundance of its specific phage vB_PhoS_XC. These results demonstrate the dynamic relationship between prokaryotic and eukaryotic components of the coastal ecological system. The abundance of this phage significantly increased might be with the accumulation of high amounts of algal-derived DOM after the bloom arrival (Han et al., 2023). By the end of the bloom termination stage, most of the labile DOM had been utilized by bacteria, it might result the decrease of viral abundance like phage vB_PhoS_XC. Although there was no obvious correlation between the phage vB_PhoS_XC and environment factors (Supplementary Figure S2), further implied that there might be a complex interaction between the phage vB_PhoS_XC and its host.
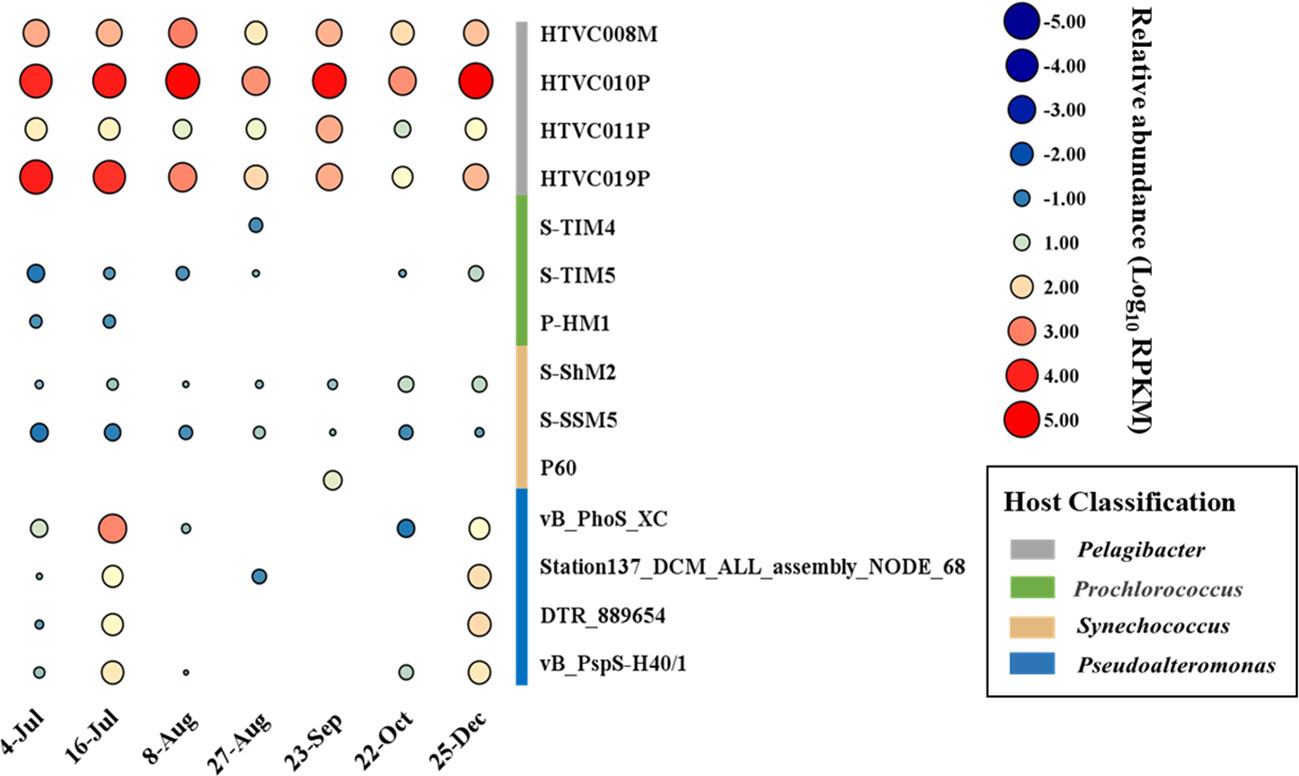
Figure 4 Temporal changes of Pseudoalteromonas phage vB_PhoS_XC. Bubble size shows relative abundance on Pseudoalteromonas phage vB_PhoS_XC of different time series, expressed by RPKM (reads per kilobase per million mapped reads) values.
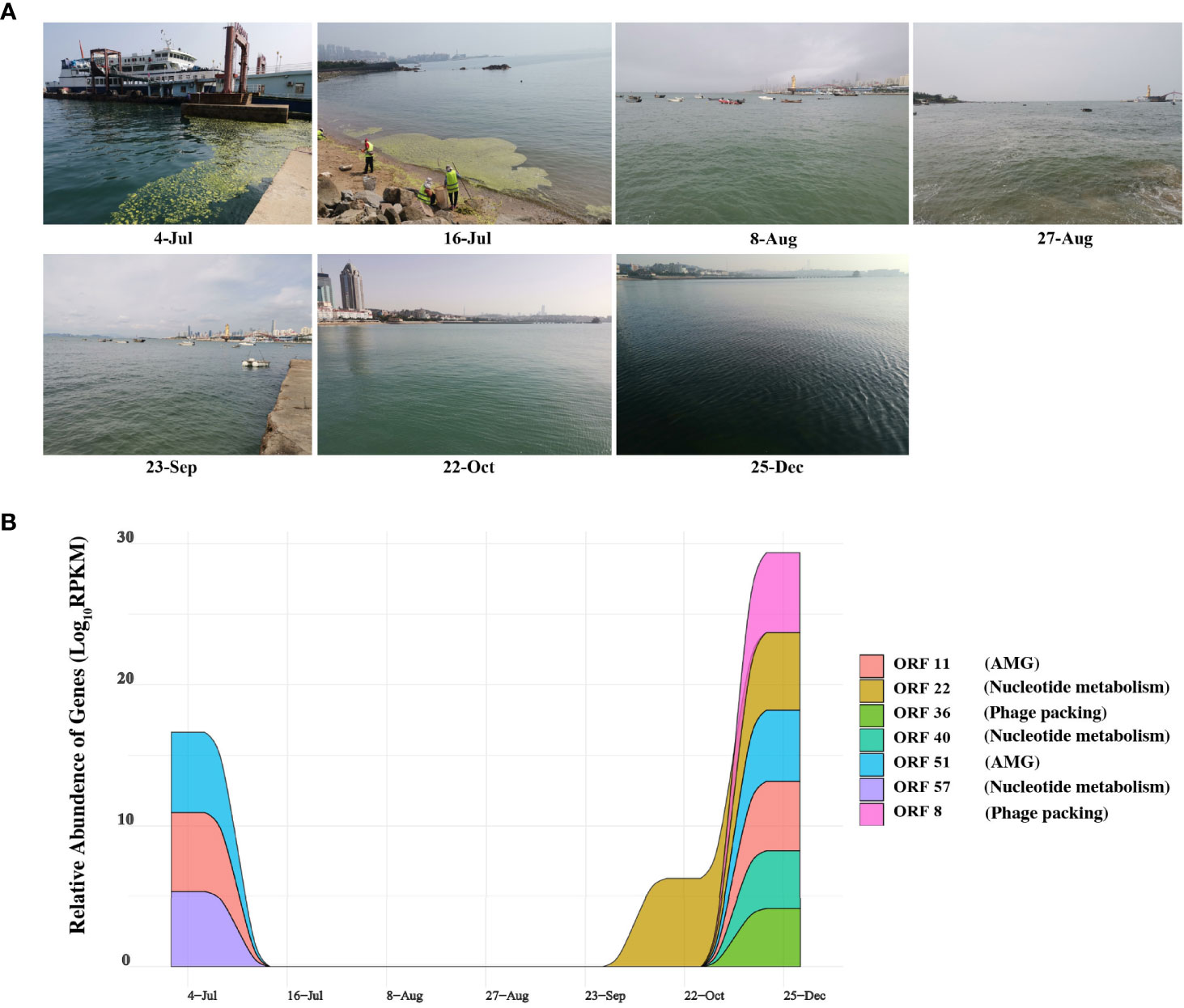
Figure 5 Expression of genes and sampling scene pictures. (A) The real scene pictures of different sample taken time. (B) Auxiliary metabolic genes (AMGs) and nucleotide metabolic Genes with Temporal changes. Colored area shows the relative abundance on the ORFs encoding AMGs and nucleotide metabolic of different time series, expressed by RPKM (reads per kilobase per million mapped reads) values.
Thus, we also analyzed the expression of AMGs and other functional genes in the corresponding metatranscriptome data to explore the correlation between the phage vB_PhoS_XC and its host. Figure 5B demonstrates that ORF 11 and ORF 51 and ORF 57 (AMGs or accessory metabolic genes) had a high relative abundance in the phage on July 4th and December 25th (Figure 5B). ORF 11 is largely involved in proliferation, stress response, and increasing the survival rate of the host. This may assist the host cells in preventing programmed cell death resulting from rapid changes in abiotic factors caused by U. prolifera blooms. ORF 51 regulates the activity of proteins associated with a variety of metabolic pathways. Every year from June to July, a large-scale U. prolifera bloom typically occurs in Qingdao. This bloom rapidly causes a decline in inorganic carbon, increasing the pH of the seawater (Han et al., 2023). When the bloom terminates, pH, DOM, DOC, and nutrients are significantly altered to an extent that disrupts the dynamic balance of bacterial community structure and function (Li H et al., 2016; Zhang and Wang, 2017; Chen et al., 2020; Liang et al., 2021). Environmental changes result in the increased expression of these AMGs, which helps the host to survive and maximizes their own replication.
Interestingly, the relative abundance of some genes had a higher expression in December (ORF 22, ORF 36, ORF 40, ORF57 and ORF 8) (Figure 5B). At this time, concentration of nutrients is poor, the water temperature is low and the abundance of bacterioplankton are significantly reduced (Uyà et al., 2017; Guo et al., 2020; Zhao et al., 2022). We speculated that during this period the phage vB_PhoS_XC is manipulating the host through AMGs to improve metabolic efficiency and survival. It may also improve its replication efficiency by repacking genes and increasing nucleotide metabolism, helping create a “virus factory” in the host cells to maximize the output when favorable conditions return.
Isolating and culturing viruses that infect bacteria provides a new and valuable insight into novel viral sequences from metagenomic and metatranscriptomic data. Few Pseudoalteromonas phages have been isolated from the bacterial hosts and research on them is limited. This study aimed to isolate the phage vB_PhoS_XC from coastal seawaters of Qingdao during a U. prolifera bloom. Results indicate that the phage has a short latent period and a large burst size, as well as being active in a wide range of environmental conditions. Additionally, abundance of the phage was observed to be higher during U. prolifera blooms and winter, two AMGs and nucleotide metabolism system were also expressed a higher abundance in these periods. This suggests that the phage may be able to improve the host’s survival rate and enhance its metabolic efficiency to improve its own replication. Consequently, further research will enable a better understanding of the role of phages in marine ecosystems, as well as their interactions with their hosts.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.cncb.ac.cn, GSA: CRA010505.
YTL, AM and MW: conceptualization, revision, project administration, supervision and funding acquisition. JS, XZ, KZ, MH, GL, YDL and FK: methodology, formal analysis, writing, and original draft preparation. JS, XZ and GL: sample collection and expedition organization. YS, WM, LW and FK: review and editing. All authors contributed to the article and approved the submitted version.
This work was supported by the Laoshan Laboratory (No. LSKJ202203201), National Key Research and Development Program of China (2022YFC2807500), Natural Science Foundation of China (No. 42120104006, 41976117 and 42176111), and the Fundamental Research Funds for the Central Universities (202172002, 201812002, 202072001 and AM).
We sincerely appreciate the Center for High Performance Computing and System Simulation, Pilot National Laboratory for Marine Science and Technology (Qingdao). We appreciate the computing resources provided by IEMB-1, a high-performance computing cluster operated by the Institute of Evolution and Marine Biodiversity, and Marine Big Data Center of Institute for the Advanced Ocean Study of the Ocean University of China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1201434/full#supplementary-material
Supplementary Figure 1 | Phylogenetic tree of the Pseudoalteromonas genus.
Supplementary Figure 2 | Pearson correlation coefficients among the phage vB_PhoS_XC, Picoplankton (Pico), Synechococcus (Syn), Bacterioplankton (Bac), NH4: Ammonium (NH3-N), Nitrite (NO2-N), Phoshate (PO43-), pH value, Nitrate (NO3-N), Salinity and Dissolved Oxygen (DO). *Correlation is significant at the 0.05 level, ** Correlation is significant at the 0.01 level, *** Correlation is significant at the 0.001 level.
Supplementary Table 1 | Homologous genes of auxiliary metabolic genes (AMGs).
Supplementary Table 2 | The sampling environmental parameters.
filter_no_single.pl.zip | The perl script can be used for removing N from the data.
filter_no_single2030.pl.zip | The perl script can be used for removing less than 20% and 30% bases with a quality score.
Akter L., Ullah M. A., Hossain M. B., Karmaker A. R., Hossain M. S., Albeshr M. F., et al. (2022). Diversity and assemblage of harmful algae in homestead fish ponds in a tropical coastal area. Biol. (Basel) 11 (9), 1335. doi: 10.3390/biology11091335
Al-Zahrani A. S., Kondabagil K., Gao S., Kelly N., Ghosh-Kumar M., Rao V. B. (2009). The small terminase, gp16, of bacteriophage T4 is a regulator of the DNA packaging motor. J. Biol. Chem. 284 (36), 24490–24500. doi: 10.1074/jbc.M109.025007
Aravind L., Anand S., Iyer L. M. (2013). Novel autoproteolytic and DNA-damage sensing components in the bacterial SOS response and oxidized methylcytosine-induced eukaryotic DNA demethylation systems. Biol. Direct 8, 20. doi: 10.1186/1745-6150-8-20
Aravind L., Koonin E. V. (1998). The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 23 (12), 469–472. doi: 10.1016/S0968-0004(98)01293-6
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Boulanger P., Jacquot P., Plançon L., Chami M., Engel A., Parquet C., et al. (2008). Phage T5 straight tail fiber is a multifunctional protein acting as a tape measure and carrying fusogenic and muralytic activities. J. Biol. Chem. 283 (20), 13556–13564. doi: 10.1074/jbc.M800052200
Bowman J. P. (2007). Bioactive compound synthetic capacity and ecological significance of marine bacterial genus pseudoalteromonas. Mar. Drugs 5 (4), 220–241. doi: 10.3390/md504220
Bryan M. J., Burroughs N. J., Spence E. M., Clokie M. R., Mann N. H., Bryan S. J. (2008). Evidence for the intense exchange of MazG in marine cyanophages by horizontal gene transfer. PloS One 3 (4), e2048. doi: 10.1371/journal.pone.0002048
Chan P. P., Lin B. Y., Mak A. J., Lowe T. M. (2021). tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49 (16), 9077–9096.
Chartron J., Shiau C., Stout C. D., Carroll K. S. (2007). 3'-Phosphoadenosine-5'-phosphosulfate reductase in complex with thioredoxin: a structural snapshot in the catalytic cycle. Biochemistry 46 (13), 3942–3951. doi: 10.1021/bi700130e
Chen J., Li H., Zhang Z., He C., Shi Q., Jiao N., et al. (2020). DOC dynamics and bacterial community succession during long-term degradation of ulva prolifera and their implications for the legacy effect of green tides on refractory DOC pool in seawater. Water Res. 185, 116268. doi: 10.1016/j.watres.2020.116268
Chen L. X., Zhao Y., McMahon K. D., Mori J. F., Jessen G. L., Nelson T. C., et al. (2019). Wide distribution of phage that infect freshwater SAR11 bacteria. mSystems 4(5), e00410-19. doi: 10.1128/mSystems.00410-19
Culley A. I., Lang A. S., Suttle C. A. (2006). Metagenomic analysis of coastal RNA virus communities. Science 312 (5781), 1795–1798.
Dixit A. B., Ray K., Black L. W. (2019). A viral small terminase subunit (TerS) twin ring pac synapsis DNA packaging model is supported by fluorescent fusion proteins. Virology 536, 39–48. doi: 10.1016/j.virol.2019.07.021
Dixon P. (2003). VEGAN, a package of r functions for community ecology. J. Vegetation Sci. 14 (6), 927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x
Duhaime M. B., Wichels A., Waldmann J., Teeling H., Glöckner F. O. (2011). Ecogenomics and genome landscapes of marine pseudoalteromonas phage H105/1. Isme J. 5 (1), 107–121. doi: 10.1038/ismej.2010.94
Dunin-Horkawicz S., Feder M., Bujnicki J. M. (2006). Phylogenomic analysis of the GIY-YIG nuclease superfamily. BMC Genomics 7, 98. doi: 10.1186/1471-2164-7-98
Esterman E. S., Wolf Y. I., Kogay R., Koonin E. V., Zhaxybayeva O. (2021). Evolution of DNA packaging in gene transfer agents. Virus Evol. 7 (1), veab015. doi: 10.1093/ve/veab015
Fuhrman J. A. (1999). Marine viruses and their biogeochemical and ecological effects. Nature 399 (6736), 541–548.
Geibel S., Banchenko S., Engel M., Lanka E., Saenger W. (2009). Structure and function of primase RepB′ encoded by broad-host-range plasmid RSF1010 that replicates exclusively in leading-strand mode. Proc. Natl. Acad. Sci. 106 (19), 7810–7815. doi: 10.1073/pnas.0902910106
Gonçalves A. M. D., de Sanctis D., McSweeney S. M. (2011). Structural and functional insights into DR2231 protein, the MazG-like nucleoside triphosphate pyrophosphohydrolase from deinococcus radiodurans. J. Biol. Chem. 286 (35), 30691–30705. doi: 10.1074/jbc.M111.247999
Gong Z., Wang M., Yang Q., Li Z., Xia J., Gao Y., et al. (2017). Isolation and complete genome sequence of a novel pseudoalteromonas phage PH357 from the Yangtze river estuary. Curr. Microbiol. 74 (7), 832–839. doi: 10.1007/s00284-017-1244-8
Gregory A. C., Zayed A. A., Conceição-Neto N., Temperton B., Bolduc B., Alberti A., et al. (2019). Marine DNA viral macro- and microdiversity from pole to pole. Cell 177 (5), 1109–1123.e1114. doi: 10.1016/j.cell.2019.03.040
Guo J., Yuan H., Song J., Li X., Duan L. (2020). Hypoxia, acidification and nutrient accumulation in the yellow Sea cold water of the south yellow Sea. Sci. Total Environ. 745, 141050. doi: 10.1016/j.scitotenv.2020.141050
Halgasova N., Mesarosova I., Bukovska G. (2012). Identification of a bifunctional primase-polymerase domain of corynephage BFK20 replication protein gp43. Virus Res. 163 (2), 454–460. doi: 10.1016/j.virusres.2011.11.005
Han M., Sun J., Yang Q., Liang Y., Jiang Y., Gao C., et al. (2023). Spatiotemporal dynamics of coastal viral community structure and potential biogeochemical roles affected by an ulva prolifera green tide. mSystems 8 (2), e0121122. doi: 10.1128/msystems.01211-22
Hu B., Margolin W., Molineux I. J., Liu J. (2015). Structural remodeling of bacteriophage T4 and host membranes during infection initiation. Proc. Natl. Acad. Sci. U.S.A. 112 (35), E4919–E4928. doi: 10.1073/pnas.1501064112
Hurwitz B. L., U'Ren J. M. (2016). Viral metabolic reprogramming in marine ecosystems. Curr. Opin. Microbiol. 31, 161–168. doi: 10.1016/j.mib.2016.04.002
Hyman P., Abedon S. T. (2010). Bacteriophage host range and bacterial resistance. Adv. Appl. Microbiol. 70, 217–248. doi: 10.1016/S0065-2164(10)70007-1
Ivanova E. P., Bakunina I. Y., Nedashkovskaya O. I., Gorshkova N. M., Alexeeva Y. V., Zelepuga E. A., et al. (2003). Ecophysiological variabilities in ectohydrolytic enzyme activities of some pseudoalteromonas species, p. citrea, p. issachenkonii and p. nigrifaciens. Curr. Microbiol. 46 (1), 6–10.
Jacob A., Lancaster J., Buhler J., Harris B., Chamberlain R. D. (2008). Mercury BLASTP: accelerating protein sequence alignment. ACM Trans. Reconfigurable Technol. Syst. 1 (2), 9. doi: 10.1145/1371579.1371581
Jain A., Krishnan K. P., Begum N., Singh A., Thomas F. A., Gopinath A. (2020). Response of bacterial communities from kongsfjorden (Svalbard, Arctic ocean) to macroalgal polysaccharide amendments. Mar. Environ. Res. 155, 104874. doi: 10.1016/j.marenvres.2020.104874
Jamalludeen N., Johnson R. P., Friendship R., Kropinski A. M., Lingohr E. J., Gyles C. L. (2007). Isolation and characterization of nine bacteriophages that lyse O149 enterotoxigenic escherichia coli. Vet. Microbiol. 124 (1-2), 47–57. doi: 10.1016/j.vetmic.2007.03.028
Kamada R., Kudoh F., Ito S., Tani I., Janairo J. I. B., Omichinski J. G., et al. (2020). Metal-dependent Ser/Thr protein phosphatase PPM family: evolution, structures, diseases and inhibitors. Pharmacol. Ther. 215, 107622. doi: 10.1016/j.pharmthera.2020.107622
Kang I., Oh H. M., Kang D., Cho J. C. (2013). Genome of a SAR116 bacteriophage shows the prevalence of this phage type in the oceans. Proc. Natl. Acad. Sci. U.S.A. 110 (30), 12343–12348. doi: 10.1073/pnas.1219930110
Katoh K., Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780. doi: 10.1093/molbev/mst010
Kechin A., Boyarskikh U., Kel A., Filipenko M. (2017). cutPrimers: a new tool for accurate cutting of primers from reads of targeted next generation sequencing. J. Comput. Biol. 24 (11), 1138–1143. doi: 10.1089/cmb.2017.0096
Kopylova E., Noé L., Touzet H. (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28 (24), 3211–3217. doi: 10.1093/bioinformatics/bts611
Kredich N. M., Tomkins G. M. (1966). The enzymic synthesis of l-cysteine in escherichia coli and salmonella typhimurium. J. Biol. Chem. 241 (21), 4955–4965. doi: 10.1016/S0021-9258(18)99657-2
Kumar P., Meghvansi M. K., Kamboj D. V. (2021). "Phenotypic characterization and whole-genome analysis of a novel bacteriophage HCF1 infecting citrobacter amalonaticus and c. freundii." Front. Microbiol. 12, 644013. doi: 10.3389/fmicb.2021.644013
Labrie S. J., Frois-Moniz K., Osburne M. S., Kelly L., Roggensack S. E., Sullivan M. B., et al. (2013). Genomes of marine cyanopodoviruses reveal multiple origins of diversity. Environ. Microbiol. 15 (5), 1356–1376. doi: 10.1111/1462-2920.12053
Lammers T., Lavi S. (2007). Role of type 2C protein phosphatases in growth regulation and in cellular stress signaling. Crit. Rev. Biochem. Mol. Biol. 42 (6), 437–461. doi: 10.1080/10409230701693342
Li Y., Wang M., Liu Q., Song X., Wang D., Ma Y., et al. (2016). Complete genomic sequence of bacteriophage H188: a novel vibrio kanaloae phage isolated from yellow Sea. Curr. Microbiol. 72 (5), 628–633. doi: 10.1007/s00284-015-0984-6
Li H., Zhang Y., Han X., Shi X., Rivkin R. B., Legendre L. (2016). Growth responses of ulva prolifera to inorganic and organic nutrients: implications for macroalgal blooms in the southern yellow Sea, China. Sci. Rep. 6, 26498. doi: 10.1038/srep26498
Liang J., Liu J., Zhan Y., Zhou S., Xue C. X., Sun C., et al. (2021). Succession of marine bacteria in response to ulva prolifera-derived dissolved organic matter. Environ. Int. 155, 106687. doi: 10.1016/j.envint.2021.106687
Lipps G., Röther S., Hart C., Krauss G. (2003). A novel type of replicative enzyme harbouring ATPase, primase and DNA polymerase activity. EMBO J. 22 (10), 2516–2525. doi: 10.1093/emboj/cdg246
Liu Z., Li H., Wang M., Jiang Y., Yang Q., Zhou X., et al. (2018). Isolation, characterization and genome sequencing of the novel phage SL25 from the yellow Sea, China. Mar. Genomics 37, 31–34. doi: 10.1016/j.margen.2017.09.008
Lokareddy R. K., Hou C. D., Doll S. G., Li F., Gillilan R. E., Forti F., et al. (2022a). Terminase subunits from the pseudomonas-phage E217. J. Mol. Biol. 434 (20), 167799. doi: 10.1016/j.jmb.2022.167799
Lokareddy R. K., Hou C. D., Li F., Yang R., Cingolani G. (2022b). Viral small terminase: a divergent structural framework for a conserved biological function. Viruses 14 (10), 2215. doi: 10.3390/v14102215
Lu L. D., Sun Q., Fan X. Y., Zhong Y., Yao Y. F., Zhao G. P. (2010). Mycobacterial MazG is a novel NTP pyrophosphohydrolase involved in oxidative stress response. J. Biol. Chem. 285 (36), 28076–28085. doi: 10.1074/jbc.M109.088872
Lu G., Wang Y. (2008). Functional diversity of mammalian type 2C protein phosphatase isoforms: new tales from an old family. Clin. Exp. Pharmacol. Physiol. 35 (2), 107–112. doi: 10.1111/j.1440-1681.2007.04843.x
Ma R., Lai J., Chen X., Wang L., Yang Y., Wei S., et al. (2021). A novel phage infecting alteromonas represents a distinct group of siphophages infecting diverse aquatic copiotrophs. mSphere 6 (3), e0045421. doi: 10.1128/mSphere.00454-21
Mahony J., Alqarni M., Stockdale S., Spinelli S., Feyereisen M., Cambillau C., et al. (2016). Functional and structural dissection of the tape measure protein of lactococcal phage TP901-1. Sci. Rep. 6, 36667. doi: 10.1038/srep36667
Middelboe M., Chan A., Bertelsen S. (2010). Isolation and life cycle characterization of lytic viruses infecting heterotrophic bacteria and cyanobacteria. In S. W. Wilhelm, M. G. Weinbauer, & C. A. Suttle (Eds.), Manual of aquatic viral ecology (pp. 118–133). doi: 10.4319/mave.2010.978-0-9845591-0-7.118
Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G. A., Sonnhammer E. L. L., et al. (2021). Pfam: the protein families database in 2021. Nucleic Acids Res. 49 (D1), D412–d419. doi: 10.1093/nar/gkaa913
Mistry J., Finn R. D., Eddy S. R., Bateman A., Punta M. (2013). Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41 (12), e121. doi: 10.1093/nar/gkt263
Murray-Rust J., Leiper J., McAlister M., Phelan J., Tilley S., Santa Maria J., et al. (2001). Structural insights into the hydrolysis of cellular nitric oxide synthase inhibitors by dimethylarginine dimethylaminohydrolase. Nat. Struct. Biol. 8 (8), 679–683. doi: 10.1038/90387
Nadalin F., Vezzi F., Policriti A. (2012). GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinf. 13 Suppl 14 (Suppl 14), S8. doi: 10.1186/1471-2105-13-S14-S8
Pedersen M., Ostergaard S., Bresciani J., Vogensen F. K. (2000). Mutational analysis of two structural genes of the temperate lactococcal bacteriophage TP901-1 involved in tail length determination and baseplate assembly. Virology 276 (2), 315–328. doi: 10.1006/viro.2000.0497
Pesant S., Not F., Picheral M., Kandels-Lewis S., Le Bescot N., Gorsky G., et al. (2015). Open science resources for the discovery and analysis of Tara oceans data. Sci. Data 2, 150023.
Qu T., Hou C., Zhao X., Zhong Y., Guan C., Lin Z., et al. (2021). Bacteria associated with ulva prolifera: a vital role in green tide formation and migration. Harmful Algae 108, 102104. doi: 10.1016/j.hal.2021.102104
Qu T., Zhao X., Hao Y., Zhong Y., Guan C., Hou C., et al. (2020). Ecological effects of ulva prolifera green tide on bacterial community structure in qingdao offshore environment. Chemosphere 244, 125477. doi: 10.1016/j.chemosphere.2019.125477
Rawlings N. D., Barrett A. J. (2004). “67 - introduction: metallopeptidases and their clans,” in Handbook of proteolytic enzymes (Second edition). Eds. Barrett A. J., Rawlings N. D., Woessner J. F. (London: Academic Press), 231–267.
Ren L., Lu Z., Xia X., Peng Y., Gong S., Song X., et al. (2022). Metagenomics reveals bacterioplankton community adaptation to long-term thermal pollution through the strategy of functional regulation in a subtropical bay. Water Res. 216, 118298. doi: 10.1016/j.watres.2022.118298
Rong J. C., Cha Q. Q., Ren X. B., Xie B. B., Song X. Y. (2018). Complete genome sequence of pseudoalteromonas espejiana DSM9414(T), an amino-acid-requiring strain from seawater. Mar. Genomics 38, 21–23. doi: 10.1016/j.margen.2017.08.006
Salazar G., Paoli L., Alberti A., Huerta-Cepas J., Ruscheweyh H. J., Cuenca M., et al. (2019). Gene expression changes and community turnover differentially shape the global ocean metatranscriptome. Cell 179 (5), 1068–1083.e1021. doi: 10.1016/j.cell.2019.10.014
Sharma R., Pielstick B. A., Bell K. A., Nieman T. B., Stubbs O. A., Yeates E. L., et al. (2019). A novel, highly related jumbo family of bacteriophages that were isolated against erwinia. Front. Microbiol. 10, 1533. doi: 10.3389/fmicb.2019.01533
Stewart F. J., Ulloa O., DeLong E. F. (2012). Microbial metatranscriptomics in a permanent marine oxygen minimum zone. Environ. Microbiol. 14 (1), 23–40. doi: 10.1111/j.1462-2920.2010.02400.x
Tamura S., Toriumi S., Saito J., Awano K., Kudo T. A., Kobayashi T. (2006). PP2C family members play key roles in regulation of cell survival and apoptosis. Cancer Sci. 97 (7), 563–567. doi: 10.1111/j.1349-7006.2006.00219.x
Truglio J. J., Rhau B., Croteau D. L., Wang L., Skorvaga M., Karakas E., et al. (2005). Structural insights into the first incision reaction during nucleotide excision repair. EMBO J. 24 (5), 885–894. doi: 10.1038/sj.emboj.7600568
Uyà M., Maggi E., Mori G., Nuccio C., Gribben P. E., Bulleri F. (2017). Carry over effects of nutrient addition on the recovery of an invasive seaweed from the winter die-back. Mar. Environ. Res. 126, 37–44. doi: 10.1016/j.marenvres.2017.02.006
Wang C., Chen Q., Zhang C., Yang J., Lu Z., Lu F., et al. (2017). Characterization of a broad host-spectrum virulent salmonella bacteriophage fmb-p1 and its application on duck meat. Virus Res. 236, 14–23. doi: 10.1016/j.virusres.2017.05.001
Wang Q., Liu Y., Liu Q., Liu X., Yang F., Wang M., et al. (2020). Isolation and complete genome of the marine pseudoalteromonas phage C7 from coastal seawater of yellow Sea, China. Curr. Microbiol. 77 (2), 279–285. doi: 10.1007/s00284-019-01815-4
Wang F., Marshall C. B., Ikura M. (2013). Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cell Mol. Life Sci. 70 (21), 3989–4008. doi: 10.1007/s00018-012-1254-4
Wang D. B., Sun M. Q., Shao H. B., Li Y., Meng X., Liu Z. Y., et al. (2015). Characterization and genome sequencing of a novel bacteriophage PH101 infecting pseudoalteromonas marina BH101 from the yellow Sea of China. Curr. Microbiol. 71 (5), 594–600. doi: 10.1007/s00284-015-0896-5
Wang Z., Zhang F., Liang Y., Zheng K., Gu C., Zhang W., et al. (2021). Genome and ecology of a novel alteromonas podovirus, ZP6, representing a new viral genus, mareflavirus. Microbiol. Spectr. 9 (2), e0046321.
Xiao J., Wang Z., Liu D., Fu M., Yuan C., Yan T. (2021). Harmful macroalgal blooms (HMBs) in china's coastal water: green and golden tides. Harmful Algae 107, 102061. doi: 10.1016/j.hal.2021.102061
Xu J., Cai Y., Ma Z., Jiang B., Liu W., Cheng J., et al. (2021). DEAD/DEAH-box helicase 5 is hijacked by an avian oncogenic herpesvirus to inhibit interferon beta production and promote viral replication. Dev. Comp. Immunol. 119, 104048. doi: 10.1016/j.dci.2021.104048
Xu M., Guo L., Gu S., Wang O., Zhang R., Peters B. A., et al. (2020). TGS-GapCloser: a fast and accurate gap closer for large genomes with low coverage of error-prone long reads. Gigascience 9 (9), giaa094. doi: 10.1093/gigascience/giaa094
Yang Q., Gao C., Jiang Y., Wang M., Zhou X., Shao H., et al. (2019). Metagenomic characterization of the viral community of the south Scotia ridge. Viruses 11 (2), 95. doi: 10.3390/v11020095
Yuan Y., Li Q., Zhang S., Gu J., Huang G., Qi Q., et al. (2022). Enhancing thermal stability and lytic activity of phage lysin PlyAB1 from acinetobacter baumannii. Biotechnol. Bioengineering 119 (10), 2731–2742. doi: 10.1002/bit.28187
Zhang Y., He P., Li H., Li G., Liu J., Jiao F., et al. (2019). Ulva prolifera green-tide outbreaks and their environmental impact in the yellow Sea, China. Natl. Sci. Rev. 6 (4), 825–838. doi: 10.1093/nsr/nwz026
Zhang X., Liang Y., Zheng K., Wang Z., Dong Y., Liu Y., et al. (2023). Characterization and genomic analysis of phage vB_ValR_NF, representing a new viral family prevalent in the ulva prolifera blooms. Front. Microbiol. 14, 1161265. doi: 10.3389/fmicb.2023.1161265
Zhang T., Wang X. (2017). Release and microbial degradation of dissolved organic matter (DOM) from the macroalgae ulva prolifera. Mar. pollut. Bull. 125 (1-2), 192–198. doi: 10.1016/j.marpolbul.2017.08.029
Zhang R., Weinbauer M. G., Qian P. Y. (2007). Viruses and flagellates sustain apparent richness and reduce biomass accumulation of bacterioplankton in coastal marine waters. Environ. Microbiol. 9 (12), 3008–3018. doi: 10.1111/j.1462-2920.2007.01410.x
Zhang L., Yin W., Wang C., Zhang A., Zhang H., Zhang T., et al. (2021). Untangling microbiota diversity and assembly patterns in the world's largest water diversion canal. Water Res. 204, 117617. doi: 10.1016/j.watres.2021.117617
Zhao G., He H., Wang H., Liang Y., Guo C., Shao H., et al. (2022). Variations in marine bacterial and archaeal communities during an ulva prolifera green tide in coastal qingdao areas. Microorganisms 10 (6), 1204. doi: 10.3390/microorganisms10061204
Zhao Y., Qin F., Zhang R., Giovannoni S. J., Zhang Z., Sun J., et al. (2019). Pelagiphages in the podoviridae family integrate into host genomes. Environ. Microbiol. 21 (6), 1989–2001. doi: 10.1111/1462-2920.14487
Zhao Y., Temperton B., Thrash J. C., Schwalbach M. S., Vergin K. L., Landry Z. C., et al. (2013). Abundant SAR11 viruses in the ocean. Nature 494(7437), 357–360.
Zheng K., Dong Y., Liang Y., Liu Y., Zhang X., Zhang W., et al. (2023). Genomic diversity and ecological distribution of marine Pseudoalteromonas phages. Mar. Life Sci. Technol 5 (2), 271–285. doi: 10.1007/s42995-022-00160-z
Zhu B., Wang L., Mitsunobu H., Lu X., Hernandez A. J., Yoshida-Takashima Y., et al. (2017). Deep-sea vent phage DNA polymerase specifically initiates DNA synthesis in the absence of primers. Proc. Natl. Acad. Sci. 114 (12), E2310–E2318. doi: 10.1073/pnas.1700280114
Keywords: bacteriophage, Pseudoalteromonas, Ulva prolifera blooms, genomic and meta-omics analysis, ecological footprint
Citation: Sun J, Zhang X, Liang Y, Zheng K, Kennedy F, Han M, Liu G, Liu Y, Wang Z, Chen X, Sung YY, Mok WJ, Wong LL, McMinn A and Wang M (2023) Abundance and ecological footprint of Pseudoalteromonas phage vB_PhoS_XC in the Ulva prolifera green tide. Front. Mar. Sci. 10:1201434. doi: 10.3389/fmars.2023.1201434
Received: 06 April 2023; Accepted: 03 July 2023;
Published: 18 July 2023.
Edited by:
Zhiyong Li, Shanghai Jiao Tong University, ChinaReviewed by:
Wang Minxiao, Chinese Academy of Sciences (CAS), ChinaCopyright © 2023 Sun, Zhang, Liang, Zheng, Kennedy, Han, Liu, Liu, Wang, Chen, Sung, Mok, Wong, McMinn and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yantao Liang, bGlhbmd5YW50YW9Ab3VjLmVkdS5jbg==; Andrew McMinn, YW5kcmV3Lm1jbWlubkB1dGFzLmVkdS5hdQ==; Min Wang, bWluZ3dhbmdAb3VjLmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.