 Juanjo Rodríguez
Juanjo Rodríguez- Department of Ecology and Environmental Sciences, Umeå University, Umeå, Sweden
Methylmercury (MeHg) formation is a concerning environmental issue described in waters and sediments from multiple aquatic ecosystems. The genetic and metabolic bases of mercury (Hg) methylation have been well described in anoxic environments, but a number of factors seem to point towards alternative pathways potentially occurring in pelagic waters under oxic conditions. Boreal aquatic ecosystems are predicted to undergo increasing concentrations of dissolved organic matter (DOM) as a result of higher terrestrial runoff induced by climate change, which may have important implications in the formation of MeHg in the water column. In this review, different Hg methylation mechanisms postulated in the literature are discussed, with particular focus on potential pathways independent of the hgcAB gene pair and occurring under oxic conditions. Potential effects of DOM on Hg methylation and MeHg bioaccumulation are examined in the context of climate in boreal aquatic ecosystems. Furthermore, the implementation of meta-omic technologies and standardized methods into field measurements and incubation experiments is discussed as a valuable tool to determine taxonomic and functional aspects of Hg methylation in oxic waters and under climate change-induced conditions.
Introduction
Mercury (Hg) is a heavy metal with no physiological role for any known life form. It can be naturally found in geological deposits and coal, but human activities have greatly enhanced the mobilisation and transport of Hg into natural environments (Streets et al., 2011; Mason et al., 2012). Anthropogenic activities, such as coal combustion, mining, and technology industries, release large quantities of mercury into the atmosphere every year (~ 2000 metric tons), which is mainly transported as elemental mercury (Hg0) by atmospheric currents (Pirrone et al., 2010; Zhang et al., 2016; Kuss et al., 2018; Ghimire et al., 2019). In the global biogeochemical cycle of mercury (Figure 1), Hg0 is oxidized by different molecules present in the atmosphere (e.g., halogen compounds, nitrate radicals or ozone) and eventually converted into divalent inorganic mercury (Hg2+), which is then deposited into aquatic and terrestrial environments (Ariya and Peterson, 2005; Kuss et al., 2018; Bowman et al., 2020). Thereafter, part of the Hg2+ is reduced and recirculated back to the atmosphere as Hg0, but, due to its high reactivity, Hg2+ tends to rapidly bind to more stable molecules present in waters and sediments, remaining in these ecosystems for long periods (Krabbenhoft and Sunderland, 2013). Once in aquatic environments, mercury is transformed and uptaken by the resident biota as methylmercury (MeHg), which is bioaccumulated and biomagnified as it is transferred up to the food chain (Boening, 2000; Ullrich et al., 2001; Jonsson et al., 2014). This organic form of mercury is known to be a potent neurotoxin with severe effects for ecosystem health. On humans, MeHg can cause multiple adverse effects, such as neurological and endocrine disorders, cardiovascular problems, or fetal death (Tsubaki and Irukayama, 1977; Grandjean et al., 2004; Tan et al., 2009).
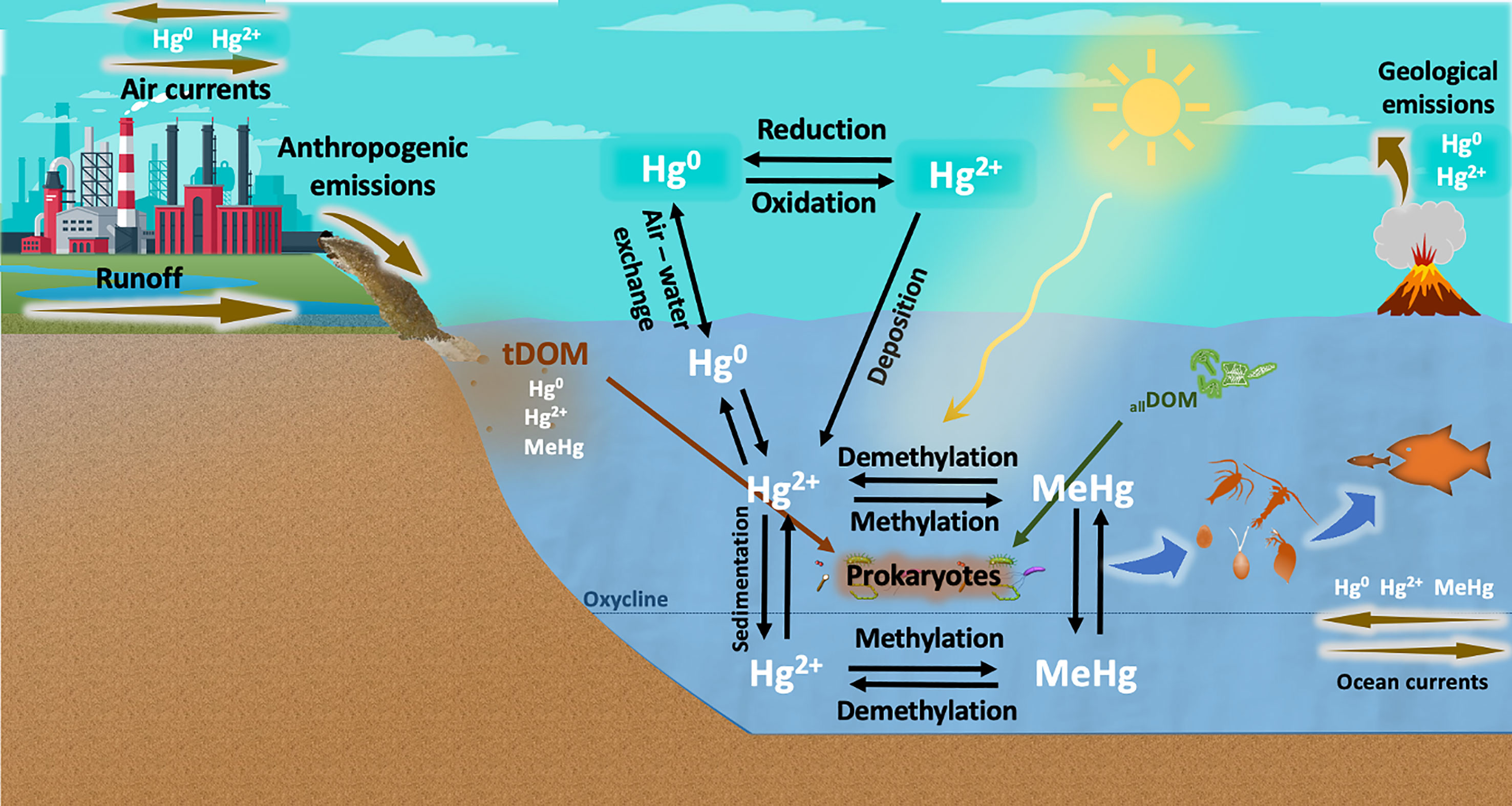
Figure 1 Geobiochemical cycle of mercury (Hg) in aquatic environments. Hg is released by anthropogenic activities and geological processes. Elemental mercury (Hg0) is transported by atmospheric currents and oxidized to divalent inorganic mercury (Hg2+), which is eventually deposited in aquatic ecosystems. Part of this Hg2+ is reduced to Hg0 and recirculated back into the atmosphere, but a substantial portion of Hg2+ is bound to DOM molecules and transformed into methylmercury (MeHg) by abiotic processes (e.g., UV radiation) and, to a greater extent, by prokaryotic communities. MeHg formation can be enhanced by DOM from terrestrial origin (tDOM) and DOM freshly produced by phytoplankton communities (allochthonous DOM, allDOM), and is then bioaccumulated up to the food web. Hg methylation has been well described to occur in anoxic layers of the water column through metabolic pathways involving hgcAB genes. However, the biotic methylation of Hg under oxic conditions is not well understood and is currently under debate.
MeHg problematic and early studies
The problematic of MeHg as environmental pollutant dates back to 1960s, where Minamata Disease was postulated to be caused by MeHg poisoning (Minamata Disease Research Group, 1966). Similarly, Westoo (1967) reported abnormally high concentrations of mercury in fish and other foodstuff in Sweden, where 80% to 100% of the mercury was present as MeHg. Considering these and other similar major public health outbreaks worldwide, the microbial participation in MeHg formation in natural aquatic ecosystems was postulated for the first time in a series of studies by Jernelov (1969), where mercury chloride was added into bottom lake sediments and MeHg was observed to be formed after 5-10 days. MeHg was not detected when the sediments were previously sterilized, which led to the conclusion that this process must involve microbial activity, although no microbial species or genes were postulated at the time (Jensen and Jernelöv, 1969; Jernelov, 1969). In addition, one year earlier, Wood et al. (1968) had shown that MeHg could be formed from methylcobalamin when an extract of a methanogenic bacterium (isolated from a canal mud) was incubated with ATP and Hg+2 under a hydrogen atmosphere (anoxic conditions).
In light of the emerging knowledge of biotic methylation as the primary source of MeHg, important progress was made by further studies identifying microbial species capable of Hg methylation, particularly in anoxic conditions. Initially, several studies reported microbial methylators under culture-based conditions, isolates and cell extracts (Wood et al., 1968; Taira, 1975; Ridley et al., 1977; Robinson and Tuovinen, 1984), which left yet unresolved the methylation of Hg by complex communities under natural conditions. Compeau and Bartha (1985) was the first comprehensive study identifying and isolating a particular bacterial species capable of Hg methylation in natural aquatic environments. They demonstrated Desulfovibrio desulfuricans capability to methylate Hg2+ in low-salinity anoxic estuarine sediments, postulating, for the first time, sulfate-reducing bacteria as major methylators in anoxic aquatic environments. Multiple later studies continued reporting new bacterial species and entire functional groups responsible for MeHg formation in natural environments, from sulfate- and iron-reducing bacteria (e.g., Geobacter sulfurreducens, Desulfuromonas palmitatis, Desulfosporosinus acidiphilus) to methanogenic Archaea (e.g., Methanolobus tindarius, Methanobrevibacter smithii, Methanoculleus bourgensis) (Kerin et al., 2006; Ranchou-Peyruse et al., 2009; Gilmour et al., 2013; Podar et al., 2015; Bravo et al., 2018; Jones et al., 2019; Capo et al., 2020b; Gionfriddo et al., 2020). However, the molecular bases (genes and enzymes) behind Hg methylation remained unclear, which implied a substantial gap towards a full understanding of this process. This gap would be in turn fulfilled with the discovery of the hgcAB gene cluster.
HgcAB pathway and the emerging paradigm shift
Since its discovery in 2013 (Parks et al., 2013), the hgcAB gene cluster has captured most of the attention in studies on Hg methylation, as hgcA and hgcB genes have been proven to be involved in the formation of the bulk of MeHg found in aquatic environments. In a series of experiments, Parks et al. (2013) demonstrated that the biotic formation of MeHg requires the presence of both hgcA and hgcB genes in the methylator’s genome. The deletion of either of these genes supresses the ability to methylate Hg through the reductive acetyl–coenzyme A pathway, but it does not hamper cell growth, which suggests that hgcA and hgcB are not essential for survival (Parks et al., 2013). HgcA has been described to encode the putative protein HgcA, which is responsible for the transfer of methyl groups to Hg2+. On the other hand, hgcB encodes a 2[4Fe-4S] ferredoxin (HgcB protein) capable of reducing a corrinoid cofactor present in HgcA, which enables the acceptance of a methyl group by HgcA. Therefore, the redox potential is an important physicochemical factor that can control Hg uptake and MeHg production by regulating the enzymatic activity of the methylators (Beckers et al., 2019; Regnell and Watras, 2019; Wang et al., 2021).
After a decade of numerous studies on the mechanism of Hg methylation, hgcAB gene cluster has been found in every known anaerobic Hg methylator and in metagenomes from multiple anoxic sediments and waters containing substantial MeHg concentrations (Paranjape and Hall, 2017; Gionfriddo et al., 2019; Ma et al., 2019; Regnell and Watras, 2019; Capo et al., 2020a). This fact has led to the use of hgcAB genes as indicators of potential Hg methylation occurring in anoxic aquatic environments. However, recent studies have reported environmentally high concentrations of MeHg in surface waters (Lehnherr et al., 2011; Kirk et al., 2012; Paranjape and Hall, 2017; Villar et al., 2020), where the concentration of oxygen would not allow Hg methylators to perform through the acetyl-CoA/hgcAB pathway. These observations were hypothesised by early studies to be caused by MeHg production in anoxic coastal sediments, which would be then transported to the overlying water column and open waters (Kraepiel et al., 2003; Hammerschmidt and Fitzgerald, 2006). However, this hypothesis conflicts with results from more recent studies showing high demethylation rates and short MeHg lifetime (Monperrus et al., 2007; Whalin et al., 2007; Lehnherr et al., 2011; Ortiz et al., 2015; Wang et al., 2020), which would prevent MeHg to remain in substantial quantities throughout such journey (Cossa et al., 2017). Furthermore, this hypothesis also contrasts with MeHg profiles reported by numerous studies, where the highest concentrations are found in subsurface layers, while deeper waters show lower concentrations (Mason and Fitzgerald, 1993; Cossa et al., 2009; Sunderland et al., 2009; Heimbürger et al., 2015; Munson et al., 2015). External sources, such as atmospheric MeHg deposition and riverine transport, could also be regarded as significant factors, but they are currently considered to be minor sources for the MeHg concentrations found in most marine pelagic environments (Mason et al., 2012; Liu et al., 2021; Wang et al., 2022).
Alternatively, in situ methylation of inorganic Hg has been suggested by numerous studies to be the main source of MeHg in the water column. In an incubation experiment, Lehnherr et al. (2011) reported water-column Hg methylation as a significant source of monomethylmercury in Arctic pelagic marine food webs, estimating this process to account for up to 47% of the total monomethylmercury found in these seawaters. Similar studies have also reported MeHg formation in oxygenated marine waters where no known anaerobic methylators or hgcAB genes were detected (Malcolm et al., 2010; Podar et al., 2015; Bowman et al., 2019; Rodríguez et al., 2022). These observations pose an apparent paradox where MeHg is produced in non-anoxic waters through a process where anaerobic methylators are considered the main players. As a result, four main explanations can be postulated (Figure 2): 1) Hg methylation by abiotic processes; 2) Biotic MeHg production within anoxic micro-environments; 3) Biotic MeHg production under oxic conditions through metabolic pathways involving hgcAB (or hgcAB-like) genes or 4) independent of hgcAB genes.
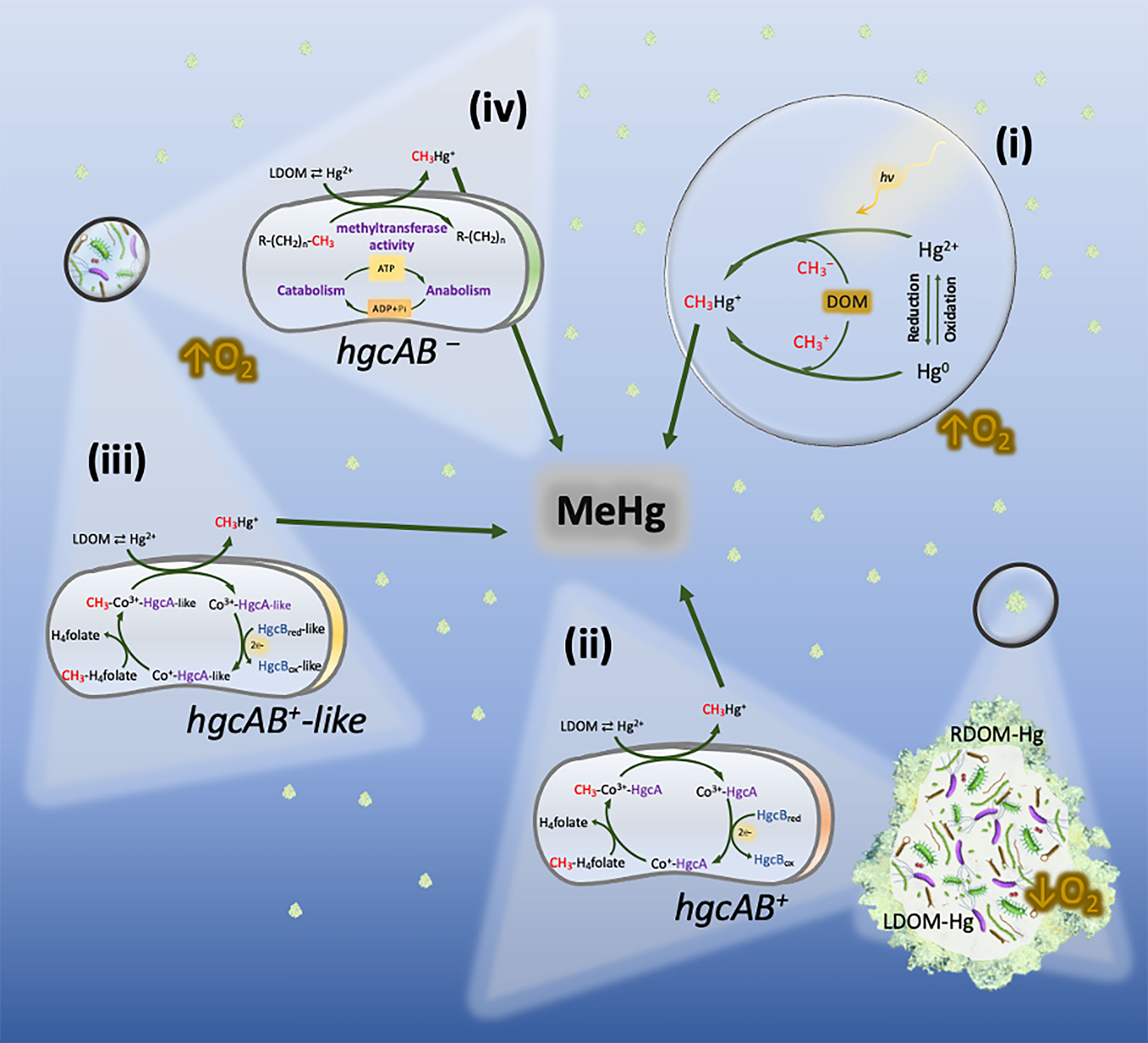
Figure 2 Hg methylation pathways potentially co-occurring in oxic layers of the water column. (i) Abiotic Hg methylation, with photochemical and non-photochemical (involving DOM molecules) processes taking place simultaneously. (ii) Biotic Hg methylation by hgcAB+ anaerobes (e.g., Shewenella, Desulfobacula, or Desulforhopalus) inhabiting settling marine snow. The formation of methylmercury (CH3Hg+) is carried out under anoxic micro-environments through the acetyl- CoA/hgcAB pathway. (iii) Hg methylation potentially carried out through metabolic pathways similar to the acetyl-CoA/hgcAB pathway by aerobic prokaryotes carrying hgcAB-like genes, such as bacteria belonging to the genus Nitrospina. (iv) Hg methylation postulated to take place under truly oxic conditions by unknown aerobic methylators. The formation of MeHg may respond to housekeeping genes which increase their overall activity when carbon, nutrients and Hg2+ are present.
The abiotic methylation of Hg has been repeatedly addressed in numerous studies (Celo et al., 2006; Li and Cai, 2013; Regnell and Watras, 2019; Chetelat et al., 2022). While photochemical and non-photochemical methylation are partially responsible for the production of MeHg in oxic waters, these abiotic mechanisms do not generally account for a substantial fraction of the MeHg found in pelagic environment (Monperrus et al., 2007; Lehnherr et al., 2011; Munson et al., 2018; Wang et al., 2022). Therefore, this review will mainly focus on the biotic mechanisms potentially occurring in oxic waters.
Anoxic micro-environments as Hg methylation hotspots in oxic waters
The existence of settling particles in the pelagic zone, enriched in organic matter and showing marked oxygen and pH gradients has been documented since the 1980s (Alldredge and Cohen, 1987; Decho, 1990; Shanks and Reeder, 1993). These particles can range in size from colloids (< 0.2 µm) to aggregates (> 300 µm). In particular, marine snow has been widely described as aggregated mixtures of large particles (> 300 µm) mostly composed by organic matter from plankton fecal pellets, living cells, detritus, and exudates (Alldredge and Silver, 1988; Turner, 2015). Alldredge and Cohen 1987 was one of the first studies reporting oxygen-depleted microenvironments within and around such particles, where microbial processes requiring low oxygen conditions, such as denitrification, may occur. Respiratory and photosynthetic activities were measured in light and dark conditions. Their results showed significant oxygen depletion (up to 45%) particularly during dark conditions, when respiration by the bacterial communities inhabiting these particles overtakes the photosynthetic production of oxygen carried out by phytoplankton communities. This respiratory activity is enhanced by the presence of organic matter and essential nutrients present in these particles, which further stimulates bacterial activity (Azam and Long, 2001). These observations were supported by later studies (Ploug et al., 1997; Tang et al., 2011; Decho and Gutierrez, 2017; Bianchi et al., 2018). In a combination of laboratory experiments and field observations, Shanks and Reeder 1993 also found oxygen-depleted conditions within marine snow particles, where sulfide was detected in concentrations far exceeding the values in the surrounding oxic waters. The production of sulfide was also attributed to microbial activity supported by the organic matter within the particles, and shed light to a similar paradox on the production of sulfide in oxygenated layers of the water column.
Marine snow particles are not the only structures where anoxic microenvironments can be formed within oxic waters. The interior of planktonic biota, particularly zooplankton, has also been described to enclose anoxic conditions where anaerobic bacteria can thrive. Glud et al. (2015) reported anoxic microenvironments within carcasses of the copepod Calanus finmarchicus in fully oxygenated seawater from Godthåbsfjord (Greenland). They detected denitrification activity by the bacterial communities naturally inhabiting the interior of these copepods, suggesting sinking C. finmarchicus carcasses as hotspots of pelagic denitrification. The gut of living zooplankton has also been reported to show anoxic conditions where different bacterial processes take place (PROCTOR, 1997; Braun et al., 1999). Tang et al. (2011) measured oxygen levels inside the guts of the copepods Calanus hyperboreusand and C. glacialis from arctic and subarctic waters. The recorded oxygen profiles indicated microbial respiration, which can deplete oxygen levels inside the guts and maintain anoxic conditions.
Considering previous studies reporting the existence of these anoxic microenvironments, the hypothesis of such microenvironments being responsible for MeHg production in oxic pelagic waters started to gain importance as an explanation for the MeHg paradox (Monperrus et al., 2007; Cossa et al., 2009; Sunderland et al., 2009; Lehnherr et al., 2011; Sonke et al., 2013; Schartup et al., 2015). Ortiz et al. (2015) was one of the first studies directly measuring the production of MeHg within settling marine particles. In a microcosm experiment, marine aggregates ranging in size (0.2 µm to > 300 µm; including marine snow) were produced from sieved estuarine seawater and spiked with MeHg (CH3199Hg) and inorganic Hg (200Hg+2). Methylation and demethylation rates were measured based on changes in isotopic composition of 199Hg and 200Hg. The results pointed to a net Hg methylation particularly in larger particles such as marine snow, which was comparable to rates found in benthic sediments. Gascón Díez et al. (2016) carried out a similar study in the largest freshwater lake in Western Europe (Lake Geneva). Over a period of two years, sediments and settling particles from this lake were collected monthly. Total Hg/MeHg concentrations and methylation rates were measured in the upper first cm of the sediments and in settling particles. Interestingly, MeHg concentrations were 10-fold higher in settling particles compared to sediments, whereas total Hg concentrations were similar in both compartments. Furthermore, while demethylation rates were similar in sediments and particles, methylation rate constants (km) were 2-fold greater in settling particles, which suggests a net MeHg formation in settling particles 10 orders of magnitudes higher than sediments. In order to determine the biological origin of this MeHg production, they amended the sediments and particles with molybdate, a known inhibitor of sulfate reducing metabolism, which resulted in a reduction of MeHg production by 80%. In a more recent study, Capo et al. (2020a) addressed this question by a genetic and phylogenetic approach. They analysed 81 metagenomes collected from Baltic Sea waters ranging in oxygen levels from normoxic (2 mL O2 L-1) to hypoxic (< 2 mL O2 L-1) and anoxic (no detectable O2). The hgcAB genes were mainly detected in settling particles (marine snow) from anoxic waters, with lower presence under hypoxic and normoxic conditions. However, higher hgcAB abundance was found in marine snow compared to filtered water with no aggregates, confirming that settling particles can be hotspots of MeHg production. In addition, a phylogenetic study revealed a Hg methylator community predominantly composed by sulfate reducing bacteria affiliated with Deltaproteobacteria.
Hg methylation under oxic conditions: is there an alternative methylation pathway?
The production of MeHg in oxic waters by anaerobic methylators (microenvironments) and, to a lesser extent, by abiotic processes may represent a substantial fraction of the total MeHg pool in certain pelagic environments, but it does not satisfactorily resolve the MeHg paradox in oxic waters where no hgcAB genes or anaerobic Hg methylator are detected. Are there Hg methylation mechanisms other than the hgcAB pathway? Or do we need higher-resolution molecular tools to detect hgcAB abundances below the current detection thresholds?
Podar et al. (2015) carried out the first comprehensive study on the distribution and prevalence of hgcAB genes and unknown Hg methylators across different environments on Earth (>3500 microbial metagenomes), including oxic pelagic waters. The hgcAB gene pair was found in every anoxic environment, whereas these genes were only detected in 7 of the 138 metagenomes from pelagic marine water columns. These observations are supported by a more recent study (Bowman et al., 2019) where hgcAB genes were screened but not detected in the upper water column (< 800 m) from Arctic Ocean seawater. These findings support the hypothesis of alternative Hg methylation pathways carried out under true oxic conditions. On the contrary, other studies have reported the presence of hgcAB-like genes in surface waters. Gionfriddo et al. (2016) identified for the first time hgcAB-like genes in two Nitrospina genomes from waters of the East Antarctic Sea. These microaerophilic bacteria were therefore proposed to play an important role as Hg methylators in pelagic waters, although the authors still referred to microenvironments, such as brine pockets and periphytic biofilms associated with settling organic matter, as the most probable niches where Hg methylation may occur. A more recent study on the distribution of the hgcAB genes and Hg methylators in the global ocean (Villar et al., 2020) detected the presence and expression of hgcAB homologues in seawaters from most of ocean basins worldwide (except in the Arctic Ocean). These genes were linked to taxonomic relatives of known Hg methylators belonging to Deltaproteobacteria, Firmicutes, Chloroflexi, and particularly Nitrospina, which was suggested to be the predominant and widespread bacteria carrying and expressing hgcAB-like genes. However, as in Gionfriddo et al. (2016), the methylating activity of Nitrospina was not tested under true oxic conditions, and the authors attributed its methylating capacity to be likely perform in oxygen-deficient microenvironments within sinking marine particles. Moreover, although hgcAB-like genes are predicted to encode corrinoid iron-sulphur and transmembrane domains distinctive of HgcA and a 4Fe-4S ferredoxin motif characteristic of HgcB, to date organisms carrying these genes have not been proven to produce MeHg (Podar et al., 2015; Gilmour et al., 2018).
To address this controversy, Rodríguez et al. (2022) carry out a microcosm experiment (37 L aquaria) where surface water from the Baltic Sea was filtered through 0.7 µm to remove sinking particles potentially creating anoxic microenvironments, while preserving the natural prokaryotic communities. The water was exposed to increased concentration of inorganic Hg (250 pM Hg+2) and an air-bubbling system ensured continuous oxygen saturation. Interestingly, MeHg production was detected above ambient levels after addition of inorganic Hg. Using 16S amplification and shotgun metagenomics, neither hgcAB genes nor known Hg methylators were detected in this study (including Nitrospina). Concentration of MeHg was highly correlated with bacterial activity, which strongly suggest the existence of an alternative Hg methylation pathway under truly oxic conditions where hgcAB gene pair is not involved.
MeHg formation and climate change
The potential magnification in MeHg formation and bioaccumulation as a consequence of climate change is currently of great concern for national and international environmental authorities (Stern et al., 2012; Krabbenhoft and Sunderland, 2013; Marnane, 2018; Chetelat et al., 2022). But what is the link between climate change and MeHg formation in boreal aquatic systems? The key factor is organic matter. Prediction models point to an increase of precipitation regimes in the Northern hemisphere as a result of climate change, which in turn will lead to higher terrestrial runoff and river inflows into boreal aquatic ecosystems (Meier et al., 2011; Andersson et al., 2015). These terrestrial inflows contain high concentrations of dissolved organic matter (DOM) swept from the catchment area, thus increasing the overall concentration of DOM in the water column. The reactive nature of organic matter to interact with different environmental pollutants has been object of study for decades, and Hg has been particularly targeted by numerous studies (Hsu-Kim et al., 2013; Ripszam et al., 2015b; Schartup et al., 2015; Alava et al., 2017; Jiang et al., 2018; Rodríguez et al., 2018). Thus, evidence of the role of DOM on Hg methylation and bioaccumulation has exponentially increased over the last years, pointing to several levels at which DOM can interact with Hg (Figure 3): 1) As a complexing agent on Hg+2 speciation (Hsu-Kim et al., 2013; Chiasson-Gould et al., 2014; Jiang et al., 2018) and providing methyl groups required for methylation (Nagase et al., 1982, 1984; Weber et al., 1985; from Wang et al., 2022); 2) by enhancing microbial metabolism (Hall et al., 2004; Paranjape and Hall, 2017); and 3) by promoting changes in the physiology and permeability of the bacterial cell membrane (Campbell et al., 1997; Vigneault et al., 2000).
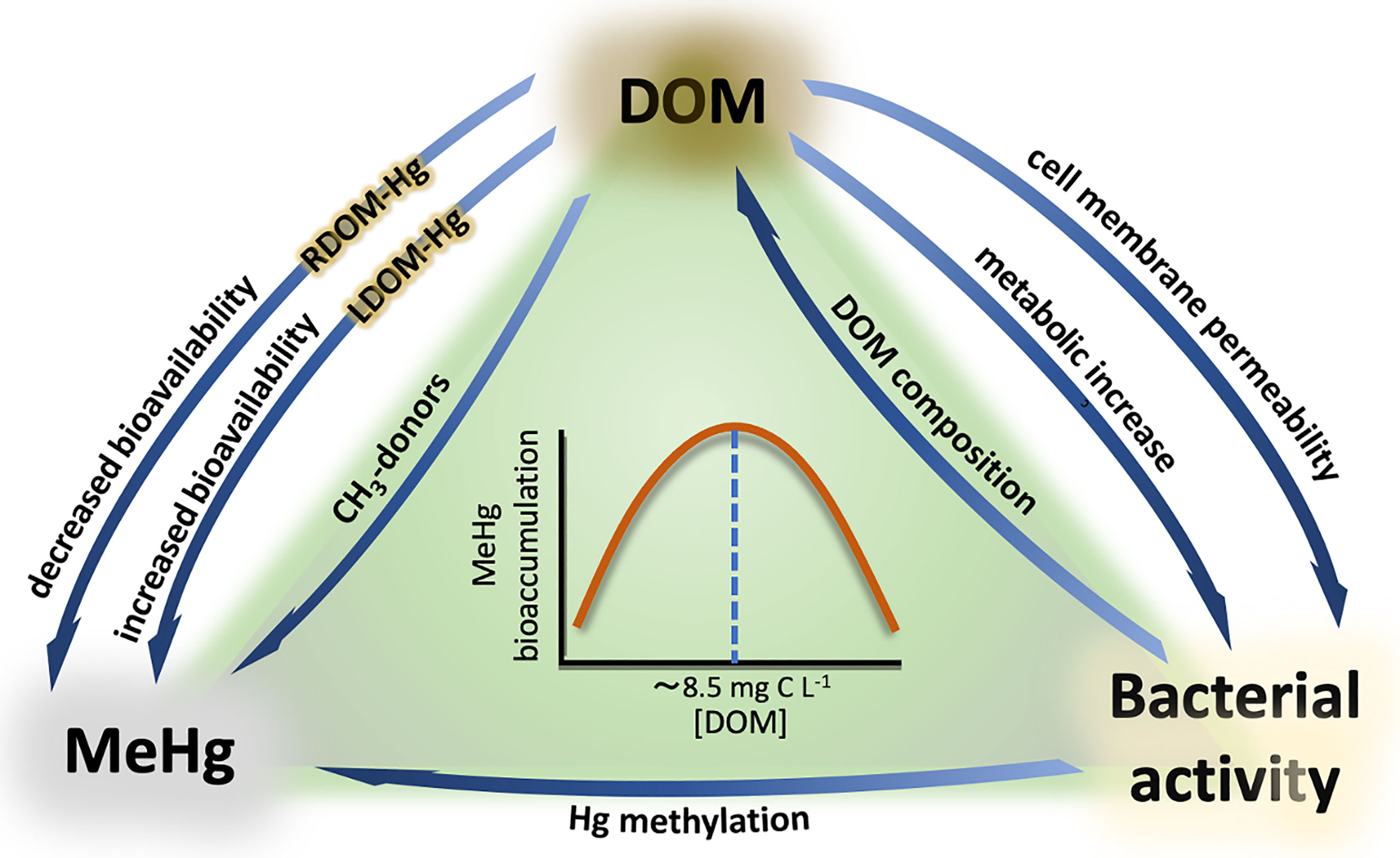
Figure 3 Interactions between DOM, MeHg, and bacterial activity. RDOM-Hg and LDOM-Hg refer to the aggregates formed by Hg and refractory compounds (RDOM) and labile compounds (LDOM). The relationship between MeHg bioaccumulation and DOM concentration is represented by a bell-shaped pattern, where MeHg bioaccumulation increases with increasing DOM concentration until a threshold (~8.5 C L-¹), from which Hg bioavailability starts to progressively decrease with increasing DOM concentration.
DOM-Hg chemical interactions
The chemical interactions between DOM and Hg largely depend on both DOM concentration and composition. Different ligands within the DOM molecular pool strongly bind to Hg, which can have a dual effect on Hg bioavailability and MeHg production. On the one hand, Hg+2 can rapidly bind to biologically labile DOM (LDOM), forming LDOM-Hg aggregates that are highly bioavailable for prokaryotic uptake (Hsu-Kim et al., 2013; Chiasson-Gould et al., 2014; Jiang et al., 2018). This higher bioavailability increases the internalization of Hg into the cytoplasm, where it is transformed into MeHg. A typical example of LDOM is the autochthonous DOM produced by phytoplankton, also known as exudates (Seymour et al., 2017; Mühlenbruch et al., 2018; Eigemann et al., 2022). These molecules can be easily degraded or directly uptaken by prokaryotes, and consequently they have been linked to high MeHg production rates (Kim et al., 2011; Lázaro et al., 2013; Bravo et al., 2017). On the other hand, the bioavailability of Hg can be reduced when refractory compounds (RDOM) act as the main ligands for Hg binding, forming RDOM-Hg aggregates. These molecules are more recalcitrant for biodegradation and, therefore, Hg uptake and MeHg formation are reduced in these situations (Chiasson-Gould et al., 2014; French et al., 2014; Bravo et al., 2017). Furthermore, more complex interactions have been suggested to occur between LDOM-Hg and RDOM-Hg aggregates, where Hg+2 can be dissociated from RDOM under photochemical oxidation or microbial degradation and bind to LDOM, thus becoming more bioavailable (Chiasson-Gould et al., 2014). The opposite situation has also been described, where Hg+2 bioavailability is reduced due to the progressive degradation of the most labile fraction of DOM by heterotrophic activity, which leads to higher RDOM : LDOM ratios and thus more abundant RDOM-Hg aggregates. These complex interactions between different fractions of DOM and Hg were hypothesized in two companion studies by Chiasson-Gould et al. (2014) and French et al. (2014). In both studies, a bell-shaped relationship between DOM concentration and Hg bioavailability was observed, where Hg bioavailability increased with increasing DOM concentration until a threshold (8.5 – 10 mg C L-1), from which Hg bioavailability started to progressively decrease with increasing DOM concentration. Each of these studies focused on different aspects of the Hg pathway. Whereas French et al. (2014) examined MeHg bioaccumulation in littoral amphipods collected from 26 Arctic lakes (Canada), Chiasson-Gould et al. (2014) used a modified strain of Escherichia coli as bioreporter under oxic conditions. This bacterium contains a merlux construct that emits bioluminescence when Hg+2 is actively transported through the cell membrane, which is used to quantify intracellular Hg+2 levels. Interestingly, both studies observed similar patterns in DOM-Hg interactions.
In addition, Chiasson-Gould et al. (2014) also studied Hg+2 bioavailability under two conditions: pseudoequilibrium and nonequilibrium at different DOM concentrations. They observed a decrease in bioavailability when Hg+2 was pre-incubated with DOM over 24 h (i.e., DOM-Hg pseudoequilibrium), which was explained by Hg+2 being mostly bound to strong binding sites in humic and fluvic acids. However, bioavailability increased when Hg+2 was freshly added (i.e., DOM-Hg nonequilibrium) to solutions already containing cells and humic/fluvic acids at increasing concentrations (0 to 10 mg C L−1), after which bioavailability started to decrease (bell-shaped pattern).
These observations suggest that the formation of DOM-Hg aggregates is a slow process involving competitive ligand exchange dynamics with multiple functional groups within the DOM pool, which may have important implications in a changing climate where DOM is expected to increase in the water column of boreal aquatic systems. DOM chemical structure and content should be considered of capital importance when studying effects on Hg bioavailability, MeHg production and bioaccumulation, particularly considering that organic matter in aquatic ecosystems is a heterogeneous mixture of molecules derived from terrestrial (allochthonous) and internal (autochthonous) sources, as well as the fact that DOM present in aquatic systems is usually more diverse in molecular structure and primary sources than in terrestrial environments (Jaffe et al., 2008; Schartup et al., 2015).
MeHg production enhanced by increasing microbial metabolism
As discussed above, Hg bioavailability and MeHg formation can be increased through chemical interactions with different DOM molecules, which ultimately interact with the Hg methylators. However, since Hg bio-methylation is an enzymatic process, it can also be enhanced by direct stimulation of the microbial metabolic activity, which has been observed to take place in pelagic waters (French et al., 2014; Paranjape and Hall, 2017; Rodríguez et al., 2022). Numerous studies have provided large evidence of the effects of DOM on the overall increase of microbial metabolism and the importance of organic matter remineralization in the methylation of Hg (Hall et al., 2004; Heimbürger et al., 2010; Bowman et al., 2016; Bravo et al., 2017; Kim et al., 2017; Herrero Ortega et al., 2018; Regnell and Watras, 2018). In fact, the role of settling particles on Hg methylation can be understood to operate at two different levels: 1) By providing low-oxygen micro-environments; 2) By providing organic matter as fuel to increase microbial metabolism. In line with this, Lehnherr et al. (2011) found significant correlations between POC remineralization and MeHg concentrations in the Arctic Sea, but referred to poor POC availability as probable cause for the low methylation rates detected in their experiments, which may have been higher if the water samples had been collected during the seasonal phytoplankton blooms. This relationship between phytoplankton production and Hg methylation is also supported by other studies where peak MeHg concentrations were detected in oxic subsurface euphotic zones during chlorophyll maximum (Bowman et al., 2015; Wang et al., 2018).
Oxygen deficient zones (ODZ) have been suggested as major sources of MeHg linked to organic matter remineralization (Wang et al., 2012; Bowman et al., 2015; Cossa et al., 2017; Kim et al., 2017; Gallorini and Loizeau, 2021). A common ground hypothesis is that POC (such as phytoplankton exudates) and DOM originated in surface and subsurface waters gradually sink to deeper layers, scavenging Hg in its path and forming aggregates that are later metabolized by prokaryotic communities under anoxic or hypoxic conditions. However, while ODZ may be the main source of MeHg in the water column, MeHg production has also be linked to DOM degradation under fully oxygenated waters. Rodríguez et al. (2022) found strong positive correlations between bacterial production and MeHg formation in oxygenated coastal waters from the Baltic Sea under increasing DOM concentrations, where micro-particles larger than 0.7 μm were discarded. Considering projected climate change scenarios for the Northern hemisphere (Meier et al., 2011; Andersson et al., 2015), the addition of terrestrial DOM led to an increase of the overall bacterial activity and diversity, as well as the relative abundance of genes related to enzymatic activity and energy generation. Furthermore, MeHg formation was detected shortly after the addition of Hg+2 (13 h), pointing to a rapid response of bacterial communities to methylate Hg. This quick metabolic response was also suggested by the rapid increase in relative abundance of genes related to cellular activity, with no significant changes in taxonomic composition. These observations may indicate that Hg methylation under oxic conditions just responds to housekeeping genes which increase their overall activity when carbon, nutrients or temperature are provided, but further studies are required to determine the exact mechanisms.
The rapid response of microbial communities to methylate Hg+2 may have important implications under a changing climate where DOM concentration in the water column is expected to increase. Although terrestrial DOM flushed from the catchment area in boreal systems is usually rich in refractory organic carbon (Asmala et al., 2013; Bravo et al., 2017), MeHg formation could potentially be increased through a rapid methylation of newly deposited Hg by the local prokaryotic communities boosted by high DOM concentrations, before Hg+2 becomes complexed with recalcitrant compounds in the DOM pool (Chiasson-Gould et al., 2014; Rodríguez et al., 2022). In addition, the overall MeHg concentration may be further increased by inputs of allochthonous MeHg derived from runoff (Jonsson et al., 2012; Krabbenhoft and Sunderland, 2013; Jonsson et al., 2014), as well as by reducing MeHg photo-demethylation due to water brownification and subsequent light attenuation typically induced by terrestrial dissolved organic matter (tDOM) (Poste et al., 2015; Wu et al., 2021).
Impacts of DOM on cell membrane permeability
Hg bioavailability can also increase by changes in the physiology of the bacterial cell membrane resulting in higher permeability. Humic and fluvic acids from natural DOM have been shown to act as surfactants, affecting the chemical properties of biological membranes and particularly the level of permeability to neutrally charged chemical species (Vigneault et al., 2000; Slaveykova et al., 2003; Ojwang’ and Cook, 2013; Graham et al., 2017), which could constitute a potential pathway for Hg+2 internalization (Benoit et al., 1999; Benoit et al., 2001). Chiasson-Gould et al. (2014) studied changes in the bacterial membrane permeability under different DOM concentrations and oxic conditions using bile salts, which reduce cell growth due to increased membrane permeability. They observed no significant increase in membrane permeability, and attributed the higher internalization of Hg+2 to interactions between DOM and the bacterial cell wall that resulted in the destabilization of the lipopolysaccharide layer, which could make Hg transport sites more accessible. Nonetheless, these conclusions should be taken with caution as other studies have reported decreased uptake and toxicity of charged metals by humic substances (Koukal et al., 2003; Kostić et al., 2013; Perelomov et al., 2018).
Temperature is also a substantially important factor
The central aspect from which all climate change effects derive is global warming. The increasing temperatures have multiple impacts on a variety of biogeochemical systems, from ice melting in polar and alpine regions to changes in hydrological patterns, increased sea levels, and frequency of extreme atmospheric events (i.e., droughts and deluges), among others (Stern et al., 2012; Andersson et al., 2015; Stott, 2016). Therefore, raising temperatures can have indirect and direct effects on Hg methylation. Permafrost thaw in polar and sub-polar regions has been observed to mobilize significant amounts of Hg accumulated over decades, which can be ultimately transported into aquatic systems (Schuster et al., 2018; Schaefer et al., 2020; Dastoor et al., 2022). Particularly in boreal regions, increased precipitation regimes are predicted to import higher organic matter from catchment areas, with effects on Hg methylation already discussed in the above sections. This increased terrestrial runoff will also import higher amounts of both Hg+2 and MeHg into aquatic ecosystems (Stern et al., 2012; Jonsson et al., 2014). Together with nutrients and organic carbon from tDOM, prokaryotic communities have all the necessary elements to potentially carry out Hg methylation (Rodríguez et al., 2022). In addition, a more direct impact of increasing temperatures is the positive effects on microbial activity, further stimulating the overall microbial metabolism, including the methylation activity (Monperrus et al., 2007; Johnson et al., 2016). On the other hand, negative effects on Hg methylation should be also considered by Hg cycling prediction models. For instance, higher rates of demethylation may be caused by the overall increase of microbial activity (Lu et al., 2016; Lu et al., 2017; Paranjape and Hall, 2017), as well as by higher exposure to solar radiation due to loss of sea ice (Stern et al., 2012).
Other indirect effects may account for a potential increase of Hg methylation as a result of increasing temperatures. Thermal stratification of the water column is associated with warmer air temperatures, and it has been shown to promote hypoxic conditions as oxygenated surface waters are prevented from mixing with the bottom layers (Cloern, 2001; Altieri and Keryn, 2015). This may lead to higher Hg methylation rates under anoxic conditions. Furthermore, oxygen levels can also be depleted by heterotrophic activity boosted by both higher temperatures and higher DOM concentration, where the eutrophication levels may increase Hg methylation (Soerensen et al., 2016; Ji et al., 2020; Yao et al., 2020).
Experimental approaches to study MeHg formation in oxic waters
With so many confounding factors affecting the Hg methylation process, how do we conduct suitable experimental strategies and measurements? Studying biological systems is a particularly complex task due to the outstanding complexity derived from numerous interdependent metabolic pathways, number of biological and chemical species involved, and bidirectional interactions with multiple physicochemical factors.
Field measurements and laboratory incubation experiments are the most employed approaches in the study of Hg methylation (Paranjape and Hall, 2017; Ma et al., 2019; Regnell and Watras, 2019; Gallorini and Loizeau, 2021). Since MeHg is a potent neurotoxin naturally bioaccumulated in natural environments, field experiments within aquatic systems are very constrained due to the exposure of Hg or MeHg to the natural biota. Furthermore, both field measurements and incubation experiments are restricted to a limited framework of variables, as it is virtually impossible to account for all the variables that operate in natural ecological systems. However, a combination of field measurements and controlled experiments is a valuable approach that has been used in a number of studies on Hg methylation in pelagic environments (Lehnherr et al., 2011; French et al., 2014; Paranjape and Hall, 2017; Wang et al., 2022). In some instances, either field measurements or controlled experiments may be sufficient to advance our knowledge towards a particular research question, but both approaches are subjected to their own limitations (Wang et al., 2020).
Field measurements
Field measurements typically consist of the collection of water and sediments to determine physical variables (e.g., temperature and light intensity), chemical variables (e.g., Hg/MeHg concentrations, organic matter content, oxygen, pH, salinity, etc.) and/or biological variables (e.g., metabolic activity, taxonomic composition, gene expression, etc.). Since Hg methylation is primarily a biochemical process, comprehensive studies should include a combination of such bio-physico-chemical variables. However, until recently, field measurements generally considered only chemical factors such as organic matter, oxygen, or relevant chemical species such as thiols (in addition to Hg/MeHg) (Monperrus et al., 2007; Merritt and Amirbahman, 2009; Li and Cai, 2013). Following the breakthrough of meta-omic technologies, more recent studies have introduced taxonomic and metabolic parameters principally through the sequencing of environmental DNA (eDNA). Bowman et al. (2019) and Villar et al. (2020) are among the most recent studies that have greatly contributed to survey the occurrence of Hg/MeHg concentrations and relevant microbial genes in oxic waters from the global oceans. In Bowman et al. (2019), water samples (<800 m depth) were collected from multiple areas ranging from Arctic to equatorial Pacific oceans, where detailed Hg speciation measurements were conducted, as well as targeted and shotgun metagenomics to evaluate the presence of Hg-cycling genes (hgcAB and mer) and other relevant genes potentially involved in Hg transportation and methylation. On the other hand, Villar et al. (2020) carried out a compilation of metagenomic and metatranscriptomic data obtained from locations covering most of the global ocean basins, with the purpose of addressing the paradox between MeHg production in the upper water column and absence of known anaerobic Hg methylating prokaryotes. Similarly, Podar et al. (2015) also conducted a compilation study using data generated by several metagenome sequencing projects. The compiled sequences were assembled and annotated to study the occurrence of hgcAB genes and Hg methylators across different environments, including boreal aquatic systems. Interestingly, these studies reached similar conclusions, where no hgcAB genes were generally detected in the oxic water column. Although hgcAB-like genes were found in Bowman et al. (2019) and Villar et al. (2020), to date these genes have not been proven to be involved in the methylation of Hg.
Incubation experiments
Incubation experiments have traditionally relied on experimental models consisting of one or few microbial species (Benoit et al., 2001; Kerin et al., 2006; Gilmour et al., 2013; Parks et al., 2013). Microbial isolates have been used to target specific aspects of the Hg methylation mechanism by simplifying the bio-physico-chemical system. Although this approach has been extensively used under anoxic conditions, only a few experiments have been conducted to investigate Hg methylation under oxic conditions. Chiasson-Gould et al. (2014) is one of the few examples, where the experimental system was reduced to a single bacterial strain (E. coli HMS174) genetically modified to study Hg+2 bioavailability. A natural DOM extract was used for incubation at different DOM concentrations, which allowed the identification of biochemical interactions between DOM, Hg+2 and the bacterial membrane that determined Hg+2 bioavailability and methylation. In a similar study, Golding et al. (2002) carried out an incubation experiment where the same E. coli strain (HMS174), as well as Vibrio anguillarum (a natural aquatic species), were exposed to both anaerobic and aerobic conditions under increased Hg+2 concentrations. By using this simplified experimental system, they observed higher uptake of Hg+2 under aerobic conditions, mediated by facilitated uptake mechanisms. In Cao et al. (2021), the aerobic methylation of Hg by two γ-proteobacteria strains (P. fluorescens TGR-B2 and P. putida TGR-B4) was evaluated under different oxygen concentrations, where they detected MeHg formation under both conditions, although Hg methylation under anaerobic conditions appeared to be more efficient.
As new molecular tools have been progressively developed in the field of Microbial Ecology, more integrated experimental designs have shown to be more valuable to study the complex interactions among the vast consortium of microbial species. Fundamental ecological functions are typically carried out by entire microbial communities, where syntrophic interactions between different members provide the necessary physicochemical conditions (Kato and Watanabe, 2010; Morris et al., 2013; Kouzuma et al., 2015). Therefore, whereas highly simplified experimental systems have the potential to unravel specific aspects of the genetic and metabolic bases of Hg methylation, the absence of certain microbial members can significantly alter the natural functioning of this process (Kerin et al., 2006; Ranchou-Peyruse et al., 2009; Bravo and Cosio, 2019). Mesocosm model ecosystems have been used in an attempt to reproduce natural conditions while isolating the system to apply controlled conditions. Combined with recently developed Hg isotope tracer methodologies (Jonsson et al., 2012; Jonsson et al., 2014) and high-throughput sequencing techniques, these experimental designs constitute powerful tools for comprehensive studies of how Hg methylation works in natural environments, and particularly under climate change-induced conditions. In a series of recent studies, Jonsson et al. (2017; 2022) used a 2000 L mesocosm setup to determine MeHg production and bioaccumulation in brackish waters and sediments collected from the Bothnian Sea, where multiple variables were included: increasing concentrations of DOM, nutrients, different Hg isotope tracers, a pelagic food web model (native heterotrophic bacteria, phytoplankton, protozoa, and mesozooplankton), light conditions, and controlled temperature. In a similar incubation experiment, Rodríguez et al. (2022) combined shotgun metagenomics with Hg/MeHg measurements to study Hg methylation by natural bacterial communities in oxygen-saturated pelagic waters, where variables such as DOM, Hg+2 concentration, oxygen level, and light intensity were considered.
In order to further improve our experimental models, it is important to point out potential biases and limitations from incubation studies based on the addition of isotopically enriched Hg species. In a recent critique article, Wang et al. (2020) evaluated the validity of this type of experimental approach as a method to study Hg methylation and demethylation rates in seawater. Based on field measurements and incubation experiments conducted along the Canadian Arctic Archipelago, they drew attention to potential reliability issues derived from methylation and demethylation at time zero, as well as the widely accepted assumption of first-order kinetics to calculate methylation and demethylation constants. Although these observations may respond to particular experimental procedures (Tsui et al., 2020; Zhang et al., 2021), and therefore generalizations should be taken with caution, future holistic studies should rely on an integration of field measurements and incubation experiments that allows the identification of potential experimental biases. Thus, field surveys can be highly valuable to guide subsequent incubation experiments where environmental parameters can be tuned to investigate hidden and emergent properties within the complex biological systems, as well as to generate prediction models for future scenarios under a changing climate.
Meta-omic technologies as tools to address the MeHg formation problematic
Until recently, simplified experimental systems were the main source of knowledge to study complex environmental microbial processes such as Hg methylation. After the discovery of the microbial bases of Hg methylation (Parks et al., 2013), early studies essentially relied on culture-dependent approaches to further advance our knowledge on the metabolic pathways, genes and species involved (Benoit et al., 2001; Kerin et al., 2006; Gilmour et al., 2013). The irruption of sequencing-based meta-omic technologies gave rise to a new kind of approach based on the generation of large quantities of sequencing data from entire microbial assemblages. Being culture-independent, this approach has allowed the implementation of more sophisticated experimental designs to disentangle the complex interactions between Hg methylators, their syntrophs, and relevant environmental variables (such as DOM) under natural or modelled conditions (Quince et al., 2017; Pérez-Cobas et al., 2020; Cho, 2021). In particular, metagenetics, metagenomics and metatranscriptomics (sequencing-based meta-omics) have been the meta-omic approaches most frequently used in the field of Microbial Ecology (Table 1), mainly due to: 1) cost-time effectiveness (simultaneous analyses of multiple samples); 2) analytical depth to cover even rare members of microbial communities; and 3) direct applicability in both field surveys and controlled experiments.

Table 1 Description of the main sequencing-based meta-omic approaches.
Metagenetics (also known as amplicon sequencing, marker-gene metagenomics, or targeted metagenomics) is a targeted approach where specific genes (called marker genes) present in the community gene pool are amplified by PCR, sequenced and aligned against an existing reference database for taxonomic and/or functional characterization of the community. The most frequently used gene marker for taxonomic characterization of bacterial communities is the 16S rRNA gene, which is universally found in bacteria with enough variability (nine hypervariable regions) as to allow taxonomic classification and biodiversity profiling (Větrovský and Baldrian, 2013). As discussed in previous sections, the marker genes in Hg methylation studies have consistently been the hgcAB gene pair. In a recent study, Capo et al. (2022) presented a hgc gene database (Hg-MATE) covering Hg-cycling microorganisms from terrestrial and aquatic ecosystems. This catalogue compiles isolated, single-cell and metagenome-reconstructed genomes, which can be used as a reference database to identify hgcAB genes from complex meta-omic datasets. Due to the lack of knowledge about alternative Hg methylation pathways potentially operating under truly oxic conditions, studies only relying on hgcAB genes may be limiting our ability to detect important Hg methylators in pelagic waters (Podar et al., 2015; Jones et al., 2019; Lin et al., 2021).
Shotgun metagenomics (or simply metagenomics) is a non-targeted approach where fragments of the metagenome (i.e., collection of all genomes present in a community) are randomly sequenced and, therefore, taxonomically relevant genes (such as 16S rRNA) as well as functional genes (i.e., involved in physiological processes) can be represented in the dataset, including bacteria, archaea, eukaryotes, and viruses (Quince et al., 2017). One advantage of shotgun metagenomics is the possibility to create biodiversity and functional profiles simultaneously from the very same environmental sample, which can be a valuable tool to detect intercorrelations between the presence of Hg-cycling genes and abundance of potential methylators, as well as other functional genes potentially involved in the Hg methylation process (Rodríguez et al., 2022). Another advantage is the recovery of metagenome-assembled genomes (MAGs), which can provide taxonomic and functional information on specific taxa (down to strain-level diversity) potentially involved in Hg methylation (Bowers et al., 2017; Gionfriddo et al., 2019; McDaniel et al., 2020; Peterson et al., 2020; Lin et al., 2021; Capo et al., 2022). On the other hand, some drawback must be also pointed out, such as the lower sequencing depth compared to specific targeted approaches, where the latter can direct the totality of the sequencing effort towards detecting marker genes such as 16S rRNA or hgcAB. In addition, although this technique can offer valuable insights about the functional potential of microbial communities (i.e., presence of genes involved in different metabolic pathways), it does not reflect the actual metabolic activity, as genes need to be transcribed to carry out their biological functions. Other meta-omic techniques are employed to address these aspects, such as metatranscriptomics or metaproteomics (Yap et al., 2022). Overall, shotgun metagenomics represents a cost-time-effective approach to obtain an integral picture of complex microbial communities where detailed resolution is not required.
Metatranscriptomics refers to the non-targeted sequencing of the mRNA pool from biological samples, which offers a higher level of functional resolution as compared to shotgun metagenomics. Multiple studies on Hg methylation have used this approach, particularly to determine levels of hgcAB gene expression and thus estimate Hg methylation activity (Vishnivetskaya, 2018; Christensen et al., 2019; Lin et al., 2021). However, the association between these two variables must be taken with caution, as increases in hgcAB transcripts may be caused by higher abundance of Hg methylators, and not necessarily by higher cell-specific hgcAB expression (Lin et al., 2021). In addition, the expression of hgcAB does not confer the capacity for Hg methylation on its own, as the cellular uptake of Hg (i.e., Hg bioavailability) is a necessary requirement. In fact, a number of studies have shown lack of correlation between hgcAB expression and concentration of MeHg or Hg methylation rates (Goñi-Urriza et al., 2015; Bravo et al., 2016; Vishnivetskaya, 2018; Christensen et al., 2019), although demethylation activity must be considered as a potential confounding factor for the lack of correlation. Despite these drawbacks, metatranscriptomics is one of the most promising tools in our repertory to potentially elucidate novel Hg methylation pathways occurring in oxic waters. In addition, the analysis of RNA is a measure of the active microbial fraction, allowing the distinction between viable and nonviable cells (Villar et al., 2020; Lin et al., 2021). DNA-based meta-omics (i.e., metagenetics and metagenomics) are still significantly more employed in Hg methylation studies, mainly due to more simplified and cost/time-effective sample preparation procedures. As new methods are developed to improve technical aspects of mRNA isolation, library preparation and sequencing, metatranscriptomics is becoming an extremely useful approach to disentangle complex microbial processes in natural environments.
Considering the advantages and disadvantages of the above-discussed sequencing-based meta-omic techniques, an increasingly popular approach is the combination of metagenomics and metatranscriptomics (multi-omics) to obtain a more comprehensive overview of natural microbial communities (Lin et al., 2021). This approach can facilitate the study of how relevant climate change-related variables (e.g., DOM and temperature) may affect the interconnections between community structure (i.e., occurrence of Hg methylators and syntrophs) and functioning (i.e., Hg methylation activity and overall metabolism). Functional redundancy is a typical property of natural prokaryotic communities that confers high levels of resistance and resilience under environmental disturbance, which can lead to the decoupling of community composition, functional potential, and the actual metabolic activity (Allison and Martiny, 2008; Bissett et al., 2013; Rodríguez et al., 2022). In this context, multi-omics offers an excellent tool for incubation experiments aiming the study of how the metabolic potential to perform Hg methylation and, more importantly, the actual ability to methylate Hg, may be affected under climate change scenarios.
Need for standardized methods and current challenges
A common issue frequently reported is the lack of standardization among different Hg methylation studies employing meta-omic approaches, which may lead to rather disparate results even from samples from the same sampling areas (Capo et al., 2022; Yap et al., 2022). Previous studies have addressed the standardization of analytical methods related to Hg and MeHg concentration and characterization (Creswell et al., 2013; Wen et al., 2017). This review will briefly address potential ways to standardize the analysis of microbial communities, from sample collection to bioinformatic data processing.
Meta-omic studies on aquatic environments usually begin with the collection of water, which is commonly carried out by filtration methods where the cellular fraction is captured onto a fine-pored matrix such as membrane filters (e.g., Lin et al., 2021; Rodríguez et al., 2022). The preservation of the matrix is a crucial step to maintain the integrity of the DNA and, especially, the RNA molecules. Inadequate preservation methods can easily result in loss of genetic material and hence can reduce the accuracy and resolution of the sequencing techniques. In addition, the degradation rate of DNA/RNA molecules can be related to their molecular size and nucleotide composition, which can vary among different taxonomic groups (Mitchell and Takacs-Vesbach, 2008; Kumar et al., 2020; Pavlovska et al., 2021). Therefore, deficient preservation can lead to dissimilar degradation of different DNA/RNA species, resulting in an artificial selection towards certain taxonomic groups over others, and potential loss of low-abundant members of the communities. Similar to hgcAB+ Hg methylators, other potential methylators using different pathways in oxic waters may be rare members of the communities and, therefore, a proper sample preservation is required to ensure detection and sufficient representation for subsequent analyses. Although preservation buffers have been frequently used (e.g., DNA/RNA Shield™ and RNAlater®), the most prevalent method in meta-omic studies is the use of deep freezing, which is usually achieved by liquid nitrogen (Anchordoquy and Molina, 2007; Pavlovska et al., 2021). The use of pre-fixative solutions can also improve the preservation of the original DNA/RNA pool, particularly when the filtration process takes prolonged periods of times (i.e., up to hours). In these cases, microbial communities may experience changes in community composition and gene expression while enclosed in collection bottles, which can be partially prevented by the addition of fixative alcoholic solutions (such as phenol-ethanol solution) to stop all biological activity while maintaining the original taxonomic and functional composition (Charvet et al., 2019; Rodríguez et al., 2022). Furthermore, the processing of samples immediately after collection is an emerging approach promoted by the development of the portable sequencers MinION (Oxford Nanopore Technologies), which eliminates the need for preservation and thus ensures a greater integrity of DNA and RNA molecules (Tyler et al., 2018; Runtuwene et al., 2019).
The detection of Hg methylators using hgcAB primers is also a controversial aspect widely discussed in Hg methylation studies (Capo et al., 2020a; Gionfriddo et al., 2020; McDaniel et al., 2020). The characterization of hgcAB genes can be biased by primer design, sequence length, or by the use of different classification methods. Since the first characterization of the hgcAB gene pair by Parks et al., 2013, a number of hgcAB variants have been described for different Hg methylators (Jones et al., 2019; Villar et al., 2020; Capo et al., 2022). While the design of hgcAB primers in early studies was based on the limited knowledge of a reduced number of Hg methylators (Liu et al., 2014; Bae et al., 2014; Schaefer et al., 2014), newly developed primers increasingly capture a wider diversity (Christensen et al., 2019; Bravo and Cosio, 2019; Jones et al., 2019 Gionfriddo et al., 2020). More efficient primers may help to detect Hg methylators in oxic pelagic environments, where hgcAB genes are scarce and sequencing depths can be insufficient for the detection of low-abundant genes. On the other hand, due to the lack of knowledge of the genetic bases and species involved, the study of alternative Hg methylation pathways potentially occurring under oxic conditions will require the design of more universal 16S rRNA primers to cover a wider range of prokaryotic diversity, as well as the use of non-targeted metagenomic approaches (such as MAGs by shotgun metagenomics) for a comprehensive functional characterization of specific taxa potentially involved.
Bioinformatic analysis of sequencing data is another aspect with little consensus among the literature. The existence of multiple bioinformatic pipelines to analyse metagenomic and metranscriptomic data has been shown to cause contrasting results (Cho, 2021; Yap et al., 2022). Bioinformatic analyses typically include a series of steps such as cleaning, assembly, read mapping, gene prediction, gene identification, and gene counting. However, within every step, multiple bioinformatic programs have been developed over the years (Pérez-Cobas et al., 2020). In particular, the use of common, standardized reference databases (either for taxonomic identification or for hgcAB genes) has been reiterated in the literature as an important step to facilitate comparisons across different studies (Gallorini and Loizeau, 2021; Capo et al., 2022; Wang et al., 2022). Normalization of gene counts is another key aspect to determine the prevalence of different members of the microbial communities, as well as to allow comparative analyses with similar studies. For instance, in meta-omic studies considering hgcAB genes, number of mapped reads and expression levels of housekeeping genes (such as rpoB, gyrB, and recA) are frequently used to normalize hgcAB read counts (Tada et al., 2020; Vigneron et al., 2021; Lin et al., 2021; Capo et al., 2022). Rarefaction to a common library size and normalization to gene length and metatranscriptomic sequencing depth are also normalization approaches frequently used in microbial community studies (Rodríguez et al., 2018; Pérez-Cobas et al., 2020). Considering that different normalization approaches can cope with different analytical biases, it is recommended to employ several normalization methods when reporting meta-omic data, which may facilitate data interpretation and inter-study comparisons.
Summary and concluding remarks
The study of Hg methylation in the water column under oxic conditions is still in its infancy. The formation of MeHg in pelagic areas where no anaerobic methylators are detected poses an apparent paradox that may be explained by different processes: 1) Abiotic Hg methylation; 2) Biotic MeHg production within anoxic micro-environments; 3) Biotic MeHg production under oxic conditions through metabolic pathways involving hgcAB genes or 4) independent of hgcAB genes. Anoxic microenvironments within settling particles have been frequently postulated as the main source of MeHg in the oxic water column, but recent studies suggest that Hg methylation can take place in fully oxygenated waters where anoxic microenvironments are not present. Given the current controversy, more studies on Hg methylation under truly oxic conditions are required to clarify whether Hg methylation is also performed through metabolic pathways different to the reductive acetyl–CoA pathway involving hgcAB genes.
Boreal aquatic ecosystems are predicted to be impacted by global warming through increases in temperatures and DOM concentration in the water column. In particular, DOM is known to affect Hg methylation at different levels: 1) Through chemical interactions affecting Hg+2 speciation and providing methyl groups; 2) Enhancing microbial metabolism; 3) Promoting changes in the bacterial cell membrane which affect Hg uptake. These aspects have been poorly studied in boreal aquatic systems under oxic conditions, and, therefore, comprehensive studies including the main biochemical factors involved are required to predict potential ecosystem effects under climate change scenarios.
Meta-omic approaches represent promising tools to accomplish this task. A combination of metagenetics, metagenomics, and metatranscriptomics is advised to simultaneously study taxonomic and functional aspects of Hg methylation, which is particularly valuable in the study of unknown pathways not involving hgcAB genes. Due to the wide variety of techniques for sample processing, DNA/RNA sequencing, and data analysis, standardization of meta-omic approaches may improve data interpretation and inter-study comparisons. In addition, an integration of field measurements and incubation experiments is recommended to improve our experimental models when studying how Hg methylation works in natural aquatic environments under oxic conditions, and particularly under climate change-induced conditions.
Author contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This study was supported by Swedish Environmental Protection Agency (Naturvårdsverket): Project: 2020-00032.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alava J. J., Cheung W. W., Ross P. S., Sumaila U. R. (2017). Climate change–contaminant interactions in marine food webs: Toward a conceptual framework. Global Change Biol. 23 (10), 3984–4001. doi: 10.1111/gcb.13667
Alldredge A. L., Silver M. W. (1988). Characteristics, dynamics and significance of marine snow. Prog. Oceanography 20.1, 41–82. doi: 10.1016/0079-6611(88)90053-5
Alldredge A. L., Cohen Y. (1987). Can microscale chemical patches persist in the sea? Microelectrode study of marine snow, fecal pellets. Science 235 (4789), 689–691. doi: 10.1126/science.235.4789.689
Allison S. D., Martiny J. B. (2008). Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. 105, 11512–11519. doi: 10.1073/pnas.0801925105
Altieri A. H., Keryn B. G. (2015). Climate change and dead zones. Global Change Biol. 21.4, 1395–1406. doi: 10.1111/gcb.12754
Anchordoquy T. J., Molina M. C. (2007). Preservation of DNA. Cell Preserv. Technol. 5 (4), 180–188. doi: 10.1089/cpt.2007.0511
Andersson A., Meier H. M., Ripszam M., Rowe O., Wikner J., Haglund P., et al. (2015). Projected future climate change and Baltic Sea ecosystem management. Ambio 44, 345–356. doi: 10.1007/s13280-015-0654-8
Ariya P. A., Peterson K. A. (2005). “Chemical transformation of gaseous elemental Hg in the atmosphere,” in Dynamics of Mercury Pollution on Regional and Global Scales (Boston, MA: Springer US), 261–294. doi: 10.1007/0-387-24494-8_12
Asmala E., Autio R., Kaartokallio H., Pitkänen L., Stedmon C. A., Thomas D. N. (2013). Bioavailability of riverine dissolved organic matter in three baltic sea estuaries and the effect of catchment land use. Biogeosciences 10 (11), 6969–6986. doi: 10.5194/bg-10-6969-2013
Bae H. S., Dierberg F. E., Ogram A. (2014). Syntrophs dominate sequences associated with the mercury methylation-related gene hgcA in the water conservation areas of the florida everglades. Appl. Environ. Microbiol. 80 (20), 6517–6526. doi: 10.1128/AEM.01666-14
Beckers F., Mothes S., Abrigata J., Zhao J., Gao Y., Rinklebe J. (2019). Mobilization of mercury species under dynamic laboratory redox conditions in a contaminated floodplain soil as affected by biochar and sugar beet factory lime. Sci. Total Environ. 672, 604–617. doi: 10.1016/j.scitotenv.2019.03.401
Benoit J. M., Gilmour C. C., Mason R.P. (2001). The influence of sulfide on solid phase mercury bioavailability for methylation by pure cultures of Desulfobulbus propionicus (1pr3). Environ. Sci. Technol. 35 (1), 127–132. doi: 10.1021/es001415n
Benoit J. M., Gilmour C. C., Mason R. P., Heyes A. (1999). Sulfide controls on mercury speciation and bioavailability to methylating bacteria in sediment pore waters. Environ. Sci. Technol. 33 (6), 951–957. doi: 10.1021/es9808200
Bianchi D., Weber T. S., Kiko R., Deutsch C. (2018). Global niche of marine anaerobic metabolisms expanded by particle microenvironments. Nat. Geosci. 11 (4), 263–268. doi: 10.1038/s41561-018-0081-0
Bissett A., Brown M. V., Siciliano S. D., Thrall P. H. (2013). Microbial community responses to anthropogenically induced environmental change: towards a systems approach. Ecol. Lett. 16, 128–139. doi: 10.1111/ele.12109
Boening D. W. (2000). Ecological effects, transport, and fate of mercury: a general review. Chemosphere 40, 1335–1351. doi: 10.1016/S0045-6535(99)00283-0
Bowers R. M., Kyrpides N. C., Stepanauskas R., Harmon-Smith M., Doud D., Reddy T. B. K., et al. (2017). Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35 (8), 725–731. doi: 10.1038/nbt.3893
Bowman K. L., Collins R. E., Agather A. M., Lamborg C. H., Hammerschmidt C. R., Kaul D., et al. (2019). Distribution of mercury-cycling genes in the arctic and equatorial pacific oceans and their relationship to mercury speciation. Limnology Oceanography 65, S310–S320. doi: 10.1002/lno.11310
Bowman K. L., Lamborg C. H., Agather A. M. (2020). A global perspective on mercury cycling in the ocean. Sci. Total Environ. 710, 136166. doi: 10.1016/j.scitotenv.2019.136166
Bowman K. L., Hammerschmidt C. R., Lamborg C. H., Swarr G. (2015). Mercury in the north atlantic ocean: The US GEOTRACES zonal and meridional sections. Deep Sea Res. Part II: Topical Stud. Oceanography 116, 251–261. doi: 10.1016/j.dsr2.2014.07.004
Bowman K. L., Hammerschmidt C. R., Lamborg C. H., Swarr G. J., Agather A. M. (2016). Distribution of mercury species across a zonal section of the eastern tropical South Pacific Ocean (US GEOTRACES GP16). Marine Chemistry 186, 156–166. doi: 10.1016/j.marchem.2016.09.005
Braun S. T., Proctor L. M., Zani S., Mellon T. M., Zehr P. J. (1999). Molecular evidence for zooplankton-associatednitrogen-fixing anaerobes based on amplification of the nifHgene. FEMS Microbiol. Ecol. 28, 273–279. doi: 10.1111/j.1574-6941.1999.tb00582.x
Bravo A. G., Loizeau J. L., Dranguet P., Makri S., Bjoürn E., Ungureanu V. G., et al. (2016). Per- sistent Hg contamination and occurrence of Hg- methylating transcript (hgcA) downstream of a chlor-alkali plant in the Olt River (Romania). Environ. Sci. pollut. Res. 23, 10529–10541. doi: 10.1007/s11356-015-5906-4
Bravo A. G., Zopfi J., Buck M., Xu J., Bertilsson S., Schaefer J. K., et al. (2018). Geobacteraceae are important members of mercury-methylating microbial communities of sediments impacted by waste water releases. ISME J. 12 (3), pp.802–pp.812. doi: 10.1038/s41396-017-0007-7
Bravo A. G., Bouchet S., Tolu J., Björn E., Mateos-Rivera A., Bertilsson S. (2017). Molecular composition of organic matter controls methylmercury formation in boreal lakes. Nat. Commun. 8, 1–9. doi: 10.1038/ncomms14255
Bravo A. G., Cosio C. (2019). Biotic formation of methylmercury: A bio–physico–chemical conundrum. Limnology Oceanography 65 (5), 1010–1027. doi: 10.1002/lno.11366
Campbell P. G., Twiss M. R., Wilkinson K. J. (1997). Accumulation of natural organic matter on the surfaces of living cells: implications for the interaction of toxic solutes with aquatic biota. Can. J. Fish. Aquat. Sci. 54, 2543–2554. doi: 10.1139/f97-161
Cao D., Chen W., Xiang Y., Mi Q., Liu H., Feng P., et al. (2021). The efficiencies of inorganic mercury bio-methylation by aerobic bacteria under different oxygen concentrations. Ecotoxicol. Environ. Saf. 207, 111538. doi: 10.1016/j.ecoenv.2020.111538
Capo E., Bravo A. G., Soerensen A. L., Bertilsson S., Pinhassi J., Feng C., et al. (2020a). Marine snow as a habitat for microbial mercury methylators in the Baltic Sea. bioRxiv, 2020–2003. doi: 10.1101/2020.03.04.975987
Capo E., Bravo A. G., Soerensen A. L., Bertilsson S., Pinhassi J., Feng C., et al. (2020b). Deltaproteobacteria and spirochaetes-like bacteria are abundant putative mercury methylators in oxygen-deficient water and marine particles in the Baltic Sea. Front. Microbiol. 11, 574080. doi: 10.3389/fmicb.2020.574080
Capo E., Peterson B. D., Kim M., Jones D. S., Acinas S. G., Amyot M., et al. (2022). A consensus protocol for the recovery of mercury methylation genes from metagenomes. Mol. Ecol. Resour. 23 (1), 190–204. doi: 10.1101/2022.03.14.484253
Celo V., Lean D. R., Scott S. L. (2006). Abiotic methylation of mercury in the aquatic environment. Sci. Total Environ. 368 (1), 126–137. doi: 10.1016/j.scitotenv.2005.09.043
Charvet S., Riemann L., Alneberg J., Andersson A. F., von Borries J., Fischer U., et al. (2019). AFISsys-An autonomous instrument for the preservation of brackish water samples for microbial metatranscriptome analysis. Water Res. 149, 351–361. doi: 10.1016/j.watres.2018.11.017
Chetelat J., McKinney M. A., Amyot M., Dastoor A., Douglas T. A., Heimbürger-Boavida L. E., et al. (2022). Climate change and mercury in the Arctic: Abiotic interactions. Sci. Total Environ. 824, 153715. doi: 10.1016/j.scitotenv.2022.153715
Chiasson-Gould S. A., Blais J. M., Poulain A. J. (2014). Dissolved organic matter kinetically controls mercury bioavailability to bacteria. Environ. Sci. Technol. 48, 3153–3161. doi: 10.1021/es4038484
Cho J. C. (2021). Omics-based microbiome analysis in microbial ecology: from sequences to information. J. Microbiol. 59, 229–232. doi: 10.1007/s12275-021-0698-3
Christensen G. A., Gionfriddo C. M., King A. J., Moberly J. G., Miller C. L., Somenahally A. C., et al, et al. (2019). Determining the reliabil- ity of measuring mercury cycling gene abundance with cor- relations with mercury and methylmercury concentrations. Environ. Sci. Technol. 53, 8649–8663. doi: 10.1021/acs.est.8b06389
Cloern J. E. (2001). Our evolving conceptual model of the coastal eutrophication problem. Mar. Ecol. Prog. Ser. 210, 223–253. doi: 10.3354/meps210223
Compeau G. C., Bartha R. (1985). Sulfate-reducing bacteria: principal methylators of mercury in anoxic estuarine sediment. Appl. Environ. Microbiol. 50 (2), 498–502. doi: 10.1128/aem.50.2.498-502.1985
Cossa D., Averty B., Pirrone N. (2009). The origin of methylmercury in open Mediterranean waters. Limnol. Oceanography 54 (3), 837–844. doi: 10.4319/lo.2009.54.3.0837
Cossa D., de Madron X. D., Sch€afer J., Lanceleur L., Guedron S., Buscail R., et al. (2017). The open sea as the main source of methylmercury in the water column of the Gulf of Lions (Northwestern Mediterranean margin). Geochim. Cosmochim. Acta 199, 222–237. doi: 10.1016/j.gca.2016.11.037
Creswell J. E., Engel V., Carter A., Davies C. (2013). “Results from three years of the world's largest interlaboratory comparison for total mercury and methylmercury: Method performance and best practices,” in AGU Fall Meeting Abstracts 2013, B41C–0405. doi: 2013AGUFM.B41C0405C
Dastoor A., Angot H., Bieser J., Christensen J. H., Douglas T. A., Heimbürger-Boavida L. E., et al. (2022). Arctic mercury cycling. Nat. Rev. Earth Environ. 3 (4), 270–286. doi: 10.1038/s43017-022-00269-w
Decho A. W. (1990). Microbial exopolymer secretions in ocean environments: their role (s) in food webs and marine processes. Oceanogr. Mar. Biol. Annu. Rev. 28 (7), 73–153, doi: 10.4319/lo.1990.35.5.1039
Decho A. W., Gutierrez T. (2017). Microbial extracellular polymeric substances (EPSs) in ocean systems. Front. Microbiol. 8, 922. doi: 10.3389/fmicb.2017.00922
Eigemann F., Rahav E., Grossart H. P., Aharonovich D., Sher D., Vogts A., et al. (2022). Phytoplankton exudates provide full nutrition to a subset of accompanying heterotrophic bacteria via carbon, nitrogen and phosphorus allocation. Environ. Microbiol. 24 (5), pp.2467–2483. doi: 10.1111/1462-2920.15933
French T. D., Houben A. J., Desforges J. P. W., Kimpe L. E., Kokelj S. V., Poulain A. J., et al. (2014). Dissolved organic carbon thresholds affect mercury bioaccumulation in Arctic lakes. Environ. Sci. Technol. 48, 3162–3168. doi: 10.1021/es403849d
Gallorini A., Loizeau J. L. (2021). Mercury methylation in oxic aquatic macro-environments: a review. J. Limnol. 80 (2). doi: 10.4081/jlimnol.2021.2007
Gascón Díez E, Loizeau J. L., Cosio C., Bouchet S., Adatte T., Amouroux D., et al. (2016). Role of settling particles on mercury methylation in the oxic water column of freshwater systems. Environ. Sci. Technol. 50 (21), 11672–11679. doi: 10.1021/acs.est.6b03260
Ghimire P. S., Tripathee L., Zhang Q., Guo J., Ram K., Huang J., et al. (2019). Microbial mercury methylation in the cryosphere: Progress and prospects. Sci. Total Environ. 697, 134150. doi: 10.1016/j.scitotenv.2019.134150
Gilmour C. C., Podar M., Bullock A. L., Graham A. M., Brown S. D., Somenahally A. C., et al. (2013). Mercury methylation by novel microorganisms from new environments. Environ. Sci. Technol. 47 (20), pp.11810–11820. doi: 10.1021/es403075t
Gilmour C. C., Bullock A. L., McBurney A., Podar M., Elias D. A. (2018). Robust mercury methylation across diverse methanogenic archaea. MBio 9 (2), 10–1128. doi: 10.1128/mbio.02403-17
Gionfriddo C., Podar M., Gilmour C., Pierce E., Elias D. (2019). ORNL compiled mercury methylator database (No. MCI533). ORNLCIFSFA (Critical Interfaces Science Focus Area) (TN (United States: Oak Ridge National Lab. (ORNL), Oak Ridge).
Gionfriddo C. M., Wymore A. M., Jones D. S., Wilpiszeski R. L., Lynes M. M., Christensen G. A., et al. (2020). An improved hgcAB primer set and direct high-throughput sequencing expand Hg-methylator diversity in nature. Front. Microbiol. 11, 541554. doi: 10.3389/fmicb.2020.541554
Gionfriddo C. M., Tate M. T., Wick R. R., Schultz M. B., Zemla A., Thelen M. P. (2016). MicrobialmercurymethylationinAntarctic sea ice. Nat. Microbiol. 1, 16127–16112. doi: 10.1038/nmicrobiol.2016.127
Glud R. N., Grossart H. P., Larsen M., Tang K. W., Arendt K. E., Rysgaard S., et al. (2015). Copepod carcasses as microbial hot spots for pelagic denitrification. Limnol. Oceanography 60 (6), 2026–2036. doi: 10.1002/lno.10149
Golding G. R., Kelly C. A., Sparling R., Loewen P. C., Rudd J. W., Barkay T. (2002). Evidence for facilitated uptake of Hg (II) by Vibrio anguillarum and Escherichia coli under anaerobic and aerobic conditions. Limnol. Oceanography 47 (4), pp.967–pp.975. doi: 10.4319/lo.2002.47.4.0967
Goñi-Urriza M. Y., Corsellis L., Lanceleur E., Tessier J., Gury M., Monperrus, et al. (2015). Relationships between bacterial energetic metabolism, mercury methyla- tion potential, and hgcA/hgcB gene expression in Des- ulfovibrio dechloroacetivorans BerOc1. Environ. Sci. pollut. Res. 22, 13764–13771. doi: 10.1007/s11356-015-4273-5
Graham A. M., Cameron-Burr K. T., Hajic H. A., Lee C., Msekela D., Gilmour C. C. (2017). Sulfurization of dissolved organic matter increases Hg–sulfide–dissolved organic matter bioavailability to a Hg-methylating bacterium. Environ. Sci. Technol. 51 (16), 9080–9088. doi: 10.1021/acs.est.7b02781
Grandjean P., Murata K., Budtz-Jørgensen E., Weihe P. (2004). Cardiac autonomic activity in methylmercury neurotoxicity: 14-year follow-up of a Faroese birth cohort. J. Pediatr. 144 (2), 169–176. doi: 10.1016/j.jpeds.2003.10.058
Hall B. D., Louis V. L. S., Bodaly R. D. (2004). The stimulation of methylmercury production by decomposition of flooded birch leaves and jack pine needles. Biogeochemistry 68, 107–129. doi: 10.1023/B:BIOG.0000025745.28447.8b
Hammerschmidt C. R., Fitzgerald W. F. (2006). Methylmercury cycling in sediments on the continental shelf of southern New England. Geochim. Cosmochim. Acta 70 (4), 918–930. doi: 10.1016/j.gca.2005.10.020
Heimbürger L.-E., Cossa D., Marty J.-C., Migon C., Averty B., Dufour A., et al. (2010). Methyl mercury dis- tributions in relation to the presence of nano-and picophytoplankton in an oceanic water column (Ligurian Sea, north-western Mediterranean). Geochim. Cosmochim. Acta 74 (19), 5549–5559. doi: 10.1016/j.gca.2010.06.036
Heimbürger L.-E., Sonke J. E., Cossa D., Point D., Lagane C., Laffont L., et al. (2015). Shallow methylmercury production in the marginal sea ice zone of the central Arctic Ocean. Sci. Rep. 5 (1), 1–6. doi: 10.1038/srep10318
Herrero Ortega S., Catalaín N., Bjoürn E., Groüntoft H., Hilmarsson T. G., Bertilsson S., et al. (2018). High methylmercury formation in ponds fueled by fresh humic and algal derived organic matter. Limnol. Oceanogr. 63, S44–S53. doi: 10.1002/lno.10722
Hsu-Kim H., Kucharzyk K. H., Zhang T., Deshusses M. A. (2013). Mechanisms regulating mercury bioavailability for methylating microorganisms in the aquatic environment: a critical review. Environ. Sci. Technol. 47, 2441–2456. doi: 10.1021/es304370g
Jaffe R., McKnight D., Maie N., Cory R., McDowell W. H. ;., Campbell, et al. (2008). Spatial and temporal variations in DOM composition in ecosystems: The importance of long-term monitoring of optical properties. J. Geophys. Res. 113, 1–15. doi: 10.1029/2008JG000683
Jensen Sören, Jernelöv A. (1969). Biological methylation of mercury in aquatic organisms. Nature 223, 753–754, 5207. doi: 10.1038/223753a0
Jernelov A. (1969). Are there any differences between" biologically" and "chemically" synthesized methyl mercury. Vatten 3, 304.
Ji X., Liu C., Zhang M., Yin Y., Pan G. (2020). Mitigation of methylmercury production in eutrophic waters by interfacial oxygen nanobubbles. Water Res. 173, 115563. doi: 10.1016/j.watres.2020.115563
Jiang T., Bravo A. G., Skyllberg U., Björn E., Wang D., Yan H., et al. (2018). Influence of dissolved organic matter (DOM) characteristics on dissolved mercury (Hg) species composition in sediment porewater of lakes from Southwest China. Water Res. 146, 146–158. doi: 10.1016/j.watres.2018.08.054
Johnson N. W., Mitchell C. P. J., Engstrom D. R., Bailey L. T., Coleman Wasik J. K., Berndt M. E. (2016). Methylmercury production in a chronically sulfate-impacted sub-boreal wetland. Environ. Sci.: Processes Impacts 18 (6), 725–734. doi: 10.1039/c6em00138f
Jones D. S., Walker G. M., Johnson N. W., Mitchell C. P., Coleman Wasik J. K., Bailey J. V. (2019). Molecular evidence for novel mercury methylating microorganisms in sulfate-impacted lakes. ISME J. 13 (7), 1659–1675. doi: 10.1038/s41396-019-0376-1
Jonsson S., Skyllberg U., Nilsson M. B., Lundberg E., Andersson A., Björn E. (2014). Differentiated availability of geochemical mercury pools controls methylmercury levels in estuarine sediment and biota. Nat. Commun. 5, 4624. doi: 10.1038/ncomms5624
Jonsson S., Skyllberg U., Nilsson M. B., Westlund P.-O., Shchukarev A., Lundberg E., et al. (2012). Mercury methylation rates for geochemically relevant HgII species in sediments. Environ. Sci. Technol. 46, 11653–11659. doi: 10.1021/es3015327
Jonsson S., Andersson A., Nilsson M. B., Skyllberg U., Lundberg E., Schaefer J. K., et al. (2017). Terrestrial discharges mediate trophic shifts and enhance methylmercury accumulation in estuarine biota. Sci. Adv. 3 (1), e1601239. doi: 10.1126/sciadv.1601239
Jonsson S., Liem-Nguyen V., Andersson A., Skyllberg U., Nilsson M. B., Lundberg E., et al. (2022). Geochemical and dietary drivers of mercury bioaccumulation in estuarine benthic invertebrates. Environ. Sci. Technol. 56 (14), 10141–10148. doi: 10.1021/acs.est.2c03265
Kato S., Watanabe K. (2010). Ecological and evolutionary interactions in syntrophic methanogenic consortia. Microbes Environments 25 (3), 145–151. doi: 10.1264/jsme2.ME10122
Kerin E. J., Gilmour C. C., Roden E., Suzuki M. T., Coates J. D., Mason R. P. (2006). Mercury methylation by dissimilatory Iron-reducing bacteria. Appl. Environ. Microbiol. 72, 7919–7921. doi: 10.1128/AEM.01602-06
Kim M., Han S., Gieskes J., Deheyn D. D. (2011). Importance of organic matter lability for monomethylmercury production in sulfate-rich marine sediments. Sci. Total Environ. 409 (4), 778–784. doi: 10.1016/j.scitotenv.2010.10.050
Kim H., Soerensen A. L., Hur J., Heimburger L. E., Hahm D., Rhee T. S., et al. (2017). Methylmercury mass budgets and distribution characteristics in the Western Pacific Ocean. Environ. Sci. Technol. 51 (3), 1186–1194. doi: 10.1021/acs.est.6b04238
Kirk J. L., Lehnherr I., Andersson M., Braune B. M., Chan L., Dastoor A. P., et al. (2012). Mercury in Arctic marine ecosystems: sources, pathways and exposure. Environ. Res. 119, 64–87. doi: 10.1016/j.envres.2012.08.012
Kostić I. S., Anđelković T. D., Nikolić R. S., Cvetković T. P., Pavlović D. D., Bojić A. L. (2013). Comparative study of binding strengths of heavy metals with humic acid. Hemijska Industrija 67 (5), 773–779. doi: 10.2298/HEMIND121107002K
Koukal B., Gueguen C., Pardos M., Dominik J. (2003). Influence of humic substances on the toxic effects of cadmium and zinc to the green alga Pseudokirchneriella subcapitata. Chemosphere 53 (8), 953–961. doi: 10.1016/S0045-6535(03)00720-3
Kouzuma A., Kato S., Watanabe K. (2015). Microbial interspecies interactions: recent findings in syntrophic consortia. Front. Microbiol. 6, 477. doi: 10.3389/fmicb.2015.00477
Krabbenhoft D. P., Sunderland E. M. (2013). Global change and mercury. Science 341 (6153), 1457–1458. doi: 10.1126/science.1242838
Kraepiel A. M., Keller K., Chin H. B., Malcolm E. G., Morel F. M. (2003). Sources and variations of mercury in tuna. Environ. Sci. Technol. 37 (24), 5551–5558. doi: 10.1021/es0340679
Kumar G., Eble J. E., Gaither M. R. (2020). A practical guide to sample preservation and pre-PCR processing of aquatic environmental DNA. Mol. Ecol. Resour. 20 (1), 29–39. doi: 10.1111/1755-0998.13107
Kuss J., Krüger S., Ruickoldt J., Wlost K. P. (2018). High-resolution measurements of elemental mercury in surface water for an improved quantitative understanding of the Baltic Sea as a source of atmospheric mercury. Atmospheric Chem. Phys. 18 (6), 4361–4376. doi: 10.5194/acp-18-4361-2018
Lázaro W. L., Guimarães J. R. D., Ignácio A. R., Da Silva C. J., Díez S. (2013). Cyanobacteria enhance methylmercury production: a hypothesis tested in the periphyton of two lakes in the Pantanal floodplain, Brazil. Sci. Total Environ. 456, 231–238. doi: 10.1016/j.scitotenv.2013.03.022
Lehnherr I., Louis V. L. S., Hintelmann H., Kirk J. L. (2011). Methylation of inorganic mercury in polar marine waters. Nat. Geosci. 4, 298–302. doi: 10.1038/ngeo1134
Li Y., Cai Y. (2013). Progress in the study of mercury methylation and demethylation in aquatic environments. Chin. Sci. Bull. 58, 177–185. doi: 10.1007/s11434-012-5416-4
Lin H., Ascher D. B., Myung Y., Lamborg C. H., Hallam S. J., Gionfriddo C. M., et al. (2021). Mercury methylation by metabolically versatile and cosmopolitan marine bacteria. ISME J. 15 (6), 1810–1825. doi: 10.1038/s41396-020-00889-4
Liu M., Zhang Q., Maavara T., Liu S., Wang X., Raymond P. A. (2021). Rivers as the largest source of mercury to coastal oceans worldwide. Nat. Geosci. 14 (9), 672–677. doi: 10.1038/s41561-021-00793-2
Liu Y. R., Yu R. Q., Zheng Y. M., He J. Z. (2014). Analysis of the microbial community structure by monitoring an hg methylation gene (hgcA) in paddy soils along an hg gradient. Appl. Environ. Microbiol. 80, 2874–2879. doi: 10.1128/AEM.04225-13
Lu X., Gu W., Zhao L., Farhan Ul Haque M., DiSpirito A. A., Semrau J. D., et al. (2017). Methylmercury uptake and degradation by methanotrophs. Sci. Adv. 3 (5), e1700041. doi: 10.1126/sciadv.1700041
Lu X., Liu Y., Johs A., Zhao L., Wang T., Yang Z., et al. (2016). Anaerobic mercury methylation and demethylation by Geobacter bemidjiensis Bem. Environ. Sci. Technol. 50 (8), 4366–4373. doi: 10.1021/acs.est.6b00401
Ma M., Du H., Wang D. (2019). Mercury methylation by anaerobic microorganisms: A review. Crit. Rev. Environ. Sci. Technol. 49 (20), 1893–1936. doi: 10.1080/10643389.2019.1594517
Malcolm E. G., Schaefer J. K., Ekstrom E. B., Tuit C. B., Jayakumar A., Park H., et al. (2010). Mercury methylation in oxygen deficient zones of the oceans: no evidence for the predominance of anaerobes. Mar. Chem. 122, 11–19. doi: 10.1016/j.marchem.2010.08.004
Marnane I. (2018). Mercury in Europe's environment a priority for European and global action. European Environmental Agency; EEA Report 2018 No.11/2018 pp.72 pp. Record Number doi: 20219947452
Mason R. P., Choi A. L., Fitzgerald W. F., Hammerschmidt C. R., Lamborg C. H., Soerensen A. L., et al. (2012). Mercury biogeochemical cycling in the ocean and policy implications. Environ. Res. 119, 101–117. doi: 10.1016/j.envres.2012.03.013
Mason R. P., Fitzgerald W. F. (1993). The distribution and biogeochemical cycling of mercury in the equatorial Pacific Ocean. Deep Sea Res. Part I.: Oceanographic Res. Papers 40 (9), 1897–1924. doi: 10.1016/0967-0637(93)90037-4
McDaniel E. A., Peterson B. D., Stevens S. L., Tran P. Q., Anantharaman K., McMahon K. D. (2020). Expanded phylogenetic diversity and metabolic flexibility of mercury-methylating microorganisms. Msystems 5 (4), e00299–e00220. doi: 10.1128/mSystems.00299-20
Meier M., Andersson H., Dieterich C., Eilola K., Gustafsson B., Hoüglund A., et al. (2011). Transient scenario simulations for the Baltic Sea Region during the 21st century. In SMHI, 2011, p. 81. DiVA id: diva2:947527
Merritt K. A., Amirbahman A. (2009). Mercury methylation dynamics in estuarine and coastal marine environments—a critical review. Earth-Sci. Rev. 96 (1-2), 54–66. doi: 10.1016/j.earscirev.2009.06.002
Minamata Disease Research Group (1966). [Minamata Disease: Studies About Methyl-Mercury Poisoning] (in Japanese) (Kumamoto, Japan: Kumamoto University School of Medicine).
Mitchell K. R., Takacs-Vesbach C. D. (2008). A comparison of methods for total community DNA preservation and extraction from various thermal environments. J. Ind. Microbiol. Biotechnol. 35 (10), 1139–1147. doi: 10.1007/s10295-008-0393-y
Monperrus M., Tessier E., Amouroux D., Leynaert A., Huonnie P., Donald O. F. X. (2007). Mercury methylation, demethylation and reduction rates in coastal and marine surface waters of the Mediterranean Sea. Mar. Chem. 107, 49–63. doi: 10.1016/j.marchem.2007.01.018
Morris B. E., Henneberger R., Huber H., Moissl-Eichinger C. (2013). Microbial syntrophy: interaction for the common good. FEMS Microbiol. Rev. 37 (3), 384–406. doi: 10.1111/1574-6976.12019
Mühlenbruch M., Grossart H. P., Eigemann F., Voss M. (2018). Mini-review: Phytoplankton-derived polysaccharides in the marine environment and their interactions with heterotrophic bacteria. Environ. Microbiol. 20 (8), pp.2671–2685. doi: 10.1111/1462-2920.14302
Munson K. M., Lamborg C. H., Boiteau R. M., Saito M. A. (2018). Dynamic mercury methylation and deme- thylation in oligotrophic marine water. Biogeosciences 15 (21), 6451–6460. doi: 10.5194/bg-15-6451-2018
Munson K. M., Lamborg C. H., Swarr G. J., Saito M. A. (2015). Mercury species concentrations and fluxes in the central tropical Pacific Ocean. Global Biogeochem. Cycles 29 (5), 656–676. doi: 10.1002/2015GB005120
Nagase H., Ose Y., Sato T., Ishikawa T. (1982). Methylation of mercury by humic substances in an aquatic environment. Sci. Total Environ. 25 (2), 133–142. doi: 10.1016/0048-9697(82)90082-1
Nagase H., Ose Y., Sato T., Ishikawa T. (1984). Mercury methylation by compounds in humic material. Sci. total Environ. 32 (2), 147–156. doi: 10.1016/0048-9697(84)90127-X
Ojwang’ L. M., Cook R. L. (2013). Environmental conditions that influence the ability of humic acids to induce permeability in model biomembranes. Environ. Sci. Technol. 47 (15), pp.8280–8287. doi: 10.1021/es4004922
Ortiz V. L., Mason R. P., Ward J. E. (2015). An examination of the factors influencing mercury and methylmercury particulate distributions, methylation and demethylation rates in laboratory-generated marine snow. Mar. Chem. 177, 753–762. doi: 10.1016/j.marchem.2015.07.006
Paranjape A. R., Hall B. D. (2017). Recent advances in the study of mercury methylation in aquatic systems. Facets 2, 85–119. doi: 10.1139/facets-2016-0027
Parks J. M., Johs A., Podar M., Bridou R., Hurt R. A., Smith S. D., et al. (2013). The genetic basis for bacterial mercury methylation. Sci. (New York N.Y.) 339 (6125), 1332–1335. doi: 10.1126/science.1230667
Pavlovska M., Prekrasna I., Parnikoza I., Dykyi E. (2021). Soil sample preservation strategy affects the microbial community structure. Microbes Environments 36 (1), ME20134. doi: 10.1264/jsme2.ME20134
Perelomov L. V., Sarkar B., Sizova O. I., Chilachava K. B., Shvikin A. Y., Perelomova I. V., et al. (2018). Zinc and lead detoxifying abilities of humic substances relevant to environmental bacterial species. Ecotoxicol. Environ. Saf. 151, 178–183. doi: 10.1016/j.ecoenv.2018.01.018
Pérez-Cobas A. E., Gomez-Valero L., Buchrieser C. (2020). Metagenomic approaches in microbial ecology: an update on whole-genome and marker gene sequencing analyses. Microbial. Genomics 6 (8). doi: 10.1099/mgen.0.000409
Peterson B. D., McDaniel E. A., Schmidt A. G., Lepak R. F., Janssen S. E., Tran P. Q., et al. (2020). Mercury methylation genes identified across diverse anaerobic microbial guilds in a eutrophic sulfate-enriched lake. Environ. Sci. Technol. 54 (24), 15840–15851. doi: 10.1021/acs.est.0c05435
Pirrone N., Cinnirella S., Feng X., Finkelman R. B., Friedli H. R., Leaner J., et al. (2010). Global mercury emissions to the atmosphere from anthropogenic and natural sources. Atmospheric Chem. Phys. 10 (13), 5951–5964. doi: 10.5194/acp-10-5951-2010
Ploug H., Buchholz B., Jørgensen B. (1997). Anoxic aggregates an ephemeral phenomenon in the ocean. Aquat. Microbial. Ecol. 13, 285–294. doi: 10.3354/ame013285
Podar M., Gilmour C. C., Brandt C. C., Soren A., Brown S. D., Crable B. R., et al. (2015). Global prevalence and distribution of genes and microorganisms involved in mercury methylation. Sci. Adv. 1, e1500675. doi: 10.1126/sciadv.1500675
Poste A. E., Braaten H. F. V., de Wit H. A., Sørensen K., Larssen T. (2015). Effects of photodemethylation on the methylmercury budget of boreal Norwegian lakes. Environ. Toxicol. Chem. 34 (6), 1213–1223. doi: 10.1002/etc.2923
PROCTOR L. M. (1997). Nitrogen-fixing, photosynthetic, anaerobicbacteria associated with pelagic copepods. Aquat. Microb. Ecol. 12, 105–113. doi: 10.3354/ame012105
Quince C., Walker A. W., Simpson J. T., Loman N. J., Segata N. (2017). Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 35 (9), 833–844. doi: 10.1038/nbt.3935
Ranchou-Peyruse M., Monperrus M., Bridou R., Duran R., Amouroux D., Salvado J. C., et al. (2009). Over- view of mercury methylation capacities among anaerobic bacteria including representatives of the sulphate-reducers: implications for environmental studies. Geomicrobiol. J. 26, 1–8. doi: 10.1080/01490450802599227
Regnell O., Watras C. J. (2018). Microbial mercury methylation in aquatic environments: a critical review of published field and laboratory studies. Environ. Sci. Technol. 53, 4–19. doi: 10.1021/acs.est.8b02709
Regnell O., Watras C. J. (2019). Microbial mercury methylation in aquatic environments: A critical review of published field and laboratory studies. Environ. Sci. Technol. 53 (1), 4–19. doi: 10.1021/acs.est.8b02709
Ridley W. P., Dizikes L. J., Wood J. M. (1977). Biomethylation of toxic elements in the environment. Science. Science 197, 4301, 329–332. doi: 10.1126/science.877556
Ripszam M., Paczkowska J., Figueira J., Veenaas C., Haglund P. (2015b). Dissolved organic carbon quality and sorption of organic pollutants in the Baltic Sea in light of future climate change. Environ. Sci. Technol. 49, 1445–1452. doi: 10.1021/es504437s
Robinson J. B., Tuovinen O. (1984). Mechanisms of microbial resistance and detoxification of mercury and organomercury compounds: physiological, biochemical, and genetic analyses. Microbiol. Rev. 48 (2), 95–124. doi: 10.1128/mr.48.2.95-124.1984
Rodríguez J., Andersson A., Björn E., Timonen S., Brugel S., Skrobonja A., et al. (2022). Inputs of terrestrial dissolved organic matter enhance bacterial production and methylmercury formation in oxic coastal water. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.809166
Rodríguez J., Gallampois C. M., Timonen S., Andersson A., Sinkko H., Haglund P., et al. (2018). Effects of organic pollutants on bacterial communities under future climate change scenarios. Front. Microbiol. 9, 2926. doi: 10.3389/fmicb.2018.02926
Runtuwene L. R., Tuda J. S., Mongan A. E., Suzuki Y. (2019). On-site minION sequencing. In: ingle Molecule and Single Cell Sequencing; Advances in Experimental Medicine and Biology. Singapore: Springer, 1129, 143–150. doi: 10.1007/978-981-13-6037-4_10
Schaefer K., Elshorbany Y., Jafarov E., Schuster P. F., Striegl R. G., Wickland K. P., et al. (2020). Potential impacts of mercury released from thawing permafrost. Nat. Commun. 11 (1), 4650. doi: 10.1038/s41467-020-18398-5
Schaefer J. K., Kronberg R. M., Morel F. M., Skyllberg U. (2014). Detection of a key hg methylation gene, hgcA, in wetland soils. Environ. Microbiol. Rep. 6 (5), 441–447. doi: 10.1111/1758-2229.12136
Schartup A. T., Ndu U., Balcom P. H., Mason R. P., Sunderland E. M. (2015). Contrasting effects of marine and terrestrially derived dissolved organic matter on mercury speciation and bioavailability in seawater. Environ. Sci. Technol. 49 (10), 5965–5972. doi: 10.1021/es506274x
Schuster P. F., Schaefer K. M., Aiken G. R., Antweiler R. C., Dewild J. F., Gryziec J. D., et al. (2018). Permafrost stores a globally significant amount of mercury. Geophysical Res. Lett. 45 (3), 1463–1471. doi: 10.1002/2017GL075571
Seymour J. R., Amin S. A., Raina J. B., Stocker R. (2017). Zooming in on the phycosphere: the ecological interface for phytoplankton–bacteria relationships. Nat. Microbiol. 2 (7), 1–12. doi: 10.1038/nmicrobiol.2017.65
Shanks A. L., Reeder M. L. (1993). Reducing microzones and sulfide production in marine snow. Mar. Ecol. Prog. Ser. 96 (1), 43–47. doi: 10.3354/meps096043
Slaveykova V. I., Wilkinson K. J., Ceresa A., Pretsch E. (2003). Role of fulvic acid on lead bioaccumulation by Chlorella kesslerii. Environ. Sci. Technol. 37 (6), 1114–1121. doi: 10.1021/es025993a
Soerensen A. L., Schartup A. T., Gustafsson E., Gustafsson B. G., Undeman E., Bjoürn E. (2016). Eutrophication increases phytoplankton methylmercury concentrations in a coastal sea: a baltic sea case study. Environ. Sci. Technol. 50 (21), 11787–11796. doi: 10.1021/acs.est.6b02717
Sonke J. E., Heimbürger L.-E., Dommergue A. (2013). Mercury biogeochemistry: paradigm shifts, outstanding issues and research needs. Comptes Rendus Geosci. 345, 213–224. doi: 10.1016/j.crte.2013.05.002
Stern G. A., Macdonald R. W., Outridge P. M., Wilson S., Chetelat J., Cole A., et al. (2012). How does climate change influence arctic mercury? Sci. Total Environ. 414, 22–42. doi: 10.1016/j.scitotenv.2011.10.039
Stott P. (2016). How climate change affects extreme weather events. Science 352 (6293), 1517–1518. doi: 10.1126/science.aaf7271
Streets D. G., Devane M. K., Lu Z., Bond T. C., Sunderland E. M., Jacob D. J. (2011). All-time releases of mercury to the atmosphere from human activities. Environ. Sci. Technol. 45 (24), 10485–10491. doi: 10.1021/es202765m
Sunderland E. M., Krabbenhoft D. P., Moreau J. W., Strode S. A., Landing W. M. (2009). Mercury sources, distribution, and bioavailability in the north Pacific Ocean: Insights from data and models. Global Biogeochem. Cycles 23 (2), n/a–n14. doi: 10.1029/2008GB003425
Tada Y., Marumoto K., Takeuchi A. (2020). Nitrospina-like bacteria are potential mercury methylators in the mesopelagic zone in the east china sea. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.01369
Taira M. (1975). Studies on microbial synthesis and decomposition of organomercury compounds. Nippon Eiseigaku Zasshi (Japanese J. Hygiene) 30 (4), 461–489. doi: 10.1265/jjh.30.461
Tan S. W., Meiller J. C., Mahaffey K. R. (2009). The endocrine effects of mercury in humans and wildlife. Crit. Rev. Toxicol. 39 (3), 228–269. doi: 10.1080/10408440802233259
Tang K. W., Glud R. N., Glud A., Rysgaard S., Nielsen T. G. (2011). Copepod guts as biogeochemical hot- spots in the sea: Evidence from microelectrode profiling of Calanus spp. Limnol. Oceanography 56 (2), 666–672. doi: 10.4319/lo.2011.56.2.0666
Tsubaki T., Irukayama K. (1977). Minamata disease. Methylmercury poisoning in Minamata and Niigata, Japan (Amsterdam, The Netherlands: North-Holland Publishing Company, PO Box 211).
Tsui M. T. K., Blum J. D., Kwon S. Y. (2020). Review of stable mercury isotopes in ecology and biogeochemistry. Sci. Total Environ. 716, 135386. doi: 10.1016/j.scitotenv.2019.135386
Turner, Jefferson T. (2015). Zooplankton fecal pellets, marine snow, phytodetritus and the ocean’s biological pump. Prog. Oceanography 130, 205–2485. doi: 10.1016/j.pocean.2014.08.005
Tyler A. D., Mataseje L., Urfano C. J., Schmidt L., Antonation K. S., Mulvey M. R., et al. (2018). Evaluation of Oxford Nanopore’s MinION sequencing device for microbial whole genome sequencing applications. Sci. Rep. 8 (1), 1–12. doi: 10.1038/s41598-018-29334-5
Ullrich S. M., Tanton T. W., Abdrashitova S. A. (2001). Mercury in the aquatic environment: a review of factors affecting methylation. Crit. Rev. Environ. Sci. Technol. 31, 241–293. doi: 10.1080/20016491089226
Větrovský T., Baldrian P. (2013). The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PloS One 8 (2), e57923. doi: 10.1371/journal.pone.0057923
Vigneault B., Percot A., Lafleur M., Campbell P. G. (2000). Permeability changes in model and phytoplankton membranes in the presence of aquatic humic substances. Environ. Sci. Technol. 34 (18), pp.3907–3913. doi: 10.1021/es001087r
Vigneron A., Cruaud P., Aubé J., Guyoneaud R., Goñi-Urriza M. (2021). Transcriptomic evidence for versatile metabolic activities of mercury cycling microorganisms in brackish microbial mats. NPJ Biofilms Microbiomes 7 (1), 83. doi: s41522-021-00255-y
Villar E., Cabrol L., Heimbürger-Boavida. L.-E. (2020). Widespread microbial mercury methylation genes in the global ocean. Environ Microbiol Rep 12 (3), 277–287. doi: 10.1111/1758-2229.12829
Vishnivetskaya T. A. (2018). Microbial community structure with trends in methylation gene diversity and abundance in mercury-contaminated rice paddy soils in Guizhou, China. Environ. Sci. Process. Impacts 20, 673–685. doi: 10.1039/C7EM00558J
Wang K., Liu G., Cai Y. (2022). Possible pathways for mercury methylation in oxic marine waters. Crit. Rev. Environ. Sci. Technol. 52 (22), 3997–4015. doi: 10.1080/10643389.2021.2008753
Wang F., Macdonald R. W., Armstrong D. A., Stern G. A. (2012). Total and methylated mercury in the Beaufort Sea: The role of local and recent organic remineralization. Environ. Sci. Technol. 46 (21), 11821–11828. doi: 10.1021/es302882d
Wang K., Munson K. M., Armstrong D. A., Macdonald R. W., Wang F. (2020). Determining seawater mercury methylation and demethylation rates by the seawater incubation approach: A critique. Mar. Chem. 219, 103753. doi: 10.1016/j.marchem.2020.103753
Wang K., Munson K. M., Beaupre-Laperriere A., Mucci A., Macdonald R. W., Wang F. (2018). Subsurface seawater methylmercury maximum explains biotic mercury concentrations in the canadian arctic. Sci. Rep. 8 (1), 1–5. doi: 10.1038/s41598-018-32760-0
Wang J., Shaheen S. M., Jing M., Anderson C. W., Swertz A.-C., Wang S.-L., et al. (2021). Mobilization, methylation, and demethylation of mercury in a paddy soil under systematic redox changes. Environ. Sci. Technol. 55 (14), 10133–10141. doi: 10.1021/acs.est.0c07321
Weber J. H., Reisinger K., Stoeppler M. (1985). Methylation of mercury (II) by fulvic acid. Environ. Technol. 6 (1-11), 203–208. doi: 10.1080/09593338509384337
Wen Q., Su Z., Cai W., Huang W., Fan J., Huang H., et al. (2017). Recent progress on detection methods of methyl mercury. J. Food Saf. Qual. 8 (3), 845–853. doi: 20173134599
Westoo G. (1967). Determination of methylmercury compounds in foodstuffs. Acta Chem. Scand. 21, 1790–1800. doi: 10.3891/acta.chem.scand.21-1790
Whalin L., Kim E.-H., Mason R. (2007). Factors influencing the oxidation, reduction, methylation and deme- thylation of mercury species in coastal waters. Mar. Chem. 107 (3), 278–294. doi: 10.1016/j.mar-chem.2007.04.002
Wood J. M., Kennedy F. S., Wolfe R. S. (1968). Reaction of multihalogenated hydrocarbons with free and bound reduced vitamin B12. Biochemistry 7 (5), 1707–1713. doi: 10.1021/bi00845a013
Wu P., Kainz M. J., Valdés F., Zheng S., Winter K., Wang R., et al. (2021). Elevated temperature and browning increase dietary methylmercury, but decrease essential fatty acids at the base of lake food webs. Sci. Rep. 11 (1), 16859. doi: 10.1038/s41598-021-95742-9
Yao C., He T., Xu Y., Ran S., Qian X., Long S. (2020). Mercury bioaccumulation in zooplankton and its relationship with eutrophication in the waters in the karst region of Guizhou Province, Southwest China. Environ. Sci. pollut. Res. 27, 8596–8610. doi: 10.1007/s11356-019-07479-8
Yap M., Ercolini D., Álvarez-Ordóñez A., O'Toole P. W., O'Sullivan O., Cotter P. D. (2022). Next-generation food research: use of meta-omic approaches for characterizing microbial communities along the food chain. Annu. Rev. Food Sci. Technol. 13, .361–.384. doi: 10.1146/annurev-food-052720-010751
Zhang Y., Jacob D. J., Horowitz H. M., Chen L., Amos H. M., Krabbenhoft D. P., et al. (2016). Observed decrease in atmospheric mercury explained by global decline in anthropogenic emissions. Proc. Natl. Acad. Sci. 113, 526–531. doi: 10.1073/pnas.1516312113
Keywords: mercury methylation, oxic waters, climate change, omic technologies, microbial communities
Citation: Rodríguez J (2023) Mercury methylation in boreal aquatic ecosystems under oxic conditions and climate change: a review. Front. Mar. Sci. 10:1198263. doi: 10.3389/fmars.2023.1198263
Received: 31 March 2023; Accepted: 10 July 2023;
Published: 02 August 2023.
Edited by:
Jacob Carstensen, Aarhus University, DenmarkCopyright © 2023 Rodríguez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juanjo Rodríguez, amoucnNlcnJhbm9AZ21haWwuY29t