 Yuqian Li
Yuqian Li Tifeng Shan
Tifeng Shan Shaojun Pang
Shaojun Pang- 1CAS and Shandong Province Key Laboratory of Experimental Marine Biology, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China
- 2Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao, China
- 3Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
- 4University of Chinese Academy of Sciences, Beijing, China
According to historical documents, China has its own native Undaria pinnatifida populations in Zhejiang and Fujian provinces, and they are potentially valuable breeding materials for this economically important kelp. However, their distribution range has been declining northward and we cannot find it now in Fujian province and southern Zhejiang province. They are currently confirmed to be distributed only on some small islands distant from the mainland, likely facing the anthropogenic threats such as habitat deterioration and marine heatwave. Thus, they are of conservation concern. Herein, we used the mitochondrial sequences of cox3 and tatC–tLeu regions, and 10 nuclear microsatellites to analyze the genetic diversity and genetic structure of three temporal populations from Gouqi Island and one population from Yushan Island. A total of eight haplotypes of the combined cox3 and tatC–tLeu sequences were identified with five of them being unique in these populations. Based on microsatellites, we estimated a minimum expected heterozygosity of 0.671 and a minimum effective number of alleles of 5.3 within each population, and revealed shallow genetic structure among populations by Bayesian model-based STRUCTURE analysis and low Fst values. We did not detect signs of recent bottleneck in all populations. The high genetic diversity identified in the southern native populations will provide an advantageous basis for them to adapt to the future environmental changes. A thorough field survey about the detailed distribution of the native populations is needed in order to estimate their population size and better protect the natural resources of this important alga.
Introduction
Undaria pinnatifida is an economically important kelp (Laminariales) species native to the Northwest Pacific and extensively farmed in Japan, Korea and China (Yamanaka and Akiyama, 1993). Its annual yield in China has been maintained ca. 200 thousand tonnes in recent years, and the major farmed regions are Liaoning and Shandong provinces in northern China (Fishery Bureau of Ministry of Agriculture and Rural Affairs, 2022).
Despite the large-scale production of this alga in China, the cultivated stock has originated from Japan and Korea since its small-scale cultivation experiments in Dalian in the 1940s and subsequent large-scale commercial farming in the 1980s in northern China (Qu, 1993; Tseng, 2001). A large proportion of U. pinnatifida products in China are exported to Japan, and thus, the industry of this alga is largely oriented towards the requirement of Japanese market. As a consequence, introduction of the cultivated stock from Japan has been frequently conducted by Chinese kelp-farming companies to maintain desirable economic traits of the farmed populations (Qu, 1993). It has been documented that there is no native U. pinnatifida in northern China although at present its natural populations are extensively distributed on rocky coasts of the northern Yellow Sea and Bohai Sea such as in Dalian, Qingdao, Yantai and Weihai (Tseng and Zhang, 1952; Tseng, 2001). The natural populations from rocky shores in Dalian and Qingdao originated from intentional transplantation from Korea in the 1930s, and those in Yantai and Weihai were secondarily derived from the transplanted populations in Dalian and Qingdao (Li, 1991). Previous genetic analysis based on mitochondrial sequences indicated that the natural populations of U. pinnatifida from rocky shores in Dalian and Qingdao belonged to the Continental type, and the farmed populations were classified as the Northern Japan type (Shan et al., 2022), which is consistent with the above-mentioned historical documents about the sources of farmed and natural populations (Li, 1991; Qu, 1993; Tseng, 2001). In addition, previous studies based on mitochondrial sequences and microsatellite markers revealed high genetic diversity within the farmed and subtidal natural populations in northern China and high genetic differentiation between these two kinds of populations (Shan et al., 2018; Li et al., 2020; Shan et al., 2022).
Different from the situation in northern China, it was documented that southern China has its native distribution of U. pinnatifida off the coasts of Zhejiang and Fujian provinces, although details about its distribution range in Fujian were not elaborated (Tseng, 2001). The historically documented distribution of this alga in Zhejiang province included a narrow and long chain of small islands from the north in Shengsi Islands to the south in Qixing Island which are all distant from the main land (Sun and Hang, 1976; Zhang et al., 1983). However, natural U. pinnatifida individuals has not been observed recently in Fujian and southern Zhejiang such as Nanji Island which previously had distribution of this alga (Zhang et al., 1983). Thus, U. pinnatifida populations have been declining in its southern distribution range, as have its populations in Japan and other kelp forests around the world (Tamaki et al., 2009; Diehl et al., 2021). Native natural populations of U. pinnatifida are currently affirmed to be distributed on Gouqi Island in Shengsi county (i.e. Shengsi Islands) and Yushan Island in Xiangshan county, with the latter probably being its current southernmost distribution limit in China (Figure 1). The previous studies showed that the natural population from Gouqi Island harbored haplotypes belonging to both the Continental and the Northern Japan types according to mitochondrial sequences, and it is genetically differentiated from the farmed and natural populations in northern China (Shan et al., 2022). The southern native populations are therefore potentially useful as breeding materials to obtain heterosis, although they have not yet been exploited for breeding purposes. Theoretically, isolated island populations with limited gene exchange with other populations tend to have small effective population size, and are therefore more easily impacted by genetic drift, resulting in decrease in genetic variation. Moreover, negative impacts associated with human activity such as deterioration of their habitats and marine heatwave may bring about local bottleneck, further reducing their population size and genetic diversity. Given the above considerations, the southern native populations of U. pinnatifida need to be protected from the perspective of conservation. Evaluation of genetic diversity is a crucial step towards making conservation strategies as the level of genetic variations within a population usually determines its adaptive potential to changing environment in the future.
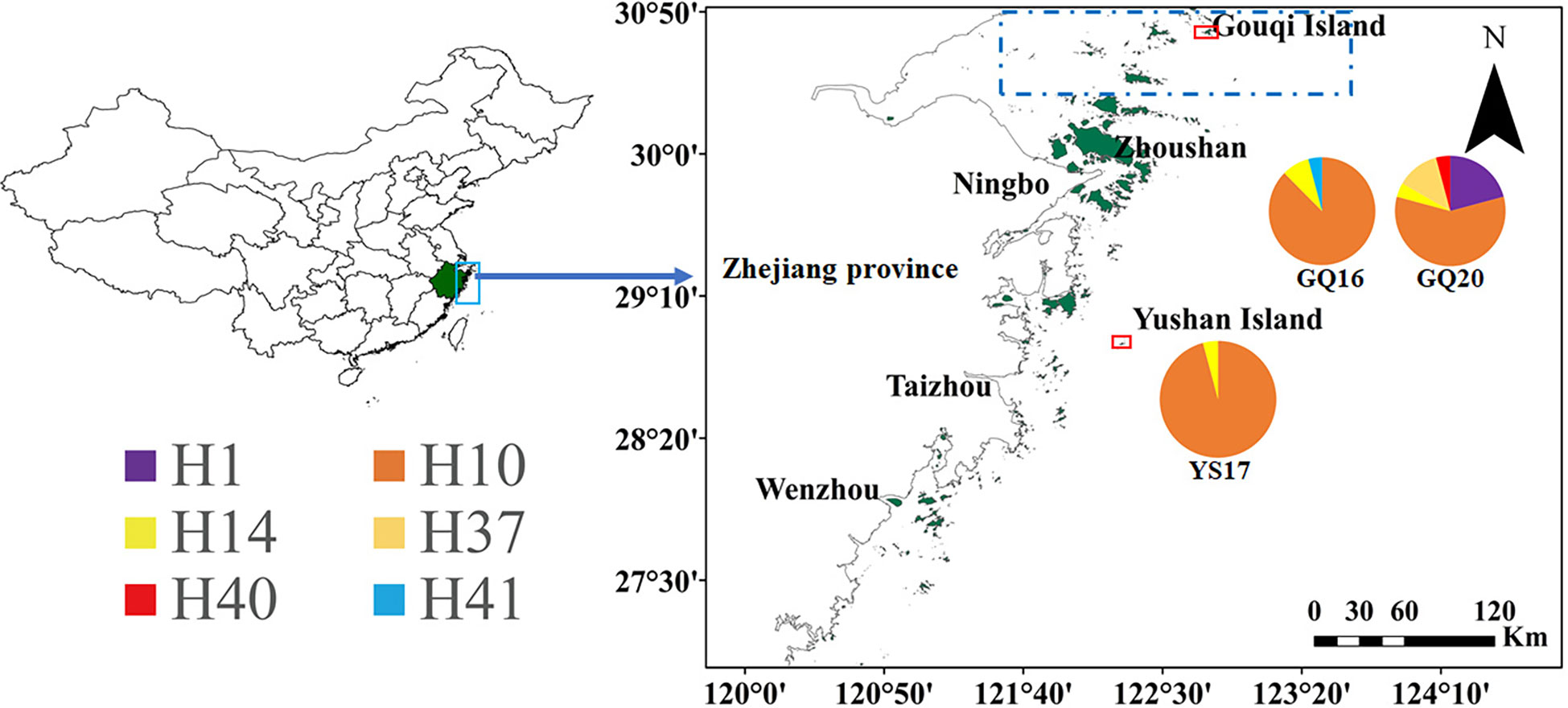
Figure 1 Geographic locations of Gouqi and Yushan Islands (indicated by red rectangle) in Zhejiang province, China, and the haplotypes of the combined cox3 and tatC–tLeu sequences in two temporal populations of Undaria pinnatifida from Gouqi Island (GQ16 and GQ20) and one population from Yushan Island (YS17). The color areas in the pie charts are proportional to the haplotype frequency. Dashed rectangle indicates the rough range of Shengsi Islands.
We previously analyzed the genetic diversity of two temporal populations of U. pinnatifida sampled from Gouqi Island, one in 2016 using microsatellites (Shan et al., 2018), and the other in 2021 using mitochondrial and nuclear DNA sequences (Shan et al., 2022). High genetic diversity was detected in both studies. However, genetic diversity and differentiation among temporal populations have not been compared due to different methods being used, and the population from Yushan Island has not yet been analyzed. In the present study we evaluated the genetic diversity and genetic structure of three temporal populations of U. pinnatifida from Gouqi Island and one population from Yushan Island using both mitochondrial sequences and nuclear microsatellite loci, aiming at guiding future plans on conservation of the native stock resources and their rational exploitation in breeding programs for this economically important alga.
Materials and methods
Sample collection and DNA extraction
Three temporal natural populations of Undaria pinnatifida were sampled from Gouqi Island (30°42’N, 122°45’E) in 2016, 2020 and 2021 (designated GQ16, GQ20 and GQ21), and one natural population was sampled from Yushan Island (N28°53.3′, E122°16.5′) in 2017 (designated YS17) in Zhejiang province, China (Figure 1). The direct distance between Gouqi and Yushan Islands was about 200 km. GQ20 and GQ21 were collected from subtidal rocky reefs at the same locality, and GQ16 was from the nearby plastic buoys (about 50 m from the locality of subtidal population) that were used to float the rafts for mussel farming (Shan et al., 2018). Because there was no cultivated U. pinnatifida in this island, GQ16 was considered to be derived from the nearby subtidal population. We planned on sampling the populations at different years in order to investigate their temporal changes in genetic diversity. However, our efforts to collect samples from Yushan Island in 2020 and 2021 were hindered by the COVID-19 pandemic which led to limited traffic to this island. Thus, the temporal populations sampled in these two islands were uneven. Individuals were sampled with a distance between any two of them being at a minimum of three meters to reduce the probability of collecting relatives. We had performed microsatellite genotyping for GQ16 and mitochondrial DNA sequencing for GQ21 (Shan et al., 2018; Shan et al., 2022). Hence, GQ16, GQ20 and YS17 were used for mitochondrial DNA sequencing, and GQ20, GQ21 and YS17 were used for microsatellite genotyping in the present study. A small part of the frond (about two grams in wet weight) from each individual was cleaned and preserved in silica gels. Genomic DNA was extracted from 24 dry specimens of each population using a plant genomic DNA isolation kit (DP305, Tiangen Biotech Co., Ltd, China). The present and former data were combined together for integrated analysis, which allowed us to compare the genetic diversity retrieved between 2016 and 2021 in Gouqi Island.
Analysis of mitochondrial DNA sequences
The partial cox3 gene and the tatC–tLeu region were amplified using the primers and PCR programs of Uwai et al. (2006). The primers were CAF4A (5’-ATGTTTACTTGGTGRAGRGA-3’) and CAR4A (5’-CCCCACCARTAWATNGTNAG-3’) for the cox3 gene, and tatCEF (5’-AAATAATATATTGAGATTTTAAGTCTATTCAT-3’) and tLeuR (5’-AACCTAAACACCGCGTGTATACC-3’) for the tatC-tLeu region. The PCR reaction was performed using a kit of Taq Master Mix (Accurate Biology, China) with a T-gradient thermocycler (Biometra, Germany). The PCR products were sequenced in both directions with forward and reverse primers on an ABI 3730XL automated sequencer (Applied Biosystems, USA) at Beijing Tsingke Biotechnology Co., Ltd.
Consensus sequences were obtained using the BioEdit v7.0.9.0 according to the forward and reverse sequencing results for the cox3 and tatC–tLeu regions, respectively. They were aligned with the sequences in the previous studies (Uwai et al., 2006; Shan et al., 2022) to determine the haplotypes present in the southern native populations from China. The aligned sequences of cox3 and tatC–tLeu regions were then joined to form a concatenated alignment. Haplotypes were examined using DnaSP v5.10 (Rozas et al., 2003). The number of haplotypes (Nh), haplotype diversity (h), and nucleotide diversity (π) were analyzed with ARLEQUIN v3.11 (Excoffier et al., 2005). The pairwise Fst coefficients among populations were calculated using ARLEQUIN with 1000 permutations to assess the levels of genetic differentiation. The “BH” method (Benjamini and Hochberg, 1995) in the p.adjust function in R was used to adjust the false discovery rate for multiple comparisons. The adjusted P-value < 0.05 was considered significant.
Microsatellite genotyping and data analysis
Ten microsatellites (UPN1528, 3197, 5085, 5354, 5463, 5577, 5613, 7150, 9919 and 10532) were used for genetic diversity and genetic structure analyses. PCR reaction and microsatellite genotyping were performed according to the procedures of Shan et al. (2018).
The numbers of alleles (Na) and effective alleles (Ne), observed and expected heterozygosity (Ho and He), and inbreeding coefficient Fis at each microsatellite locus were computed using GenAlEx 6.5 (Peakall and Smouse, 2012). The probability for Hardy-Weinberg equilibrium (HWE) at each locus was tested using ARLEQUIN. The population selfing rate g2 was estimated in inbreedR (Stoffel et al., 2016). The Fst values between pairs of populations were computed using ARLEQUIN with 1,000 permutations. In order to evaluate the genetic relationship between the four southern native populations (GQ16, GQ20, GQ21 and YS17) and the subtidal natural populations in northern China, two populations namely QD16 and SW18 from the previous studies (Shan et al., 2018; Li et al., 2020) were included for genetic structure analysis. A Bayesian-algorithm clustering analysis was run in STRUCTURE v2.3.4 to assess the most likely number of genetic clusters based on allele frequencies (Pritchard et al., 2000). The admixture model was implemented and the number of clusters (K value) was set from 1 to 6 with 20 independent runs for each K value. Each run consisted of a burn-in length of 100,000 followed by 1,000,000 Monte Carlo Markov Chain repetitions. The results files were submitted to STRUCTURE HARVESTER (Earl and Vonholdt, 2012) to estimate the most likely K value. Results of the 20 repetitive runs for the most likely K value were combined in CLUMPP v1.1.2 and represented as bar plots using the software DISTRUCT 1.1 (Rosenberg, 2004). The STRUCTURE analysis was also only run for the four native populations to further analyze their genetic structure with K value set from 1 to 4 and all other parameters the same as mentioned above. The genetic relationship among the four southern populations as well as their relationship with the two northern natural populations were also evaluated with Principal Coordinates Analysis (PCoA) according to Nei’s standard genetic distance (Nei, 1972) between populations using GenAlEx 6.5.
The Bottleneck software v1.2.02 (Piry et al., 1999) was used to estimate whether the populations have experienced a recent reduction in effective population size. The estimation is based on comparison between He and the expected equilibrium heterozygosity (Heq), and an excess of He (He > Heq) suggests a recent bottleneck occurring in the population. A TPM model (two-phase model) was applied with 95% single-step mutations and 5% multiple-step mutations (with a variance of 12) as recommended for microsatellites (Piry et al., 1999), and 1000 simulation iterations were performed. A Wilcoxon’s test was used to estimate whether there was a significant excess of He at P < 0.05 level.
Results
Genetic diversity
After alignment with the haplotypes in Uwai et al. (2006), the length of cox3 sequences was 470 bp for the populations GQ16, GQ20 and YS17. Two haplotypes were found in GQ20, with the values of h and π being 0.083 and 0.00018, respectively (Table S1). One of them was identical to the haplotype with a GenBank accession number of AB213030, which was dominant in the population (in 23 of 24 specimens). The other was novel with an accession number of ON809500. Only one haplotype was detected in GQ16 and YS17, being identical to the haplotype with the accession number of AB213030. After further alignment with the haplotypes used in Shan et al. (2022), the sequences of cox3 were 431 bp in length.
The length of aligned sequences of the tatC–tLeu region was 410 bp. Two to four haplotypes were detected in each of the three populations, with the values of h ranging from 0.083 to 0.573 and π from 0.0002 to 0.0176 (Table S1). A total of five haplotypes were detected, with four of them being the same as the haplotypes with the accession numbers of AB240644, AB240652, AB240655 and OL580829, respectively, and the other one being novel with an accession number of ON809501.
The alignment of the concatenated sequences of cox3 and tatC–tLeu comprised 841 bp. A total of six haplotypes were detected in the three native populations (Figure 1), with two to five haplotypes being detected in each population. The h ranged from 0.083 to 0.623 and π from 0.0001 to 0.00827, with values being lowest in YS17 and highest in GQ20 (Table 1). In addition to four haplotypes (H1, H10, H14 and H37) present in the previous studies (Uwai et al., 2006; Shan et al., 2022), two novel haplotypes were detected and accordingly designated H40 and H41 (Table 2), with each of them appearing in one specimen of GQ20 and GQ16, respectively. H10 was the dominant haplotype in all the three populations, appearing in a total of 58 individuals (81%). H40 and H41 had a 20-bp insertion in the intergenic region between tTrp and tIle within the tatC-tLeu region, and thus, they belonged to the Continental type (Uwai et al., 2006). Each of them had a base mutation in comparison with H10.
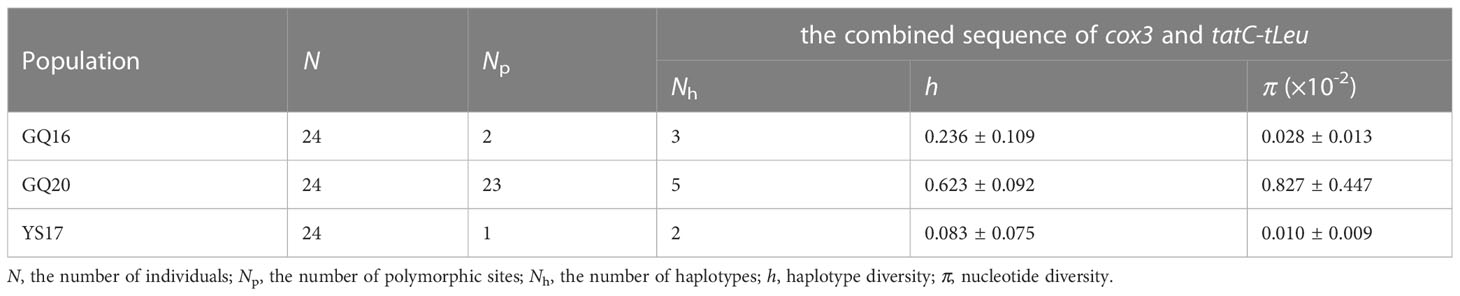
Table 1 Genetic diversity of three Chinese native populations of Undaria pinnatifida estimated by using the combined sequences of cox3 gene and tatC-tLeu region.
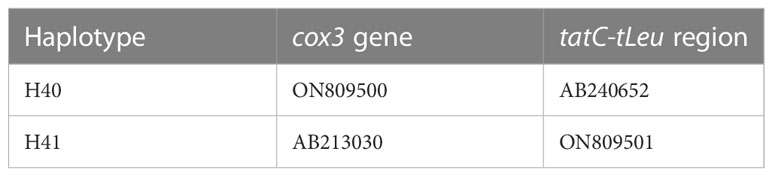
Table 2 Novel haplotypes of the combined sequences of cox3 and tatC-tLeu detected in Chinese native populations of Undaria pinnatifida and the corresponding combination of the accession numbers.
The values of Na, Ho, He and Fis for each population at each microsatellite locus are shown in Table 3. Significant departure from the HWE (P < 0.05) was detected at three loci of GQ20 (UpN5354, UpN5463 and UpN5613), two loci of GQ21 (UpN5354 and UpN7150) and three loci of YS17 (UpN5354, UpN5577 and UpN5613), all with a positive Fis value indicating heterozygosity deficiency. The average values of Na, Ho and He across the 10 microsatellites were highest in YS17 and lowest in GQ21. In order to compare the allelic diversity with those populations already investigated in the previous study, the Ne values were also estimated, which ranged from 5.3 to 7.4 in the four native populations (Table 4). Among all the natural populations analyzed, only GQ16 had a selfing rate (g2) significantly deviating from zero (P < 0.05).
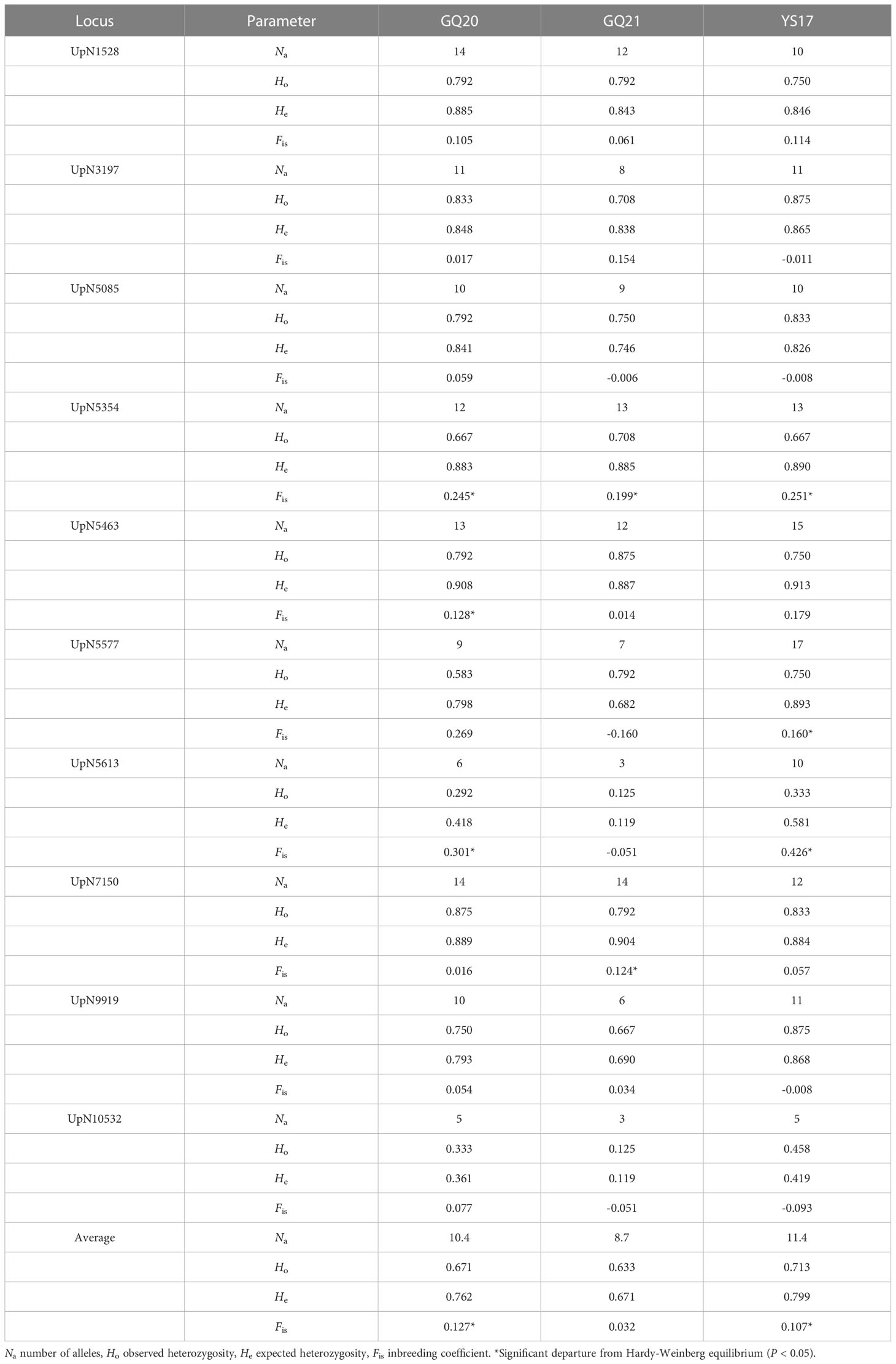
Table 3 Genetic diversity of native Undaria pinnatifida populations from southern China at 10 microsatellite loci.
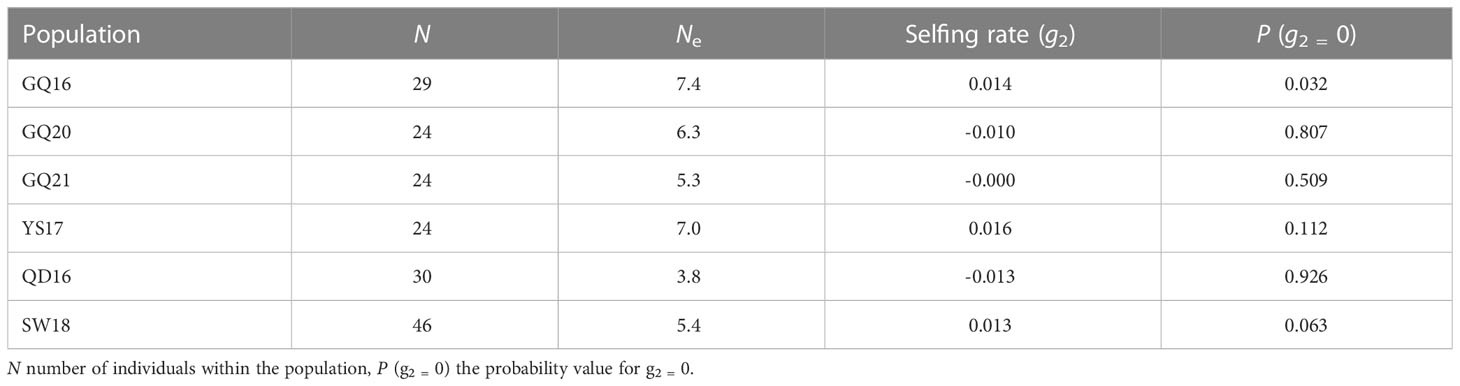
Table 4 Number of effective alleles and selfing rates of the native populations of Undaria pinnatifida from southern China and the control subtidal natural populations from northern China.
The P values of one-tailed Wilcoxon’s tests were all larger than 0.05 for the four populations (Table 5). Hence, the null hypothesis of no significant heterozygosity excess across loci could not be rejected, suggesting no signs of bottleneck for the four native populations of U. pinnatifida.
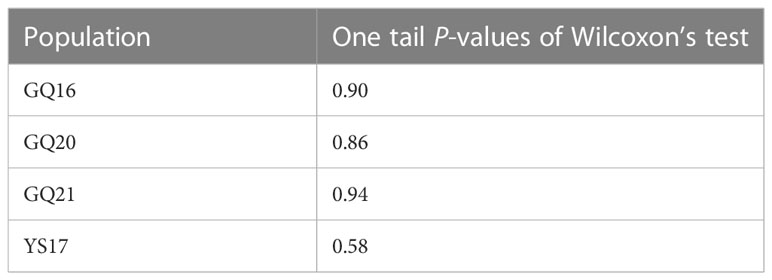
Table 5 One tail probability values of Wilcoxon’s tests for He excess across 10 microsatellite loci in four native populations of Undaria pinnatifida from China.
Genetic structure
According to the combined sequences of cox3 gene and tatC-tLeu region, no genetic differentiation (Fst = 0) was detected between GQ16 and YS17, or between GQ20 and GQ21 (Table 6). Significant pairwise Fst values were found between all other pairs of populations, with the Fst value being highest between YS17 and GQ21.
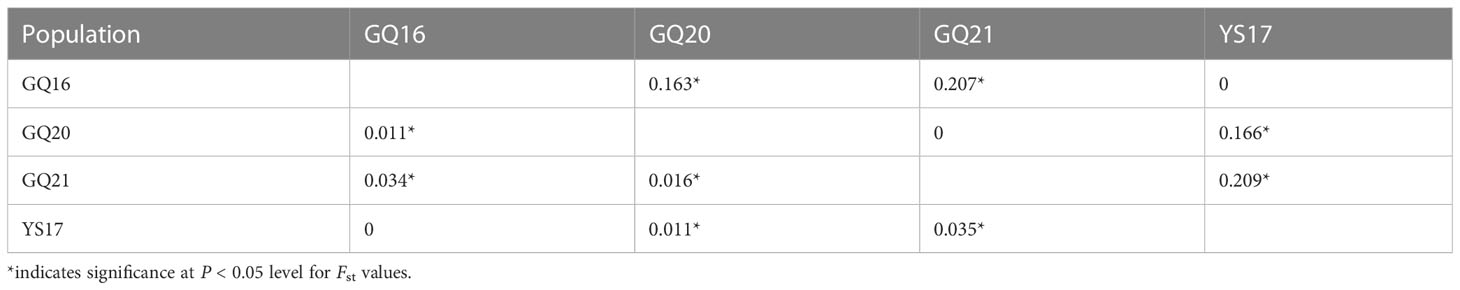
Table 6 Pairwise Fst values between four native populations of Undaria pinnatifida from China estimated using the combined sequences of cox3 gene and tatC-tLeu region (above diagonal) and microsatellites (below diagonal).
Based on microsatellite analysis, there was either no genetic differentiation (Fst = 0) between GQ16 and YS17, and the Fst value between YS17 and GQ21 was also highest (Table 6). Except for the Fst between GQ16 and YS17, Fst values between all other pairs of populations were statistically significant. The Pearson correlation coefficient between the Fst values estimated from mitochondrial sequences and microsatellites was 0.69, suggesting that the results obtained by the two methods are generally consistent.
When using QD16 and SW18 as the controls to analyze the genetic structure, the most likely number of K was determined to be four by the use of STRUCTURE HARVESTER according to the ΔK values. Individuals of the two northern populations QD16 and SW18 were classified as two respective clusters, distinctively separated from the southern populations (Figure 2A). The four native populations were divided into other two clusters, with GQ21 dominated by one cluster, and the other three populations showing marked admixture, in particular GQ16 and YS17. When only the four southern native populations were analyzed, the most likely number of K was two, and all the four populations showed marked admixture of the two clusters (Figure 2B).
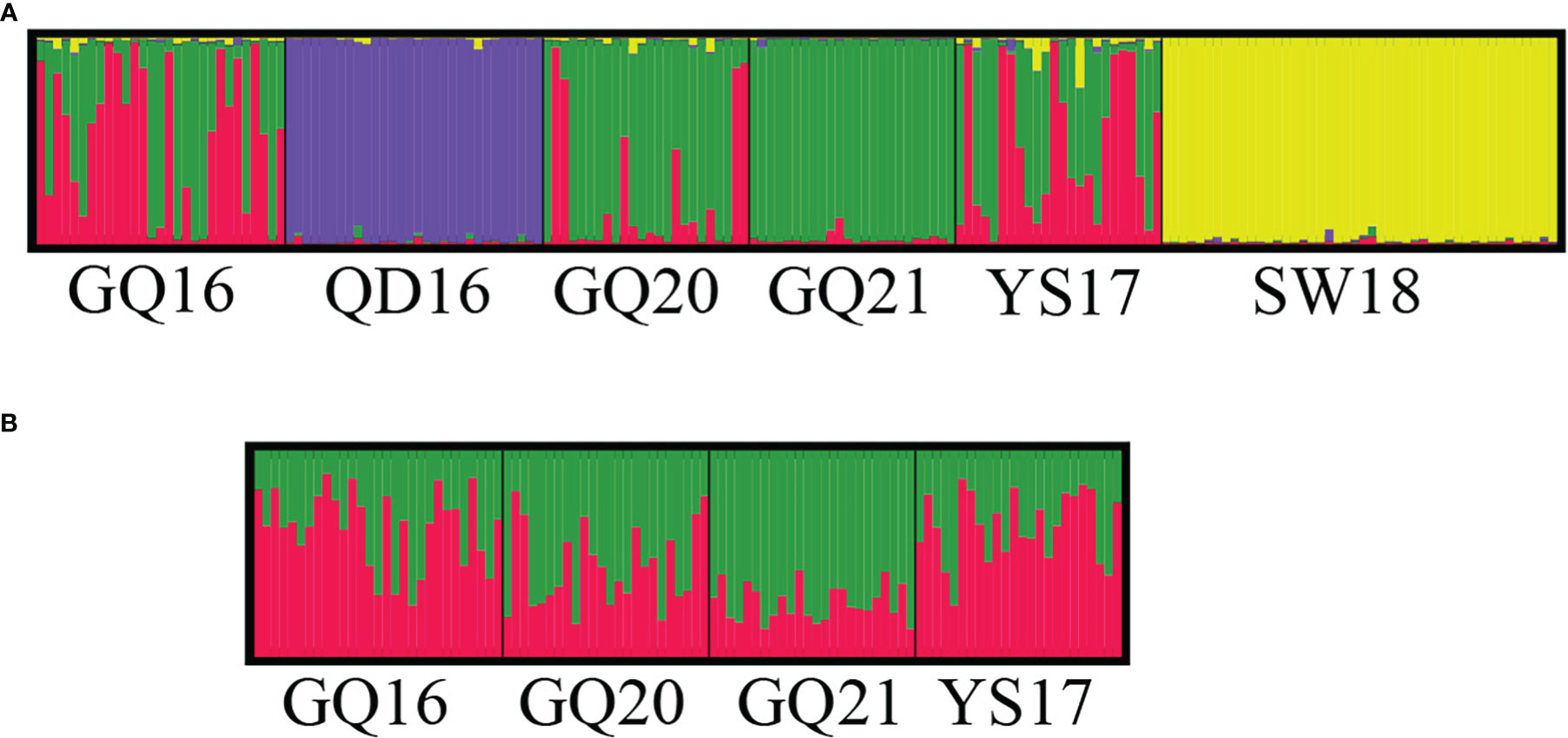
Figure 2 The genetic structure of (A) four native populations of Undaria pinnatifida from southern China and two subtidal natural populations from northern China, and (B) only four southern native populations, estimated using the STRUCTURE program. The vertical colored bars represent individuals within the population and the proportion of the color of each bar indicates the probability of membership in the corresponding cluster.
Three distinct clusters were revealed by the PCoA analysis, corresponding to SW18, QD16 and the four native populations, respectively (Figure 3). Within the native populations, GQ16 and YS17 were closest to each other, GQ21 was the farthest from others, and GQ20 was in the middle of them.
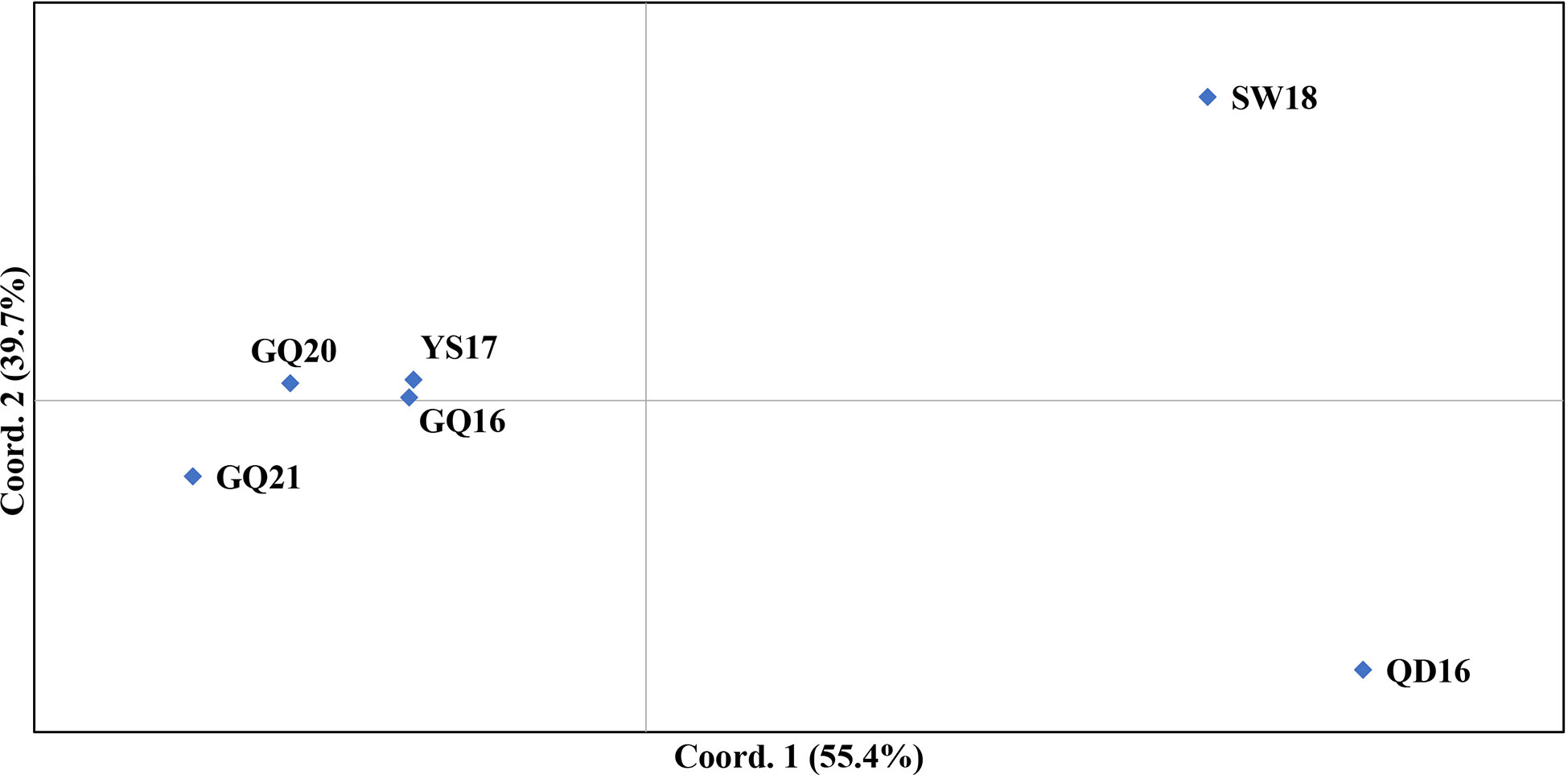
Figure 3 PCoA analysis result of the four native populations of Undaria pinnatifida from southern China and two subtidal natural populations from northern China. Each population is represented by a diamond.
Discussion
In the previous study, five haplotypes of the combined sequences of cox3 and tatC-tLeu were identified in GQ21 with an h value of 0.580 (Shan et al., 2022). In comparison with GQ21, the values of Nh and h were similar in GQ20, and lower in GQ16 and YS17. Three novel haplotypes (H37, H38 and H39) were previously detected in GQ21 compared to the haplotypes reported in Uwai et al. (2006), and another two novel haplotypes were identified in the populations from Gouqi Island in the present study (H40 and H41). Therefore, the total Nh value in the four native populations of China was eight with five of them being unique. By contrast, only four haplotypes were detected in natural populations on rocky coasts of northern China, and none of them was unique. The large number of unique haplotypes (H37-H41) found in the southern populations likely resulted from their long-term independent evolution on islands that are distant from other distribution ranges, which again is consistent with the document that these populations are native to China. A haplotype network containing H1-H39 had been constructed based on the concatenated sequences of cox3 and tatC–tLeu in the previous study, which clearly showed the genetic relationship between Chinese populations and Japanese and Korean populations (Shan et al., 2022). Chinese natural populations were grouped with the Continental type or the Northern Japan type. The Northern Japan type and the Continental type differed by an indel of 20 bp in the intergenic region between tTrp and tIle within the tatC-tLeu region, with the Northern Japan type having a 20-bp deletion (alternatively speaking, the Continental type having a 20-bp insertion). The Continental type was found in Korea and China, and the Northern Japan type was distributed in Hokkaido and Pacific northern Honshu of Japan according to Uwai et al. (2006). The two novel haplotypes H40 and H41 have the 20-bp insertion and each of them had one base mutation compared to H10. Hence, they belong to the Continental type. Taken together, only the Continental type and the Northern Japan type were detected in the four southern native populations (GQ16, GQ20, GQ21 and YS17), with the Continental type accounting for 88.5% of the total samples, and thus being dominant in these populations. Their haplotype component is similar to those detected in the western coast of Korea, suggesting that native populations from southern China and western Korea likely have the same origin.
Besides the native populations of U. pinnatifida from China, only one native population from other native countries (Nagasaki, Japan) has been analyzed with microsatellites (Daguin et al., 2005), which revealed low genetic diversity as shown by the average Na and He values (Na = 2.47 and He = 0.179 across 20 loci). Because such a level of genetic diversity was even lower than that of the introduced countries, it could not represent the level of genetic diversity in Japan. Hence, we are not able to compare the genetic diversity between native populations from China and those from Japan or Korea based on microsatellites. We previously detected high genetic diversity in terms of Na, Ho and He in natural and farmed populations in northern China using microsatellites (Shan et al., 2018; Li et al., 2020). The average He across the 10 loci ranged from 0.721 to 0.795 in subtidal natural populations, from 0.821 to 0.874 in natural populations on the cultivation rafts, and from 0.737 to 0.791 in farmed populations. The He of GQ16 was estimated to be 0.802 in the previous study (Shan et al., 2018). By comparison, the He values of the four native populations were similar to those of subtidal natural populations in northern China, and the Ne values of the four native populations were higher than or equal to those of subtidal natural populations in northern China. The genetic diversity of the four native populations is higher than that detected in all the introduced populations in Europe, although different sets of microsatellite loci were used (Guzinski et al., 2018; Shan et al., 2019).
Genetic diversity is the ultimate basis of all evolutionary change, and thus plays a critical role in the long-term persistence of a population and usually determines its adaptability to the future environmental changes (Alsuwaiyan et al., 2021). Small and isolated populations are susceptible to genetic drift, which can result in low genetic diversity (reduced allelic richness and heterozygosity) and increased inbreeding (Frankham et al., 2014). Fitness of such populations are more likely to be impaired by inbreeding depression, making them more vulnerable to anthropogenic stressors. The endangered brown macroalga Nereia lophocladia in Australia is an example of such populations (Mamo et al., 2021). This alga was shown to have low genetic diversity (He = 0.055) and high inbreeding rate (Fis = 0.721) within populations and high genetic differentiation among populations by using SNP markers. It was suggested that the low genetic diversity and high level of inbreeding render this species vulnerable to predicted ocean change (Mamo et al., 2021). By comparison, we revealed high genetic diversity in the Chinese native populations of U. pinnatifida based on mitochondrial and nuclear markers. Also, we did not detect signs of bottleneck in all the four native populations, suggesting that there has been no significant recent reduction of effective population size in these populations. According to these findings, we suggest that the effective population size of U. pinnatifida from Gouqi and Yushan Islands is either large, or otherwise, they have enough gene flow with other neighboring populations to counterbalance the negative impact of genetic drift on genetic diversity. Zhang et al. (1983) reported the existence of U. pinnatifida on Langgang Island and Zhongjieshan Islands in addition to Shengsi Islands and Yushan Island. We have also confirmed its distribution on Lühua Island and certain reefs near it in the northwest of Gouqi Island (K. Wang, personal communication, 2022). Thus, there is likely a continuous distribution of U. pinnatifida on a number of small islands and reefs within the long and narrow range between Shengsi Islands and Yushan Island. Such a continuous distribution along these islands and the northward and southward currents within this region may make gene flow among populations possible, thus accounting for the shallow genetic structure (i.e., low genetic differentiation revealed by microsatellites) between the populations from Gouqi and Yushan Islands. The small amount of genetic difference among the three temporal populations from Gouqi Island revealed by microsatellites was likely due to genetic drift. A field survey is needed to elucidate the detailed distribution range and assess the census size of the native populations of U. pinnatifida in this region, and it will be useful to help us understand the genetic diversity and population structure revealed in the present study. Despite the high He values of all the four native populations, the mean Fis values were significantly higher than zero (heterozygosity deficiency) across the 10 microsatellite loci in GQ20 and YS17, suggesting probable occurrence of inbreeding within these two populations. The selfing rate was significant for GQ16, suggesting that selfing likely existed in this population.
The genetic structure revealed by mitochondrial and microsatellite markers is generally consistent. The genetic structure and the genetic relationship estimated by programs of STRUCTURE and PCoA based on microsatellites are also consistent, both revealing that the four native populations are genetically close but different than the natural populations from northern China. The comparatively higher Fst values between GQ16 or YS17 and GQ20 or GQ21 estimated by mitochondrial sequences was largely attributed to the difference of the 20-bp indel between the Continental and Northern Japan types because haplotypes of the Northern Japan type were present in GQ20 (H1) and GQ21(H1 and H38) but absent in GQ16 and YS17. When we regarded the 20-bp indel as a single mutational event, the Fst values became much smaller (data not shown). Moreover, the temporal populations sampled later (GQ20 and GQ21) contained several more haplotypes than that sampled earlier (GQ16) from Gouqi Island. Because it was impossible for the population to evolve these new haplotypes in such a short period, the earlier sample size proved to be not big enough to encompass the haplotypes identified later. Hence, Fst values among the temporal populations were likely overestimated by the mitochondrial sequences due to small sample size. There was discrepancy between mitochondrial and microsatellite markers for evaluation of genetic diversity. Mitochondrial sequences revealed GQ20 and GQ21 having higher genetic diversity, while microsatellites revealed GQ16 and YS17 having higher genetic diversity. This might be due to the different inheritance pattern between the two kinds of markers. Because the haploid mitochondrial genome is maternally inherited in kelps (Li et al., 2015), the genetic diversity estimated by mitochondrial sequences may be more easily impacted by a small sample size. A larger sample size is needed for future study to reduce the biases caused by the small sample size in the present study.
In conclusion, we found high genetic diversity, shallow genetic structure and no signs of recent bottleneck in native populations of U. pinnatifida from southern China. The high genetic diversity will provide an advantageous basis for these populations to adapt to the future environmental changes. Caution will still be needed especially for the populations in the southernmost limit because they must bear the brunt of ocean warming and heatwave. Ex situ establishment of gametophyte stock is suggested to be prioritized to reduce the risk of possible devastating damage of extreme weather on the native populations of this important alga.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
TS conceived the study. YL extracted DNA and conducted the PCR. TS and YL performed the genetic analysis, and wrote the draft. SP reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the National Natural Science Foundation of China (No. 42176100), Biological Resources Program from Chinese Academy of Sciences (KFJ-BRP-017-27), China Agriculture Research System (CARS-50), and the Foundation for Huiquan Scholar of Institute of Oceanology, Chinese Academy of Sciences.
Acknowledgments
We would like to thank K. Wang and Q. Sun for their help in sample collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1122058/full#supplementary-material
References
Alsuwaiyan N. A., Vranken S., Filbee-Dexter K., Cambridge M., Coleman M. A., Wernberg T. (2021). Genotypic variation in response to extreme events may facilitate kelp adaptation under future climates. Mar. Ecol. Prog. Ser. 672, 111–121. doi: 10.3354/meps13802
Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Daguin C., Voisin M., Engel C., Viard F. (2005). Microsatellites isolation and polymorphism in introduced populations of the cultivated seaweed Undaria pinnatifida (Phaeophyceae, laminariales). Conserv. Genet. 6, 647–650. doi: 10.1007/s10592-005-9020-x
Diehl N., Roleda M. Y., Bartsch I., Karsten U., Bischof K. (2021). Summer heatwave impacts on the European kelp Saccharina latissima across its latitudinal distribution gradient. Front. Mar. Sci. 8. doi: 10.3389/fmars.2021.695821
Earl D. A., Vonholdt B. M. (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the evanno method. Conserv. Genet. Resour. 4, 359–361. doi: 10.1007/s12686-011-9548-7
Excoffier L., Laval G., Schneider S. (2005). Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol. Bioinf. Online 1, 47.
Fishery Bureau of Ministry of Agriculture and Rural Affairs. (2022). China fishery statistical yearbook 2022. (Beijing: China Agriculture Press), p. 23. (in Chinese).
Frankham R., Bradshaw C. J. A., Brook B. W. (2014). Genetics in conservation management: Revised recommendations for the 50/500 rules, red list criteria and population viability analyses. Biol. Conserv. 170, 56–63. doi: 10.1016/j.biocon.2013.12.036
Guzinski J., Ballenghien M., Daguin-Thiebaut C., Leveque L., Viard F. (2018). Population genomics of the introduced and cultivated pacific kelp Undaria pinnatifida: Marinas-not farms-drive regional connectivity and establishment in natural rocky reefs. Evol. Appl. 11, 1582–1597. doi: 10.1111/eva.12647
Li H. J. (1991). Progress of research on cultivation techniques of Undaria pinnatifida (Harv.) suringar in China. Mod. Fish. Inf. 6, 1–4.
Li Q., Shan T., Wang X., Su L., Pang S. (2020). Evaluation of the genetic relationship between the farmed populations on a typical kelp farm and the adjacent subtidal spontaneous population of Undaria pinnatifida (Phaeophyceae, laminariales) in China. J. Appl. Phycol. 32, 653–659. doi: 10.1007/s10811-019-01917-3
Li Q., Wang X., Zhang J., Yao J., Duan D. (2015). Maternal inheritance of organellar DNA demonstrated with DNA markers in crosses of Saccharina japonica (Laminariales, phaeophyta). J. Appl. Phycol. 28, 2019–2026. doi: 10.1007/s10811-015-0687-1
Mamo L. T., Wood G., Wheeler D., Kelaher B. P., Coleman M. A. (2021). Conservation genomics of a critically endangered brown seaweed. J. Phycol. 57, 1345–1355. doi: 10.1111/jpy.13177
Peakall R., Smouse P. E. (2012). GenAlEx 6.5: genetic analysis in excel. population genetic software for teaching and research–an update. Bioinformatics 28, 2537–2539. doi: 10.1093/bioinformatics/bts460
Piry S., Luikart G., Cornuet J. M. (1999). Computer note. BOTTLENECK: a computer program for detecting recent reductions in the effective size using allele frequency data. J. Hered. 90, 502–503. doi: 10.1093/jhered/90.4.502
Pritchard J. K., Stephens M., Donnelly P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. doi: 10.1093/genetics/155.2.945
Qu S. S. (1993). Raising of Undaria pinnatifida introduced from Japan and investigations on its key aquaculture techniques. Aquacul. Sci. 2, 14–18.
Rosenberg N. A. (2004). DISTRUCT: a program for the graphical display of population structure. Mol. Ecol. Notes 4, 137–138. doi: 10.1046/j.1471-8286.2003.00566.x
Rozas J., Sánchez-Delbarrio J. C., Messeguer X., Rozas R. (2003). DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19, 2496–2497. doi: 10.1093/bioinformatics/btg359
Shan T. F., Li Y. Q., Pang S. J. (2022). Genetic diversity of Undaria pinnatifida populations from China and their genetic relationship with those from Japan and Korea as revealed by mitochondrial and nuclear DNA sequences. Bot. Mar. 65, 185–195. doi: 10.1515/bot-2021-0100
Shan T., Pang S., Wang X., Li J., Su L. (2018). Assessment of the genetic connectivity between farmed and wild populations of Undaria pinnatifida (Phaeophyceae) in a representative traditional farming region of China by using newly developed microsatellite markers. J. Appl. Phycol. 30, 2707–2714. doi: 10.1007/s10811-018-1449-7
Shan T. F., Pang S. J., Wang X. M., Li J., Su L., Schiller J., et al. (2019). Genetic analysis of a recently established Undaria pinnatifida (Laminariales: Alariaceae) population in the northern wadden Sea reveals close proximity between drifting thalli and the attached population. Eur. J. Phycol. 54, 154–161. doi: 10.1080/09670262.2018.1532116
Stoffel M. A., Esser M., Kardos M., Humble E., Nichols H., David P., et al. (2016). inbreedR: an r package for the analysis of inbreeding based on genetic markers. Methods Ecol. Evol. 7, 1331–1339. doi: 10.1111/2041-210x.12588
Sun J., Hang J. (1976). A preliminary survey of seaweeds in the region of nanji islands. Acta Phytotaxonom. Sin. 14, 51–56.
Tamaki H., Kusaka K., Fukuda M., Arai S., Muraoka D. (2009). Undaria pinnatifida habitat loss in relation to sea urchin grazing and water flow conditions, and their restoration effort in ogatsu bay, Japan. J. Water Environ. Technol. 7, 201–213. doi: 10.2965/jwet.2009.201
Tseng C. K. (2001). Algal biotechnology industries and research activities in China. J. Appl. Phycol. 13, 375–380. doi: 10.1023/A:1017972812576
Tseng C. K., Zhang J. F. (1952). Economic seaweeds in the north of China. J. Shandong Univer. 2, 57–82.
Uwai S., Nelson W., Neill K., Wang W. D., Aguilar-Rosas L. E., Boo S. M., et al. (2006). Genetic diversity in Undaria pinnatifida (Laminariales, phaeophyceae) deduced from mitochondria genes – origins and succession of introduced populations. Phycologia 45, 687–695. doi: 10.2216/05-66.1
Yamanaka R., Akiyama K. (1993). Cultivation and utilization of Undaria pinnatifida (wakame) as food. J. Appl. Phycol. 5, 249–253. doi: 10.1007/BF00004026
Keywords: kelp, climate change, brown macroalga, genetic structure, conservation genetics, seaweed cultivation
Citation: Li Y, Shan T and Pang S (2023) High genetic diversity revealed in the native populations of Undaria pinnatifida from China by mitochondrial sequences and nuclear microsatellites. Front. Mar. Sci. 10:1122058. doi: 10.3389/fmars.2023.1122058
Received: 12 December 2022; Accepted: 13 February 2023;
Published: 22 February 2023.
Edited by:
Kwang-Sik Albert Choi, Jeju National University, Republic of KoreaReviewed by:
Christophe William Vieira, Ghent University, BelgiumWei Lun Ng, Xiamen University, Malaysia, Malaysia
Copyright © 2023 Li, Shan and Pang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tifeng Shan, c2hhbnRpZmVuZ0BxZGlvLmFjLmNu