 Sathish Kumar Thangaraj1†
Sathish Kumar Thangaraj1† Suganya Panjan Nathamuni2†
Suganya Panjan Nathamuni2† Vinaya Kumar Katneni2
Vinaya Kumar Katneni2 Ashok Kumar Jangam2*
Ashok Kumar Jangam2* Satheesha Avunje1
Satheesha Avunje1 Devika Neelakantan Thulasi2Monendra Grover3Jesudhas Raymond Jani Angel2
Devika Neelakantan Thulasi2Monendra Grover3Jesudhas Raymond Jani Angel2 Mudagandur Shashi Shekhar2
Mudagandur Shashi Shekhar2- 1Aquatic Animal Health and Environment Division, ICAR-Central Institute of Brackishwater Aquaculture, Chennai, Tamil Nadu, India
- 2Centre for Bioinformatics, Nutrition Genetics and Biotechnology Division, ICAR-Central Institute of Brackishwater Aquaculture, Chennai, Tamil Nadu, India
- 3Division of Computer Science, ICAR-Indian Agricultural Statistics Research Institute, New Delhi, India
Introduction
Shrimp aquaculture is witnessing production losses due to several emerging diseases in Penaeus vannamei farming both at hatchery and grow-out stages. Diseases are caused due to multiple factors, which include biotic, abiotic and managemental factors (Alavandi et al., 2019; Millard et al., 2021) and in many cases, the etiology is unknown. One such disease commonly observed in hatcheries is zoea-2 syndrome.
Zoea-2 syndrome (ZS) reported since 1990 has become a significant challenge to shrimp larviculture. Recently in India, especially after the introduction of P. vannamei, Zoea-2 syndrome has emerged as a serious threat and severely affected the shrimp larval production. This syndrome is characterized by reduced feeding and impaired metamorphosis and causes delayed molting and mass mortality. Zoea-2 syndrome affected larvae observed to stop feeding suddenly after 36-48 hrs of zoea I stage, then exhibit clinical symptoms and mortality. The clinical signs of Zoea-2 syndrome include loss of appetite, empty gut with no fecal strands, arrested peristaltic movement of gut, inflammation in the intestinal epithelium and white balls or white spheres like structures in the gut. The loss due to zoea-2 syndrome in a 100 million nauplii stocking capacity hatchery was reported to be approximately ₹ 1.2 to 4.0 million Indian rupees in 2016 (Sathish Kumar et al., 2017). So far, incidences of zoea-2 syndrome were only associated with Vibrio harveyi and Vibrio alginolyticus (Vandenberghe et al., 1999). In our earlier study, among other vibrio isolates V. alginolyticus was found to be significantly associated and no known pathogen was found to be causally associated with this syndrome (Sathish Kumar et al., 2017). Thus, etiology and pathology of zoea 2 syndrome endure to be unresolved for almost three decades. Eventually, this syndrome results in mass mortality with a pattern of gradual, progressive manner increasing with day-by-day stocking to the maximum of 70-90%.
Another important disease of grow-out ponds that has become a concern in recent years is white feces syndrome (WFS) which affects the juvenile and adult stages of shrimp in aquaculture farms. Reduced feeding, growth retardation, white gut and loose carapace with floating white feces on the surface of the pond are the symptoms of WFS (Tang et al., 2016). The disease occurs commonly after 30-40 days of culture which may also be accompanied by mortality of animals (Sriurairatana et al., 2014). WFS occurrences were reported to be associated with aggregated transformed microvilli (ATM) structures resembling gregarine worms, vibriosis, Enterocytozoon hepatopenaei (EHP), blue-green algae, and fungi (Sathish Kumar et al., 2022). Disease manifestations are noticed even in the absence of these agents (Wang et al., 2020). Studies on intestinal bacterial communities of WFS shrimp indicated less diverse (Hou et al., 2018), more abundant V. sinaloensis and V. parahaemolyticus (Wang et al., 2020) and other potential role of pathogenic bacteria (Zeng et al., 2020).
The 16s rRNA amplicon sequencing offers to provide diversity and dynamics of both culturable and unculturable microorganisms of a habitat, which is not possible through traditional laboratory approaches (Martínez-Porchas and Vargas-Albores, 2017). In this study, 16s rRNA amplicon sequencing was carried out for ZS and WFS which affect hatcheries and grow-out ponds, respectively. This is the first report on the 16s amplicon sequencing data of zoea-2 syndrome and the recovered sample profiles of white feces Syndrome. The sequencing information generated from this study is useful for understanding microbial communities associated with two important diseases and forms basis for possible future designing of preventives and treatments.
Materials and methods
Sample collection
The Zoea-II syndrome samples were obtained from hatcheries located near Marakkanam Tamilnadu, India (12.19° N, 79.92° E). The larvae of zoea stage 1 (50 zoeas per sample) displaying the clinical symptoms as stated above were collected at 2-, 4-, 6- and 8-days post-infection (ZIIS2, ZIIS4, ZIIS6 and ZIIS8). The control group is devoid of ZS infection (ZIISZN). With regard to white feces syndrome, a total of five samples, comprising two diseased (D1 and D2), two recovered (R1 and R2) and one healthy sample (C1) were collected from the shrimp farms located near Nellore of Andhra Pradesh India (14.47° N, 80.09° E). The samples were identified based on the symptoms displayed for each condition, and collected at 45 to 80 days of culture, covering different stages of infection. Pooled gut contents from three biological replicates were collected from each sample, representing all three selected health/disease conditions. All the collected shrimp samples of ZS and WFS groups were stored at -80°C for further analysis.
DNA extraction and sequencing
Samples were subject to CTAB and Phenol: chloroform method followed by RNase A treatment to isolate the total DNA and the quality was checked using NanoDrop 2000 (Thermo Scientific, Brea, CA, United States) followed by 16s rRNA gene amplification. The primers targeting the 16s V3-V4 regions (F -GCCTACGGGNGGCWGCAG; R- ACTACHVGGGTATCTAATCC) designed by Eurofins (Bisht et al., 2018) were used for PCR amplification, and 3 µl of the PCR product was checked using 1.2% agarose gel. This was followed by library preparation using the amplicon library preparation kit Nextera XT Index by Illumina Inc. (San Diego, CA, United States) in accordance with the 16s metagenomic Sequencing Library preparation guide Part #15044223 Rv. B. The amplicon libraries were purified by AMPure XP beads quantified using Qubit Fluorometer and analyzed using the Agilent 4200 TapeStation (Santa Clara, CA, United States) with D1000 Screen tape. The libraries were subject to 2x300 paired ends sequencing using the Illumina MiSeq platform (San Diego, CA, United States).
Read quality control
The quality of the raw reads was assessed using the FastQC v.0.11.8 tool (https://www.bioinformatics.babraham.ac.uk/projects/fastqc ). Further, the raw reads with inadequate quality standards were filtered using Trimmomatic ver. 0.39 (Bolger et al., 2014). The adapters were removed with ILLUMINACLIP option using built-in NextEra PE adapter file, and the low-quality bases were trimmed using the SLIDINGWINDOW option with a phred score threshold of 25 for every 5 bases. Finally, the MINLEN option was used to remove all the sequences that had a length below 50 bases.
Taxonomic profiling and diversity analysis
The 16s amplicon sequence analyses of the samples were performed using the Quantitative Insights Into Microbial Ecology (QIIME) software, an open source microbiome analysis pipeline (Caporaso et al., 2010). The trimmed high-quality data were subjected to sequence preprocessing such as joining paired ends, converting from fastq to fasta files and combining the sequence with QIIME identifiable label. The pre-processed sequences were further subjected to OTU classification (operational taxonomic units) with open referencing method against the SILVA v132 database using default parameters. Further, taxonomy summarization of the OTU table was carried out followed by alpha and beta diversity analysis (https://github.com/biocore/qiime). The calculated alpha diversity indices include Shannon, Chao1, Simpson’s diversity and evenness, fisher alpha and goods coverage, while the beta diversity indices include the weighted and unweighted unifrac distances (Supplementary Figures 1, 2).
Functional analysis
The function prediction was carried out using the PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States, Langille et al., 2013) tool in three steps. The PICRUSt predictions were based on the OTUs generated using closed referencing method with default parameters (Supplementary Table 5). At first, the OTU table is normalized based on copy number abundance, while the second step involves prediction of Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000) and Cluster of Orthologues Groups (COG) (Tatusov et al., 2003) functional groups. Finally, the categorization of KEGG and COG functions at different levels was carried out (https://github.com/picrust/picrust). The results of QIIME and PICRUSt pipelines are tabulated and visualized using vegan, ggplot2, reshape2, RColorBrewer, and vennDiagram libraries of R (Oksanen, 2007; Wickham, 2007; Chen and Boutros, 2011; Neuwirth and Neuwirth, 2014; Wickham, 2016). Heatmaps on KEGG annotations of PICRUSt are generated using the online tool Morpheus (https://software.broadinstitute.org/morpheus/)
Results and discussions
Read quality control
An average of 418 K and 205 K paired-end reads were obtained for ZS and WFS respectively. On trimming, these were reduced to 341 K and 180 K. Finally, high quality reads of 81.16% - 82.31% for ZS and 87.03% – 88.83% for WFS are retained from the raw reads. The Q30 stats for ZS and WFS samples were observed to be in the ranges of 95.65 - 95.71 and 95.19 - 95.44 percentages respectively (Supplementary Table 1).
Taxonomic profiling and diversity analysis
Amplicon sequence analysis results revealed an average read classifications of 87100 and 190371 for WFS and ZS respectively. The taxonomic profile revealed that all the samples were dominated by Proteobacteria and Firmicutes irrespective of disease state and life stages. In addition, the phyla Bacteroidetes and Actinobacteria were found to be high in ZS samples, while Tenericutes, and Cyanobacteria were high in WFS samples.
Family level abundances revealed a dominance of Planococcaceae, Moraxellaceae, Flavobacteriaceae and Rhodobacteraceae in ZS, while Mycoplasmataceae, Vibrionaceae, and Pirellulaceae were found to be predominant in all WFS samples. In addition, Streptococcaceae and Enterococcaceae were observed to be high in recovered and healthy samples of WFS respectively.
Psychrobacter, Planomicrobium, Pedobacter, and Glutamicibacter are found to be predominant genera of ZS samples, whereas Candidatus Bacilloplasma, Photobacterium, Lactococcus, Enterococcus, and Vibrio were predominant in WFS (Figure 1).
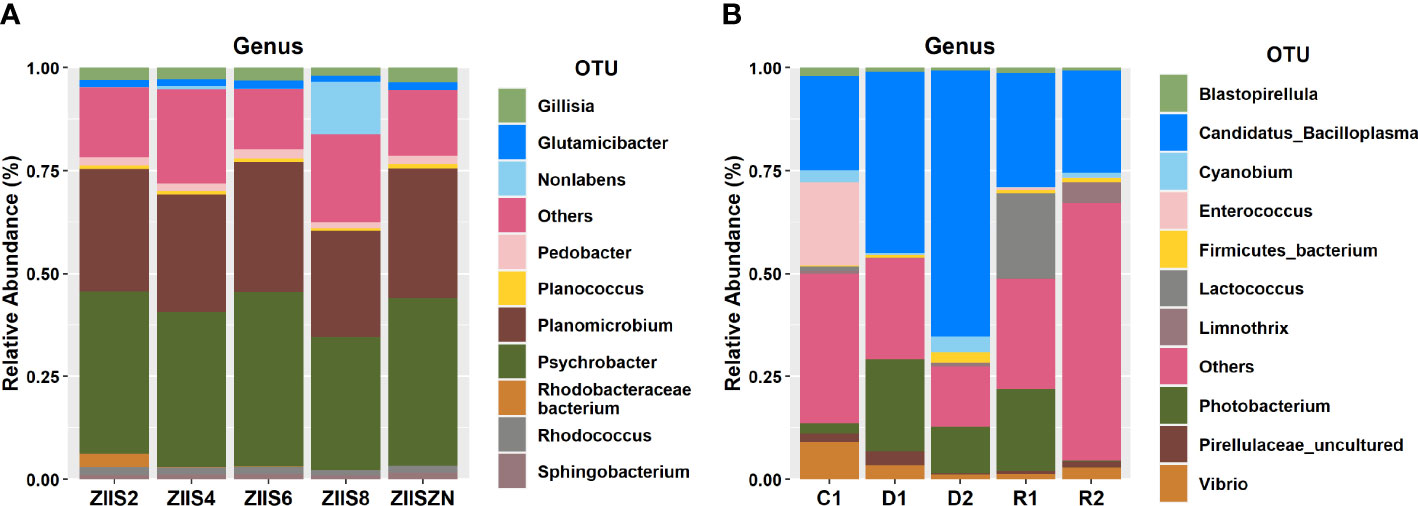
Figure 1 Taxonomic classifications at genus for (A) ZS and (B) WFS.
The complete taxonomic profile for both the sample groups are provided in Supplementary Tables 2 and 3 for ZS and WFS respectively. Additionally, sample-wise microbial profiles are depicted in the form of krona charts and are made available in Supplementary Data Sheets 1 and 2. The diversity analysis results revealed high diversity indices for healthy groups than diseased groups for ZS and WFS samples (Supplementary Table 4). Among the diversity indices calculated for ZS, richness and the fisher’s alpha indices, were high in control sample and gradually declined with the increase in days after infection. However, a sudden rise in diversity was noticed on the 8th day of infection (Supplementary Figure 1A). The richness and fisher alpha indices depicted for WFS samples clearly indicated the higher diversity of healthy and recovered samples compared to diseased samples (Supplementary Figure 1B).
In concurrence with earlier studies, genus Candidatus Bacilloplasma and Photobacterium were observed to be high in the diseased samples and Lactococcus were high in recovered sample group of WFS.
Candidatus Bacilloplasma is a lineage of the Mollicutes which are identified as opportunistic pathogens and have earlier been reported to be high in shrimp affected with WFS (Hou et al., 2018). Candidatus Bacilloplasma was first described by Kostanjsek et al. (2007) colonising the cuticular surface of the terrestrial isopod Porcellio scaber (Crustacea: Isopoda), but it had also been found in several marine species. In the Chinese mitten crab Eriocheir sinensis, Candidatus Bacilloplasma was the dominant genus in the midgut (Dong et al., 2018). As this is where food digestion primarily occurs, the presence of Candidatus Bacilloplasma suggests a possible relationship with the function of the digestive tract. In general, Mollicutes represent a common bacterium class in the intestinal tract of arthropods which is beneficial to the host, with some species associated with an increased performance in the adaptive response to food-limiting conditions. Therefore, the increase in abundance of Candidatus Bacilloplasma in diseased crustaceans may represent a compensatory strategy to enhance the absorption of nutrients when they stop feeding.
On the other hand, many species of Vibrio are commensal microorganisms and are involved in the digestion of a wide range of recalcitrant biopolymers, such as chitin, cellulose and alga-derived compounds, as well as lipids and carbohydrates (Gatesoupe et al., 1997). But also, many species of Vibrio are opportunistic pathogens that impact the physiology and health of their hosts.
Similarly, Photobacterium damselae was found to be associated with many marine animal diseases (Terceti et al., 2018). While, Lactococcus was reported to be among the healthy animals’ gut contents which indicates the recovering health status of the shrimps (Piamsomboon and Han, 2022). In the ZS samples, the genus Psychrobacter was consistently high irrespective of the disease stage, in an earlier study Psychrobacter has been reported to have evolved from a pathobiont ancestor (Welter et al., 2021). This genus has been observed in marine mammals’ skin, respiratory, and gut and has also been reported to cause opportunistic infection in mammals (Welter et al., 2021).
Functional analysis
The Functional analysis revealed the abundant KEGG categories related to cellular processes, environmental information and processing, metabolism and organismal system in both WFS and ZS samples (Supplementary Table 6). In case of WFS samples, the functional pathways were highly abundant in the recovered samples followed by control samples in comparison to the diseased samples (Supplementary Table 6). Similar observations have been made in earlier studies where the pathways, especially the ones related to metabolism and biological processes, were high in healthy samples and low in disease-affected samples (Wang et al., 2019). Similar trend was observed for ZS with an exception at 8 days post-infection.
KEGG ontology terms (level2) for all the samples of both the diseases are depicted in Figure 2. Though there is no clear trend observed with regard abundance of functional categories with disease progression, eight days after infection in case of Zoea II syndrome and recovered samples in case of white feces syndrome have reveled higher abundances. The abundant functional categories of previously mentioned disease stages include Carbohydrate Metabolism, Amino Acid Metabolism, Energy Metabolism, Replication and Repair and Membrane Transport. Among the sub-functional classes’ (level 3) transporters, ABC transporters, DNA repair and recombination proteins, purine metabolism and ribosomes are found to be commonly observed in both the diseases with a variation in the abundances. (Supplementary Figures 3, 4).
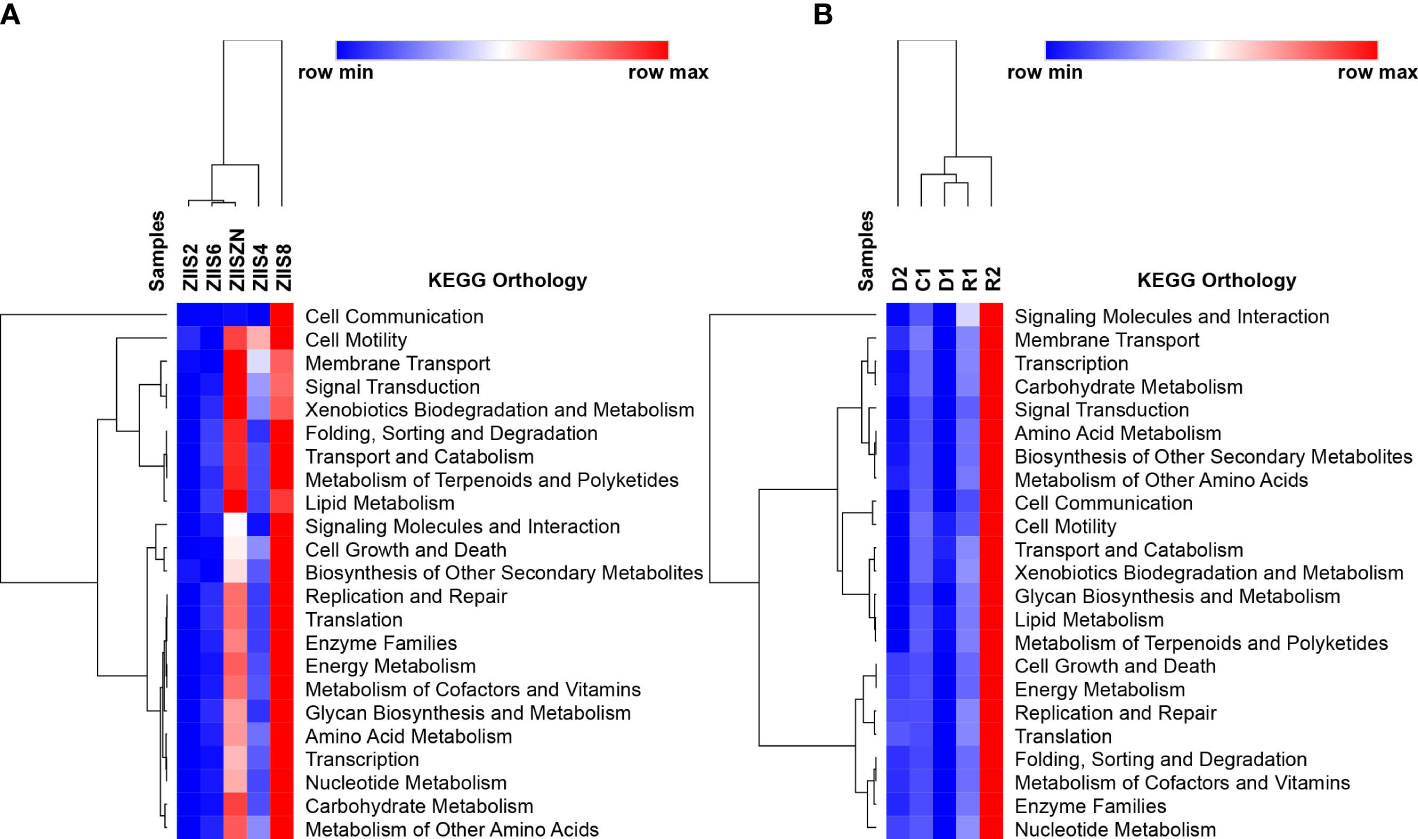
Figure 2 Identified KEGG ontology terms (Level 2) for (A) Zoea II syndrome and (B) White feces syndrome.
Conclusion
Emerging diseases are major concern in P. vannamei hatcheries and grow-out ponds. Here we report microbial profiling of ZS and WFS for which the exact causative agents are still unknown and affect hatcheries and grow-out ponds respectively. This is the first 16s amplicon report on Zoea-2 syndrome. In addition, 16s amplicon sequence data from WFS recovered samples are new addition to the existing datasets of WFS. The 16s amplicon data generated from this study are useful for future attempts on designing preventives and treatment for the reported diseases.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA832951 https://www.ncbi.nlm.nih.gov/, PRJNA832944.
Author contributions
ST, VK, AJ and SA conceived the idea and collected the animal samples. SN and AJ performed the bioinformatics analysis. AJ wrote the manuscript by taking inputs from ST, SN, VK, DT, MG, JA, and MS. All authors contributed to the article and approved the submitted version.
Funding
This research was supported under the Network Project on Agricultural Bioinformatics and Computational Biology, ICAR, New Delhi.
Acknowledgments
The authors are thankful to the funding agency for financial support. The authors are also thankful to the Director, ICAR-CIBA, for providing the necessary approval for conducting this research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1120004/full#supplementary-material
Supplementary Table 1 | Amplicon sequence statistics of WFS and ZS samples.
Supplementary Table 2 | Relative Abundance of ZS at Phylum, Class, Order, Family, Genus, and Species level.
Supplementary Table 3 | Relative Abundance of WFS samples at Phylum, Class, Order, Family, Genus, and Species level.
Supplementary Table 4 | Alpha Diversity results of WFS and ZS samples.
Supplementary Table 5 | OTU Table referenced using closed referencing method.
Supplementary Table 6 | KEGG and COG ontologies of ZS and WFS samples at levels 1, 2 and 3.
Supplementary Figure 1 | Graph representing fisher alpha and Chao’s indices of ZS (A) and WFS (B) samples respectively.
Supplementary Figure 2 | (A) PCoA plot representing Unweighted Unifrac Distance for ZS; (B) PCoA plot representing Weighted Unifrac Distance for ZS; (C) PCoA plot representing Unweighted Unifrac Distance for WFS; (D) PCoA plot representing Weighted Unifrac Distance for WFS.
Supplementary Figure 3 | Level 3 KEGG ontologies in ZS samples.
Supplementary Figure 4 | Level 3 KEGG ontologies in WFS samples.
Supplementary Data Sheet 1 | Krona charts of ZS samples.
Supplementary Data Sheet 2 | Krona charts of WFS samples.
References
Alavandi S. V., Muralidhar M., Syama Dayal J., Rajan J. S., Ezhil Praveena P., Bhuvaneswari T., et al. (2019). Investigation on the infectious nature of running mortality syndrome (RMS) of farmed pacific white leg shrimp, penaeus vannamei in shrimp farms of India. Aquaculture 500, 278–289. doi: 10.1016/J.AQUACULTURE.2018.10.027
Bisht G., Sourirajan A., Baumler D. J., Dev K. (2018). 16S rRNA gene amplicon data set-based bacterial diversity in a water-soil sample from pangong tso lake, a high-altitude grassland lake of the Northwest Himalayas. Microbiol. Resour. Announc. 7 (17), e01192-18. doi: 10.1128/MRA.01192-18
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/BIOINFORMATICS/BTU170
Caporaso J. G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F. D., Costello E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010 7 (5), 335–336. doi: 10.1038/nmeth.f.303
Chen H., Boutros P. C. (2011). VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in r. BMC Bioinf. 12, 1–7. doi: 10.1186/1471-2105-12-35/TABLES/1
Dong J., Li X., Zhang R., Zhao Y., Wu G., Liu J., et al. (2018). Comparative analysis of the intestinal bacterial community and expression of gut immunity genes in the Chinese mitten crab (Eriocheir sinensis). AMB Express 8, 1–12. doi: 10.1186/s13568-018-0722-0
Gatesoupe F.-J., Infante J.-L. Z., Cahu C., Quazuguel P. (1997). Early weaning of seabass larvae, dicentrarchus labrax: the effect on microbiota, with particular attention to iron supply and exoenzymes. Aquaculture 158, 117–127. doi: 10.1016/S0044-8486(97)00179-8
Hou D., Huang Z., Zeng S., Liu J., Wei D., Deng X., et al. (2018). Intestinal bacterial signatures of white feces syndrome in shrimp. Appl. Microbiol. Biotechnol. 102, 3701–3709. doi: 10.1007/S00253-018-8855-2/FIGURES/5
Kanehisa M., Goto S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/NAR/28.1.27
Kostanjsek R., Strus J., Avgustin G. (2007). “Candidatus bacilloplasma,” a novel lineage of mollicutes associated with the hindgut wall of the terrestrial isopod porcellio scaber (Crustacea: isopoda). Appl. Environ. Microbiol. 73, 5566–5573. doi: 10.1128/AEM.02468-06
Langille M. G. I., Zaneveld J., Caporaso J. G., McDonald D., Knights D., Reyes J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013 319 31, 814–821. doi: 10.1038/nbt.2676
Martínez-Porchas M., Vargas-Albores F. (2017). Microbial metagenomics in aquaculture: a potential tool for a deeper insight into the activity. Rev. Aquac. 9, 42–56. doi: 10.1111/RAQ.12102
Millard R. S., Ellis R. P., Bateman K. S., Bickley L. K., Tyler C. R., van Aerle R., et al. (2021). How do abiotic environmental conditions influence shrimp susceptibility to disease? a critical analysis focussed on white spot disease. J. Invertebr. Pathol. 186, 107369. doi: 10.1016/J.JIP.2020.107369
Oksanen J. (2007). Vegan: community ecology package. R Package version, 1, 15–4. https://CRAN.R-project.org/package=vegan.2009
Piamsomboon P., Han J. E. (2022). White feces syndrome, a multifactorial syndrome of cultured shrimp: a mini review. Fishes 2022 7, 339. doi: 10.3390/FISHES7060339
Sathish Kumar T., Makesh M., Alavandi S. V., Vijayan K. K. (2022). Clinical manifestations of white feces syndrome (WFS), and its association with enterocytozoon hepatopenaei in penaeus vannamei grow-out farms: a pathobiological investigation. Aquaculture 547, 737463. doi: 10.1016/J.AQUACULTURE.2021.737463
Sathish Kumar T., Vidya R., Kumar S., Alavandi S. V., Vijayan K. K. (2017). Zoea-2 syndrome of penaeus vannamei in shrimp hatcheries. Aquaculture 479, 759–767. doi: 10.1016/J.AQUACULTURE.2017.07.022
Sriurairatana S., Boonyawiwat V., Gangnonngiw W., Laosutthipong C., Hiranchan J., Flegel T. W. (2014). White feces syndrome of shrimp arises from transformation, sloughing and aggregation of hepatopancreatic microvilli into vermiform bodies superficially resembling gregarines. PloS One 9, e99170. doi: 10.1371/JOURNAL.PONE.0099170
Tang K. F. J., Han J. E., Aranguren L. F., White-Noble B., Schmidt M. M., Piamsomboon P., et al. (2016). Dense populations of the microsporidian enterocytozoon hepatopenaei (EHP) in feces of penaeus vannamei exhibiting white feces syndrome and pathways of their transmission to healthy shrimp. J. Invertebr. Pathol. 140, 1–7. doi: 10.1016/J.JIP.2016.08.004
Tatusov R. L., Fedorova N. D., Jackson J. D., Jacobs A. R., Kiryutin B., Koonin E. V., et al. (2003). The COG database: an updated vesion includes eukaryotes. BMC Bioinf. 4, 1–14. doi: 10.1186/1471-2105-4-41/FIGURES/5
Terceti M. S., Vences A., Matanza X. M., Dalsgaard I., Pedersen K., Osorio C. R. (2018). Molecular epidemiology of photobacterium damselae subsp. damselae outbreaks in marine rainbow trout farms reveals extensive horizontal gene transfer and high genetic diversity. Front. Microbiol. 9. doi: 10.3389/FMICB.2018.02155/BIBTEX
Vandenberghe J., Verdonck L., Robles-Arozarena R., Rivera G., Bolland A., Balladares M., et al. (1999). Vibrios associated with litopenaeus vannamei larvae, postlarvae, broodstock, and hatchery probionts. Appl. Environ. Microbiol. 65, 2592–2597. doi: 10.1128/AEM.65.6.2592-2597.1999/ASSET/13662F32-0996-4762-AA62-44FCE1F7C1E6/ASSETS/GRAPHIC/AM0691868002.JPEG
Wang J., Huang Y., Xu K., Zhang X., Sun H., Fan L., et al. (2019). White spot syndrome virus (WSSV) infection impacts intestinal microbiota composition and function in litopenaeus vannamei. Fish Shellfish Immunol. 84, 130–137. doi: 10.1016/J.FSI.2018.09.076
Wang H., Wan X., Xie G., Dong X., Wang X., Huang J. (2020). Insights into the histopathology and microbiome of pacific white shrimp, penaeus vannamei, suffering from white feces syndrome. Aquaculture 527, 735447. doi: 10.1016/J.AQUACULTURE.2020.735447
Welter D. K., Ruaud A., Henseler Z. M., De Jong H. N., van Coeverden de Groot P., Michaux J., et al. (2021). Free-living, psychrotrophic bacteria of the genus psychrobacter are descendants of pathobionts. mSystems 6 (2), e00258-21. doi: 10.1128/MSYSTEMS.00258-21/SUPPL_FILE/MSYSTEMS.00258-21-ST004.XLSX
Wickham H. (2007). Reshaping data with the reshape package. J. Stat. Software 21, 1–20. doi: 10.18637/jss.v021.i12
Keywords: Zoea-2 syndrome, white feces syndrome, metagenomics, pacific white shrimp, taxonomic profiles, functional profiles
Citation: Thangaraj SK, Nathamuni SP, Katneni VK, Jangam AK, Avunje S, Thulasi DN, Grover M, Angel JRJ and Shekhar MS (2023) Microbial communities associated with Zoea-2 syndrome and White feces syndrome in P. vannamei farming. Front. Mar. Sci. 10:1120004. doi: 10.3389/fmars.2023.1120004
Received: 09 December 2022; Accepted: 20 April 2023;
Published: 05 May 2023.
Edited by:
Ylenia Carotenuto, Stazione Zoologica Anton Dohrn, ItalyReviewed by:
Rossanna Rodriguez-Canul, Center for Research and Advanced Studies - Mérida Unit, MexicoXimena Dubinsky-Velasquez, Israel Oceanographic and Limnological Research Institute (IOLR), Israel
Copyright © 2023 Thangaraj, Nathamuni, Katneni, Jangam, Avunje, Thulasi, Grover, Angel and Shekhar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ashok Kumar Jangam, YXNob2suamFuZ2FtQGljYXIuZ292Lmlu
†These authors have contributed equally to this work