 Morgane Eléouët
Morgane Eléouët Ni Zhang1†
Ni Zhang1† Fanna Kong
Fanna Kong Zhenghong Sui
Zhenghong Sui- 1Key Laboratory of Marine Genetics and Breeding (Ocean University of China), Ministry of Education, Qingdao, China
- 2Synbio Technologies, Chinese site, Suzhou Hongxun Biotechnologies CO., LTD, Suzhou, China
Gracilariopsis lemaneiformis (Gp. lemaneiformis) is an economically important agar-producing red alga applicable in the food and cosmetic industries. The genetic knowledge of this species is, however, limited, and genetic tools for studying and engineering it are lacking. This has limited the understanding of its developmental genetics and hindered the development of new strains, and developing genetic tools would allow to tackle these problems. Here, transient DNA transformation via microparticle bombardment is reported for the first time in this species, as well as efficient exogenous gene expression driven by the CaMV35S promoter, the endogenous GlAct1 promoter, and the Pyropia yezoensis PyAct1 promoter in the transformed branches. Moreover, the Blue Fluorescent Protein (BFP) is demonstrated to be a suitable reporter gene for studies in Gp. lemaneiformis. Screening of antibiotic sensitivity is needed for the development of transient DNA transformation, and selection of transformants is also reported in the alga. Hygromycin B (Hyg) is determined to be the most effective antibiotic for Gp. lemaneiformis selection. The Hyg resistance gene driven by the CaMV35S promoter is shown to confer resistance to Hyg at a concentration of 1 mg.ml-1, but no transformed individual could be regenerated so far. These results are promising for future refining of the experimental conditions, for instance, by using different promoters and developing techniques for facilitating the penetration of the DNA in the cells.
1. Introduction
Gracilariopsis lemaneiformis (Gp. lemaneiformis) (Gracilariaceae, Rhodophyta) is a red macroalga among those most cultivated in various countries and is one of the most widely cultivated malcroalgae in China (Tseng, 2001; Yang et al., 2006). It has an important economic value, as species of the Gracilariaceae family and Gp. lemaneiformis, in particular, have a high agar polysaccharides content (Marinho-Soriano, 2001; Freile-Pelegrín and Murano, 2005; González-Leija et al., 2009; Shi et al., 2017), which is used as a gelling and stabilizing agent in the food, cosmetic, and biotechnology industries (Tseng, 2001; Saha and Bhattacharya, 2010; Lee et al., 2017). They also contain various compounds found to have anti-bacterial and anti-inflammatory properties, which are promising for new pharmaceutical applications and mariculture disease control (Chakraborty and Antony, 2020; Ghosh et al., 2021). Additionally, Gracilariaceae species can be used for human food (Wen et al., 2006), abalone mariculture, and preservation of crab’s habitat (Qi et al., 2010; Wood and Lipcius, 2022) and bioremediation (Yang et al., 2006; Zhou et al., 2006).
The genetic diversity of Gp. lemaneiformis is quite low, partly due to human selection and asexual propagation (Wang et al., 2007; Pang et al., 2010). Moreover, few cultivars have been developed so far (Wang et al., 2014). Developing genetic manipulation systems for Gp. lemaneiformis is therefore necessary for obtaining new desirable traits and new strains and for studying genes and pathways involved in macroalgal growth and development. Although this is still hampered by the lack of genetic data, recent sequencing studies are changing the situation. Gp. lemaneiformis organelle DNA has already been sequenced (Zhang et al., 2012; Du et al., 2016), and nuclear genome (Zhou et al., 2013; Sun et al., 2018) and transcriptome (Huang et al., 2017; Wang et al., 2017) sequencing data are now available. As a result, new genetic manipulation tools are starting to appear, such as CRISPR/Cas12a site-directed mutagenesis (Zhang et al., 2022), which explored a different approach to achieve genetic modification. However, these tools are still limited, and transgene expression and genome integration have not been described yet in Gp. lemaneiformis. Developing efficient DNA transformation methods, approaches to select transformants, and genetic analysis tools, such as promoters and terminators, is still a challenge in red macroalgae including Gp. lemaneiformis.
DNA transformation via particle bombardment has successfully been used in some red macroalgae (Mikami et al., 2011; Huddy et al., 2012; Uji et al., 2014) but has not been tested systematically in Gp. lemaneiformis. Stable transformation has only been achieved in Pyropia/Porphyra yezoensis (P. yezoensis) so far (Uji et al., 2014; Hirata et al., 2014). A high-efficiency plastid transformation platform for P. yezoensis was reported (Kong et al., 2017), which is different from genomic transformation. To develop a reproducible transformation method via particle bombardment in Gp. lemaneiformis, a selection system that enables to specifically select the transformed cells is needed. Such a selection system is still unavailable in Gp. lemaneiformis.
So far, several algae species have been found to be sensitive to a few antibiotics that could be tested on Gp. lemaneiformis before being used for transformation with a resistance gene. First, chloramphenicol (Cm) was shown to affect the red macroalga P. yezoensis (Kong et al., 2017) and the red microalga Cyanidioschyzon merolae (Zienkiewicz et al., 2015; Fujiwara et al., 2017). Second, the antibiotic hygromycin B (Hyg) was shown to affect the red macroalgae Griffithsia japonica (Lee et al., 2000) and P. yezoensis (Takahashi et al., 2011; Uji et al., 2014). Third, the antibiotic paromomycin (PMM) is also efficient on the red macroalga P. yezoensis (Takahashi et al., 2011). Finally, zeocin (Zeo) has been used in the green microalgae C. reinhardtii and Dunaliella salina (Fuhrmann et al., 1999; Sun et al., 2005), in Nannochloropsis sp. (Kilian et al., 2011), and in the red unicellular alga Porphyridium purpureum (Li & Bock, 2018).
For the transformed constructs to be efficiently expressed, the choice of the promoter and terminator is crucial. The 35SCaMV promoter is widely used in higher plants and is active in Gracilaria gracilis (Huddy et al., 2012) and could therefore be efficient in Gp. lemaneiformis as well. As summarized in Mikami et al. (2011), however, the CaMV35S promoter is not always efficient in algae. It was shown to have a low activity in P. yezoensis (Fukuda et al., 2008), and GFP fluorescence was not observed when the EGFP gene was expressed under this promoter in the red microalga C. merolae (Watanabe et al., 2011). Strong endogenous promoters such as GAPDH or Act1 could therefore be more efficient. P. yezoensis’ Actin promoter (PyAct1) was for instance found to be effective in expression vectors in P. yezoensis and a few other red algae species (Mikami et al., 2009; Uji et al., 2010; Hirata et al., 2011). The Arabidopsis Ubiquitin 6 (U6) promoter was also successfully used in P. yezoensis (unpublished data from Kong laboratory, Ocean University of China, Qingdao). Four different promoters could therefore be tested to express reporter genes in Gp. lemaneiformis: CaMV35S, the Gp. lemaneiformis Actin1 (GlAct1) promoter, the PyAct1 promoter, and the U6 promoter. The Nopaline synthase terminator (NosT) of Agrobacterium tumefasciens was also successfully used in vectors transformed into P. yezoensis and P. haitanensis (Uji et al., 2010; Wang et al., 2010a) and could therefore be tested in Gp. lemaneiformis.
The choice of an observable reporter gene is also important to easily track gene expression in the tissues. LacZ had successfully been used in Gracilaria changii (Gan et al., 2004), Gracilaria gracilis (Huddy et al., 2012), Porphyra haitanensis, and Kappaphycus alvarezii (2010b; Wang et al., 2010a). However, its use is time consuming and requires the tissues to be fixed before observation, so selecting a fluorescent reporter gene could be more suited for observation in live cells. The EGFP expression in Gp. lemaneiformis is difficult to distinguish from the natural green background fluorescence. However, red algae’s tissues appear red under violet light excitation, which is also the excitation wavelength for the Blue Fluorescent Protein (BFP) (Subach et al., 2011). The BFP gene mTagBFP2 (Subach et al., 2011) was therefore selected as a reporter gene in Gp. lemaneiformis, so that the red light emission from the tissues could easily be distinguishable from the blue fluorescence of mTagBFP2 under violet light excitation.
Species belonging to the Gracilaiaceae family are clonal macroalgae that can reproduce asexually by vegetative propagation of the haploid individuals. New branch tips are formed by the division of their apical meristem and can detach themselves from the whole individual to form a new individual and grow their own thallus (Fredericq and Hommersand, 1989; Collado-Vides, 2002). Female haploid gametophytes are therefore ideal for transformation, as they could propagate clonally to give rise to a haploid transformed strain. Haploid tetraspores are also ideal targets as a single-transformed tetraspore would give rise to a whole-transformed gametophyte. Transient transformation could therefore be tested on gametophytic branch tips and on tetraspores. Here, it was first tested on haploid and diploid gametophytic branch tips.
This article reveals that Gp. lemaneiformis is sensitive to hygromycin B, and a DNA transformation method by particle bombardment is described in this species. The CaMV35S, GlAct1, and PyAct1 promoters are shown to efficiently drive the expression of the mTagBFP2, LacZ, and hygromycin resistance genes after transient transformation. The sequence of the GlAct1 promoter is also thereby identified for the first time. Additionally, the efficiency of the mTagBFP2 reporter gene for observation under the fluorescence microscope is demonstrated in this red alga. This work sets a basis for genetic transformation in Gp. lemaneiformis, and it can be refined in the future for genetic research in this species.
2. Materials and methods
2.1. Algae material and growth conditions
2.1.1. Gp. lemaneiformis
The diploid individuals used for the antibiotic sensitivity test as well as for particle bombardment of pGFPGUSPlus and GUS staining were collected from Rongcheng, Shandong Province, China in November 2018, and the WT tetrasporophytes were collected in Ao Shanwei in September 2019. The haploid individuals used for the antibiotic sensitivity test were WT female gametophytes collected in Taiping Jiao bay, Qingdao, during their reproductive period in October 2018.
The diploid individuals used for particle bombardment and BFP observation of GBFP-PA7 and PBFP-PA7 were collected in Lidao, Weihai, in October 2020.
The algae were brushed in sterilized seawater until they were free from epiphytes. They were cultivated in 5-L flasks containing sterile seawater complemented with Provasoli (Pro) medium (Provasoli, 1966), and closed with permeable lids permitting gas exchange. They were grown under a 12h light/12h dark cycle and under a light intensity of 30 μmol·m-2·s-1, at 20 ± 1°C. The seawater containing Pro medium was renewed every 5 days.
2.1.2. P. yezoensis
The P. yezoensis RZ line was cultivated in sterile seawater complemented with Pro medium, under a 12h light/12h dark cycle and under a light intensity of 35–45 μmol·m-2·s-1, at 10 ± 1°C. The seawater containing Pro medium was renewed every 3 days.
2.2. Antibiotics treatment
Gp. lemaneiformis tertiary branch tips were cut out (about 1-cm long) and grown under the same temperature and light conditions as described above. They were grown in 12-well plastic plates with lid (Corning Inc., USA) in 5 ml of seawater with Pro medium per well. For preliminary screening, the wells contained 1; 0.5 or 0.1 mg.ml-1 of Cm, Hyg, PMM or Zeo (Solarbio Science & Technology Co., Ltd, Beijing, China). For the determination of working conditions, they contained 1; 0.5 or 0.25 mg.ml-1 of Hyg or PMM. Six branch tips from different individuals per genotype were put in presence of each antibiotic (three tips per well) during 15 days. The control wells contained seawater with Pro medium without any antibiotic. All the different media were adjusted to Ph 8.8 ± 0.1 using NaOH. The medium was changed every 5 days.
The viability of the branch tips was determined by their color. The color of Gp. lemaneiformis ZC and WT individuals is dark red when they are alive, but becomes white or yellow when they die as the concentration of photosynthetic pigments decreases. The tips were scored as dead when they became completely white or yellow.
2.3. IC50 calculation
The inhibitory concentration 50 (IC50), used to determine the concentrations of Hyg required to inhibit the growth of Gp. lemaneiformis, was calculated using the online tool ICEstimator (biostat.fr) (Kaddouri et al., 2006; Le Nagard et al., 2020). The 95% interval was obtained from the same online tool. The method used for estimating IC50 applies the following model:
where RE is the relative effect of the antibiotic, C is the antibiotic concentration, and γ is the sigmoidicity factor.
2.4. Plasmid construction, preparation, and verification
The maps of the plasmids purchased from external sources are presented on Supplementary Figure S7, and those of the plasmid cloned during this project are presented on Supplementary Figure S8 (designed with Snapgene Viewer 5.1, GSL Biotech LLC, USA).
The pGFPGUSPlus plasmid (Vickers et al., 2007) was ordered from Addgene (Watertown, MA, USA). Its sequence was verified by Sanger sequencing using the M13F20, GFP-F/R, and Hpt-F/R primers (see Supplementary Table S1) and by enzymatic digestion using the EcoRI and HindIII restriction enzymes (Thermo Fisher Scientific, USA) (see Supplementary Table S4, Supplementary Figure S9).
The JRH0647 plasmid was kindly provided by Pr Fanna Kong (Ocean University of China). Its quality was verified by simple gel electrophoresis (Supplementary Figure S10) and by Sanger sequencing using the JRH0647-F/R primers.
The mTagBFP2-N and pA7-GFP expression vectors were purchased from Miaoling Biology (Hangzhou, China). They were verified by enzymatic digestion (Supplementary Figure S11) and Sanger sequencing using the GFP-F/R and BFP-F/R primers.
The R0097-07N plasmid containing the EGFP sequence under the control of the GlAct1 promoter was cloned by Synbio Technologies (Suzhou Hongxun Biotechnologies CO., LTD) using the pGFPGUSPlus vector backbone, synthesizing the GlAct1 promoter sequence and inserting it in front of EGFP instead of the CaMV35S promoter. It was verified by enzymatic digestion (Supplementary Figure S11) and Sanger sequencing using the GFP-F/R and Hpt-F/R primers.
The GBFP-pA7 plasmid containing the mTagBFP2 reporter gene under the control of the GlAct1 promoter and the PBFP-pA7 plasmid containing the mTagBFP2 reporter gene under the control of the PyAct1 promoter were constructed by first obtaining intermediate GGFP-pA7 and PGFP-pA7 vectors. Briefly, these intermediate vectors were obtained by amplifying the GlAct1 and PyAct1 promoters from the R0097-07N plasmid and the P.yezoensis genome, respectively, using primers introducing HindIII and XhoI restriction sites upstream and downstream the promoter fragments (GlAct1-F/R and PyAct1-F/R primers, see Supplementary Table S1, Supplementary Figure S12A). The resulting PCR products and the pA7-GFP vector were then simultaneously digested with HindII and XhoI (Supplementary Figure S12B) and ligated with the T4 DNA ligase to obtain the GGFP-pA7 and PGFP-pA7 intermediate vectors containing the EGFP gene under the control of either the GlAct1 or the PyAct1 promoters instead of the CaMV35S promoter. The two vectors were then verified by colony PCR (Supplementary Figure S12C) and Sanger sequencing.
To obtain the final GBFP-pA7 and PBFP-pA7 plasmids, the mTagBFP2 reporter gene sequence was amplified from the mTagBFP2-N expression vector template, using primers introducing XhoI and BamHI restriction sites upstream and downstream the target fragment (BFP-F/R primers, see Supplementary Table S1, Supplementary Figure S12A). The mTagBFP2 PCR fragment and the GGFP-PA7 and PGFP-PA7 intermediate vectors were then double digested with XhoI and BamHI (Supplementary Figures S12B, S13A), and the digested mTagBFP2 fragment was ligated to respectively the GGFP-PA7 and PGFP-PA7 digested vectors, following the same procedure as above. The final GBFP-pA7 and PBFP-pA7 plasmids expressing the mTagBFP2 reporter gene under the control of either the GlAct1 or the PyAct1 promoters were thereby obtained before being verified by colony PCR (Supplementary Figure S13B, S13C) and Sanger sequencing.
All DNA concentrations were measured using a nanodrop (Implen, Munich, Germany). DH5α E. coli transformed with the different plasmids were grown at 37°C in LB medium containing the appropriate antibiotics (pGFPGUSPlus, R0097-07N, pmTagBFP2-N: 50 μg/ml Kanamycin [Kan], JRH0647: 25 μg/ml Chloramphenicol [Cm], pA7-GFP, GGFP-pA7, PGFP-pA7, GBFP-pA7, PBFP-pA7: 50 μg/ml Ampicillin [Amp]). Fifty percent glyerol stocks were kept. The plasmids were extracted using the TIANprep Mini Plasmid Kit (Tiangen Biotech, Beijing, China).
The plasmid sequences are presented in the Supplementary Data (Supplementary Sequences S1–S5).
2.5. Identification of the Gp. lemaneiformis Actin1 promoter
As no Actin promoter sequence was known in Gracilariopsis lemaneiformis, a BLAST search on the Gp. lemaneiformis genomic database (Zhou et al., 2013) was carried out using the Gracilaria textorii beta-actin mRNA sequence (GenBank: EU095962.1). The SRR1011281.38131 sequence was identified on the Gp. lemaneiformis chromosome 3, with a 63% query cover and 72% identity (Supplementary Sequence S8). After having translated the putative Actin sequence using the ExPAsy Translate tool (https://web.expasy.org/translate/) (Supplementary Sequence 9), a BLAST query on the NCBI protein database yielded the following sequences (the top three sequences are described): Actin (Chondrus crispus), Ref. Seq. XP_005717262.1, 100% query cover, 94% identity; Actin (Gracilariopsis chorda) Ref. Seq. PXF4927.1, 100% query cover, 90% identity; Actin 1 (Nemalionopsis shawii) Ref. Seq. CAI56221.1, 96% query cover, 95% identity.
Another BLAST search on Gp. lemaneiformis genome database was carried out using the P. yezoensis Act1 mRNA sequence (GenBank: AB455256.1). The same sequence as obtained with the Gracilaria textorii beta-actin mRNA sequence (Supplementary Sequence S8) was found, with a 93% query cover and 76.16% identity. Another sequence, SRR1011281.3103 (Supplementary Sequence S10) was identified with a 98% query cover and 73.13% identity and positioned on chromosome 22. After having translated the putative Actin sequence using the ExPAsy Translate tool (Supplementary Sequence S11), a BLAST query on the NCBI protein database yielded the following sequences (the top three sequences are described): Actin (Gracilariopsis chorda) Ref. Seq. PXF4927.1, 100% query cover, 100% identity; Actin (Chondrus crispus), Ref. Seq. XP_005717262.1, 99% query cover, 92.45% identity; Hypothetical protein BU14_0031s0095 (Porphyra umbilicalis), 100% query cover, 86.10% identity.
To select the most suitable Gp. lemaneiformis Actin promoter, the sequences upstream the Actin coding sequences identified were analyzed with the TSSP Prediction of plant promoters’ tool (http://linux1.softberry.com/berry.phtml?topic=tssp&group=programs&subgroup=promoter), but no significant promoter sequence was identified. Upstream (200 bp) the coding sequences were therefore selected and compared with the CGI gene analysis tool (http://218.4.117.30:3000/cgi-bin/gene_analysis.cgi). The SRR1011281.38131 promoter sequence had a total GC content of 53.30%, seven direct and seven reverse repeats, whereas the SRR1011281.3103 promoter sequence had a total GC content of 58.13%, six direct and 13 reverse repeats. The SRR1011281.38131 promoter sequence was thus less complex and was chosen as it was assumed to be easier to clone. The 200 bp upstream the coding sequence was named Gp. lemaneiformis Actin 1 (GlAct1) promoter and was cloned upstream the EGFP sequence in the pGFPGUSPlus vector by Synbio Technologies.
2.6. Microparticle bombardment and transformation test
Thirty milligram of 0.6-μm diameter gold particles was suspended in 1 ml of 70% ethanol and vortexed for 5 min. After letting them stand in the solution for 15 min at room temperature, they were centrifuged at 1500 rpm for 5 min. The supernatant was then discarded and the particles were washed three times by adding 1 ml of sterile water, vortexing for 1 min, and centrifuging at 1,500 rpm for 5 min. The supernatant was then discarded and the particles were resuspended in 500 μl of 50% glycerol. They were stored at −20°C. The plasmids to be bombarded were extracted with the TIANprep Mini Plasmid Kit (Tiangen Biotech, Beijing, China) and concentrated to 1 μg/μl using a vacuum concentrator centrifuge (MSLNC01). To coat the plasmids on the gold particles, 17 μl of the gold particle suspension were added to 6 μl of plasmid, 17 μl of 2.5 M CaCl2, and 6.8 μl of 0.1 M spermidine, before being sequentially vortexed 1 min and kept on ice 1 min 10 times in a row. The suspension was then kept on ice for 30 min before being centrifuged at 10,000 rpm for 10 s. The supernatant was removed, and the coated gold paticles were sequencially washed with 140 μl of 70% ethanol, 140 μl of absolute ethanol, and resuspended in 60 μl of absolute ethanol. The same protocol was repeated for the negative control without plasmid. One centimeter–long Gp. lemaneiformis tertiary branch tips were placed in the center of polystyrene 10-cm-diameter petri dishes. The tips were organized on a single layer, forming a circle of 2-cm diameter. DNA transformation was carried out by particle bombardment using the Biolistic PDS-1000/He particle acceleration device (Bio-Rad, USA), according to the manufacturer’s instructions. Twenty-four microliter of coated gold particles was used for each shot at a 6-cm target distance at 28 in. Hg of vacuum pressure and 1,100 psi of helium pressure, through a rupturable membrane of 1,350 psi. The transformed tips were kept in the dark for 1h without water, and sterile seawater complemented with Pro medium was then added. The tips were kept in the dark for 24h after bombardment, before being grown under normal conditions (see above). Pro medium was changed every day.
For the DNA transformation, 150 tips were placed in each dish; three dishes were transformed with the pGFPGUSPlus plasmid and three with the control uncoated gold particles. Fifty tips of each were randomly selected 2–8 days after bombardment to extract DNA.
2.7. Promoter efficiency test
One hundred fifty tips bombarded with the JRH0647 plasmid or the control uncoated gold particles were randomly picked 36h and 48h after bombardment and kept at −80°C during a few weeks before extracting RNA. Similarly, 60 tips bombarded with the pGFPGUSPlus plasmid or with the control uncoated gold particles were collected every day from the 2nd to 8th day after bombardment. Fifty tips bombarded with either the GBFP-PA7 plasmid, the PBFP-PA7 plasmid, or the control uncoated gold particles were collected every day 2, 4, and 6 days after bombardment. Fifty tips of each group were kept at −80°C during a few weeks before carrying out RNA extraction, whereas the remaining tips were used for GUS staining or Bfp fluorescence observation on the day of collection.
2.8. Antibiotic selection
Two hundred tips were placed in petri dishes and transformed with the pGFPGUSPlus plasmid and 200 with the control uncoated gold particles. One hour after bombardment, the algal tips were grown in sterile seawater complemented with Pro medium. The bombarded dishes were grown in presence of 0.5 mg.ml-1 Hyg during 20 days. The tips were kept in the dark for 24h after bombardment.
The same experiment was then repeated with 1 mg.ml-1 Hyg. Five hundred tips transformed with the pGFPGUSPlus plasmid (125 tips in each of the four dishes) and 500 with the control uncoated gold particles. One hour after bombardment, the algal tips were grown in sterile seawater complemented with Pro medium. The tips were kept in the dark for 24h, and 1 mg.ml-1 Hyg Hyg was added 24h after bombardment. The tips were treated with Hyg during 10 days and transferred to normal seawater complemented with Pro until they all died after 15 days. The dead tips were scored every day.
2.9. DNA and RNA extraction
One hundred milligram of Gp lemaneiformis branch tips were grinded using the Jingxin Tissuelyser-24 (Shanghai Jingxin Industrial Development, Shanghai, China), after having added liquid Nitrogen, following the manufacturer’s instructions. RNA was then extracted using the E.Z.N.A Total RNA kit (Omega Bio-Tek, USA) following the manufacturer’s instructions. DNA was extracted using the TIANGEN Plant Genomic DNA kit (Tiangen Biotech, Beijing, China) following the manufacturer’s instructions, and treated with DNAse (Supplementary Table S4).
Before extracting DNA after microparticle bombardment to check the presence of the plasmids, it was necessary to ensure that there was no plasmid in vitro outside the tips, to avoid false positives. The tips were then treated with DNAse I (Thermo Fisher Scientific, USA) directly in Eppendorff tubes (Supplementary Table S4).
2.10. Reverse transcription
cDNA was generated from total RNA using the Evo M-MLV reverse-transciption kit (Accurate Biotechnology, Hunan, China) following the manufacturer’s instructions. RNA (1.5 μg) was used in each 20-μl reaction.
2.11. PCR and RT-PCR
Primers described here are presented on Supplementary Table S1. PCR reaction components are presented on Supplementary Table S2. Thermal cycling conditions are presented on Supplementary Table S3. To check the presence of the pGFPGUSPlus plasmid in the tip cells after microparticle bombardment, PCR was carried out using the HygR-F/R and GFP-F/R primers on total DNA extracted from bombarded tips.
The expression of the Hpt gene from the pGFPGUSPlus plasmid, and the mTagBFP2 gene from the GBFP-pA7 and PBFP-pA7 plasmids in the bombarded tips, was checked by RT-PCR on their cDNA. The cDNA was amplified using the HygR-F/R, GFP-F/R and BFP-F/R primers. The Gapdh gene was used as standard; it was amplified using the primers GAPDH-F/R. The gRNA expression from the JRH0647 plasmid was checked on the cDNA extracted from bombarded tips using the JRH0647-F/R primers.
2.12. GUS staining
Ten algal tips were randomly collected 5 days after microparticle bombardment with the pGFPGUSPlus plasmid or the control uncoated particles, and 2-mm thick-cross sections were performed on the tips before proceeding to the GUS staining reaction. Ten 1 cm2 onion inner epidermis sections were also bombarded and used as a positive control for GUS expression. The staining solution was obtained with a GUS staining kit (Solarbio G3060, Beijing, China) following the manufacturer’s instructions. Twenty microliter of X-Gluc solution was mixed with 1 ml of GUS buffer. It was protected from light and stored at 4°C for 1 day before staining. Three hundred thirty microliter of GUS staining solution was used to completely submerge the tips and the onion epidermis in 1.5-ml EP tubes. The tubes were wrapped in aluminum foil to protect the samples from light and were incubated at 37°C for 20h. The material was then incubated for 1.5h in 70% ethanol for decolorizing the tissues. The tissues were then observed under an Olympus BX53 microscope using 40× magnification, and pictures were taken with a Mshot MD30 camera; the sites of GUS activity appeared blue.
2.13. Fluorescence observation of the bombarded tips
Fifty tips bombarded with the GBFP-PA7 and PBFP-PA7 plasmids and 50 unbombarded tips (negative controls) were collected on the 2nd, 4th, and 6th days after transformation. They were observed under a Nikon 80i fluorescence microscope under blue fluorescence excitation filter using DAPI mode (380–420 nm) to observe the expression of the mTagBFP2 gene in the cells.
3. Results
3.1. Antibiotic sensitivity test
To develop a selection system for genetically transformed cells, the sensitivity of Gp lemaneiformisto various antibiotics was tested.
Branch tips were cut out and grown in the presence of 0.1, 0.5, or 1 mg.ml-1 of Cm, Hyg, PMM, or Zeo during 15 days (Figure 1). Six branch tips from different individuals per genotype were put in the presence of each antibiotic (three tips per well). The tips were considered dead when they became completely yellow and were counted every day to determine the mortality rate.
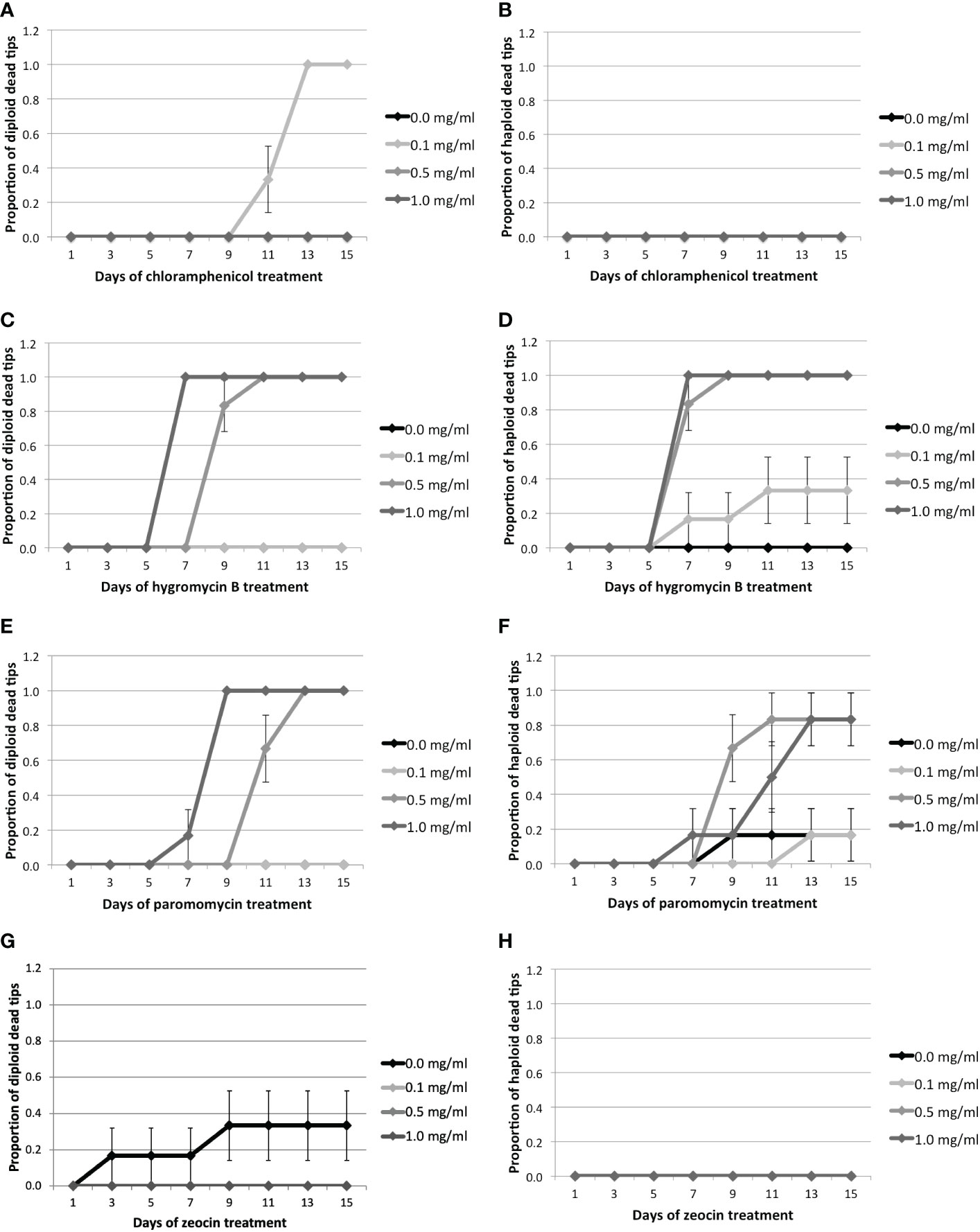
Figure 1 Proportion of dead tips 1–15 days after antibiotic treatment for 0.0, 0.1, 0.5, or 1.0 mg/ml of antibiotics.Cm treatment: (A) Proportion of diploid and (B) haploid dead tips after Cm treatment. Hyg treatment: (C) Proportion of diploid and (D) haploid dead tips after Hyg treatment. PMM treatment: (E) Proportion of diploid and (F) haploid dead tips after PMM treatment. Zeo treatment: (G) Proportion of diploid and (H) haploid dead tips after Zeo treatment. Error bars represent the standard error of the mean.
Cm did not have any significant effect on Gp. lemaneiformis tips, as only diploid tips started to die after 11 days at the lowest concentration (Figures 1A, B, Supplementary Table S5, Supplementary Figure S1). On the other hand, the diploid and haploid tips treated with 0.5 mg.ml-1 of Hyg started to die after 9 and 7 days, respectively (Figures 1C, D). They started to die after 7 days when treated with 1 mg.ml-1 Hyg, and all the tips treated with 0.5 or 1 mg.ml-1 were dead after fifteen days (Supplementary Table S5, Supplementary Figure S3). The mortality of the tips treated with 0.1 mg.ml-1 Hyg did not increase significantly, as only two haploid tips were dead after fifteen days (Figures 1C, D, Supplementary Table S5, Supplementary Figure S3). PMM also seemed to affect the tips, as the diploid individuals treated with 0.5 and 1 mg.ml-1 PMM started to die after 11 and 7 days, respectively, and after 9 and 7 days for haploid tips (Figures 1E, F). There was no significant increase in mortality at 0.1 mg.ml-1. All the diploid tips were dead after 15 days at 0.5 and 1 mg.ml-1, whereas only one haploid tip survived (Supplementary Table S5, Supplementary Figure S4). Finally, Zeo did not have any significant effect on the tips’ survival, as only two control diploid tips died (Figures 1H, Table 1, Supplementary Figure S2).
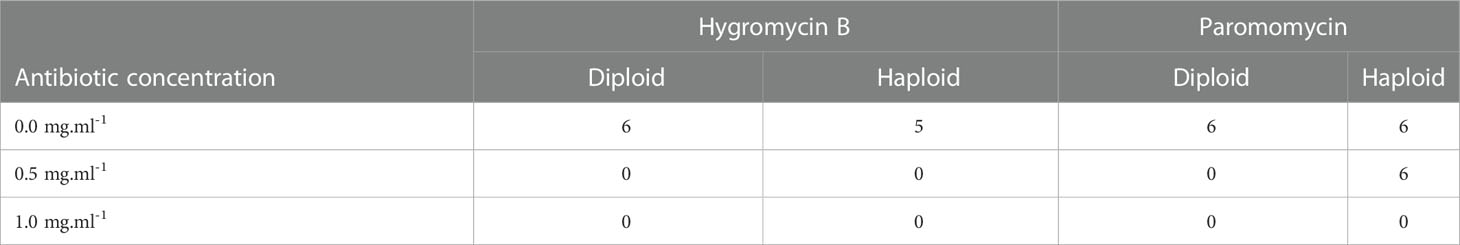
Table 1 Total number of live tips after fifteen days of antibiotic treatment, for each antibiotic concentration (mg.ml-1), during the second set of treatments.
This test suggests that Hyg and PMM negatively affect the survival of Gp. lemaneiformis branch tips at concentrations of 0.5 and 1 mg.ml-1. They were therefore selected for further screening.
3.2. Determination of antibiotic concentration for screening
After Hyg and PMM were selected for screening, a new set of treatment was performed to confirm the results and choose between the two antibiotics. As a concentration of 0.1 mg.ml-1 did not show any significant effect on the tips’ survival, the treatment was only repeated with concentrations of 0.5 and 1 mg.ml-1. Hyg was confirmed to negatively affect Gp. lemaneiformis branch tip survival as all the treated tips were dead after fifteen days of treatments, whereas only one non-treated haploid tip had died (Table 1, Supplementary Figure S5). PMM was also found to affect tip survival at 1 mg.ml-1 but less strongly than Hyg at 0.5 mg.ml-1 as all the diploid tips had died, whereas all the haploid survived (Table 1, Supplementary Figure S6).
The results of the two sets of treatments were pooled together to increase the statistical power. It can be seen that diploid tips treated with 0.5 and 1 mg.ml-1 Hyg respectively started to die after 7 and 5 days, whereas none of the controls had died after 15 days (Figure 2, Supplementary Table S6). The haploid tips treated with 0.5 and 1 mg.ml-1 Hyg started to die after 5 days, whereas only one haploid control had died (Figure 2, Supplementary Table S6). This confirms that Hyg negatively affects the survival of both diploid and haploid tips, at 0.5 and 1 mg.ml-1. For PMM, the diploid tips treated with 0.5 and 1 mg.ml-1 respectively started to die after 7 and 5 days, whereas none of the controls had died after 15 days (Figure 2, Supplementary Table S6). The haploid tips treated with 0.5 and 1 mg.ml-1 PMM also had an increased mortality, although lower than the diploids, as respectively five and 11 of them had died after 15 days, whereas only one control tip had died (Figure 2, Supplementary Table S6). It can therefore be concluded that PMM negatively affects the survival of both diploid and haploid tips at 0.5 and 1 mg.ml-1, but less strongly than Hyg.
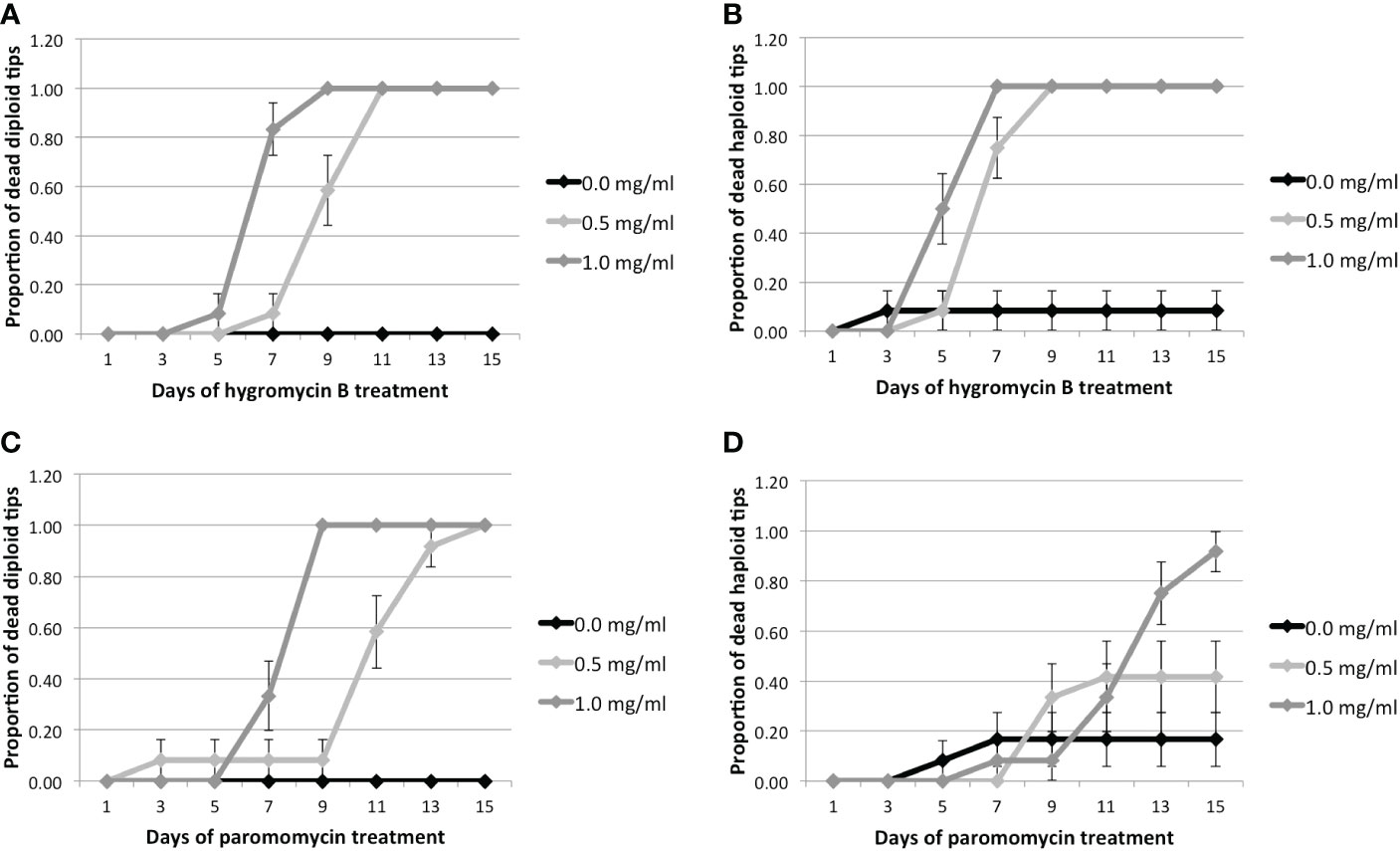
Figure 2 Proportion of dead tips 1–15 days after antibiotic treatment for 0.0, 0.5, or 1.0 mg/ml of antibiotics. Hyg treatment: (A) Proportion of diploid and (B) haploid dead tips after Hyg treatment. PMM treatment: (C) Proportion of diploid and (D) haploid dead tips after PMM treatment. Error bars represent the standard error of the mean.
These results suggest that Gp. lemaneiformis branch tips are sensitive to both Hyg and PMM, but that higher concentrations of PMM are required to have a negative effect on survival. Hyg was therefore chosen as a selection antibiotic for transformation experiments.
To use Hyg as a selection antibiotic for future genetic transformation experiments, the suitable Hyg concentration needed for selection should be determined. The inhibitory concentration 50 (IC50) was therefore calculated. The IC50 measures the Hyg concentration needed for 50% loss of Gp. leamaneiformis viability. For the diploid tips, it could be determined for 5, 7, and 9 days of Hyg treatment (not for the other time points as the proportion of dead tips was either of 0 or 100%) (Table 2). The IC50 was 0.71 mg.ml-1 after 5 days of Hyg treatment and fell to 0.48 mg.ml-1 after 9 days of treatment. As the range of Hyg concentration was small, the IC50 95% intervals varied between 0.52 and 0.87 at 5 days of treatment, so the determination of the IC50 was not very sensitive. For the haploid tips, IC50 was 0.69 mg.ml-1 after 5 days of Hyg treatment and 0.45 mg.ml-1 after 7 days. A concentration of 0.5 mg.ml-1 was finally chosen to be tested on diploids (see below), with which 50% loss of viability should be expected after 9 days and therefore 50% of survivors should be expected to be transformants.
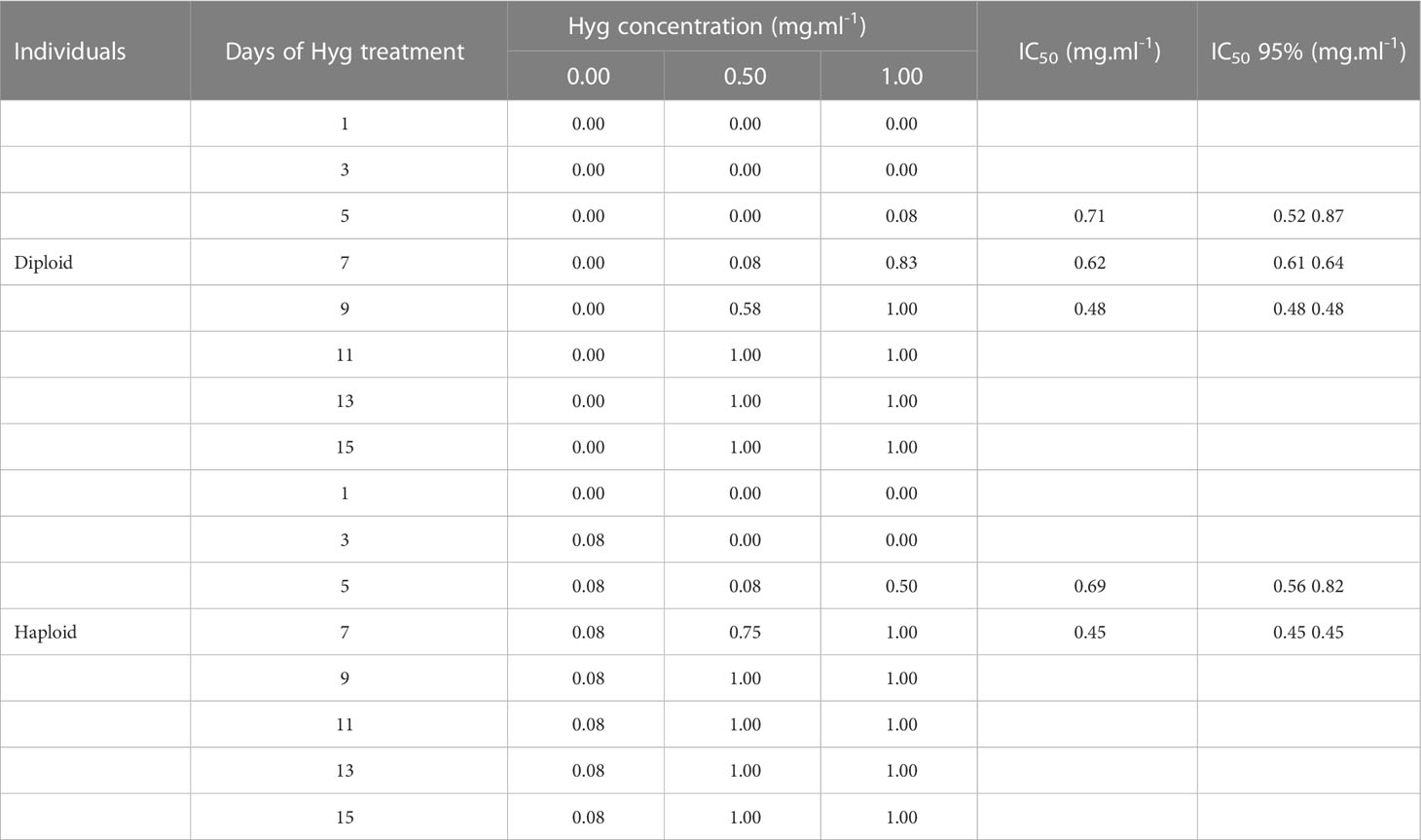
Table 2 Proportion of dead tips after several days of Hyg treatment and calculation of IC50 with a 95% confidence interval.
3.3. Transient DNA transformation
After having selected the most suitable antibiotic and antibiotic concentration for Gp. lemaneiformis selection, transient DNA transformation was tested. A plasmid containing the Hyg resistance gene (Hygromycin phosphotranspherase or Hpt) was therefore chosen to select resistant transformed cells. The pGFPGUSPlus plasmid containing the Hpt gene, as well as the EGFP and β-Glucuronidase (GUS) reporter genes, all under the P35S promoter, was used (Supplementary Figure S7A). To first determine whether the pGFPGUSPlus plasmid could be successfully transformed into branch tip cells. The pGFPGUSPlus plasmid was coated on microparticles and bombarded on Gp. lemaneiformis tertiary branch tips without antibiotic selection. After having removed potential plasmid contamination outside the branch tips by DNAse treatment, PCR with primers specific for the Hpt sequence was carried out on total DNA extracted from bombarded tips, 2–8 days after bombardment. The gel electrophoresis results showed that amplification of the Hpt fragment (514 bp) was detectable in the tips from 2 to 8 days after microparticle bombardment (Figure 3), although the signal was weaker after 5 days. It can hence be concluded that the pGFPGUSPlus plasmid was present in the tips and therefore that it was indeed transiently transformed in the bombarded tips, at least until 8 days after bombardment. Microparticle bombardment is thus a suitable method for transient DNA transformation in Gp. lemaneiformis under the conditions tested here. Before performing the transformation with Hyg selection, efficient expression of the Hpt gene in the tips needed to be checked, so transgene expression was verified first.
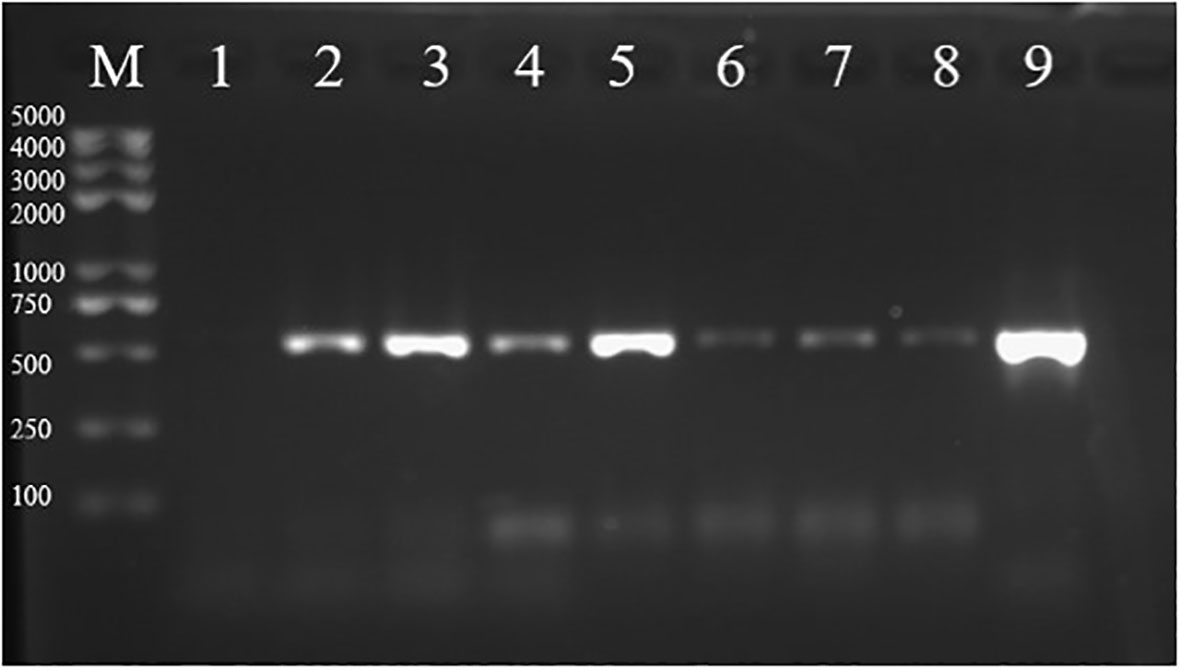
Figure 3 Analysis of the pGFPGUSPlus plasmid presence in transformed tips. PCR amplification products of the Hpt gene from tips transformed with the pGFPGUSPlus plasmid were run on a 1% agarose gel (expected size: 514 bp). (M) marker; (1) amplification from tips bombarded with uncoated gold particles; (2–8) amplification from tips bombarded with the pGFPGUSPlus plasmid for which total DNA was extracted respectively 2, 3, 4, 5, 6, 7, and 8 days after bombardment; (9) positive control of the Hpt fragment amplified from the pGFPGUSPlus plasmid.
3.4. Transgene expression
3.4.1. RT-PCR
After having verified that DNA transformation was possible in Gp. lemaneiformis, efficient expression of the transformed genes in the alga should be tested. Tips were therefore bombarded with either the pGFPGUSPlus or JRH0647 plasmid, and the efficiency of the CaMV35S and U6 promoters to drive respectively the Hpt and gRNA expressions was tested by RT-PCR. To this aim, 150 tips bombarded with the JRH0647 plasmid were randomly picked 36h and 48h after bombardment, and 50 tips bombarded with the pGFPGUSPlus plasmid were randomly picked from 2 to 8 days after bombardment. RNA was extracted and used as a template for cDNA synthesis.
Control Gapdh PCR amplification from total cDNA yielded the expected fragments in all samples bombarded with the JRH0647 and collected either 36h or 48h after bombardment (Figure 4). The gRNA under the U6 promoter, however, did not yield any PCR amplification. This suggests that the gRNA was not expressed under the U6 promoter and, therefore, that this promoter is not efficient in Gp. lemaneiformis.
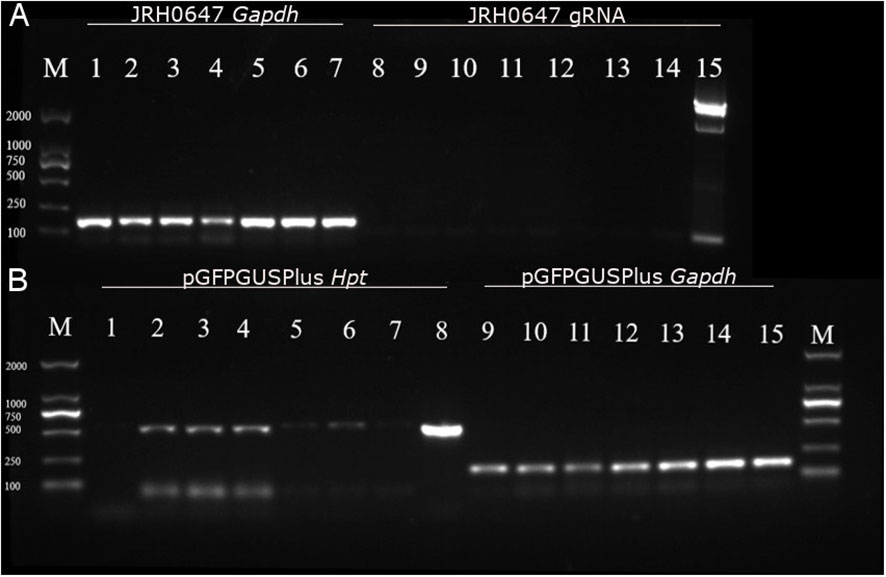
Figure 4 Analysis of gRNA and Hpt expressions in tips tranformed with JRH0647 or pGFPGUSPlus. PCR products were run on a 1.5% electrophoresis gel. (A) Tips bombarded with the JRH0647 plasmid. M = marker; lanes 1–7 = PCR reaction carried out on the control Gapdh cDNA (expected size: 163 bp); lane 1 = control tips bombarded with uncoated gold particles; lanes 2, 3, and 4 = tips for which RNA was extracted 36h after bombardment; lanes 5–7 = tips for which RNA was extracted 48h after bombardment; lanes 8–14 = PCR reaction carried out on the gRNA sequence (expected size: 84 bp); lane 8 = control tips bombarded with uncoated gold particles; lanes 9–11 = tips for which RNA was extracted 36h after bombardment; lanes 12–14 = tips for which RNA was extracted 48h after bombardment; lane 15 = positive control of gRNA amplification from the JRH0647 plasmid template. (B) Tips bombarded with the PGFPGusPlus plasmid. M = marker; lanes 1–8 = PCR reaction carried out on the Hpt sequence (expected size: 514 bp); lane 1 = control tips bombarded with uncoated gold particles; lanes 2–7 = tips for which RNA was extracted respectively 2–7 days after bombardment; lane 8 = positive control of Hpt amplification from the pGFPGusPlus plasmid template; lanes 9–15 = PCR reaction carried out on the control Gapdh cDNA; lane 9 = control tips bombarded with uncoated gold particles; lanes 10–15 = tips for which RNA was extracted respectively 2 to 7 days after bombardment.
For the tips bombarded with pGFPGUSPlus, on the other hand, amplification of the Hpt fragment could be detected for all samples collected between 2 and 7 days after bombardment (Figure 4), although the signal was relatively weak and weaker after 5 days.
These results suggest that the Hpt gene was efficiently expressed under the CaMV35S promoter in bombarded tips, but that the expression was not very strong. It can therefore be assumed that microparticle bombardment is efficient for delivering DNA into the cells, and that the CaMV35S promoter can drive gene expression in Gp. lemaneiformis, but that it may be necessary to look for a stronger promoter to drive exogenous gene expression.
3.4.2. GUS staining
To confirm the above results, 10 tips were randomly selected 5 days after microparticle bombardment with the pGFPGUSPlus plasmid or the control uncoated particles, and 2-mm-thick cross sections were performed on the tips before proceeding to the GUS staining reaction. Ten 1 cm2 onion inner epidermis sections were also bombarded and used as a positive control for GUS staining.
Blue coloring could be seen in some of the onion cells bombarded with the pGFPGUSPlus plasmid (Figure 5), suggesting that the GUS gene was indeed expressed under the CaMV35S promoter in onion cells and that onion epidermis could be used as a positive control for GUS staining. No blue staining could be observed in the negative control (Figure 5), whereas it could be observed on Gp. lemaneiformis branch tips transformed with pGFPGUSPlus (Figure 5). Cross sections of the tips showed that no cell was stained in the negative control (Figure 5), but that several cells were stained in blue in the transformed tips (Figures 5E, F).
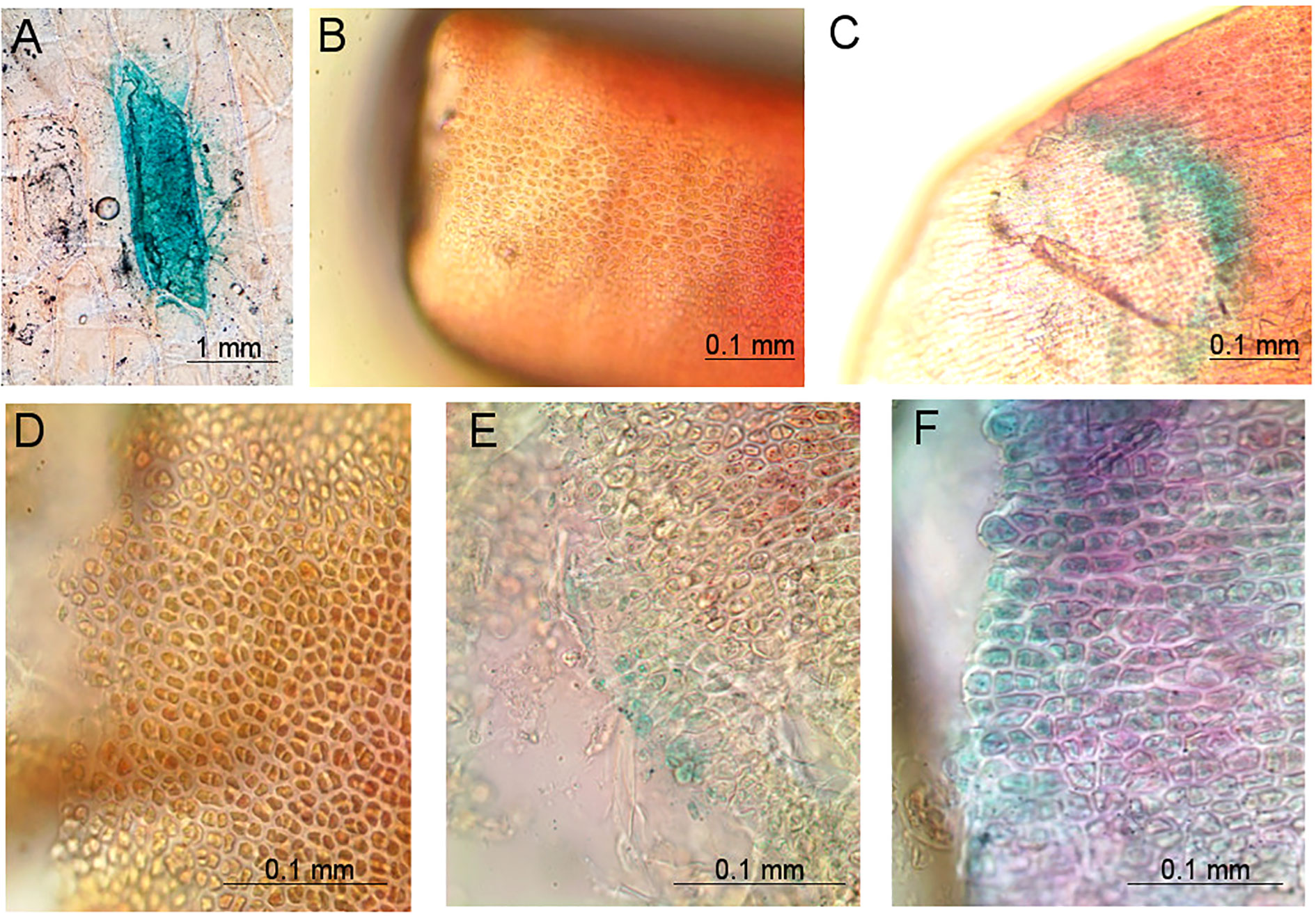
Figure 5 Pictures of tissues bombarded with the pGFPGUSPlus plasmid for which GUS staining reactions were performed 5 days after bombardment, observed under a compound light microscope at 20× and 40× magnification. (A) Positive control of an onion cell bombarded with pGFPGUSPlus, showing blue coloring after GUS staining; (B) Negative control of a Gp. lemaneiformis branch tip bombarded with uncoated gold particles and for which GUS staining reaction was performed; (C) Gp. lemaneiformis branch tip bombarded with the pGFPGUSPlus plasmid showing blue coloring after GUS staining; (D) Negative control of a cross section of a Gp. lemaneiformis branch tip bombarded with uncoated gold particles and for which GUS staining reaction was performed; (E, F) Cross sections of Gp. lemaneiformis branch tips bombarded with the pGFPGUSPlus plasmid showing blue coloring after GUS staining.
These results suggest that the GUS gene was expressed under the CaMV35S promoter in the Gp. lemaneiformis branch tips transformed with pGFPGUSPlus. It confirms that microparticle bombardment is efficient for transient DNA transformation in Gp. lemaneiformis cells, and that the CaMV35S promoter can be used for driving foreign gene expression in this species.
3.5. Selection of transformants
After having confirmed that the Hpt gene was expressed in the transformed tips, efficient resistance against Hyg should be tested. Two hundred tips were transformed with either the pGFPGUSPlus plasmid or with no plasmid for the control and treated with 0.5 mg.ml-1 Hyg. At this concentration, 50% loss of viability should be expected after 9 days, and therefore 50% of survivors should be expected to be transformants (Table 2). The dead tips were counted every day (Figure 6). As no significant difference in survival could be observed between the tips transformed with the plasmid and the controls after 9 days, the treatment was continued during 20 days to keep the selection pressure and determine whether a different survival could be observed. The tips transformed with the plasmid only had a significant lower death rate than the control 13 days after bombardment (Figure 6, Supplementary Table S7). After 20 days, all the control tips had died. Five tips transformed with the plasmid had survived, that is, some parts of the tip’s tissue had conserved a red color, but they died shortly after having been transferred to seawater without antibiotic, so they could not be regenerated.
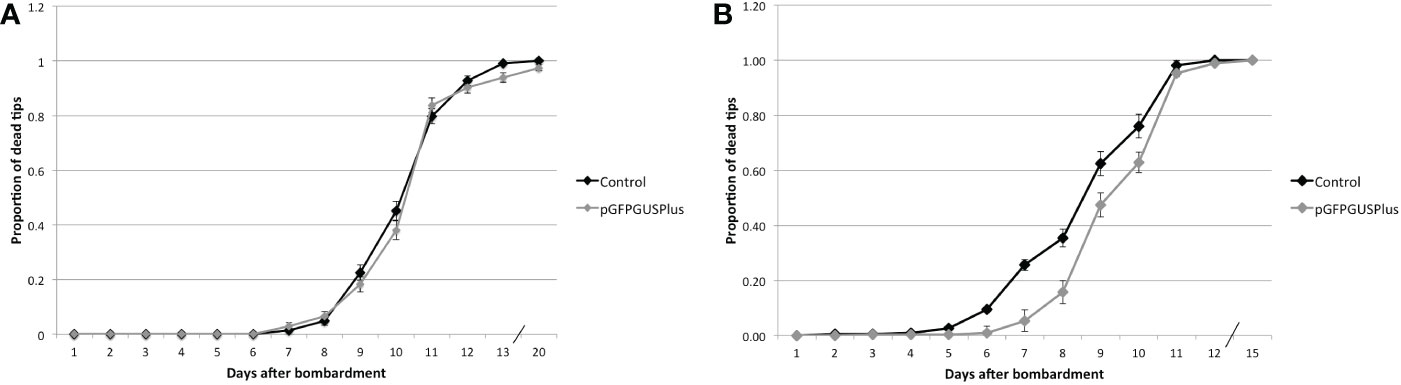
Figure 6 Proportion of dead tips after bombardment with pGFPGUSPlus and treated with different Hyg concentrations. (A) Tips transformed with either pGFPGUSPlus or no plasmid (Control) and treated with 0.5 mg.ml-1 Hyg. (B) Tips transformed with either pGFPGUSPlus or no plasmid (Control) and treated with 1 mg.ml-1 Hyg. Error bars represent the standard error of the proportion. Not statistically sign, put p-value table.
The experiment was repeated with 1 mg.ml-1 Hyg instead of 0.5. Five hundred tips were bombarded with pGFPGUSPlus and 500 with uncoated gold particles, and the number of dead tips was counted every day. After 10 days, the surviving tips were transferred to normal seawater to see whether they could recover, but they died after a few days (Figure 6). The survival of tips transformed with the pGFPGUSPlus plasmid was however significantly different from the control from days 6 to 10 (Figure 6, Supplementary Table S8), suggesting that the transfection of the pGFPGUSPlus plasmid in the tips had a positive impact on their survival.
3.6. Transgene expression using an endogenous promoter
3.6.1. Identification of the GlAct1 promoter
Although the Hpt gene was expressed in the transformed tips, the RT-PCR bands and GUS staining intensities were not very high. An endogenous promoter was therefore used to check whether the gene expression could be increased. As no strong endogenous promoter sequence was known yet in Gp. lemaneiformis, a nucleotide BLAST search on the Gp. lemaneiformis genomic database was performed, using Actin mRNA sequences of the related species Gracilaria textorii and P. yezoensis available on the NCBI as queries to identify Gp. lemaneiformis’s Actin genes. Two putative Actin gene sequences were thereby identified (Supplementary Sequences 8, 10) and confirmed by a protein BLAST on the NCBI database, which yielded Actin protein sequences of related species. Both Actin sequences identified were thus good candidates for selecting an Actin promoter to drive exogenous gene expression in Gp. lemaneiformis. Upstream (200 bp) the coding sequences were selected as Actin promoter sequences, and the promoter sequence with the lowest complexity was selected for cloning, and named Gp. lemaneiformis Actin1 (GlAct1) promoter.
3.6.2. Construction of reporter plasmids using the GlAct1 and PyAct1 promoter sequences
To see whether the expression of the reporter gene in Gp. lemaneiformis could be increased, the newly identified GlAct1 promoter was selected to build a reporter construct. The Actin 1 promoter from P. yezoensis (PyAct1) was also selected, as it had previously been identified and successfully used in P. yezoensis to transiently and stably express several reporter genes (2014; Uji et al., 2010; Hirata et al., 2011). As P. yezoensis and Gp. lemaneiformis are related species, it was speculated that the PyAct1 promoter could also be efficient in Gp. lemaneiformis, and possibly more so than the CaMV35S promoter. The GlAct1 promoter was first cloned in front of the EGFP reporter gene in the pGFPGUSPlus vector, and the resulting plasmid was called R0097-07N (Supplementary Figure S8A, Supplementary Sequence S3).
Several attempts were made to observe EGFP expression in Gp. lemaneiformis’s tips, under the CaMV35S and GlAct1 promoters. However, these attempts were not successful, as it was difficult to distinguish the EGFP fluorescence from the green natural background fluorescence of the algal tissues (data not shown). The BFP reporter gene mTagBFP2 was therefore used as a reporter for the GlAct1 and PyAct1 promoters, as Gp. lemaneiformis was not observed to have natural blue fluorescence.
Two reporter constructs GBFP-PA7 (Supplementary Figure S8B, Supplementary Sequence S4) and PBFP-PA7 (Supplementary Figure S8C, Supplementary Sequence S5) were constructed, expressing the mTagBFP2 reporter gene under respectively the GlAct1 and PyAct1 promoters. They were then transfected into Gp. lemaneiformis tips to test the efficiency of the two promoters to drive the expression of the mTagBFP2 reporter gene.
3.6.3. Transient expression of the mTagBFP2 gene under the GlAct1 and PyAct1 promoters
On the 2nd, 4th, and 6th day after microparticle bombardment, the Gp. lemaneiformis tips transformed with either the GBFP-PA7 plasmid, PBFP-PA7 plasmid or no plasmid for negative control were collected. The expression of the mTagBFP2 reporter was then checked by RT-PCR and fluorescence microscopy, to verify the efficiency of the GlAct1 and PyAct1 promoters.
3.6.3.1. Determination of the mTagBFP2 expression level through RT-PCR
Total RNA was extracted 2, 4, and 6 days after transformation with either the GBFP-PA7 or the PBFP-PA7 plasmid, and PCR was performed on the cDNA using the BFP primers (Figure 7).
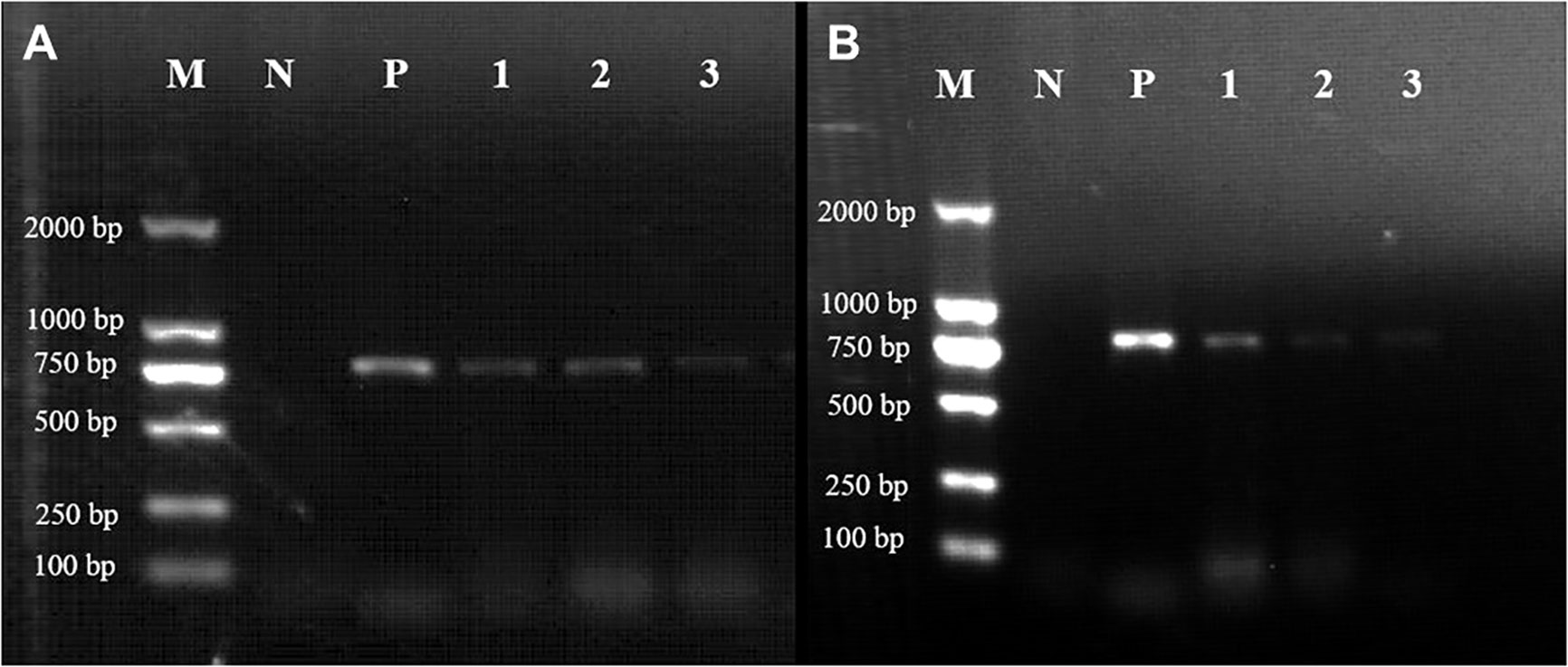
Figure 7 Expression of the mTagBFP2 reporter in Gp. lemaneiformis tips after transformation of the GBFP-PA7 and PBFP-PA7 plasmids. Gel electrophoresis showing the RT-PCR results of the tips transformed with either (A) the GBFP-PA7 plasmid or (B) the PBFP-PA7 plasmid. M: 2000bp marker; N: negative control (untransformed tips); P: positive control (amplification from the mTagBFP2-N plasmid template); 1, 2, and 3: amplification of the cDNA from tips collected respectively 2, 4, and 6 days after transformation. Expected band size: 780 bp.
The expected 780-bp-long band could be observed in tips transformed with GBFP-PA7, 2, 4, and 6 days after bombardment (Figure 7, lanes 1, 2, and 3, respectively). The expression seemed to be weaker after 6 days. The expected band could also be observed in tips transformed with PBFP-PA7, but the expression seemed to be weaker 4 and 6 days after bombardment (Figure 7).
It can be seen that the expression of mTagBFP2 in tips transformed with PBFP-PA7 was lower than that in tips transformed with GBFP-PA7, especially 4 and 6 days after transformation. Based on these results, it can be concluded that the mTagBFP2 gene was successfully expressed in the two plasmids in Gp. lemaneiformis, until 6 days after transformation. The expression under the GpAct1 promoter seemed stronger than under the PyAct1 promoter, suggesting that the first promoter is more efficient in this species, as expected because it is endogenous. However, the expression did not seem stronger than under the CaMV35S promoter, suggesting that these three promoters could be used interchangeably in Gp. lemaneiformis in future work, preferentially CaMV35S and GlAct1.
3.6.3.2. Observation of the BFP fluorescence in Gp. lemaneiformis tips after transformation with the GBFP-PA7 and PBFP-PA7 vectors
To further verify the expression of mTagBFP2, the tips transformed with the GBFP-PA7 plasmid were observed under the fluorescence microscope. The autofluorescence was orange-red, and while no fluorescence could be observed in the negative control (Figures 8A), blue fluorecence could be detected at the surface of the tips transformed with the GBFP-PA7 plasmid 2, 4 and 6 days after bombardment (Figures 8B–D). The mTagBFP2 reporter was therefore efficiently expressed under the GlAct1 promoter 2, 4, and 6 days after bombardment.
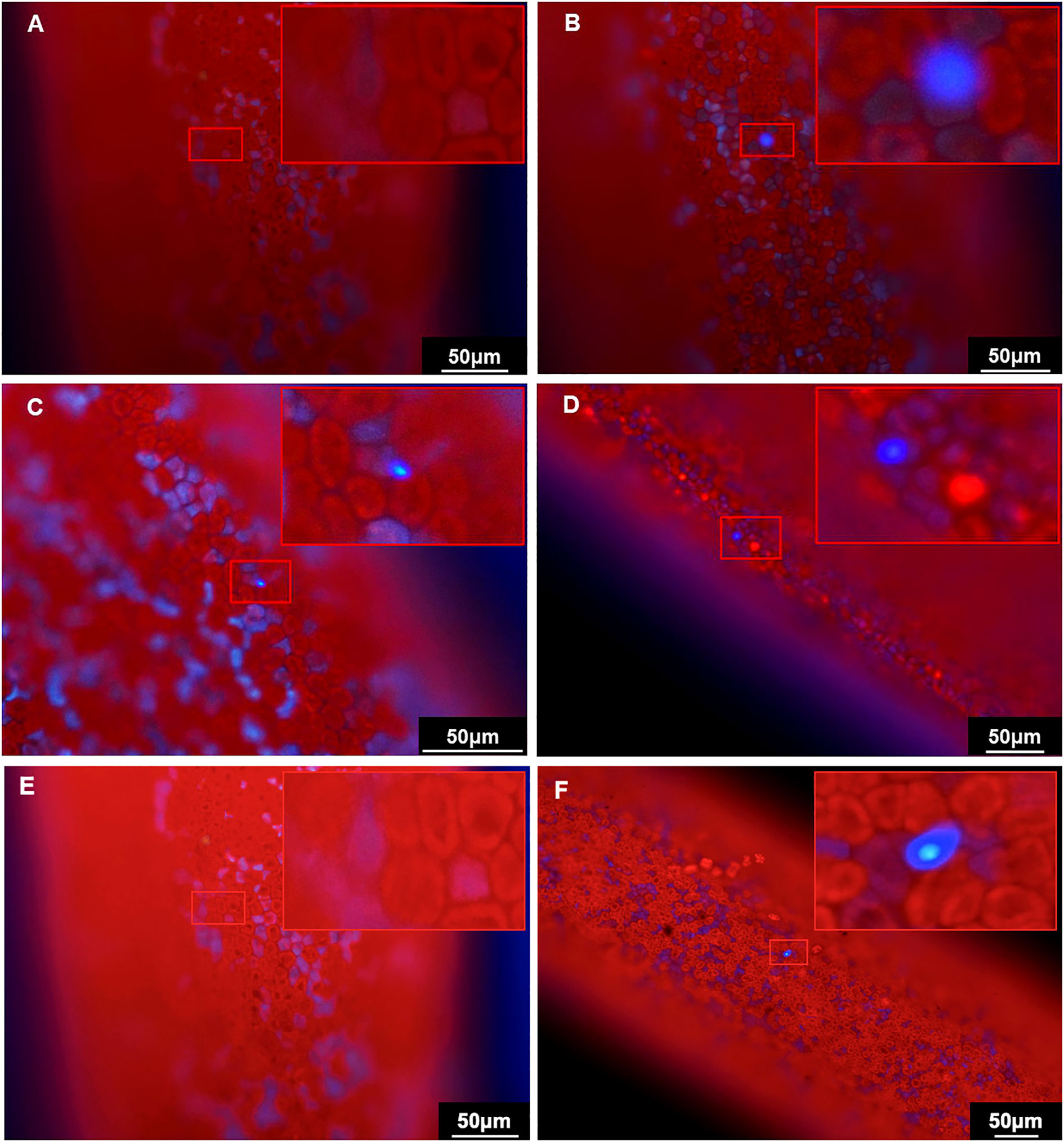
Figure 8 Observation of the BFP fluorescence in the Gp. lemaneiformis tips after transformation of the GBFP-PA7 and PBFP-PA7 plasmids by particle bombardment. Gp. lemaneiformis tips transformed with (A, E) uncoated gold particles (negative control), (B) GBFP-PA7 plasmid observed 2 days after bombardment, (C) GBFP-PA7 plasmid observed 4 days after bombardment, (D) GBFP-PA7 plasmid observed 6 days after bombardment, (F) PBFP-PA7 plasmid observed 6 days after bombardment. The tips were observed under blue fluorescence excitation. (A, B, D-F): 20× magnification, (C): 40× magnification. The upper right corner of each picture is an enlargement of the red box indicating the presence of BFP fluorescence.
The tips transformed with the PBFP-PA7 plasmid were also observed under the fluorescence microscope. Blue fluorescence could be detected, but only 6 days after bombardment, whereas no fluorescence was observed in the negative control (Figures 8E, F). Fewer fluorescent cells could be observed compared with tips transformed with GBFP-PA7. It can be concluded that the mTagBFP2 gene was successfully expressed in the algal tips under the PyAct1 promoter 6 days after bombardment, but that the expression was weaker than under the GpAct1 promoter.
Although the number of cells expressing mTagBFP2 was relatively low, the blue fluorescence was clear and contrasted greatly with the red autofluorescence from the algal tissue. The mTagBFP2 gene is therefore an ideal fluorescence reporter for this species, which could be used for further expression studies in the future.
4. Discussion
4.1. Transgene expression
The CaMV35S promoter was shown to have various efficiencies in red algae. It was indeed shown to efficiently drive gene expression in G. gracilis (Huddy et al., 2012), but it was found to have a low activity in P. yezoensis and Cyanidioschyzon merolae (Fukuda et al., 2008; Watanabe et al., 2011). This could be due to differences in the promoter structure and transcriptional regulation of protein-coding genes between red algae and dicot plants. This article showed that the CaMV35S promoter could efficiently drive the expression of the Hpt and LacZ genes in Gp. lemaneiformis, but potential higher efficiency of other promoters needed to be checked. Promoters taken from the genome of other species can usually serve as efficient heterologous promoters to drive the expression of foreign genes in algae. Porphyra’s actin promoter (PyAct1) has for instance been found effective in expression vectors in various red algae species (Uji et al., 2010; Hirata et al., 2011). The current study showed that it was also efficient in Gp. lemaneiformis but less so than the endogenous GlAct1 promoter. In conclusion, the CaMV35S, PyAct1, and GlAct1 promoters could be used alternatively for transgene expression in this species, although gene expression under the PyAct1 promoter seemed to be the weakest. CaMV35S would be more suited for stable genomic integration and expression of a transgene to avoid any risk of recombination of the GlAct1 promoter at the endogenous Actin1 locus.
Other promoters could be tried to see if gene expression could still be improved, such as promoters of other ubiquitous genes like GAPDH or Ubiquitin. The putative promoter region upstream the GAPDH and Ubiquitin GUBQ1 genes can indeed be found in the Gp. lemaneiformis genomic SRA database, and 200 bp of these regions could be cloned upstream reporter genes. However, for stable DNA integration experiments, a promoter sequence from a related species could be more favorable to avoid integration at the promoter site caused by sequence homology. GAPDH and Ubiquitin promoter sequences from G. gracilis, C. crispus or P. yezoensis could therefore be tested.
The PCR and RT-PCR results of the plasmid detection and the expression of the Hpt and mTagBFP2 genes in the transformants showed very weak PCR amplification 1 week after transformation (Figures 3, 4B, 7). This suggests that the plasmid was present in lower amounts and that the transgene was not expressed anymore or very weakly in the tips after that time, which could be due to the plasmid being diluted because of cell division, or being degraded. The antibiotic selection process should therefore be done within this timeframe of one week for future experiments.
4.2. Reporter gene
The GUS staining and BFP observation results also confirm that microparticle bombardment is a reliable method for DNA transformation in Gp. lemaneiformis branch tips. Moreover, they show that the two reporter genes are suited for this species. The GUS staining and BFP fluorescence, however, could not be observed in many cells, indicating that transformation efficiency was not high. This may be due to a rather low efficiency of DNA coating on the gold particles or to plasmid damage during the bombardment process. Different coating conditions could be tested in the future to tackle this problem, with different buffers and DNA concentrations. The mTagBFP2 sequence could also be optimized to the codon usage of Gp. lemaneiformis to see whether the expression could be improved. The Cyan Fluorescent Protein (CFP) could also be tested, as it has been successfully used in Pyropia (Lim et al., 2013).
4.3. Selection of transformants
There was a discrepancy between the viability that was expected from the IC50 results (Table 2) and the observed viability after treatment with 0.5 mg.ml-1 Hyg (Figure 6). Fifty percent viability was expected for diploid tips after nine days of 0.5 mg.ml-1 Hyg treatment (Table 2), but about 80% viability was observed in both transformed and non-transformed tips (Figure 6). This could be caused by the fact that a smaller number of tips were tested in the IC50 experiment or by the way the dead tips were screened. As the tips that had some red pigments left were not considered dead, there could be variability among individual tips, in particular if their size varied slightly.
Resistant tips transformed with the Hpt resistance gene could still not be recovered after treatment with Hyg, suggesting either that the transformation rate was too low, or that the Hyg treatment was inadequate. It was determined that a concentration of 0.5 mg.ml-1 Hyg was not enough to screen the tips transformed with the resistance gene from the control tips (Figure 6). With a concentration of 1 mg.ml-1, although the tips could not be regenerated, their survival was increased compared with controls between 6 and 10 days (Figure 6). This suggests that the Hpt gene conferred Hyg resistance to the tips, but that the regeneration setup has to be improved. To be able to regenerate transformed tips, shortening the treatment period could for instance be tested. The RT-PCR results (Figure 4) suggest that the transgene was almost not expressed anymore in the transformed tips 7 days after transformation. The 10 days Hyg treatment of tips transformed with pGFPGUSPlus (Figure 6) was therefore too much, as the tip cells would probably not be able to resist against Hyg from 7 to 10 days. A shorter Hyg treatment of 7 days or less could thus be tested, and the tips could be transferred to seawater without antibiotic after the short Hyg treatment to test whether they could be regenerated. Regeneration of the tips might also be improved by increasing the oxygenation of the culture flasks.
To tackle the problem of low transformation rate, the number of individuals to be transformed could be increased, for instance at least 500 tips or more could be used for each bombardment. Second, the tips could be slightly cut with a blade before microparticle bombardment to facilitate the penetration of the coated gold particles in the tissue, as the Gp. lemaneiformis cell wall is thick. Enzymes degrading the cell wall could also be used to facilitate the transformation, such as glycoside hydrolases, polysaccharide lyases or carbohydrate esterases, which have successfully been used in green macroalga to degrade the cell wall (Costa et al., 2022). Third, transformation could be tried in spores instead of branch tips, for example, via electroporation as what was done in the unicellular alga Chlamydomonas reinhardtii (Ferenczi et al., 2017). A transformed spore could therefore give rise to a fully transformed organism. However, to obtain stably transformed organisms, strategies for obtaining stable DNA integration in the genome will have to be developed.
5. Conclusion
In conclusion, this study showed that microparticle bombardment is an efficient DNA transformation technique in Gp. lemaneiformis and that the CaMV35S, GlAct1, and PyAct1 promoters can efficiently drive gene expression in this species. mTagBFP2 also appeared to be a well-suited reporter gene for this alga. The selective antibiotic and its working concentration for transformant selection were determined, although experimental conditions should be refined to be able to select transformants. The work presented here constitutes the first step toward genetic transformation and transformant selection in Gp. lemaneiformis and opens the door to more genetic applications and discoveries.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
ME wrote the main manuscript text. She prepared Figures 1, 2, and 6 and performed the experimental research for antibiotic sensitivity test, antibiotic concentration determination for screening, selection of transformants, identification of the GlAct1 promoter and Cas12a gene editing. NZ prepared Figures 7 and 8 and performed the experimental research for transient transformation and mTagBFP gene expression test under the Actin promoters, including BFP fluorescence observation. PG edited Figures 3, 4, and 5 and performed the experimental research for transient transformation and gene expression test under the CaMV35S promoter, including GUS staining. YL participated in the Hpt and EGFP gene PCR experiments. FK helped with the culture of P. yezoensis, and ZS supervised the project. ME and ZS reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the China Agriculture Research System of MOF and MARA (CARS-50) and the National Natural Science Foundation of China (N° 32072953). It was also supported by the Shandong Province Key Research and Development Program (Grant N° 2021LZGC004).
Conflict of interest
Author ME was employed by Suzhou Hongxun Biotechnologies CO., LTD.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1112180/full#supplementary-material
References
Chakraborty K., Antony T. (2020). Salicornolides a-c from gracilaria salicornia attenuate pro-inflammatory 5-lipoxygense: Prospective natural anti-inflammatory leads. Phytochemistry 172, 112259. doi: 10.1016/j.phytochem.2020.112259
Collado-Vides L. (2002). Clonal architecture in marine macroalgae: Ecological and evolutionary perspectives. Evol. Ecol. 15, 531–545. doi: 10.1023/A:1016009620560
Costa M. M., Pio L. B., Bule P., Cardoso V. A., Duarte M., Alfaia C. M., et al. (2022). Recalcitrant cell wall of ulva lactuca seaweed is degraded by a single ulvan lyase from family 25 of polysaccharide lyases. Anim. Nutr. 9, 184–192. doi: 10.1016/j.aninu.2022.01.004
Du Q., Bi G., Mao Y., Sui Z. (2016). The complete chloroplast genome of gracilariopsis lemaneiformis (Rhodophyta) gives new insight into the evolution of family gracilariaceae. J. Phycol. 52, 441–450. doi: 10.1111/jpy.12406
Ferenczi A., Pyott D. E., Xipnitou A., Molnar A. (2017). Efficient targeted DNA editing and replacement in chlamydomonas reinhardtii using Cpf1 ribonucleoproteins and single-stranded DNA. Proc. Nat. Acad. Sci. 114, 13567–13572. doi: 10.1073/pnas.1710597114
Fredericq S., Hommersand M. (1989). Comparative morphology and taxonomic status of gracilariopsis (Gracilariales, rhodophyta). J. Phycol. 25, 228–241. doi: 10.1111/j.1529-8817.1989.tb00117.x
Freile-Pelegrín Y., Murano E. (2005). Agars from three species of gracilaria (Rhodophyta) from Yucatán Peninsula. Bioresour. Technol. 96, 295–302. doi: 10.1016/j.biortech.2004.04.010
Fuhrmann M., Oertel W., Hegemann P. (1999). A synthetic gene coding for the green fluorescent protein (GFP) is a versatile reporter in chlamydomonas reinhardtii. Plant J. 19 (3), 353–361. doi: 10.1046/j.1365-313X.1999.00526.x
Fujiwara T., Ohnuma M., Kuroiwa T., Ohbayashi R., Hirooka S., Miyagishima S.-Y. (2017). Development of a double nuclear gene-targeting method by two-step transformation based on a newly established chloramphenicol-selection system in the red alga cyanidioschyzon merolae. Front. Plant Sci. 8, 343. doi: 10.3389/fpls.2017.00343
Fukuda S., Koji M., Uji T., Park E. J., Ohba T., Asada K., et al. (2008). Factors influencing efficiency of transient gene expression in the red macrophyte porphyra yezoensis. Plant Sci. 174, 329–339. doi: 10.1016/j.plantsci.2007.12.006
Gan S. Y., Qin S., Othman R. Y., Phang S. M. (2004). “Development of a transformation system for gracilaria changii (Gracilariales, rhodophyta), a Malaysian red alga via microparticle bombardment,” in The 4th annual seminar of national sience fellowship 2004. http://web.usm.my/nsf/proceedings/pBIO08.pdf
Ghosh A. K., Panda S. K., Luyten W. (2021). Anti-vibrio and immune-enhancing activity of medicinal plants in shrimp: A comprehensive review. Fish Shellfish Immunol. 117, 192–210. doi: 10.1016/j.fsi.2021.08.006
González-Leija J. A., Hernández-Garibay E., Pacheco-Ruíz I., Guardado-Puentes J., Espinoza-Avalos J., López-Vivas J. M., et al. (2009). Optimization of the yield and quality of agar from gracilariopsis lemaneiformis (Gracilariales) from the gulf of California using an alkaline treatment. J. Appl. Phycol. 21, 321–326. doi: 10.1007/s10811-008-9370-0
Hirata R., Takahashi M., Saga N., Mikami K. (2011). Transient gene expression system established in porphyra yezoensis is widely applicable in bangiophycean algae. Mar. Biotechnol. 13, 1038–1047. doi: 10.1007/s10126-011-9367-6
Hirata R., Uji T., Fukuda S. (2014). Development of a stable transformation system with a codon-optimized selection marker and reporter genes in pyropia yezoensis (Rhodophyta). J. Appl. Phycol. 26, 1863–1868. doi: 10.1007/s10811-013-0234-x
Huang X., Zang X., Wu F., Jin Y., Wang H., Liu C., et al. (2017). Transcriptome sequencing of gracilariopsis lemaneiformis to analyze the genes related to optically active phycoerythrin synthesis. PloS One 12 (1), e0170855. doi: 10.1371/journal.pone.0170855
Huddy S. M., Meyers A. E., Coyne V. E. (2012). Transformation of LacZ using different promoters in the commercially important red alga, gracilaria gracilis. Afr. J. Biotechnol. 11 (8), 1879–1885. doi: 0.5897/AJB11.2641
Kaddouri H., Nakache S., Houzé S., Mentré F., Le Bras J. (2006). Drug susceptibility of plasmodium falciparum clinical isolates from Africa using plasmodium lactate dehydrogenase immunodetection assay and inhibitory emax model for precise IC50 measurement. Antimicrobial Agents Chemotherapy 50, 3343–3349.
Kilian O., Benemann C. S. E., Niyogi K. K., Vick B. (2011). High-efficiency homologous recombination in the oil-producing alga nannochloropsis sp. Proc. Natl. Acad. Sci. U.S.A. 108 (52), 21265–21269. doi: 10.1073/pnas.1105861108
Kong F., Zhao H., Liu W., Li N., Mao Y. (2017). Construction of plastid expression vector and development of genetic transformation system for the seaweed pyropia yezoensis. Mar. Biotechnol. 19 (2), 147–156. doi: 10.1007/s10126-017-9736-x
Lee Y. K., An G., Lee I. K. (2000). Antibiotics resistance of a red alga, griffithsia japonica. J. Plant Biol. 43 (3), 179–182. doi: 10.1007/BF03030496
Lee W. K., Lim Y. Y., Thean-Chor Leow A., Namasivayam P., Ong Abdullah J., Ho C. L. (2017). Biosynthesis of agar in red seaweeds: A review. Carbohydr. Polymers 164, 23–30. doi: 10.1016/j.carbpol.2017.01.078
Le Nagard H., Vincent C., Mentré F., Le Bras J. (2011). Online analysis of in vitro resistance to antimalarial drugs through nonlinear regression. Comput. Methods Programs BioMed. 104 (1), 10–18.
Li Z., Bock R. (2018). Replication of bacterial plasmids in the nucleus of the reg alga porphyridium purpureum. Nat. Commun. 9 (1), 3451. doi: 10.1038/s41467-018-05651-1
Lim J., Ahn J., Hwangbo K., Choi D. W., Park E. J., Hwang M. S., et al. (2013). Development of cyan fluorescent protein (CFP) reporter system in green alga chlamydomonas reinhardtii and macroalgae pyropia sp. Plant Biotechnol. Rep. 7, 407–414. doi: 10.1007/s11816-013-0274-3
Marinho-Soriano E. (2001). Agar polysaccharides from gracilaria species (Rhodophyta, gracilariaceae). J. Biotechnol. 89, 81–84. doi: 10.1016/S0168-1656(01)00255-3
Mikami K., Hirata R., Takahashi M., Uji T., Saga N. (2011). “Transient transformation of red algal cells: Breakthrough toward genetic transformation of marine crop porphyra species,” in Genetic transformation. Ed. Alvarez M., 241–258. https://www.intechopen.com/chapters/18822
Mikami K., Uji T., Li L., Takahashi M., Yasui H., Saga N. (2009). Visualization of phosphoinositides via the development of the transient expression system of a cyan fluorescent protein in the red alga porphyra yezoensis. Mar. Biotechnol. (NY). 11 (5), 563–569. doi: 10.1007/s10126-008-9172-z
Pang Q., Sui Z., Kang K. H., Kong F., Zhang X. (2010). Application of SSR and AFLP to the analysis of genetic diversity in gracilariopsis lemaneiformis (Rhodophyta). J. Appl. Phycol. 22, 607–612. doi: 10.1007/s10811-009-9500-3
Provasoli L. (1966). “Media and prospects of the cultivation of marine algae,” in Cultures and collections of algae. proceedings of the US–Japan conference hakone. Eds. Watanabe A., Hattori A. (Tokyo: Japanese Society of Plant Physiology), 1–26.
Qi Z. H., Liu H. M., Li B., Mao Y., Jiang Z. J., Zhang J. H., et al. (2010). Suitability of two seaweeds, gracilaria lemaneiformis and sargassum pallidum, as feed for the abalone haliotis discus hannai ino. Aquaculture 300, 189–193. doi: 10.1016/j.aquaculture.2010.01.019
Saha D., Bhattacharya S. (2010). Hydrocolloids as thickening and gelling agents in food: a critical review. J. Food Sci. Technol. 47 (6), 587–597. doi: 10.1007/s13197-010-0162-6
Shi C. S., Sang Y. X., Sun G. Q., Li T. Y., Gong Z. S., Wang X. H. (2017). Characterization and bioactivities of a novel polysaccharide obtained from gracilariopsis lemaneiformis. An. Acad. Bras. Cienc. 89, 175–189. doi: 10.1590/0001-3765201720160488
Subach O. M., Cranfill P. J., Davidson M. W., Verkhusha V. V. (2011). An enhanced monomeric blue fluorescent protein with the high chemical stability of the chromophore. PloS One 6 (12), e28674. doi: 10.1371/journal.pone.0028674
Sun X., Wu J., Wang G., Kang Y., Ooi H.S., Shen T., et al. (2018). Genomic analyses of unique carbohydrate and phytohormone metabolism in the macroalga gracilariopsis lemaneiformis (Rhodophyta). BMC Plant Biol. 18, 94. doi: 10.1186/s12870-018-1309-2
Sun Y., Yang Z., Gao X., Li Q., Zhang Q., Xu Z. (2005). Expression of foreign genes in dunaliella by electroporation. Mol. Biotechnol. 30 (3), 185–192. doi: 10.1385/MB:30:3:185
Takahashi M., Mikami K., Mizuta H., Saga N. (2011). Identification and efficient utilization of antibiotics for the development of a stable transformation system in porphyra yezoensis (Bangiales, rhodophyta). J. Aquac. Res. Dev. S3, 002. doi: 10.4172/2155-9546.1000118
Tseng C. K. (2001). Algal biotechnology industries and research activities in China. Appl. Phycol. 13 (4), 375–380. doi: 10.1023/A:1017972812576
Uji T., Hirata R., Fukuda S. (2014). A codon-optimized bacterial antibiotic gene used as selection marker for stable nuclear transformation in the marine red alga pyropia yezoensis. Mar. Biotechnol. 16, 251–255. doi: 10.1007/s10126-013-9549-5
Uji T., Takahashi M., Saga N., Mikami K. (2010). Visualization of nuclear localisation of transcription factors with cyan and green fluorescent proteins in the red alga porphyra yezoensis. Mar. Biotechnol. 12, 150–159. doi: 10.1007/s10126-009-9210-5
Vickers C. E., Schenk P. M., Li D., Mullineaux P. M., Gresshoff P. M. (2007). pGFPGUSPlus, a new binary vector for gene expression studies and optimising transformation systems in plants. Biotechnol. Lett. 29 (11), 1793–1796. doi: 10.1007/s10529-007-9467-6
Wang J., Jiang P., Cui Y., Guan X., Qin S.. (2010a). Gene transfer into conchospores of porphyra haitanensis (Bangiales, rhodophyta) by glass bead agitation. Phycologia 49 (4), 355–360. doi: 10.2216/09-91.1
Wang J., Jiang P., Cui Y., Deng X., Li F., Liu J., et al. (2010b). Genetic transformation in kappaphycus alvarezii using micro-particle bombardment: A potential strategy for germplasm improvement. Aquacult. Int. 18, 1027–1034. doi: 10.1007/s10499-010-9320-0
Wang J. G., Sui Z. H., Zhou W., Chang L. P., Zhang S., Ma J. H., et al. (2014). The determination of growth, physiological, biochemical parameters and the development of SCAR marker of the new strain ZC of gracilariopsis lemaneiformis (Rhodophyta). Period Ocean Univ. China 44 (11), 037–044. doi: 10.16441/j.cnki.hdxb.2014.02.010
Wang W., Wang G., Gao Z., Lin X., Xu P. (2007). Characterization of gracilaria lemaneiformis bory (Gracilariaceae, rhodophyta) cultivars in China using the total soluble proteins and RAPD analysis. Bot. Mar. 50, 177–184. doi: 10.1515/BOT.2007.021
Wang F., Wang C., Zou T., Xu N., Sun X. (2017). Comparative transcriptional profiling of gracilariopsis lemaneiformis in response to salicylic acid- and methyl jasmonate-mediated heat resistance. PloS One 12 (5), e0176531. doi: 10.1371/journal.pone.0176531
Watanabe S., Ohnuma M., Sato J., Yoshikawa H., Tanaka K. (2011). Utility of a GFP reporter system in the red alga cyanidioschyzon merolae. J. Gen. Appl. Microbiol. 57, 69–72. doi: 10.2323/jgam.57.69
Wen X., Peng C., Zhou H., Lin Z., Lin G., Chen S., et al. (2006). Nutritional composition and assessment of gracilaria lemaneiformis bory. J. Integr. Plant Biol. 48, 1047–1053. doi: 10.1111/j.1744-7909.2006.00333.x
Wood M. A., Lipcius R. N. (2022). Non-native red alga gracilaria vermiculophylla compensates for seagrass loss as blue crab nursery habitat in the emerging Chesapeake bay ecosystem. PloS One 17 (5), e0267880. doi: 10.1371/journal.pone.0267880
Yang Y. F., Fei X. G., Song J. M., Hu H. Y., Wang G. C., Chung I. K., et al. (2006). Growth of gracilaria lemaneiformis under different cultivation conditions and its effects on nutrient removal in Chinese coastal waters. Aquaculture 254, 248–255. doi: 10.1016/j.aquaculture.2005.08.029
Zhang L., Wang X., Qian H., Chi S., Liu C., Liu T. (2012). Complete sequences of the mitochondrial DNA of the wild gracilariopsis lemaneiformis and two mutagenic cultivated breeds (Gracilariaceae, rhodophyta). PloS One 7 (6), e40241. doi: 10.1371/journal.pone.0040241
Zhang J., Wu Q., Eléouët M., Chen R., Chen H., Zhang N., et al. (2022). CRISPR/LbCas12a-mediated targeted mutation of gracilariopsis lemaneiformis (Rhodophyta). Plant Biotechnol. J. doi: 10.1111/pbi.13949
Zhou W., Hu Y., Sui Z., Fu F., Wang J., Chang L., et al. (2013). Genome survey sequencing and genetic background characterization of gracilariopsis lemaneiformis (Rhodophyta) based on next-generation sequencing. PloS One 8 (7), e69909. doi: 10.1371/journal.pone.0069909
Zhou Y., Yang H. S., Hu H. Y. (2006). Bioremediation potential of the macroalga gracilaria lemaneiformis (Rhodophyta) integrated into fed fish culture in coastal waters of north China. Aquaculture 252, 264–276. doi: 10.1016/j.aquaculture.2005.06.046
Keywords: Gracilariopsis lemaneiformis, Rhodophyta, antibiotic selection, DNA transformation, promoter
Citation: Eléouët M, Zhang N, Guo P, Lei Y, Kong F and Sui Z (2023) Setting the basis for transient DNA transformation and transformant selection in the red macroalga Gracilariopsis lemaneiformis. Front. Mar. Sci. 10:1112180. doi: 10.3389/fmars.2023.1112180
Received: 30 November 2022; Accepted: 09 January 2023;
Published: 26 January 2023.
Edited by:
Carole Anne Llewellyn, Swansea University, United KingdomReviewed by:
Yanhui Bi, Shanghai Ocean University, ChinaKit Wayne Chew, Nanyang Technological University, Singapore
Copyright © 2023 Eléouët, Zhang, Guo, Lei, Kong and Sui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenghong Sui, c3VpemhlbmdoQG91Yy5lZHUuY24=
†These authors have contributed equally to this work