 Hiroki Kise1†‡
Hiroki Kise1†‡ Akira Iguchi1,2*‡
Akira Iguchi1,2*‡ Naoki Saito1
Naoki Saito1 Yuki Yoshioka3
Yuki Yoshioka3 Koji Uda4
Koji Uda4 Tomohiko Suzuki4
Tomohiko Suzuki4 Atsushi J. Nagano5,6
Atsushi J. Nagano5,6 Atsushi Suzuki1,2
Atsushi Suzuki1,2 Nozomu Iwasaki7
Nozomu Iwasaki7- 1Geological Survey of Japan, National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba, Ibaraki, Japan
- 2Research Laboratory on Environmentally-conscious Developments and Technologies (E-code), National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba, Japan
- 3Department of Bioresources Engineering, National Institute of Technology, Okinawa College, Nago, Okinawa, Japan
- 4Faculty of Science, Kochi University, Kochi, Japan
- 5Faculty of Agriculture, Ryukoku University, Otsu, Shiga, Japan
- 6Institute for Advanced Biosciences, Keio University, Tsuruoka, Yamagata, Japan
- 7Faculty of Geo-Environmental Science, Rissho University, Saitama, Japan
Population sizes of the Japanese red coral Corallium japonicum have been severely affected by poaching and overfishing. Although genetic structure and connectivity patterns are considered important parameters for conservation strategies, there are few studies focusing on the population genetics of C. japonicum in the Northwest Pacific. We examined the genetic population structure of C. japonicum, in the Northwest Pacific. We used restriction-site-associated DNA sequencing (RAD-seq), which can be used to identify genome-wide single-nucleotide polymorphism (SNPs), to reveal detailed within-species genetic variations. Using the variable SNP loci identified from this analysis, we successfully evaluated the population-level genetic diversity and patterns of gene flow among multiple populations of C. japonicum around Japan. The results of genetic analysis basically showed that gene flow is widely maintained in the geographic range examined in this study, but the analysis in combination with larval dispersal simulations revealed several populations that were genetically distinct from the other populations, suggesting geographically limited gene flows. The information obtained from this study will be useful for the design of effective management schemes for C. japonicum, which is under threat from overfishing.
Introduction
Precious red corals in the family Coralliidae are a commercially important deep-sea resource, and are used for jewelry production in China and Taiwan (Tsounis et al., 2010). Three Coralliidae species Corallium japonicum Kishinouye, 1903 (Japanese red coral), Pleurocorallium elatius (Ridley, 1882) (pink coral), and Pleurocorallium konojoi (Kishinouye, 1903) (white coral) have been harvested in Japanese waters for 150 years (Iwasaki et al., 2022). However, population sizes of these corals have been highly impacted by poaching and overfishing (Iwasaki, 2019). Consequently, the Japanese Ministry of the Environment has designated these three species as “Near Threatened”. Although genetic structure and patterns of connectivity are considered as important parameters for conservation strategies (Palumbi, 2004), there are few studies focused on population genetics of precious red corals in Japanese waters.
Because the adult stage of corals is sessile, these corals rely on larval dispersal to maintain populations and expand into new habitats. Understanding this larval dispersal is quite challenging, however, because although ocean currents are a major determinant of larval dispersal patterns, data on deep-sea currents are often quite limited. A promising alternative approach is the use of DNA markers to evaluate patterns of connectivity and genetic diversity among populations (e.g., Baco et al., 2016; Taylor and Roterman, 2017). This method can also be combined with ocean-current-based larval dispersal simulations to better assess inter-population connectivity (Fujita et al., 2016; Nakabayashi et al., 2019).
Because haplotype sequences are especially easy to identify in mitochondrial DNA, mitochondrial DNA markers have historically been used to evaluate the population structures of many animal species (e.g., Avise, 2000). However, these markers are of limited use for corals and other Anthozoa, because the extremely slow mutation rates of mitochondrial DNA in these taxa (Hellberg, 2006) reduce the ability to detect genetic differences between populations (Nakajima et al., 2012a; Takata et al., 2019). Fortunately, recent advances in high-throughput DNA sequencing have enabled the use of variable DNA markers in non-model organisms (Andrews et al., 2016). In particular, single nucleotide polymorphisms (SNPs) in genomic DNA can be easily identified by using several methods such as restriction-site-associated DNA sequencing (RAD-seq; Baird et al., 2008) or multiplexed ISSR genotyping by sequencing (MIG-seq; Suyama and Matsuki, 2015). MIG-seq has attracted particular interest; although only a small number of SNP loci can be genotyped with this method, it has been used to identify population-level genetic differences and is increasingly being used to study marine organisms (Takata et al., 2019; Yorisue et al., 2020; Takata et al., 2021).
Numerous population genetic analyses of coastal benthic organisms have been conducted in Japanese waters (e.g., Kojima et al., 1997; Kojima et al., 2004). However, although several of these studies have used high-resolution markers such as microsatellites and SNPs to examine shallow-water scleractinian corals (e.g., Nakajima et al., 2012a; Nakajima et al., 2012b; Nakajima et al., 2017; Iguchi et al., 2019; Nakabayashi et al., 2019), few have examined deep-water species such as precious corals. In the Mediterranean Sea, the population genetics of the precious coral Corallium rubrum (Linnaeus, 1758) are well studied in shallow water (Costantini et al., 2007; Ledoux et al., 2010; Aurelle et al., 2011; Costantini et al., 2018) as well as in mesophotic to deep-sea (Costantini et al., 2010; Costantini et al., 2013; Cannas et al., 2015; Cannas et al., 2016).
In Japanese waters, two studies have used MIG-seq to examine the population genetics of Corallium japonicum (Takata et al., 2019; Takata et al., 2021). However, these studies only examined colonies in Kochi Prefecture, and information on other regions of Japan is lacking. The Kuroshio Current, which flows along the coast of Japan, strongly affects the population structure of many marine organisms (e.g. Nakabayashi et al., 2019), but little is known about its effects on deep-water species such as precious corals. In this study, we used double-digest restriction-site-associated DNA sequencing (ddRAD-seq) following the protocol described by Sakaguchi et al. (2015) to identify the detailed genetic population structure of C. japonicum in the Northwest Pacific. These data were then compared against an ocean-current simulation to examine the formation and patterning of C. japonicum populations in this region.
Materials and methods
Sampling, DNA extraction, and library preparation
Fragments of Corallium japonicum in the Northwest Pacific were collected with remotely operated vehicles or obtained from fishers (Figure 1 and Table 1). Sample collection was conducted with permission from Okinawa Prefecture (Oki 21-131) and other regional authorities. A total of 84 C. japonicum samples were used for this study. The samples were preserved by immersion in CHAOS solution (guanidine thiocyanate 50 g, N-lauroyl sarcosin sodium 0.5 g, 2-mercaptoethanol 0.7 mL, 1M Tris pH 8 2.5 mL, sterile distilled water 100 mL; Fukami et al., 2004), and genomic DNA was extracted with the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). We measured the DNA concentration of each sample by using a Qubit Fluorometer (ThermoFisher, Waltham, MA, USA). A RAD-seq library was prepared by using ddRAD-seq as described by Sakaguchi et al. (2015). For the library preparation, we used EcoRI and BglII and performed size selection at around 320 bp. We sequenced the ddRAD-seq library by using an Illumina HiSeq 2500 sequencer (Illumina, San Diego, CA, USA) with 50-bp single-end reads.
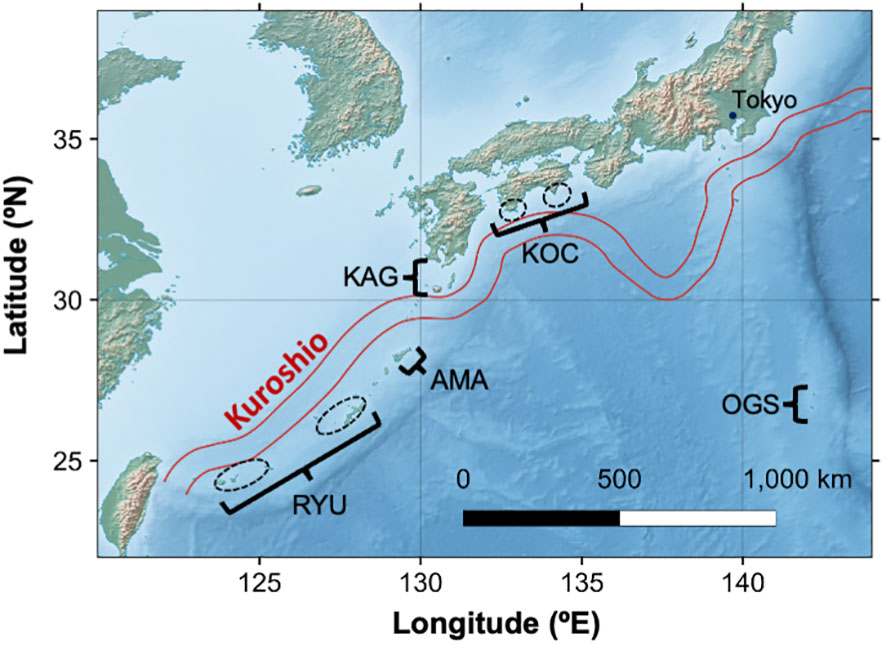
Figure 1 Map of sampling sites of Corallium japonicum surveyed in this study. The map was made with Natural Earth (https://www.naturalearthdata.com). Flow path of Kuroshio between Dec 28, 2018 and Jan 3, 2019 is shown using a shapefile provided by Japan Coast Guard (https://www1.kaiho.mlit.go.jp/KANKYO/KAIYO/qboc/kurosio-num.html).

Table 1 Summary of samples used in this study.
Bioinformatics
Raw Illumina reads were de-multiplexed by using the process_radtags module in Stacks (Catchen et al., 2013). These reads were then processed by using the pipeline ipyrad (Eaton and Overcast, 2020) with the reference-based assembly under default settings. The draft genomes of Corallium japonicum reported in Takata et al. (2021) were used for the reference genome. Low-quality bases (Q < 20), adapters, and restriction sites were filtered out from each read prior to mapping to the draft genome. Loci present in at least 70% of individuals were retained. We removed individuals that had less than 1000 loci according to the mapping statistics. In this manner, 76 individuals of C. japonicum were selected for downstream analyses (Table 1). From the “.vcf” output file generated by ipyrad, we filtered the dataset using VCFtools (Danecek et al., 2011) to obtain the final SNP dataset including biallelic SNPs and one SNP per RAD locus. We further filtered the SNP dataset to remove SNPs out of Hardy–Weinberg equilibrium (p < 0.001) within each population using the script filter_hwe_by_pop.pl from the dDocent package (Puritz et al., 2014: https://github.com/jpuritz/dDocent/blob/master/scripts/filter_hwe_by_pop.pl) and to exclude SNPs with >30% missing genotypes in at least one of five populations with pop_missing_filter.sh in dDocent ver. 2.2.7 (Puritz et al., 2014: https://github.com/jpuritz/dDocent/blob/master/scripts/pop_missing_filter.sh).
We used BayeScan (Foll and Gaggiotti, 2008) to identify possible SNPs under natural selection (outlier loci) based on the FST outlier method with default settings. No outlier loci were detected, and therefore all loci were used for downstream analyses. The final SNP dataset consisted of 961 loci from 76 individuals.
Population structure was analyzed with STRUCTURE (Pritchard et al., 2000) by using an admixture model without priors. We ran STRUCTURE with 500,000 Markov chain Monte Carlo replications, with a burn-in period of 250,000 generations with K = 1–7, repeated 10 times for each value of K. The optimal K was evaluated using two statistics as suggest by Janes et al. (2017); we used log probabilities of Pr(X|K) and ΔK statistic (Evanno et al., 2005) in Structure Harvester (Earl and VonHoldt, 2012). The mean of the permuted matrices across replicates was calculated for the optimal K in Clumpp (Jakobsson and Rosenberg, 2007) implemented in Clumpak (Kopelman et al., 2015) with the Greedy search option (2000 repeats). Structure results were visualized with Distruct (Rosenberg, 2004). We conducted a principal component analysis (PCA) of the genotype covariance matrix by using the “glpca” function in the R package adegenet (Jombart, 2008) to characterize genetic relationships among all individuals. Prior to perform PCA, we imputed missing data based on linkimpute algorithm (Money et al., 2015) with the default settings implemented in Tassel 5 (Bradbury et al., 2007). The imputed dataset was used for PCA only.
Genetic diversity metrics including the average number of alleles, observed and expected heterozygosity, and GIS inbreeding coefficient (an analogue of FIS) were estimated by using GenoDive v3.0 (Meirmans, 2020). We calculated pairwise FST between populations as described by Weir and Cockerham (1984), and calculated 95% confidence intervals using 10,000 bootstrap replications with the “stamppFst” function in the R package StAMPP (Pembleton et al., 2013), with the adjustment of the p-value by the Benjamini-Hochberg FDR procedure. Isolation by distance was tested to identify any correlations between genetic distance (linearized FST: FST/[1 – FST]) and geographic distance by applying Mantel tests. Geographic distance that was defined as the shortest distance between populations was calculated by using the “distGeo” function in the R package Geosphere (Hijmans, 2015). Mantel tests were performed using the mantel.randtest function in the R package ade4 (Dray and Dufour, 2007). We also performed analysis of molecular variance (AMOVA) based on allele frequency data among sites (999 permutations) by using the “poppr.amova” function in the R package poppr (Kamvar et al., 2014).
Contemporary migration among populations was estimated by using BayesAss v.3.04 (Mussmann et al., 2019). Delta values (-m, -a, and -f) were each set to 1.0. We ran the analysis for 8,000,000 iterations with a burn in of 2,000,000 steps, and 8000 sampling intervals. Convergence was inspected by performing two independent analyses using different seed numbers.
Larval dispersal simulation
To investigate larval dispersal patterns and understand the effect of ocean currents on regional connectivity, we performed a larval dispersal simulation using Lagrangian particle tracking analysis. The analytical framework used was Parcels (Delandmeter and van Sebille, 2019), with input values of horizontal flow velocities from the data-assimilative ocean general circulation model JCOPE2M (Miyazawa et al., 2017; Miyazawa et al., 2019). JCOPE2M has a horizontal resolution of 1/12°, a vertical resolution of 46 layers in sigma coordinates, and a data interval of 1 day. Because JCOPE2M can accurately reproduce the Kuroshio path (Miyazawa et al., 2017), it is a suitable model for simulating the influence of the Kuroshio current on our study area. All sampling sites released one particle per day between 1 January 2019 and 31 December 2021, and a total of over 80,000 particle dispersion pathways were calculated. The release depth was 200 m, which is the depth at which the majority of samples were collected. Particles were assumed to be neutrally buoyant and vertical migration was not considered. The period of larval suspension was assumed to be 60 days, since the maximum life span of most coral larvae is between 50 and 70 days (Markey et al., 2016). The simulation domain extended from 121°E to 147°E and from 22°N to 36°N, and particle tracking was terminated when particles left the domain.
Results
The ddRAD-seq libraries yielded on average 1.05 million 51-bp reads per individual. After filtration by ipyrad, we obtained a total of 2602 loci including 8950 SNPs (with 26.36% missing sites) that were present in at least 70% of 76 individuals (Table 1). Three samples from Kochi (KOC), four samples from Kagoshima (KAG), and one sample from Ogasawara Island (OGS) were removed from analyses due to insufficient data (fewer than 1000 loci per individual). The filtered SNP data used for downstream analyses included 961 unlinked SNPs including 7.44% of missing data (one SNP per RAD locus). The proportion of missing data for each population was listed in Table S3.
The first principal component obtained from our PCA analysis explained 5.315% of the overall variance, and the second explained 2.895% (Figure 2B). There was no clear geographic grouping of the sampling sites along the two PCA axes (Figure 2); the KAG specimens formed two clusters and the OGS samples were somewhat separate from the others.
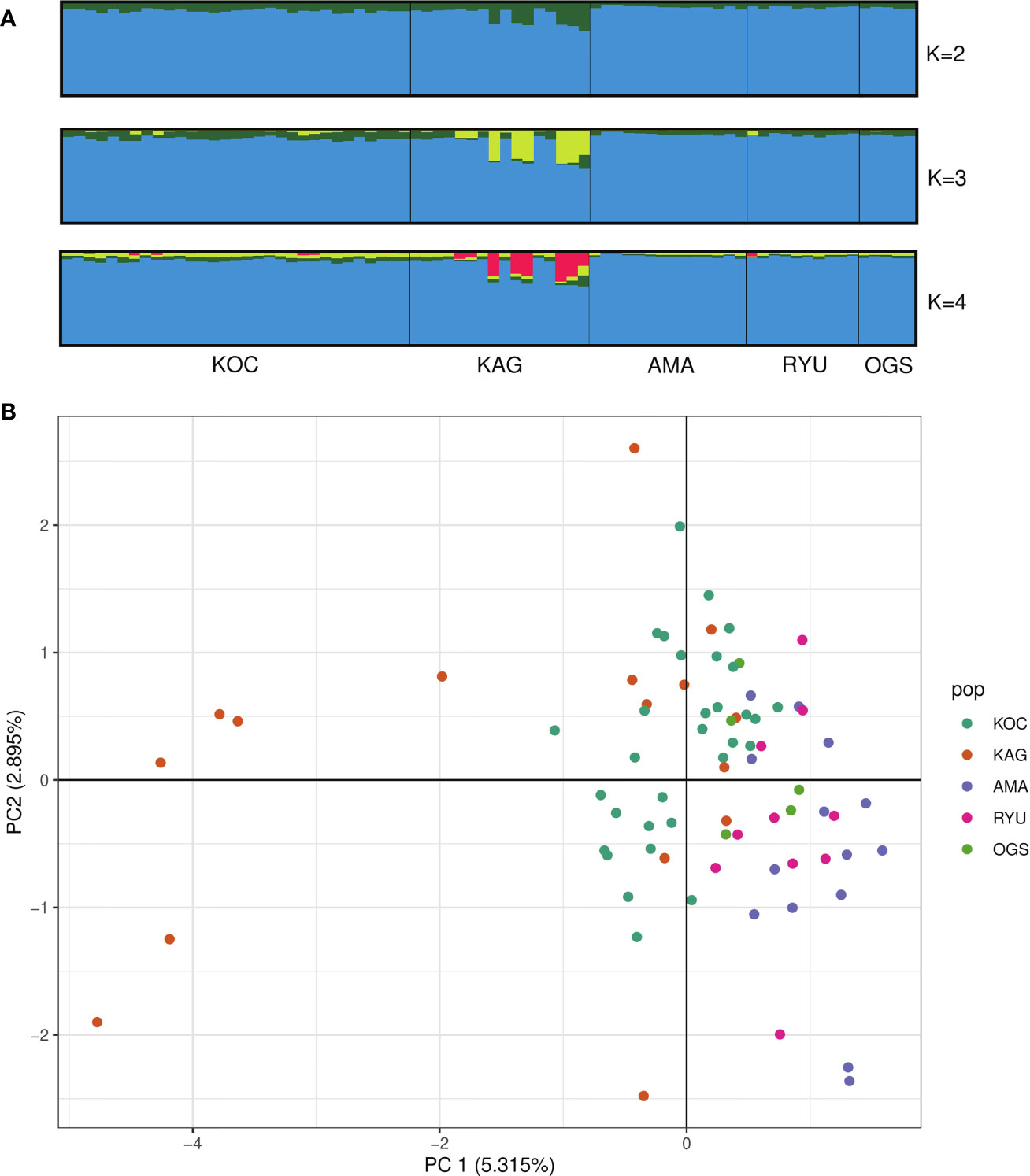
Figure 2 (A) STRUCTURE results at K = 2–4 with individuals on the x axis (sorted by site) and assignment probability on the y axis. (B) Principal component analysis of 1st and 2nd axes for a matrix of individual genotype frequencies. KOC, Kochi; KAG, Kagoshima; AMA, Amami; RYU, Okinawa; OGS, Ogasawara.
We detected significant variations among sites by AMOVA analysis (Table S1; Phi-sites-total = 0.045, p = 0.001). STRUCTURE analysis based on unlinked SNPs identified the most likely number of genetic populations of Corallium japonicum as K = 2 based on mean log likelihood and ΔK (Figure S1). With K = 2, all individuals from each population tend to assign to the same cluster (Figure 2; blue cluster). The green cluster found at K = 2 was further divided into two and three clusters at K = 3 and K = 4 respectively (Figure 2).
The greatest genetic distance between populations in this study was that between KAG and AMA (pairwise FST = 0.0446, adjusted p-value < 0.001), and the most genetically similar populations were AMA and RYU (FST = 0.0048, adjusted p-value = 0.1723) (Figure 3 and Table S2). Observed and expected heterozygosities were similar for all populations except OGS (Table S3), which had low observed heterozygosity.
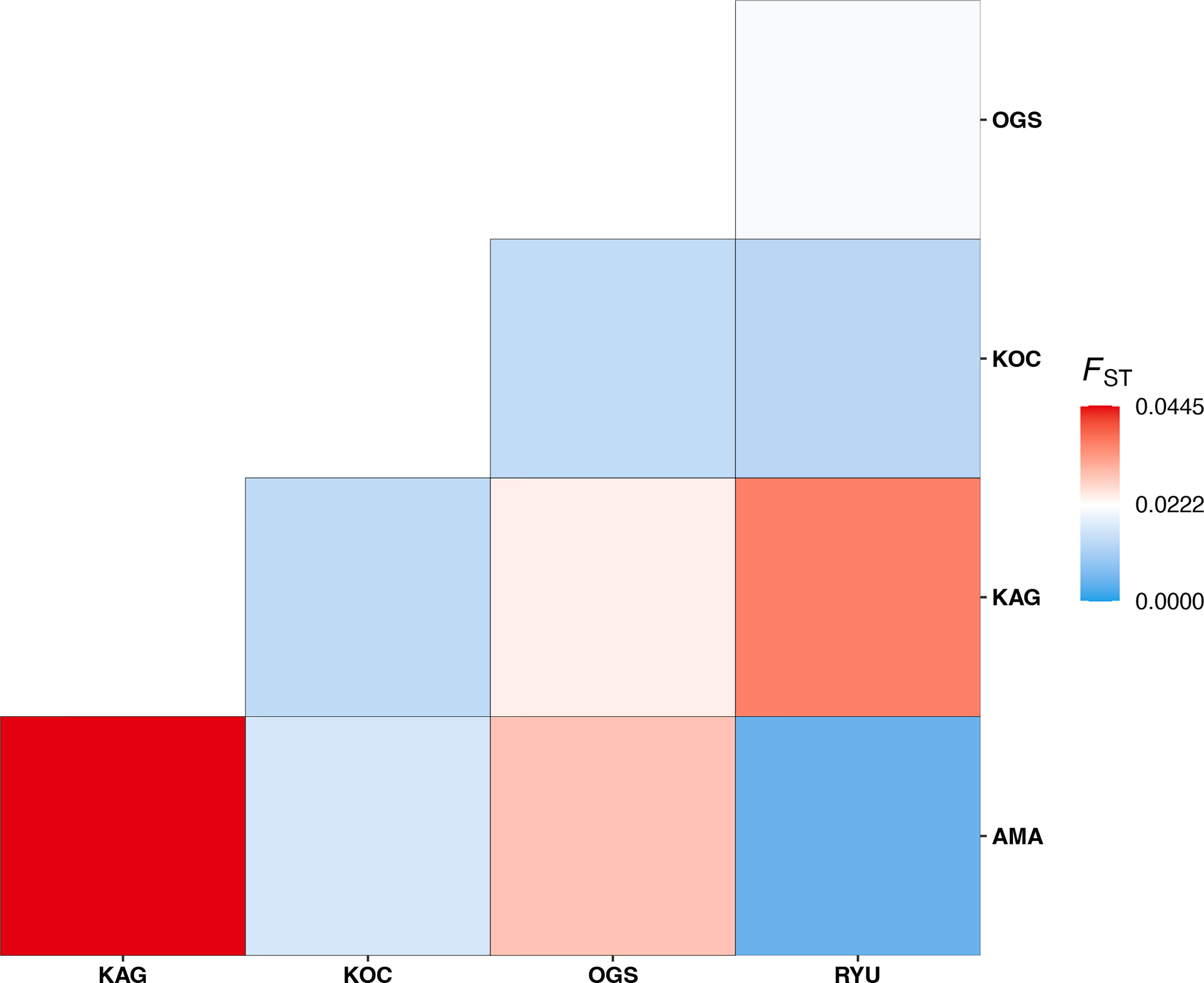
Figure 3 Heatmap showing FST values between each population.
BayesAss analysis indicated high migration rates from AMA to KOC, KAG, RYU, and OGS (>17%; Table S4), and a higher self-recruitment rate at AMA (93%; Table S4) than at the other sites (68–70%). Isolation-by-distance testing indicated that the observed correlation between populations was nearly zero (r = 0.0003) and statistically insignificant (p = 0.526) (Figure 4).
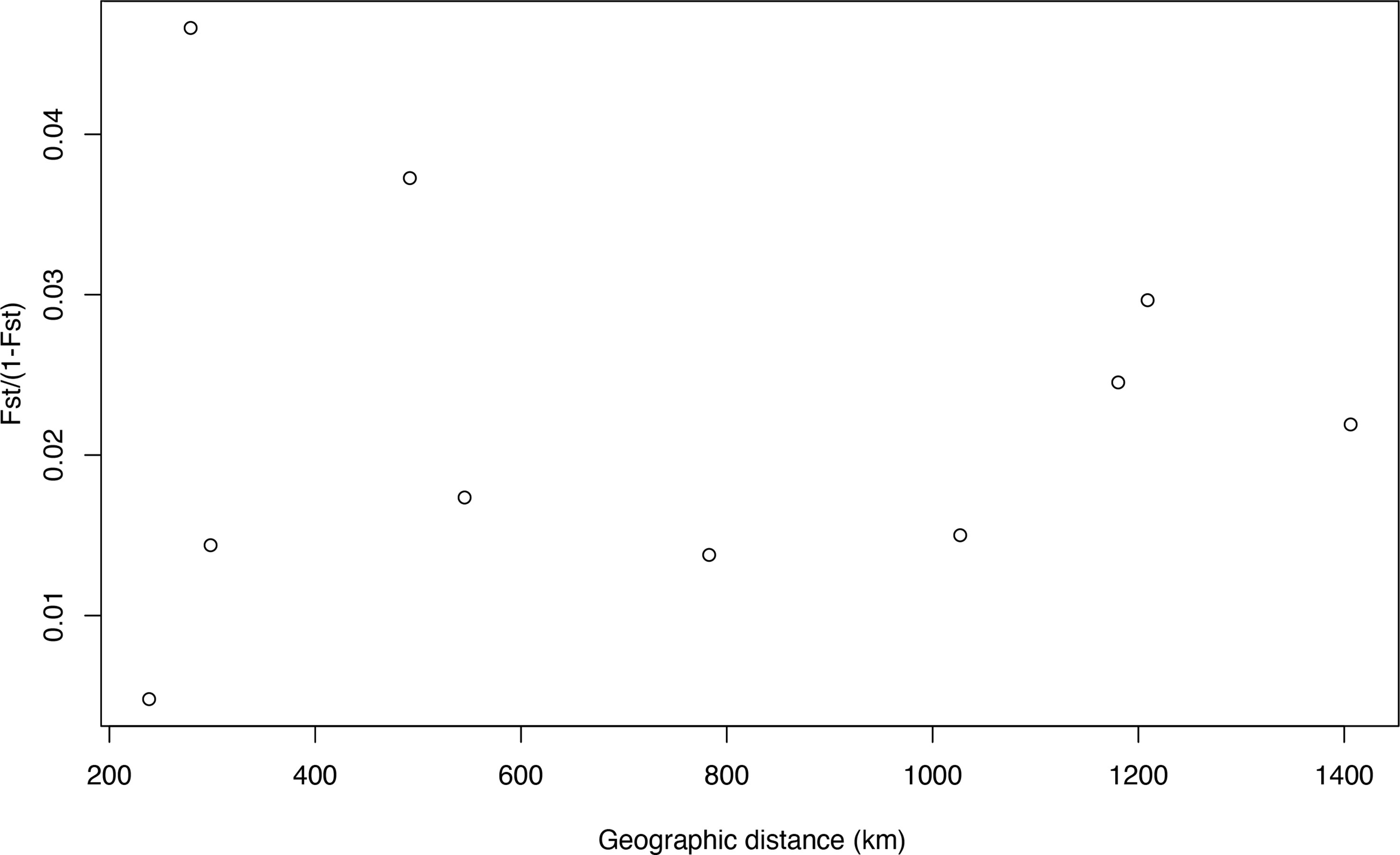
Figure 4 Relationship between genetic differentiation (FST/[1 – FST]) and geographic distance among each population for Corallium japonicum.
The results of our simulation of the dispersal of larvae over 60 days showed that the particles released by RYU and AMA, respectively tending to reach each other (Figures 5B, C), while particles released from KGS tending not to reach the other sites except for KOC (Figures 5D, E). In addition, particles released from OGS seldom reaching the other sites (Figure 5F).
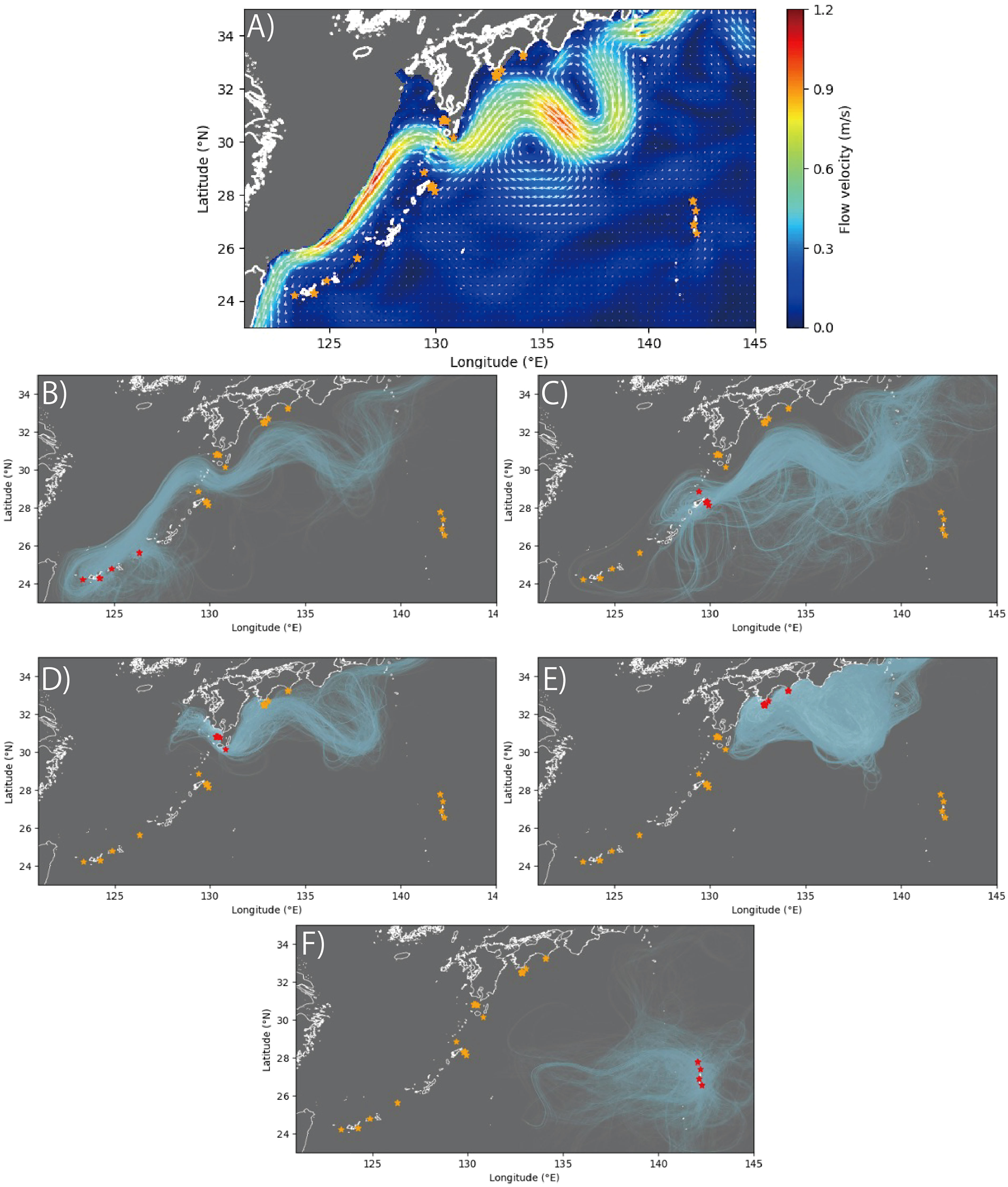
Figure 5 Mean flows from JCOPE2M at (A) 200-m depth and (B–F) results of larval dispersal simulations. (A) is the three-year average from January 1, 2019 to December 31, 2021. The results of particle release are shown for sampling sites: (B) Ryukyu (RYU), (C) Amami (AMA), (D) Kagoshima (KAG), (E) Kochi (KOC), and (F) Ogasawara (OGS). The light blue lines indicate the trajectories of the particles. Orange stars indicate sampling sites, and red stars indicate sampling sites where particle were released.
Discussion
In this study, we evaluated the genetic population structures of the precious red coral Corallium japonicum in the Northwest Pacific. Using variable SNP markers obtained by using the ddRAD-seq approach, we succeeded in evaluating the genetic diversity of and gene flow patterns among populations of C. japonicum. Among the C. japonicum populations examined in this study, none had especially low genetic diversity and inter-population gene flows tended to be maintained, although genetic diversity was somewhat lower at OGS than at the other sites. This reduced genetic diversity at OGS can be explained by its geographic isolation. Populations of other coral species in Ogasawara also tend to be isolated from those of the Ryukyu Archipelago (Nakajima et al., 2012b; Wepfer et al., 2022). Because of the difficulty of sample collection from the deep sea, sample sizes at AMA, KAG, RYU, and OGS were low (n < 20). Therefore, the lack of genetic differentiation among populations and sampling sites in our study could reflect the small number of samples and/or SNPs. Further sampling and a larger SNP dataset are needed for a comprehensive evaluation of C. japonicum population structure.
In contrast to Corallium rubrum, which can sustain genetic differentiation even at fine spatial scales in the Mediterranean (Ledoux et al., 2010; Costantini et al., 2018; Cannas et al., 2019), C. japonicum does not appear to have fine-scale changes in population structure. This is likely related to differences in the two species’ reproductive mode (C. rubrum, brooding; C. japonicum, broadcast spawning) (Takata et al., 2021).
The pelagic larval duration (PLD) of C. rubrum is relatively short, ranging from 16 to 42 days (Martínez-Quintana et al., 2015). Although there is no information available on the PLD of C. japonicum, broadcast spawners generally have greater dispersal potential than brooders (Nishikawa, 2008). Therefore, the lack of genetic differentiation between populations observed in our study may imply C. japonicum has a longer PLD than that of C. rubrum. Further investigations are needed to clarify the PLD of C. japonicum.
The three populations located along the flow path of the Kuroshio Current (KOC, AMA, and RYU) had no detectable genetic differentiation, suggesting that high gene flow was maintained among them and that the Kuroshio facilitated larval dispersal. Broadcast-spawning corals in shallow waters show slight genetic differentiation between these two regions, probably because there are no islands between them (Nakajima et al., 2012b). In the case of C. japonicum, however, it is possible that deep-water stepping-stone habitats help maintain gene flow between the regions. Around Japan, C. japonicum has been reported from depths of up to 300 m (Nonaka and Muzik, 2009), and several seamounts with relatively shallow summits are located between the Japanese archipelago and the Ogasawara Islands.
Our analysis also identified some genetic differentiation measured by pairwise FST among populations that are not separated by large geographic distances (i.e., KAG vs AMA). Dispersal limitation by ocean currents could explain the relative genetic isolation of the KAG population, as the KAG population was isolated from the other populations in our larval dispersal simulation (Figures 5A, D). Genetic differentiation across ocean current boundaries (i.e., the Kuroshio and Tsushima Currents) has been reported for some coastal organisms around Japan (Kojima et al., 1997; Kojima et al., 2004). The Kuroshio Current is especially likely to affect the dispersal of C. japonicum because of its deep current flow, with its flow axis reaching depths of several hundred meters (Oka and Kawabe, 2003; Kawabe, 2005). Data from population-level genetic analyses are likely to be useful for the establishment of marine protected areas for precious corals in the future.
Our study is the first to investigate population genetics and patterns of connectivity of C. japonicum in the Northwest Pacific and its relationship to the flow path of the Kuroshio Current through both RAD-seq analysis and larval dispersal simulations. Our results provide fundamental information needed for the monitoring and conservation of C. japonicum in this region.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ddbj.nig.ac.jp/, DRA014858.
Author contributions
HK, AI, and NI designed the study. YY, KU, and TS performed the molecular experiments. HK, AI, NS, and AN performed the analyses. AS and NI contributed the materials and reagents. HK, AI, and NS wrote the main manuscript text. All authors contributed to the writing and editing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by a KAKENHI Grant-in-Aid (grant no. 17K07274) from the Japan Society for the Promotion of Science as well as by the Research Laboratory on Environmentally Conscious Developments and Technologies at the National Institute of Advanced Industrial Science and Technology and the Research and Development Projects for Application in Promoting New Policy of Agriculture, Forestry and Fisheries (Ministry of Agriculture and Fisheries, Japan), 22032. This study was also supported by the Research and Assessment Program for Internationally Managed Fisheries Resources, Fisheries Agency of Japan.
Acknowledgments
We thank Fukada Salvage & Marine Works Co., Ltd.; the Kochi Prefectural Fisheries Experimental Station, Department of Fisheries, Kochi Prefecture; the Sukumo-wan Japan Fisheries Cooperative; and the Fisheries Agency of Japan for their help in sampling.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1052033/full#supplementary-material
Supplementary Figure 1 | STRUCTURE HARVESTER plots for Corallium japonicum (K=1–7 with 10 replicates per K).
References
Andrews K. R., Good J. M., Miller M. R., Luikart G., Hohenlohe P. A. (2016). Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 17, 81–92. doi: 10.1038/nrg.2015.28
Aurelle D., Ledoux J. B., Rocher C., Borsa P., Chenuil A., Féral J. P. (2011). Phylogeography of the red coral (Corallium rubrum): inferences on the evolutionary history of a temperate gorgonian. Genetica 139, 855–869. doi: 10.1007/s10709-011-9589-6
Avise J. C. (2000). Phylogeography: the history and formation of species (Cambridge, Massachusetts: Harvard University Press).
Baco A. R., Etter R. J., Ribeiro P. A., Von der Heyden S., Beerli P., Kinlan B. P. (2016). A synthesis of genetic connectivity in deep-sea fauna and implications for marine reserve design. Mol. Ecol. 25, 3276–3298. doi: 10.1111/mec.13689
Baird N. A., Etter P. D., Atwood T. S., Currey M. C., Shiver A. L., Lewis Z. A., et al. (2008). Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3, e3376. doi: 10.1371/journal.pone.0003376
Bradbury P. J., Zhang Z., Kroon D. E., Casstevens T. M., Ramdoss Y., Buckler E. S. (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1093/bioinformatics/btm308
Cannas R., Follesa M. C., Cau A., Cau A., Friedman K. (2019). Global report on the biology, fishery and trade of precious corals Vol. 118 (Rome: FAO).
Cannas R., Sacco F., Cau A., Coluccia E., Follesa M. C., Cau A. (2015). New insights into connectivity patterns of mesophotic red coral (Corallium rubrum) populations. Hydrobiologia 759, 63–73. doi: 10.1007/s10750-015-2198-0
Cannas R., Sacco F., Cau A., Cuccu D., Follesa M. C., Cau A. (2016). Genetic monitoring of deep-water exploited banks of the precious Sardinia coral Corallium rubrum (L1758): useful data for a sustainable management. Aquat. Cons. 26, 236–250. doi: 10.1002/aqc.2522
Catchen J., Hohenlohe P. A., Bassham S., Amores A., Cresko W. A. (2013). Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140. doi: 10.1111/mec.12354
Costantini F., Carlesi L., Abbiati M. (2013). Quantifying spatial genetic structuring in mesophotic populations of the precious coral Corallium rubrum. PLoS 8, e61546. doi: 10.1371/journal.pone.0061546
Costantini F., Fauvelot C., Abbiati M. (2007). Fine-scale genetic structuring in Corallium rubrum: evidence of inbreeding and limited effective larval dispersal. Mar. Ecol. Prog. Ser. 340, 109–119. doi: 10.3354/meps340109
Costantini F., Rugiu L., Cerrano C., Abbiati M. (2018). Living upside down: patterns of red coral settlement in a cave. PeerJ 6, e4649. doi: 10.7717/peerj.4649
Costantini F., Taviani M., Remia A., Pintus E., Schembri P. J., Abbiati M. (2010). Deep-water Corallium rubrum (L., 1758) from the Mediterranean Sea: preliminary genetic characterization. Mar. Ecol. 31, 261–269. doi: 10.1111/j.1439-0485.2009.00333.x
Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27 (15), 2156–2158.
Delandmeter P., van Sebille E. (2019). The parcels v2.0 Lagrangian framework: new field interpolation schemes. Geosci. Model Dev. 12, 3571–3584. doi: 10.5194/gmd-12-3571-2019
Dray S., Dufour A. (2007). The ade4 package: Implementing the duality diagram for ecologists. J. Stat. Software 22, 1–20. doi: 10.18637/jss.v022.i04
Earl D. A., VonHoldt B. M. (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the evanno method. Conserv. Genet. Resour. 4, 359–361. doi: 10.1007/s12686-011-9548-7
Eaton D. A., Overcast I. (2020). Ipyrad: Interactive assembly and analysis of RADseq datasets. Bioinformatics 36, 2592–2594. doi: 10.1093/bioinformatics/btz966
Evanno G., Regnaut S., Goudet J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Foll M., Gaggiotti O. (2008). A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180, 977–993. doi: 10.1534/genetics.108.092221
Fujita J., Zenimoto K., Iguchi A., Kai Y., Ueno M., Yamashita Y. (2016). Comparative phylogeography to test for predictions of marine larval dispersal in three amphidromous shrimps. Mar. Ecol. Prog. Ser. 560, 105–120. doi: 10.3354/meps11911
Fukami H., Budd A. F., Levitan D. R., Jara J., Kersanach R., Knowlton N. (2004). Geographic differences in species boundaries among members of the Montastraea annularis complex based on molecular and morphological markers. Evolution 58, 324–337. doi: 10.1111/j.0014-3820.2004.tb01648.x
Hellberg M. E. (2006). No variation and low synonymous substitution rates in coral mtDNA despite high nuclear variation. BMC Evol. Biol. 6, 24. doi: 10.1186/1471-2148-6-24
Hijmans R. J. (2015) Geosphere: spherical trigonometry. r package version. 1.5-14. Available at: https://CRAN.Rproject.org/package=geosphere.
Iguchi A., Yoshioka Y., Forsman Z. H., Knapp I. S., Toonen R. J., Hongo Y., et al. (2019). RADseq population genomics confirms divergence across closely related species in blue coral (Heliopora coerulea). BMC Evol. Biol. 19, 187. doi: 10.1186/s12862-019-1522-0
Iwasaki N. (2019). “Asian Regional report on the biology, fishery and trade of precious and semi-precious corals,” in Global report on the biology, fishery and trade of precious corals. Eds. Cannas R., Follesa M. C., Cau A., Cau A., Friedman K. (Rome: FAO Fisheries and Aquaculture Circulation), 191–254.
Iwasaki N., Hasegawa H., Tamenori Y., Kikunaga M., Yoshimura T., Sawai H. (2022). Synchrotron µ-XRF mapping analysis of trace elements in in-situ cultured Japanese red coral, Corallium japonicum. PeerJ 10, e13931. doi: 10.7717/peerj.13931
Jakobsson M., Rosenberg N. A. (2007). CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806. doi: 10.1093/bioinformatics/btm233
Janes J. K., Miller J. M., Dupuis J. R., Malenfant R. M., Gorrell J. C., Cullingham C. I., et al. (2017). The k= 2 conundrum. Mol. Ecol. 26 (14), 3594–3602. doi: 10.1111/mec.14187
Jombart T. (2008). Adegenet: a r package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405. doi: 10.1093/bioinformatics/btn129
Kamvar Z. N., Tabima J. F., Grünwald N. J. (2014). Poppr: an r package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2, e281. doi: 10.7717/peerj.281
Kawabe M. (2005). Variations of the kuroshio in the southern region of Japan: Conditions for large meander of the kuroshio. J. Oceanogr. 61, 529–537. doi: 10.1007/s10872-005-0060-0
Kishinouye K. (1903). Preliminary note on the coralliidae of Japan. Zoologischer Anzeiger 26, 623–626. Available at: https://www.biodiversitylibrary.org/page/30125595.
Kojima S., Hayashi I., Kim D., Iijima A., Furota T. (2004). Phylogeography of an intertidal direct-developing gastropod Batillaria cumingi around the Japanese islands. Mar. Ecol. Prog. Ser. 276, 161–172. doi: 10.3354/meps276161
Kojima S., Segawa R., Hayashi I. (1997). Genetic differentiation among populations of the Japanese turban shell Turbo (Batillus) cornutus corresponding to warm currents. Mar. Ecol. Prog. Ser. 150, 149–155. doi: 10.3354/meps150149
Kopelman N. M., Mayzel J., Jakobsson M., Rosenberg N. A., Mayrose I. (2015). Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 15, 1179–1191. doi: 10.1111/1755-0998.12387
Ledoux J. B., Garrabou J., Bianchimani O., Drap P., Feral J. P., Aurelle D. (2010). Fine-scale genetic structure and inferences on population biology in the threatened Mediterranean red coral, Corallium rubrum. Mol. Ecol. 19, 4204–4216. doi: 10.1111/j.1365-294X.2010.04814.x
Linnaeus C. (1758). Systema naturae per regna tria naturae, secundum classes, ordines, genera, species, cum characteribus, differentiis, synonymis, locis. editio decima, reformata, 10th revised edition, Vol. 1. 824, Laurentius Salvius: Holmiae.
Markey K. L., Abdo D. A., Evans S. N., Bosserelle C. (2016). Keeping it local: dispersal limitations of coral larvae to the high latitude coral reefs of the houtman abrolhos islands. PLoS One 11 (1), e0147628. doi: 10.1371/journal.pone.0147628
Martínez-Quintana A., Bramanti L., Viladrich N., Rossi S., Guizien K. (2015). Quantification of larval traits driving connectivity: the case of corallium rubrum (L. 1758). Mar. Biol. 162, 309–318. doi: 10.1007/s00227-014-2599-z
Meirmans P. G. (2020). GENODIVE version 3.0: Easy-to-use software for the analysis of genetic data of diploids and polyploids. Mol. Ecol. Res. 20, 1126–1131. doi: 10.1111/1755-0998.13145
Miyazawa Y., Kuwano-Yoashida A., Doi T., Nishikawa H., Narazaki T., Fukuoka T., et al. (2019). Temperature profiling measurements by sea turtles improve ocean state estimation in the kuroshio-oyashio confluence region. Ocean Dynam. 69, 267–282. doi: 10.1007/s10236-018-1238-5
Miyazawa Y., Varlamov S. M., Miyama T., Guo X., Hihara T., Kiyomatsu K., et al. (2017). Assimilation of high-resolution sea surface temperature data into an operational nowcast/forecast system around Japan using a multi-scale three-dimensional variational scheme. Ocean Dynam. 67, 713–728. doi: 10.1007/s10236-017-1056-1
Money D., Gardner K., Migicovsky Z., Schwaninger H., Zhong G. Y., Myles S. (2015). LinkImpute: fast and accurate genotype imputation for nonmodel organisms. G3: Gen. Genom. Genet. 5, 2383–2390. doi: 10.1534/g3.115.021667
Mussmann S. M., Douglas M. R., Chafin T. K., Douglas M. E. (2019). BA3-SNPs: Contemporary migration reconfigured in BayesAss for next-generation sequence data. Methods Ecol. Evol. 10, 1808–1813. doi: 10.1111/2041-210X.13252
Nakabayashi A., Yamakita T., Nakamura T., Aizawa H., Kitano Y. F., Iguchi A., et al. (2019). The potential role of temperate Japanese regions as refugia for the coral Acropora hyacinthus in the face of climate change. Sci. Rep. 9, 1892. doi: 10.1038/s41598-018-38333-5
Nakajima Y., Nishikawa A., Iguchi A., Nagata T., Uyeno D., Sakai K., et al. (2017). Elucidating the multiple genetic lineages and population genetic structure of the brooding coral Seriatopora (Scleractinia: Pocilloporidae) in the Ryukyu archipelago. Coral Reefs 36, 415–426. doi: 10.1007/s00338-017-1557-x
Nakajima Y., Nishikawa A., Iguchi A., Sakai K. (2012a). The population genetic approach delineates the species boundary of reproductively isolated corymbose acroporid corals. Mol. Phylogenet. Evol. 63, 527–531. doi: 10.1016/j.ympev.2012.01.006
Nakajima Y., Nishikawa A., Iguchi A., Sakai K. (2012b). Regional genetic differentiation among northern high-latitude island populations of a broadcast-spawning coral. Coral Reefs 31, 1125–1133. doi: 10.1007/s00338-012-0932-x
Nishikawa A. (2008). Degree and pattern of gene flow in several scleractinian corals in the Ryukyu archipelago, southern Japan. Pac. Sci. 62, 413–421. doi: 10.2984/1534-6188(2008)62[413:DAPOGF]2.0.CO;2
Nonaka M., Muzik K. (2009). Recent harvest records of commercially valuable precious corals in the Ryukyu archipelago. Mar. Ecol. Prog. Ser. 397, 269–278. doi: 10.3354/meps08396
Oka E., Kawabe M. (2003). Dynamic structure of the kuroshio south of Kyushu in relation to the kuroshio path variations. J. Oceanogr. 59, 595–608. doi: 10.1023/B:JOCE.0000009589.28241.93
Palumbi S. R. (2004). Marine reserves and ocean neighborhoods: the spatial scale of marine populations and their management. Annu. Rev. Environ. Resour. 29, 31–68. doi: 10.1146/annurev.energy.29.062403.102254
Pembleton L. W., Cogan N. O., Forster J. W. (2013). StAMPP: An r package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol. Ecol. Res. 13, 946–952. doi: 10.1111/1755-0998.12129
Pritchard J. K., Stephens M., Donnelly P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. doi: 10.1093/genetics/155.2.945
Puritz J. B., Hollenbeck C. M., Gold J. R. (2014). dDocent: a RADseq, variant-calling pipeline designed for population genomics of non-model organisms. PeerJ 2, e431. doi: 10.7717/peerj.431
Ridley S. O. (1882). On the arrangement of the coralliidae, with descriptions of new or rare species. Proc. Zool. Soc. London 60, 221–234. Available at: https://www.biodiversitylibrary.org/page/30825505.
Rosenberg N. A. (2004). DISTRUCT: a program for the graphical display of population structure. Mol. Ecol. Notes. 4, 137–138. doi: 10.1046/j.1471-8286.2003.00566.x
Sakaguchi S., Sugino T., Tsumura Y., Ito M., Crisp M. D., Bowman D. M., et al. (2015). High-throughput linkage mapping of Australian white cypress pine (Callitris glaucophylla) and map transferability to related species. Tree Genet. Genom. 11, 121. doi: 10.1007/s11295-015-0944-0
Suyama Y., Matsuki Y. (2015). MIG-Seq: an effective PCR-based method for genome-wide single-nucleotide polymorphism genotyping using the next-generation sequencing platform. Sci. Rep. 5, 16963. doi: 10.1038/srep16963
Takata K., Iwase F., Iguchi A., Yuasa H., Taninaka H., Iwasaki N., et al. (2021). Genome-wide SNP data revealed notable spatial genetic structure in the deep-Sea precious coral Corallium japonicum. Front. Mar. Sci. 8. doi: 10.3389/fmars.2021.667481
Takata K., Taninaka H., Nonaka M., Iwase F., Kikuchi T., Suyama Y., et al. (2019). Multiplexed ISSR genotyping by sequencing distinguishes two precious coral species (Anthozoa: Octocorallia: Coralliidae) that share a mitochondrial haplotype. PeerJ 7, e7769. doi: 10.7717/peerj.7769
Taylor M. L., Roterman C. N. (2017). Invertebrate population genetics across earth's largest habitat: The deep-sea floor. Mol. Ecol. 26, 4872–4896. doi: 10.1111/mec.14237
Tsounis G., Rossi S., Grigg R., Santangelo G., Bramanti L., Gili J. M. (2010). “The exploitation and conservation of precious corals,” in Oceanography and marine biology: An annual review. Eds. Gibson R. N., Atkinson R. J. A., Gordon J. D. M. (Boca Raton, FL: CRC Press), 161–212.
Weir B. S., Cockerham C. C. (1984). Estimating f-statistics for the analysis of population structure. Evolution 38, 1358–1370. doi: 10.2307/2408641
Wepfer P. H., Nakajima Y., Fujimura A., Mikheyev A. S., Economo E. P., Mitarai S. (2022). The oceanographic isolation of the ogasawara islands and genetic divergence in a reef-building coral. J. Biogeo. 49, 1978–1990. doi: 10.1111/jbi.14475
Keywords: precious coral, population genetic structure, RAD-seq, snps, ocean currents
Citation: Kise H, Iguchi A, Saito N, Yoshioka Y, Uda K, Suzuki T, Nagano AJ, Suzuki A and Iwasaki N (2023) Genetic population structure of the precious coral Corallium japonicum in the Northwest Pacific. Front. Mar. Sci. 10:1052033. doi: 10.3389/fmars.2023.1052033
Received: 23 September 2022; Accepted: 15 March 2023;
Published: 27 April 2023.
Edited by:
Marco Casu, University of Sassari, ItalyCopyright © 2023 Kise, Iguchi, Saito, Yoshioka, Uda, Suzuki, Nagano, Suzuki and Iwasaki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Akira Iguchi, aWd1Y2hpLmFAYWlzdC5nby5qcA==
†Present address: Hiroki Kise, Environmental Management Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba, Ibaraki, Japan
‡These authors have contributed equally to this work