 Greta Reintjes
Greta Reintjes
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 07 March 2023
Sec. Ocean Observation
Volume 10 - 2023 | https://doi.org/10.3389/fmars.2023.1051510
This article is part of the Research Topic The Atlantic Meridional Transect Programme (1995-2023) View all 12 articles
Particulate organic matter plays a significant role in the marine carbon cycle. Its sinking exports organic carbon from the surface to deep oceans. Using fractionated filtration, we analysed particles of 3 – 10 µm and >10 µm and their microbiomes in thirty-five stations along a latitudinal transect of the Atlantic Ocean and provide new insights into the composition, community dynamics, and catabolic potential of particle-attached bacteria. Samples were taken during an Atlantic Meridional Transect (AMT22), which traversed six distinctive ocean provinces. Using 16S rRNA amplicon sequencing and fluorescence in situ hybridisation, we could show a strong variation between particle-attached and free-living bacterial communities at each station and across the biogeographical provinces – a dynamic likely driven by chlorophyll a concentrations, temperature, and the oxygen content of the respective biogeographical provinces. Whereas the <3 µm fraction was primarily composed of SAR11, SAR86, Prochlorococcus and Bacteroidetes of the NS9 and NS5 clades, particle-attached communities were dominated by other Bacteroidetes (Polaribacter spp.), diverse Gammaproteobacteria including members of the genera Alteromonas and Vibrio, Alphaproteobacteria, Planctomycetes, OM27 and Verrucomicrobia. In three provinces, we quantified particle abundance and analysed their glycan composition using four lectins targeting fucose, galactose, N-acetylgalactosamine and mannose. Particles were mainly composed of fucose glycans with only a minor abundance of the other glycans, and particle abundance was directly correlated with the chlorophyll a concentrations. Functional analysis of 54 metagenome-assembled genomes retrieved from bacterial communities attached to small particles showed that particle-attached Bacteroidetes, Planctomycetes and Verrucomicrobia displayed key roles in the degradation of sulfated fucose-containing polysaccharides. We also identified gene clusters potentially encoding the utilisation of mannan and laminarin, suggesting an adaptation to the glycan composition of the particles, potentially resulting in niche diversification. Together, our results provide insights into particle-attached bacteria and their ecological strategies in the Atlantic.
In the vast nutrient-limited expanses of the world’s oceans, there exist temporary nutrient-rich “hot spots”, namely marine particles. Marine particles are the primary vehicles of organic carbon flux from the surface to the deep sea (Azam and Malfatti, 2007). As a point source of organic matter in an otherwise largely oligotrophic environment, they are rapidly colonised by specialised bacteria from the surrounding water column (Datta et al., 2016). Particle-attached bacteria show high hydrolytic activity of extracellular enzymes - a prerequisite to the extracellular degradation of particles - and are therefore important for the reduction of particle half-life, significantly impacting global nutrient and carbon cycling (Huston and Deming, 2002; Simon et al., 2002; Grossart et al., 2007; Ziervogel and Arnosti, 2008; Ziervogel et al., 2010; Lyons and Dobbs, 2012).
However, whether and how fast a particle is broken down depends not only on the associated bacteria but also on the particle’s composition. Particles are composed predominantly of organic material produced by photosynthetic organisms, such as phytoplankton. Hence, they are more abundant during and after phytoplankton blooms in upwelling areas and close to the coast (Passow, 2002; Behrenfeld et al., 2005). Soluble glycans like laminarin and mannan are more readily degradable compared to particles rich in fucoidan. Whereas the former are degradable within days to weeks, the latter, the highly complex and sulfated polysaccharide fucoidan, can take months to be fully broken down (Sichert et al., 2020; Vidal-Melgosa et al., 2021).
Due to the scarcity of particles, attached bacteria are low in abundance and often make up only 1% of the total community (Alldredge et al., 1986; Heins et al., 2021). Nevertheless, attached bacteria show a high respiration rate (Grossart et al., 2007), have large cells and large genomes (Smith et al., 2013), and show extensive gene repertoires for polysaccharide degradation (Smith et al., 2013; Rieck et al., 2015; Kappelmann et al., 2019; Schultz et al., 2020). Particle-attached bacteria can function both as particle degraders and as their builders, thereby supporting both carbon sequestration and carbon remineralisation (Smith et al., 1992; Heissenberger and Herndl, 1994; Azam and Malfatti, 2007).
Alphaproteobacteria, especially of the family Rhodobacteraceae, Bacteroidetes, Gammproteobacteria, and Planctomycetes are typically the most dominant bacteria attached to particles (Salazar et al., 2015). In-depth analyses suggest that they fill different niches within the particle microenvironment provided by the substrate’s complexity. Alphaproteobacteria are more efficient in the incorporation of monomers and amino acids, whereas Bacteroidetes, especially Flavobacteriia, can utilise a selfish uptake mechanism and degrade high molecular weight compounds without losing energy to their surroundings and to scavenging bacteria (Cottrell and Kirchman, 2000; Reintjes et al., 2019). Genomes of particle-attached Bacteroidetes showed genes potentially involved in the degradation of complex organic matter (Kappelmann et al., 2019). In contrast, in metagenome-assembled genomes of free-living Bacteroidetes these genes were rare (Krüger et al., 2019), indicating different adaption between members of the particle-attached and free-living fraction.
Gammaproteobacteria possess homologs to the selfish uptake mechanism. Like Flavobacteriia they can upregulate TonB-dependent transporters, when the nutrient concentration rises, for example during phytoplankton blooms (Reintjes et al., 2020b; Francis et al., 2021).
Planctomycetes are predominantly present in the larger particle fractions (DeLong et al., 1993; Fuchsman et al., 2012) and especially the classes Rhodopirellula, Blastopirellula, Pirellula, and Planctomyces were shown to be capable of breaking down complex organic matter (Wegner et al., 2013). They are part of the Planctomycetes-Verrucomicrobia-Chlamydia (PVC) superphylum, which contains a large number of bacteria, capable of degrading complex sugars like fucoidan (Glockner et al., 2003; Van Vliet et al., 2019; Orellana et al., 2022). Since it is hypothesised that these complex sulfated sugars are mostly remineralised through these bacteria, Planctomycetes and other members of the PVC serve an important ecological function (Glockner et al., 2003; Wegner et al., 2013; Spring et al., 2018; Orellana et al., 2022).
In this study, we pursued a genomic, glycobiological, and ecological investigation of marine particles and their attached microbial communities in a north-south transect of the Atlantic Ocean (AMT22). The study included samples from six Longhurst provinces (Longhurst, 2010), the North Atlantic Drift (NADR), North Atlantic Subtropical (NAST), North Atlantic Tropical Gyre (NATR), Western Tropical Atlantic (WTRA), South Atlantic Gyre (SATL), and South Subtropical Convergence (SSTC) (Figure 1). In these provinces, we investigated particle-attached microbial communities using a combination of 16S tag sequencing and fluorescence in situ hybridisation. Furthermore, in three provinces, we assessed the prevailing glycans in particles using fluorescent-lectin-binding-analysis, (Bennke et al., 2013), and investigated the potential of attached bacteria to degrade these glycans using metagenomic analysis. We hypothesised that glyco-conjugate distributions in marine aggregates will shift across the different provinces of the Atlantic Ocean and directly affect the composition of the particle-attached bacterial community. Our study aims to advance the knowledge of the ecological functioning of the ocean carbon cycle as mediated by particle-attached bacteria.
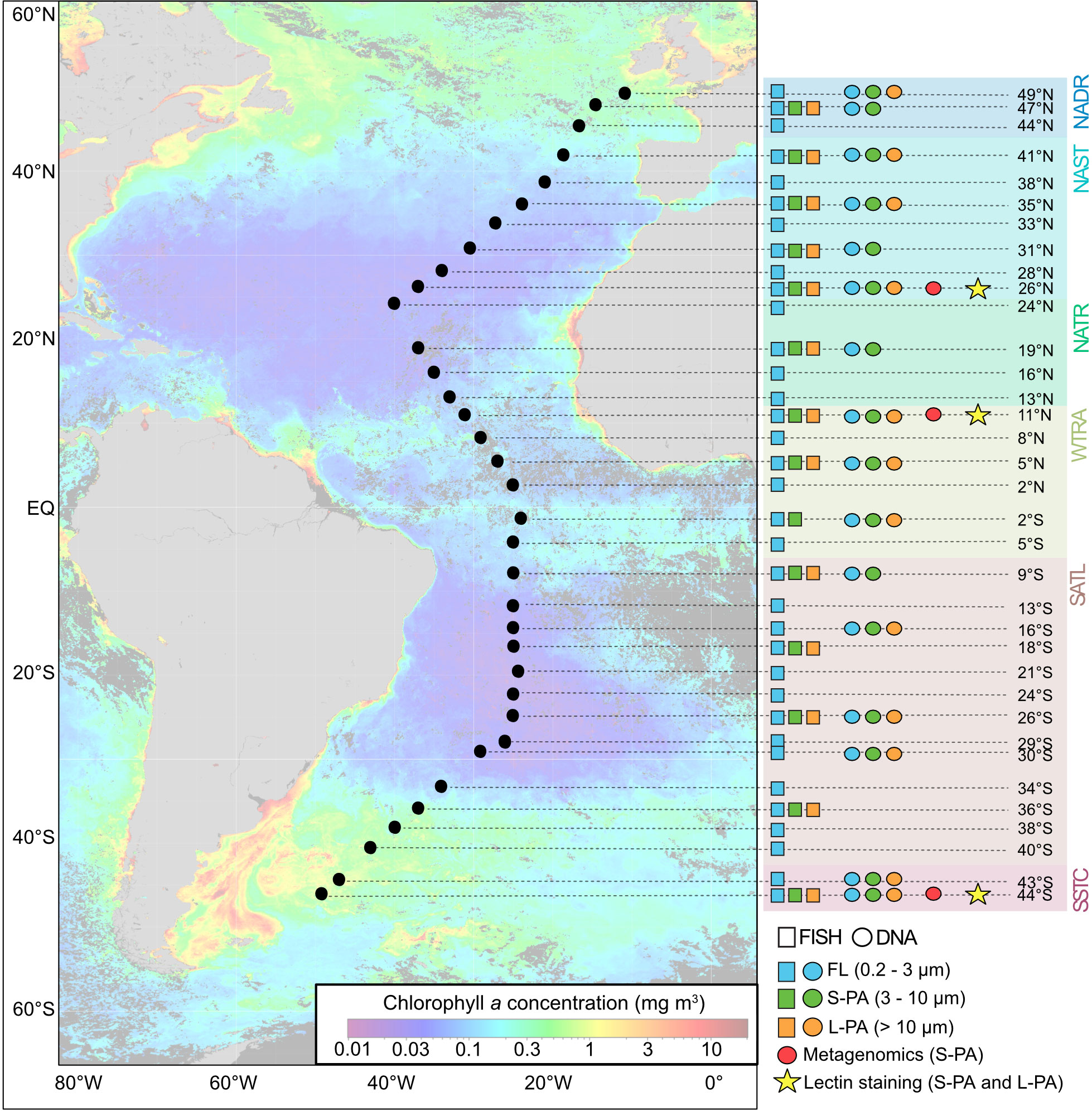
Figure 1 Sampling map and analysis list from AMT22. Map shows satellite-based chlorophyll a concentration during the time of sampling (Oct-Nov 2012) obtained from Aqua MODIS (Ocean Biology Processing Group, 2014). Stations are labeled according to latitude 49°N to 44°S and indicated as black dots across the map. Individual samples are shown in blue (FL), green (S-PA fraction), and orange (L-PA fraction) and indicate with FISH (boxes) and DNA (circles) analyses. Red dots indicate metagenomics samples from the S-PA fraction, stars indicate lectin staining from the S-PA and L-PA fraction. NADR North Atlantic Drift, NAST North Atlantic Subtropical, NATR North Atlantic Tropical Gyre, WTRA Western Tropical Atlantic, SATL South Atlantic Gyre, and SSTC South Subtropical Convergence.
Samples were taken along the 22nd Atlantic Meridional Transect (AMT22) cruise on the Royal Research Ship James Cook (October-November 2012) from Southampton, United Kingdom, to Punta Arenas, Chile. Seawater was collected from 35 stations at solar noon with 20 L Niskin bottles mounted on the sampling rosette of a conductivity-temperature-depth (CTD) profiler (Sea-Bird Electronics, Washington, USA) from a depth of 20 m (Figure 1).
For microbial cell counts and CARD-FISH, 1 L of surface seawater was sampled from 35 stations for the free-living fraction (FL, 0.2 – 3 µm), 14 stations for the small particle fraction (S-PA, 3 – 10 µm) and 13 stations for the large particle fraction (L-PA, > 10 µm). All samples were fixated using formaldehyde to a final concentration of 1% for 1 h at room temperature and subsequently filtered in triplicate through a 47 mm diameter polycarbonate filter with a pore size of 10 µm, 3 µm, and 0.2 µm, respectively, applying a gentle vacuum of < 200 mbar. These filters were left to air dry and stored at -20°C until further analysis.
For microbial diversity analysis, between 15 L to 45 L of seawater were collected from 16 stations and sequentially filtered onto 142 mm diameter polycarbonate filters with pore sizes of 10 µm, 3 µm and 0.2 µm (Supplementary Table 1D). Different volumes of seawater were sampled to prevent filter clogging, the volume was determined from previous cell counts (Zubkov et al., 2000; Schattenhofer et al., 2009). All filters were stored at -80°C until further analysis.
The AMT22 passed through several oceanic provinces (Longhurst, 2010). The biogeographical provinces were identified using their physical, chemical and biological characteristics (Supplementary Table 1). Chlorophyll-a (Chl a) fluorescence was measured on board by a CTG FAST track Fast Repetition Rate fluorometer (Chelsea Technologies Group, UK) and calibrated against extracted Chl a measurements of seawater samples collected from 9 depths at each station. The main nutrient analyser was a 5-channel Bran and Luebbe AAIII segmented flow autoanalyser. The analytical chemical methodologies used were according to Brewer and Riley (1965) for nitrate, Grasshoff (1976) for nitrite, Kirkwood (1996) for phosphate and silicate. Salinity (PSU) was measured using a Guideline Autosal 8400B salinometer (OSIL, UK) and calibrated against bench salinometer measurements from 4 samples collected from each cast. Dissolved oxygen (ml L-1) was measured using the Sea-Bird 43 dissolved oxygen sensor (Sea Bird Scientific) and calibrated against Winkler titration measurements from 9 samples collected at the pre-dawn CTD. Temperature (°C) was measured using a Sea-Bird 3 premium temperature sensor (Sea Bird Scientific) (all metadata is available via the BODC website (https://www.bodc.ac.uk/data/documents/cruise/11427/). The physico-chemical data were analysed using the ODV4 software (www.odv.awi.de).
The total cellular abundance and abundance of specific bacterial phylogenetic groups (Supplementary Table 2) was determined using the CARD-FISH procedure according to (Pernthaler et al., 2004). Hybridisations were done with horseradish peroxidase-labelled oligonucleotides probes (Biomers, Ulm, Germany) at varying formamide concentrations depending on the probe used (Supplementary Table 2). Working solutions of probes and competitors (both at 50 ng μl−1) were mixed with hybridisation buffer in a 1:1:300 proportion and hybridisation was carried out 2.5 h at 46°C. The probe-delivered horseradish peroxidase was detected with tyramides that were custom labelled with fluorescein (Molecular Probes, Eugene, OR, USA). After the procedure the samples were counterstained with 4’,6-diamidino-2-phenylindole (1 μg ml–1).
Cell quantification was done using an automated image acquisition and cell enumeration system (Bennke et al., 2016). For our evaluation FISH positive signals for each probe were determined by an overlapping (30% minimum overlap) signal of both DAPI (360 nm) and FISH (488 nm), with a minimum area of 17 (DAPI) or 30 (FISH) pixels (0.17 - 0.3 µm2) and minimal signal background ratio of 1 (DAPI) or 2.5 (FISH). Specific cellular abundance of aggregate associated samples were manually enumerated on a Zeiss Axioskop 2 motplus fluorescence microscope.
Within this study we designed new FISH probes targeting the Sphingopyxis, Erythrobacter, Opitutae and OM27 clade (Supplementary Table 2), using the probe design tool of the ARB software (Ludwig et al., 2004). Probe specificity was checked using the actual data set and SILVA release_119. For the newly designed subgroup-specific probes, optimal conditions in FISH were established by evaluating the fluorescence intensities of the target cells after hybridisation with Cy3-labeled probes at increasing concentrations of the formamide in the hybridisation buffer (Pernthaler et al., 2001).
To quantify particle abundance and identify the particles carbohydrate composition we performed lectin staining (Bennke et al., 2013). We applied the lectins Aleuria Aurantia Lectin (AAL), Concanavalin A (ConA), Wheat Germ Agglutinin (WGA) and Soybean Agglutinin (SBA). We tested varying lectin concentrations ranging from 1 to 100 µg µl-1. Optimised dilutions were determined microscopically and defined as strong fluorescent specific binding signals in aggregates without nonspecific background staining. The sugar specificity and working concentration of all lectins used in this study are given in Supplementary Table 3.
For glyco-conjugate staining of the aggregates, filters with formaldehyde-fixed cells were washed with filter-sterilised tap water and subsequently incubated with lectins for 20 min at room temperature. Afterwards, stained samples were carefully washed three times with filter-sterilised tap water to remove unbound lectins. For combined visualisations with particle-attached bacterial cells, CARD-FISH was performed prior to lectin staining (see protocol above).
Particles were visualised using a Zeiss ELYRA PS.1 (Carl Zeiss) with 561, 488 and 405 nm lasers and BP 573-613, BP 502-538 and BP 420-480+LP 750 optical filters (Carl Zeiss, Jena, Germany). Z-stack images were taken with a Plan-Apochromat 63 ×/1.4 Oil objective using an iXON897 EM-CCD camera. The images were reconstructed using ZEN software (Black edition, 2011, Carl Zeiss, Germany). Further analysis was performed on reconstructed super-resolution images in ZEN software blue edition (2012, Carl Zeiss, Germany).
Microbial DNA was extracted using the MoBio Ultra Clean Soil DNA Extraction Kit (MoBio Laboratories) as recommended by the manufacturer with the following alterations. A 150 mm x 250 mm piece of polycarbonate filter was directly added to the Bead Solution Tubes. Sequencing was carried out on a 454 Titanium FLX (ROCHE, CT, USA) and Ion Torrent PGM (Thermo Fisher). Two sequencing platforms were used to reduce possible biases between the two systems. The 454 Titanium FLX is a pyrosequencing method. In contrast, the Ion Torrent PGM measures pH changes from the release of a proton during the incorporation of a dNTP into a DNA polymer. Where possible, samples were sequenced on both platforms to increase the accuracy (reduce sequencing bias) and yield per sample.
PCR was carried out for both platforms, using the primers S-D-Bact-0341-b-S-17 (5′-CCTACGGGNGGCWGCAG-3′) and S-D-Bact-0785-a-A-21 (5′-GACTACHVGGGTATCTAATCC -3’) targeting the V3 - V4 variable region of the 16S rRNA, evaluated by (Klindworth et al., 2013). For 454 Titanium FLX sequencing, PCR was carried out in a total volume of 50 µl. The PCR products were visualised by gel electrophoresis (1% LE agarose, Biozyme), the amplicon bands were cut out with a sterile scalpel and purified using the QiagenMinElute kit (Qiagen). If bimodal amplicon bands were detected, both bands were cut out of the gel and combined (range 430 – 490). The purified PCR products were pooled into libraries with a minimum DNA concentration of 1 µg DNA as measured using a Qubit assay (Invitrogen, Darmstadt, Germany), and sequenced on a ROCHE 454 titanium FLX (ROCHE) at the Max Planck Institute for Plant Breeding Research in Cologne.
PCR for Ion Torrent PGM was carried out using the Platinum PCR SuperMix High Fidelity polymerase kit (Thermo Fisher). PCR amplicons were size selected on 2% E-Gel size select gels using the E-Gel iBase Power System and E-Gel Safe Imager Real Time Transilluminator (Thermo Fisher), and cleaned up and concentrated over silica column using the Qiagen QIAquick PCR purification kit (Qiagen). Amplicon concentrations and quality were quantified using a Fragment Analyser (AATI) and the DNF - 472 standard sensitivity NGS fragment analysis kit (1 bp – 6,000 bp). Subsequently, the amplicons were pooled as described in the Ion Amplicon Library Preparation (Fusion Method) Manual (Thermo Fisher).
Ion Torrent sequencing was carried out as recommended by the manufacturer using an ION 314 v2 chips (Thermo Fisher). Briefly, emulsion PCR and enrichment of template-positive ion sphere particles (ISP) was done using the Ion PGM Hi-Q OT2 Kit (Thermo Fisher) on the Ion OneTouch 2 Instrument (Thermo Fisher) and Ion OneTouch ES instrument (Thermo Fisher) following the Ion Torrent user manual. Subsequently, the ISP were sequenced using the Ion PGM Hi-Q Sequencing Kit (Thermo Fisher) following the user manual on an Ion PGM system (Thermo Fisher) with a total of 1200 flows. The Torrent Suite software, which converts the raw signals (raw pH values) into incorporation measurements and ultimately into basecalls for each read, was used for initial quality trimming. We applied the following settings for base calling: Basecaller –barcode-mode 1 –barcode-cutoff 0 –trim-qual-cutoff 15 –trim-qual-window-size 10 –trim-min-read-len 250.
The sequence reads for each sample from the Ion Torrent PGM (Thermo Fisher) and 454 Titanium FLX (Roche) were further processed using the bioinformatics pipeline of the SilvaNGS project (Quast et al., 2013). This involved quality controls for sequence length (> 200 bp) and the presences of ambiguities (< 2%) and homopolymers (< 2%). The remaining reads were aligned against the SSU rRNA seed of the SILVA database release 125 (Quast et al., 2013). The classification was done by a local BLAST search against the SILVA SSURef 123 NR database using blast -2.2.22 + with standard settings.
Statistical analyses were carried out using normalised read abundances and classification to genus level. Normalised read abundances were calculated using within-sample relative abundances. These were calculated using the R (R Development Core Team) function decostand(method=total) from the Vegan package (Oksanen et al., 2013). Community alpha diversity (Simpson Index) and beta diversity (dissimilarity calculated using Bray-Curtis) was calculated using R and subsequently plotted using NMDS plots. Simpsons Index was chosen for alpha diversity calculation as it provides more weight to evenness and accounts for differences in units. The Bray-Curtis Index was used for beta diversity analysis as it gives weight to species’ presence and absence, and abundance. Significance tests, analyses of site-specific community composition differences and correlations to environmental factors, were done using ANOSIM and Mantel tests.
High-molecular-weight genomic DNA from three S-PA representative samples in the Northern Gyre (NAST, N26°), Equator (WTRA, N11°) and Southern Temperate (SSTC, S44°) region were shotgun-sequenced on an Illumina HiSeq2500 sequencer at the Max Planck Genome Center (MPGC, Cologne, Germany) after library construction using the Ovation Ultralow Library system kit (NuGen, San Carlos CA, USA). Approximately 54.7, 60.6 and 58.8 million reads were obtained for one Gyre, Equator and Temperate sample, respectively (Supplementary Table 4). Phylogenetic analysis of the reads by MetaPhlAn indicated that 56 - 68% were associated to Bacteria, 13-26% to Eukaryote, and only ~2% of the reads were Archaea related (Beghini et al., 2021).
Raw sequence reads were quality-trimmed and error-corrected using BBtools (BBmap package v. 33.57 http://sourceforge.net/projects/bbmap/) with default parameters. Bulk assembly of the metagenomes was separately performed with IDBA_UD v1.1.1 with k-mer sizes from 21 to 124 in steps of 10, and SPAdes v3.9 with k-mer sizes from 21 to 127 in steps of 10. This yielded a total of 561,913 scaffolds from all three aggregates, and the largest scaffold length was 545,657 bp (Supplementary Table 4). To obtain coverage profile of contigs from each aggregate metagenomic assembly, the trimmed reads were mapped back to contigs using BWA-MEM (v. 0.7.12) (Li, 2013). Full-length 16S rRNA genes were reconstructed from the raw reads using PhyloFlash 2.0 (http://github.com/HRGV/phyloFlash).
Genome binning was performed using CONCOCT (Alneberg et al., 2014) within the Anvi’o package (v. 2.0.2) (Eren et al., 2015). The metagenomic workflow employed here is described online (merenlab.org/2015/05/02/anvio-tutorial). CheckM was used to evaluate the accuracy of the binning approach by determining the percentage of completeness and contamination (Parks et al., 2015) using the lineage-specific workflow. The statistics of each MAG recovered from aggregates-associated microbial community is given in Supplementary Table 5. These metagenome-assembled genomes included 35 - 1,065 scaffolds with a scaffold largest length between 15,377 and 545,657 bp. Average nucleotide identities (ANIs) between the assemblies and to the next sequenced relative were calculated with JSpeciesWS web service (Richter et al., 2016). Genes were called using Prodigal (Hyatt et al., 2010). The generated assemblies were automatically annotated with the standard RAST annotation pipeline (Aziz et al., 2008) and the functions of predicted genes were curated and revised by a comparison of homology between databases including KEGG (release 94.2), Pfam-A (version 32.0), and NCBI-nr database (version of 25 August 2020). Specifically, the results of the KEGG annotations using DIAMOND (version 2.0.11) and BLASTP were compared to hidden Markov models-based HMMER3 searches against Pfam-A database and BLASTP searches against the NCBI-nr database. All predicted genes were used to query the TransportDB database (Elbourne et al., 2017), and matches were assigned to transporter families within the TransportDB database (www.membranetransport.org).
For phylogenetic analyses, the reconstructed genomes were placed within the reference genome tree of CheckM (v. 0.9.7) (Parks et al., 2015) and then visualised in ARB (Ludwig et al., 2004). In addition to analysing ribosomal proteins, partial 16S rRNA genes were retrieved from reconstructed genomes and then aligned by SINA (v. 1.3.0) (Pruesse et al., 2012) to a curated SILVA SSU123 NR99 database, where all sequences with a pintail value below 50 and alignment quality below 70 were excluded from further analyses. Phylogenetic trees were calculated with various algorithms: neighbour-joining (Ludwig et al., 2004) and PhyML (v. 3.1) (Guindon, 2010) to check the stability of the basic topology. The phylogeny of the assembled metagenomic bins were determined according to both the ribosomal protein and 16S rRNA genes alignments.
Annotation for CAZymes were performed as described in (Liu et al., 2013). Briefly, protein coding genes identified in each genomes were searched against the HMM profile-based database of carbohydrate-active enzymes obtained from dbCAN (Yin et al., 2012) in December 2012 using hmmsearch in the HMMER software package (v.3.0; http://hmmer.janelia.org/help) (Finn et al., 2011). Results were filtered using an e-value cut-off < 10−5. Additionally, all returned hits were manually evaluated based on their functional annotation in RAST and pfam. Sulfatase encoding genes were identified with HMMER scans versus the PFAM database 33.1 (Mistry et al., 2021) using an e-value cut-off < 10−5. Presence of extracellular peptidases was evaluated by MEROPS using an e-value cut-off < 10−10 (Rawlings et al., 2012).
The metagenomic data from this project can be found in ENA under the BioProject accession no. PRJNA421797 and drafts of genomes are available with accession no. PKCH00000000-PKEK00000000. The raw metagenomic reads were deposited to NCBI SRA under accession number SRP126598. The 16S rRNA sequencing and FISH data was deposited using the GFBio platform (Diepenbroek et al., 2014) and can be found under the INSDC accession number: PRJEB2516 and in Pangaea https://doi.pangaea.de/10.1594/PANGAEA.878060 and https://doi.pangaea.de/10.1594/PANGAEA.891265, respectively.
During the AMT22 we performed a comprehensive analysis of the free-living (FL 0.2 – 3 µm) and particle-attached (S-PA 3 – 10 µm and L-PA > 10 µm) bacterioplankton across a north-south Atlantic transect (Figure 1). Thirty-five stations were sampled, covering six Longhurst ocean provinces (Longhurst, 2010). The biogeographical provinces varied in their physical, chemical, and biological characteristics (Supplementary Table 1). The primary production was generally low, especially in the gyre regions and increased in the temperate provinces, especially the SSTC, where an active phytoplankton bloom was occurring. Across the transect, Chl a concentrations ranged from 0.03 mg m3 (gyres) to up to 1.51 mg m3 (SSTC) (Figure 1; Supplementary Table 1). Oxygen concentration remained with 231 ± 28 µmol L-1 more similar across the transect, but increased amidst the active bloom at S44°, alongside nitrate, nitrite and phosphate (Supplementary Table 1).
In three contrasting stations, N26°, N11°, and S44°, chosen based on differences in productivity and Chl a concentrations (N26°: 0.09 mg m3 NATR, N11°: 0.28 mg m3 WTRA, and S44°: 1.51 mg m3 SSTC), we performed particle quantification and biochemical identification by lectin staining. Many particles were strongly stained with the fucose-binding lectin AAL, and showed only minor staining with the lectins ConA, WGA, and SBA (Supplementary Table 3), indicating a high presence of fucose-containing glycans (Figure 2).
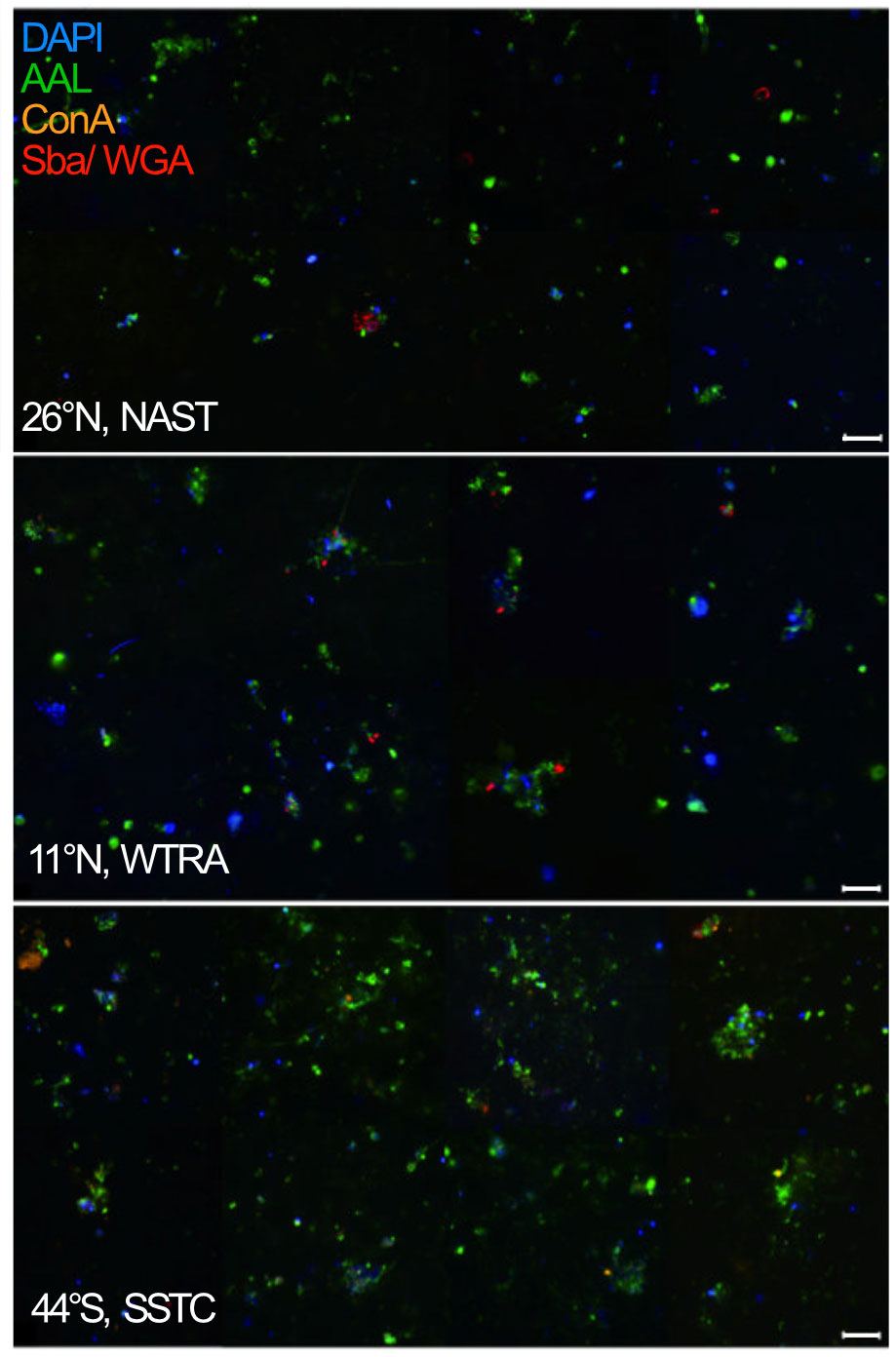
Figure 2 Epifluorescence microscope image of marine particles obtained from the S-PA fraction in the North Atlantic Subtropical (NAST), Western Tropical Atlantic (WTRA), and South Subtropical Convergence (SSTC) province of the Atlantic Ocean at 40 × magnification with the super-resolution microscope. Each sample was simultaneously stained by DAPI (blue), specific to DNA, and the lectins AAL (green), specific to fucose, ConA (orange), specific to a- mannopyranosyl and a-glucopyranosyl residues, as well as SBA and WGA (red), specific to Galactose/N-acetylgalactosamine. Scale bars: 20 μm.
Particle abundance correlated with the Chl a concentration and was 4-times as high in the SSTC (206 particles L-1), compared to 48 particles L-1 in the NATR and 58 particles L-1 in the WTRA. Additionally, the number of bacterial cells per particle was higher in the more productive region, with 1435 cells particle-1 in the SSTC and 47 cells particle-1 in the WTRA (Figure 2).
Absolute bacterial cellular abundances were quantified in all size fractions across the 35 stations (Figure 1). The counts in the FL fraction were three to four orders of magnitude higher than those in the S-PA and L-PA, with a mean of 7.6×105 ± 3.1x105 and 9.3x102 ± 1.4x103 cells ml-1, respectively (Figure 3). Concentrations of the FL and the S-PA fraction were lowest in the gyres (average 5.9x105 and 4.8x102 cells ml-1, respectively), increased in the temperate and equatorial regions (average 8.2x105 and 9.2x102 cells ml-1) and peaked in the phytoplankton bloom encountered in SSTC (average 1.5x106 and 4.1x103 cells ml-1). The L-PA fraction was less affected by the bloom condition and remained the lowest across all stations (3.2x102 ± 2.5x102 cells ml-1, Figure 3).
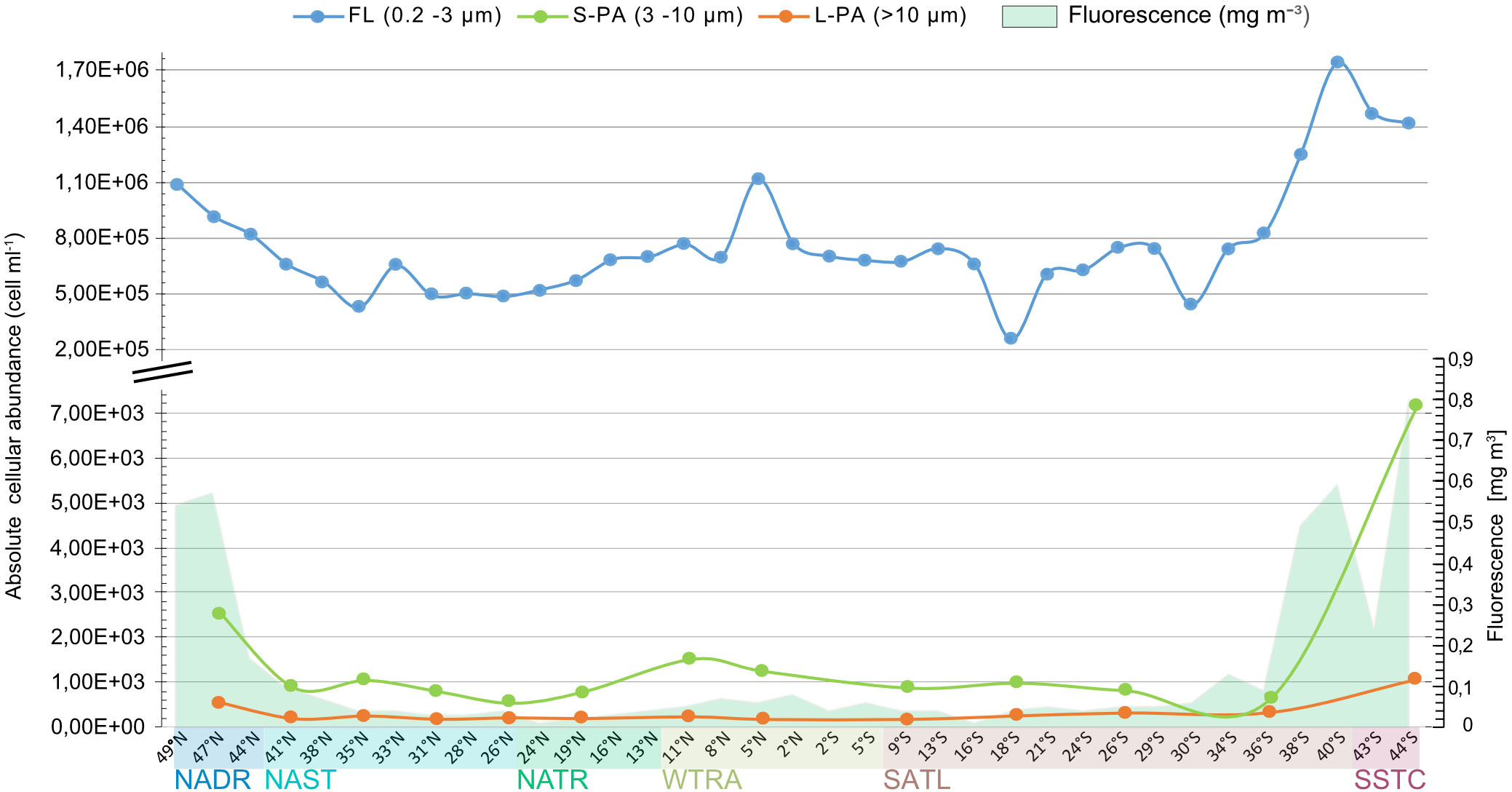
Figure 3 Total bacterial cellular abundance determined by CARD-FISH using the EUB I-III general bacteria probes of the free-living (FL, blue),small particle(S-PA, green),and large particle (L-PA, orange) fraction across the Atlantic Ocean sampled during the AMT22 cruise in 2012. Fluorescence is shown in green and is based on chlorophyll a calibration. NADR North Atlantic Drift, NAST North Atlantic Subtropical, NATR North Atlantic Tropical Gyre, WTRA Western Tropical Atlantic, SATL South Atlantic Gyre, and SSTC South Subtropical Convergence.
The community composition varied considerably between the FL and PA fractions. In the FL (0.2 - 3 µm) fraction, the most abundant clades were the Prochlorococcus, SAR11, SAR116, AEGEAN 169 marine group, and uncultured Rhodobacteraceae. The SAR86 clade was the most significant gammaproteobacterial group and Ca. Actinomarina the most prominent Actinobacteria (Figure 4).
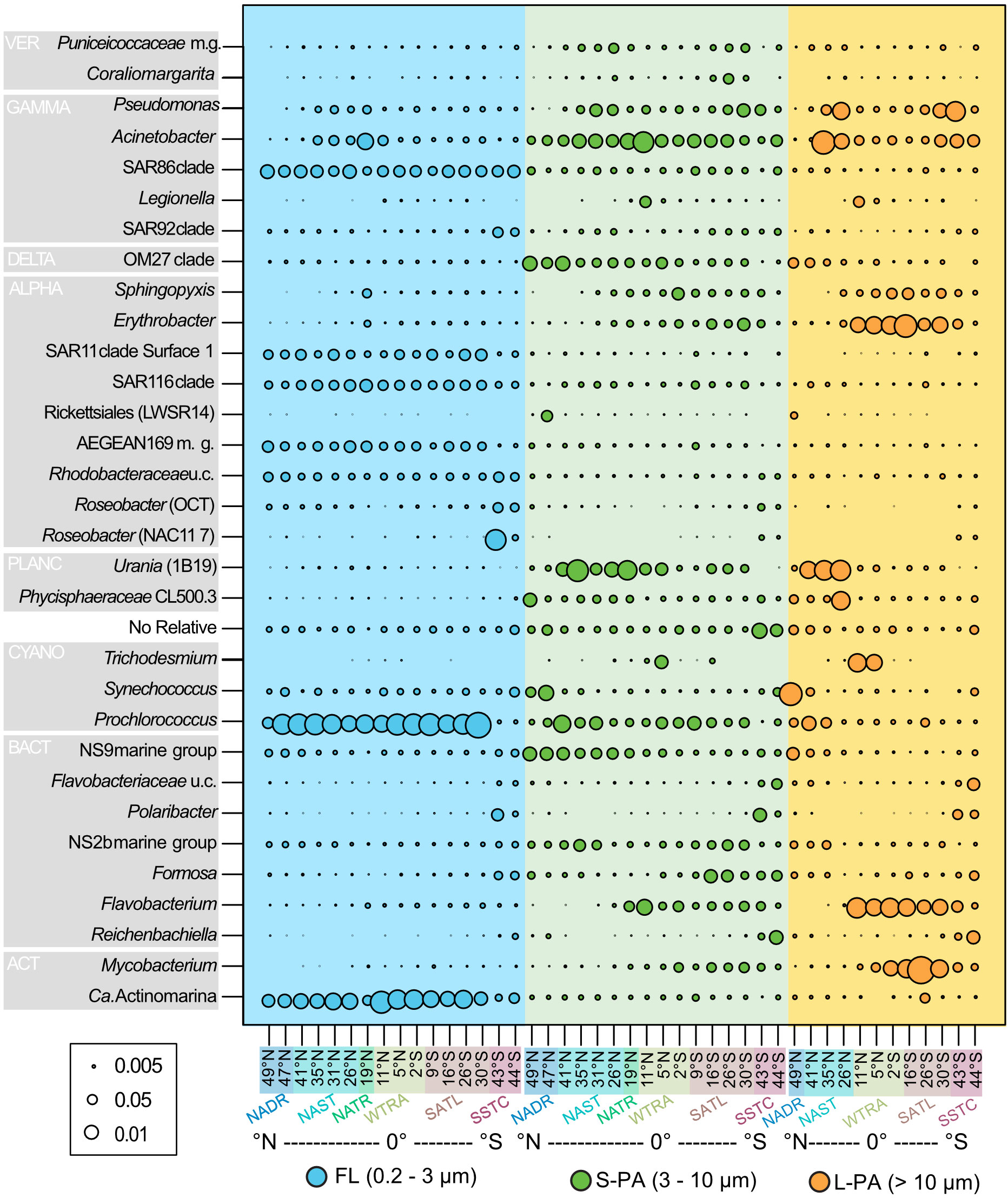
Figure 4 Bubble plot of bacterial taxa that reached a minimum of 5% relative read abundance from samples taken across the Atlantic Ocean during the AMT22 cruise in 2012. Colors indicate the size fraction: Blue free-living (FL), green small particle-attached (S-PA), and orange large particle-attached (L-PA) fraction. ACT Actinobacteria, ALPHA Alphaproteobacteria, BACT Bacteroidetes, CYANO Cyanobacteria, DELTA Deltaproteobacteria, GAMMA Gammaproteobacteria, PLANC Planctomycetes, VER Verrucomicrobia. NADR North Atlantic Drift, NAST North Atlantic Subtropical, NATR North Atlantic Tropical Gyre, WTRA Western Tropical Atlantic, SATL South Atlantic Gyre, and SSTC South Subtropical Convergence.
The most significant biogeographical distribution pattern across latitudes was in the highly productive southern temperate region. At the last two stations, sampled during the phytoplankton bloom, the relative read abundance of SAR11 and SAR116, Prochlorococcus, and Ca. Actinomarina were lower. Comparatively, the gammaproteobacterial SAR92 clade, uncultured Rhodobacteraceae, NAC11-7 and OCT Roseobacters had a higher abundance (Figure 4). Additionally, the Bacteroidetes showed a distinct response to the bloom, in both the FL and PA community, with an increased abundance of Ulvibacter (now Ca. Prociliicoccus (Francis et al., 2019)), Polaribacter, Formosa, Reichenbachiella, Owenweeksia, NS10, Fluviicola and uncultured Flavobacteriaceae (Supplementary Figure 1).
Other bacterial groups showed smaller biogeographical distribution patterns; for example, Acinetobacter was more abundant at the equator in the S-PA. Comparatively, they showed a bimodal distribution in the L-PA, with a higher abundance in both gyres. A similar bimodal pattern in the L-PA was also seen in Pseudomonas. The Planctomycetes group Urania was more abundant in the S-PA and L-PA fraction of the northern gyre. Erythrobacter was higher in abundance in the equator of the L-PA fraction. Finally, several Bacteroidetes groups showed distinct distribution patterns in addition to their increase in the southern temperate region. NS9 was more abundant in both particle fractions in the northern samples, whereas Flavobacterium was more abundant in the equator and southern samples.
Across the Atlantic, members of the Bacteroidetes (e.g., NS9, Flavobacterium and Formosa), Planctomycetes (e.g. Urania and Phycisphaerae), and Verrucomicrobia (e.g. Puniceicoccaceae marine group and Coraliomargarita) were more prevalent on particles (Figure 4). Other taxa with higher read abundances in the PA fraction, than in the FL communities, were yet uncultured bacteria related to the genera Pseudomonas, Acinetobacter, and Legionella (all Gammaproteobacteria), OM27 (Deltaproteobacteria), Shingopyxis, Erythrobacter and Rickettsiales (LWSR 14) (all Alphaproteobacteria), and Mycobacterium (Actinobacteria).
Based on the 16S rRNA sequencing data we chose specific FISH probes to quantify the absolute abundance of the key bacterial groups. Samples clearly corresponding to a specific province were chosen. Their averaged cell count was considered representative for each respective province.
FISH counts and 16S tag sequencing were largely consistent. However, some groups showed discrepancies, most strikingly SAR11 that comprised less than 13 ± 3% of the reads in the FL fraction, but more than half of the absolute bacterial abundance, in line with repeated counter-selection by the PCR primers used (Parada et al., 2016).
The FL and PA communities had different bacterial compositions. The majority of the bacteria in the FL community, across all provinces, were Alphaproteobacteria, specifically SAR11 and Roseobacter, followed by Cyanobacteria, specifically Prochlorococcus and Synechococcus, and diverse Bacteroidetes (Figure 5). Comparatively, the PA bacteria were composed of Bacteroidetes, specifically NS5, Polaribacter and Formosa, Gammaproteobacteria, specifically Pseudoalteromonas, Alteromonas, Vibrio and Balneatrix, diverse Alphaproteobacteria, as well as Planctomycetes, specifically Phycisphaeraceae and Rhodopirellula. In the highly productive southern temperate station (S44°, SSTC) there was a high abundance of Bacteroidetes in both the FL and PA community. Gammaproteobacteria were more abundant in the L-PA community, compared to the S-PA.
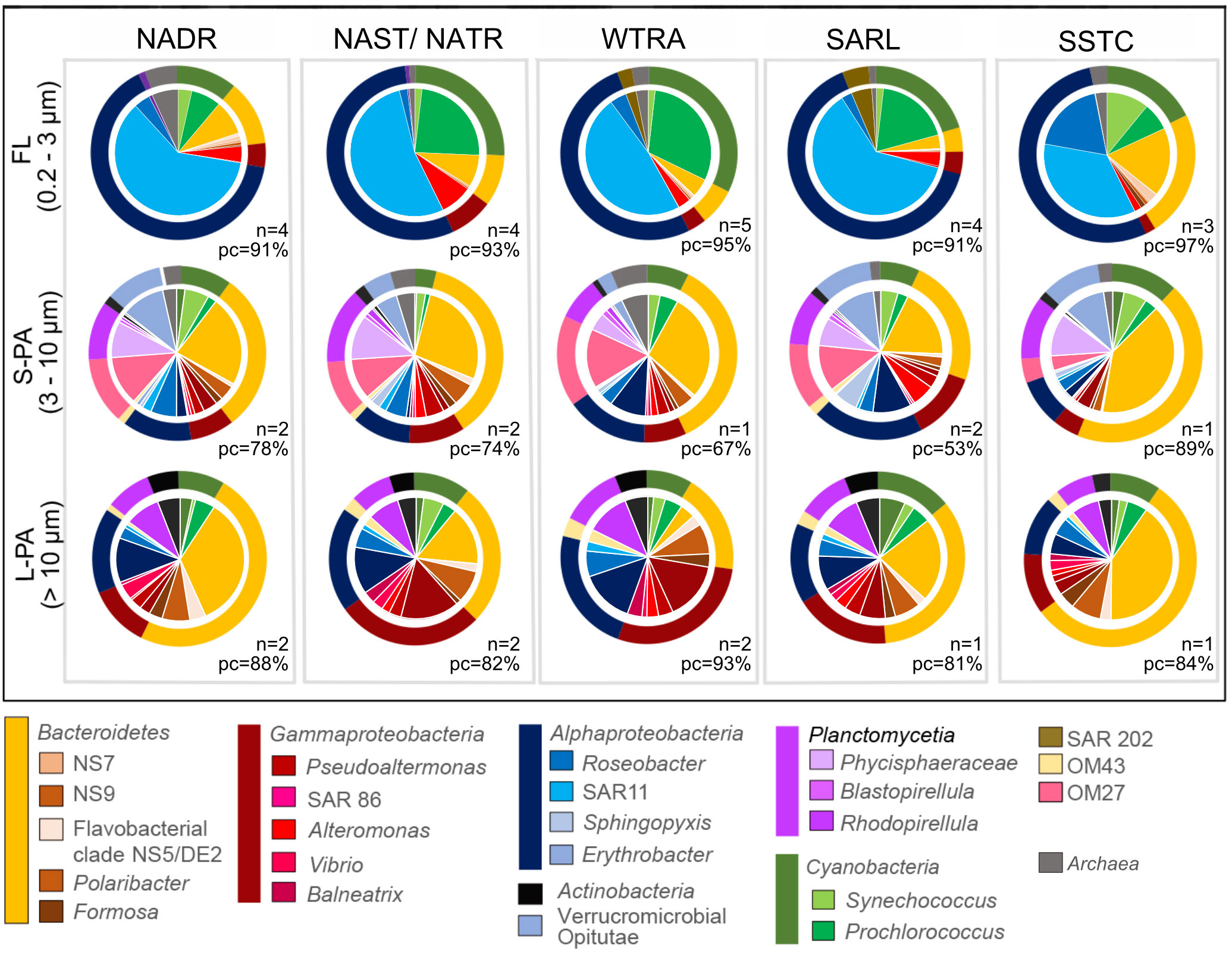
Figure 5 Pie chart of total bacterial cellular abundances counted using specific CARD-FISH probes (Supplement Table 2) during the AMT22 cruise in 2012. The abundance of representative stations was averaged from multiple samples for each region. All fish data is available in Pangaea. The outer rings of the chart indicate the bacterial phylum, and the inner part indicates the taxon to genus level where determined. The samples were taken across five regions, NADR North Atlantic Drift, NAST North Atlantic Subtropical, NATR North Atlantic Tropical Gyre, WTRA Western Tropical Atlantic, SATL South Atlantic Gyre, and SSTC South Subtropical Convergence, and fractionated into a free-living (FL), small particle-attached (S-PA) and large particle-attached (L- PA) fraction. n number of stations, PC probe coverage of total bacterial cell counts (EUBI-III).
Cells were counted with specific probes of the subphyla and genera to determine the absolute cell number in cases where cells showed high relative read abundance in a specific fraction. OM27 for example comprised between 6.8x101 to 2.6x102 cells ml-1 (3 - 9% relative to EUBI-III counts) of the total cell counts in the S-PA community and was also abundant in relative read abundance (Figure 4). Other important groups, based on abundances, in the S-PA were members of the family Puniceicoccaceae (Verrucomicrobia, Opitutae) with 1.8x101 - 6.6x102 cells ml-1 (1 - 9%), as well as members of the genus Phycisphaera (Planctomycetes, Phycisphaerae) with 3.4x101 - 6.9x102 cells ml-1 (3 - 10%).
To understand the finer-scale spatial organisation of PA bacteria, we used probes targeting the Bacteroidetes, Planctomycetes and Cyanobacteria to visualise cells directly attached to particles with a super-resolution microscope (Supplementary Figure 2D-F). The probes showed a striking degree of spatial organisation within the particle with brightly stained cells not only as surface colonisers, but also embedded within the particles (Supplementary Figure 2D-F). Dual hybridisation also showed that bacteria of at least two different taxa were intermingled within the fucose-enriched particles instead of forming large single-taxon clusters (Supplementary Figure 2E, F).
The FL, S-PA, and L-PA bacterial communities had a similarly high within-sample diversity (alpha diversity, Supplementary Table 1D). The alpha diversity only showed a slight decrease in the L-PA and FL within the Southern Gyre. Cross-community analysis (beta diversity) showed a significant difference between size fractions (ANOSIM: r2 = 0.67, P = 0.001, Figure 6A). Additionally, there was a significant biogeographical difference across the oceanic provinces (ANOSIM: r2 = 0.23, P = 0.001), with the highly productive southern temperate stations being separated from the others independent of the size fraction (Figure 6A). The FL and PA bacterial communities of the northern and southern temperate sites were more similar to each other, as were the L-PA communities of the two gyre regions and the equator. The similarity within FL and PA communities became more prominent with increasing Chl a concentrations (Figure 6B).
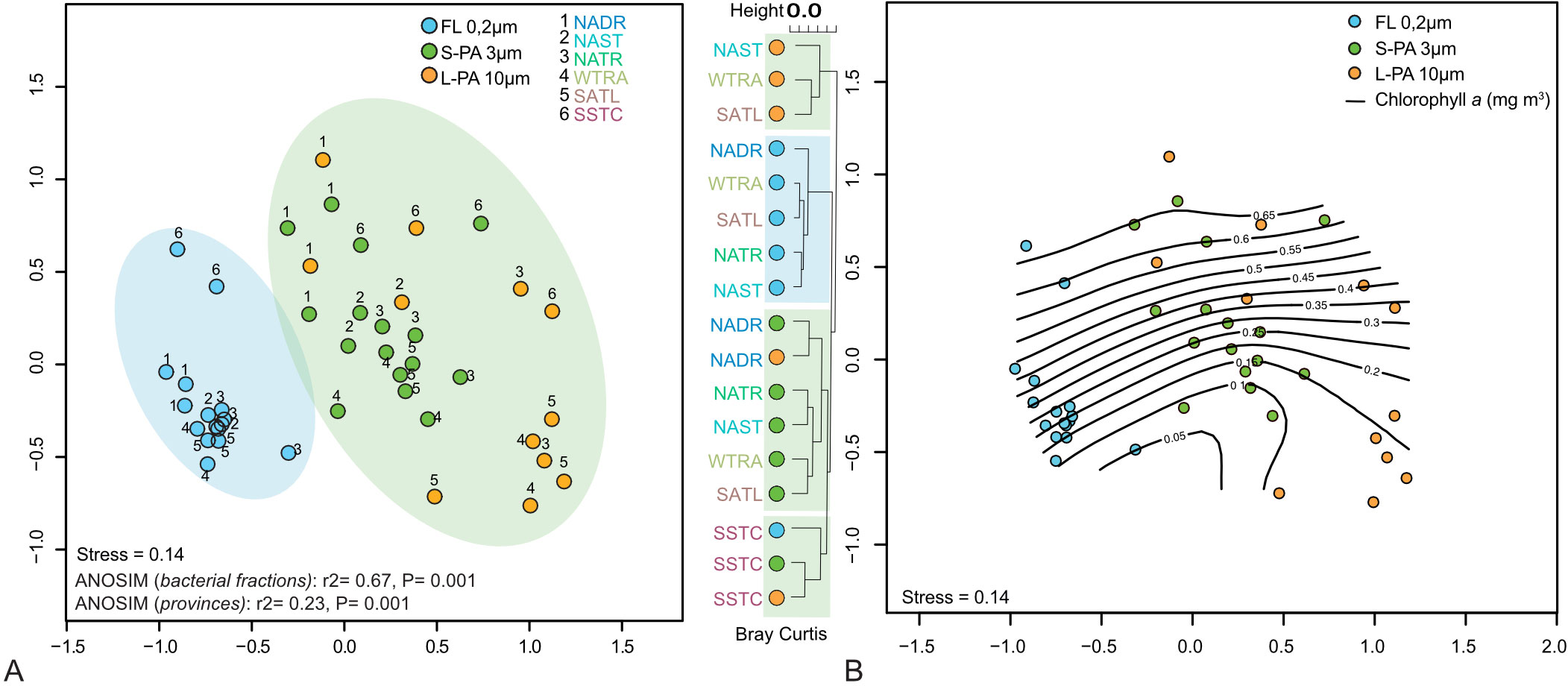
Figure 6 NMDS plots of 16S tag data showing bacterial communities sampled during the AMT22 cruise in 2012. (A) Communities clustering into significantly different groups of free-living (FL, blue background) and a small and large particle-attached (S-PA, L-PA, green background) fraction. (B) Communities shown across chlorophyll a as isotherm. NADR North Atlantic Drift, NAST North Atlantic Subtropical, NATR North Atlantic Tropical Gyre, WTRA Western Tropical Atlantic, SATL South Atlantic Gyre, and SSTC South Subtropical Convergence.
Sequencing of whole community DNA extracted from three S-PA samples from three stations (N26°, N11°, S44°) (Figure 1) yielded a total of 151,454,079 reads after quality filtering. De novo genomic assembly and binning resulted in the reconstruction of 54 draft bacterioplankton metagenome-assembled-genomes (MAGs) (Supplement Table 4). Fifty-four MAGs had predicted completeness > 80% and only four had predicted contamination > 6%. The genomic bins were 1.4 – 6.7 Mb in size and contained 1,276 – 6,560 genes. Phylogenies based on concatenated marker genes and 16S rRNA genes showed that these MAGs were assigned to six bacterial phyla. MAGs reconstructed from PA microbial communities were taxonomically diverse and included members of the Actinobacteria, Planctomycetes (Urania-1B-19 and CL500-3, both Phycisphaerae, and Planctomycetaceae), Verrucomicrobia (Opitutae and Verrucomicrobiaceae), Bacteroidetes (Flavobacterium, NS4 and NS9 marine groups), Gammaproteobacteria (Acinetobacter and Legionella lineage), and Alphaproteobacteria (Sphingopyxis) (Supplementary Figure 3). Most of the recovered MAGs, like Opitutae, NS9, or members of the OM27 had also been identified as prevalent clades in the PA fractions by amplicon sequencing (Figure 4), and CARD-FISH (Figure 5).
The metabolic potential of the PA communities to utilise polysaccharides was assessed by screening the MAGs of the Actinobacteria, Planctomycetes, Verrucomicrobia, Bacteroidetes, Alpha- and Gammaproteobacteria against the dbCAN database (Yin et al., 2012) and classified according to the carbohydrate-active enzymes (CAZy) database (Cantarel et al., 2009). All genomic bins contained genes relevant for carbohydrate degradation (Figure 7), mostly glycosyl transferases (GTs), glycoside hydrolases (GHs) and carbohydrate esterases (CEs), while carbohydrate binding modules (CBMs), auxiliary activities (AAs) and polysaccharide lyases (PLs) made up for a smaller proportion (Figure 7). GHs and GTs showed the highest diversity with, respectively, up to 20 and 18 different kinds across all MAGs. The most numerous glycoside hydrolase across all MAGs was GH109. Also highly abundant were GH13 (second most numerous), as well as GH23 and GH74 (Supplementary Figure 4).
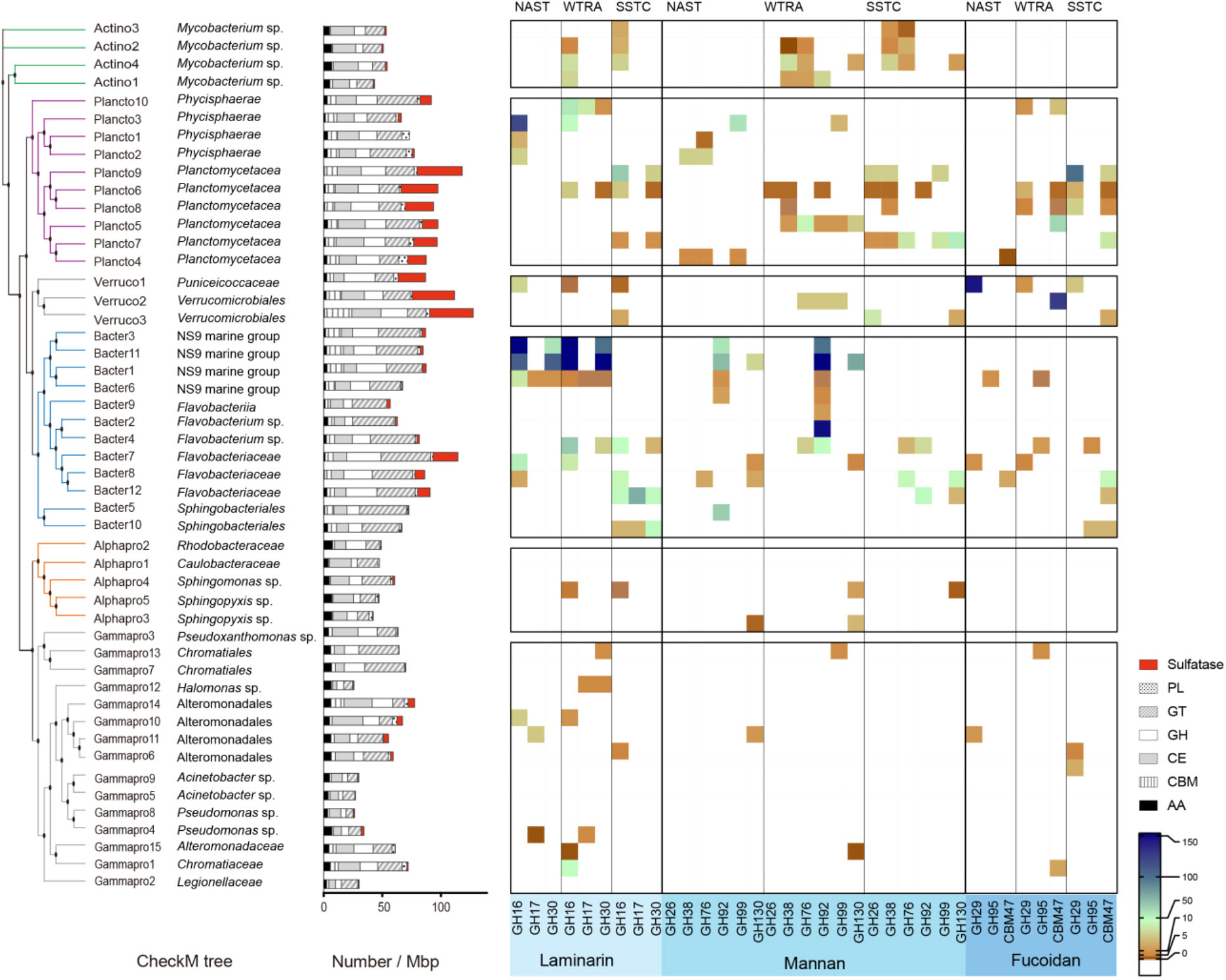
Figure 7 Carbohydrate-degradation enzyme profile across reconstructed genomes (MAGs). Heatmap visualises the abundance profiles, the number of CAZymes found in a particular MAG related to bin coverage, of glycoside hydrolases responsible for degrading various glycans in each MAG. GT glycosyl transferases, GH glycoside hydrolases, CE carbohydrate esterases, CBM carbohydrate binding modules, AA auxiliary activities, PL polysaccharide lyases.
Verrucomicrobia, Planctomycetaceae (Planctomycetes) and Flavobacteriaceae (Bacteroidetes) had a high frequency of sulfatases (Figure 7). These phyla also showed a GHs preference with on average 2.0 - 2.4% GH genes per MAG (Supplementary Table 5). Annotation indicated that the encoded enzymes likely targeted a diverse array of glycans (Figure 7, Supplementary Figure 3). GH16 and 30, used for the degradation of laminarin, were particularly high in the MAGs of Bacter3, 11, and 1 all affiliating with NS9 marine group. The highest abundance for the degradation of mannan (e.g. GH92) was found in Bacter3, 11 (both NS9 marine group), and Bacter2 (Flavobacterium sp.). The degradation capability for fucoidan was highest in the MAGs of Verruco1 and 2 (Puniceicoccaceae and Verrucomicrobiales, respectively), as well as Plancto9 (Planctomycetaceae) (Figure 7).
Since single GH genes provide only a preliminary view of degradation potential, we also extracted from the MAGs the polysaccharide utilisation loci (PULs), PUL-like structures, as well as co-localized genes for the degradation of polysaccharides of three substrates of interest: laminarin, mannan, and fucoidan. PULs and PUL-like structures rarely comprised all GHs, binding sites and transporters associated with the degradation and incorporation of oligo- or polysaccharides (Supplementary Figure 5). Bacter1 (NS9 marine group) for example was the only PUL that contained genes for the SusCD heteromer, including the porin type TonB-dependent transporter (SusC) and the glycan binding site/lid of the transporter protein (SusD). Most MAGs contained only the gene for the SusD protein in close proximity to the other genes important for polysaccharide degradation. More than one copy of a particular GH family was seen in several MAGs, for example Bacter1, 4, 7 (Supplementary Figure 5.1), Bacter5 (Supplementary Figure 5.2), and Plancto6, 8, and Verruco1 (Supplementary Figure 5.3).
In the marine environment, particulate organic matter (POM) is a primary vector of carbon export to the deep sea, resulting in long-term storage. The chemical composition of particles and the associated microbial community, specifically their degradation potential, are critical factors for the level of carbon export. In this study, we present a comprehensive analysis of the microbial particle-attached community, in diversity and abundance, from diverse open ocean regions across the Atlantic Ocean (49°N-44°S). Our findings decipher ecological functioning in marine carbon cycling by expanding our understanding of the particle-attached microbiomes.
Similar to previous research, we found that PA bacteria make up only a small fraction of the microbial community (Alldredge et al., 1986; Heins et al., 2021). However, despite a lower cellular abundance, PA bacteria showed a high diversity and distinct dissimilarity from the free-living community, indicating that particles offer a high number of selective niches.
In line with previous studies, the particle-attached bacterial communities consisted primarily of OM27 (Deltaproteobacteria), Opitutae (Verrucomicrobia), Phycisphaera (Planctomycetes), Gammaproteobacteria, and several Bacteroidetes groups (e.g. NS9, Flavobacterium and Formosa). Members of these groups have been associated with attachment and organic matter degradation before (Buchan et al., 2014; Milici et al., 2017), except for OM27, which is considered a member of the oligotrophic gyre community (Yilmaz et al., 2012).
Despite higher abundance in the particle fractions, some taxa, like Roseobacter (Alphaproteobacteria), the flavobacterial NS5 clade (Bacteroidetes), and Alteromonas (Gammaproteobacteria), were present in both fractions. Reasons for this could be a “stick-or-swim” lifestyle switch, as shown for Roseobacter (Michael et al., 2016), Gammaproteobacteria and Flavobacteriia (Fernández-Gómez et al., 2013; Marín, 2014); as well as stickiness of the particles (Passow, 2002); or micro-niching within clades (Dadon-Pilosof et al., 2017; Mestre et al., 2020), genera (Thompson et al., 2005; Hunt et al., 2008), and species (Moormann et al., 1997). We show, using FISH, that the abundant attached bacterial groups were not only attached to the particle surface but also embedded within the particle, indicating a tight linkage.
Particles were analysed across a broad geographic range and showed differences in abundance, as well as bacterial colonisation density. This finding could be related to the particle production time point or age (inferred by the level of primary production, e.g., within a gyre compared to an active phytoplankton bloom) and the glycan composition of the particles. We found that most particles were largely fucose-based, with only a minor fraction of mannose, galactose and N-acetylgalactosamine residues. It should be noted that lectin staining requires washing steps during the preparation of the particle and cell staining, which can lead to the unspecific removal of less persistent sugars.
Our finding corresponds with the finding from Huang et al. (2021), who showed that only a fraction of the secreted polysaccharides by microalgae promote particle formation. Specifically, 1,4-xylan and β-1,4-mannan are predominant in POM. At the same time, fucose-containing polysaccharides are mainly secreted but subsequently tend to enrich in POM, indicating that they promote particle formation. Furthermore, it has been proposed that fucoidans are quite resistant to degradation (Sichert et al., 2020), and that through their accumulation and aggregation, they drive carbon sequestration (Huang et al., 2021; Vidal-Melgosa et al., 2021). Sequestration is also affected by the particle sinking rate, because it affects whether particle-associated bacteria can react to the particle’s nutrient plume and stay in its proximity long enough for degradation to set in (Stocker et al., 2008; Seymour et al., 2017). How fast particles sink is related to a particle’s shape, size, composition and density (Bach et al., 2012; Turner, 2015), however, keeping the 3D particle structure intact requires other means for particle extraction than filtration, for example, with syringes, which was not done in this study.
The presence of bacteria potentially capable of using fucose-containing sulfated polysaccharides in the particle fraction in our study – 24% of the MAG’s contain GH29 or GH95 – supports a prevalence of these glycans in particles. However, their degradation must be slower than the production because these organisms showed moderately high cellular abundance. Verrucomicrobia and Planctomycetes had 7 - 16% and 1 – 9% relative abundance in the S-PA fraction, respectively, and Planctomycetes had an abundance of 9 – 16%in the L-PA fraction. Potentially the complexity of the required enzymes (Sichert et al., 2020), and the compositional complexity of the particles are preventing the degradation. Based on previous results, Verrucomicrobia are particularly well-adapted for fucose-containing polysaccharide degradation(Orellana et al., 2022).
Several MAGs in this study showed a partial mannan degradation pathway by targeting α-mannosidic linkages, including lineages of Actinobacteria, Planctomycetes, NS9, Flavobacteriaceae as well as Deltaproteobacteria. We found that of 54 MAGs 30 had predictive α-mannan glycoside hydrolases (GH38, GH76, GH92, and GH99). GH38 (α-mannosidase) and GH76 (α-1,6-mannanase) were mainly enriched in Mycobacterium and Planctomycetes clades, while GH92 (α-1,2/3-mannosidase) was abundantly found in most genomes in Bacteroidetes. Only the Planctomycetes, Verrucomicrobia and Gammaproteobacteria genomes encoded the gene for endo-α-1,2-mannosidase (GH99). Notably, one of the Planctomycetes genomes (Plancto5) contained the whole subset of glycoside hydrolases for the complete degradation of mannan. The incomplete pathways indicate a potential partitioning of mannan degradation pathways to individual community members and suggest that a complete degradation of mannan could be mediated through synergistic interactions. However, the particles 3D structure must be considered in the hypothesis. The individual organisms must be located near each other to profit from the degradation potential of others. Such cross-feeding on particles has been experimentally shown by (Enke et al., 2018). Although we did not visualise diverse organisms in co-localisation with mannan on a single particle, we could show that some groups, with partial mannan degrading potential, are located on and within the particles (Supplementary Figure 2D-F). Some of this degradation could also appear as selfish uptake – surface binding, partial hydrolysis, and direct uptake of hydrolysis products without loss to the environment –, driven by Bacteroidetes and Gammaproteobacteria as was shown for yeast α-mannan in rumen bacteria (Klassen et al., 2021). Recent research also showed the degradation of fungal α-mannan by a Salegentibacter sp. (Bacteroidota) strain isolated during a phytoplankton bloom in the North Sea (Solanki et al., 2022).
In the PA bacteria a lineage-specific pattern for GHs was observed; Verrucomicrobia, Planctomycetes and Bacteroidetes generally contained more GHs (on average 2.0-2.4% genes per genome), while Proteobacteria, Actinobacteria and Cyanobacteria showed the relatively lower GHs (0.8-1.1% genes per genome). The most numerous glycoside hydrolase across all genomes was GH109 (Supplementary Figure 4). An α-N-acetylgalactosaminidase activity is described for GH109, despite more functions assigned to this family. α-N-acetylgalactosaminidase can cleave N-acetylgalactosamine residues from glycoproteins and glycolipids (Desnick, 2001). GH13 is the second most abundant and contains many hydrolyzing enzymes with diverse functions, and it was originally established as the α-amylase family (Jespersen et al., 1993). GH23 and GH74 also appear to be abundantly present within the reconstructed genomes. These two enzymes generally act on the β-1,4-linkages in peptidoglycans and glucans, respectively. Together, these observations suggest that the most apparent nutrient sources might be α-glucan storage molecules such as glycogen or peptidoglycans.
Newly formed biomass, which contains more simple sugars like the storage polysaccharide laminarin, was encountered at the SSTC stations where an active phytoplankton bloom occurred. There was a high concentration of laminarin in the particle of the SSTC and WTRA (4mg/L) comparatively; the NAST/NTRA had only 0-1mg/L (Becker et al., 2020). Compared to fucose-containing sulfated polysaccharides, bacteria quickly break down laminarin when available (Arnosti et al., 2018; Reintjes et al., 2020a; Vidal-Melgosa et al., 2021). Correspondingly, the formation of fresh algal material in the SSTC supported a surge in bacterial cell numbers for both the free-living and the particle-attached bacterial community. These communities were highly similar, indicating that free-living bacteria were colonizing the POM, and that either similar selection forces were acting on all bacteria or an active exchange between communities occurred.
Equally, as mentioned above, bacteria are not necessarily fixed to a particle but can exhibit hop-on, hop-off behavior (McCarter, 1999; Kiorboe et al., 2003) or alternate between lifestyles, as was shown for some Bacteroidetes species (Polaribacter dokdonensis (MED 134) and Leeuwenhoekiella blandensis (MED217)) and are therefore present in multiple size fractions. They can attach to surfaces and use complex organic matter such as polysaccharides and proteins (Fernández-Gómez et al., 2013), and during times of organic matter limitation, they can switch to a free-living lifestyle using proteorhodopsins to obtain energy from light (Béjà et al., 2000; González et al., 2008; Fernández-Gómez et al., 2013). The apparent interchangeability of bacteria between different size fractions would explain the overlap in community composition between different size fractions found in our study and multiple others (Hollibaugh et al., 2000; Crespo et al., 2013; Mestre et al., 2017; Milici et al., 2017).
The potential differences in the nature of particles, whether they are of an abiotic or biotic source (live cells, diatomaceous earth, sand, chitin and cellulose), affect the colonisation by bacteria (López-Pérez et al., 2016). The “new” particles produced at the SSTC stations were predominantly phytoplankton-derived organic matter and selected for specific heterotrophs in both the free-living and particle-attached fraction. Specifically, there was an increase in the abundance of Bacteroidetes, which are often associated with phytoplankton-derived organic matter (Teeling et al., 2012). The high dissimilarity between the SSTC particle-attached community and the particle-attached communities of the other stations indicated that particles in different oceanic provinces may vary in chemical composition and therefore select for different bacterial groups.
Another reason for the high variability between the particle-attached communities could be due to succession patterns occurring during particle colonization (Datta et al., 2016). Analysis of bacterial colonization of chitin particles demonstrated that particle-attached bacteria undergo rapid succession patterns (Datta et al., 2016). Motile bacteria that can use the particles as a resource are the initial colonizers. Subsequently, secondary consumers colonize the particle, likely because they are attracted by the metabolites produced by the primary colonizers rather than the particle composition (Datta et al., 2016). The colonization of “new” particles at the SSTC stations by predominantly Bacteroidetes may represent an initial colonization by organisms using the particle as a resource (i.e. polysaccharides, proteins). The communities of particles in other regions, such as in the gyres, represent a more established but variable community of secondary colonizers.
Our study shows biogeographical differences in bacterial communities, especially the PA fraction, caused by differences in age and composition of particles. The results stress the importance of transect campaigns, such as the AMT22.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/ena, PRJNA421797, https://www.ebi.ac.uk/ena, PKCH00000000-PKEK00000000, https://www.ncbi.nlm.nih.gov/, SRP126598, https://www.ebi.ac.uk/ena, PRJEB2516, https://doi.pangaea.de/10.1594/PANGAEA.878060, PANGAEA.878060, https://doi.pangaea.de/10.1594/PANGAEA.891265, PANGAEA.891265, https://www.bodc.ac.uk/data/documents/cruise/11427/, 11427.
The project was designed by GR and RA. GR performed the sampling, FISH of the FL and 16S rRNA sequencing of all samples. CW performed the particle-associated FISH, lectin staining and metagenomic analysis. Statistical analysis was performed by GR and CW. The figures were prepared by GR and AH. The manuscript was written and reviewed by GR, AH, CW and RA. All authors contributed to the article and approved the submitted version.
This work was supported by the Max Planck Society. This study is a contribution to the international IMBER project and was also supported by the National Oceanography Centre, Southampton. The Atlantic Meridional Transect is funded by the UK Natural Environment Research Council through its National Capability Long-term Single Centre Science Programme, Climate Linked Atlantic Sector Science (grant number NE/R015953/1). This study contributes to the international IMBeR project and is contribution number 389 of the AMT programme.
We thank Jörg Wulf, Martha Schattenhoffer, Dimitri Meier, Karen Krüger, and Andreas Ellrott for technical assistance and helpful discussions. We thank the captain and crew of the RRS James Cook, as well as the principle scientist Glen Tarran (Plymouth Marine Laboratories) for assistance at sea. We are grateful to Bernhard M. Fuchs for helpful suggestions during the planning and realization of this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1051510/full#supplementary-material
Alldredge A. L., Cole J. J., Caron D. A. (1986). Production of heterotrophic bacteria inhabiting macroscopic organic aggregates (marine snow) from surface waters. Limnol. Oceanogr. 31 (1), 68–78. doi: 10.4319/lo.1986.31.1.0068
Alneberg J., Bjarnason B. S., de Bruijn I., Schirmer M., Quick J., Ijaz U. Z., et al. (2014). Binning metagenomic contigs by coverage and composition. Nat Methods 11, 1144–1146. doi: 10.1038/nmeth.3103
Arnosti C., Reintjes G., Amann R. (2018). A mechanistic microbial underpinning for the size-reactivity continuum of dissolved organic carbon degradation. Mar. Chem. 206, 93–99. doi: 10.1016/j.marchem.2018.09.008
Azam F., Malfatti F. (2007). Microbial structuring of marine ecosystems. Nat. Rev. Microbiol. 5 (10), 782–791. doi: 10.1038/nrmicro1747
Aziz R. K., Bartels D., Best A. A., DeJongh M., Disz T., Edwards R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9 (1), 1–15. doi: 10.1186/1471-2164-9-75
Bach L. T., Riebesell U., Sett S., Febiri S., Rzepka P., Schulz K. G. (2012). An approach for particle sinking velocity measurements in the 3–400 μm size range and considerations on the effect of temperature on sinking rates. Mar. Biol. 159 (8), 1853–1864. doi: 10.1007/s00227-012-1945-2
Becker S., Tebben J., Coffinet S., Wiltshire K., Iversen M. H., Harder T., et al. (2020). Laminarin is a major molecule in the marine carbon cycle. PNAS. 117. 6599–6607.
Beghini F., McIver L. J., Blanco-Míguez A., Dubois L., Asnicar F., Maharjan S., et al. (2021). Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 10, e65088. doi: 10.7554/eLife.65088.sa2
Behrenfeld M. J., Boss E., Siegel D. A., Shea D. M. (2005). Carbon-based ocean productivity and phytoplankton physiology from space. Global Biogeochem. Cycles 19 (1), GB1006. doi: 10.1029/2004GB002299
Béjà O., Suzuki M. T., Koonin E. V., Aravind L., Hadd A., Nguyen L. P., et al. (2000). Construction and analysis of bacterial artificial chromosome libraries from a marine microbial assemblage. Environ. Microbiol. 2 (5), 516–529. doi: 10.1046/j.1462-2920.2000.00133.x
Bennke C. M., Neu T. R., Fuchs B. M., Amann R. (2013). Mapping glycoconjugate-mediated interactions of marine Bacteroidetes with diatoms. Syst. Appl. Microbiol. 36 (6), 417–425. doi: 10.1016/j.syapm.2013.05.002
Bennke C. M., Reintjes G., Schattenhofer M., Ellrott A., Wulf J., Zeder M., et al. (2016). Modification of a high-throughput automatic microbial cell enumeration system for shipboard analyses. Appl. Environ. Microbiol. 82 (11), 3289–3296. doi: 10.1128/AEM.03931-15
Brewer P.G., Riley J.P. (1965). The automatic determination of nitrate in sea water. Deep Sea Research and Oceanographic Abstracts 12 (6), 765–772. doi: 10.1016/0011-7471(65)90797-7
Buchan A., LeCleir G. R., Gulvik C. A., González J. M. (2014). Master recyclers: Features and functions of bacteria associated with phytoplankton blooms. Nat. Rev. Microbiol. 12 (10), 686–698. doi: 10.1038/nrmicro3326
Cantarel B. L., Coutinho P. M., Rancurel C., Bernard T., Lombard V., Henrissat B. (2009). The carbohydrate-active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 37 (Database issue), 233–238. doi: 10.1093/nar/gkn663
Cottrell M. T., Kirchman D. L. (2000). Natural assemblages of marine Proteobacteria and members of the Cytophaga-flavobacter cluster consuming low- and high-molecular-weight dissolved organic matter. Appl. Environ. Microbiol. 66 (4), 1692–1697. doi: 10.1128/AEM.66.4.1692-1697.2000
Crespo B. G., Pommier T., Fernandez-Gomez B., Pedros-Alio C. (2013). Taxonomic composition of the particle-attached and free-living bacterial assemblages in the Northwest Mediterranean Sea analyzed by pyrosequencing of the 16S rRNA. Microbiologyopen 2 (4), 541–552. doi: 10.1002/mbo3.92
Dadon-Pilosof A., Conley K. R., Jacobi Y., Haber M., Lombard F., Sutherland K. R., et al. (2017). Surface properties of SAR11 bacteria facilitate grazing avoidance. Nat. Microbiol. 2 (12), 1608–1615. doi: 10.1038/s41564-017-0030-5
Datta M. S., Sliwerska E., Gore J., Polz M. F., Cordero O. X. (2016). Microbial interactions lead to rapid micro-scale successions on model marine particles. Nat. Commun. 7 (1), 11965. doi: 10.1038/ncomms11965
DeLong E. F., Franks D. G., Alldredge A. L. (1993). Phylogenetic diversity of aggregate-attached vs free-living marine bacterial assemblages. Limnol. Oceanogr. 38 (5), 924–934. doi: 10.4319/lo.1993.38.5.0924
Desnick R. (2001). α-n-acetylgalactosaminidase deficiency: Schindler disease. Metab. Mol. bases inherit. Dis., 3483–3505.
Diepenbroek M., Glöckner F. O., Grobe P., Güntsch A., Huber R., König-Ries B., et al. (2014). “Towards an integrated biodiversity and ecological research data management and archiving platform: The German federation for the curation of biological data (GFBio),” in Informatik 2014. Eds. Plödereder E., Grunske L., Schneider E., Ull D. (Bonn: Gesellschaft für Informatik e.V), S.1711–1721.
Elbourne L. D., Tetu S. G., Hassan K. A., Paulsen I. T. (2017). TransportDB 2.0: A database for exploring membrane transporters in sequenced genomes from all domains of life. Nucleic Acids Res. 45 (1), 320–324. doi: 10.1093/nar/gkw1068
Enke T. N., Leventhal G. E., Metzger M., Saavedra J. T., Cordero O. X. (2018). Microscale ecology regulates particulate organic matter turnover in model marine microbial communities. Nat. Commun. 9, 2743. doi: 10.1038/s41467-018-05159-8
Eren A. M., Esen Ö.C., Quince C., Vineis J. H., Morrison H. G., Sogin M. L., et al. (2015). Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3, e1319. doi: 10.7717/peerj.1319
Fernández-Gómez B., Richter M., Schüler M., Pinhassi J., Acinas S. G., González J. M., et al. (2013). Ecology of marine Bacteroidetes: A comparative genomics approach. ISME J. 7 (5), 1026–1037. doi: 10.1038/ismej.2012.169
Finn R. D., Clements J., Eddy S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39 (2), 29–37. doi: 10.1093/nar/gkr367
Francis T. B., Bartosik D., Sura T., Sichert A., Hehemann J.-H., Markert S., et al. (2021). Changing expression patterns of TonB-dependent transporters suggest shifts in polysaccharide consumption over the course of a spring phytoplankton bloom. ISME J. 15, 2336–2350. doi: 10.1038/s41396-021-00928-8
Francis B., Krüger K., Fuchs B. M., Teeling H., Amann R. I. (2019). Candidatus prosiliicoccus vernus, a spring phytoplankton bloom associated member of the Flavobacteriaceae. Syst. Appl. Microbiol. 42 (1), 41–53. doi: 10.1016/j.syapm.2018.08.007
Fuchsman C. A., Staley J. T., Oakley B. B., Kirkpatrick J. B., Murray J. W. (2012). Free-living and aggregate-associated Planctomycetes in the black Sea. FEMS Microbiol. Ecol. 80 (2), 402–416. doi: 10.1111/j.1574-6941.2012.01306.x
Glockner F. O., Kube M., Bauer M., Teeling H., Lombardot T., Ludwig W., et al. (2003). Complete genome sequence of the marine planctomycete pirellula sp. strain 1. PNAS. 100. 8298–8303.
González J. M., Fernández-Gómez B., Fernàndez-Guerra A., Gómez-Consarnau L., Sánchez O., Coll-Lladó M., et al. (2008). Genome analysis of the proteorhodopsin-containing marine bacterium polaribacter sp. MED152 (Flavobacteria). PNAS. 105, 8724–8729.
Grasshoff K. (1976). Determination of nitrate and nitrite. methods of seawater analysis (Weinheim, New York: Verlag Chemie).
Grossart H. P., Tang K. W., Kiorboe T., Ploug H. (2007). Comparison of cell-specific activity between free-living and attached bacteria using isolates and natural assemblages. FEMS Microbiol. Lett. 266 (2), 194–200. doi: 10.1111/j.1574-6968.2006.00520.x
Guindon S., Lethiec F., Duroux P., Gascuel O. (2005). PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Research 33(suppl_2), W557–W559. doi: 10.1093/nar/gki352
Heins A., Reintjes G., Amann R. I., Harder J. (2021). Particle collection in imhoff sedimentation cones enriches both motile chemotactic and particle-attached bacteria. Front. Microbiol. 12 (619), 1–17. doi: 10.3389/fmicb.2021.643730
Heissenberger A., Herndl G. J. (1994). Formation of high molecular weight material by free-living marine bacteria. Mar. Ecol. Prog. series. Oldendorf. 111 (1), 129–135. doi: 10.3354/meps111129
Hollibaugh J. T., Wong P. S., Murrell M. C. (2000). Similarity of particle-associated and free-living bacterial communities in northern San Francisco bay, California. Aquat. Microbial. Ecol. 21 (2), 103–114. doi: 10.3354/ame021103
Huang G., Vidal-Melgosa S., Sichert A., Becker S., Fang Y., Niggemann J., et al. (2021). Secretion of sulfated fucans by diatoms may contribute to marine aggregate formation. Limnol. Oceanogr. 66 (10), 3768–3782. doi: 10.1002/lno.11917
Hunt D. E., David L. A., Gevers D., Preheim S. P., Alm E. J., Polz M. F. (2008). Resource partitioning and sympatric differentiation among closely related bacterioplankton. Science 320 (5879), 1081–1085. doi: 10.1126/science.1157890
Huston A., Deming J. (2002). Relationships between microbial extracellular enzymatic activity and suspended and sinking particulate organic matter: Seasonal transformations in the north water. Deep Sea Res. Part II: Topical Stud. Oceanogr. 49 (22-23), 5211–5225. doi: 10.1016/S0967-0645(02)00186-8
Hyatt D., Chen G.-L., LoCascio P. F., Land M. L., Larimer F. W., Hauser L. J. (2010). Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11 (1), 1–11. doi: 10.1186/1471-2105-11-119
Jespersen H. M., Ann MacGregor E., Henrissat B., Sierks M. R., Svensson B. (1993). Starch- and glycogen-debranching and branching enzymes: Prediction of structural features of the catalytic (β/α)8-barrel domain and evolutionary relationship to other amylolytic enzymes. J. Protein Chem. 12 (6), 791–805. doi: 10.1007/BF01024938
Kappelmann L., Krüger K., Hehemann J.-H., Harder J., Markert S., Unfried F., et al. (2019). Polysaccharide utilization loci of north Sea Flavobacteriia as basis for using SusC/D-protein expression for predicting major phytoplankton glycans. ISME J. 13 (1), 76–91. doi: 10.1038/s41396-018-0242-6
Kiorboe T., Tang K., Grossart H. P., Ploug H. (2003). Dynamics of microbial communities on marine snow aggregates: Colonization, growth, detachment, and grazing mortality of attached bacteria. Appl. Environ. Microbiol. 69 (6), 3036–3047. doi: 10.1128/AEM.69.6.3036-3047.2003
Kirkwood D. (1996). Nutrients: Practical notes on their determination in sea water (Copenhagen, Denmark: ICES).
Klassen L., Reintjes G., Tingley J. P., Jones D. R., Hehemann J.-H., Smith A. D., et al. (2021). Quantifying fluorescent glycan uptake to elucidate strain-level variability in foraging behaviors of rumen bacteria. Microbiome 9 (1), 23. doi: 10.1186/s40168-020-00975-x
Klindworth A., Pruesse E., Schweer T., Peplies J., Quast C., Horn M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41 (1), e1. doi: 10.1093/nar/gks808
Krüger K., Chafee M., Ben Francis T., Glavina del Rio T., Becher D., Schweder T., et al. (2019). In marine Bacteroidetes the bulk of glycan degradation during algae blooms is mediated by few clades using a restricted set of genes. ISME J. 13 (11), 2800–2816. doi: 10.1038/s41396-019-0476-y
Li H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv: Genomics. 1303. doi: 10.48550/arXiv.1303.3997
Liu G., Zhang L., Wei X., Zou G., Qin Y., Ma L., et al. (2013). Genomic and secretomic analyses reveal unique features of the lignocellulolytic enzyme system of Penicillium decumbens. PloS One 8 (2), e55185. doi: 10.1371/journal.pone.0055185
López-Pérez M., Kimes N. E., Haro-Moreno J. M., Rodriguez-Valera F. (2016). Not all particles are equal: the selective enrichment of particle-associated bacteria from the mediterranean sea. Front. Microbiol. 7, 996. doi: 10.3389/fmicb.2016.00996
Ludwig W., Strunk O., Westram R., Richter L., Meier H., Yadhukumar, et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32 (4), 1363–1371. doi: 10.1093/nar/gkh293
Lyons M., Dobbs F. (2012). Differential utilization of carbon substrates by aggregate-associated and water-associated heterotrophic bacterial communities. Hydrobiologia 686 (1), 181–193. doi: 10.1007/s10750-012-1010-7
Marín I. (2014). “Proteobacteria,” in Encyclopedia of astrobiology. Eds. Amils R., Gargaud M., Cernicharo Quintanilla J., Cleaves H. J., Irvine W. M., Pinti D., Viso M. (Berlin, Heidelberg: Springer Berlin Heidelberg), 1–2.
McCarter L. (1999). The multiple identities of Vibrio parahaemolyticus. J. Mol. Microbiol. Biotechnol. 1 (1), 51–57.
Mestre M., Ferrera I., Borrull E., Ortega-Retuerta E., Mbedi S., Grossart H.-P., et al. (2017). Spatial variability of marine bacterial and archaeal communities along the particulate matter continuum. Mol. Ecol. 26 (24), 6827–6840. doi: 10.1111/mec.14421
Mestre M., Höfer J., Sala M. M., Gasol J. M. (2020). Seasonal variation of bacterial diversity along the marine particulate matter continuum. Front. Microbiol. 11, 1590. doi: 10.3389/fmicb.2020.01590
Michael V., Frank O., Bartling P., Scheuner C., Göker M., Brinkmann H., et al. (2016). Biofilm plasmids with a rhamnose operon are widely distributed determinants of the ‘swim-or-stick’ lifestyle in roseobacters. ISME J. 10 (10), 2498–2513. doi: 10.1038/ismej.2016.30
Milici M., Vital M., Tomasch J., Badewien T. H., Giebel H. A., Plumeier I., et al. (2017). Diversity and community composition of particle-associated and free-living bacteria in mesopelagic and bathypelagic southern ocean water masses: evidence of dispersal limitation in the bransfield strait. Limnol. Oceanogr. 62 (3), 1080–1095. doi: 10.1002/lno.10487
Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G. A., Sonnhammer E. L. L., et al. (2021). Pfam: The protein families database in 2021. Nucleic Acids Res. 49 (1), 412–419. doi: 10.1093/nar/gkaa913
Moormann M., Zähringer U., Moll H., Kaufmann R., Schmid R., Altendorf K. (1997). A new glycosylated lipopeptide incorporated into the cell wall of a smooth variant of Gordona hydrophobica. J. Biol. Chem. 272, 10729–10738. doi: 10.1074/jbc.272.16.10729
Oksanen J., Blanchet F. G., Kindt R., Legendre P., Minchin P. R., O’hara R., et al. (2013). “Package ‘vegan’,” in Community ecology package, version 2, 1–295.
Orellana L. H., Francis T. B., Ferraro M., Hehemann J.-H., Fuchs B. M., Amann R. I. (2022). Verrucomicrobiota are specialist consumers of sulfated methyl pentoses during diatom blooms. ISME J. 16, 630–641. doi: 10.1038/s41396-021-01105-7
Parada A. E., Needham D. M., Fuhrman J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414. doi: 10.1111/1462-2920.13023
Parks D. H., Imelfort M., Skennerton C. T., Hugenholtz P., Tyson G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Passow U. (2002). Transparent exopolymer particles (TEP) in aquatic environments. Prog. Oceanogr. 55, 287–333. doi: 10.1016/S0079-6611(02)00138-6
Pernthaler J., Glöckner F.-O., Schönhuber W., Amann R. (2001). Fluorescence in situ hybridization (FISH) with rRNA-targeted oligonucleotide probes. Methods Microbiol. 30, 207–226. doi: 10.1016/S0580-9517(01)30046-6
Pernthaler A., Pernthaler J., Amann R. (2004). “Sensitive multi-color fluorescence in situ hybridization for the identification of environmental microorganisms,” in Molecular microbial ecology manual. Eds. Kowalchuk G., De Bruijn F. J., Head I. M., Akkermans A. D., Van Elsas J. D. (Dordrecht, the Netherlands: Kluwer Academic Publishers), 711–726.
Pruesse E., Peplies J., Glöckner F. O. (2012). SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829. doi: 10.1093/bioinformatics/bts252
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Rawlings N., Barrett A., Bateman A. (2012). MEROPS. the www. jbc. org downloaded from database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 40, D343–D350. doi: 10.1093/nar/gkx1134
Reintjes G., Arnosti C., Fuchs B., Amann R. (2019). Selfish, sharing and scavenging bacteria in the Atlantic ocean: a biogeographical study of bacterial substrate utilisation. ISME J. 13, 1119–1132. doi: 10.1038/s41396-018-0326-3
Reintjes G., Fuchs B. M., Amann R., Arnosti C. (2020a). Extensive microbial processing of polysaccharides in the south pacific gyre via selfish uptake and extracellular hydrolysis. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.583158
Reintjes G., Fuchs B. M., Scharfe M., Wiltshire K. H., Amann R., Arnosti C. (2020b). Short-term changes in polysaccharide utilization mechanisms of marine bacterioplankton during a spring phytoplankton bloom. Environ. Microbiol. 22, 1884–1900. doi: 10.1111/1462-2920.14971
Richter M., Rosselló-Móra R., Oliver Glöckner F., Peplies J. (2016). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Rieck A., Herlemann D. P. R., Jürgens K., Grossart H.-P. (2015). Particle-associated differ from free-living bacteria in surface waters of the Baltic Sea. Front. Microbiol. 6, 1297. doi: 10.3389/fmicb.2015.01297
Salazar G., Cornejo-Castillo F. M., Borrull E., Díez-Vives C., Lara E., Vaqué D., et al. (2015). Particle-association lifestyle is a phylogenetically conserved trait in bathypelagic prokaryotes. Mol. Ecol. 24, 5692–5706. doi: 10.1111/mec.13419
Schattenhofer M., Fuchs B. M., Amann R., Zubkov M. V., Tarran G. A., Pernthaler J. (2009). Latitudinal distribution of prokaryotic picoplankton populations in the Atlantic ocean. Environ. Microbiol. 11, 2078–2093. doi: 10.1111/j.1462-2920.2009.01929.x
Schultz D., Zühlke D., Bernhardt J., Francis T. B., Albrecht D., Hirschfeld C., et al. (2020). An optimized metaproteomics protocol for a holistic taxonomic and functional characterization of microbial communities from marine particles. Environ. Microbiol. Rep. 12, 367–376. doi: 10.1111/1758-2229.12842
Seymour J. R., Amin S. A., Raina J.-B., Stocker R. (2017). Zooming in on the phycosphere: the ecological interface for phytoplankton–bacteria relationships. Nat. Microbiol. 2 (7), 17065. doi: 10.1038/nmicrobiol.2017.65
Sichert A., Corzett C. H., Schechter M. S., Unfried F., Markert S., Becher D., et al. (2020). Verrucomicrobia use hundreds of enzymes to digest the algal polysaccharide fucoidan. Nat. Microbiol. 5, 1026–1039. doi: 10.1038/s41564-020-0720-2
Simon M., Grossart H. P., Schweitzer B., Ploug H. (2002). Microbial ecology of organic aggregates in aquatic ecosystems. Aquat. Microbial. Ecol. 28, 175–211. doi: 10.3354/ame028175
Smith D. C., Simon M., Alldredge A. L., Azam F. (1992). Intense hydrolytic enzyme-activity on marine aggregates and implications for rapid particle dissolution. Nature 359, 139–142. doi: 10.1038/359139a0
Smith M., Zeigler Allen L., Allen A., Herfort L., Simon H. (2013). Contrasting genomic properties of free-living and particle-attached microbial assemblages within a coastal ecosystem. Front. Microbiol. 4, 120. doi: 10.3389/fmicb.2013.00120
Solanki V., Krüger K., Crawford C. J., Pardo-Vargas A., Danglad-Flores J., Hoang K. L. M., et al. (2022). Glycoside hydrolase from the GH76 family indicates that marine salegentibacter sp. Hel_I_6 consumes alpha-mannan from fungi. ISME J. 16, 1818–1830. doi: 10.1038/s41396-022-01223-w
Spring S., Bunk B., Spröer C., Rohde M., Klenk H. P. (2018). Genome biology of a novel lineage of Planctomycetes widespread in anoxic aquatic environments. Environ. Microbiol. 20, 2438–2455. doi: 10.1111/1462-2920.14253
Stocker R., Seymour J. R., Samadani A., Hunt D. E., Polz M. F. (2008). Rapid chemotactic response enables marine bacteria to exploit ephemeral microscale nutrient patches. PNAS. 105. 4209–4214.
Teeling H., Fuchs B. M., Becher D., Klockow C., Gardebrecht A., Bennke C. M., et al. (2012). Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336, 608–611. doi: 10.1126/science.1218344
Thompson J. R., Pacocha S., Pharino C., Klepac-Ceraj V., Hunt D. E., Benoit J., et al. (2005). Genotypic diversity within a natural coastal bacterioplankton population. Science 307, 1311–1313. doi: 10.1126/science.1106028
Turner J. T. (2015). Zooplankton fecal pellets, marine snow, phytodetritus and the ocean’s biological pump. Prog. Oceanogr. 130, 205–248. doi: 10.1016/j.pocean.2014.08.005
Van Vliet D. M., Palakawong Na Ayudthaya S., Diop S., Villanueva L., Stams A. J., Sánchez-Andrea I. (2019). Anaerobic degradation of sulfated polysaccharides by two novel Kiritimatiellales strains isolated from black Sea sediment. Front. Microbiol. 10, 253. doi: 10.3389/fmicb.2019.00253
Vidal-Melgosa S., Sichert A., Francis T. B., Bartosik D., Niggemann J., Wichels A., et al. (2021). Diatom fucan polysaccharide precipitates carbon during algal blooms. Nat. Commun. 12, 1150. doi: 10.1038/s41467-021-21009-6
Wegner C.-E., Richter-Heitmann T., Klindworth A., Klockow C., Richter M., Achstetter T., et al. (2013). Expression of sulfatases in rhodopirellula baltica and the diversity of sulfatases in the genus rhodopirellula. Mar. Genomics 9, 51–61. doi: 10.1016/j.margen.2012.12.001
Yilmaz P., Iversen M. H., Hankeln W., Kottmann R., Quast C., Glöckner F. O. (2012). Ecological structuring of bacterial and archaeal taxa in surface ocean waters. FEMS Microbiol. Ecol. 81, 373–385. doi: 10.1111/j.1574-6941.2012.01357.x
Yin Y., Mao X., Yang J., Chen X., Mao F., Xu Y. (2012). dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 40, 445–451. doi: 10.1093/nar/gks479
Ziervogel K., Arnosti C. (2008). Polysaccharide hydrolysis in aggregates and free enzyme activity in aggregate-free seawater from the north-eastern gulf of Mexico. Environ. Microbiol. 10, 289–299. doi: 10.1111/j.1462-2920.2007.01451.x
Ziervogel K., Steen A. D., Arnosti C. (2010). Changes in the spectrum and rates of extracellular enzyme activities in seawater following aggregate formation. Biogeosciences 7, 1007–1015. doi: 10.5194/bg-7-1007-2010
Keywords: MAGs: metagenome assembled genomes (MAGs), particle-attached and free-living microbes, particle composition, fucose-containing polysaccharide, CAZy (carbohydrate-active enzymes), bacterial cell abundance
Citation: Reintjes G, Heins A, Wang C and Amann R (2023) Abundance and composition of particles and their attached microbiomes along an Atlantic Meridional Transect. Front. Mar. Sci. 10:1051510. doi: 10.3389/fmars.2023.1051510
Received: 22 September 2022; Accepted: 17 February 2023;
Published: 07 March 2023.
Edited by:
Timothy James Smyth, Plymouth Marine Laboratory, United KingdomReviewed by:
Brandon Kieft, University of British Columbia, CanadaCopyright © 2023 Reintjes, Heins, Wang and Amann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Greta Reintjes, greintje@mpi-bremen.de
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.