 Tiancheng Zhou
Tiancheng Zhou Simin Hu
Simin Hu Nan Jia1,2,3
Nan Jia1,2,3 Hui Huang
Hui Huang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 03 October 2022
Sec. Coral Reef Research
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.993305
This article is part of the Research TopicReef-Building Coral Holobionts under Environmental Stressors: Responses and AdaptationView all 7 articles
The microbiota is an important component of the epilithic algal matrix (EAM) and plays a central role in the biogeochemical cycling of important nutrients in coral reef ecosystems. Insufficient studies on EAM microbiota diversity have led to a limited understanding of the ecological functions of EAMs in different states. To explore the microbial community of EAMs in the Luhuitou fringing reef in Sanya, China, which has undergone the incessant expansion and domination of algae over the past several decades, investigations were conducted in the reef’s intertidal zone. Five types of substrate habitats (dead branching coral, dead massive coral, dead flat coral, granite block, and concrete block) were selected, and their microbial communities were analyzed by high-throughput sequencing of EAM holobionts using the 16S rDNA V4 region. Proteobacteria was the most abundant group, accounting for more than 70% of reads of the microbial composition across all sites, followed by Cyanobacteria (15.89%) and Bacteroidetes (5.93%), respectively. Cluster analysis divided all microbial communities into three groups, namely short, medium, and long EAMs. Algal length was the most important morphological factor impacting the differences in the composition of the EAM microbiota. The three EAM groups had 52 common OTUs and 78.52% common sequences, among which the most abundant were Vibrio spp. and Photobacterium spp. The three types of EAM also had unique OTUs. The short EAMs had 238 unique OTUs and 48.61% unique sequences, mainly in the genera Shewanella and Cyanobacterium. The medium EAMs contained 130 unique OTUs and 4.36% unique sequences, mainly in the genera Pseudomonas and Bacillus. The long EAMs only had 27 unique OTUs and 4.13% unique sequences, mainly in the genus Marinobacter. Compared with short EAM, medium and long EAM had a lower proportion of autotrophic bacteria and higher proportion of potential pathogenic bacteria. It is suggested that EAMs with different phenotypes have different microbial compositions, and the ecological function of the EAM microbiota changes from autotrophic to pathogenic with an increase in algal length. As EAMs have expanded on coastal coral reefs worldwide, it is essential to comprehensively explore the community structure and ecological role of their microbial communities.
The epilithic algal matrix (EAM) is an important benthic habitat in coral reef ecosystems, which include turf algae, non-living organic components (detritus), microbial components, and inorganic material (sediment) (Wilson and Bellwood, 1997). Turf algae are heterogeneous assemblages of short filamentous algae and juvenile macroalgae (Wilson and Bellwood, 1997; Hester et al., 2016). EAM is the major contributor to benthic productivity in coral reefs, being responsible for one-third of the gross production and most of the net production (Klumpp and McKinnon, 1989; Tebbett and Bellwood, 2021). EAM is also home to diverse eukaryotic and prokaryotic microbial communities (Barott et al., 2011), including various coral pathogens (Egan et al., 2013). Direct contact between coral and algae may facilitate the invasion of opportunistic pathogens, which may lead to coral disease or bleaching due to toxicity or hypoxia in coral tissues (Barott and Rohwer, 2012). Thus, characterizing the microbial components of EAM is critical for understanding its functional diversity and the key microflora that mediate coral-algal phase shifts.
EAM seems to have a defined and highly diverse core bacterial community consisting of Cyanobacteria, Proteobacteria, Actinobacteria, Bacteroidetes, and Firmicutes species (Hollants et al., 2013; Hester et al., 2016). Cyanobacteria are considered the most important bacteria in the EAM, representing more than half of all microbes (Stal, 1995; Connell et al., 2014; Echenique-Subiabre et al., 2015). These cyanobacteria comprise the genera Anabaena, Hydrocoleum, Lyngbya, Oscillatoria, Phormidium, Schizothrix, and Symploca. Proteobacteria is the most common bacterial phylum associated with the EAM (34–65% relative abundance) (Barott et al., 2011; Hester et al., 2016; Walter et al., 2016; Meirelles et al., 2018). The Proteobacteria include the orders Rhodospirales, Rhodobacterales, and Rhizobiales (Hester et al., 2016). In addition, some studies have found that Firmicutes can also be a dominant group in some EAM, with a relative abundance exceeding 80% (Roach et al., 2020). In particular, these microbes play a role in photosynthesis and the metabolism of nitrogen and sulfur in the EAM due to their complementary synergistic functions (Walter et al., 2016). In benthic communities, EAM microbiota that are dominated by Cyanobacteria significantly contribute to photosynthesis and nitrogen cycling (Echenique-Subiabre et al., 2015). Moreover, the EAM microbiota indirectly promotes disease and mortality in adjacent corals because they include several Cyanobacteria genera that are associated with coral black band disease, including Phormidium, Leptolyngbya, Oscillatoria, Geitlerinema, and Cyanobacterium (Myers et al., 2007). However, it is currently unclear what factors dominate the differences in EAM microbiota, especially considering that its morphology is complex and varied (Connell et al., 2014).
The bacterial communities associated with the EAM significantly differ among the various types of substrate habitats. Cyanobacteria, which are the dominant microbial group in the EAM, grow on sandy sediments or hard rocky substrates, or are epiphytic on sea grasses in coral reefs and tropical lagoons (Stal, 1995; Connell et al., 2014; Echenique-Subiabre et al., 2015). In contrast, approximately 50% of EAM microbiota existing on massive porous substrates are Proteobacteria (Barott et al., 2011). Thus, it is hypothesized that substrate differences play a major role in shaping the diversity of the EAM algae and microbiota (Tebbett and Bellwood, 2021), but a lack of knowledge and evidence about the deeper reasons that lead to these differences leaves a significant gap in our understanding of EAM ecological functions and coral reef dynamics.
The Luhuitou fringing reef is located adjacent to the Sanya urban area along the southern coast of Hainan Island in China. However, almost 80% of the fringing reef in this area was damaged due to intensive human activities between the 1970s and 1990s, leaving the current living coral coverage at only 12.8–23.38% (Huang et al., 2019). The combination of coral bleaching and high nutrient levels in the Luhuitou fringing reef (Table S1) has provided opportunities for benthic algae to reproduce, causing the reef to be replaced by abundant benthic macroalgae and EAM. The number of algal species found in Luhuitou between 2008 and 2015 increased by 26% compared to the 1990s (Titlyanov et al., 2018), with the more abundant filamentous turf algae forming EAM under high sediment loads. Additionally, different EAM-forming algal compositions can be found on different hard substrates (Titlyanov et al., 2019). For example, the upper intertidal zone is mainly occupied by Polysiphonia howei, Ulva prolifera, and U. clathrata. In the lower intertidal zone, the surfaces of dead coral have been overgrown by Centroceras clavulatum and Jania adhaerens (Titlyanov et al., 2019). The composition of the algae in the EAM framework has been studied for many years; however, the components of the EAM microbiota remain unclear.
Therefore, we explored the core groups of the EAM microbiota in the Luhuitou fringing reef and tested their stability and shift patterns. The objectives of this study were to 1) assess the microbiota composition of different types of EAM and 2) analyze the relationships between EAM microbiota composition, algal morphological characteristics, and substrate heterogeneity. The results of this study will provide information on the fundamental aspects of coral-algae interactions, as well as reef conservation and management.
Investigations were conducted in the Luhuitou fringing reef, Sanya Bay, Hainan Island, China (18°12′45″ N; 109°28′31″ E) (Figure 1A) from July 23 to 25, 2021. The complexity of the substrate was measured at first. To reduce the influence of tidal changes, EAM sampling was carried out during the flat tide period when the hydrodynamic force was weak, and the sampling was completed within 3 hours. The mean annual air temperature of the study area is 25.8°C (ranging from 24.7 to 27.0°C), the mean monthly seasonal sea surface temperature (SST) is 27°C (ranging from 22.8°C to 29.8°C), and the mean sea surface salinity (SSS) is 33.1‰ (ranging from 32.1‰ to 33.8‰) (Yu et al., 2010). Water quality parameters showed that the Luhuitou coastal reef was in a state of high nutrient content and turbidity at the time of this study (Table S1).

Figure 1 Study sites on Hainan Island. (A) Location of the Luhuitou fringing reef; (B) diagram of the study area; (C) representative photograph of the five types of substrate (dead branching coral [BC], dead massive coral [MC], dead flat coral [FC], granite [GB], and concrete block [CB]).
A 50-m wide area dividing the upper and lower intertidal zones was taken as the research area (Figure 1B). This zone was mainly composed of dead skeletons of massive and branched corals intermixed with sand. Abundant and complex EAM communities have been identified in this region (Titlyanov et al., 2018; 2019). Based on differences in substrate complexity and porosity, five types of substrate habitats were obtained in this study (Figure 1C). The five types of substrates were dead branching coral (BC), dead massive coral (MC), dead flat coral (FC), granite block (GB), and concrete block (CB). Depending on the field conditions, 5-9 duplicate samples were collected for each substrate, and ensuring that each sample was more than 5m apart. Data were collected to quantify the structural characteristics of the substrate microhabitats (Table 1). Structural complexity was determined based on the rugosity index of the coral skeletons (Alvarez-Filip et al., 2011); a completely flat surface had a rugosity index of 1, and higher values indicated greater complexity. In addition, photographs of 25 cm × 25 cm sample frames were taken. Coral Point Count with Excel extensions (CPCe) (Kohler and Gill, 2006) was used to analyze the images by manually annotating 20 randomly generated points and calculating the EAM coverage. The EAMs were collected in two pieces (each piece approximately 40 cm2) with a chisel and hammer, and stored in separate sealed bags at -20° for DNA extraction the next day and microscopies a few days later. One of each paired sample was randomly selected for DNA extraction and the other for assessing algae morphology.

Table 1 Classification and characteristics of the five studied substrates (Subscripts after substrate code are used to distinguish different tidal levels, with u and l representing the upper and lower intertidal zones respectively).
To evaluate the morphological differences of the EAM, three key parameters including algal length, biomass, and sediment load were selected for analysis. To measure the biomass and sediment load, the EAM was scraped with a sterile scraper to a depth of 2 mm at the bottom layer, and then the scraped EAM was transferred into a sealed bag. Seawater filtered through a 0.2 μm filter membrane was added, and then the EAM was shaken well and passed in turn through 200 and 75 μm screens. Algal and detrital components were sequentially intercepted by the screens, and the sediment components were filtered through a 0.2 μm membrane as turbidite. The water was drained and a 0.0001 g precision scale analytical balance (Sartorius® SQP, Goettingen, Germany) was used to obtain the wet weight of the algal, detrital, and sediment components. The sum of the three components was considered the total EAM biomass. The surface area of the substrate after removing the EAM was measured using the aluminum foil embedding method (Marsh, 1970). To measure the algal length, the algal components were placed in separate Petri dishes, and the lengths of 10 intact algal filaments from each sample were randomly measured using anatomical and graduated lenses (OLYMPUS® SZ61, Tokyo, Japan). The average value of each sample was calculated.
A sterile scalpel was used to quickly scrape approximately 1 cm2 of EAM, and 5-9 duplicate samples of the same substrate and tide level were mixed into a tube, which was then evenly ground with a 5 mL glass homogenizer to collect approximately 400 mg of homogenate. Total genomic DNA was extracted from the samples using the FastDNA® Spin Kit for Feces (MP Biomedicals, Santa Ana, USA) according to the manufacturer’s instructions. This kit can crush and homogenize samples faster and more efficiently than traditional extraction methods, and without the need to grind the samples in advance.
We used Illumina sequencing of dual-indexed polymerase chain reaction (PCR) amplicons spanning the taxonomically informative V4 region of the 16S rRNA gene to analyze the microbial community composition. See Pratte et al. (2018) for details on the methods of primer selection, the PCR program, and 16S rDNA sequencing (Caporaso et al., 2011; Pratte et al., 2018). Moreover, indexed adapters were added to the ends of the amplicons by limited cycle PCR. The DNA concentration was detected using a microplate reader (Infinite 200 Pro, Tecan, Männedorf, Switzerland), and the fragment size (~600 bp) was determined via 1.5% agarose gel electrophoresis. Next-generation sequencing was conducted on an Illumina MiSeq platform (Illumina, San Diego, USA) at Genewiz, Inc. (South Plainfield, NJ, USA). Sequencing was performed using automated cluster generation with 250 paired-end reads.
Double-end sequencing of positive and negative reads was conducted, and the sequencing results were filtered to retain sequences with lengths greater than 200 bp. After filtering for quality and purifying the chimeric sequences, the resulting sequences were used for operational taxonomic unit (OTU) clustering in VSEARCH (version 1.9.6; sequence similarity was set to 97%) (Rognes et al., 2016) with Silva-138 (Quast et al., 2013) as the 16 s rRNA reference database. We then used the Ribosomal Database Program (RDP) classifier Bayesian algorithm for OTU species taxonomy analysis on representative sequences (Cole et al., 2014). Finally, the microbial community composition of each sample was determined according to different species classification level statistics. OTUs that could not be identified at the genus level were labeled as “u. family level names”.
Under the condition that the sample reached sufficient sequencing depth, each sample was randomly rarefied to a uniform depth of 57,264 sequences to avoid the bias caused by different sample data sizes (Weiss et al., 2017). After removing eukaryotes and chloroplasts, the OTUs with sequence numbers greater than 10 were defined as effective OTUs for further analysis.
Using SAS 9.4, a general linear model (GLM) and Duncan’s multiple range test were used to test for significant differences in substrate and EAM morphology, and regression and stepwise selection were used to identify variables that were important to variation in the EAM microbiota. Before the significance test and correlation analysis, the Shapiro-Wilk Test was used to test whether the data conforms to the Gaussian distribution, and the RANK process was used to transform the data that did not conform to the Gaussian distribution.
The Shannon and Chao1 alpha diversity indices and rarefaction curves graph were calculated to reflect the species richness and evenness of the samples. We used clustering heatmaps based on Euclidean Distance and Group Average clustering methods to show differences in relative abundance at the genus level among different samples. We compared beta diversity between samples using unweighted UNIFRAC distance matrices (Unifrac- PCoA) (Lozupone and Knight, 2005), However, due to insufficient sequencing samples, no significant difference analysis was performed in the microbial community. We also analyzed the relationship between habitat characteristics and microbiota through RDA based on Hellinger Distance matrices, to demonstrate the influence of different environmental factors on microbial adaptability.
To predict the function of the microbial communities across habitats, individual effective OTUs were assigned to three categories: autotrophs, heterotrophs, and potential pathogens based on a literature review (Which have been documented to cause disease in hermatypic corals, other marine animals, humans, and livestock) (Supplementary Table S2).
Graphpad Prism 8 and Tutools (https://www.cloudtutu.com) were used for correlation analysis and mapping, and Origin 2017 was used to draw heat cluster maps.
Microscopic examination showed that the EAM was mainly composed of filamentous red algae (mainly Polysiphonia spp.). The morphological characteristics of EAM significantly differed depending on the type of substrate. The coverage rate (F = 11.56, P < 0.0001), biomass (F = 5.21, P = 0.0093), and algal length (F = 3.67, P = 0.0339) of EAM significantly differed depending on the substrate material. The EAM on the porous dead coral reef substrata (BCu, BCl, MCu, MCl, FCl, FCu) had higher coverage (mean ± SD: 68.8 ± 26.7%), higher biomass (mean ± SD: 596.5 ± 632.4 g/m2), and longer algal length (mean ± SD: 1.921 ± 0.986 mm) compared to the EAM on the concrete substrate (CBu) (mean ± SD: coverage: 21.2 ± 21.7%; biomass: 38.8 ± 60.2 g/m2; algal length: 0.588 ± 0.351 mm). There were also significant differences in the complexity of the five substrates (F = 4.24, P = 0.0039). EAM on flat substrates (FCl, FCu) had higher coverage (mean ± SD: 97.8 ± 3.4%), higher biomass (mean ± SD: 1145.3 ± 707.0g/m2) and longer algal length (mean ± SD: 2.564 ± 0.365 mm) compared to the branching substrate of the same material (BCu, BCl) (mean ± SD: coverage: 37.8 ± 17.0%; biomass: 62.0 ± 68.6 g/m2; algal length: 1.375 ± 0.813 mm).
According to the three studied morphological parameters, the EAM was divided into three categories: short, medium, and long EAM. Moreover, the coverage rate (F = 36.67, P < 0.0001), biomass (F = 15.49, P < 0.0001), and algal length (F = 7.83, P = 0.0013) of EAM significantly differed among the three categories (Figure 2). Short EAM existed only on dense concrete blocks (CBu) and was characterized by a low coverage rate (mean ± SD: 21.3 ± 21.7%), low biomass (mean ± SD: 38.80 ± 60.18 g/m2), and short algal length (mean ± SD: 0.588 ± 0.351 mm). Medium EAM were widely distributed on a variety of substrates (GBu, BCu, BCl, MCu, MCl), and the EAM coverage rate ranged from 21.2 ± 21.7% (GBu, mean ± SD) to 69.3 ± 24.8% (BCl, mean ± SD) (Figure 2A), the EAM biomass ranged from 78.33 ± 79.13 g/m2 (BCu, mean ± SD) to 461.33 ± 351.40 g/m2 (MCl, mean ± SD) (Figure 2B), and the algal length ranged from 1.302 ± 0.917 mm (BCu, mean ± SD) to 1.866 ± 0.706 mm (GBu, mean ± SD) (Figure 2C). Long EAM existed only on dead flat coral reef substrates (FCu, FCl) and was characterized by the highest coverage (mean ± SD: 97.8 ± 3.4%), highest biomass (mean ± SD: 1145.3 ± 707.0g/m2), and the longest algal length (mean ± SD: 2.564 ± 0.365 mm).
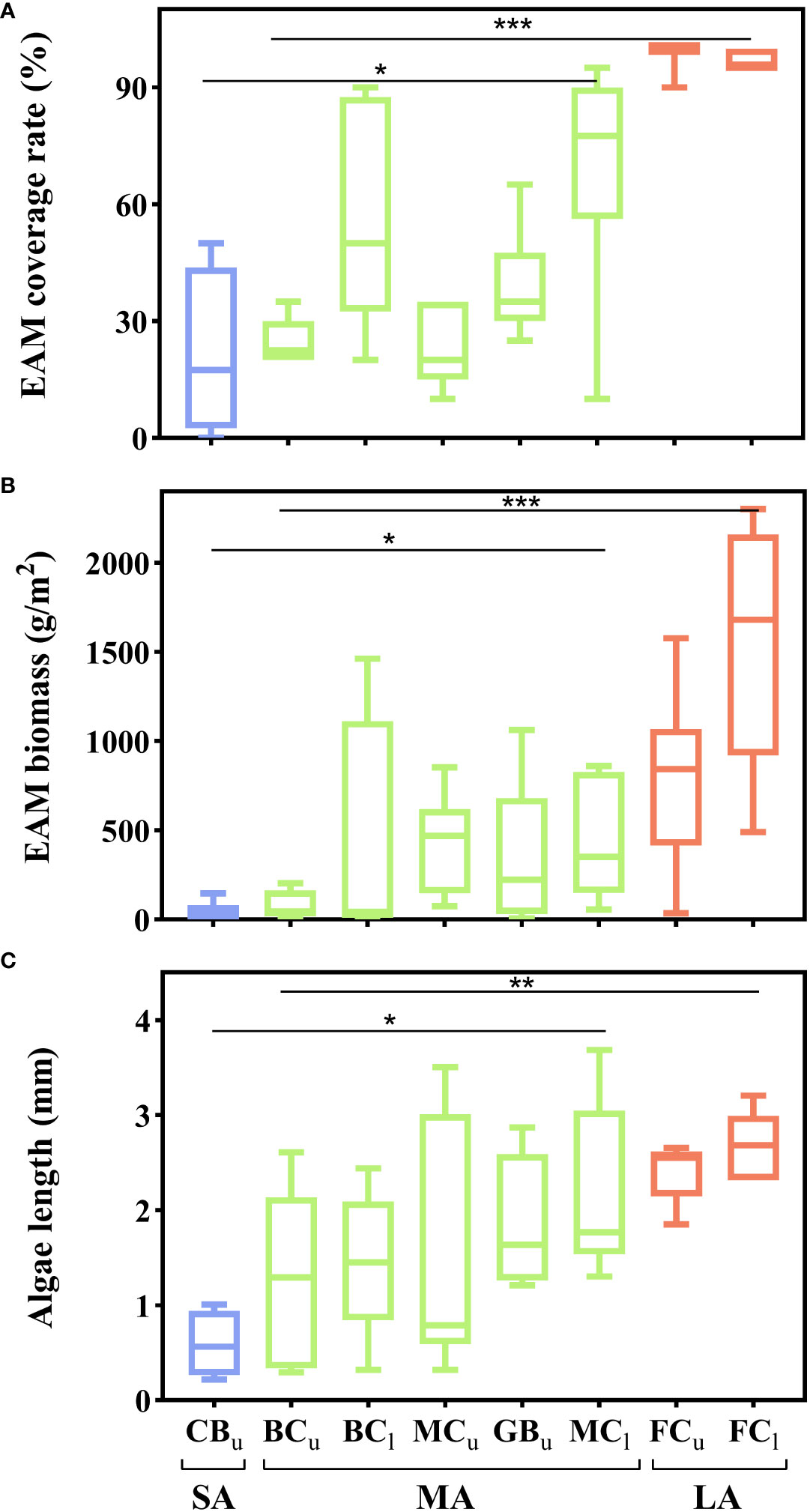
Figure 2 EAM morphological characteristics on different substrates. (A) EAM coverage rate; (B) EAM biomass; (C) algal length (The different substrates were divided into three categories: short EAM [SA], medium EAM [MA], and long EAM [LA]. Subscripts indicate different tidal levels, with u and l representing the upper and lower intertidal zones respectively. Asterisks indicate the level of significance, *: P < 0.05, **: P < 0.01, ***: P < 0.001).
A total of 458,112 high-quality sequences were retrieved from all the samples, which were divided into 3414 OTUs. The rarefaction curves based on Richness, Chao1 Index, and Shannon Index showed that the existing sequencing depths had reached the plateau (Supplementary Figure S1), and the effective sequence ratios of all samples were above 95% (Supplementary Table S3). A total of 57 bacterial phyla were detected, among which Proteobacteria was the most abundant group, accounting for more than 70% of reads for the microbial composition across all sites. Other groups like Cyanobacteria, Bacteroidetes, and Desulfobacterota were also abundant at some sites (Figure 3A). The number of OTUs in all bacterial phyla was consistent with the sequence abundance, and the phyla with a number of OTUs greater than 50 were Proteobacteria (418), Cyanobacteria (111), and Bacteroidetes (68) (Figure 3B).
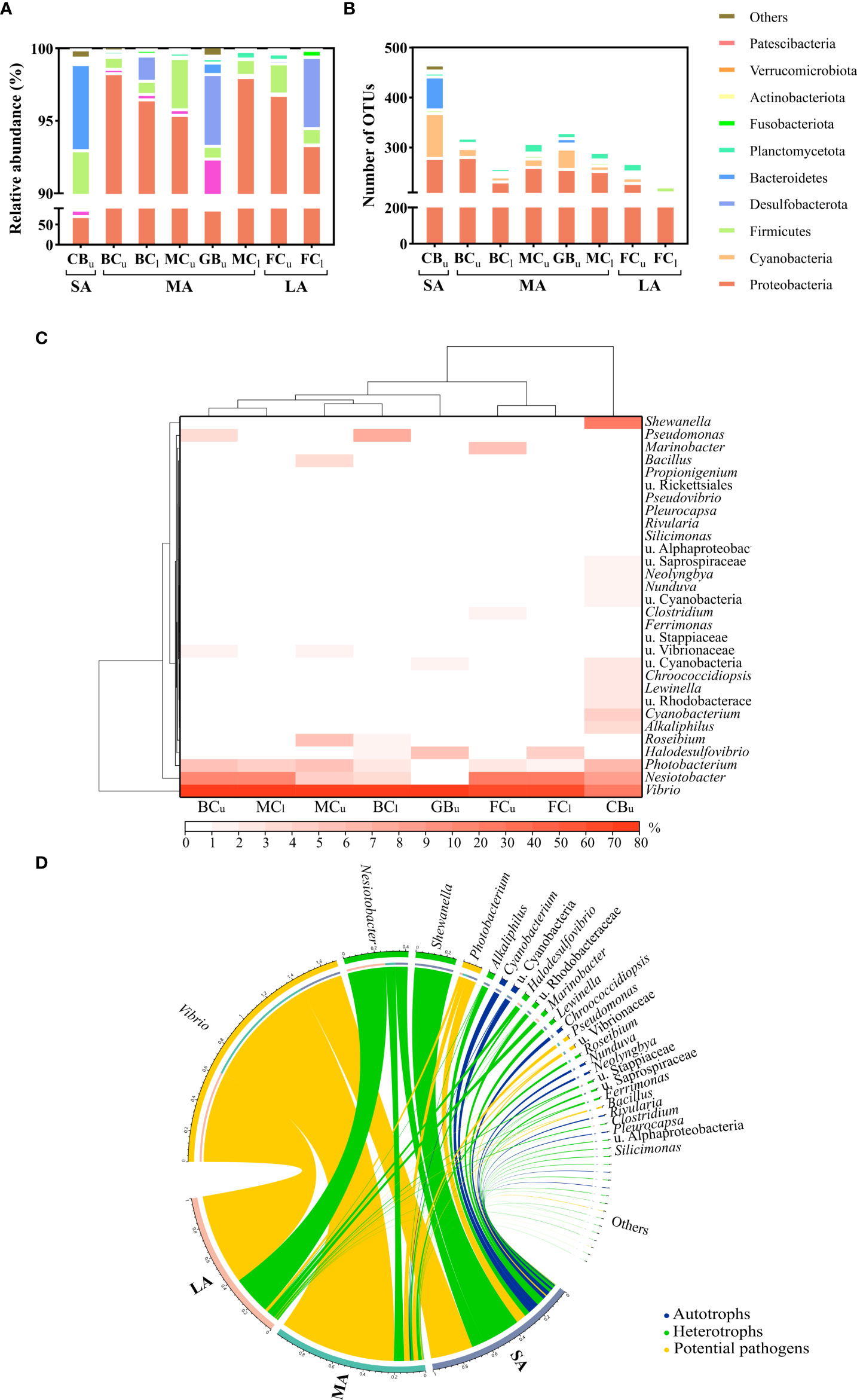
Figure 3 Composition of the microbiota of different EAM samples. (A) Relative abundance of the phyla; (B) number of OTUs per phylum; (C) cluster heat maps of the top 30 most abundant bacterial genera; (D) distribution of the microbiota functional categories (The different substrates were divided into three categories: short EAM [SA], medium EAM [MA], and long EAM [LA]. Subscripts indicate different tidal levels, with u and l representing the upper and lower intertidal zones respectively. u.: unclassified).
At the genus level, 130 bacterial genera were detected, of which the top 10 were the Proteobacteria Vibrio, Nesiotobacter, Photobacterium, Shewanella, Pseudomonas, Roseibium, u. Vibrionaceae, and Marinobacter; the Desulfobacterota Halodesulfovibrio; and the Firmicutes Alkaliphilus (Figure 3C; Specific differences in microbiota between groups can be seen in Supplementary Figure S2). Cluster analysis divided the microbial communities into three groups, which were consistent with the differences in their algal length. A Venn diagram analysis of the OTUs showed that there were 52 common OTUs shared across the three groups, occupying 78.52% of the sequences (Supplementary Figure S3). These shared microflora mainly belonged to the genera Vibrio, Nesiotobacter, Photobacterium, and Alkaliphilus, including V. diabolicus, N. exalbescens, and P. gaetbulicola. In at least one of the samples in each of the three groups, the relative abundance of these OTUs was greater than 1%. In addition, each group had numerous unique species. Specifically, the short EAM had 238 unique OTUs (48.61% of all sequences), mainly of the genera Shewanella and Cyanobacterium, most of which showed a high abundance of >1%. The medium EAM contained 130 unique OTUs (4.36% of all sequences), mainly including the genera Pseudomonas and Bacillus. However, only one of these OTUs was abundant (>1%). The long EAM only had 27 unique OTUs (4.13% of all sequences), mainly including the genus Marinobacter. Only OTU59 (M. hydrocarbonoclasticus, 99.32% similarity) had a high abundance (>1%).
To better understand the functional role of these microbial communities, individual OTUs were assigned as autotrophs, heterotrophs, and potential pathogens according to previous studies (Table S2). The autotrophs were mainly composed of the genera u. Cyanobacteria, Cyanobacterium, and Chroococcidiopsis, and they were most abundant in the short EAM (20.68% abundance). Heterotrophs were mainly composed of the genera Nesiotobacter and Shewanella and were most abundant in the short and long EAM (44.92% and 34.12%, respectively). Potential pathogens mainly included the genera Vibrio and Photobacterium and were most abundant in the medium and long EAM (85.36% and 63.11%, respectively) (Figure 3D).
Based on the UniFrac-PCoA analysis, the microbiota composition was distinguished among EAMs with different algal characteristics (Figure 4A). Meanwhile, OTU number, diversity, and evenness were significantly negatively correlated with algal length. The contributions of algal morphology and other factors (e.g., substrate complexity) to microbiota variation were illustrated using a modified variance partitioning diagram. The complete set of all variables together explained 59.43% of the variation in the EAM microbiota communities, with algal length being the single property that contributed the most (24.02%). Cyanobacteria, Bacteroidetes, and Firmicutes responded positively to the EAM morphological indices (coverage rate, biomass, and algal length), whereas Proteobacteria responded positively to substrate complexity (Figure 4B).
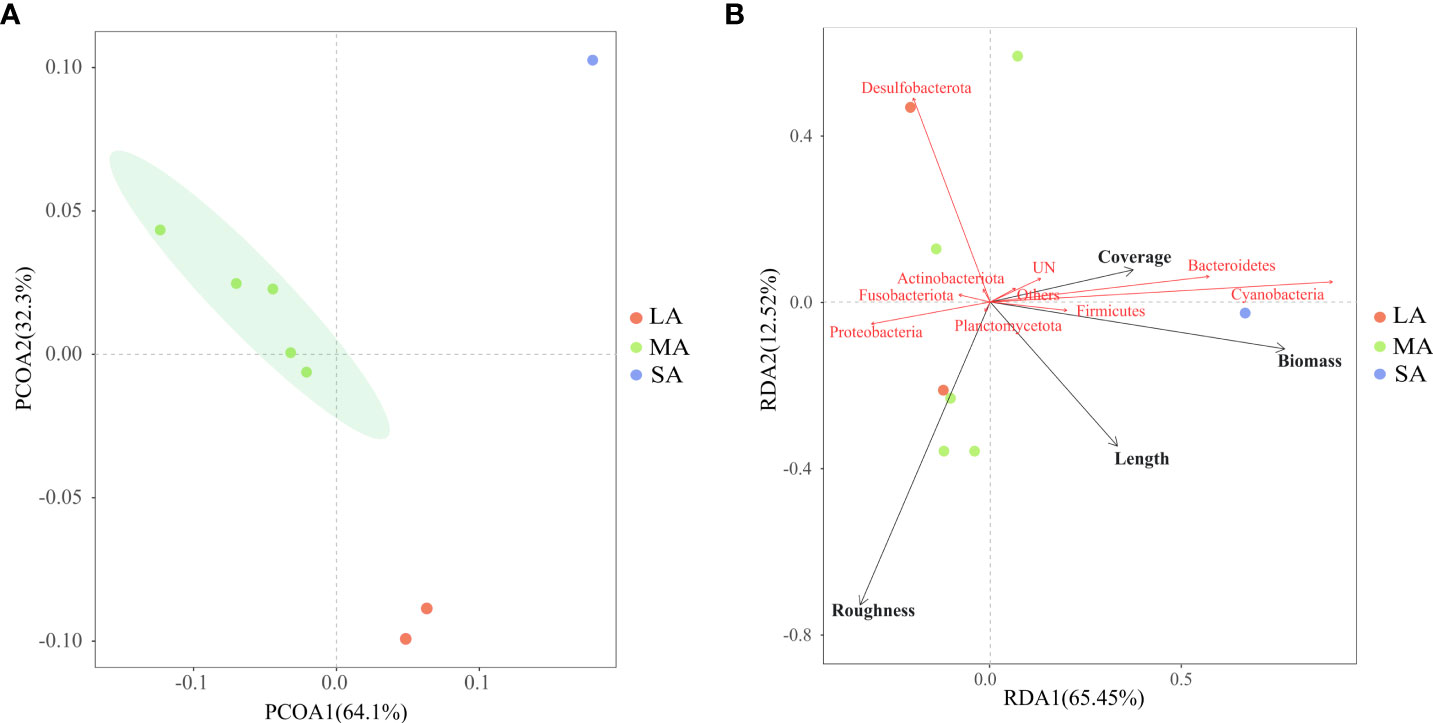
Figure 4 Beta diversity analysis of EAM microbiota. (A) UniFrac-PCoA analysis of EAM microbiota based on unweighted UNIFRAC distance; (B) redundancy analysis of EAM microbiota and potential influencing factors based on Hellinger distance matrices (The percentage on the coordinate axis represents the interpretation of the total variation of that axis. The different substrates were divided into three categories: short EAM [SA], medium EAM [MA], and long EAM [LA]).
In terms of microbial function, autotrophic groups were significantly negatively correlated with algal length (r = -0.9051, P = 0.0020), whereas potential pathogens were positively but insignificantly correlated with algal length (r = 0.2345, P = 0.5761) (Figure 5). The stepwise regression analysis results (Table 2) also showed that among the multiple candidate factors, potential pathogens were positively correlated with algal length and EAM coverage. Autotrophic bacteria were negatively correlated with EAM coverage and substrate complexity, and heterotrophic bacteria were negatively correlated with substrate complexity.
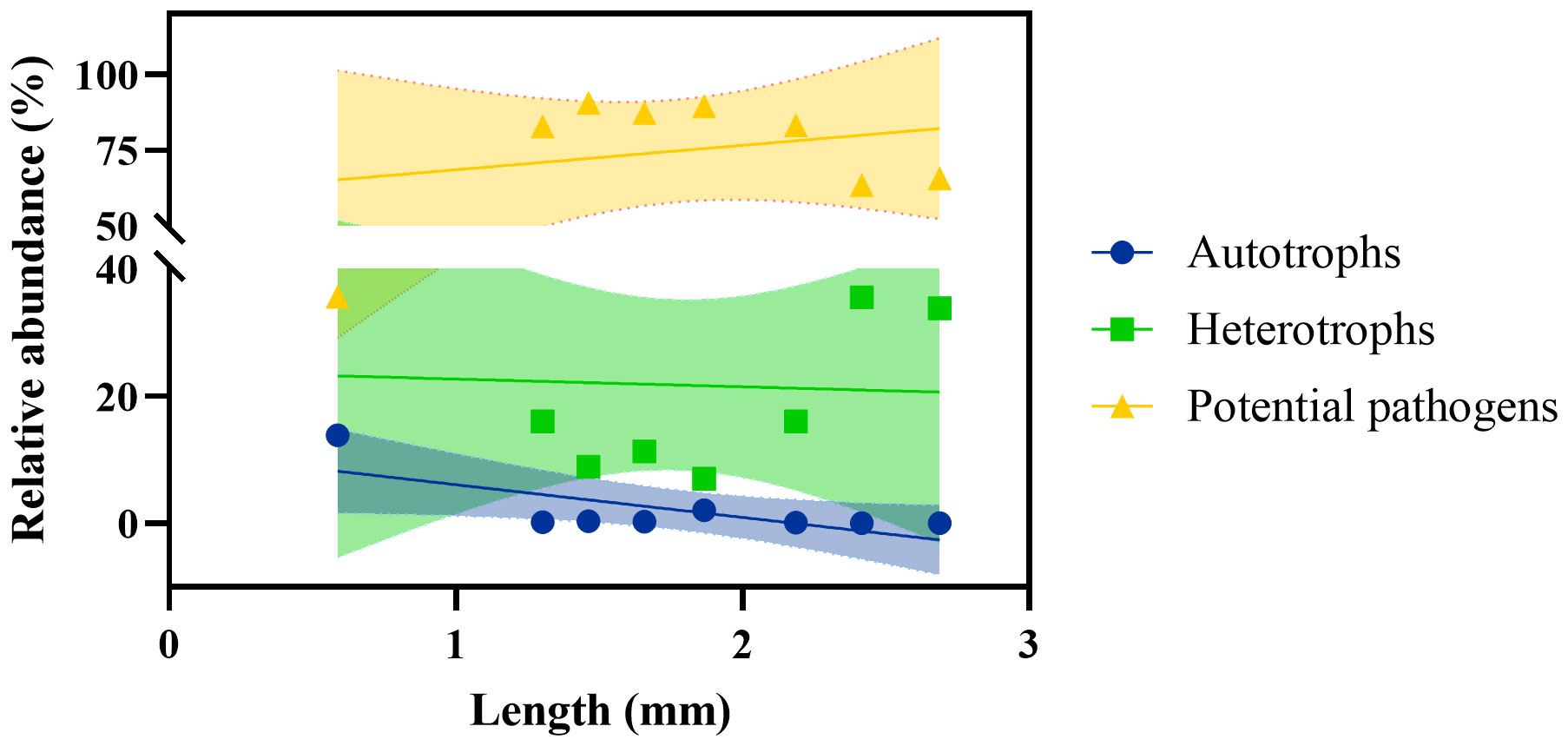
Figure 5 Correlation between EAM microbial function and algal length. Dashed lines indicate 95% confidence intervals.
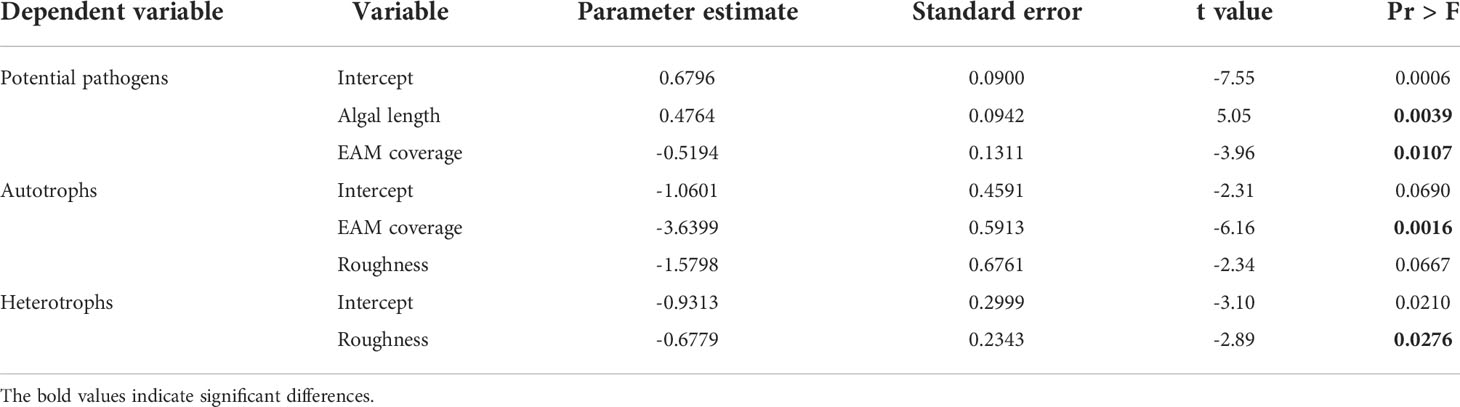
Table 2 Summary of stepwise regression analysis parameter estimation results.
This study found that EAMs with different morphologies had different microbial compositions, and algal length was the most important morphological factor impacting the differences in EAM microbiota composition and function. Compared with short EAM, the diversity and evenness of the bacterial community of medium and long EAM decreased, and the bacterial community showed a trend from Cyanobacteria, Bacteroidetes, and Patescibacteria to Proteobacteria. Simultaneously, the abundance of autotrophic bacteria of medium and long EAM also decreased, and the number of potential pathogenic bacteria increased. Considering the negative correlation between substrate complexity and algal length, future simplifications of substrate habitats may facilitate the expansion of EAM and increase susceptibility to coral diseases.
Proteobacteria, Bacteroidetes and Cyanobacteria were abundant phyla in our samples, with Proteobacteria occupying a dominant position in all samples (73–98% relative abundance). This was consistent with another study conducted on Ishigaki Island, which has the same level of eutrophication and similar coverage of EAM in its reefs as our study area, showing that Proteobacteria were the most abundant at all sites (at least 48.53%), followed by Cyanobacteria (at least 7.12%) and Bacteroidetes (at least 6.05%) (Meirelles et al., 2018). Proteobacteria was also reported to be the most regnant bacterial phylum associated with seaweeds (34–65% relative abundance) (Hollants et al., 2013; Egan et al., 2013; Meirelles et al., 2018). This suggests that EAMs have homogeneous microbial compositions across space, at least at the phylum level (Walter et al., 2016). Cyanobacteria are important early pioneer species of dead coral reef substrata. Some filamentous cyanobacteria also form the framework of some EAM, with Oscillatoriales and Nostocales being dominant (Stal, 1995; Connell et al., 2014; Echenique-Subiabre et al., 2015). In this study, a high abundance (15.89%) of cyanobacterial sequences was detected only in the EAM with the shortest algal length. Although cyanobacterial sequences were detected in other types of EAM, their relative abundance was lower than 0.5%, indicating that cyanobacteria may be abundant only in early immature EAM. Thus, their importance may decline as the EAM matures. Most of the cyanobacterial sequences identified in this study were not matched, which is similar to the results of other studies; for example, approximately a third of OTUs in the La Réunion lagoon could not be matched to specific cyanobacteria species (Echenique-Subiabre et al., 2015). Therefore, the high abundance of Cyanobacteria suggests that undefined cyanobacterial OTUs may be an indispensable functional group in the functioning of early immature EAM. Firmicutes and Bacteroidetes are also dominant groups in some EAM, and the Bacteroidetes-to-Firmicutes ratio can predict the outcome of coral-algal competition (Roach et al., 2020). Firmicutes were dominant in EAM, whereas Bacteroidetes were dominant at the interface between algae and coral (Roach et al., 2020). The present study also found that the relative abundance of Firmicutes in different types of EAM was higher than that of Bacteroidetes, except for the EAM with the shortest algal length. This result indicates a decrease in the abundance and diversity of Bacteroidetes and, by contrast, an increase in the abundance and diversity of Firmicutes during the EAM algal growth process.
The effects of EAM are generally detrimental to coral, and the microbiota associated with EAM is an important contributor to this process. Potential coral disease pathogens have been found in the genera Geitlerinema, Leptolyngbya, Oscillatoria, Sphingomonas, Bartonella, Cardiobacterium, and Inquilinus, with the first three thought to be linked to black band disease (Myers et al., 2007). In contrast to previous studies, the potential pathogens found in this study belonged to Vibrio, Photobacterium, Pseudomonas, u. Vibrionaceae, Bacillus, u. Rickettsiales, and u. Rhizobiales, which are mainly related to the diseases and zoonoses of aquaculture animals (Rosenberg et al., 2007; Austin, 2010). Vibrio was the main genus of Proteobacteria and also the dominant genus in all samples in this study, which is consistent with previous studies showing that Vibrio strains are generally highly abundant in red algae (Hollants et al., 2013), especially in Polysiphonia urceolata (Wang et al., 2009), one of the major algal constituents of EAM. Vibrio has been associated with diseases in aquatic animals (Austin, 2010), but thus far, only V. shiloi has been linked to coral diseases (Rosenberg et al., 2007). The most abundant species in this study was V. diabolicus (99.66% similarity), which was found both in deep sea and aquaculture areas. V. diabolicus has been found in a variety of economically important shellfish (e.g., Ruditapes philippinarum, Tegillarca granosa, Mytilus coruscus, Callista chinensis) in Korea and China (Song et al., 2021), and it is evolutionarily similar to a number of animal disease pathogens, such as V. parahaemolyticus and V. alginolyticus (Turner et al., 2018). The high levels of Vibrio in the study area may be linked to the once-thriving aquaculture industry in Luhuitou, as previous research has found that wastewater from grouper farms was responsible for algal blooms (Li et al., 2016). It is speculated that Vibrio bacteria in aquacultural waste may also be captured and accumulated by neighboring EAM. Even though the farm has long been dismantled, the collective action of EAM can nevertheless lead to the long-term presence of pathogens in the neighboring ecosystem.
Changes in the EAM microbiota was significantly correlated with morphological characteristics of EAM, such as algal length and biomass. Algal length is an important factor for classifying the type and function of the EAM (Connell et al., 2014; Tebbett and Bellwood, 2019; Tebbett and Bellwood, 2020; Tebbett and Bellwood, 2021). For example, algal length is highly correlated with the sediment capture capacity of EAM (Tebbett and Bellwood, 2019). The captured detritus also provides a rich source of organic matter for heterotrophic bacteria. In this study, EAMs with a short length (< 1 mm) had the most abundant diversity and evenness of bacteria, with the core bacteria being Shewanella, Vibrio, Nesiotobacter, Photobacterium, and Cyanobacterium. More than 50% of the OTUs in this type of EAM were unique. Species in the genus Shewanella possess broad organic and inorganic substrate metabolism (e.g., of nitrate, nitrite, and iron [III]), which may have helped the EAM to occupy bare and barren substrates during early evolution (Beliaev and Saffarini, 1998; Miller et al., 2018; Tebbett and Bellwood, 2019). Furthermore, autotrophic bacteria were in the greatest proportion, up to 13.83%, allowing for the efficient use of light sources. Specifically, cyanobacteria utilize phycobilisomes to capture photons between the blue and red regions of the light spectrum that are not efficiently captured by chlorophyll (Steunou et al., 2006). For EAMs of medium length (1–2mm), the most abundant group consisted of potential pathogens, reaching as high as 86.6%, with the core bacteria being Vibrio, Nesiotobacter, Photobacterium, and Pseudomonas. Bacterial species of Nesiotobacter have been reported to have nitrogenase activity and are capable of degrading aromatic hydrocarbons (Dashti et al., 2015). Species in the genus Photobacterium are widely distributed in the marine environment, occurring in seawater, marine sediments, marine surfaces, and the intestines of marine animals (Urbanczyk et al., 2011). Furthermore, these species have unique organic decomposition abilities, which may be beneficial for detritus decomposition and organic matter release in EAM. P. mendocina can produce cellulase, which is beneficial for nematode feeding activities and is conducive to the circulation of materials throughout the ecosystem (Dai et al., 2019). B. horikoshii has been isolated from marine environments, and it possesses genes associated with the metabolism of cyanophycin, a reserve compound and spore matrix material that is potentially relevant for survival in oligotrophic environments (Zarza et al., 2017).
EAMs with a long length (>2 mm) exhibited the lowest diversity and evenness and contained the lowest number of OTUs. The core bacteria were Vibrio and Nesiotobacter. In addition, the proportion of Nesiotobacter increased from less than 10% to more than 25%, which may be related to the anaerobic growth capacity of Nesiotobacter and its strong adaptability to harsh environments (Donachie et al., 2006). The unique species (e.g., M. hydrocarbonoclasticus, B. kexueae, and V. Toranzoniae) may play a role in the EAM nitrogen cycle (Li et al., 2013). Long filamentous algae interact with high sediment loads to form long sediment-laden algal turfs; this might be the reason for the low microbiota diversity in EAMs with long algal length in the Luhuitou fringing reef (Tebbett and Bellwood, 2019). These sediment-dominated components are mainly deposited and accumulated in the EAM owing to the complex algal turf structure, which can slow down water movement (Latrille et al., 2019). Preliminary results have highlighted that increased algae length and sediment load in the EAM can influence the microbes within algal turfs and ultimately compromise reef health (Bourne et al., 2016; Meirelles et al., 2018). The mechanisms of this phenomenon have not yet been defined, but they are likely to include (1) the release of dissolved organic matter by algae, stimulating microbial reproduction (Barott and Rohwer, 2012; Smith et al., 2016); (2) sediment enrichment, which can lead to changes in the availability of dissolved gases, light, and nutrients within the EAM (Chapman and Fletcher, 2002); and (3) differences in tolerance among different strains (Bourne et al., 2016).
Increasing evidence suggests that with continued global change and anthropogenic influences, coral reefs will emerge as low-complexity systems dominated by EAM, especially in coastal ecosystems (Smith et al., 2016; Bellwood et al., 2018). One example is the Luhuitou fringing reef, which was once regarded as one of the best-developed fringing reefs on Hainan Island but then experienced an extreme degradation of live coral coverage from 80–90% in the 1960s to approximately 12% in 2009 (Zhao et al., 2012). This decline may be mainly due to human activities, such as mariculture and coastal construction, which lead to eutrophication and increased sediment input (Zhao et al., 2012). However, EAMs are highly stress-tolerant, and future climate change conditions actually appear to be conducive to them (Johnson et al., 2017). Currently, the live coral coverage of Luhuitou is approximately 20% (Huang et al., 2019), whereas the average coverage rate of EAM is almost 56.17% and may further increase due to future intense disturbance.
EAMs exist in a spectrum of forms from short to long EAMs, and there are significant differences in terms of algae length, biomass, sediment load, and productivity (Tebbett and Bellwood, 2020). The effects of EAM on coral may depend on the presence and nature of the EAM occurring on the substratum (Birrell et al., 2005), and EAMs with different compositions may potentially lead to very different outcomes. The productivity of EAM has been found to be over twice as high on flattened coral blocks/tiles (0.44 ± 0.02 mm/day) relative to highly complex natural substrata (0.19 ± 0.01 mm/day) (Tebbett and Bellwood, 2021). In the present study, we found that the low-complexity substrates of dead flat coral have the longest length, largest biomass, and highest EAM coverage. Considering the negative correlation between substrate complexity and algal length, the role of the EAM microbiota is likely to be further increased through the loss of topographic complexity and the expansion of EAM in Anthropocene coral reefs (Alvarez-Filip et al., 2009). Because microbes are key players in maintaining coral health, it is imperative to consider EAM microbiota communities when interpreting the mechanisms of coral reef degradation. It should be noted that this study only inferred functional differences in the EAM based on microbial composition information; thus, further controlled trial work is needed to test hypotheses about how diverse types of EAM affect corals.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, The DNA sequencing data can be found in the National Center for Biotechnology Information (NCBI) under the BioProject accession number PRJNA846109.
Conceived and designed the experiments: TZ, SH, and SL. Field survey and sampling: TZ, NJ, and CZ. Analyzed the data: TZ. Contributed reagents/materials/analysis tools: SL and HH. Wrote the paper: TZ and SH. All authors contributed to the article and approved the submitted version.
This research was funded by the Natural Science Foundation of China [contract Nos. 42176118 and 41806188]; the Science and Technology Planning Project of Guangdong Province [contract No. 2020B1212060058]; the Hainan Provincial Natural Science Foundation of China [contract No. 422QN442]; and the Science and Technology Program of Guangzhou, China [contract No. 202201010674].
The authors thank the staff of the Tropical Marine Biological Research Station in Hainan for their logistical support. Dr. Xianzhi Lin is greatly acknowledged for his help in the drawing of figures and the organization of the manuscript. We would also like to thank Dr. Yong Luo for assistance with the calculation of environmental parameters and Editage (www.editage.cn) for English language editing. We also thank two anonymous reviewers for helpful comments that improved the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.993305/full#supplementary-material
Alvarez-Filip L., Dulvy N. K., Cote I. M., Watkinson A. R., Gill J. A. (2011). Coral identity underpins architectural complexity on Caribbean reefs. Ecol. Appl. 21, 2223–2231. doi: 10.1890/10-1563.1
Alvarez-Filip L., Dulvy N. K., Gill J. A., Cote I. M., Watkinson A. R. (2009). Flattening of Caribbean coral reefs: region-wide declines in architectural complexity. Proc. R. Soc B.-Biol. Sci. 276, 3019–3025. doi: 10.1098/rspb.2009.0339
Austin B. (2010). Vibrios as causal agents of zoonoses. Vet. Microbiol. 140, 310–317. doi: 10.1016/j.vetmic.2009.03.015
Barott K. L., Rodriguez-Brito B., Janouskovec J., Marhaver K. L., Smith J. E., Keeling P., et al. (2011). Microbial diversity associated with four functional groups of benthic reef algae and the reef-building coral montastraea annularis. Environ. Microbiol. 13, 1192–1204. doi: 10.1111/j.1462-2920.2010.02419.x
Barott K. L., Rohwer F. L. (2012). Unseen players shape benthic competition on coral reefs. Trends Microbiol. 20, 621–628. doi: 10.1016/j.tim.2012.08.004
Beliaev A. S., Saffarini D. A. (1998). Shewanella putrefaciens mtrB encodes an outer membrane protein required for Fe(III) and Mn(IV) reduction. J. Bacteriol. 180, 6292–6297. doi: 10.1128/jb.180.23.6292-6297.1998
Bellwood D. R., Tebbett S. B., Bellwood O., Mihalitsis M., Morais R. A., Streit R. P., et al. (2018). The role of the reef flat in coral reef trophodynamics: Past, present, and future. Ecol. Evol. 8, 4108–4119. doi: 10.1002/ece3.3967
Birrell C. L., McCook L. J., Willis B. L. (2005). Effects of algal turfs and sediment on coral settlement. Mar. pollut. Bull. 51, 408–414. doi: 10.1016/j.marpolbul.2004.10.022
Bourne D. G., Morrow K. M., Webster N. S. (2016). Insights into the coral microbiome: Underpinning the health and resilience of reef ecosystems. Annu. Rev. Microbiol. 70, 317–340. doi: 10.1146/annurev-micro-102215-095440
Caporaso J. G., Lauber C. L., Walters W. A., Berg-Lyons D., Lozupone C. A., Turnbaugh P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. PNAS 108, 4516–4522. doi: 10.1073/pnas.1000080107
Chapman A. S., Fletcher R. L. (2002). Differential effects of sediments on survival and growth of fucus serratus embryos (Fucales, phaeophyceae). J. Phycol. 38, 894–903. doi: 10.1046/j.1529-8817.2002.t01-1-02025.x
Cole J. R., Wang Q., Fish J. A., Chai B., Mcgarrell D. M., Sun Y., et al. (2014). Ribosomal database project: data and tools for high throughput rRNA analysis. Nucl. Acids Res. 42, 633–642. doi: 10.1093/nar/gkt1244
Connell S. D., Foster M. S., Airoldi L. (2014). What are algal turfs? towards a better description of turfs. Mar. Ecol. Prog. Ser. 495, 299–307. doi: 10.3354/meps10513
Dai Y., Qiu Y., Jin J., Jia Q., Sarsaiya S., Wang Z., et al. (2019). Improving the properties of straw biomass rattan by corn starch. Bioengineered 10, 659–667. doi: 10.1080/21655979.2019.1688127
Dashti N., Ali N., Eliyas M., Khanafer M., Sorkhoh N. A., Radwan S. S. (2015). Most hydrocarbonoclastic bacteria in the total environment are diazotrophic, which highlights their value in the bioremediation of hydrocarbon contaminants. Microbes Environ. 30, 70–75. doi: 10.1264/jsme2.ME14090
Donachie S. P., Bowman J. P., Alam M. (2006). Nesiotobacter exalbescens gen. nov., sp nov., a moderately thermophilic alphaproteobacterium from an Hawaiian hypersaline lake. Int. J. Syst. Evol. Microbiol. 56, 563–567. doi: 10.1099/ijs.0.63440-0
Echenique-Subiabre I., Villeneuve A., Golubic S., Turquet J., Humbert J.-F., Gugger M. (2015). Influence of local and global environmental parameters on the composition of cyanobacterial mats in a tropical lagoon. Microb. Ecol. 69, 234–244. doi: 10.1007/s00248-014-0496-0
Egan S., Harder T., Burke C., Steinberg P., Kjelleberg S., Thomas T. (2013). The seaweed holobiont: understanding seaweed-bacteria interactions. FEMS Microbiol. Rev. 37, 462–476. doi: 10.1111/1574-6976.12011
Hester E. R., Barott K. L., Nulton J., Vermeij M. J. A., Rohwer F. L. (2016). Stable and sporadic symbiotic communities of coral and algal holobionts. ISME J. 10, 1157–1169. doi: 10.1038/ismej.2015.190
Hollants J., Leliaert F., De Clerck O., Willems A. (2013). What we can learn from sushi: a review on seaweed-bacterial associations. FEMS Microbiol. Ecol. 83, 1–16. doi: 10.1111/j.1574-6941.2012.01446.x
Huang R., Yu K., Huang X., Zou W., Wang Y. (2019). Combining landsat images with historic records to estimate the live coral cover of luhuitou fringing reef in northern south China Sea. Image Vis. Comput. 92, 103812. doi: 10.1016/j.imavis.2019.09.003
Johnson M. D., Comeau S., Lantz C. A., Smith J. E. (2017). Complex and interactive effects of ocean acidification and temperature on epilithic and endolithic coral-reef turf algal assemblages. Coral Reefs 36, 1059–1070. doi: 10.1007/s00338-017-1597-2
Klumpp D. W., McKinnon A. D. (1989). Temporal and spatial patterns in primary production of a coral-reef epilithic algal community. J. Exp. Mar. Biol. Ecol. 131, 1–22. doi: 10.1016/0022-0981(89)90008-7
Kohler K. E., Gill S. M. (2006). Coral point count with excel extensions (CPCe): A visual basic program for the determination of coral and substrate coverage using random point count methodology. Comput. Geosci. 32, 1259–1269. doi: 10.1016/j.cageo.2005.11.009
Latrille F. X., Tebbett S. B., Bellwood D. R. (2019). Quantifying sediment dynamics on an inshore coral reef: Putting algal turfs in perspective. Mar. pollut. Bull. 141, 404–415. doi: 10.1016/j.marpolbul.2019.02.071
Li X., Titlyanov E. A., Zhang J., Titlyanova T. V., Zhang G., Huang H. (2016). Macroalgal assemblage changes on coral reefs along a natural gradient from fish farms in southern hainan island. Aquat. Ecosyst. Health Manage. 19, 74–82. doi: 10.1080/14634988.2016.1140952
Li R., Zi X., Wang X., Zhang X., Gao H., Hu N. (2013). Marinobacter hydrocarbonoclasticus NY-4, a novel denitrifying, moderately halophilic marine bacterium. Springerplus 2, 346. doi: 10.1186/2193-1801-2-346
Lozupone C., Knight R. (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005
Marsh (1970). Primary productivity of reef-building calcareous red algae. Ecology 51, 255–263. doi: 10.2307/1933661
Meirelles P. M., Soares A. C., Oliveira L., Leomil L., Appolinario L. R., Francini-Filho R. B., et al. (2018). Metagenomics of coral reefs under phase shift and high hydrodynamics. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.02203
Miller R. B. II, Lawson K., Sadek A., Monty C. N., Senko J. M. (2018). Uniform and pitting corrosion of carbon steel by shewanella oneidensis MR-1 under nitrate-reducing conditions. Appl. Environ. Microbiol. 84, 2203. doi: 10.1128/aem.00790-18
Myers J. L., Sekar R., Richardson L. L. (2007). Molecular detection and ecological significance of the cyanobacterial genera geitlerinema and leptolyngbya in black band disease of corals. Appl. Environ. Microbiol. 73, 5173–5182. doi: 10.1128/aem.00900-07
Pratte Z.A., Longo G.O., Burns A.S., Hay and Stewart M. E.F.J. (2018). Contact with turf algae alters the coral microbiome: Contact versus systemic impacts. Coral Reefs 37, 113. doi: 10.1007/s00338-017-1615-4
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Roach T. N. F., Little M., Arts M. G. I., Huckeba J., Haas A. F., George E. E., et al. (2020). A multiomic analysis of in situ coral-turf algal interactions. PNAS 117, 13588–13595. doi: 10.1073/pnas.1915455117
Rognes T., Flouri T., Nichols B., Quince C., Mahé F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584. doi: 10.7717/peerj.2584
Rosenberg E., Koren O., Reshef L., Efrony R., Zilber-Rosenberg I. (2007). The role of microorganisms in coral health, disease and evolution. nat. rev. Microbiol. 5, 355–362. doi: 10.1038/nrmicro1635
Smith J. E., Brainard R., Carter A., Grillo S., Edwards C., Harris J., et al. (2016). Re-evaluating the health of coral reef communities: baselines and evidence for human impacts across the central pacific. Proc. R. Soc B-Biol. Sci. 283, 20151985. doi: 10.1098/rspb.2015.1985
Song J., Liu X., Wu C., Zhang Y., Fan K., Zhang X., et al. (2021). Isolation, identification and pathogenesis study of vibrio diabolicus. Aquaculture 533, 736043. doi: 10.1016/j.aquaculture.2020.736043
Stal L. J. (1995). Physiological ecology of cyanobacteria in microbial mats and other communities. New Phytol. 131, 1–32. doi: 10.1111/j.1469-8137.1995.tb03051.x
Steunou A. S., Bhaya D., Bateson M. M., Melendrez M. C., Ward D. M., Brecht E., et al. (2006). In situ analysis of nitrogen fixation and metabolic switching in unicellular thermophilic cyanobacteria inhabiting hot spring microbial mats. PNAS 103, 2398–2403. doi: 10.1073/pnas.0507513103
Tebbett S. B., Bellwood D. R. (2019). Algal turf sediments on coral reefs: what's known and what's next. Mar. pollut. Bull. 149, 110542. doi: 10.1016/j.marpolbul.2019.110542
Tebbett S. B., Bellwood D. R. (2020). Sediments ratchet-down coral reef algal turf productivity. Sci. Total Environ. 713, 136709. doi: 10.1016/j.scitotenv.2020.136709
Tebbett S. B., Bellwood D. R. (2021). Algal turf productivity on coral reefs: A meta-analysis. Mar. Environ. Res. 168, 105311. doi: 10.1016/j.marenvres.2021.105311
Titlyanov E. A., Titlyanova T. V., Huang H., Scriptsova A. V., Xu H., Li X. (2019). Seasonal changes in the intertidal and subtidal algal communities of extremely and moderately polluted coastal regions of sanya bay (Hainan island, China). J. Mar. Sci. Eng. 7, 93. doi: 10.3390/jmse7040093
Titlyanov E. A., Titlyanova T. V., Li X., Huang H. (2018). An inventory of marine benthic macroalgae of hainan island, China. Russ. J. Mar. Biol. 44, 175–184. doi: 10.1134/s1063074018030112
Turner J. W., Tallman J. J., Macias A., Pinnell L. J., Elledge N. C., Azadani D. N., et al. (2018). Comparative genomic analysis of vibrio diabolicus and six taxonomic synonyms: A first look at the distribution and diversity of the expanded species. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.01893
Urbanczyk H., Ast J. C., Dunlap P. V. (2011). Phylogeny, genomics, and symbiosis of photobacterium. FEMS Microbiol. Rev. 35, 324–342. doi: 10.1111/j.1574-6976.2010.00250.x
Walter J. M., Tschoeke D. A., Meirelles P. M., De Oliveira L., Leomil L., Tenorio M., et al. (2016). Taxonomic and functional metagenomic signature of turfs in the abrolhos reef system (Brazil). PloS One 11, e0161168. doi: 10.1371/journal.pone.0161168
Wang Z., Xiao T., Pang S., Liu M., Yue H. (2009). Isolation and identification of bacteria associated with the surfaces of several algal species. Chin. J. Oceanol. Limnol. 27, 487–492. doi: 10.1007/s00343-009-9165-4
Weiss S., Xu Z., Peddada S., Amir A., Bittinger K., Gonzalez A., et al. (2017). Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 5, 27. doi: 10.1186/s40168-017-0237-y
Wilson S., Bellwood D. R. (1997). Cryptic dietary components of territorial damselfishes (Pomacentridae, labroidei). Mar. Ecol. Prog. Ser. 153, 299–310. doi: 10.3354/meps153299
Yu K.-F., Zhao J.-X., Lawrence M. G., Feng Y. (2010). Timing and duration of growth hiatuses in mid Holocene massive porites corals from the northern south China Sea. J. Quat. Sci. 25, 1284–1292. doi: 10.1002/jqs.1410
Zarza E., Alcaraz L. D., Aguilar-Salinas B., Islas A., Olmedo-Alvarez G. (2017). Complete genome sequence of bacillus horikoshii strain 20a from cuatro cienegas, coahuila, Mexico. Microbiol. Resour. Announc. 5, e00592-17. doi: 10.1128/genomeA.00592-17
Keywords: microbiota, community structure, epilithic algal matrix, coral reef, morphological difference
Citation: Zhou T, Hu S, Jia N, Zhang C, Huang H and Liu S (2022) Microbial communities associated with epilithic algal matrix with different morphological characters in Luhuitou fringing reef. Front. Mar. Sci. 9:993305. doi: 10.3389/fmars.2022.993305
Received: 13 July 2022; Accepted: 20 September 2022;
Published: 03 October 2022.
Edited by:
Cinzia Corinaldesi, Marche Polytechnic University, ItalyReviewed by:
Giorgia Palladino, University of Bologna, ItalyCopyright © 2022 Zhou, Hu, Jia, Zhang, Huang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simin Hu, aHVzaW1pbkBzY3Npby5hYy5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.