 Yifei Yu1†
Yifei Yu1† Wenyan Nong1†
Wenyan Nong1† Wai Lok So1
Wai Lok So1 Yichun Xie1
Yichun Xie1 Ho Yin Yip1Jasmine Haimovitz2Thomas Swale2
Ho Yin Yip1Jasmine Haimovitz2Thomas Swale2 David M. Baker3
David M. Baker3 William G. Bendena4
William G. Bendena4 Ting Fung Chan5
Ting Fung Chan5 Apple P. Y. Chui5
Apple P. Y. Chui5 Kwok Fai Lau5
Kwok Fai Lau5 Pei-Yuan Qian6,7
Pei-Yuan Qian6,7 Jian-Wen Qiu6,8
Jian-Wen Qiu6,8 Benoit Thibodeau5
Benoit Thibodeau5 Fei Xu9
Fei Xu9 Jerome H. L. Hui1*
Jerome H. L. Hui1*- 1School of Life Sciences, Simon F.S. Li Marine Science Laboratory, State Key Laboratory of Agrobiotechnology, The Chinese University of Hong Kong, Hong Kong, Hong Kong SAR, China
- 2Dovetail Genomics, California, CA, United States
- 3School of Biological Science, The University of Hong Kong, Hong Kong, Hong Kong SAR, China
- 4Department of Biology, Queen’s University, Kingston, ON, Canada
- 5School of Life Sciences, The Chinese University of Hong Kong, Hong Kong, Hong Kong SAR, China
- 6Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou), Guangzhou, China
- 7Department of Ocean Science and Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou), The Hong Kong University of Science and Technology, Hong Kong, Hong Kong SAR, China
- 8Department of Biology, Hong Kong Baptist University, Hong Kong, Hong Kong SAR, China
- 9CAS and Shandong Province Key Laboratory of Experimental Marine Biology, Center for Ocean Mega-Science, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China
Coral reefs are under stress throughout the world. To better understand the molecular mechanisms underlying coral biology and their genomic evolution, here we sequenced the genome and transcriptomes of elegance coral Catalaphyllia jardinei (Euphylliidae). This monotypic genus stony coral is widespread but rare, being found across the Indo-West Pacific, from the northern Indian Ocean, Australia, Philippines, to the South China Sea. Due to its popularity among aquarium hobbyists, it is an overexploited species collected in large quantities from the wild for aquarium trade. The assembled genome is ~ 651.3 Mb in total length and of high physical contiguity with a scaffold N50 size of 28.9 Mb. The gene copy numbers of abiotic stress regulator (heat shock protein family genes) and neuropeptides (GLWamide, GRFamide, PRGamide and HIRamide) are similar to other sequenced anthozoans, and we have also identified the first set of sesquiterpenoid biosynthetic pathway genes in coral. Sequencing of small RNAs allows us to identify 35 microRNAs in C. jardinei and update the number of conserved microRNAs in cnidarians. This study established a foundation for further investigation into the roles of sesquiterpenoids and microRNAs in development of coral and understand their responses to climate change. Due to the easiness to culture C. jardinei in reef tanks and the established resources in this study, we propose this species be adopted as a new laboratory model in environmental and ecological experiments aiming to understand coral biology and responses to environmental stressors.
Introduction
The elegance coral Catalaphyllia jardinei was first described as Pectinia jardinei found in the shallow water on the reefs at the Warrior Reef (latitude 9° 45’ S), Thursday Island (latitude 10° 35’ S), and Albany Pass (latitude 10° 45’ S) (Saville-Kent, 1893). Based on the morphological and color identification, the species was later determined to be a new genus under Euphylliidae (Yonge and Nicholls, 1931; Catala, 1964). Given that its skeleton is homomorphic to that of Euphyllia, and in recognition of Catala’s contribution to tropical marine fauna, Pectinia was changed to Catalaphyllia (Wells, 1971). Today, the genus Catalaphyllia is considered monospecific with C. jardinei as its only member (Fujii et al., 2020).
Coral reef ecosystems are important in providing a favorable habitat for its associated organisms (Bellwood et al., 2004). Throughout the world coral reefs are under stresses that include habitat destruction (Yeung et al., 2021), overexploitation (Jackson et al., 2001; Pandolfi et al., 2003), pollution (Williams et al., 2002; McCulloch et al., 2003), diseases (Harvell et al., 2002), predation (Qiu et al., 2014), and climate change (Wilkinson, 2002; Hughes et al., 2003; Gardener et al., 2003; Schlager, 1981; Dullo, 2005; Xie et al., 2020). The reef-building coral C. jardinei has a broad geographical distribution and can be found both in the Indian Ocean and Pacific Ocean (Veron and Stafford-Smith, 2000), including Australia, Indonesia, Japan, Maldives, Malaysia, Papua New Guinea, Philippines, Seychelles and Vietnam (UNEP, 2005). Nevertheless, given its small population size and overexploitation, it is considered by the IUCN Red List as having a “Vulnerable” status (IUCN, 2020).
Since the late 1980s, the popularity of keeping live corals at home has increased rapidly (Delbeek, 2001). Large-scale culture of C. jardinei is a current business in Indonesia and corals are exported globally (Akbar and Asadi, 2021). In addition to its appearance as described in its common name, C. jardinei is a popular coral species due to its easiness to culture ex situ. Naturally found in deep flats (greater than 35 meters) with sandy/silty bottoms, lagoons, and nearshore mudflats (Richard, 2005), C. jardinei can tolerate and survive in fluctuating or less favorable environmental conditions (Richard, 2005; http://www.edgeofexistence.org/species/elegance-coral/), making it a potential new model in the laboratory to study coral biology and their responses to environmental stressors.
To facilitate the use of C. jardinei to study the molecular mechanisms underlying coral biology, this study sequenced and provided the first genome and transcriptomic resources of this coral species.
Materials and methods
Sample collection and genome sequencing
Adult Catalaphyllia jardinei (Euphylliidae) (Figure 1A) were purchased from mainland China (original source from Indonesia) and Hong Kong suppliers (original source from Australia), and were cultured in an aquarium tank with circulating artificial seawater (salinity: 30-35 ppt) at around 23 ± 1°C and other equipment to simulate pristine waters at The Chinese University of Hong Kong. Corals were allowed to acclimatize under the laboratory conditions for 15 days prior to be used for DNA and RNA extraction.
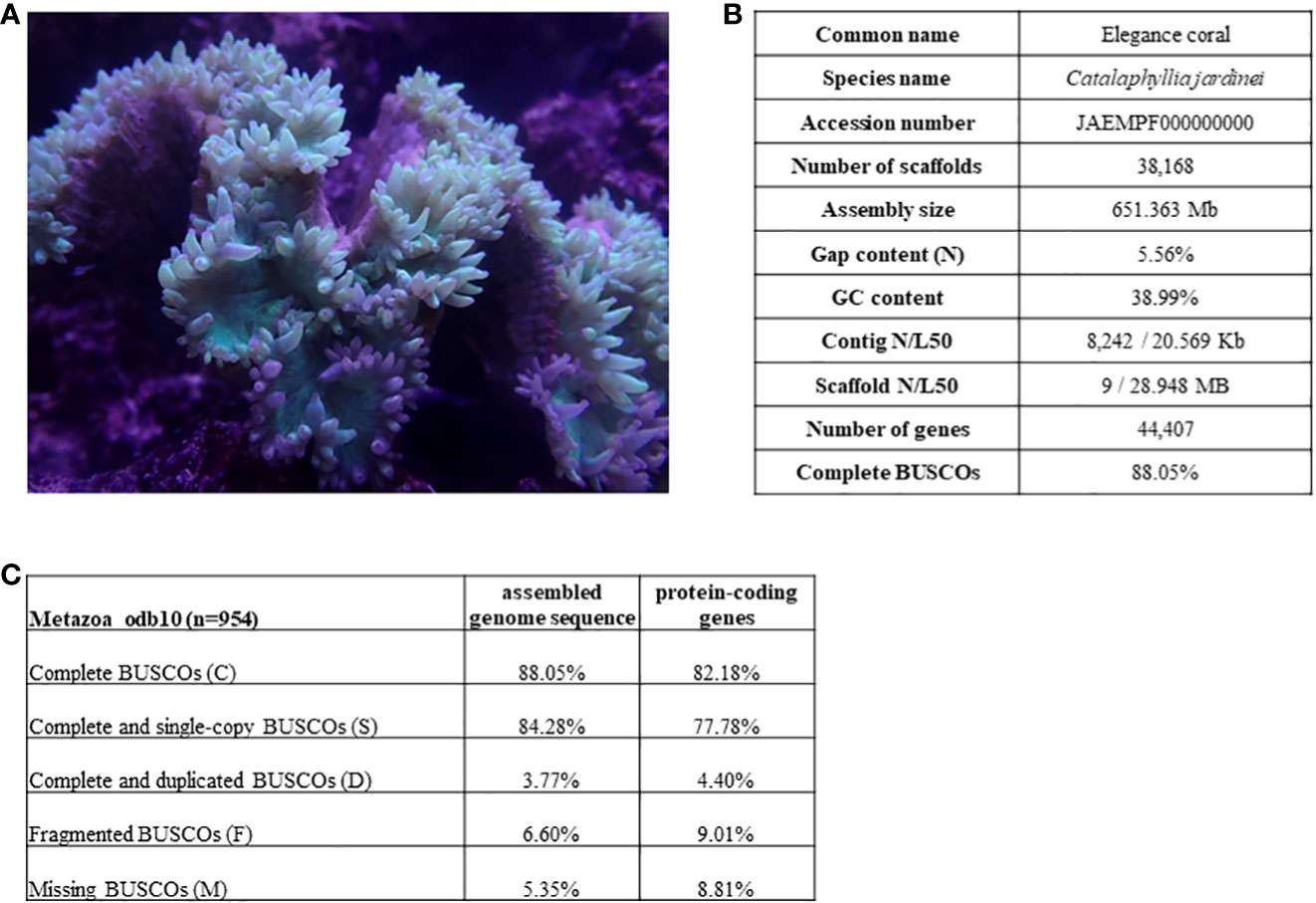
Figure 1 (A) Picture of elegance coral Catalaphyllia jardinei; (B) Genome assembly statistics of C jardinei; (C) BUSCO scores.
Sample for genome sequencing originated from tentacles from a single individual (Figure 1A). Genomic DNA (gDNA) was extracted using the QIAamp DNA minikit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. Extracted gDNA was subjected to quality control using a Nanodrop spectrophotometer (Thermo Scientific) and gel electrophoresis. Qualified samples were sent to Novogene and Dovetail Genomics for library preparation and sequencing. The resulting library was sequenced on an Illumina HiSeq X platform to produce 2 × 150 paired-end sequences. The length-weighted mean molecule length was 17.9 Kb, and details of the sequencing data were highlighted in Supplementary Information S1.
Dovetail Omni-C library preparation and sequencing
For each Dovetail Omni-C library, chromatin was fixed with formaldehyde and extracted. Fixed chromatin was digested with DNase I, and chromatin ends were repaired and ligated to a biotinylated bridge adapter followed by proximity ligation of adapter containing ends. After proximity ligation, crosslinks were reversed and the DNA was purified. Purified DNA was treated to remove biotin that was not internal to ligated fragments. Sequencing libraries were generated using NEBNext Ultra enzymes and Illumina-compatible adapters. Biotin-containing fragments were isolated using streptavidin beads before PCR enrichment of each library. The library was sequenced on an Illumina HiSeqX platform to produce 196.9 millions of 150 bp reads, and the raw data were listed in Supplementary Information S1.
Transcriptome sequencing
Total RNA, including messenger RNA and small RNA, from tentacles from single individual was isolated using mirVana™ miRNA Isolation Kit (Ambion) according to the manufacturer’s instructions, and subjected to quality control using a Nanodrop spectrophotometer (Thermo Scientific), gel electrophoresis, and Agilent 2100 Bioanalyzer (Agilent RNA 6000 Nano Kit). Qualified samples underwent library construction and 150 bp paired-end (PE) sequencing at Novogene; polyA-selected RNA-Sequencing libraries were prepared using TruSeq RNA Sample Prep Kit v2. Insert sizes and library concentrations of final libraries were determined using an Agilent 2100 bioanalyzer instrument (Agilent DNA 1000 Reagents) and real-time quantitative PCR (TaqMan Probe) respectively. Small RNA was submitted to BGI Hong Kong for HiSeq Small RNA library construction and 50 bp single-end (SE) sequencing. Details of the sequencing data can be found in Supplementary Information S1.
Genome assembly
K-mers of the HiseqXten 150PE (Chromium WGS) were counted using jellyfish version 2.2.5 (Marçais and Kingsford, 2011) with k-mers = 21, and estimation of genome size, repeat content, and heterozygosity were analyzed based on a k-mer-based statistical approach in the GenomeScope2 webtool (Ranallo-Benavidez et al., 2020, Supplementary Information S3). Kraken2 (Wood et al., 2019) (k2_standard_20210517 (kraken2 standard database) from https://benlangmead.github.io/aws-indexes/k2, which contains archaea, bacteria, viral, plasmid, human1, UniVec_Core) was used to mark and remove the contamination from the gDNA and transcriptome reads, 3.75% and 0.29% of contaminated reads were removed respectively. Processed Chromium WGS reads were used to construct a de novo assembly using Supernova (v 2.1.1) with default parameters (raw coverage = 78.86x), which generates phased, whole-genome de novo assemblies from a Chromium-prepared library (Weisenfeld et al., 2017) (https://support.10xgenomics.com/de-novo-assembly/software/pipelines/latest/using/running). The supernova output pseudohap assembly were used to generate the dedupe assembly by dedupe.sh (version 19Feb, 2020) of BBMap (Bushnell B. - sourceforge.net/projects/bbmap/), and then scaffolded using Dovetail Omni-C library reads by HiRise, a software pipeline designed specifically for using proximity ligation data to scaffold genome assemblies (Putnam et al., 2016). Dovetail Omni-C library sequences were aligned to the draft input assembly using bwa (https://github.com/lh3/bwa). The separations of Dovetail Omni-C read pairs mapped within draft scaffolds were analyzed by HiRise to produce a likelihood model for genomic distance between read pairs, and the model was used to identify and break putative misjoins, to score prospective joins, and make joins above a threshold. To further check for potential symbiotic contamination, 11 symbiodinium genomes were downloaded from NCBI (Supplementary Information S2) and BLASTN (Altschul et al., 1990) were carried out on the assembled genomes (e-value 1e-3). Scaffolds with alignment lengths >= 500 bp were further searched on nt database with BLASTN, and no symbiotic contamination was found in the final assembly genome.
Gene model prediction
Gene models were predicted as described in the jellyfish genomes (Nong et al., 2020). Briefly, the gene models were trained and predicted using funannotate (v1.8.9, https://github.com/nextgenusfs/funannotate) (Palmer and Stajich, 2020) with the following parameters: “–protein_evidence uniprot_sprot.fasta –genemark_mode ET –busco_seed_species fly –optimize_augustus –busco_db arthropoda –organism other –max_intronlen 350000”. In the “funannotate predict” step, GeneMark-ES v4.32 (Lomsadze et al., 2005) was used for ab initio gene prediction and Augustus (Keller et al., 2011) was trained using BUSCO. The gene models from the prediction sources “Augustus”, high-quality Augustus predictions (HiQ) with exon evidence, “pasa” (Haas et al., 2008)_, “snap” (Korf, 2004), “GlimmerHMM” (Majoros et al., 2004), and “GeneMark” were passed to Evidence Modeler (EVM Weights: {‘GeneMark’: 1, ‘HiQ’: 2, ‘pasa’: 6, ‘proteins’: 1, ‘Augustus’: 1, ‘GlimmerHMM’: 1, ‘snap’: 1, ‘transcripts’: 1}) to generate the final annotation files. Finally, followed by PASA to update the EVM consensus predictions, and add UTR annotations and models for alternatively spliced isoforms. Protein-coding genes were searched with BLASTP against the nr and SwissProt databases by diamond (v0.9.24) (Buchfink et al., 2015) with parameters “–more-sensitive –evalue 1e-3”, and mapped by HISAT2 (version 2.1.0) (Kim et al., 2019) with transcriptome reads. Gene models with no similarity to any known proteins and no mRNA support were removed from the final version. The gene models were screened by CGAT (Sims et al., 2014) and only the longest genes were retained.
Synteny analysis
Orthologues and orthogroups in C. jardinei and other 33 cnidarians proteomes were identified and inferred using OrthoFinder v2.5.2 (Emms and Kelly, 2019) with “-M msa” activated and default values to other parameters. To symbolise the gene families, the longest protein of each gene was taken as the representative in orthologues analysis. The species tree was constructed with fasttree using STAG algorithm and Hydra vulgaris was identified as the best outgroup species to construct the rooted tree using STRIDE method by OrthoFinder (Emms and Kelly, 2017; Emms and Kelly, 2018). Tree supporting value were the proportion of species tree derived from gene tree supporting each bipartition. The species tree was then converted into divergence tree using r8s (Sanderson, 2003). The tree structure and annotations were visualised using R package ‘ggtree’ (Wickham, 2016; Yu et al., 2017) in R v4.1.2 (R Core Team, 2022). Orthologues anchored by mutual best Diamond blastp hits (e-value cut-off 0.001) between C. jardinei and other 33 cnidarians as mentioned above were used in macrosynteny analyses. Oxford synteny plot were generated following previously described methods (So et al., 2022; Yichun, 2022). Detailed analytical scripts and materials were distributed at https://github.com/xieyichun50/coral_genome_Catalaphyllia_jardinei.
microRNA annotation
Adaptor sequences were trimmed from small RNA sequencing reads and Phred quality score less than 20 were removed. Processed reads of length within 18 bp and 27 bp were then mapped to the genomes using mapper.pl module of the mirDeep2 package (Friedländer et al., 2012). To identify known miRNA, the predicted coral microRNA hairpins were compared against metazoan microRNA precursor sequences from miRBase (Kozomara and Griffiths-Jones, 2014) using BLASTN (e-value <1e-2). For miRNAs with no significant sequence similarity to any of the miRNAs in miRBase were then checked manually. Novel microRNAs were defined when they fulfilled the criteria of microRNAs (Fromm et al., 2020). The expression levels of different arms of a miRNA were calculated based on the number of sequencing reads mapped to the respective arm region in the predicted miRNA hairpin by bowtie. In addition to the above, the precursor sequences of microRNAs from other corals in previous studies (Liew et al., 2014; Praher et al., 2021) were also used to carry out BLASTN searches for identification of any missed microRNA annotations.
Annotation of sesquiterpenoid pathway and neuropeptide genes
17 gene sequences involved in the sesquiterpenoid biosynthetic pathway retrieved from Nematostella vectensis and 4 gene sequences of neuropeptide (GLWamide, GRFamide, PRGamide and HIRamide) sequences from Hydra vulgaris were used for searching against the gene models and genome using BLASTP and TBLASTN, respectively. Putatively identified orthologues with a threshold of E-value equals to 10E-3 were tested by reciprocal searches in the NCBI nr database using BLASTP. For neuropeptide preprohormones, amino acid sequences were translated manually and aligned with MEGA 7.0 (Kumar et al., 2016), while the signaling sequences were analysed with SignalP 3.0 (Dyrløv Bendtsen et al., 2004). The cleavage sites were predicted by software ProP 1.0 (Duckert et al., 2004) and checked manually accordingly to the criteria suggested by Veenstra (2000). The potential mature neuropeptides were aligned using MEGA 7.0 (Kumar et al., 2016).
Results and discussion
High quality genome
Here, we presented a high-quality chromosome-level genome assembly of C. jardinei (2n = 28) (Heyward, 1985; Kenyon, 1997; Flot et al., 2006), the assembled genome size was 651 Mb, which is compatible with the kmer 21 estimate of a genome size of 685.5 Mb (Figure 1, Supplementary Information S3). 69.64% of the genomic sequences were contained on 14 pseudomolecules (Supplementary Information S1.3). The BUSCO score for complete genes is 88.05% (BUSCO version 5.1.3, metazoa_odb10), with complete and single-copy BUSCOs (S) being 84.28%, complete and duplicated BUSCOs (D) being 3.77%, fragmented BUSCOs (F) being 6.60% and Missing BUSCOs (M) being 5.35% (Figure 1C). Its scaffold N50 size (28.9Mb) represents the highest physical contiguity among the published coral genomes (Table 1). Using the transcriptomes, we predicted a total of 44,407 gene models including 37,003 protein coding genes and 7,404 tRNA genes, in the C. jardinei genome, which is within the range of 21,369 to 39,160 for other published coral genomes (Table 1).
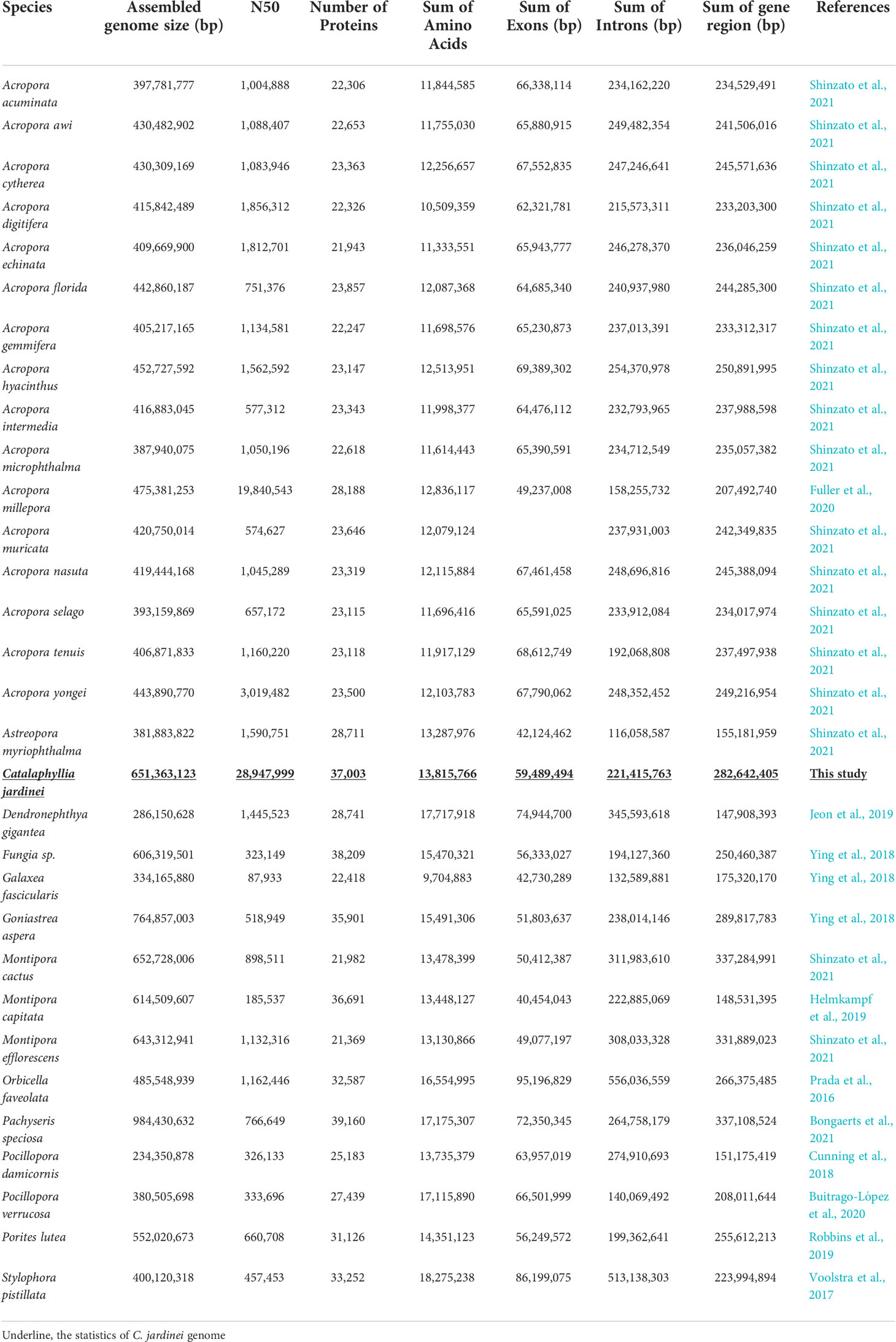
Table 1 Statistics of published coral genomes.
Synteny analysis
To clarify the syntenic relationship and gene linkage blocks between C. jardinei and other cnidarians, we generated the Oxford synteny plots of species pairs using the orthologues anchored by mutual best BLAST hit (e-value 1e-3) (Figure 2, Supplementary Information S4). Global gene orthogroup inference identified 26,260 orthogroups. The phylogenetic tree was constructed using 1,029 single-copy orthogroups presented in 31 out of 34 species, where single-copy indicated only one gene was assigned to specific orthogroup for each species. The chromosomal organisations are conserved in most soft corals, and each pseudo-chromosome in C. jardinei was associated with a different chromosome in another soft coral Acropora millipora. Further comparisons of C. jardinei against the more distant species, such as the sea anemone (Nematostella vectensis) and the true jellyfish (Rhopilema esculentum), displayed the one-to-one relationship on most synteny blocks, and a few one-to-many blocks (Supplementary Figures 1–3). Evidence on breakage, fusion or large-scale duplication was rarely found in these investigated cnidarians, suggesting that the genomes of these cnidarians have not undergone massive evolutionary changes.
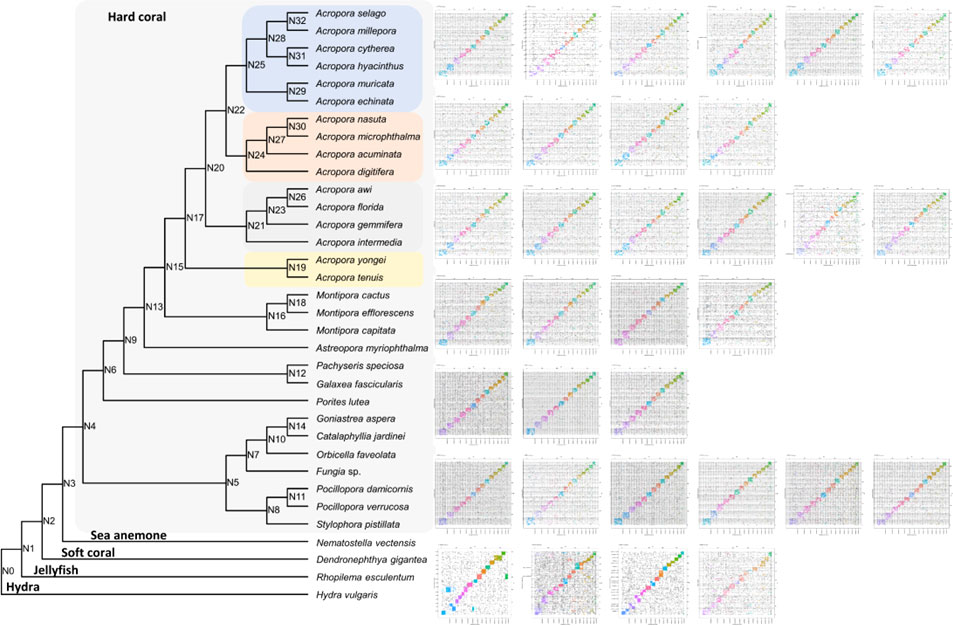
Figure 2 Oxford synteny plot of orthologous genes between Catalaphyllia jardinei and other 33 cnidarians. The divergence tree was constructed based on the concatenated alignment of single-copy orthogroups. Supporting values were inferred from STAG algorithm, and all were equal to 1 except that N26 = 0.583 and N28 = 0.713. Subplots were listed from left to right according to the top to bottom order of the phylogenetic tree subclades.
Homeobox genes
Homeobox genes are important developmental genes in animals and have been used as markers to understand genomic changes in animal evolution (e.g. Nong et al., 2020, Nong et al., 2021, Nong et al., 2022; Li et al., 2020; So et al., 2022; Qu et al., 2020; Shum et al., 2022). A total of 93 ANTP-class homeobox genes have been identified in the C. jardinei genome, including clusters of Hox-like and NK-like genes, and a ParaHox gene cluster (Figure 3; Supplementary Information S5). The data here further strengthens the idea that if distinct Hox and NK genes clusters are associated with ectodermal and mesodermal patterning, it could be a bilaterian-lineage specific event (Simakov et al., 2013; Nong et al., 2020), and the three gene ParaHox gene cluster could only be retained in the medusozoans than anthozoans among the extant cnidarians.

Figure 3 ANTP-class homeobox genes arrangement in high-quality cnidarian genomes. Double slash denotes genomic distance >100 kb and <1 Mb; triple slash denotes genomic distance over 1 Mb.
Heat shock protein family genes
Corals experiences abiotic stresses such as temperature, salinity, and oxygen concentration, and heat shock proteins (HSP) can assist and reconstruct the structure of damaged proteins under stresses (Collier and Benesch, 2020; Matambo et al., 2004; Whitley et al., 1999; Supplementary Figure 4). In general, HSP can be divided into six subfamilies, including HSPE (GroEL, cpn10), small heat shock proteins, HSP40, HSP70, HSP90, and HSP110, and HSP70 has been shown as chaperone proteins participated in response to heat stress in reef-building scleractinian corals (Franzellitti et al., 2018), (Haslbeck et al., 2016). In C. jardinei, we identified members of six HSP subfamilies, and their numbers were similar to those identified in the stony coral Acropora millepora chromosomal-level genome (Supplementary Figure 5, Supplementary Information S6). The identified heat shock protein gene families will be useful for future investigation of their roles in C. jardinei under different environmental conditions.
Neuropeptide genes
Neuropeptides play essential roles as endocrine factors and neurotransmitters, and are involved in the cellular differentiation and development of cnidarians (Takahashi, 2020). Here, Scleractinia conserved neuropeptides GLWamide, GRFamide, PRGamide, HIRamide, RWamide, RPamide and pQITRFamide were identified in C. jardinei (Figures 4A, B, Supplementary Information S7). A total of 6 mature peptides were identified from the preprohormone sequences of GLWamide in C. jardinei, and sequence alignment showed a conserved C-terminal GXW motif contained in these sequences (Figures 4C). Two of the GLWamides had “-GIW” replacement and such mutations were also identified in hard coral Euphyllia ancora (Shikina et al., 2020) (Figure 4C, Supplementary Information S8). For GRFamide preprohormones, 16 mature peptides were predicted. Among the identified PRGamide genes in C. jardinei, a single preprohormone gene was found to yield 46 different mature peptides (Figure 4B, Supplementary Information S8). For HIRamides, it was found that there was no glutamine residue identified from the predicted HIRamides in C. jardinei, where this phenomenon is also observable in other cnidarian HIRamides (Koch and Grimmelikhuijzen, 2019; Supplementary Information S8). For RWamide and RPamide, only 1 and 2 mature peptide(s) are yielding from the precursor respectively. Meanwhile, the precursor of pQITRFamide could be identified from the genome, but there was no expression shown in the sequenced transcriptomes, thus the number and form of mature peptides could not be identified (Figures 4B, C). The identified neuropeptides will be useful for future investigation of their developmental roles in C. jardinei.
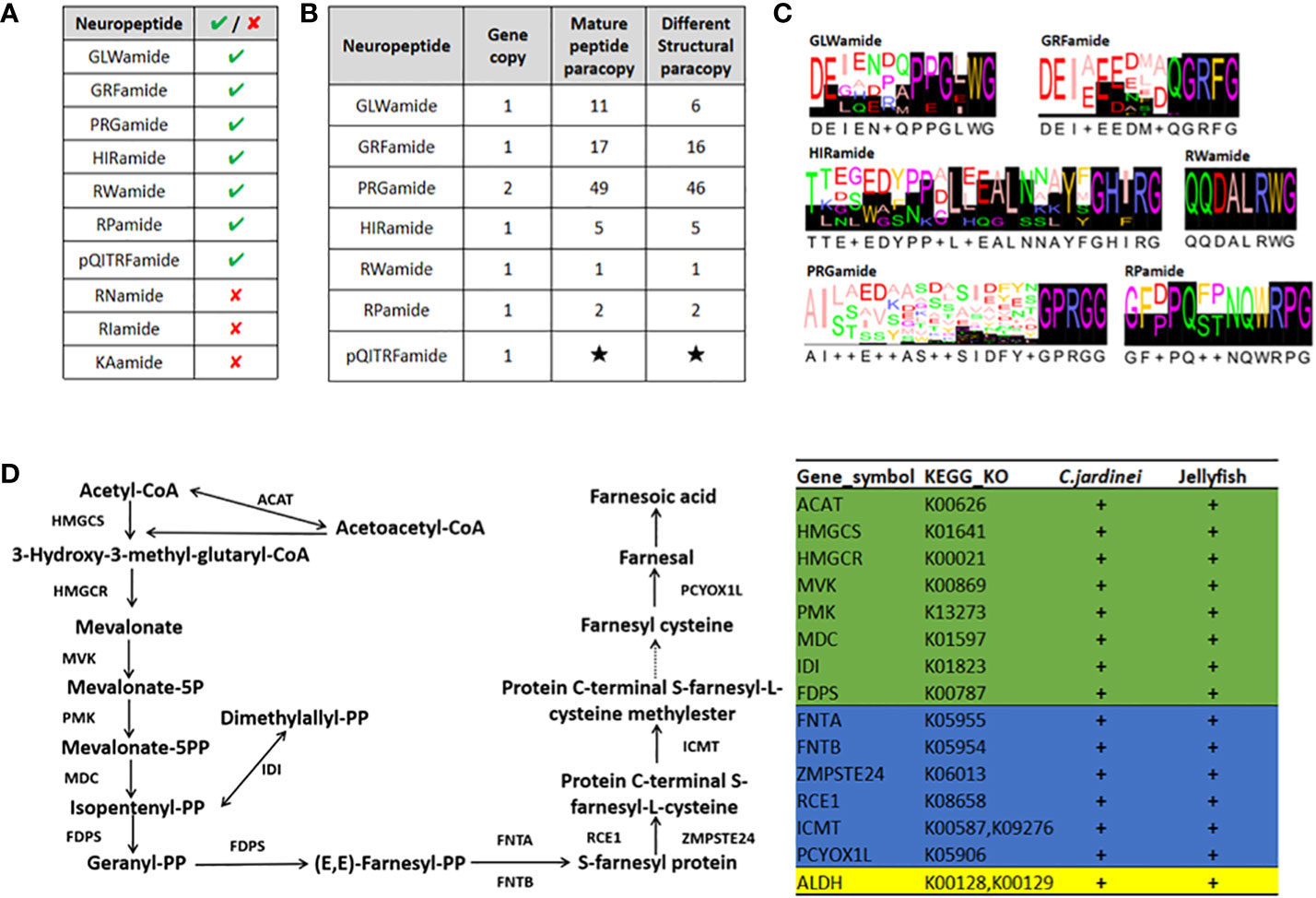
Figure 4 (A) The presence of anthozoan neuropeptides identified in C jardinei; (B) The numbers of mature peptides identified in each Scleratinian neuropeptides. Black star indicates unknown number due to its absence in the transcriptome. (C) The sequence logos of the Scleratinian conserved mature neuropeptides identified in C jardinei. (D) Schematic diagram showing the sesquiterpenoid hormone biosynthetic pathway in cnidarians. “+” sign denotes the presence of particular gene in respective class. The green background refers to genes in the mevalonate pathway, while the blue and yellow backgrounds refer to genes in the different downstream pathways. ACAT, Acetyl-CoA acetyltransferase, HMGCS, 3-hydroxy-3-methylglutaryl-CoA synthase, HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase, MVK, Mevalonate kinase, PMK, Phosphomevalonate kinase, MDC, Mevalonate (Diphospho) decarboxylase, IDI, Isopentenyl-diphosphate delta isomerase, FDPS, Farnesyl diphosphate synthase, FNTA, Farnesyltransferase, CAAX box, alpha, FNTB, Farnesyltransferase, CAAX box, beta, ZMPSTE24, STE24 endopeptidase, RCE, prenyl protein protease, ICMT, isoprenylcysteine carboxyl methyltransferase, PCYOX1L, Prenylcysteine oxidase 1, ALDH, Aldehyde dehydrogenase.
Sesquiterpenoid biosynthetic pathway genes
Sesquiterpenoid hormones, such as juvenile hormone, regulate the development, metamorphosis, and reproduction in insects, and have long been thought to be confined to the arthropods (e.g. Qu et al., 2018; Tsang et al., 2020). A recent study has unexpectedly identified the sesquiterpenoid hormonal system in jellyfishes, suggesting that the sesquiterpenoid hormone system is established at the bilaterian-cnidarian ancestor (Nong et al., 2020). To test whether coral species contain a sesquiterpenoid hormone system, we searched the sesquiterpenoid biosynthetic pathway genes in C. jardinei genome (Figure 4D) and identified the mevalonate pathway, aldehyde dehydrogenase (ALDH), farnesyl transferase (FNT), Ste 24 endopeptidase (ZMPSTE24), prenyl protein peptidase (RCE1), isoprenylcysteine carboxymethyl transferase (ICMT) and prenylcysteine oxidase (PCYOXIL) in a similar manner as the jellyfishes (Figure 4D, Supplementary Information S9). This result suggests that coral also has the sesquiterpenoid hormone system, and the functional roles of these hormones remain to be seriously tested in different cnidarians.
microRNA gain and loss in corals
Unlike the situation in bilaterians which contain lots of conserved microRNAs across different lineages, the cnidarians and bilaterians are well known to share only one conserved microRNA miR-100 (Griffiths-Jones et al., 2011). Information of small RNA sequences among different cnidarians reveals limited number of conserved microRNAs among different cnidarian lineages (Nong et al., 2020; Praher et al., 2021). Here, we sequenced the small RNA from C. jardinei, and annotated a total of 35 microRNAs in this species (Figure 5, Supplementary Information S10). In comparison with other published cnidarian microRNAs, in congruent to previous studies (e.g. Nong et al., 2020), there were more lineage-specific microRNAs than conserved microRNAs among different cnidarian lineages. We also identified conserved microRNAs at different key nodes in the cnidarian phylogeny, including miR-2022 and miR-2030 that are conserved across all cnidarians, six other microRNAs (miR-2023, miR-2025, miR-2036, miR-2037, miR-2050, miR-9425) that are conserved across all anthozoans, and 4 and 1 other microRNAs that are conserved in the Actiniaria and Scleractinia, respectively (Figure 5). Our findings established the foundation for further investigation of the different roles of these conserved microRNAs in cnidarians.

Figure 5 (A) Evolutionary gains of microRNAs in the Cnidaria. Numbers shown at nodes of the phylogenetic tree represent the gains of shared microRNAs, while numbers at the end of the branches describe the total numbers of identified microRNAs in each species. (B) Cnidarian microRNAs that are shared between at least four species. “+” and “-” signs denote the presence and absence of microRNAs in respective species. Nve, Nematostella vectensis; Eca, Edwardsiella carnea; Epa, Exaiptasia pallida, Sca, Scolanthus callimorphus; Mse, Metridium senile; Avi, Anemonia viridis; Spi, Stylophora pistillata; Ami, Acropora millepora; Adi, Acropora digitifera; Hma, Hydra magnipapillata; Cja, Catalaphyllia jardinei.
Conclusion
In the present study, we provided a high-quality genome assembly and transcriptome resources of the elegance coral Catalaphyllia jardinei (Euphylliidae). Abiotic stress regulator, hormonal gene families, and microRNAs were annotated, which will serve as a foundation for further investigation into their roles in development and responses to climate change. Due to the easiness to culture C. jardinei in reef tanks and the established resources in this study, we propose this species to be adopted as a new laboratory model.
Data availability statement
The final chromosome assembly was submitted to NCBI Assembly under accessionnumber JAEMPF000000000 in NCBI. The raw reads generated in this study havebeen deposited to the NCBI database under the BioProject accessions:PRJNA687327, the genome annotation files were deposited in the Figshare (https://Emms, D. M., & Kelly, S. (2018). STAG:species tree inference from all genes. BioRxiv, 267914.doi.org/10.6084/m9.figshare.19619700).
Author contributions
JHLH conceived and supervised the study. YY performed the extraction of DNA and RNA, and analyses of homeobox gene, heat shock protein, and microRNA. WN, JH, and TS performed genome assembly. WN performed gene model prediction, and transcriptomic analyses of mRNA and sRNA. WLS performed the neuropeptide analyses. YY and WLS performed the sesquiterpenoid analyses. YX performed synteny analyses. HYY and YY maintained the animal culture. DMB, WGB, TFC, APYC, KFL, PYQ, JWQ, BT, FX, JHLH obtained the funding. YY, WN, WLS, YX, JHLH wrote the first draft of the manuscript. All authors approve the final version of the manuscript.
Funding
This work was supported by the TUYF Charitable Trust, Hong Kong Research Grant Council Collaborative Research Grant (C4015-20EF, C7013-19G), General Research Fund (14100919, 14100420), the Open Collaborative Research Fund and Operation Fund from the Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (HKB L20200008), Center for Ocean Research in Hong Kong and Macau (CORE) (CORE is a joint research center for ocean research between QNLM and HKUST), and the Direct Grant of The Chinese University of Hong Kong (4053489, 4053547). YY was supported by the PhD studentship of The Chinese University of Hong Kong.
Conflict of interest
JHa and TS are employees of Dovetail Genomics, but have no roles in experimental design.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.991391/full#supplementary-material
Supplementary Information 1 | Genome and transcriptome sequencing data statistics.
Supplementary Information 2 | Symbiodinium genomes.
Supplementary Information 3 | Reports of GenomeScope2.
Supplementary Information 4 | Species used in orthologues searches.
Supplementary Information 5 | ANTP-class homeobox genes.
Supplementary Information 6 | Heat shock proteins.
Supplementary Information 7 | Anthozoan conserved neuropeptides
Supplementary Information 8 | Mature neuropeptides.
Supplementary Information 9 | Sesquiterpenoid biosynthetic pathway genes.
Supplementary Information 10 | MicroRNAs.
References
Akbar D., Asadi M. A. (2021). PERKEMBANGAN PENYAKIT KARANG HIAS KOLANG KALING KEMBANG (Catalaphyllia jardinei) OLEH CACING ACOEL FLATWORM (Waminoa sp.) DI PERAIRAN LAUT GILIMANUK, JEMBRANA BALI. J. Fish. Mar. Res. 5, 270–275. doi: 10.21776/ub.jfmr.2021.005.02.12
Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Bellwood D. R., Hughes T. P., Folke C., Nyström M. (2004). Confronting the coral reef crisis. Nature 429, 827–833. doi: 10.1038/nature02691
Bongaerts P., Cooke I. R., Ying H., Wels D., den Haan S., Hernandez-Agreda A., et al. (2021). Morphological stasis masks ecologically divergent coral species on tropical reefs. Curr. Biol. 31, 2286–2298.e8. doi: 10.1016/j.cub.2021.03.028
Buchfink B., Xie C., Huson D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Buitrago-López C., Mariappan K. G., Cárdenas A., Gegner H. M., Voolstra C. R. (2020). The genome of the cauliflower coral pocillopora verrucosa. Genome Biol. Evol. 12, 1911–1917. doi: 10.1093/gbe/evaa184
Catala R. (1964). Carnival under the sea. EdSicard R. [ie Éditions R. Sicard. Available at: https://archive.org/details/carnivalundersea00cata
Collier M. P., Benesch J. L. P. (2020). Small heat-shock proteins and their role in mechanical stress. Cell Stress Chaperones. 25, 601–613. doi: 10.1007/s12192-020-01095-z
Cunning R., Bay R. A., Gillette P., Baker A. C., Traylor-Knowles N. (2018). Comparative analysis of the Pocillopora damicornis genome highlights role of immune system in coral evolution. Sci. Rep. 8, 16134. doi: 10.1038/s41598-018-34459-8
Delbeek J. C. (2001). Coral farming: Past, present and future trends. Aquarium. Sci. Conserv. 3, 171–181. doi: 10.1023/A:1011306125934
Duckert P., Brunak S., Blom N. (2004). Prediction of proprotein convertase cleavage sites. Protein Eng. Des. Sel. 17, 107–112. doi: 10.1093/protein/gzh013
Dullo W.-C. (2005). Coral growth and reef growth: a brief review. Facies 51, 33–48. doi: 10.1007/s10347-005-0060-y
Dyrløv Bendtsen J., Nielsen H., von Heijne G., Brunak S. (2004). Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340, 783–795. doi: 10.1016/j.jmb.2004.05.028
Emms D. M., Kelly S. (2017). STRIDE: Species tree root inference from gene duplication events. Mol. Biol. Evol. 34, 3267–3278. doi: 10.1093/molbev/msx259
Emms D. M., Kelly S. (2018). STAG: Species tree inference from all genes 267914. BioRxiv 267914. doi: 10.1101/267914
Emms D. M., Kelly S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238. doi: 10.1186/s13059-019-1832-y
Flot J.-F., ozouF-cosTaz C., Tsuchiya M., Woesik R. (2006). Comparative coral cytogenetics. In. Proc. 5th. Int. Coral. Reef. Symposium. 1, 4–8. Available at: https://www.semanticscholar.org/paper/Comparative-coral-cytogenetics-Flot-ozouF-cosTaz/9c0679897fd7e6f84069b8e2600bcbed47715268 [Accessed May 31, 2022].
Franzellitti S., Airi V., Calbucci D., Caroselli E., Prada F., Voolstra C. R., et al. (2018). Transcriptional response of the heat shock gene hsp70 aligns with differences in stress susceptibility of shallow-water corals from the Mediterranean Sea. Mar. Environ. Res. 140, 444–454. doi: 10.1016/j.marenvres.2018.07.006
Friedländer M. R., Mackowiak S. D., Li N., Chen W., Rajewsky N. (2012). miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 40, 37–52. doi: 10.1093/nar/gkr688
Fromm B., Keller A., Yang X., Friedlander M. R., Peterson K. J., Griffiths-Jones S. (2020). Quo vadis microRNAs? Trends Genet. 36, 461–463. doi: 10.1016/j.tig.2020.03.007
Fujii T., Kitano Y., Tachikawa H. (2020). New distributional records of three species of euphylliidae (Cnidaria, anthozoa, hexacorallia, scleractinia) from the Ryukyu islands, Japan. Species. Diversity 25, 275–282. doi: 10.12782/specdiv.25.275
Fuller Z. L., Mocellin V. J. L., Morris L. A., Cantin N., Shepherd J., Sarre L., et al. (2020). Population genetics of the coral Acropora millepora: Toward genomic prediction of bleaching. Science 369, eaba4674. doi: 10.1126/science.aba4674
Gardener M. R., Whalley R. D. B., Sindel B. M. (2003). Ecology of nassella neesiana, Chilean needle grass, in pastures on the northern tablelands of new south wales. II. seedbank dynamics, seed germination, and seedling recruitment. Aust. J. Agric. Res. 54, 621–626. doi: 10.1071/AR01076
Griffiths-Jones S., Hui J. H. L., Marco A., Ronshaugen M. (2011). MicroRNA evolution by arm switching. EMBO Rep. 12, 172–177. doi: 10.1038/embor.2010.191
Haas B. J., Salzberg S. L., Zhu W., Pertea M., Allen J. E., Orvis J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7. doi: 10.1186/gb-2008-9-1-r7
Harvell C. D., Mitchell C. E., Ward J. R., Altizer S., Dobson A. P., Ostfeld R. S., et al. (2002). Climate warming and disease risks for terrestrial and marine biota. Science 296, 2158–2162. doi: 10.1126/science.1063699
Haslbeck M., Peschek J., Buchner J., Weinkauf S. (2016). Structure and function of α-crystallins: Traversing from in vitro to in vivo. biochimica et biophysica acta (BBA) -. Gen. Subj. 1860, 149–166. doi: 10.1016/j.bbagen.2015.06.008
Helmkampf M., Bellinger M. R., Geib S. M., Sim S. B., Takabayashi M. (2019). Draft genome of the rice coral montipora capitata obtained from linked-read sequencing. Genome Biol. Evol. 11, 2045–2054. doi: 10.1093/gbe/evz135
Heyward A. J. (1985). Chromosomes of the coral goniopora lobata (Anthozoa: Scleractinia). Heredity 55, 269–271. doi: 10.1038/hdy.1985.101
Hughes T. P., Baird A. H., Bellwood D. R., Card M., Connolly S. R., Folke C., et al. (2003). Climate change, human impacts, and the resilience of coral reefs. Science 301, 929–933. doi: 10.1126/science.1085046
IUCN (2020). IUCN 2019 International union for conservation of nature annual report 2019 (IUCN). #Available at: https://portals.iucn.org/library/node/49096.
Jackson J. B. C., Kirby M. X., Berger W. H., Bjorndal K. A., Botsford L. W., Bourque B. J., et al. (2001). Historical overfishing and the recent collapse of coastal ecosystems. Science 293, 629–637. doi: 10.1126/science.1059199
Jeon Y., Park S. G., Lee N., Weber J. A., Kim H.-S., Hwang S.-J., et al. (2019). The Draft Genome of an Octocoral, Dendronephthya gigantea. Genome Biol. Evol. 11, 949–953. doi: 10.1093/gbe/evz043
Keller O., Kollmar M., Stanke M., Waack S. (2011). A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27, 757–763. doi: 10.1093/bioinformatics/btr010
Kenyon J. C. (1997). Models of reticulate evolution in the coral genus acropora based on chromosome numbers: Parallels with plants. Evolution 51, 756–767. doi: 10.1111/j.1558-5646.1997.tb03659.x
Kim D., Paggi J. M., Park C., Bennett C., Salzberg S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi: 10.1038/s41587-019-0201-4
Koch T. L., Grimmelikhuijzen C. J. P. (2019). Global neuropeptide annotations from the genomes and transcriptomes of cubozoa, scyphozoa, staurozoa (Cnidaria: Medusozoa), and octocorallia (Cnidaria: Anthozoa). Front. Endocrinol. 10. doi: 10.3389/fendo.2019.00831
Kozomara A., Griffiths-Jones S. (2014). miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68–D73. doi: 10.1093/nar/gkt1181
Kumar S., Stecher G., Tamura K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Li Y., Nong W., Baril T., Yip H. Y., Swale T., Hayward A., et al. (2020). Reconstruction of ancient homeobox gene linkages inferred from a new high-quality assembly of the Hong Kong oyster (Magallana hongkongensis) genome. BMC Genomics 21, 713. doi: 10.1186/s12864-020-07027-6
Liew Y. J., Aranda M., Carr A., Baumgarten S., Zoccola D., Tambutté S., et al. (2014). Identification of MicroRNAs in the coral stylophora pistillata. PloS One 9, e91101. doi: 10.1371/journal.pone.0091101
Lomsadze A., Ter-Hovhannisyan V., Chernoff Y. O., Borodovsky M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33. 6494–6506. doi: 10.1093/nar/gki937
Majoros W. H., Pertea M., Salzberg S. L. (2004). TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879. doi: 10.1093/bioinformatics/bth315
Marçais G., Kingsford C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Matambo T. S., Odunuga O. O., Boshoff A., Blatch G. L. (2004). Overproduction, purification, and characterization of the Plasmodium falciparum heat shock protein 70. Protein Expression Purification 33, 214–222. doi: 10.1016/j.pep.2003.09.010
McCulloch M., Fallon S., Wyndham T., Hendy E., Lough J., Barnes D. (2003). Coral record of increased sediment flux to the inner great barrier reef since European settlement. Nature 421, 727–730. doi: 10.1038/nature01361
Nong W., Cao J., Li Y., Qu Z., Sun J., Swale T., et al. (2020). Jellyfish genomes reveal distinct homeobox gene clusters and conservation of small RNA processing. Nat. Commun. 11, 3051. doi: 10.1038/s41467-020-16801-9
Nong W., Qu Z., Li Y., Barton-Owen T., Wong A. Y. P., Yip H. Y., et al. (2021). Horseshoe crab genomes reveal the evolution of genes and microRNAs after three rounds of whole genome duplication. Commun. Biol. 4, 1–11. doi: 10.1038/s42003-020-01637-2
Nong W., Yu Y., Aase-Remedios M. E., Xie Y., So W. L., Li Y., et al. (2022). Genome of the ramshorn snail Biomphalaria straminea—an obligate intermediate host of schistosomiasis. GigaScience 11, giac012. doi: 10.1093/gigascience/giac012
Palmer J. M., Stajich J. (2020). Funannotate v1.8.1: Eukaryotic genome annotation (Zenodo). Avaialble at: https://github.com/nextgenusfs/funannotate.
Pandolfi J. M., Bradbury R. H., Sala E., Hughes T. P., Bjorndal K. A., Cooke R. G., et al. (2003). Global trajectories of the long-term decline of coral reef ecosystems. Science 301, 955–958. doi: 10.1126/science.1085706
Prada C., Hanna B., Budd A. F., Woodley C. M., Schmutz J., Grimwood J., et al. (2016). Empty niches after extinctions increase population sizes of modern corals. Curr. Biol. 26, 3190–3194. doi: 10.1016/j.cub.2016.09.039
Praher D., Zimmermann B., Dnyansagar R., DJ M., Moya A., Modepalli V., et al. (2021). Conservation and turnover of miRNAs and their highly complementary targets in early branching animals. Proc. R. Soc. B.: Biol. Sci. 288, 20203169.
Putnam N. H., O’Connell B. L., Stites J. C., Rice B. J., Blanchette M., Calef R., et al. (2016). Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350. doi: 10.1101/gr.193474.115
Qiu J.-W., Lau D. C. C., Cheang C., Chow W. (2014). Community-level destruction of hard corals by the sea urchin Diadema setosum. Marine Pollution Bull. 85, 783–788. doi: 10.1016/j.marpolbul.2013.12.012
Qu Z., Bendena W. G., Tobe S. S., Hu J. H. L. (2018). Juvenile hormone and sesquiterpenoids in arthropods: Biosynthesis, signaling, and role of MicroRNA. J. Steroid Biochem. Mol. Biol. 184, 69–76. doi: 10.1016/j.jsbmb.2018.01.013
Qu Z., Nong W., Yu Y., Baril T., Yip H. Y., Hayward A., et al. (2020). Genome of the four-finger threadfin Eleutheronema tetradactylum (Perciforms: Polynemidae). BMC Genomics 21, 726. doi: 10.1186/s12864-020-07145-1
Ranallo-Benavidez T. R., Jaron K. S., Schatz M. C. (2020). GenomeScope 2.0 and smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432. doi: 10.1038/s41467-020-14998-3
Richard D. (2005). “Notes from the trenches: Asexual reproduction of catalaphyllia jardinei (Elegance coral),” in Reefkeeping magazine™ reef central, LLC-copyright © (2008).
Robbins S. J., Singleton C. M., Chan C. X., Messer L. F., Geers A. U., Ying H., et al. (2019). A genomic view of the reef-building coral Porites lutea and its microbial symbionts. Nat. Microbiol. 4, 2090–2100. doi: 10.1038/s41564-019-0532-4
Sanderson M. J. (2003). r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19, 301–302. doi: 10.1093/bioinformatics/19.2.301
Saville-Kent W. (1893). The great barrier reef of Australia: Its products and Potentialities : Containing an account, with copious coloured and photographic illustrations (the latter here produced for the first time), of the corals and coral reefs, pearl and pearl-shell, beche-de-mer, other fishing industries, and the marine fauna of the Australian great barrier region. Allen.
Schlager W. (1981). The paradox of drowned reefs and carbonate platforms. GSA. Bull. 92, 197–211. doi: 10.1130/0016-7606(1981)92<197:TPODRA>2.0.CO;2
Schlager W. (2003). Benthic carbonate factories of the phanerozoic. Int. J. Earth Sci. (Geologische. Rundschau). 92, 445–464. doi: 10.1007/s00531-003-0327-x
Shikina S., Chiu Y.-L., Zhang Y., Yao Y.-C., Liu T.-Y., Tsai P.-H., et al. (2020). Involvement of GLWamide neuropeptides in polyp contraction of the adult stony coral euphyllia ancora. Sci. Rep. 10, 9427. doi: 10.1038/s41598-020-66438-3
Shinzato C., Khalturin K., Inoue J., Zayasu Y., Kanda M., Kawamitsu M., et al. (2021). Eighteen coral genomes reveal the evolutionary origin of acropora strategies to accommodate environmental changes. Mol. Biol. Evol. 38, 16–30. doi: 10.1093/molbev/msaa216
Shum C., Nong W., So W. L., Li Y., Qu Z., Yip H. Y., et al. (2022). Genome of the sea anemone exaiptasia pallida and transcriptome profiles during tentacle regeneration. Front. Cell Dev. Biol. 10, 1558. doi: 10.3389/fcell.2022.900321
Simakov O., Marletaz F., Cho S.-J., Edsinger-Gonzales E., Havlak P., Hellsten U., et al. (2013). Insights into bilaterian evolution from three spiralian genomes. Nature 493, 526–531. doi: 10.1038/nature11696
Sims D., Ilott N. E., Sansom S. N., Sudbery I. M., Johnson J. S., Fawcett K. A., et al. (2014). CGAT: computational genomics analysis toolkit. Bioinformatics 30, 1290–1291. doi: 10.1093/bioinformatics/btt756
So W. L., Nong W., Xie Y., Baril T., Ma H., Qu Z., et al. (2022). Myriapod genomes reveal ancestral horizontal gene transfer and hormonal gene loss in millipedes. Nat. Commun. 13, 3010. doi: 10.1038/s41467-022-30690-0
Takahashi T. (2020). Comparative aspects of structure and function of cnidarian neuropeptides. Front. Endocrinol. 11. doi: 10.3389/fendo.2020.00339
Tsang S. S. K., Law S. T. S., Li C., Qu Z., Bendena W. G., Tobe S. S., et al. (2020) Diversity of Insect Sesquiterpenoid Regulation. . Available at: https://www.frontiersin.org/articles/10.3389/fgene.2020.01027 (Accessed September 11, 2022).
UNEP. (2005) World conservation monitoring centre:United Nations Environment Programme (2005). UNEP 2004 annual report.
Veenstra J. A. (2000). Mono- and dibasic proteolytic cleavage sites in insect neuroendocrine peptide precursors. Arch. Insect Biochem. Physiol. 43, 49–63. doi: 10.1002/(SICI)1520-6327(200002)43:2<49::AID-ARCH1>3.0.CO;2-M
Veron J. E. N., Stafford-Smith M. (2000). Corals of the world (Townsville MC, Qld, Australia: Australian Institute of Marine Science).
Voolstra C. R., Li Y., Liew Y. J., Baumgarten S., Zoccola D., Flot J.-F., et al. (2017). Comparative analysis of the genomes of Stylophora pistillata and Acropora digitifera provides evidence for extensive differences between species of corals. Sci. Rep. 7, 17583. doi: 10.1038/s41598-017-17484-x
Weisenfeld N., Kumar V., Shah P., Church D., Jaffe D. (2017). Direct determination of diploid genome sequences. Genome Res. 27. doi: 10.1101/gr.214874.116
Wells J. W. (1971). Notes on indo-pacific scleractinian corals. part 7 catalaphyllia, a new genus of reef corals.
Whitley D., Goldberg S. P., Jordan W. D. (1999). Heat shock proteins: A review of the molecular chaperones. J. Vasc. Surg. 29, 748–751. doi: 10.1016/S0741-5214(99)70329-0
Wickham H. (2016). ggplot2: Elegant graphics for data analysis (Springer). Available at: https://link.springer.com/book/10.1007/978-0-387-98141-3.
Williams B. K., Nichols J. D., Conroy M. J. (2002). Analysis and management of animal populations (Academic Press).
Wood D. E., Lu J., Langmead B. (2019). Improved metagenomic analysis with kraken 2. Genome Biol. 20, 257. doi: 10.1186/s13059-019-1891-0
Xie J. Y., Yeung Y. H., Kwok C. K., Kei K., Ang P., Chan L. L., et al. (2020). Localized bleaching and quick recovery in Hong Kong's coral communities. Marine Pollution Bull. 153, 110950. doi: 10.1016/j.marpolbul.2020.110950
Yeung Y. H., Xie J. Y., Kwok C. K., Kei K., Ang P., Chan L. L., et al. (2021). Hong Kong's subtropical scleractinian coral communities: Baseline, environmental drivers and management implications. Marine Pollution Bull. 167, 112289. doi: 10.1016/j.marpolbul.2021.112289
Yichun. (2022). xieyichun50/Myriapod-genomes: Myriapod-genomes (v1.0.0). Zenodo10.5281/zenodo.6482625 xieyichun50/Genome-macrosynteny-gene-family-evolution: Genome-macrosynteny-gene-family-evolution-v1.0. Zenodo
Ying H., Cooke I., Sprungala S., Wang W., Hayward D. C., Tang Y., et al. (2018). Comparative genomics reveals the distinct evolutionary trajectories of the robust and complex coral lineages. Genome Biol. 19, 175. doi: 10.1186/s13059-018-1552-8
Yonge C. M., Nicholls A. G. (1931). “Studies on the physiology of corals: IV. The structure, distribution and physiology of THE ZOOXANTHELLAE,” in Scientific Reports/Great barrier reef expedition 1928-29, vol. 1. , 135–176.
Keywords: coral, genome, Catalaphyllia, microRNA, sesquiterpenoid
Citation: Yu Y, Nong W, So WL, Xie Y, Yip HY, Haimovitz J, Swale T, Baker DM, Bendena WG, Chan TF, Chui APY, Lau KF, Qian P-Y, Qiu J-W, Thibodeau B, Xu F and Hui JHL (2022) Genome of elegance coral Catalaphyllia jardinei (Euphylliidae). Front. Mar. Sci. 9:991391. doi: 10.3389/fmars.2022.991391
Received: 11 July 2022; Accepted: 29 August 2022;
Published: 27 September 2022.
Edited by:
Yong Wang, Tsinghua University, ChinaReviewed by:
Thomas Lund Koch, University of Copenhagen, DenmarkMiriam Ruocco, Anton Dohrn Zoological Station, Italy
Copyright © 2022 Yu, Nong, So, Xie, Yip, Haimovitz, Swale, Baker, Bendena, Chan, Chui, Lau, Qian, Qiu, Thibodeau, Xu and Hui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jerome H. L. Hui, amVyb21laHVpQGN1aGsuZWR1Lmhr
†These authors have contributed equally to this work