 Na Zhao1,2†
Na Zhao1,2† Zhongdian Dong
Zhongdian Dong Bo Zhang
Bo Zhang- 1College of Fishery, Guangdong Ocean University, Zhanjiang, China
- 2Southern Marine Science and Engineering Guangdong Laboratory (Zhanjiang), Zhanjiang, China
- 3Beijing Genomics Institute (BGI)-Qingdao, BGI-Shenzhen, Qingdao, China
- 4Tianjin Fisheries Research Institute, Tianjin, China
- 5Guangdong Provincial Key Laboratory of Aquatic Animal Disease Control and Healthy Culture, College of Fishery, Guangdong Ocean University, Zhanjiang, China
Introduction
Flathead grey mullet Mugil cephalus (FishBase ID: 785, NCBI Taxonomy ID: 48193) is a highly commercial fish and has been farmed as an important aquaculture resource in many countries for decades. Mugil cephalus possesses strong salinity adaptability, which enables this fish to spread worldwide in fresh water, salty fresh water, and salty water environments (frequently found coastally in estuaries and freshwater environments) (Gorski et al., 2015). Adult mullets have been found in waters ranging from zero salinity to 75‰ (Li et al., 2017). Mugil cephalus can also be employed as an excellent biomonitor for its adaptability towards hypoxia and pollutants, such as heavy metals and chlorpyrifos (Waltham et al., 2013). The large demands of M. cephalus and its roe are reminding us of the increasing genetic risks of this species (Dor et al., 2020; Cossu et al., 2021). The in-depth research of this species, nevertheless, was restricted due to a lack of genomic data. Some studies on the transcriptome of M. cephalus were based on de novo sequencing and homologous annotation using the genome of Nile tilapia (Oreochromis niloticus) as a reference (Byadgi et al., 2016; Dor et al., 2020), which is limited and inefficient to some extent. Under this background, in this study, we successfully assembled a genome of flathead grey mullet M. cephalus for the first time using single-tube long fragment read (stLFR) technology, providing a necessary reference for further research to achieve the long-term persistence of Mugil cephalus.
Currently, most genome projects rely on long-read sequencing technologies such as PacBio or Oxford Nanopore. In the meantime, the data of whole genome shotgun (WGS) sequencing, which are sequenced on second-generation platform (BGISEQ or Illumina) are used only in the process of k-mer analysis or the polishing of the genome during assembly. This is mainly due to the phenomenon that regular WGS data could not support a satisfying genome assembly result, but the high cost of long-read sequencing technologies limited many genome projects, especially those that aimed to fill the blank of genome resources on large-scale species surveys. Here, we employed stLFR technology, which enables the access of long DNA fragments based on an economical second-generation sequencing platform (Wang et al., 2019). The principle of stLFR was that it could create millions of miniaturized barcoding reactions in a single tube, and these fragments marked with the same barcode belonged to the same molecule, making it possible to obtain a more consecutive assembly. The stLFR technology could offer a relatively high-quality genome at an economical cost, making it possible to conduct zoological research.
Value of the data
In the present study, we assembled the flathead grey mullet (Mugil cephalus) genome. We also conducted a comparative genomic analysis and inferred the phylogeny of M. cephalus with eight other teleosts and analyzed the gene family expansion and contraction, aiming to give a clue to the evolution of the specific characteristics of this fish species.
Method and results
Genome sequencing
The genomic DNA of M. cephalus was extracted using the muscle of a single fish (Figure 1A) obtained from the Bohai Sea by the Tianjin Fisheries Research Institute, China. A stLFR sequencing library was established based on high-quality purified and complete DNA (A260/A280 = ~1.8, A260/A230 = 2.0–2.2), and all experimental protocols were the same as the technology manuscript suggested (Wang et al., 2019). After sequencing, barcode sequences were extracted using a custom program (GitHub https://github.com/stLFR/stLFR_read_demux) that was provided by the stLFR technology pipeline. All demultiplexed read data were then used in genome analysis. All sequenced reads were edited to trim stLFR barcodes and filtered by SOAPnuke, and the final clean paired-end data were 128.67 Gb with a Q30 value of 90.24%. Jellyfish v2.2.6 was used to obtain the frequency distribution of 17-mers when all duplications in clean data were removed, suggesting a genome size of over 600 Mb with 0.73% heterozygosity by GenomeScope v1.0.0 (Figure 1B).
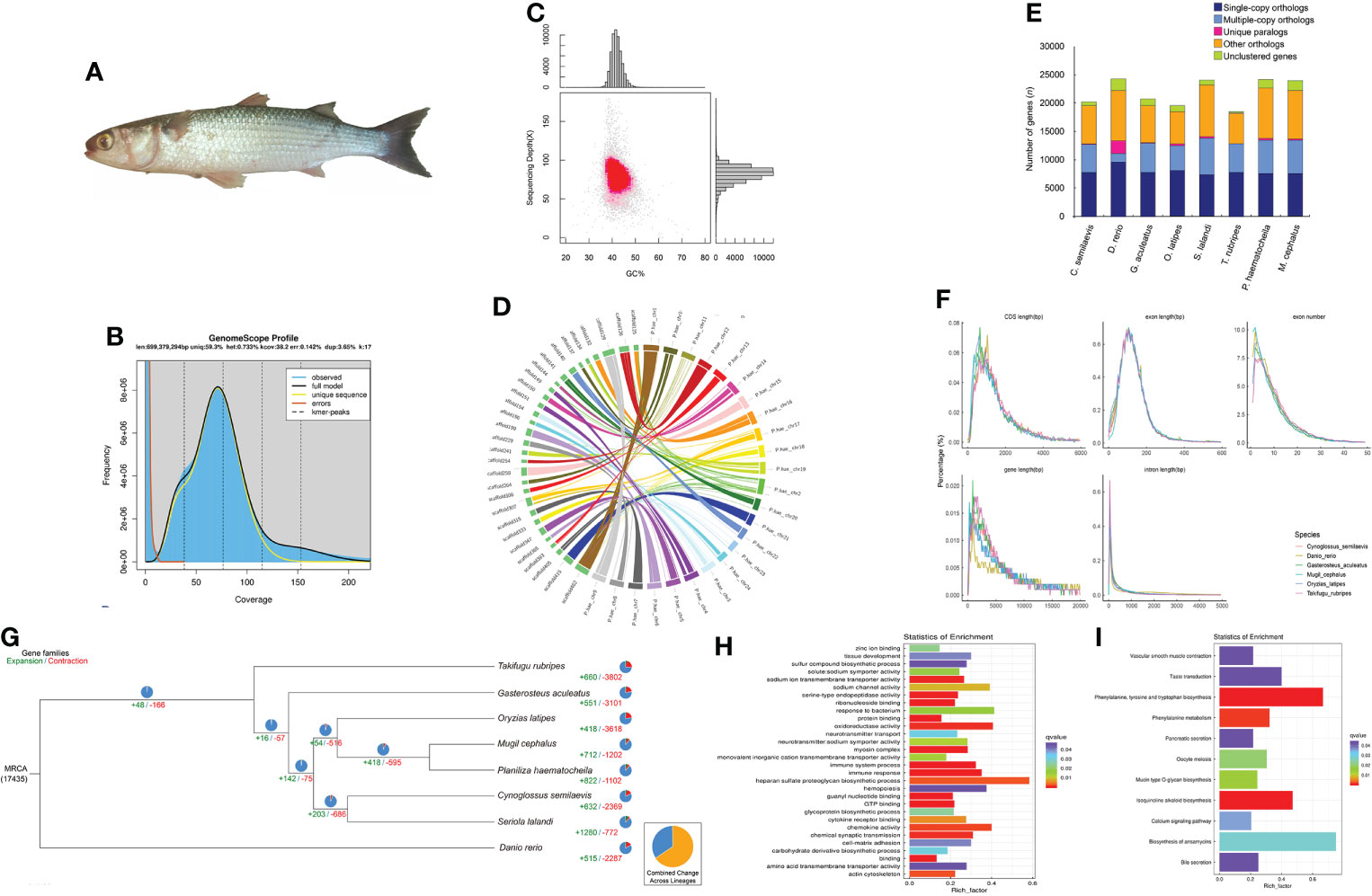
Figure 1 Statistics and data analysis of the genome assembly of the flathead grey mullet, Mugil cephalus. (A) The flathead grey mullet (Mugil cephalus). (B) Genome synteny analysis between the top 30 scaffolds of M. cephalus and the 24 chromosomes of the previously published P. haematocheila genome. (C) K-mer survey of the M. cephalus genome. (D) Distribution of GC content and sequencing depth of the M. cephalus genome. (E) Statistics of exon number and length of different gene structures in M. cephalus and several reference species. Different species are represented with different colors. (F) Gene family cluster results of M. cephalus and the other reference species. The abscissa represents species, and the ordinate represents a number of genes. (G) Phylogenetic tree based on sequence identity is indicated at each node. The estimated number of orthologous gene groups in the most recent common ancestral (MRCA) species is shown at the root. The numbers of gene families expanded or contracted in each lineage are shown on the corresponding branch; +, expansion; −, contraction. (H) GO enrichment analysis of significantly expanded gene families. (I) KEGG pathway enrichment analysis of significantly expanded gene families.
De novo genome assembly
De novo assembly was conducted with the pipeline developed and published by BGI-Qingdao (GitHub https://github.com/BGI-Qingdao/stlfr2supernova_pipeline). This pipeline could de novo assemble the stLFR raw reads using Supernova Assembler, which refers to the de novo software from 10X Genomics. The output assembly of this pipeline was then used in further scaffolding and gap closing procedures with GapCloser 1.12 in order to improve assembly quality. The final genome size was 661.32 Mb with Scaffold N50 of 6.38 Mb, and the detailed assembly indicator is shown in Table 1.
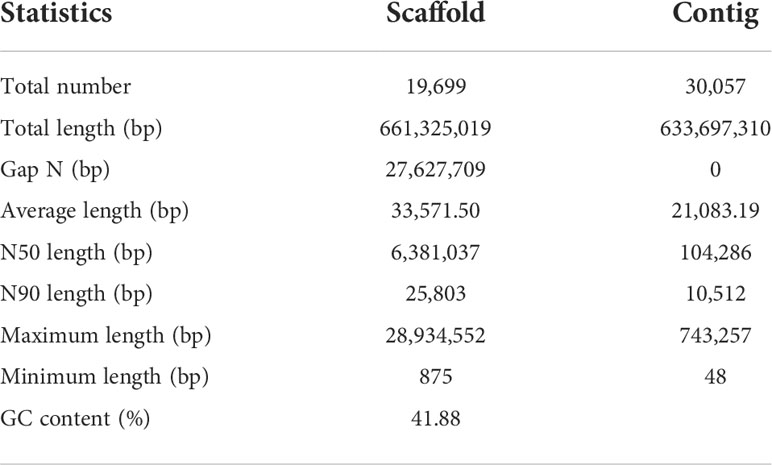
Table 1 Summary of information related to the genome assembly.
Genome evaluation
The integrity and accuracy of the assembly were evaluated by mapping all duplication-removed reads back to the genome using BWA and Samtools (Li and Durbin, 2009a; Li et al., 2009b). In total, 98.57% of reads could be aligned to the genome, covering 99.98% of the genome. According to the alignment result, we also counted and visualized the distribution of GC content, and the result showed a centralized distribution in both content value and sequencing depth, indicating a pure and reliable assembly (Figure 1C; Table 1). BUSCO was also used to evaluate the integrity of the genome with the actinopterygii_odb9 dataset, and the benchmark reached 95.4% (single-copy 92.6% and duplication 2.8%) (Simão et al, 2015). The relationship between M. cephalus and Planiliza haematocheila was closest, and the genome of P. haematocheila (Zhao et al., 2021) was newly published, offering the opportunity to validate our assembly. We chose the top 30 longest scaffolds of M. cephalus and mapped them to the chromosomes of P. haematocheila (Zhao et al., 2021) in order to observe their relationship to collinearity. The circos map showed these scaffolds could consistently map to P. haematocheila chromosomes comparatively well (Figure 1D).
Genome annotation and comparative genomics analysis
The information on repetitive elements in the genome assembly was essential before genome annotation and subsequent analysis. RepeatMasker (http://repeatmasker.org/RMDownload.html), RepeatProteinMask, RepeatModeler (http://repeatmasker.org/RepeatModeler/), LTR_FINDER, and TRF tool were all employed to identify all kinds of repetitive elements using homolog alignment based on RepBase (http://www.girinst.org/repbase) and de novo prediction, separately. A total of 152.03 Mb genome sequences were identified as repeats, accounting for 22.99% of the whole assembly. The prediction of protein-coding genes was also conducted through both de novo and homolog-based methods. Augustus, Genscan, and GlimmerHMM were used to carry out de novo prediction, and several representative fishes were used to finish homolog prediction. The coding genes were mapped to the M. cephalus genome by Blast, and the predicted gene was identified by Genewise (Birney et al., 2004). The gene sets occupied during alignment were several teleosts which frequently appeared in fish research, including Cynoglossus semilaevis, Danio rerio, Gadus morhua, Gasterosteus aculeatus, Oreochromis niloticus, Oryzias latipes, Seriola lalandi, and Takifugu rubripes. GLEAN was employed to integrate all predicted gene models into a single GFF result, and we manually filtered some unreliable predicted genes due to insufficient evidence number. For example, a predicted gene would be discarded if only one de novo evidence supports its presence but lacks evidence from the homolog-based method. In total, 23,987 genes were annotated, and the BUSCO benchmark of predicted protein sets was 91.6% (Figure 1E). We also compared the statistical characteristics of the gene sets, such as the distribution of CDS length and exon number, with the references mentioned above, which were used during BLAT, and the chart showed a consistent trend among those gene sets including our result (Figure 1F).
All valid predicted proteins were then aligned to different databases including the Kyoto Encyclopedia of Genes and Genomes (KEGG), Swissprot, TrEMBL, InterPro, and Gene Ontology (GO) using BLASTP with an E-value cutoff of 1E−5. Finally, 22,844 genes could be mapped to known functional genes among all 23,987 genes, accounting for 95.23% of the whole gene set.
Noncoding RNAs (ncRNAs) have been thought to be an important part of genome annotation since they are active in transcriptional and translational regulation of gene expression as well as in the modulation of protein function. Ribosomal RNA (rRNA) was predicted according to vertebrate rRNA data using BLASTn, and tRNAscan-SE v1.3.1 was applied with eukaryotic parameters to find transfer RNA (tRNA). MicroRNA (miRNA) and small nuclear RNA (snRNA) were discovered by mapping our genome result to the Rfam database. In total, 254 miRNAs, 1,138 tRNAs, 378 rRNAs, and 665 snRNAs were identified, and the total sequence length was 252.52 kb.
It was necessary to validate the phylogenetic position of M. cephalus since we have obtained two Mugilidae genomes. Other related species are referred to as C. semilaevis, D. rerio, G. aculeatus, O. latipes, S. lalandi, T. rubripes, and P. haematocheila. All involved CDS and protein sequences were aligned to find gene family clusters and single-copy genes among all those species. The difference in single-copy gene sequences was the base of the reconstruction of a phylogenetic tree. The differences among gene coding sequences, protein sequences, and fourfold degenerate synonymous site transversion rate (4DTV) were all taken into consideration to draw the most accurate and reasonable phylogenetic result with maximum-likelihood and Bayes methods (Figure 1G).
Except for the common gene set features among different genomes, such as single-copy gene families, the differences hidden in them were the keys to answering the phenotypic diversities. In our research, we employed Computational Analysis of Gene Family Evolution (CAFE) to discover the divergence of gene families among the species in this phylogenetic analysis. Even though it was a common trend that gene family contraction happened more than gene family expansion, the gene families that were expanded were more meaningful for phenotype or function research. A total of 712 gene families were found expanded in M. cephalus, and 243 of them were changed significantly, among which 2,535 genes were involved. GO and KEGG enrichment analyses were carried out to clarify the possible biological function or pathways they participated (Figure 1H, I).
More than 100 genes were enriched in several immune-related pathways, including immune system process, immune response, and response to the bacterium (Figure 1H). In the farming of fish, immunity occupies the top priority. Hitherto, several studies have already focused on the anti-infection immunity and stress response of M. cephalus (Waltham et al., 2013; Byadgi et al., 2016; Li et al., 2017), including the strategies for different kinds of infection, hypoxia, extreme osmotic pressure, heavy metals, and toxicants. The expansion of gene families related to immune response and sodium channel activity indicated that the source of environment adaptability of M. cephalus could be used as potential candidates during subsequent studies. Another obviously enriched pathway was the heparan sulfate proteoglycan (HSPG) biosynthetic process. HSPGs were involved in various biological processes such as bacterial and viral infection and were mainly studied in mammalian cells. The role of HSPGs in teleost is attracting attention, and they have already been identified in several fishes, including zebrafish (Filipek-Gorniok et al., 2021), Atlantic cod (G. morhua), and spotted wolffish (Anarhichas minor) (Tingbo et al., 2006). This would also be another interesting area for subsequent research on M. cephalus.
Another potentially valuable comparative genomics analysis was the identification of positively selected genes during evolution. The functions of these genes may indicate the environmental stress that the species have suffered. We employed a CodeML module from PAML with model “branch site” to find positively selected genes in M. cephalus, and a total of 1,166 genes were recognized. The presence of interferon regulatory factors (IRF1, IRF2) in the results indicated that the immune process was an important developing pathway in this species during evolution. Meanwhile, the presence of growth hormone-regulated TBC and DNA repair protein RAD51 suggested that M. cephalus could survive in a stressful environment and grow rapidly to mature in order to survive.
Data availability statement
The genome assembly was submitted to the China National Gene Bank Database (CNGBdb: CNP0002462), National Center for Biotechnology Information (NCBI: PRJNA785274), and National Genomics Data Center (GSA: CRA005518)
Ethics statement
The animal study was reviewed and approved by Southern Marine Science and Engineering Guangdong Laboratory.
Author contributions
BZ and ZD designed and supervised the study. H-BG and NZ performed computational analysis of stLFR, Hi-C, genome annotation, chromosome synteny analysis, and phylogenetic research. NZ and BZ wrote the manuscript. LJ edited the manuscript. All authors read and approved the final version of the manuscript.
Funding
This work was supported by grants from Special Funding for the Modern Agricultural Industrial Technology System (CARS-47-Z01).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Birney E., Clamp M., Durbin R. (2004). GeneWise and genomewise. Genome Res. 14 (5), 988–995. doi: 10.1101/gr.1865504
Byadgi O., Chen Y. C., Barnes A. C., Tsai M. A., Wang P. C., Chen S. C. (2016). Transcriptome analysis of grey mullet (Mugil cephalus) after challenge with lactococcus garvieae. Fish Shellfish Immunol. 58, 593–603. doi: 10.1016/j.fsi.2016.10.006
Cossu P., Mura L., Scarpa F., Lai T., Sanna D., Azzena I., et al. (2021). Genetic patterns in mugil cephalus and implications for fisheries and aquaculture management. Sci. Rep. 11, 2887. doi: 10.1038/s41598-021-82515-7
Dor L., Shirak A., Curzon A. Y., Rosenfeld H., Ashkenazi I. M., Nixon O., et al. (2020). Preferential mapping of sex-biased differentially-expressed genes of larvae to the sex-determining region of Flathead grey mullet (Mugil cephalus). Front. Genet. 11. doi: 10.3389/fgene.2020.00839
Filipek-Gorniok B., Habicher J., Ledin J., Kjellen L. (2021). Heparan sulfate biosynthesis in zebrafish. J. Histochem Cytochem. 69, 49–60. doi: 10.1369/0022155420973980
Gorski K., De Gruijter C., Tana R. (2015). Variation in habitat use along the freshwater-marine continuum by grey mullet mugil cephalus at the southern limits of its distribution. J. Fish Biol. 87, 1059–1071. doi: 10.1111/jfb.12777
Li H., Durbin R. (2009a). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25 (14), 1754–1760. doi: 10.1093/bioinformatics/btp324
Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al. (2009b). The sequence alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li L., Jiang M., Shen X. Q. (2017). Gene expressions levels of 14-3-3a, NKCCla, APO-14, and na(+)-K(+)-ATPasebeta in gill tissue of mugil cephalus acclimated to low salinity. Genet. Mol. Res. 16. doi: 10.4238/gmr16019444
Simão F. A., Waterhouse R. M., Ioannidis P., Kriventseva E. V., Zdobnov E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31 (19), 3210–3212. doi: 10.1093/bioinformatics/btv351
Tingbo M. G., Kolset S. O., Ofstad R., Enersen G., Hannesson K. O. (2006). Identification and distribution of heparan sulfate proteoglycans in the white muscle of Atlantic cod (Gadus morhua) and spotted wolffish (Anarhichas minor). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 143, 441–452. doi: 10.1016/j.cbpb.2005.12.022
Waltham N. J., Teasdale P. R., Connolly R. M. (2013). Use of flathead mullet (Mugil cephalus) in coastal biomonitor studies: Review and recommendations for future studies. Mar. pollut. Bulletin 69, 195–205. doi: 10.1016/j.marpolbul.2013.01.012
Wang O., Chin R., Cheng X., Wu M. K. Y., Mao Q., Tang J., et al. (2019). Efficient and unique cobarcoding of second-generation sequencing reads from long DNA molecules enabling cost-effective and accurate sequencing, haplotyping, and de novo assembly. Genome Res. 29, 798–808. doi: 10.1101/gr.245126.118
Keywords: Mugil cephalus, genome sequencing, de novo genome assembly, annotation, single-tube long fragment read
Citation: Zhao N, Guo H-B, Jia L, Dong Z and Zhang B (2022) The genome assembly of flathead grey mullet Mugil cephalus. Front. Mar. Sci. 9:988397. doi: 10.3389/fmars.2022.988397
Received: 07 July 2022; Accepted: 01 August 2022;
Published: 25 August 2022.
Edited by:
Marco Casu, University of Sassari, ItalyReviewed by:
Fabio Scarpa, University of Sassari, ItalySimone Peletto, Experimental Zooprophylactic Institute for Piedmont, Liguria and Valle d’Aosta (IZSTO), Italy
Copyright © 2022 Zhao, Guo, Jia, Dong and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Zhang, emI2MTEyNzNAMTYzLmNvbQ==; Zhongdian Dong, emRkb25nQGdkb3UuZWR1LmNu
†These authors have contributed equally to this work