 Jessica L. Bergman
Jessica L. Bergman Tayla Shaw
Tayla Shaw Suhelen Egan
Suhelen Egan Tracy D. Ainsworth
Tracy D. Ainsworth- Biological, Earth, and Environmental Sciences, University of New South Wales, Sydney, NSW, Australia
As sequencing techniques have advanced and become cheaper in recent years, there has been a rapid increase in the number of studies conducted into the role of the microbiome in coral health, physiology, and response to environmental change. However, there is substantial variation in the methodological approaches applied. For example, DNA extraction protocols and the types of tissues sampled from the coral meta-organism are known to influence the downstream analyses of the amplified microbial communities and subsequently the interpretation of the microbiome diversity, stability and role. Studies have generally focused on whole organisms, in which the coral sampling steps homogenize the meta-organism microhabitats, however other studies targeting specific microhabitats have identified sources of variation specific to distinct compartments of the coral’s microbial landscape. Here we present a comparative analysis of methodologies optimized for the generation of coral microbiome data from the coral tissues and whole coral fragments of two commonly studied branching coral genera with distinct tissue structure. We investigate the microbiome of the imperforate Pocillopora, where the coral tissue does not penetrate through the calcium carbonate matrix, and the perforate Acropora, where the coral tissues and skeleton are interwoven throughout the coral branch. Through comparing data generated from different DNA extraction protocols using fixed coral tissues isolated from the coral skeletal structure with fixed whole coral fragments, we identify sources of variation inherent to microbial data generated from different sample types, species, and extraction protocols.
Introduction
Environmental changes such as rising sea surface temperatures (Hoegh-Guldberg et al., 2007; Hughes et al., 2017), ocean acidification (Hoegh-Guldberg et al., 2007), and pollution (Silbiger et al., 2018) have resulted in substantial degradation to coral reefs worldwide. As reefs worldwide change in both structure and function (McFall-Ngai et al., 2013; Thompson et al., 2015; Ainsworth and Gates 2016; Apprill, 2017; Trevathan-Tackett et al., 2019), it is increasingly important to understand the contribution of the microbiome to the health, physiology, and environmental flexibility of coral species. Most studies investigating the coral microbiome have predominately focused on profiling microbial communities via next generation sequencing of 16S rRNA gene amplicons using whole fragments of individual corals, such as sections of branches collected from the colony of a branching coral species. Ultimately, the goal of these studies is to characterize coral-microbe interactions in different coral species (or morphotypes) in response to the combined challenges of environmental change (Stévenne et al., 2021). Considering the unique biotic interactions and dynamic relationships between the microbiome and the environment of morphologically, structurally, and functionally different coral hosts when assessing the response of the coral meta-organism may aid in meeting this goal.
Coral colonies harbor distinct and often complex microbial communities within the niche habitats of the colonial meta-organism (Rohwer et al., 2002; Sweet et al., 2011; Blackall et al., 2015), including the polyp tissue (Mouchka et al., 2010), surface mucus layer (Bythell and Wild, 2011; Glasl et al., 2016), and skeleton (Sweet et al., 2011). Whole coral fragments used for microbial analysis therefore include bacteria not only associated with the coral tissues but also a high number of taxa associated with coral mucus (Shnit-Orland and Kushmaro, 2009; Pereira et al., 2017), the calcium carbonate skeleton (e.g. nitrogen fixing bacteria, Williams et al., 1987; Shashar et al., 1994), and endolithic microbes (Marcelino and Verbruggen, 2016; Marcelino et al., 2017; Marcelino et al., 2018). The tissue and mucus harbor unique reservoirs of prokaryotes, with Apprill et al., 2016 finding 23-49% of sequences identified to be unique to tissues and 31-56% of sequences to be exclusive to mucus. The calcium carbonate skeleton of the coral also harbors a distinct microbial community with up to 80 taxonomic units associated with photosynthetic algae (usually Ostreobium spp: Le Campion-Alsumard et al., 1995; Massé et al., 2018; but see Marcelino and Verbruggen, 2016) and cyanobacteria within the endolithic layer (Ainsworth et al., 2010; Marcelino and Verbruggen, 2016; Marcelino et al., 2017; Marcelino et al., 2018). Interestingly, it has been suggested that in some species of coral the tissue is the microbial niche where the microbiota are responsive to different environmental conditions (Oculina patagonica, Rubio-Portillo et al., 2016; Astroides calcularis, Biagi et al., 2020), highlighting the importance of targeting the specific microbial community of the tissue in studies of coral response to anthropogenic stressors.
While there is growing awareness of the distinct microhabitats found in corals, studies often utilize whole fragments taken from the colonial coral structure to profile the coral microbiome (e.g. Pratte et al., 2018; Epstein et al., 2019). This approach results in a homogenization of the niche-specific microbial communities of the surface mucus layers, tissue, and skeleton (Rosenberg et al., 2007; Ainsworth et al., 2010). The inclusion of multiple microhabitats in whole coral fragments means that the microbial community composition of these samples includes the microbial communities of these microhabitats. Homogenization of microhabitats may therefore affect comparisons between studies describing and interpreting the role of the microbial community of the coral, depending on the objective of the study, with different methods of sample collection and processing capturing distinct compartments across the coral’s microbial landscape (Ainsworth et al., 2015, Neave et al., 2017, Weiler et al., 2018). The actual process of homogenization may also create variability depending on the method employed, e.g. between machine-programmed bead-beating and the manual variability of crushing samples with a mortar/pestle (Hernandez-Agreda et al., 2018), indicating a need for the standardization of homogenization methodology as well as targeting specific microhabitats for comparisons between species. When targeting coral tissues specifically, the effectiveness of tissue removal can vary amongst species, with tissue removal less successful for perforate coral species where the coral tissue penetrates throughout the coral skeleton (≥ 5 mm) (Edmunds, 1994; Edmunds and Gates, 2002). This can lead to potential differences in the microbial community profiles identified between species and subsequently require the application of distinct methodological approaches for different species.
Importantly, all studies comparing coral DNA extraction protocols have been conducted on homogenized whole fragments of coral, and to date there is little known about the importance of DNA extraction protocols on tissue samples. In the present study, we separated fixed coral tissue from whole coral fragments of both Pocillopora and Acropora, two morphologically distinct species, to investigate if methodological procedures for processing the microbial habitat (e.g. tissue) result in bias when interpreting the host microbiome. To further explore how different methods used in coral microbiome studies influence the microbial communities found in coral tissues versus homogenized fragments in two species, we also investigated three different sample processing approaches used in the DNA extraction steps on both coral tissue and coral whole fragments. Previous studies investigating methodological differences between DNA extraction protocols have found amplification protocols can influence the community composition (Santos et al., 2012; Baker and Kellogg, 2014; Weber et al., 2017; Bardenhorst et al., 2021), with some protocols resulting in greater microbial richness and coverage of dominant microbial groups (Weber et al., 2017) and others resulting in differing numbers of consistent amplifications (Baker and Kellogg, 2014). Of these studies, none have compared extraction methodologies on paraformaldehyde preserved tissues. A recent study also found all protocols tested suitable for comparing coral microbiomes (Pratte and Kellogg, 2021). The most common goal within coral microbiome studies is to determine if the composition of the microbiome differs significantly between two or more groups of samples, and these studies demonstrate that it is important to identify and account for sources of variation beyond variables that differ between the study groups. In the current study we aimed to determine how the microhabitat sample type and tissue processing steps, from tissue structures of differing coral genera and morphotypes, influence the resulting coral microbiome dataset. By investigating the influence of sample type/microhabitat, host species/morphotype, and DNA extraction protocols on the amplification of DNA from fixed coral tissues and the generation of microbial datasets, we found the greatest differences between the microbial communities of whole fragments versus tissue biopsies, as well as between the tissue biopsies of different species.
Materials and equipment
Sample collection
Branch fragments (~3 cm) of Acropora muricata (n = 18) and Pocillopora damicornis (n = 9) were collected on snorkel using needle-nose pliers sterilized between samples with 70% molecular-grade ethanol from the shallow fringing reef (1-2 m depth) on the southern side of Heron Island, Australia in 2020 (23.4423°S, 151.9148°E; Permit G19/41974.1). Samples were immediately placed in 50 mL conical tubes filled with preservative. Fragments (~ 3 cm) of P. damicornis (n = 9) were also collected on snorkel using needle-nose pliers sterilized between samples with 70% molecular-grade ethanol from the shallow fringing reef (1-2 m depth) of Lord Howe Island in 2019 (31.5553°S, 159.0821°E; Permit MEAA19/206) and immediately placed in 50 mL conical tubes filled with preservative. In both locations, samples were collected from colonies > 3 m apart and separated by distinct sand patches to reduce the chances that colonies were clonal. Fragments were transported in conical tubes filled with preservative (preparation detailed in the following paragraph) to the adjacent research stations following collection (Figure 1A). Species are hereafter referred to as Acropora and Pocillopora.
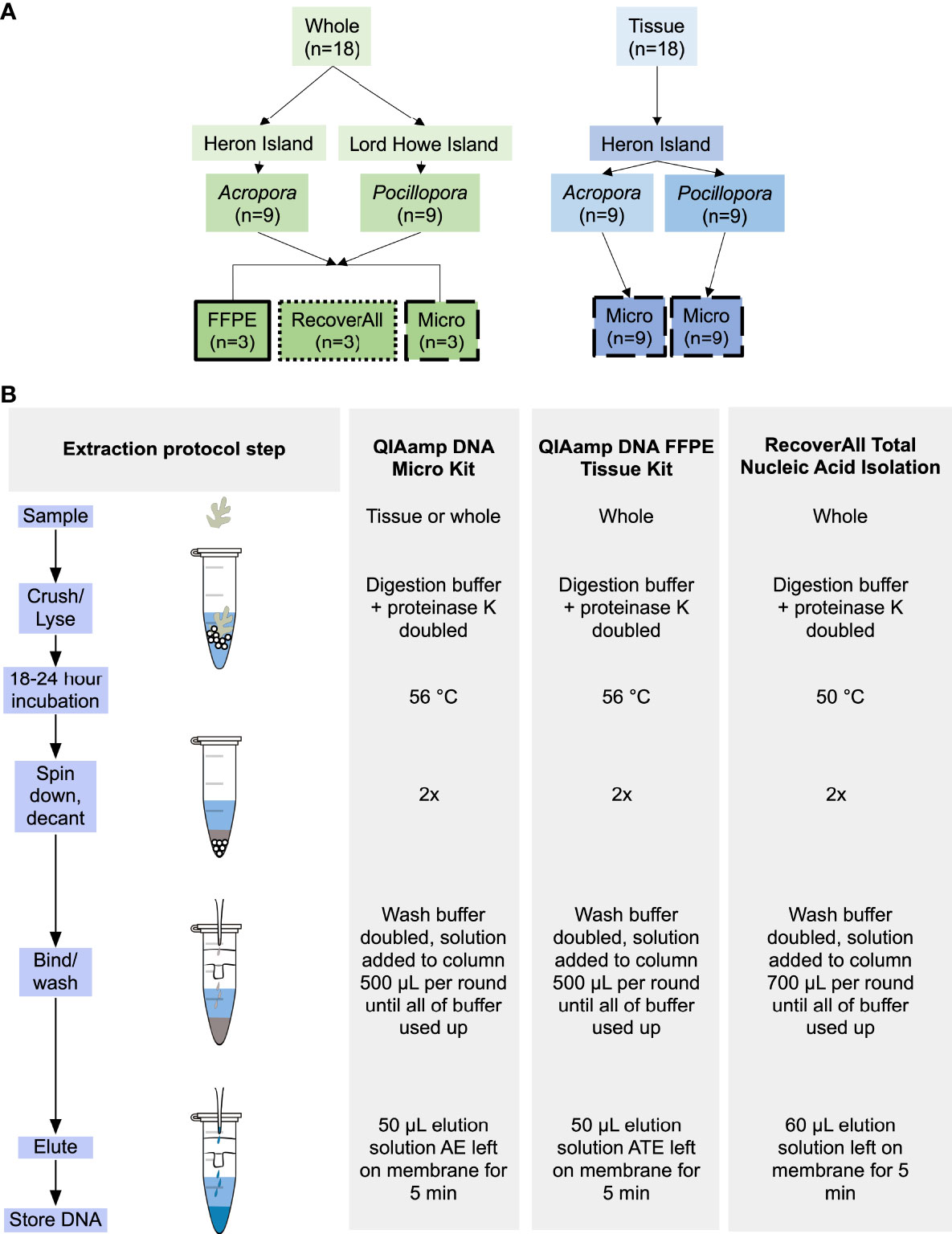
Figure 1 Schematics of (A) experimental design: highlighting sample type, species, and extraction protocol, and (B) extraction protocols used in the present study: highlighting subtle differences in methodologies between protocols.
Sample preservation
Sterile 16% paraformaldehyde ampules (Electron Microscopy Sciences, cat # 50980487) were used for the preparation of 4% paraformaldehyde by a 1:3 dilution with phosphate buffered saline (PBS) solution (PBS tablets (Invitrogen, USA) in UltraPure DNA/RNA-Free Distilled Water (ThermoFisher Scientific, USA). Coral fragments were then added to 50 mL conical tubes immediately following collection and covered with the 4% PFA solution. After 14 hours, PFA solution was removed and replaced with the DNA/RNA free PBS buffer solution for storage. Samples were stored at 4°C for ~3 – 6 months until sample processing.
Decalcification and tissue biopsies
The coral tissues were isolated from each individual branch fragment (n = 9 Acropora, 9 Pocillopora) using a DNA/RNA free solution of 20% Ethylenediaminetetraacetic acid (EDTA) (described by Hernandez-Agreda et al., 2018). When compared to other methods of preservation (e.g. snap freezing, salt-saturated dimethyl sulfoxide), samples preserved in PFA and decalcified using the following method yielded comparable results for both numbers of sequences and of generated OTUs (Hernandez-Agreda et al., 2018). Branches were submersed in EDTA solution and incubated at 4°C on a shaker tray, with a complete solution change every 3-4 days over a four-week period. Once the entire calcium carbonate structure was dissolved a tissue biopsy was collected from each coral sample using a 1.5 mm tissue biopsy punch tool (ProScitech Pty Ktd, QLD, Australia). Tissue biopsies were placed immediately into 1.5 mL centrifuge tubes and frozen at -20°C until DNA extraction (~ 1-2 weeks following decalcification).
Extraction method
DNA was extracted from coral samples using three protocols (Figure 1B):
1. QIAamp DNA FFPE Tissue Kit (cat #56404) (FFPE) – specifically designed for purifying DNA from formalin-fixed tissue sections.
2. QIAamp DNA Micro Kit (cat #56304) (Micro) – designed for purification of genomic and mitochondrial DNA from small sample sizes.
3. RecoverAll™ Total Nucleic Acid Isolation Kit for FFPE (cat #AM1975) (RecoverAll) – optimized for extraction of nucleic acid from difficult samples, e.g. formalin-preserved samples that can modify protein-protein/protein-nucleic acid crosslinks.
All three protocols were optimized from manufacturer’s protocols to produce the highest quality DNA for sequencing possible (Table S1).
Whole coral fragment sample processing with the FFPE/Micro protocol
Whole fragments of preserved Acropora and Pocillopora (1-2 cm fragments, n = 3 per species per protocol) were added to 2 mL tubes of 1.4 mm ceramic spheres (Matrix E, MP Biomedicals). Digestion buffers were doubled for all protocols, a step which greatly increased DNA yields following overnight digestion. A FastPrep-24 5G homogenizer (MP Biomedicals, Irvine, CA, USA) was programmed to run 3 rounds of 20 s each (6.0 m/s) to homogenize the sample. The duration and speed of sampling was chosen because it most effectively homogenized the coral skeleton in the shortest amount of time, e.g. homogenizing the sample fully while minimizing the amount of time the sample was at room temperature. Following homogenization, all samples were incubated overnight (18-24 hours) at 56°C. Samples were then centrifuged (3 min at 10,000 RPM) to pellet calcium carbonate remaining from the coral skeleton. The resulting supernatant was transferred to a new 2 mL tube, centrifuged again to pellet out any remaining calcium carbonate (3 min at 14,000 RPM), and supernatant was transferred to a new 2 mL tube. The centrifugation step was doubled from the manufacturer’s protocol to reduce the chances of calcium carbonate clogging the collection column. AL buffer and 100% molecular-grade ethanol were doubled to increase in proportion to the increased ATL and Proteinase K buffer volume prior to vortexing and adding to the collection column (QIAmp DNA Mini Kit, Qiagen). Following the manufacturer’s protocol for the remainder of the procedure, 500 µL of buffer solution was added to the collection column and washed until no buffer solution remained and eluted to 50 µL (Solution ATE for FFPE, Solution AE for Micro).
Whole coral fragment sample processing with RecoverAll protocol
1-2 cm fragments (n = 3 per species) were added to 2 mL tubes containing 1.4 mm ceramic spheres (Matrix E, MP Biomedicals). 400 µL of digestion buffer and 8 µL of Proteinase K were added prior to bead-beating as described previously. Following homogenization, all samples were incubated overnight (18-24 hours) at 50°C and centrifuged twice (3 min at 10,000 RPM, 3 min at 14,000 RPM) to pellet and remove calcium carbonate remaining from the coral skeleton. Isolation additive and 100% molecular-grade ethanol were doubled to increase in proportion to the increased digestion buffer and Proteinase K buffer volume prior to vortexing and adding to the collection column. The manufacturer’s protocol was followed for the remainder of the procedure and DNA eluted to 60 µL.
Tissue sample processing
1 cm diameter tissue biopsies (1.5 mm thick, n = 9 per species) were lysed following manufacturer’s protocol for RecoverAll, FFPE, and Micro protocols using corals collected from Heron Island. No mechanical lysis (e.g. bead-beating) step was required for the biopsies, as there was no calcium carbonate skeleton to break down and chemical lysis fully homogenized the sample. Following homogenization, all samples were incubated overnight for 20 hours at 56°C. The manufacturer’s protocol was followed for the remainder of the procedure. 1µg of carrier RNA was dissolved in buffer AE and added to buffer AL to increase yields for the FFPE and Micro protocols. For elution, 30 µL of elution solution was incubated at room temperature on the membrane for 5 min before centrifuging the sample for 1 min at 10,000 RPM.
Sample quality and efficiency cutoff
Extracted DNA concentration and purity were quantified using a NanoDrop 2000c Spectrophotometer (Thermo Scientific, Waltham, MA, USA). Samples with a concentration of equal to or greater than 20 ng/µL and a ratio of absorbance at 260/280 nm between 1.5 and 2.3 were sequenced. We were unable to achieve yields and 260/280 ratios that met our sequencing cutoff using the FFPE and RecoverAll protocol on tissue samples, so only the tissue biopsy samples processed using the Micro protocol were sequenced. Samples processed using the FFPE protocol yielded high 260/280 ratios (> 2.3) and poor DNA resolution, indicative of DNA shredding caused by mechanical disruption. Samples processed using the RecoverAll protocol had yields < 10 ng/µL. Extracted DNA was stored at -20°C for ~ 1 week prior to PCR amplification and sequencing.
16S rRNA gene amplicon sequencing and analysis
DNA extraction, amplification, and sequencing were performed on all samples as well as on one negative control (no sample template) prepped for each protocol and sample type cross (n = 36 samples, 4 negative controls in total). Sequencing was performed by MR DNA (Molecular Research LP, Shallowater, TX, USA) on the Illumina MiSeq platform following manufacturer’s guidelines. The 16S rRNA gene V1-V3 regions PCR primers 27F/519R were used in a 30-35 cycle PCR using the HotStarTaq Plus Master Mix Kit (Qiagen, USA) under the following conditions: 95°C for 5 minutes, followed by 30-35 cycles of 95°C for 30 seconds, 53°C for 40 seconds and 72°C for 1 minute, after which a final elongation step at 72°C for 10 minutes was performed. Samples were multiplexed using unique dual indices, pooled together in equal proportions based on molecular weight and DNA concentrations, and purified using calibrated Ampure XP beads. The pooled and purified PCR product was then used to prepare an Illumina DNA library on an Illumina MiSeq following the manufacturer’s guidelines and resulting in 2 x 300 bp paired end sequences.
Sequence data were analyzed using Quantitative Insights Into Microbial Ecology version 2022.2 (QIIME2, Bolyen et al., 2019). After denoising and primer removal using the DADA2 pipeline (Callahan et al., 2016) with parameters p-trim-left-f 6, p-trunc-len-f 300, p-trim-left-r 6, p-trunc-len-r 210, taxonomy was assigned to amplicon sequence variants (ASVs) in QIIME2 using a naïve Bayes classifier trained on the Greengenes 13_8 database (McDonald et al., 2012), a reference database commonly used in microbiome analysis (Knight et al., 2018). ASVs assigned as “chloroplasts”, “mitochondria”, or unassigned (classification absent at a phylum level) were excluded from the final ASV table. Quality control was conducted in R (version 4.1.0) using the package decontam at a threshold of 0.5, which implements a statistical classification procedure that identifies contaminants in sequencing data (Davis et al., 2018). In brief, decontam at the 0.5 threshold identifies and filters out contaminants that appear more frequently in negative controls than experimental samples (Davis et al., 2018). Data exploration was conducted in R using the package phyloseq (McMurdie and Holmes, 2013). The DESeq2 package (Love et al., 2014) was used to calculate geometric means of ASV counts prior to estimating size factors using the function estimateSizeFactors. The logfold2change value was then compared between protocols and species using the Wald Test, with the differential abundance measurements considered statistically significant at an adjusted p-value of < 0.05. The coral core microbiome was determined based on microbe presence in 100% of samples at ASV and genus level (van de Water et al., 2016). Indicator species were identified using the package indicspecies (De Cáceres and Legendre, 2009).
Statistics
All statistical analyses were performed in R, using the package vegan for multivariate statistics and ggplot2 for data visualization. Four comparisons were conducted: 1). Pocillopora whole fragments * protocol, 2). Acropora whole fragments * protocol, 3). Acropora samples extracted with the Micro protocol * sample type, and 4). Tissue samples extracted with the Micro protocol * species. Geographically distinct samples (e.g. samples from Heron Island and Lord Howe Island) were not directly compared due to previous evidence showing that bacterial community structures are different between regions and reefs (Hernandez-Agreda et al., 2016). For each comparison, a multiple analysis of variance test (MANOVA) was conducted with extraction protocol, sample type, or species as the main effect and DNA concentration, number of reads per sample, and number of taxa per sample as response variables. For community composition analysis, ASV counts were Hellinger transformed to reduce the effects of numerically large values from overly abundant taxa (Legendre and Gallagher, 2001; sensu Ricci et al., 2022). Alpha diversity metrics were analyzed using separate one-way ANOVAs for each treatment using unrarefied data (Shannon, Chao1, and Inverse Simpson). In cases where residuals were not normally distributed (determined via Shapiro test), data was log-transformed to meet assumptions of normality. For beta diversity, metrics included weighted UniFrac, unweighted Unifrac, and Bray-Curtis distance matrices on unrarefied data. A permutational multivariate analysis of variance (PERMANOVA, n = 9999, adonis function in vegan) was performed on each metric to test for dissimilarities in microbial community composition between samples. Homogeneity of dispersion around group centroids was assessed for each metric using PERMDISP (betadisper function in vegan).
Results
In total, 32,616 sequences from 36 samples and 4 negative controls were generated. Quality control and removal of chloroplasts, mitochondria, unassigned ASVs (classification absent at a phylum level), singletons, and potential contaminants resulted in the retention of 29,940 sequences with a mean of 785 ± 137 reads per sample, ranging from a minimum of 10 reads to a maximum of 4434 reads. Plotting species richness against sequence sample size for data filtered using decontam indicate that adequate depth of sequencing was achieved based on sequencing plateaus (Figure S2). Random subsampling of even numbers of sequences per sample has been criticized in studies with small sample sizes and where the number of sequences per sample vary by 2 or more orders of magnitude (McMurdie and Holmes, 2014), so rarefied data was not used in the present study.
A literature search was conducted to determine reported microbiome sequencing data for relevant coral taxa and methodological approaches used in the current study (Table 1). The total reads and average number of reads per sample were lower than what has been observed for P. damicornis and other species within its species complex (Schmidt-Roach et al., 2014), but still within the range of sequencing depth used for analysis of tissue and whole fragment samples of Pocillopora spp. (e.g. 412 – 22,900, Table 1). Clustering to 99% similarity and removing negative controls yielded 746 distinct amplicon sequence variants (ASVs) for analysis of the microbial communities present after extraction with each protocol or sample type (n = 36 samples). 11 ASVs were removed (from 22 of the 36 samples) as contaminants (Table S1; Figure S3), identified as all sequences more prevalent in negative controls than in samples and reducing total number of reads from 32,616 to 29,940. Between 1-6 ASVs identified as contaminants were removed from each sample. Contaminants were all assigned to the class Alphaproteobacteria and contained the families Caulobacteraceae, Hyphomicrobiaceae, Phyllobacteriaceae, Rhodobacteraceae, Rhodospirillaceae, Acetobacteraceae, Deinococcaceae, and Sphingomonadaceae.
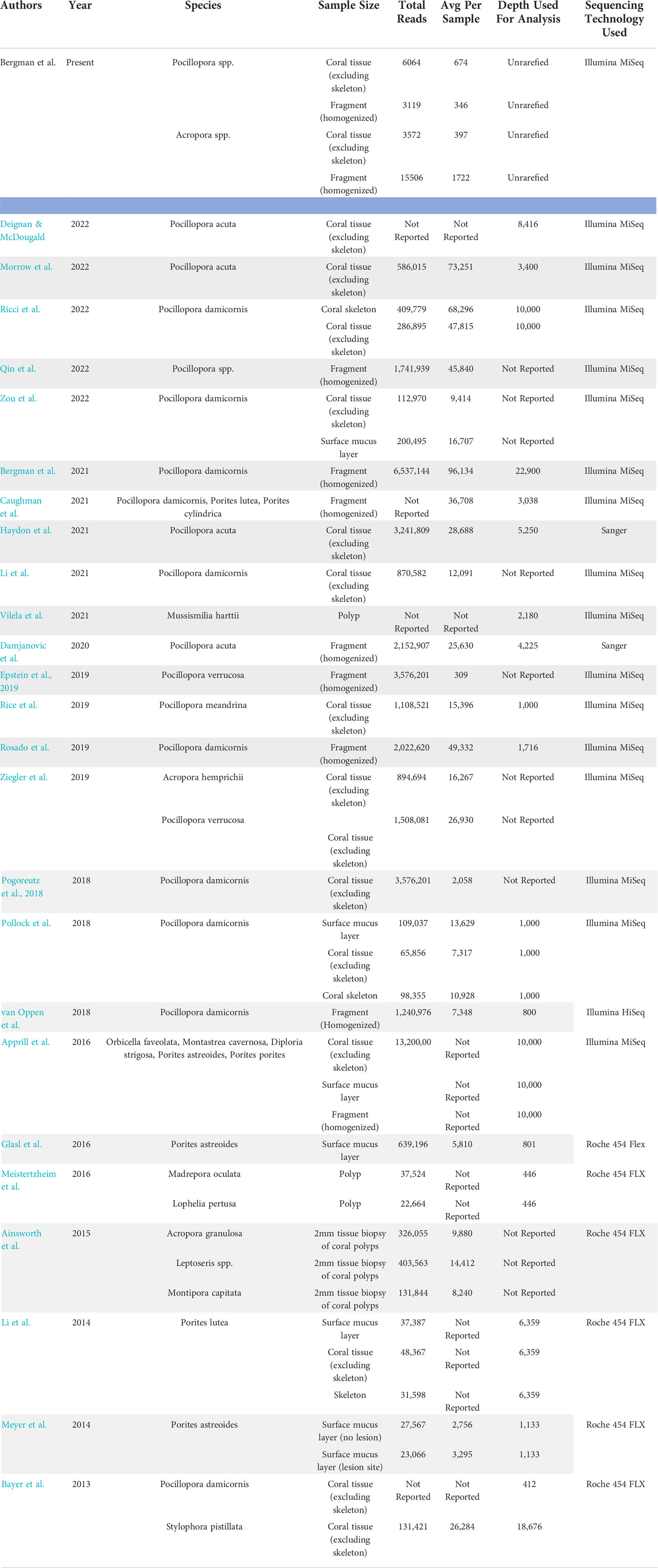
Table 1 Summary of a literature search conducted to determine reported microbiome datasets for relevant taxa and methodological approaches used in the current study.
Extraction protocol has limited influence on coral microbiome
No significant differences in alpha diversity metrics were found between extraction protocols for either Pocillopora or Acropora whole fragments (Figures 3A, B; Table 2). Overall ASV evenness (Shannon diversity) did not differ across protocols for Pocillopora samples (F = 1.750, p = 0.252) or Acropora samples (F = 1.124, p = 0.395). ASV richness (Chao1) was also similar between protocols for Pocillopora (F = 0.415, p = 0.678) and Acropora samples (F = 0.273, p = 0.770). Dominance (Inverse Simpson) also did not differ between protocols for either Pocillopora (F = 2.048, p = 0.21) or Acropora samples (F = 0.564, p = 0.597). Beta diversity and dispersion also did not differ between protocols for either Pocillopora or Acropora samples using weighted UniFrac, unweighted UniFrac, or Bray-Curtis distance matrices (Figures 4A, B; Table 2). DESeq2 did not identify significant differential abundances across protocols for Acropora samples (p > 0.97), but one ASV, an unidentified Devosia spp., was found to differ across protocols for Pocillopora samples (p < 0.05). For both Pocillopora and Acropora whole fragments, there was no difference between protocols for yield, reads, or taxa numbers (Table 3).
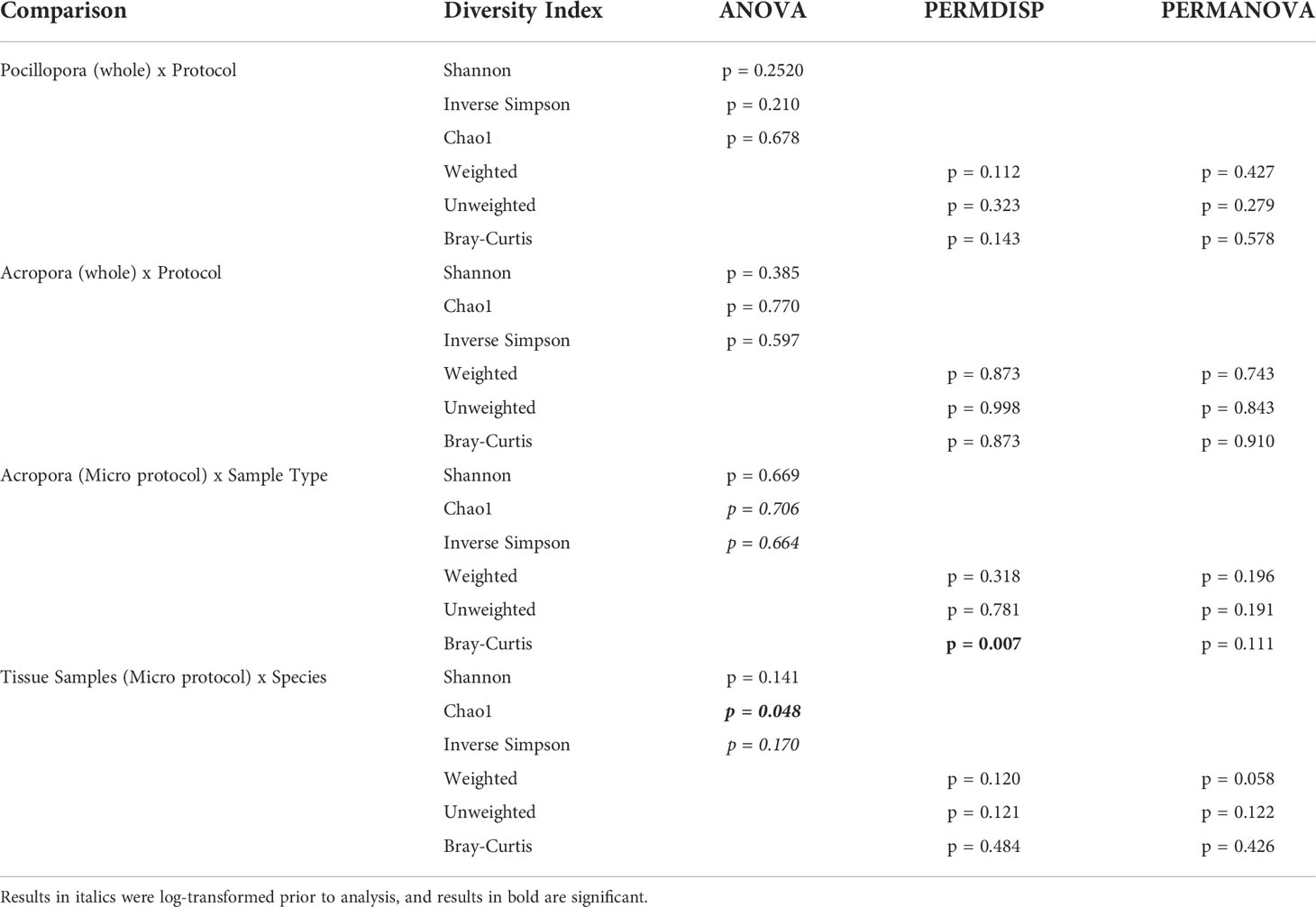
Table 2 Results for all alpha and beta diversity analyses.
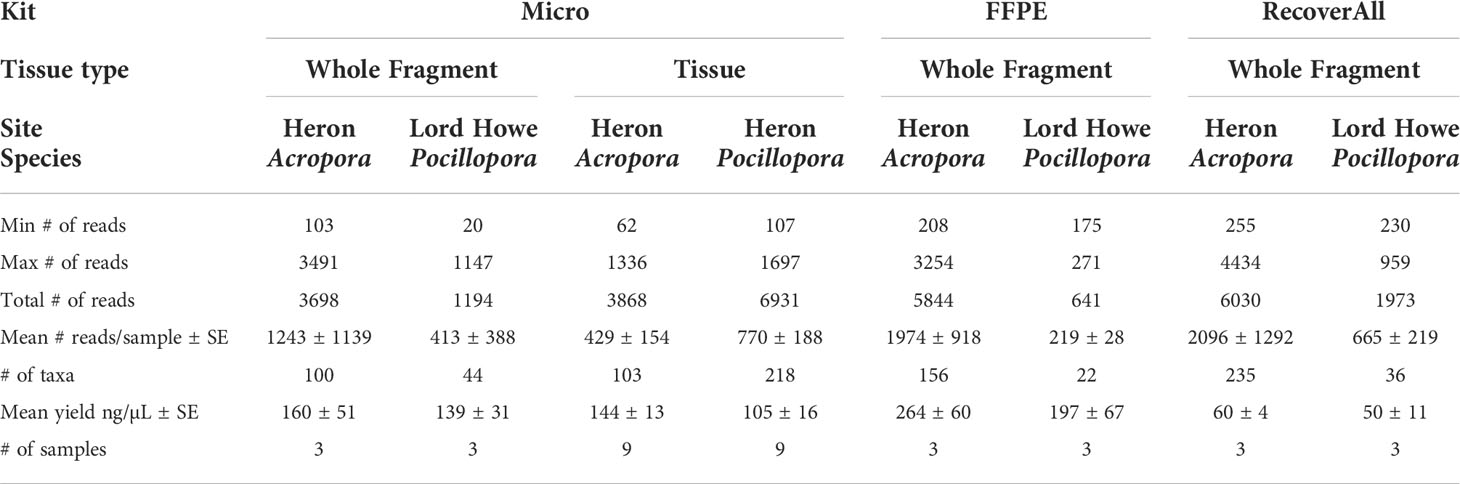
Table 4 Table of reads, taxa, and mean yields for each coral species, protocol, and sample type.
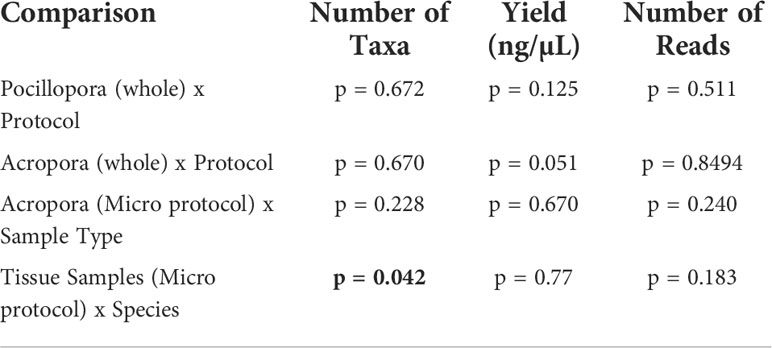
Table 3 Results of MANOVA comparing number of taxa at ASV level, yield, and number of reads for each combination of coral species, protocol, and sample type tested in the present study.
Sample type impacts differential abundance in Acropora samples
When comparing microbial communities between whole and tissue samples of Acropora samples using only the Micro protocol, both whole and tissue samples of Acropora were dominated by Alphaproteobacteria (Figure 2), with both the whole fragments and the tissue samples dominated by Rhodobacterales (relative mean abundance of 80 ± 9% and 28 ± 6%, respectively). No significant differences in alpha diversity metrics were found between whole or tissue sample types (Figure 3C). Chao1 and Inverse Simpson values were log-transformed to meet assumptions of normality. There were no differences between sample types in Shannon (F = 0.194, p = 0.669), Chao1 (F = 0.151, p = 0.706), or Inverse Simpson (F = 0.201, p = 0.664) diversity metrics (Table 2). Dispersion differed between sample types for Bray-Curtis distance (PERMDISP; p = 0.007), but no other beta diversity metrics were significantly different (Figure 4C; Table 2). 4 ASVs were significantly different in whole fragments, all of which were identified to the family Rhodobacteraceae with an average log2 fold change value of -12.84 ± 0.08. No difference was found between sample types for yield, reads, or taxa numbers (Table 3). Indicator species could not be identified between sample types, and there were no core taxa shared at 100% prevalence across whole Acropora samples or tissue Acropora samples using the Micro protocol.
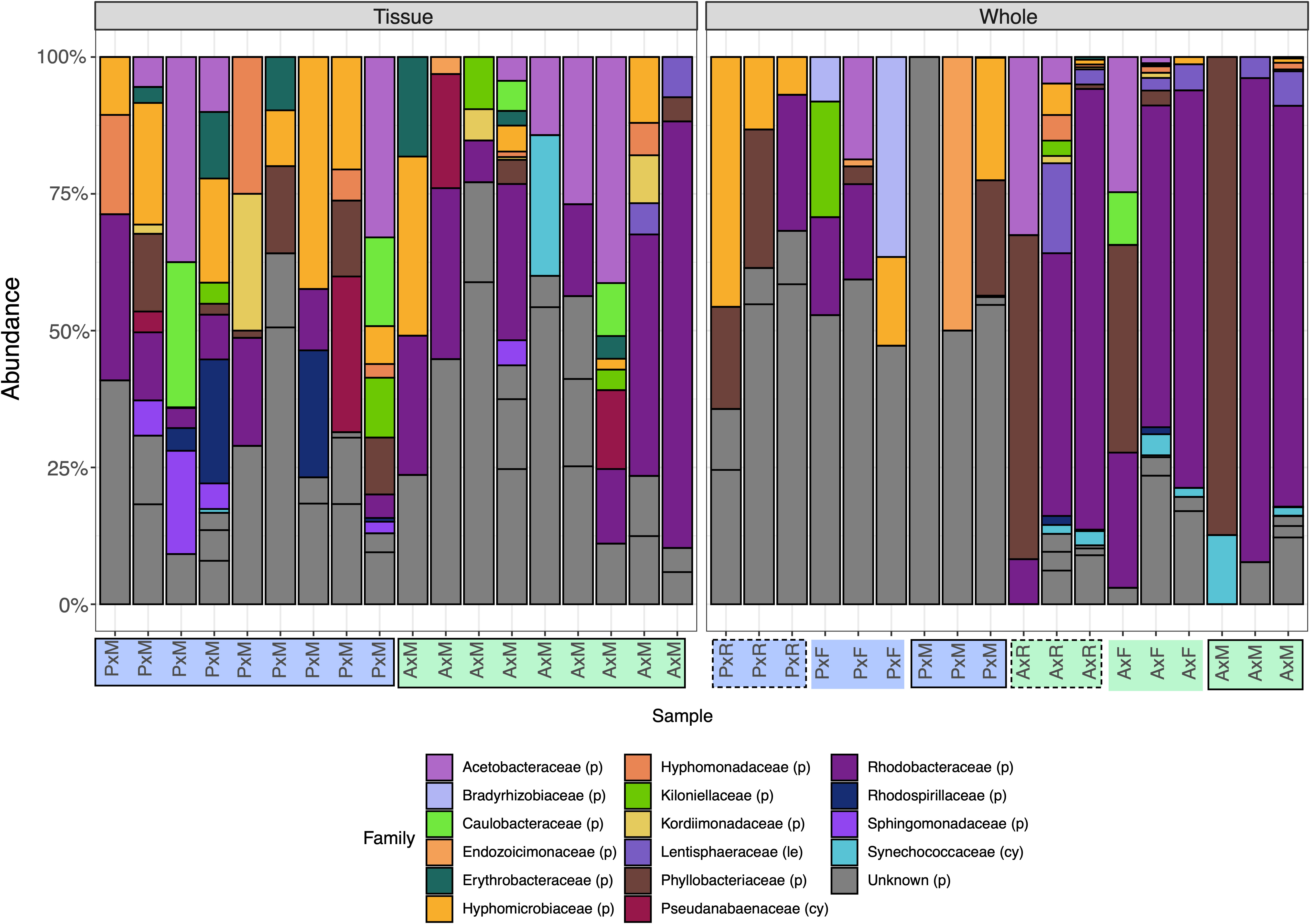
Figure 2 Relative abundance of the top 20 bacterial families present in samples from three protocols (M/solid border: Micro, R/dashed border: RecoverAll, F/no border: FFPE), two coral genera (P/blue: Pocillopora, A/green: Acropora), and two sample types (Tissue and Whole). Extractions on tissue biopsies were only successful using the Micro protocol, whereas extractions on whole fragments were successful using the RecoverAll, FFPE, and Micro protocols. Species that had the same taxonomy at the family level were merged using the tax_glom function in phyloseq. Four groups could not be resolved to the family level (“Unknown”) and are represented as distinct ASVs by subdivisions of grey columns. Phylum is indicated by either (p): Proteobacteria, (le): Lentisphaerae, or (cy): Cyanobacteria.
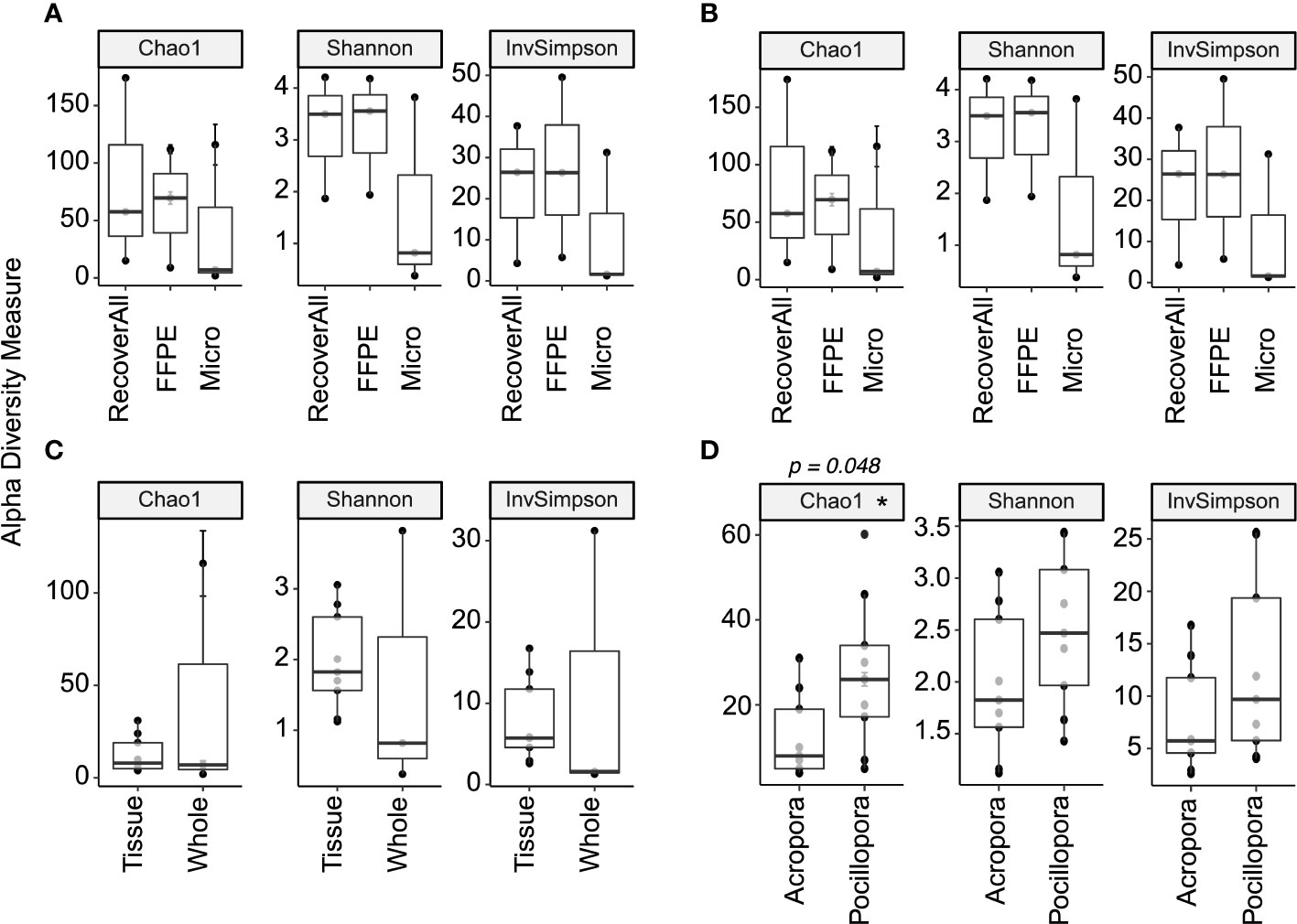
Figure 3 Alpha diversity metrics comparing extraction protocols between (A) whole Pocillopora samples and (B) whole Acropora samples, (C) sample type for Acropora using the Micro protocol, and (D) species using tissue samples extracted using the Micro protocol. A star (*) denotes significant differences (p < 0.05).
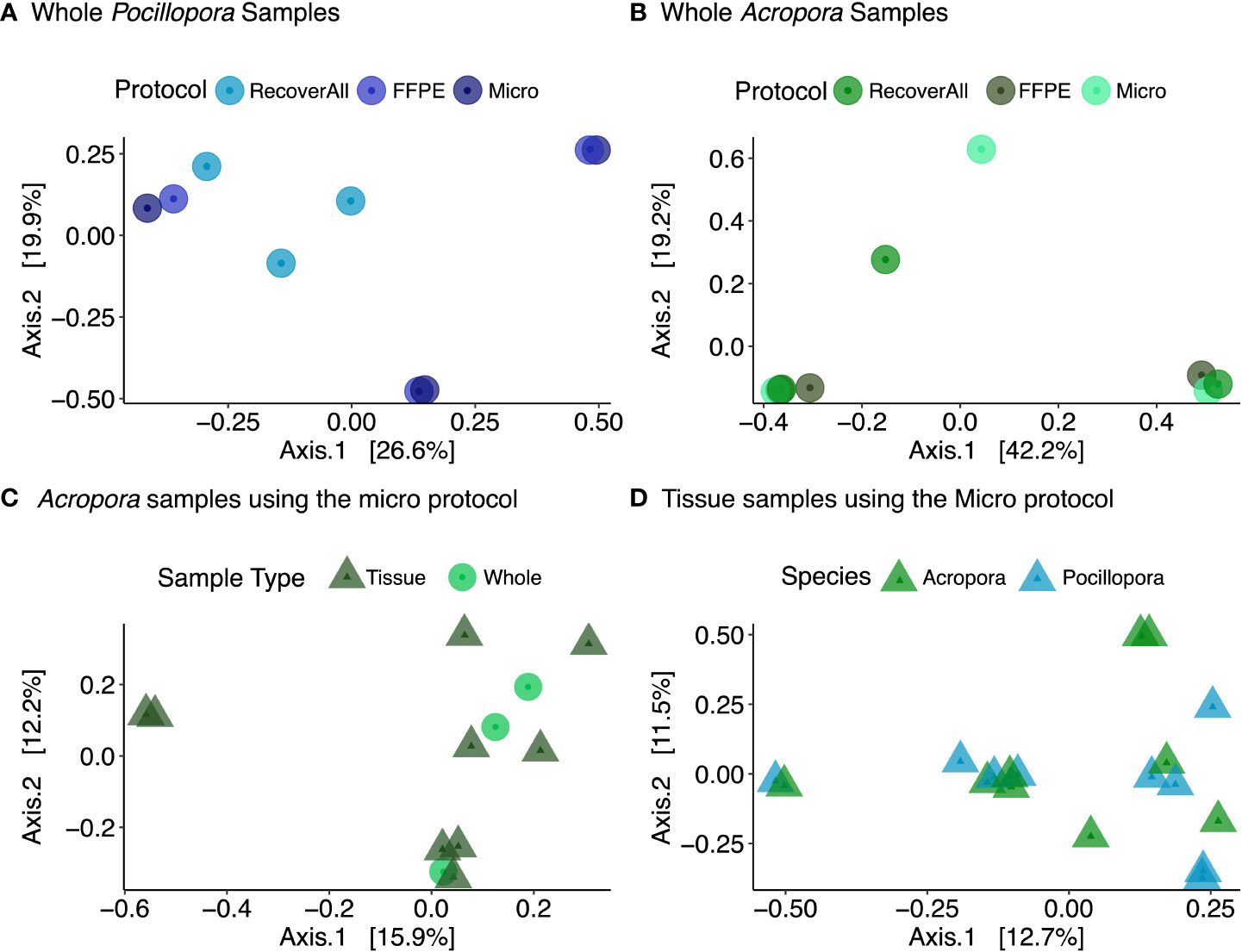
Figure 4 PCoA plots showing Bray-Curtis distance across (A) extraction protocols using whole Pocillopora samples and (B) whole Acropora samples, (C) sample type for Acropora using the Micro protocol, and (D) species using tissue samples extracted using the Micro protocol.
Host species influences diversity and taxa numbers in tissue biopsies
When comparing microbial communities between Acropora and Pocillopora tissue samples using the Micro protocol, both species were dominated mainly by Alphaproteobacteria (Figure 2). Pocillopora tissue samples were dominated by Rhiziobiales (relative mean abundance of 35 ± 8%), and Acropora tissue samples were dominated by Rhodobacterales (relative mean abundance of 28 ± 18%). Chao1 and Inverse Simpson values were log-transformed to meet assumptions of normality (Table 2). There were no differences between species in Shannon (F = 2.394, p = 0.141) and Inverse Simpson (F = 2.068, p = 0.170) metrics of diversity, but Chao1 differed between Acropora and Pocillopora samples (ANOVA; F = 4.069, p = 0.048, Figure 3D). Beta diversity metrics did not differ between species (Figure 4D; Table 2). Differential abundances were also not significant between species (p > 0.79). There was no difference found between species for yields or reads but taxa numbers were significantly higher in Pocillopora samples (p = 0.042), averaging 29 ± 6 taxa per sample for Pocillopora and 13 ± 3 taxa per sample for Acropora (Table 4). Indicator species could not be identified between sample types, and there were no core taxa shared at 100% prevalence across Acropora or Pocillopora tissue samples using the Micro protocol.
Discussion
This study aimed to identify sources of variation in microbial datasets generated from different sample types, species, and extraction protocols by comparing methodologies of coral microbiome data generation. In assessing yields, reads, taxa numbers, and diversity between protocols we find that no single extraction protocol is superior for DNA extractions when using whole coral samples, but only one protocol from the current study is suitable for tissue biopsy samples. We also find significant differences between whole fragments and tissue biopsies of Acropora, and between Acropora and Pocillopora tissue biopsies, suggesting that method of sample preparation (whole fragment versus tissue biopsy) does influence the coral microbiome based on the biology of the targeted species. We therefore suggest that future studies interpreting the response of the coral microbiome to change target communities of interest, for example by accounting for morphology when selecting species, and therefore conserve the role, function, and significance of the coral-microbe interactions across the coral meta-organism.
All extraction protocols tested met standard DNA quality standards for bacterial 16S amplification from whole fixed Pocillopora and Acropora fragments, a finding repeated in prior methodology studies (Baker and Kellogg, 2014; Weber et al., 2017; Pratte and Kellogg, 2021). However, in tissue samples from both species, low yields (< 20 ng/µL) and high 260/280 ratios (> 2.3) indicative of possible phenol contamination, DNA shredding, or poor sample quality limited extractions to the Micro protocol. It has been suggested that, in samples with low bacterial biomass in relation to overall sample size and DNA content (such as corals), contaminants may be incorrectly identified as novel experimental findings (Glassing et al., 2016; Olomu et al., 2020). Contaminants identified in blank protocol samples were therefore removed from study samples, reducing total number of reads from 32,616 to 29,940 (8% of reads associated with contaminants). Studies that have identified kit contaminants acknowledge that many of the contaminants detected can also be due to laboratory/cross-contamination or genuinely present in samples (Kellogg, 2019) so we conservatively removed only 11 taxa that were found to be more present in negative controls than in study samples. While read counts were relatively low for all samples, with 22% of samples (8 out of 36) having below 100 reads, these low reads were evenly distributed across protocols, species, and sample types and no clear pattern in read counts emerged. The number of read counts reported in this study includes samples with both large and small amounts of starting material, e.g. 1 cm diameter tissue biopsies (skeletal material removed prior to biopsy) and 3 cm branch fragments (from which 1 – 2 cm were used for DNA extraction). This is reported similarly to other studies where sample sizes with different amounts of starting material are analyzed, e.g. where surface mucus layer, tissue, and skeleton show significant differences from each other (Sweet et al., 2011). Consistently, tissue samples excluding skeletal material show lower read counts than whole fragments or mucus samples (Table 1, Meistertzheim et al., 2016; Zou et al., 2022). Specifically, for P. damicornis, read counts per sample as high as 96,134 counts have been recorded in the literature for whole fragments (Bergman et al., 2021), compared to 9,414 – 12,091 for tissue samples excluding coral skeleton (Li et al., 2021; Zou et al., 2022). Pollock et al. (2018) also found an average of 13,629 reads per sample in the P. damicornis mucus layer and 10,928 reads per sample in the P. damicornis skeleton, compared to only 7,317 reads per sample in the P. damicornis tissue, highlighting how sample size and read count per sample may bias the overall read count. In addition, similar to Clinton et al., 2021 study on fish gills, clear plateaus in the present study were seen in sequencing curves following filtration, suggesting that even where reads were < 1000 an adequate sequencing depth was reached. No significant differences were found between any alpha or beta diversity metrics across protocols for whole fragments, but one taxa differed in relative abundance in Pocillopora whole fragments across protocols. The only taxa observed to differ across protocols for Pocillopora samples, a Devosia spp. found enriched in samples using the Micro protocol, has been identified in the Clade C Symbiodinium core microbiome in corals previously (Lawson et al., 2018), suggesting that protocol choice has little bias on the bacterial community of whole Pocillopora samples but may be better suited to recover sequences of bacteria closely associated with Symbiodiniaceae.
Only the Micro protocol was found to be suitable for use with coral tissue samples. Similarly to Pratte and Kellogg, 2021, all protocols for whole fragments here used bead-beating, which has been found to result in higher DNA concentrations and higher degrees of microbial diversity than other cell lysis methods (Lim et al., 2018; Pollock et al., 2018) and for tissue samples bead beating was excluded. Pratte and Kellogg (2021) also hypothesized that the inclusion of a bead-beating step in all extraction methods contributed to very few significant differences between methods, and we therefore hypothesize that the exclusion of a bead-beating step for the tissue samples (chemical lysis only) could explain the differences in effectiveness between protocols for the tissue samples. However, the addition of a bead-beating step in preliminary trials (Table S2) did not increase yields, suggesting that the chemical lysis steps in the FFPE and RecoverAll protocols were not compatible with mechanical lysis steps for tissue samples in the present experiment. There is substantial evidence in the literature in both coral reefs (Pratte and Kellogg, 2021) and other systems that the effect of extraction protocol on bacterial sequencing result is small, and often nonexistent, when compared to variability across samples, species, and sample types (Wagner Mackenzie et al., 2015; Marotz et al., 2017). By comparing extraction protocols across different sample types, we find here that the effect of sample type (e.g. whole fragment versus tissue biopsy samples) had the greatest influence on extraction protocol success, and therefore more closely examined what influence sample type has on microbial community.
The microbiome of Acropora tissue biopsies was also found to differ from that of the whole coral fragment. The tissue and skeletal microbiomes of scleractinian corals have been found to be dominated by rare taxa and to differ in bacterial abundance (Ricci et al., 2022), so we expect to see some differences between samples including the coral skeleton and only including the tissue. Here we find a higher proportion of Rhodobacteraceae in whole fragments than tissue biopsy samples, possibly due to the inclusion of different microhabitats in the two sample types. Rhodobacteraceae have been shown to demonstrate environmental flexibility (Röthig et al., 2016) and have also been associated with thermally-stressed corals, including Porites (Pootakham et al., 2019), Montipora (van de Water et al., 2017), Acropora (Lee et al., 2017), and Pocillopora (Tout et al., 2015). However, without separating out the microhabitats from the whole fragment, it is impossible to specify what microhabitat certain bacteria are associated with or draw conclusions about the response of a specific microhabitat to possible environmental change. The advantage of tissue samples therefore is that the microbial community is limited to bacteria found in the coral tissue, not composed of a disproportionate number of bacteria from the SML, coral skeleton, and endoliths that may be included in whole fragment samples, and therefore studies can exclusively target the response of tissue-associated microbes to environmental change.
In the current study, tissue biopsies were standardized to a 1.5 mm wide biopsy punch and we find there were significantly more taxa and a higher diversity of taxa associated with Pocillopora tissue samples than Acropora. Distinct morphological, reproductive, and ecological traits between species can influence microbial associations in corals (Hernandez-Agreda et al., 2018; Ricci et al., 2022; Morrow et al., 2022). The species selected in the current study have different morphological structures: Acropora species are perforate, characterized by tissue integrated within the calcium carbonate skeletal matrix (Franzisket, 1970; Leuzinger et al., 2003; Muller-Parker et al., 2015), whereas Pocillopora are imperforate with tissues located superficially (Muller-Parker et al., 2015). Perforate corals are more likely to have concealed tissue areas shaded from a combination of temperature and light stress (Shashar et al., 1997) and have tissues up to five times thicker than imperforate corals (Yost et al., 2013). The functional traits of coral species (e.g. growth form, disease susceptibility) have also been found to have a stronger influence on tissue and skeletal microbiomes than mucus (Pollock et al., 2018), and we therefore hypothesize that distinct differences between host traits explain the variation between the taxa found in Pocillopora and Acropora tissue samples in the present study. In the present study, we observed significantly more taxa in Pocillopora tissues than in Acropora tissues. Pocillopora tissue samples were dominated by the family Rhizobiales, nitrogen-cycling bacteria capable of fixing and providing a source of organic nitrogen when in symbiosis with a plant host (Carvalho et al., 2014). On coral reefs, Rhizobiales have been reported to increase on reefs impacted by stressors such as climate change, water pollution, and overfishing (McDevitt-Irwin et al., 2017). As coral samples were collected from a shallow (1-3 m depth) reef flat during summer, one possibility in the present study is that surface-level Pocillopora tissue was more impacted by light and temperature (and therefore saw an increase in microbial taxa and diversity, e.g. Tout et al., 2015) than Acropora tissue located deeper in the skeleton. Other studies have also amplified Rhiziobiales in association with coral tissue, but not in the whole coral colony community (Ainsworth et al., 2015; Morrow et al., 2022), suggesting that characteristics of P. damicornis tissue results in higher abundances of Rhizobiales observed here. Additionally, it is possible that preservation method (e.g. fixed tissues) did not preserve communities from the surface mucus layer, which is known to be rich in Acroporid corals and has been found to host opportunistic pathogens and coral-associated commensal bacteria (Krediet et al., 2009; Lee et al., 2015; Hadaidi et al., 2017). For example, Ziegler et al. (2019) identified a mean number of 237 OTUs per sample associated with Acropora hemprichii tissues and 159 OTUs per sample associated with Pocillopora verrucosa tissues, but the airbrushed tissue slurry from frozen fragments used for16S rRNA amplicon sequencing included the mucus layer and likely accounted for the higher number of taxa in Acropora samples than Pocillopora. Liang et al. (2017) also found slightly higher mean taxa numbers associated with Acropora spp. tissues (558 OTUs) than Pocillopora spp. tissues (523 OTUs) in samples using airbrushed tissue like Ziegler et al. (2019). In the present study, we observed fewer taxa per sample, likely also due to the lower number of reads per sample. For example, in fixed tissue samples, we found an average of 11 ASVs associated with Acropora samples and an average of 24 ASVs associated with Pocillopora samples. The exclusion of the rich surface mucus layer of Acropora corals in the present study could therefore account for the lower taxa and read numbers observed overall in the present study, as well as the lower number of taxa and taxa diversity observed in Acropora corals.
The communities observed in Acropora and Pocillopora samples were likely also influenced by both preservation method and anthropogenic factors. While Alphaproteobacteria dominated the microbiome of both Acropora and Pocillopora in the present study, Bergman et al. (2021) previously found Gammaproteobacteria to dominate the microbiome of whole Pocillopora fragments. The samples in the present study were collected directly from the reef, whereas Bergman et al. (2021) collected corals and conducted an ex situ tank experiment using multiple warming trajectories, which likely influenced the differences from the microbial communities observed herein due to environmental effects. Additionally, both Acropora and Pocillopora samples were almost entirely lacking Endozoicomonas, a common bacterial associate in corals (Pogoreutz et al., 2018; Epstein et al., 2019; Glasl et al., 2019; Voolstra and Ziegler, 2020) that has been observed to be highly abundant in both Acropora and Pocillopora species (Ziegler et al., 2019). However, Ricci et al. (2022) characterized the microbiome of P. damicornis from Heron Island just a few months before the 2020 bleaching events and found a high abundance of Endozoicomonas. The difference between our results and Ricci et al. (2022) suggests that warming water can disrupt the coral-Endozoicomonas association and is also supported by Botté et al. (2022) results, who found low abundances of Endozoicomonas in P. acuta samples from the GBR. As a loss of Endozoicomonas is often recorded in bleached or diseased corals (Bayer et al., 2013; Glasl et al., 2016), one possibility for the lack of Endozoicomonas recorded herein is that repetitive and severe bleaching on the GBR has greatly reduced populations of coral tissue-associated Endozoicomonas over time.
Additionally, the present study investigated the microbiome of corals using fixed tissue, whereas many studies investigating the Pocillopora or Acropora microbiome analyze whole fragments snap-frozen in liquid nitrogen (e.g. Epstein et al., 2019, Ziegler et al., 2019, Bergman et al., 2021). While Hernandez-Agreda et al. (2018) found composition of microbial communities to be comparable between fixed and snap-frozen coral samples, some low abundance and low occurrence phylotypes were found to be variable between preservation methods. It is therefore possible that variation in the microbial community between studies, as well as number of reads and ASVs, is at least in part due to preservation method. Preservation method is a potential source of bias that should be considered when conducting experiments targeting the coral microbiome. Through investigations of the influence of sample type, host species, and DNA extraction protocols on the amplification of DNA from coral tissues, we find that differences in the microbiome are therefore potentially related to both the structure and preservation of the tissue of the tissue, skeleton, and distinct environmental niches within the two coral types.
Conclusions
All three protocols used in this study were found to be suitable for the extraction of coral DNA for sequencing from whole fragments and the Micro protocol to be suitable for use on tissue biopsies. However, greater differences were detected between the microbial communities of whole fragments versus tissue biopsies for Acropora species, as well as between the tissue biopsies of different species. Similar results have been observed in Acropora granulosa, Leptoseris spp., and Montipora capitata, where different bacterial phylotypes were observed between whole coral samples (e.g. including the surface mucus layer and the coral skeleton) and isolated coral tissues (Ainsworth et al., 2015). Ricci et al. (2022) also found that host phylogeny and microhabitat niches (e.g. tissue slurry samples versus skeletal samples) shaped the presence and relative abundance of bacterial symbionts in 12 coral species. Observation of species-specific and microhabitat-specific bacterial associations therefore highlight the need for studies to incorporate the biological and morphological features of the coral host into interpretations of the coral-microbial community interaction. Similarly, the type of microhabitat targeted can influence how the microbiome is interpreted in studies of the contribution of the microbiome to coral health and physiology as the environment changes. Future studies with a larger sample size than the present study (n = 3 coral whole fragments per species/extraction protocol) may find more differences at a community level, however studies of coral response to environmental change are often limited by permitting requirements and therefore often proceed with small sample sizes. To maximize the chances of capturing the microbial community of interest from a small sample size, we therefore suggest that studies targeting the microbial community of coral tissue exclusive of the meta-organism microbial community use tissue biopsies. Additionally, while surveying the literature for reported sequencing depths across P. damicornis tissue and whole fragments, we found that bacterial microbiomes often reported as “tissue-associated” were actually referring to homogenized whole fragments. In future studies, tissue biopsies and clear reporting of the community of interest may reduce some of the noise currently existing in coral microbial datasets. Finally, sequencing depths, average number of sequences per sample, and total number of reads for each sample variable category tested were often underreported in the literature (Table 1), pointing to a need for a consistent standard of reporting across coral microbiome studies. As DNA sequencing methods become more and more prevalent in studies of the coral microbiome, it is imperative to identify sources of bias and conduct experiments with targeted communities of interest in order to identify ecologically meaningful patterns in an often-complicated dataset. Understanding where bias may be introduced throughout different steps in the experimental design, and weighing out the costs and benefits of reducing these biases, will improve our understanding of the coral microbiome and the sequencing process as a whole.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA802894.
Author contributions
JB, TS, and TA planned the experiments. JB and TS conducted the experiments. JB, TS, and TA contributed to the experiments. JB analyzed the data and wrote the manuscript with advice, contributions, review, and editing from TA, SE, and TS.
Funding
This research was supported by an Australian Research Council Discovery Project Grant (DP180103199) and UNSW Scientia Funding to TA. JB was also supported by the UNSW Scientia PhD Scholarship.
Acknowledgments
This research was conducted at Heron Island Research Station, Lord Howe Island, and the University of New South Wales. We thank A. Fordyce, C. Lantz, T. Moriarty, and C. Page for assistance in both the field and in the lab.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.985496/full#supplementary-material
References
Ainsworth T. D., Krause L., Bridge T., Torda G., Raina J.-B., Zakrzewski M., et al. (2015). The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 9, 2261–2274. doi: 10.1038/ismej.2015.39
Ainsworth T. D., Gates R. D. (2016). Corals’ microbial sentinels. Science. 352, 1518–1519. doi: 10.1126/science.aad9957
Ainsworth T. D., Thurber R. V., Gates R. D. (2010). The future of coral reefs: a microbial perspective. Trends Ecol. Evol. 25, 233–240. doi: 10.1016/J.TREE.2009.11.001
Apprill A. (2017). Marine animal microbiomes: toward understanding host–microbiome interactions in a changing ocean. Front. Mar. Sci. 4. doi: 10.3389/fmars.2017.00222
Apprill A., Weber L. G., Santoro A. E. (2016). Distinguishing between microbial habitats unravels ecological complexity in coral microbiomes mSystems 1, e00143–e00116. doi: 10.1128/mSystems.00143-16
Baker E. J., Kellogg C. A. (2014). Comparison of three DNA extraction kits to establish maximum yield and quality of coral associated microbial DNA (US Geological Survey: St. Petersburg, Florida, USA). US Department of the InteriorOpen-File Report 1066. doi: 10.3133/ofr20141066
Bardenhorst S. K., Berger T., Klawonn F., Vital M., Karch A., Rübsamen N. (2021). Data analysis strategies for microbiome studies in human populations - a systematic review of current practice. mSystems 6, e01154–e01120. doi: 10.1128/mSystems.01154-20
Bayer T., Neave M. J., Alsheikh-Hussain A., Aranda M., Yum L. K., Mincer T., et al. (2013). The microbiome of the red sea coral stylophora pistillata is dominated by tissue-associated endozoicomonas bacteria. Appl. Env. Microbiol. 80, 4759–4762. doi: 10.1128/AEM.00695-13
Bergman J. L., Leggat W., Ainsworth T. D. (2021). The meta-organism response of the environmental generalist pocillopora damicornis exposed to differential accumulation of heat stress. Front. Mar. Sci. 8. doi: 10.3389/fmars.2021.664063
Biagi E., Caroselli E., Barone M., Pezzimenti M., Teixido N., Soverini M., et al. (2020). Patterns in microbiome composition differ with ocean acidification in anatomic compartments of the Mediterranean coral astroides calycularis living at CO2 vents. Sci. Total Env. 724, 138048. doi: 10.1016/J.scitotenv.2020.138048
Blackall L. L., Wilson B., van Oppen M. J. H. (2015). Coral - the world’s most diverse symbiotic ecosystem. Mol. Ecol. 24, 5330–5347. doi: 10.1111/mec.13400
Bolyen E., Rideout J. R., Dillon M. R., Bokulich N. A., Abnet C. C., Al-Ghalith G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Botté E. S., Cantin N. E., Mocellin V. J., O’Brien P. A., Rocker M. M., Frade P. R., et al. (2022). Reef location has a greater impact than coral bleaching severity on the microbiome of pocillopora acuta. Coral Reefs 41, 63–79. doi: 10.1007/s00338-021-02201-y
Bythell J. C., Wild C. (2011). Biology and ecology of coral mucus release. J. Exp. Mar. Biol. Ecol. 408, 88–93. doi: 10.1016/j.jembe.2011.07.028
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., Holmes S. P. (2016). DADA2: high-resolution sample inference from illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Carvalho T. L. G., Balsemão-Pires E., Saraiva R. M., Ferreira P. C. G., Hemerly A. S. (2014). Nitrogen signalling in plant interactions with associative and endophytic diazotrophic bacteria. J. Exp. Biol. 65, 5631–5642. doi: 10.1093/jxb/eru319
Caughman A. M., Pratte Z. A., Patin N. V., Stewart F. J. (2021). Coral microbiome changes over the day–night cycle. Coral Reefs 40, 921–935. doi: 10.1007/s00338-021-02097-8
Clinton M., Wyness A. J., Martin S. A., Brierley A. S., Ferrier D. E. (2021). Sampling the fish gill microbiome: a comparison of tissue biopsies and swabs. BMC Microbiol. 21, 1–15. doi: 10.1186/s12866-021-02374-0
Damjanovic K., Menéndez P., Blackall L. L., van Oppen M. J. (2020). Mixed-mode bacterial transmission in the common brooding coral Pocillopora acuta. Env. Microbiol. 22, 397–412. doi: 10.1111/1462-2920.14856
Davis N. M., Proctor D. M., Holmes S. P., Relman D. A., Callahan B. J. (2018). Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 226. doi: 10.1186/s40168-018-0605-2
Deignan L. K., McDougald D. (2022). Differential response of the microbiome of Pocillopora acuta to reciprocal transplantation within Singapore. Microb. Ecol. 83, 608–618. doi: 10.1007/s00248-021-01793-w
De Cáceres M. D., Legendre P. (2009). Associations between species and groups of sites: indices and statistical inference. Ecology 90, 3566–3574. doi: 10.1890/08-1823.1
Edmunds P. J. (1994). Evidence that reef-wide patterns of coral bleaching may be the result of the distribution of bleaching-susceptible clones. Mar. Biol. 121, 137–142. doi: 10.1007/BF00349482
Edmunds P. J., Gates R. (2002). Normalizing physiological data for scleractinian corals. Coral Reefs 21, 193–197. doi: 10.1007/s00338-002-0214-0
Epstein H. E., Torda G., van Oppen M. J. (2019). Relative stability of the pocillopora acuta microbiome throughout a thermal stress event. Coral Reefs 38, 373–386. doi: 10.1007/s00338-019-01783-y
Franzisket L. (1970). The atrophy of hermatypic reef corals maintained in darkness and their subsequent regeneration in light. Int. Rev. der gesamten Hydrobiologie und Hydrographie. 55, 1–12. doi: 10.1002/iroh.19700550102
Glasl B., Herndl G. J., Frade P. R. (2016). The microbiome of coral surface mucus has a key role in mediating holobiont health and survival upon disturbance. ISME J. 10, 2280–2292. doi: 10.1038/ismej.2016.9
Glasl B., Smith C. E., Bourne D. G., Webster N. S. (2019). Disentangling the effect of host-genotype and environment on the microbiome of the coral acropora tenuis. PeerJ 7, e6377. doi: 10.7717/peerj.6377
Glassing A., Dowd S. E., Galandiuk S., Davis B., Chiodini R. J. (2016). Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 8, 1–12. doi: 10.1186/s13099-016-0103-7
Hadaidi G., Röthig T., Yum L. K., Ziegler M., Arif C., Roder C., et al. (2017). Stable mucus-associated bacterial communities in bleached and healthy corals of porites lobata from the Arabian seas. Sci. Rep. 7, 1–11. doi: 10.1038/srep45362
Haydon T. D., Seymour J. R., Raina J. B., Edmondson J., Siboni N., Matthews J. L., et al. (2021). Rapid shifts in bacterial communities and homogeneity of Symbiodiniaceae in colonies of Pocillopora acuta transplanted between reef and mangrove environments. Front. Microbiol., 3131. doi: 10.3389/fmicb.2021.756091
Hernandez-Agreda A., Leggat W., Ainsworth T. D. (2018). A comparative analysis of microbial DNA preparation methods for use with massive and branching coral growth forms. Front. Microbiol. 9, 2146. doi: 10.3389/fmicb.2018.02146
Hernandez-Agreda A., Leggat W., Bongaerts P., Ainsworth T. D. (2016). The microbial signature provides insight into the mechanistic basis of coral success across reef habitats. MBio 4, e00560–e00516. doi: 10.1128/mBio.00560-16
Hoegh-Guldberg O., Mumby P. J., Hooten A. J., Steneck R. S., Greenfield P., Gomez E., et al. (2007). Coral reefs under rapid climate change and ocean acidification. Science 318, 1737–1742. doi: 10.1126/science.1152509
Hughes T. P., Kerry J. T., Álvarez-Noriega M., Álvarez-Romero J. G., Anderson K. D., Baird A. H., et al. (2017). Global warming and recurrent mass bleaching of corals. Nature 543, 373–377. doi: 10.1038/nature21707
Kellogg C. A. (2019). Microbiomes of stony and soft deep-sea corals share rare core bacteria. Microbiome 7, 1–13. doi: 10.1186/s40168-019-0697-3
Knight R., Vrbanac A., Taylor B. C., Aksenov A., Callewaert C., Debelius J., et al. (2018). Best practices for analysing microbiomes. Nat. Rev. Microbiol. 16, 410–422. doi: 10.1038/s41579-018-0029-9
Krediet C. J., Ritchie K. B., Cohen M., Lipp E. K., Sutherland K. P., Teplitski M. (2009). Utilization of mucus from the coral acropora palmata by the pathogen serratia marcescens and by environmental and coral commensal bacteria. Appl. Env. Microbiol. 75, 3851–3858. doi: 10.1128/aem.00457-09
Lawson C. A., Raina J.-B., Kahlke T., Seymour J. R., Suggett D. J. (2018). Defining the core microbiome of the symbiotic dinoflagellate, symbiodinium. Environ. Microbiol. Rep. 10, 7–11. doi: 10.1111/1758-2229.12599
Le Campion-Alsumard T., Golubic S., Hutchings P. (1995). Microbial endoliths in skeletons of live and dead corals: Porites lobata (Moorea, French Polynesia). Mar. Ecol. Prog. Ser. 117, 149–157. doi: 10.3354/meps117149
Lee S. T., Davy S. K., Tang S. L., Fan T. Y., Kench P. S. (2015). Successive shifts in the microbial community of the surface mucus layer and tissues of the coral acropora muricata under thermal stress. FEMS Microbiol. Ecol. 91, 1–11. doi: 10.1093/femsec/fiv142
Lee S. T., Davy S. K., Tang S. L., Kench P. S. (2017). Water flow buffers shifts in bacterial community structure in heat-stressed acropora muricata. Sci. Rep. 7, 1–13. doi: 10.1038/srep43600
Legendre P., Gallagher E. D. (2001). Ecologically meaningful transformations for ordination of species data. Oecologia 129, 271–280. doi: 10.1007/s004420100716
Leuzinger S., Anthony K. R., Willis B. L. (2003). Reproductive energy investment in corals: scaling with module size. Oecologia 136, 524–531. doi: 10.1007/s00442-003-1305-5
Liang J., Yu K., Wang Y., Huang X., Huang W., Qin Z., et al. (2017). Distinct bacterial communities associated with massive and branching scleractinian corals and potential linkages to coral susceptibility to thermal or cold stress. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00979
Li J., Long L., Zou Y., Zhang S. (2021). Microbial community and transcriptional responses to increased temperatures in coral pocillopora damicornis holobiont. Env. Microbiol. 23, 826–843. doi: 10.1111/1462-2920.15168
Lim M. Y., Song E. J., Kim S. H., Lee J., Nam Y. D. (2018). Comparison of DNA extraction methods for human gut microbial community profiling. Systematic Appl. Microbiol. 41, 151–157. doi: 10.1016/j.syapm.2017.11.008
Love M. I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome. Biol. 15, 1–21. doi: 10.1186/S13059-014-0550-8
Marcelino V. R., Morrow K. M., H van Oppen M. J., Bourne D. G., Verbruggen H. (2017). Diversity and stability of coral endolithic microbial communities at a naturally high pCO2 reef. Molec. Ecol. 26, 5344–5357. doi: 10.1111/mec.14268
Marcelino V. R., van Oppen M. J., Verbruggen H. (2018). Highly structured prokaryote communities exist within the skeleton of coral colonies. ISME J. 12, 300–303. doi: 10.1038/ismej.2017.164
Marcelino V. R., Verbruggen H. (2016). Multi-marker metabarcoding of coral skeletons reveals a rich microbiome and diverse evolutionary origins of endolithic algae. Sci. Rep. 6, 1–9. doi: 10.1038/srep31508
Marotz C., Amir A., Humphrey G., Gaffney J., Gogul G., Knight R. (2017). DNA Extraction for streamlined metagenomics of diverse environmental samples. BioTechniques 62, 290–293. doi: 10.2144/000114559
Massé A., Domart-Coulon I., Golubic S., Duché D., Tribollet A. (2018). Early skeletal colonization of the coral holobiont by the microboring ulvophyceae ostreobium sp. Sci. Rep. 8,1-11 2293. doi: 10.1038/s41598-018-20196-5
McDevitt-Irwin J. M., Baum J. K., Garren M., Vega Thurber R. L. (2017). Responses of coral-associated bacterial communities to local and global stressors. Front. Mar. Sci. 0. doi: 10.3389/fmars.2017.00262
McDonald D., Price M. N., Goodrich J., Nawrocki E. P., DeSantis T. Z., Probst A., et al. (2012). An improved greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. doi: 10.1038/ismej.2011.139
McFall-Ngai M., Hadfield M. G., Bosch T. C., Carey H. V., Domazet-Lošo T., Douglas A. E., et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Procl. Natl. Acad. Sci. 110, 3229–3236. doi: 10.1073/pnas.1218525110
McMurdie P. J., Holmes S. (2013). Phyloseq: An r package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8, e61217. doi: 10.1371/journal.pone.0061217
McMurdie P. J., Holmes S. (2014). Waste not, want not: why rarefying microbiome data is inadmissible. PloS Comp. Biol. 10, e1003531. doi: 10.1371/journal.pcbi.1003531
Meistertzheim A. L., Lartaud F., Arnaud-Haond S., Kalenitchenko D., Bessalam M., Le Bris N., et al. (2016). Patterns of bacteria-host associations suggest different ecological strategies between two reef building cold-water coral species. Deep Sea Res. Part I: Oceanographic Res. Papers 114, 12–22. doi: 10.1016/j.dsr.2016.04.013
Meyer J. L., Paul V. J., Teplitski M. (2014). Community shifts in the surface microbiomes of the coral Porites astreoides with unusual lesions. PloS One 9, e100316. doi: 10.1371/journal.pone.0100316
Morrow K. M., Pankey M. S., Lesser M. P. (2022). Community structure of coral microbiomes is dependent on host morphology. Microbiome 10, 113. doi: 10.1186/s40168-022-01308-w
Mouchka M. E., Hewson I., Harvell C. D. (2010). Coral-associated bacterial assemblages: current knowledge and the potential for climate-driven impacts. Integr. Comp. Biol. 50, 662–674. doi: 10.1093/icb/icq061
Muller-Parker G., D’elia C. F., Cook C. B. (2015). “Interactions between corals and their symbiotic algae,” in Coral reefs in the anthropocene (Dordecht: Springer), 99–116.
Neave M. J., Rachmawati R., Xun L., Michell C. T., Bourne D. G., Apprill A., et al. (2017). Differential specificity between closely related corals and abundant Endozoicomonas endosymbionts across global scales. The ISME Journal. 11, 186–200. doi: doi.org/10.1038/ismej.2016.95
Olomu I. N., Pena-Cortes L. C., Long R. A., Vyas A., Krichevskiy O., Luellwitz R., et al. (2020). Elimination of “kitome” and “splashome” contamination results in lack of detection of a unique placental microbiome. BMC Microbiol. 20, 1–19. doi: 10.1186/S12866-020-01839-Y
Pereira L. B., Palermo B. R. Z., Carlos C., Ottoboni L. M. M. (2017). Diversity and antimicrobial activity of bacteria isolated from different Brazilian coral species. FEMS Microbiol. Lett. 364, 164. doi: 10.1093/FEMSLE/FNX164
Pogoreutz C., Rädecker N., Cárdenas A., Gärdes A., Wild C., Voolstra C. R. (2018). Dominance of endozoicomonas bacteria throughout coral bleaching and mortality suggests structural inflexibility of the pocillopora verrucosa microbiome. Ecol. Evol. 8, 2240–2252. doi: 10.1002/ece3.3830
Pollock J., Glendinning L., Wisedchanwet T., Watson M. (2018). The madness of microbiome: attempting to find consensus “best practice” for 16S microbiome studies. Appl. Environ. Microbiol. 84, e02627–e02617. doi: 10.1128/AEM.02627-17
Pootakham W., Mhuantong W., Yoocha T., Putchim L., Jomchai N., Sonthirod C., et al. (2019). Heat-induced shift in coral microbiome reveals several members of the rhodobacteraceae family as indicator species for thermal stress in porites lutea. MicrobiologyOpen 8, e935. doi: 10.1002/MBO3.935
Pratte Z. A., Kellogg C. A. (2021). Comparison of preservation and extraction methods on five taxonomically disparate coral microbiomes. Front. Mar. Sci. 0. doi: 10.3389/FMARS.2021.684161
Pratte Z. A., Longo G. O., Burns A. S., Hay M. E., Stewart F. J. (2018). Contact with turf algae alters the coral microbiome: contact versus systemic impacts. Coral Reefs 37, 1–13. doi: 10.1007/s00338-017-1615-4
Qin Z., Yu K., Chen S., Chen B., Yao Q., Yu X., et al. (2022). Significant Changes in Bacterial Communities Associated with Pocillopora Corals Ingestion by Crown-of-Thorns Starfish: An Important Factor Affecting the Coral’s Health. Microorganisms 10, 207. doi: 10.3390/microorganisms10020207
Ricci F., Tandon K., Black J. R., Lê Cao K. A., Blackall L. L., Verbruggen H. (2022). Host traits and phylogeny contribute to shaping coral-bacterial symbioses. Msystems 7, 00044–00022. doi: 10.1128/msystems.00044-22
Rice M. M., Maher R. L., Thurber R. V., Burkepile D. E. (2019). Different nitrogen sources speed recovery from corallivory and uniquely alter the microbiome of a reef-building coral. PeerJ 7, e8056. doi: 10.7717/peerj.8056
Rohwer F., Seguritan V., Azam F., Knowlton N. (2002). Diversity and distribution of coral-associated bacteria. Mar. Ecol. Prog. Ser. 243, 1–10. doi: 10.3354/meps243001
Rosado P. M., Leite D. C., Duarte G. A., Chaloub R. M., Jospin G., Nunes da Rocha U., et al. (2019). Marine probiotics: increasing coral resistance to bleaching through microbiome manipulation. ISME J. 13, 921–936. doi: 10.1038/s41396-018-0323-6
Rosenberg E., Koren O., Reshef L., Efrony R., Zilber-Rosenberg I. (2007). The role of microorganisms in coral health, disease and evolution. Nat. Rev. Microbiol. 5, 355–362. doi: 10.1038/nrmicro1635
Röthig T., Ochsenkühn M. A., Roik A., van der Merwe R., Voolstra C. R. (2016). Long-term salinity tolerance is accompanied by major restructuring of the coral bacterial microbiome. Molec. Ecol. 25, 1308–1323. doi: 10.1111/mec.13567
Rubio-Portillo E., Santos F., Martínez-García M., de los Ríos A., Ascaso C., Souza-Egipsy V., et al. (2016). Structure and temporal dynamics of the bacterial communities associated to microhabitats of the coral oculina patagonica. Environ. Microbiol. 18, 4564–4578. doi: 10.1111/1462-2920.13548
Santos H. F., Carmo F. L., Leite D. C. A., Jesus H. E., de Carvalho Maalouf P., Almeida C., et al. (2012). Comparison of different protocols for the extraction of microbial DNA from reef corals. Braz. J. Microbiol. 43, 517–527. doi: 10.1590/S1517-83822012000200012
Schmidt-Roach S., Miller K. J., Lundgren P., Andreakis N. (2014). With eyes wide open: a revision of species within and closely related to the pocillopora damicornis species complex (Scleractinia; pocilloporidae) using morphology and genetics. Zoo J. Linn Soc. 170, 1–33. doi: 10.1111/zoj12092
Shashar N., Banaszak A. T., Lesser M. P., Amrami D. (1997). Coral endolithic algae: life in a protected environment. Pacific Sci. 51, 167–173.
Shashar N., Cohen Y., Loya Y., Sar N. (1994). Nitrogen fixation (acetylene reduction) in stony corals: evidence for coral-bacteria interactions. Mar. Ecol. Prog. Ser. 111, 259–264. doi: 10.3354/meps111259
Shnit-Orland M., Kushmaro A. (2009). Coral mucus-associated bacteria: a possible first line of defense. FEMS Microbiol. Ecol. 67, 371–380. doi: 10.1111/J.1574-6941.2008.00644.X
Silbiger N. J., Nelson C. E., Remple K., Sevilla J. K., Quinlan Z. A., Putnam H. M., et al. (2018). Nutrient pollution disrupts key ecosystem functions on coral reefs. Proc. R. Soc B. 285, 20172718. doi: 10.1098/rspb.2017.2718
Stévenne C., Micha M., Plumier J.-C., Roberty S. (2021). Corals and sponges under the light of the holobiont concept: how microbiomes underpin our understanding of marine ecosystems. Front. Mar. Sci. 0, 1035. doi: 10.3389/FMARS.2021.698853
Sweet M. J., Croquer A., Bythell J. C. (2011). Bacterial assemblages differ between compartments within the coral holobiont. Coral Reefs 30, 39–52. doi: 10.1007/s00338-010-0695-1
Thompson J. R., Rivera H. E., Closek C. J., Medina M. (2015). Microbes in the coral holobiont: Partners through evolution, development, and ecological interactions. Front. Cell. Infect. Microbiol. 4. doi: 10.3389/fcimb.2014.00176
Tout J., Siboni N., Messer L. F., Garren M., Stocker R., Webster N. S., et al. (2015). Increased seawater temperature increases the abundance and alters the structure of natural vibrio populations associated with the coral pocillopora damicornis. Front. Microbiol. 6. doi: 10.3389/FMICB.2015.00432/FULL
Trevathan-Tackett S. M., Sherman C. D. H., Huggett M. J., Campbell A. H., Laverock B., Hurtado-McCormick V., et al. (2019). A horizon scan of priorities for coastal marine microbiome research. Nat. Ecol. Evol. 3, 1509–1520. doi: 10.1038/s41559-019-0999-7
van de Water J. A. J. M., Chaib de Mares M., Dixon G. B., Raina J.-B., Willis B. L., Bourne D. G., et al. (2017). Antimicrobial and stress responses to increased temperature and bacterial pathogen challenge in the holobiont of a reef-building coral. Molec. Ecol. 27, 1065–1080. doi: 10.1111/mec.14489
van de Water J. A. J. M., Melkonian R., Junca H., Voolstra C. R., Reynaud S., Allemand D., et al. (2016). Spirochaetes dominate the microbial community associated with the red coral corallium rubrum on a broad geographic scale. Sci. Rep. 6, 1–7. doi: 10.1038/srep27277
van Oppen M. J., Bongaerts P., Frade P., Peplow L. M., Boyd S. E., Nim H. T., et al. (2018). Adaptation to reef habitats through selection on the coral animal and its associated microbiome. Molec. Ecol. 27, 2956–2971. doi: 10.1111/mec.14763
Vilela C. L., Villela H. D., Duarte G. A., Santoro E. P., Rachid C. T., Peixoto R. S. (2021). Estrogen induces shift in abundances of specific groups of the coral microbiome. Sci. Rep. 11, 1–10. doi: 10.1038/s41598-021-82387-x
Voolstra C. R., Ziegler M. (2020). Adapting with microbial help: microbiome flexibility facilitates rapid responses to environmental change. BioEssays 42, 2000004. doi: 10.1002/bies.202000004
Wagner Mackenzie B., Waite D. W., Taylor M. W. (2015). Evaluating variation in human gut microbiota profiles due to DNA extraction method and inter-subject differences. Front. Microbiol. 0. doi: 10.3389/FMICB.2015.00130
Weber L., DeForce E., Apprill A. (2017). Optimization of DNA extraction for advancing coral microbiota investigations. Microbiome 5, 1–14. doi: 10.1186/s40168-017-0229-y
Weiler B. A., Verhoeven J. T. P., Dufour S. C. (2018). Bacterial communities in tissues and surficial mucus of the cold-water coral paragorgia arborea. Front. Mar. Sci. 0. doi: 10.3389/FMARS.2018.00378
Williams W. M., Vinter A. B., Broughton W. J. (1987). Nitrogen fixation (acetylene reduction) associated with the living coral Acropora variabilis. Mar. Biol. 94, 531–535. doi: 10.1007/BF00431399
Yost D. M., Wang L.-H., Fan T.-Y., Chen C.-S., Lee R. W., Sogin E., et al. (2013). Diversity in skeletal architecture influences biological heterogeneity and symbiodinium habitat in corals. Zoology 116, 262–269. doi: 10.1016/J.ZOOL.2013.06.001
Ziegler M., Grupstra C. G., Barreto M. M., Eaton M., BaOmar J., Zubier K., et al. (2019). Coral bacterial community structure responds to environmental change in a host-specific manner. Nat Comm. 10, 1–11. doi: doi.org/10.1038/s41467-019-10969-5
Keywords: coral microbiome, 16S rRNA gene, amplicon sequencing, DNA extraction, microbial symbiosis, Pocillopora, Acropora
Citation: Bergman JL, Shaw T, Egan S and Ainsworth TD (2022) Assessing the coral microbiome at the scale of tissue-specific habitats within the coral meta-organism. Front. Mar. Sci. 9:985496. doi: 10.3389/fmars.2022.985496
Received: 04 July 2022; Accepted: 26 August 2022;
Published: 23 September 2022.
Edited by:
Thanos Dailianis, Hellenic Centre for Marine Research, GreeceReviewed by:
Zoe Adina Pratte, Montana State University, United StatesAna Paula B. Moreira, Federal University of Rio de Janeiro, Brazil
Copyright © 2022 Bergman, Shaw, Egan and Ainsworth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jessica L. Bergman, ai5iZXJnbWFuQHVuc3cuZWR1LmF1