 Yuan Qin*†Jiayu Li†Jiani KuangSicheng ShenJingwen JiangZhi ZhangChenhao ZhaoXiumei ZhouBiao Huang*Bingnan Han*
Yuan Qin*†Jiayu Li†Jiani KuangSicheng ShenJingwen JiangZhi ZhangChenhao ZhaoXiumei ZhouBiao Huang*Bingnan Han*- College of Life Sciences and Medicine, Zhejiang Sci-Tech University, Hangzhou, China
Okadaic acid (OA) is a biotoxin from marine microalgae and widely present in shellfish, which severely affects the seafood safety. Therefore, it is essential to establish a highly sensitive OA analysis and detection method. In this study, a new type of immunoassay technology was established on the basis of the competition method using time-resolved fluoroimmunoassay (TRFIA). OA-bovine serum albumin (OA-BSA) coated on a 96-well plate competes with OA standard or samples to bind OA antibodies. A rare-earth ion-labeled secondary antibody, which fluoresces strongly under the effect of the enhancement solution, was then added as a tracer for detection. The established linear range of OA detected by TRFIA was 2.49 × 10-3 – 50 ng/ml, and the limit of detection was 2.49 × 10-3 ng/ml. The average coefficients of variation from intra-assay and inter-assay batches were 3.34% and 5.87%, respectively, and the recovery rate was 93.04%–111.66%. The OA in shellfish samples was determined by TRFIA and high-performance liquid chromatography (HPLC), and the results showed a good correlation. This study established a TRFIA to detect OA, which has the characteristics of simplicity, sensitivity, precision, and high accuracy, far exceeding the EU or the US standards for the detection of shellfish toxins. It is expected to make proper contribution in marine biotoxin detection.
Introduction
Under the influence of factors such as climate change, some algae have the ability to produce toxic compounds, and some of them can simultaneously produce multiple algal toxins (Caron et al., 2010; Griffith and Gobler, 2020; Onofrio et al., 2020; Marampouti et al., 2021). This adversely affects the ecosystem. Toxins from harmful algal blooms (HABs) can poison or even kill shellfish and other marine organisms and can be passed on to humans through the food chain or direct contact, thus causing poisoning and even death in humans (Klijnstra et al., 2021; Hu et al., 2022). About half of all unnatural deaths in marine mammals can be attributed to ingestion of HABs or animals contaminated with them and cause about 60,000 annual poisonings worldwide (Griffith and Gobler, 2020; Klijnstra et al., 2021). Marine biotoxin pollution leads to direct economic losses and limits the development of the ocean farming and seriously affects human health and safety (Ajani et al., 2021; Tian et al., 2021). Since the 20th century, shellfish poisoning incidents have been reported in many countries. According to the symptoms of poisoning, shellfish toxins are divided into paralytic shellfish poisoning (PSP), neurotic shellfish poisoning, amnesic shellfish poisoning (ASP), and diarrheic shellfish poisoning (DSP) (Fraga et al., 2013). Among these shellfish toxins, DSP is the most frequently reported to cause poisoning worldwide (Lloyd et al., 2013).
OA is first discovered and the main kind of DSP (Verma et al., 2020). It was first identified from Halichondria melanodocia and Halichondria okadai in 1981 and is mainly produced by phytoplankton such as Prorocentrum lima, Prorocentrum concavum, Dynophysis acuta, and Dynophysis fortii, etc. (Valdiglesias et al., 2013). Like other algal toxin, OA is mainly accumulate by bivalve mollusks, such as scallop, mussel, and oyster and other shellfish through filter feeding of harmful algae, passed to humans through the food chain, causing human poisoning (Pérez et al., 2019; Yin et al., 2022). OA is colorless and odorless, making it challenging to recognize (Valdiglesias et al., 2013). Its high lipophilicity degree makes OA easy to accumulate in adipose tissue and not easily metabolized and digested (Reguera et al., 2014). The most studied toxicological mechanism of OA is its inhibitory effect on serine/threonine protein phosphatase (Fu et al., 2019). This toxin can cause diarrhea, nausea, and other typical DSP poisoning symptoms and have carcinogenesis and neurotoxicity effects (Valdiglesias et al., 2013; Ferron et al., 2014).
The general detection methods of OA are roughly divided into instrumental analysis (such as HPLC and liquid chromatography mass spectrometry) and immunoassay (such as enzyme-linked immunosorbent assay, namely ELISA and immunochromatographic assay). In recent years, some novel detection methods have also been developed. For example, the colorimetric detection system was established by fixing protein phosphatase 1 (PP1) in a microfluidic chamber modified by Al2O3 sol–gel to determine the inhibitory effect of OA on PP1; the Au@Pt/HRP immunoassay for OA detection was established by using the dual catalytic action of Au@Pt NPs and HRP on TMB-H2O2 (Castanheira et al., 2021; Tian et al., 2022). Among the detection methods, immunoassays are proven to be convenient, sensitive, cost-effective, and suitable for high-throughput screening (Wang and Xianyu, 2022).
Time-resolved fluoroimmunoassay (TRFIA) is a kind of immunoassay that has been developed in recent decades. Rare-earth metal ions, Eu3+ and Sm3+, are used as labels because of their characteristics of high fluorescence intensity, ability to overcome high-background absorption, and long relaxation time (Wang et al., 2009; Huang et al., 2009; Zhang et al., 2011). TRFIA has been used for the detection of small molecules, such as salbutamol in the environment, aflatoxin b1 in feed, and oxyfluorfen in food, and has demonstrated excellent sensitivity and accuracy (Hu et al., 2018; Fang et al., 2019; Sheng et al., 2020). In this study, an OA detection method was developed using TRFIA on the basis of competitive immunization and applied this TRFIA method on shellfish.
Materials and methods
Materials and instruments
OA-BSA conjugates, OA, and mouse monoclonal antibody to OA were purchased from China National Marine Environment Monitoring Center. Goat anti-mouse IgG was obtained from Huamei Biological Engineering (Luoyang, China). Diethylenetriaminepentaacetic (DTPA) and BSA were acquired from Saiguo Biotechnology (Guangzhou, China). Eu3+‐N1‐(p‐isothiocyanatobenzyl)‐diethylenetriamine‐N1, N2, N3, and N4‐tetraacetic acid (DTTA) were bought from Boshi Biological Technology (Hangzhou, China). Labeling buffer (50 mmol/L Na2CO3-NaHCO3, pH 9.0), coating buffer (50 mmol/L Na2CO3-NaHCO3, pH 9.6), blocking buffer (0.05% proclin-300, 1% BSA, 0.9% NaCl, 50 mmol/L Tris-HCl, pH 7.8), assay buffer (0.01% Tween-20, 0.05% NaN3, 50 mmol/L Tris-HCl, 0.9% NaCl, 0.2% BSA, 20 μM DTPA, pH 7.8), washing buffer (0.01% proclin-300, 50 mmol/L Tris-HCl, 0.02% Tween-20, 0.9% NaCl, pH 7.8), and enhancement solution (1 ml Triton X-100, 50 μmol trioctylphosphine oxide, 15 μmol/L β-NTA, pH 3.2) were prepared in our laboratory.
Ultracel-50k ultrafiltration tubes (Millipore, Massachusetts, USA) were used to change the buffer of IgG. A sephadex-G50 column (Cell Biotechnology, Shanghai, China), a peristaltic pump, and an automatic fraction collector were used to separate the reaction product, and 96-well plates were used as reaction vessels. A micro-oscillator and an electric heating incubator were used to determine immune responses. A plate washer was used to wash away materials that did not bind to the 96-well plate. Fluorescence (F) value of the reaction was measured by a time-resolved immunofluorescence analyzer (Darui Biotechnology, Foshan, China). Photodiode Array UV-Vis Detector (Shimadzu, Kyoto, Japan) and HPLC column (SilGreen, Beijing, China) were used to detect toxin samples and standards.
Preparation for europium (Eu3+)–labeled IgG
Eu3+-labeled IgG was prepared as previously described (Chen et al., 2022). The original buffer of IgG was exchanged with labeling buffer with ultrafiltration tube and centrifuged. Afterward, 125 μl of 2 mg/ml IgG was added to 30 μl of 2 mg/ml Eu3+-DTTA and shaken overnight at room temperature. A sephadex-G50 column and an automatic fraction collector were used to separate the reaction product. The eluate was collected in a 2 ml/tube. F at 340 nm was monitored. The solution with the highest fluorescence value was collected as Eu3+-labeled IgG.
Procedure of OA-TRFIA
An indirect competitive TRFIA was established in this study. In brief, 100 μl coated antigen OA-BSA was added to the 96-well plate and incubated overnight at 4°C. The washing buffer was used to wash the plate once and 150 μl/well of blocking buffer was then added for 2 h to seal the non-specific binding sites. After the blocking buffer was discarded, either 50 μl of OA standard or 50 μl of shellfish samples were added to the 96-well plate, followed by the addition of 50 μl of OA antibody to incubate. After the plate was washed twice, 100 μl of Eu3+-labeled IgG was added to the plate to incubate. After the plate was washed six times, 100 μl of enhancement solution was added to each well, followed by shaking for 3 min. The TRFIA analyzer (excitation wavelength and emission wavelength was 340 nm and 615 nm, respectively) was used to measure the F.
Optimization
The factors that have marked influences on sensitivity were optimized. Coated antigen concentration, antibody concentration, Eu3+-labeled IgG dilution ratio, reaction time, and reaction temperature were optimized on the basis of the high Fmax/IC50 values and low IC50 values (Fmax: maximum fluorescence value; in this study, it refers to the fluorescence signal without OA).
Sensitivity
After 10 standards were measured at 0 concentration, the mean and SD of F were calculated. Sensitivity was the concentration corresponding to the mean F minus twice SD, which is also the limit of detection (LOD).
Precision
The intra-assay coefficient of variation was computed through the repeated measurements of a single sample. The inter-assay coefficient of variation was computed through the repeated measurements of a single sample for several days.
Specificity
BSA and other classes of marine toxins were used as potential antigens to assess the method’s specificity under optimal operating conditions. Other common shellfish toxins which have the same toxicological mechanism as OA, namely, saxitoxin, domoic acid, and microcystin LR, were selected for this study.
Matrix effect on immunoassay
The matrix effect of the samples was studied. The extracted shellfish samples were diluted one, three, and five times with PBS, and the standard curves were drawn. The matrix effect and sample dilution ratio were determined by comparing the standard curve obtained using the standard solution without the matrix solution.
Recovery
The added recovery rate was used as the evaluation standard for TRFIA accuracy. A standard of known concentration was added to a sample of known concentration. The recovery rate was calculated by measuring the actual and theoretical concentrations.
Sample processing
Scissors and a precision knife were used to remove the soft tissues of shellfish samples (scallop, mussel, and oyster), which were then homogenized with a tissue grinder, added with a certain concentration of OA, and stored at −20°C for subsequent study. The samples were prepared as previously described (Wang et al., 2011). In brief, 200 mg of the shellfish samples were added with 1 ml of 90% methanol, and homogenization was continued for 1–2 min. The supernatant was centrifuged at 6000 r/min for 10 min and then collected. The solution was diluted with PBS and mixed evenly. Moreover, 50 μl of the sample was isolated for detection. Phytoplankton samples were processed according to the method described previously in the literature (Wu et al., 2020).
Detection OA by HPLC
The samples were prepared as previously described (Uchida et al., 2014). In brief, 0.2 g of homogenized shellfish samples were added with 0.9 ml of methanol, vortexed, and sonicated for 10 min. Centrifugation was performed at 8000 r/min for 5 min, and the supernatant was collected. Another 0.9 ml of methanol was added to the residue, and the procedure was repeated. The extract was combined, and the volume was fixed to 2 ml. Afterward, 125 μl of 2.5 mol/L NaOH was added to 1 ml of the extract, mixed and sealed, and heated at 70 °C for 1 h. After the mixture was cooled, the same amount of HCl was added and mixed. The solution was passed through 0.22 μm organic phase microporous membrane and HLB solid phase extraction column, and the volume was fixed to 1 ml. HPLC column (250×10 mm2, 5 μm), and LC-20A Photodiode Array UV-Vis Detector was used to detect toxin samples and standards. The mobile phase was water and acetonitrile, the flow rate was 2 ml/min, the wavelength was 200 nm, the detection time was 25 min, and the injection volume was 20 μl. The methanol solution was also analyzed. The concentration of the toxin sample was calculated according to chromatographic peak positions and peak areas.
Statistical analysis
The TRFIA results were compared with the HPLC results and the correlation between them was calculated by IBM SPSS Statistics 26 (SPSS Inc., Chicago, IL, USA). Pearson correlation was used to analyze the correlation between variables and was statistically significant at P<0.05.
Results
Optimization of OA-TRFIA
Preparation for Eu3+-labeled IgG
The Eu3+-labeled IgG was prepared as mentioned above. The reaction solution was separated using dextran gel and eluate. The F at 340 nm was monitored. The statistical curves of goat anti-mouse IgG antibody coupled with Eu3+-DTTA showed purified protein peaks and free peaks. The first and second peaks corresponded to the successfully labeled goat anti-mouse IgG and unlabeled free Eu3+-DTTA, respectively, as shown in Figure S1. The solution corresponding to the protein peak was collected as Eu3+-labeled IgG for subsequent studies.
Optimization of concentrations
The structural formula OA is shown in Figure 1A. The schematic diagram of OA-TRFIA is shown in Figure 1B. The concentration of OA-BSA coated antigen was optimized based on the higher Fmax/IC50 values and lower IC50 values. Figure 2A shows that under the same conditions, the largest Fmax/IC50 value and relatively low IC50 value were obtained when the concentration of OA-BSA was 0.5 μg/ml. Eu3+-labeled IgG was optimized according to sufficient specific binding while avoiding non-specific binding. Figure 2B shows that under the same conditions, a relatively large specific binding and a relatively small non-specific binding were observed when the dilution ratio of Eu3+-labeled IgG was 1:100. OA antibody was optimized at a concentration corresponding to 30%–50% of the Fmax. Figure 2C shows that when the concentration of OA antibody was 0.25 μg/ml, the corresponding fluorescence value was within 30%–50% of the Fmax. In summary, the optimum values were 0.5 μg/ml for the concentration of OA-BSA antigen, 0.25 μg/ml the concentration of OA antibody, and 1:100 for the dilution ratio of Eu3+-labeled IgG.
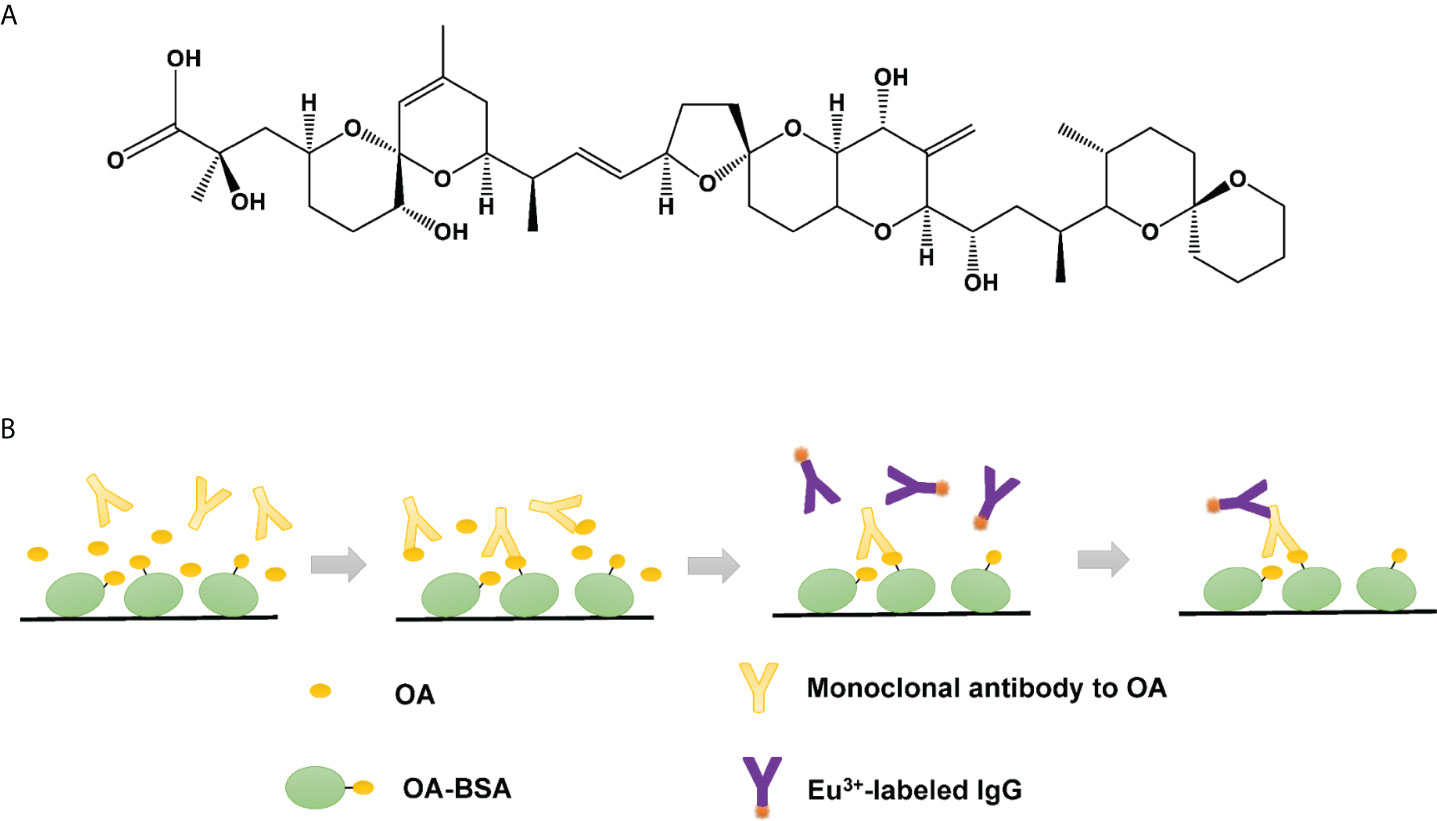
Figure 1 (A) Molecular formula of OA. (B) Schematic of TRFIA system for OA detection.
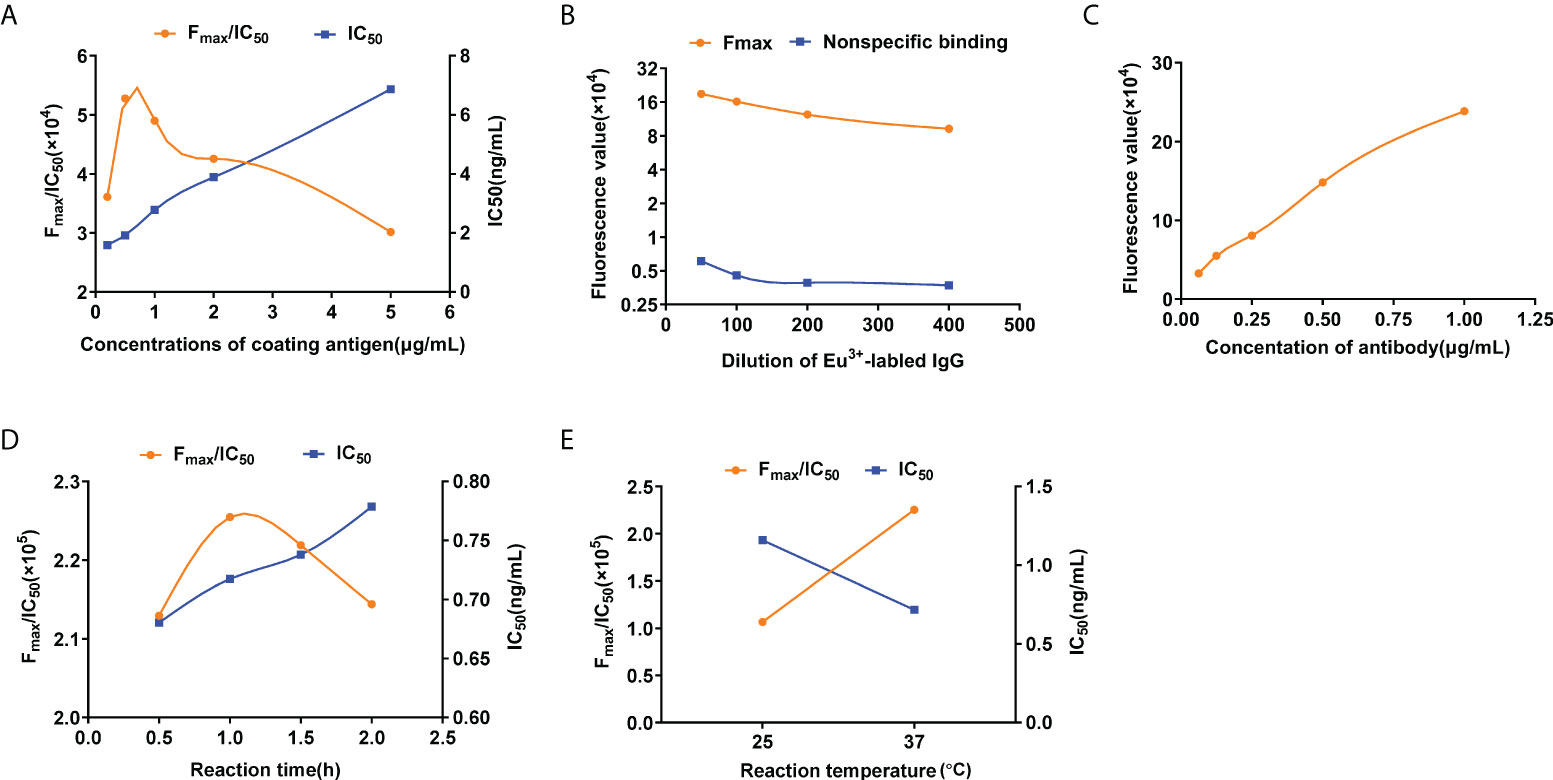
Figure 2 Optimization conditions of OA-TRFIA. (A) Fmax/IC50 values and IC50 values in different coating antigen concentration. (B) Maximum fluorescence values and non-specific binding fluorescence values at different Eu3+-labeled antibody concentrations. (C) Fluorescence valuesin different antibody concentrations. (D) Fmax/IC50 values and IC50 values in different incubation times. (E) Fmax/IC50 values and IC50 values in different incubation temperatures.
Optimization of reaction temperature and time
Incubation time and temperature were also optimized according to the high Fmax/IC50 values and low IC50 values. Four reaction times and two reaction temperatures were evaluated. Figures 2D, E show that under the same conditions, the largest Fmax/IC50 value and the smallest IC50 value were obtained when the incubation time and the incubation temperature were 1 h and 37°C, respectively. In summary, the optimum incubation temperature was 37°C, and the optimum incubation time was 1 h.
Identification of OA-TRFIA
Sensitivity, precision, and specificity
The standard curve of OA-TRFIA was established under the optimized conditions. As shown in Figure 3, the equation of the standard curve was y=-0.21391-1.08852×x, where Logit Y=ln(y/1-y), y=F/F0, and R2 = 0.99756. The results showed that the LOD and IC50 of OA were 2.49 × 10-3 ng/ml and 0.72 ng/ml, respectively. The detection range was 2.49×10-3 – 50 ng/ml. The average coefficients of variation intra-assay and inter-assay batches were 3.34% and 5.87%, respectively.
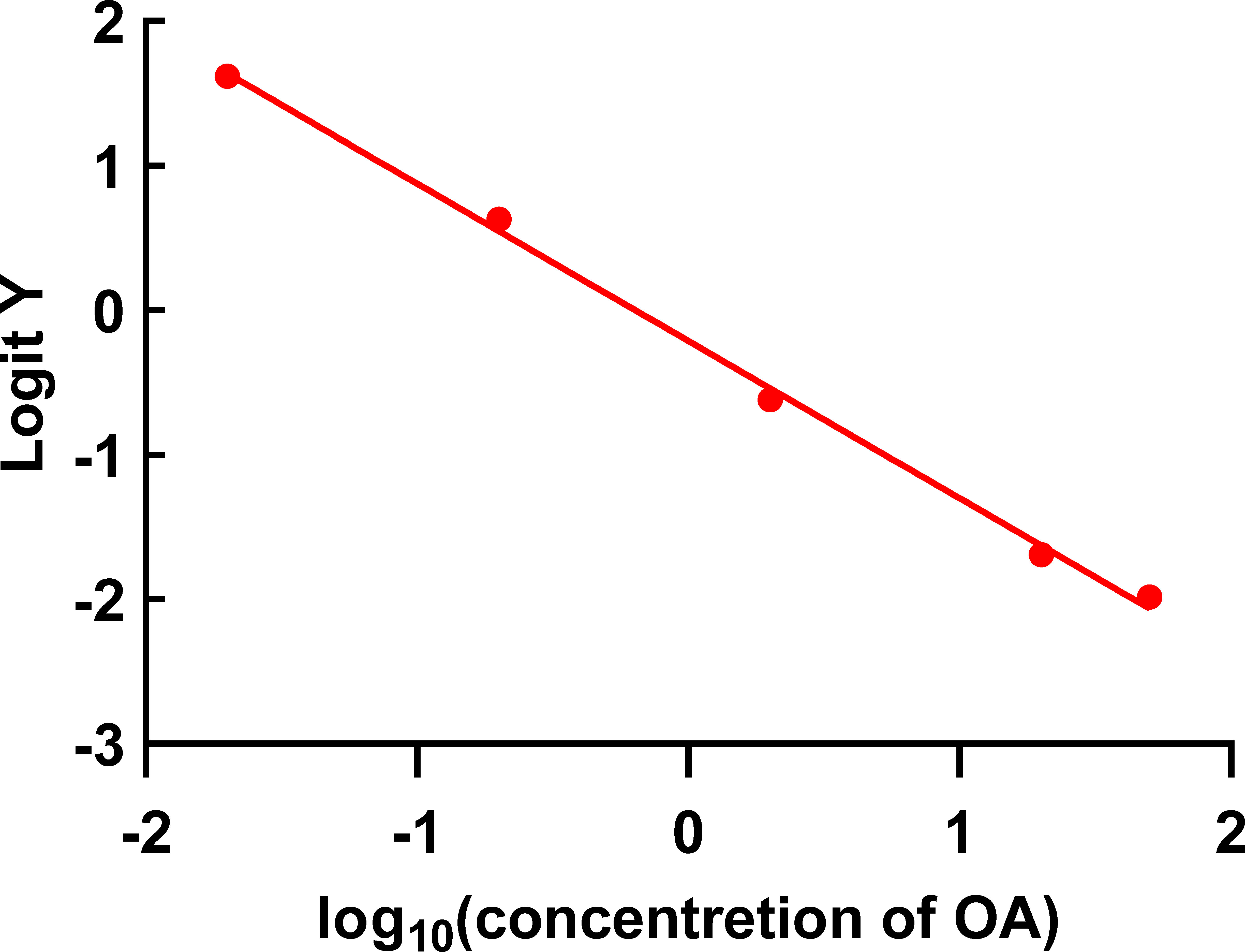
Figure 3 Standard curve of OA-TRFIA (the concentrations of OA were 0, 0.02, 0.2, 2, 20, and 50 ng/ml).
Cross reaction
TRFIA was used to detect saxitoxin, domoic acid, microcystin LR, and BSA to evaluate cross-reaction. As shown in Table 1, no cross reaction occurred.

Table 1 Cross-reactivity (CR) with other marine toxins by TRFIA.
Application of OA-TRFIA in samples
Matrix effect and recovery
The matrix effect study showed that the matrix effect did not affect TRFIA detection when the shellfish samples were diluted three times, as shown in Figure S2. Therefore, this sample dilution factor was used for subsequent studies. Table 2 shows the test results of diluted shellfish samples. Table 3 shows that the recovery rate of diluted shellfish samples was 93.04%–111.66%, indicating that the established method had high accuracy.
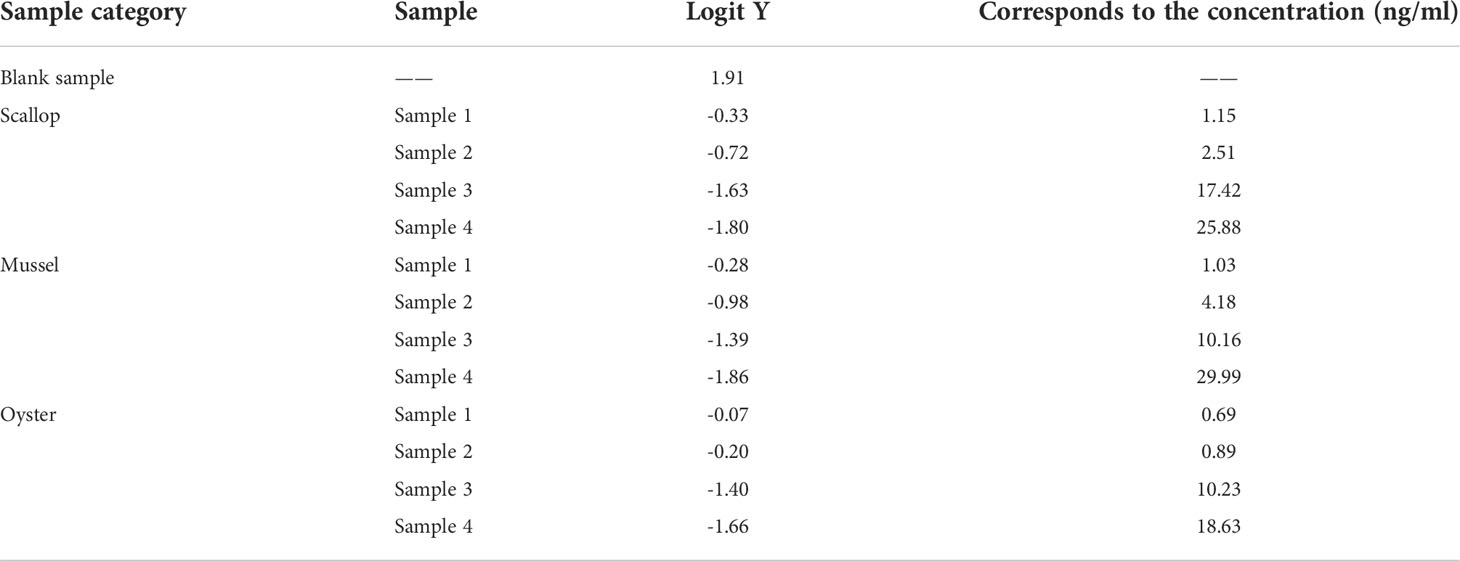
Table 2 TRFIA detection of shellfish samples.

Table 3 Recoveries of samples spiked with OA by TRFIA.
Validation of OA-TRFIA with HPLC
After OA was artificially added to the samples of mussels, scallops, and oysters, its content in the samples was determined by HPLC. As shown in Figure 4 (R2 = 0.97, P<0.01), the TRFIA established in this study has high accuracy. Moreover, phytoplankton samples were also used to detected OA by OA-TRFIA and HPLC, as shown in Table S1.
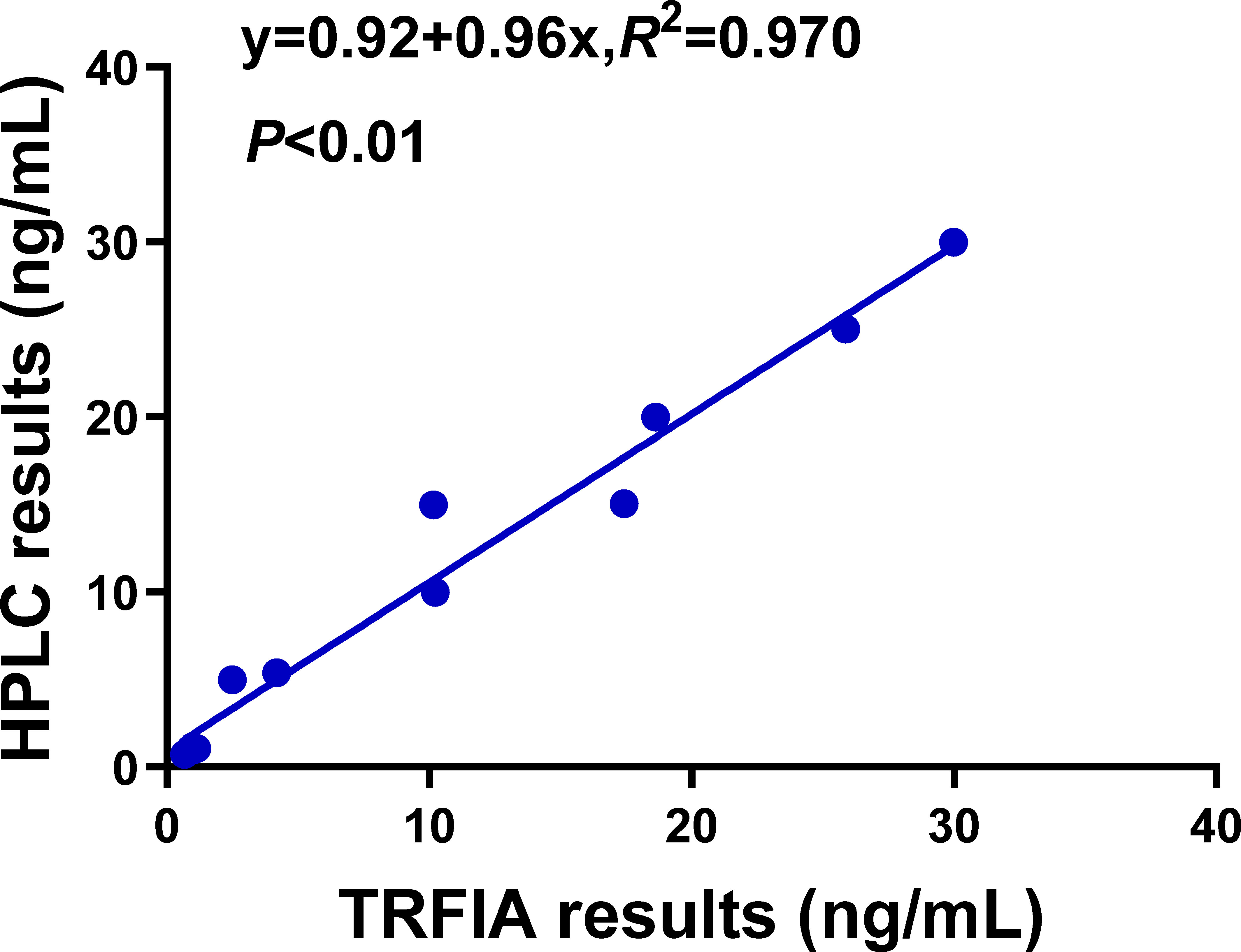
Figure 4 Correlation between TRFIA and HPLC for the concentrations of OA in shellfish samples.
Discussion and conclusion
As an important toxin from marine microalgae, OA has been detected worldwide (Lv et al., 2021). Different algal toxins are affected differently by nutrients due to their different structures. The production of carbon-rich toxins such as OA is often promoted by limiting nitrogen or phosphorus (Accoroni et al., 2018). It is mainly transmitted to humans through the consumption of seafood, and shellfish have accumulated the highest levels of this toxin due to the filter-feeding of harmful algae (Campos et al., 2020). The research to the effects of OA and other diarrheic shellfish poisoning (DSP) on aquatic animals is insufficient, and no documented mortality of bivalves associated with DSP exposure so far (Campos et al., 2020). However, there has been a growing number of publications focusing on the effects of OA on bivalves. The results of genomics and proteomics showed that OA affects the gene expression of shellfish cells, leading to cytoskeletal instability, apoptosis, stress/immune responses, and cellular energy metabolism abnormality (Madamanchi et al., 2001; Manfrin et al., 2010; Huang et al., 2015; Dou et al., 2020). It can be seen that OA affects the growth, development, and reproduction of shellfish, and seriously affects human consumption of shellfish and other seafood. Therefore, a detection method for the rapid and sensitive detection of OA in shellfish must be developed.
In this study, we established an indirect competition TRFIA for OA in shellfish. Goat anti-mouse IgG, i.e., secondary antibody, was labeled with europium for low detection limit and high sensitivity (Esteve-Turrillas et al., 2010; Sesay et al., 2013; Li et al., 2014; Sheng et al., 2020). Compared with labeling the primary antibody, labeling the secondary antibody avoids the interference of the marker to the binding site of the antigen on the first antibody. Furthermore, the secondary antibody, labeled with polyclonal antibody, can act as an amplified signal.
Under the optimized conditions, the detection range was 2.49 10-3 – 50 ng/ml, which is equivalent to 3.74×10-5 – 0.75 mg toxins per kilogram of shellfish, a value far beyond the EU or the US shellfish toxin detection limit of 0.16 mg/kg (Farrell et al., 2018). The IC50 was 0.72 ng/ml, the LOD was 2.49 × 10-3 ng/ml, the inter-batch and intra-batch precision values were 3.34% and 5.87%, respectively, and the recovery was 93.04%–111.66%. Compared with those of the previously reported indirect competition ELISA (detection limit 12 pg/ml, detection range 20–750 pg/ml), the detection limit and detection range were improved in TRFIA, which reflects its superiority (Wang et al., 2017). The unique fluorescence signal of rare-earth ions, the decay time different from that of conventional ions and the amplification of fluorescence signal by the enhancement solution enhance the sensitivity and detection range of the proposed method.
The OA-TRFIA established in this study neither cross-reacted with saxitoxin in PSP and domoic acid in ASP, which also has neurotoxicity, nor cross-reacted with microcystin LR, which has the same toxic mechanism.
The results also showed that the established method could be applied in the detection of actual shellfish samples, and the results were verified by HPLC. Compared with instrumental analysis, OA-TRFIA uses fewer organic solvents, which reduce the potential danger in the experimental process and reduce the requirements for operators and operating instruments, eliminates the need for complex sample pretreatment, reduces costs, and simplifies the operation. Moreover, besides europium (Eu3+), samarium (Sm3+) could also be used for labeling another detection antibody. Based on the results of this study, a dual-labeled TRFIA method can be established to detect OA and another toxin molecule, simultaneously, in a future study.
In conclusion, we developed OA-TRFIA, an ultrasensitive, rapid, and economic method for the wholesale screening and supervising of OA in shellfish samples. This method is expected to make proper contribution in marine biotoxin detection.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
YQ, JL, SS, JJ, and JK performed the experiments. JL, JJ, JK, ZZ, CZ, and XZ analyzed the data and made graphs. YQ and JL wrote the manuscript. YQ, BH, and BNH were responsible for project management and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Key R&D Program of Zhejiang Province (2021C02062).
Acknowledgments
We thank Prof. Yubo Liang at National Marine Environmental Monitoring Center for providing the test samples.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.961751/full#supplementary-material
References
Accoroni S., Ceci M., Tartaglione L., Romagnoli T., Campanelli A., Marini M., et al. (2018). Role of temperature and nutrients on the growth and toxin production of prorocentrum hoffmannianum (Dinophyceae) from the Florida keys. Harmful. Algae. 80, 140–148. doi: 10.1016/j.hal.2018.11.005
Ajani P. A., Sarowar C., Turnbull A., Farrell H., Zammit A., Helleren S., et al. (2021). A comparative analysis of methods (lc-ms/ms, lc-ms and rapid test kits) for the determination of diarrhetic shellfish toxins in oysters, mussels and pipis. Toxins. (Basel). 13 (8), 563. doi: 10.3390/toxins13080563
Campos A., Freitas M., de Almeida A. M., Martins J. C., Domínguez-Pérez D., Osório H., et al. (2020). OMICs approaches in diarrhetic shellfish toxins research. Toxins. (Basel). 12 (8), 493. doi: 10.3390/toxins12080493
Caron D. A., Garneau M. E., Seubert E., Howard M. D., Darjany L., Schnetzer A., et al. (2010). Harmful algae and their potential impacts on desalination operations off southern California. Water Res. 44 (2), 385–416. doi: 10.1016/j.watres.2009.06.051
Castanheira A., Dos Santos M. B., Rodriguez-Lorenzo L., Queirós R., Espiña B. (2021). A novel microfluidic system for the sensitive and cost-effective detection of okadaic acid in mussels. Analyst 146 (8), 2638–2645. doi: 10.1039/d0an02092c
Chen X., Zhou K., Xiang Z., Zhou X., Wang Y., Hong J., et al. (2022). Establishment and clinical application of time-resolved immunofluorescence assay of lipoprotein-associated phospholipase A2. Anal. Biochem., 648, 114674. doi: 10.1016/j.ab.2022.114674
Dou M., Jiao Y. H., Zheng J. W., Zhang G., Li H. Y., Liu J. S., et al. (2020). De novo transcriptome analysis of the mussel perna viridis after exposure to the toxic dinoflagellate prorocentrum lima. Ecotoxicol. Environ. Saf. 192, 110265. doi: 10.1016/j.ecoenv.2020.110265
Esteve-Turrillas F. A., Parra J., Abad-Fuentes A., Agulló C., Abad-Somovilla A., Mercader J. V. (2010). Hapten synthesis, monoclonal antibody generation, and development of competitive immunoassays for the analysis of picoxystrobin in beer. Anal. Chim. Acta 682 (1-2), 93–103. doi: 10.1016/j.aca.2010.09.042
Fang S., Zhang Y., Liu X., Qiu J., Liu Z., Kong F. (2019). Development of a highly sensitive time-resolved fluoroimmunoassay for the determination of trace salbutamol in environmental samples. Sci. Total. Environ. 679, 359–364. doi: 10.1016/j.scitotenv.2019.05.057
Farrell H., Ajani P., Murray S., Baker P., Webster G., Brett S., et al. (2018). Diarrhetic shellfish toxin monitoring in commercial wild harvest bivalve shellfish in New South Wales, Australia. Toxins. (Basel). 10 (11), 446. doi: 10.3390/toxins10110446
Ferron P. J., Hogeveen K., Fessard V., Le Hégarat L. (2014). Comparative analysis of the cytotoxic effects of okadaic acid-group toxins on human intestinal cell lines. Mar. Drugs 12 (8), 4616–4634. doi: 10.3390/md12084616
Fraga M., Vilariño N., Louzao M. C., Rodríguez P., Campbell K., Elliott C. T., et al. (2013). Multidetection of paralytic, diarrheic, and amnesic shellfish toxins by an inhibition immunoassay using a microsphere-flow cytometry system. Anal. Chem. 85 (16), 7794–7802. doi: 10.1021/ac401146m
Fu L. L., Zhao X. Y., Ji L. D., Xu J. (2019). Okadaic acid (OA): Toxicity, detection and detoxification. Toxicon 160, 1–7. doi: 10.1016/j.toxicon.2018.12.007
Griffith A. W., Gobler C. J. (2020). Harmful algal blooms: A climate change co-stressor in marine and freshwater ecosystems. Harmful. Algae. 91, 101590. doi: 10.1016/j.hal.2019.03.008
Huang B., Xiao H., Zhang J., Zhang L., Yang H., Zhang Y., et al. (2009). Dual-label time-resolved fluoroimmunoassay for simultaneous detection of aflatoxin B1 and ochratoxin a. Arch. Toxicol. 83 (6), 619–624. doi: 10.1007/s00204-009-0410-6
Huang L., Zou Y., Weng H. W., Li H. Y., Liu J. S., Yang W. D. (2015). Proteomic profile in perna viridis after exposed to prorocentrum lima, a dinoflagellate producing dsp toxins. Environ. Pollut. 196, 350–357. doi: 10.1016/j.envpol.2014.10.019
Hu L., Wang R., Wang M., Xu Y., Wang C., Liu Y., et al. (2022). Research progress of photocatalysis for algae killing and inhibition: A review. Environ. Sci. Pollut. Res. Int. 29, 47902–47914. doi: 10.1007/s11356-022-20645-9
Hu X., Yao J., Wang F., Yin M., Sun Y., Hu M., et al. (2018). Eu(3+) -labeled IgG-based time-resolved fluoroimmunoassay for highly sensitive detection of aflatoxin B(1) in feed. J. Sci. Food Agric. 98 (2), 674–680. doi: 10.1002/jsfa.8514
Klijnstra M. D., Faassen E. J., Gerssen A. (2021). A generic lc-hrms screening method for marine and freshwater phycotoxins in fish, shellfish, water, and supplements. Toxins. (Basel). 13 (11), 823. doi: 10.3390/toxins13110823
Li M., Sheng E., Yuan Y., Liu X., Hua X., Wang M. (2014). Sensitive time-resolved fluoroimmunoassay for quantitative determination of clothianidin in agricultural samples. Environ. Sci. Pollut. Res. Int. 21 (9), 5803–5809. doi: 10.1007/s11356-014-2506-7
Lloyd J. K., Duchin J. S., Borchert J., Quintana H. F., Robertson A. (2013). Diarrhetic shellfish poisoning, Washington, USA, 2011. Emerg. Infect. Dis. 19 (8), 1314–1316. doi: 10.3201/eid1908.121824
Lv J. J., Yuan K. K., Lu M. Y., He Z. B., Li H. Y., Yang W. D. (2021). Responses of JNK signaling pathway to the toxic dinoflagellate prorocentrum lima in the mussel perna viridis. Ecotoxicol. Environ. Saf. 227, 112905. doi: 10.1016/j.ecoenv.2021.112905
Madamanchi N. R., Li S., Patterson C., Runge M. S. (2001). Reactive oxygen species regulate heat-shock protein 70 via the JAK/STAT pathway. Arterioscler. Thromb. Vasc. Biol. 21 (3), 321–326. doi: 10.1161/01.atv.21.3.321
Manfrin C., Dreos R., Battistella S., Beran A., Gerdol M., Varotto L., et al. (2010). Mediterranean Mussel gene expression profile induced by okadaic acid exposure. Environ. Sci. Technol. 44 (21), 8276–8283. doi: 10.1021/es102213f
Marampouti C., Buma A. G. J., de Boer M. K. (2021). Mediterranean Alien harmful algal blooms: Origins and impacts. Environ. Sci. Pollut. Res. Int. 28 (4), 3837–3851. doi: 10.1007/s11356-020-10383-1
Onofrio M. D., Mallet C. R., Place A. R., Smith J. L. (2020). A screening tool for the direct analysis of marine and freshwater phycotoxins in organic spatt extracts from the Chesapeake Bay. Toxins. (Basel). 12 (5), 322. doi: 10.3390/toxins12050322
Pérez L., Escudero I., Cabado A. G., Molinero-Abad B., Arcos-Martínez M. J. (2019). Study of ceramic membrane behavior for okadaic acid and heavy-metal determination in filtered seawater. J. Environ. Manage. 232, 564–573. doi: 10.1016/j.jenvman.2018.11.077
Reguera B., Riobó P., Rodríguez F., Díaz P. A., Pizarro G., Paz B., et al. (2014). Dinophysis toxins: Causative organisms, distribution and fate in shellfish. Mar. Drugs 12 (1), 394–461. doi: 10.3390/md12010394
Sesay A. M., Micheli L., Tervo P., Palleschi G., Virtanen V. (2013). Development of a competitive immunoassay for the determination of cortisol in human saliva. Anal. Biochem. 434 (2), 308–314. doi: 10.1016/j.ab.2012.12.008
Sheng E. Z., Tan Y. T., Lu Y. X., Xiao Y., Li Z. X. (2020). Sensitive time-resolved fluorescence immunoassay for quantitative determination of oxyfluorfen in food and environmental samples. Front. Chem. 8. doi: 10.3389/fchem.2020.621925
Tian Y., Du L., Zhu P., Chen Y., Chen W., Wu C., et al. (2021). Recent progress in micro/nano biosensors for shellfish toxin detection. Biosens. Bioelectron. 176, 112899. doi: 10.1016/j.bios.2020.112899
Tian Y., Yuan L., Zhang M., He Y., Lin X. (2022). Sensitive detection of the okadaic acid marine toxin in shellfish by Au@Pt NPs/horseradish peroxidase dual catalysis immunoassay. Anal. Methods 14 (12), 1261–1267. doi: 10.1039/d1ay01973b
Uchida H., Watanabe R., Matsushima R., Uchida N., Nagai H., Kamio M., et al. (2014). A convenient HPLC method for detection of okadaic acid analogs as 9-anthrylmethyl esters with automated sample cleanup by column switching. J. AOAC. Int. 97 (2), 391–397. doi: 10.5740/jaoacint.sgeuchida
Valdiglesias V., Prego-Faraldo M. V., Pásaro E., Méndez J., Laffon B. (2013). Okadaic acid: More than a diarrheic toxin. Mar. Drugs 11 (11), 4328–4349. doi: 10.3390/md11114328
Verma M., Chaudhary M., Singh A., Kaur N., Singh N. (2020). Naphthalimide-gold-based nanocomposite for the ratiometric detection of okadaic acid in shellfish. J. Mater. Chem. B. 8 (36), 8405–8413. doi: 10.1039/d0tb01195a
Wang K., Huang B., Zhang J., Zhou B., Gao L., Zhu L., et al. (2009). A novel and sensitive method for the detection of deoxynivalenol in food by time-resolved fluoroimmunoassay. Toxicol. Mech. Methods 19 (9), 559–564. doi: 10.3109/15376510903380720
Wang L., Sang Y. X., Wang X. H. (2011). Enzyme-linked immunosorbent assay for okadaic acid: investigation of analytical conditions and sample matrix on assay performance. J. AOAC. Int. 94 (5), 1531–1539. doi: 10.5740/jaoacint.10-412
Wang Y., Xianyu Y. (2022). Nanobody and nanozyme-enabled immunoassays with enhanced specificity and sensitivity. Small. Methods 6 (4), e2101576. doi: 10.1002/smtd.202101576
Wang R., Zeng L., Yang H., Zhong Y., Wang J., Ling S., et al. (2017). Detection of okadaic acid (OA) using ELISA and colloidal gold immunoassay based on monoclonal antibody. J. Hazard. Mater. 339, 154–160. doi: 10.1016/j.jhazmat.2017.06.030
Wu D., Chen J., Wang J., He X., Xin M., Wang B. (2020). Monitoring and warning of lipophilic marine algal toxins in mariculture zone based on toxin profiles of phytoplankton. Ecotoxicol. Environ. Saf. 197, 110647. doi: 10.1016/j.ecoenv.2020.110647
Yin M., Wang W., Wei J., Chen X., Chen Q., Chen X., et al. (2022). Novel dual-emissive fluorescent immunoassay for synchronous monitoring of okadaic acid and saxitoxin in shellfish. Food Chem. 368, 130856. doi: 10.1016/j.foodchem.2021.130856
Keywords: okadaic acid, time-resolved fluoroimmunoassay, quantitative detection, diarrheic shellfish poisoning, phycotoxins, shellfish
Citation: Qin Y, Li J, Kuang J, Shen S, Jiang J, Zhang Z, Zhao C, Zhou X, Huang B and Han B (2022) Sensitive time-resolved fluoroimmunoassay for the quantitative detection of okadaic acid. Front. Mar. Sci. 9:961751. doi: 10.3389/fmars.2022.961751
Received: 05 June 2022; Accepted: 03 August 2022;
Published: 24 August 2022.
Edited by:
Jose Javier Fernandez, University of La Laguna, SpainReviewed by:
Tao Le, Chongqing Normal University, ChinaSteven Jing-liang Xu, Hong Kong Metropolitan University, Hong Kong SAR, China
Copyright © 2022 Qin, Li, Kuang, Shen, Jiang, Zhang, Zhao, Zhou, Huang and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bingnan Han, hanbingnan@zstu.edu.cn; Biao Huang, huangbiao@zstu.edu.cn; Yuan Qin, qinyuan@zstu.edu.cn
†These authors have contributed equally to this work