 Shanji Chen1,2†
Shanji Chen1,2† Danmei Tian1,2†
Danmei Tian1,2† Jihua Wei3Cong Li4Yihan Ma3Xiaoshuang Gou1,2Yiran Shen4Mei Chen1,2Sihao Zhang1,2Jia Li4,5
Jihua Wei3Cong Li4Yihan Ma3Xiaoshuang Gou1,2Yiran Shen4Mei Chen1,2Sihao Zhang1,2Jia Li4,5 Bin Wu3*
Bin Wu3* Jinshan Tang1,2*
Jinshan Tang1,2*- 1Institute of Traditional Chinese Medicine and Natural Products, College of Pharmacy, Jinan University, Guangzhou, China
- 2Guangdong Province Key Laboratory of Pharmacodynamic Constituents of Traditional Chinese Medicine and New Drug Research, Jinan University, Guangzhou, China
- 3Ocean College, Zhejiang University, Hangzhou, China
- 4National Center for Drug Screening, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China
- 5Open Studio for Druggability Research of Marine Natural Products, Pilot National Laboratory for Marine Science and Technology (Qingdao), Qingdao, China
Two new citrinin dimers bearing a 6,6-spiroketal moiety (1 and 2) and four known analogues (3–6), together with 18 known citrinin monomers (7–24), were isolated from the culture of hydrothermal vent-associated fungus Penicillium citrinum Y34. Their structures were identified by extensive spectroscopic analyses, 13C NMR calculation in combination with DP4+, linear correlation coefficient (R2), and mean absolute error (MAE) values analyses, and electronic circular dichroism (ECD) calculation. The α-glucosidase and ATP-citrate lyase (ACL) inhibitory activities of isolated compounds were evaluated. Compounds 1, 3, and 12 displayed moderate α-glucosidase inhibitory activities with IC50 values of 239.8, 176.2, and 424.4 μM, respectively. Enzyme kinetics investigations of 1 and 3 suggested their non-competitive inhibition of α-glucosidase with Ki values of 204.3 and 212.7 μM, respectively. Meanwhile, compound 4 showed significant ACL inhibitory potential with an IC50 value of 17.4 μM. Furthermore, the interactions of 1, 3, and 12 with α-glucosidase and 4 with ACL were investigated by molecular docking assay. This study demonstrates that citrinins, especially for their dimers, could be potential lead compounds for the development of new agents for the treatment of metabolic diseases.
Introduction
Penicillium citrinum is one of the endophytic fungi that are usually found in commensalism with plants and marine biota. Many reports showed that it can produce a plethora of secondary metabolites like polyketides, alkaloids, peptides, and meroterpenoids (El-Neketi et al., 2013; Samanthi et al., 2015; Bakhtra et al., 2019; Anas et al., 2021). Among them, citrinin, a well-known polyketide derivative first isolated from P. citrinum, represents a large family of fungal mycotoxin (Clark et al., 2006; Li et al., 2017; Li et al., 2020). Recently, this class of natural products have attracted the attention of chemists and pharmacologists due to their unique structures and broad spectrum of activities such as anticancer, antimicrobial, radical-scavenging, anti-influenza, and neuro-protective effects (Zhang et al., 2007; Chang et al., 2009; Nakajima et al., 2016; Yang et al., 2018; Cao et al., 2019; Wang et al., 2019a). Structurally, citrinin monomers often possess core skeletons including benzo-pyran, benzo-furan, monobenzene, and quinone-pyran, and can polymerize to generate diverse dimers by direct carbon/carbon bonds or by oxygen, carbon, acetone, and carbonyl bridged linkage (Wang et al., 2019b). However, the citrinin dimers bearing a 6,6-spiroketal moiety are relative rare, and only three analogues, namely, xerucitrinin A, xerucitrinic acid A, and xerucitrinic acid B, have been reported till now. Meanwhile, citrinin dimers were reported to have potential antimicrobial effect and cytotoxicity (Sadorn et al., 2016; Salendra et al., 2021).
Disorders of carbohydrate and lipid metabolism are the main reason of metabolic diseases, which show an increasing morbidity recently and has become a growing concern worldwide (Megur et al., 2022). As α-glucosidase, a carbohydrate hydrolytic enzyme, is vital in modulating blood glucose levels, and ATP-citrate lyase (ACL) is a key enzyme that links carbohydrate with lipid metabolism by producing acetyl-CoA from citrate for the biosynthesis of fatty acid and cholesterol, they have been proven to be potential targets for the treatment of carbohydrate (such as diabetes) and lipid-disorder-related disease (such as hyperlipidemia and hypercholesterolemia) (Sansenya and Payaka, 2022; Wan et al., 2022; Zhou et al., 2022). Now, three α-glucosidase inhibitors, namely, acarbose, miglitol, and voglibose, are used in clinics for the treatment of diabetes. Recently, an ACL inhibitor, bempedoic acid, was approved for the treatment of hypercholesterolemic patients as monotherapy or in combination with ezetimibe (Delevry and Gupta, 2021; Sansenya and Payaka, 2022). Additionally, it was reported that polyketides have exhibited potential α-glucosidase and ACL inhibitory activities (Chen et al., 2021; Paulose and Chakraborty, 2022; Zhong et al., 2022). Thus, discovery of new polyketides with potential α-glucosidase and ACL inhibitory activities shows great promise for the development of new agents for the treatment of metabolic diseases.
In this study, our ongoing efforts to discover new bioactive metabolites from marine microorganisms (Gou et al., 2020; Gou et al., 2021) resulted in the isolation of 24 citrinin derivatives, including six citrinin dimers (1–6), in which compounds 1 and 2 were new ones with a 6,6-spiroketal moiety, and 18 known citrinin monomers 7–24 from hydrothermal vent sediment-derived fungus P. citrinum Y34. Here, we described the isolation and structural elucidation of two new citrinin dimers (1 and 2). Meanwhile, the α-glucosidase and ATP-citrate lyase (ACL) inhibitory activities of isolated compounds were conducted. The results demonstrated that compounds 1, 3, and 12 showed moderate α-glucosidase inhibitory activities and compound 4 displayed potent ACL inhibitory activity.
Materials and Methods
General Experimental Procedure
The instruments for data acquisition of infrared (IR), ultraviolet–visible (UV/vis), high-resolution electrospray ionization mass spectroscopy (HRESIMS), NMR, optical rotation, and circular dichroism (CD) spectra were consistent with our previous reports (Gou et al., 2020; Gou et al., 2021). Signals of deuterated solvents of CD3OD at δH 3.31/δC 49.0, DMSO-d6 at δH 2.50/δC 39.5, and CDCl3 at δH 7.26/δC 77.4 were used as internal references in the NMR measurement. The analytical high-performance liquid chromatography (HPLC) was run in an LC-20AB liquid chromatography coupled with a SPD-M20A DAD detector (Shimadzu, Kyoto, Japan). The preparative HPLC was conducted on an LC-20AT liquid chromatography coupled with an SPD-20A UV/Vis detector (Shimadzu, Kyoto, Japan). Columns for analytical and preparative HPLC were YMC-Triart C18 column (5 μm, φ 4.6×250 mm) and YMC Pack ODS-A column (5 μm, φ 10×250 mm), respectively (YMC, Japan). Silica gels (200–300 mesh) for column chromatography (CC) were products of Qingdao Marine Ltd., Shandong, China. Octadecylsilane (ODS) (12 nm, 50 μm) for CC was purchased from YMC Ltd. (Tokyo, Japan).
α-Glucosidase from Saccharomyces cerevisiae (G5003-100UN) was a product of Sigma-Aldrich Chemical Co. Ltd. (St. Louis, USA). 4-Nitrophenyl α-glucopyranoside (p-NPG) was purchased from Shanghai Macklin Biochemical Technology Co. Ltd., Shanghai, China. Acarbose was a product of Aladdin Reagent Co., Ltd. (Shanghai, China), and ADP-Glo™ Kinase Assay kit was purchased from Promega, Madison, USA.
Strain Material and Identification
The marine strain Y34 was isolated from hydrothermal vent sulfur-rich sediment, which was collected from Kueishantao, Taiwan, and identified as P. citrinum according to the morphological characteristics and internal transcribed spacer (ITS) sequence (Shi et al., 2019).
Fermentation and Extraction
Strain P. citrinum Y34 was inoculated in ISP2 agar plate, which was composed of 4 g of yeast extract, 4.0 g of glucose, 10.0 g of malt extract, and 18.0 g of agar in 1 L of ddH2O in pH 7.2 at room temperature. Subsequently, the spores from the agar plate were inoculated to solid corn medium that was composed of 50.0 g of crushed corn with 0.9 g of yeast extract, 2.4 g of ammonium tartrate, 0.2 g of MgSO4, 0.2 g of KH2PO4, and 0.4 g of sea salt in 20 ml of ddH2O, and fermented for 44 days. The corn culture was extracted with EtOAc for three times, and the combined filtrate was evaporated under reduced pressure to obtain the extracts. Ten kilograms of large scaled fermentation was carried out in solid corn medium, and 88.2 g crude extracts were obtained.
Compound Isolation
The extracts were subjected to silica gel CC (φ 100×1,100 mm, 200–300 mesh, 1.2 kg) eluted with gradient petroleum ether-EtOAc (from 100:0 to 0:100) and EtOAc-MeOH (90:10 and 0:100) to get five fractions (Fr.1–5). The Fr.1 (1.1 g, eluted with Petroleum ether-EtOAc 80:20) was chromatographed by a silica gel CC (200–300 mesh, 20.0 g) with elution of Petroleum ether-EtOAc (from 98:2 to 70:30) and EtOAc-MeOH (0:100) to give nine subfractions (Fr.1-1 to Fr.1-9). Fr.1-5 (111.1 mg, eluted with Petroleum ether-EtOAc 90:10) was applied to semi-preparative RP HPLC with 62% MeOH–H2O (0.1% CH3COOH) to get compound 13 (11.5 mg). Fr.1-6 (52.7 mg, eluted by Petroleum ether-EtOAc 90:10) was further isolated by preparative RP HPLC with 55% MeOH–H2O (0.1% CH3COOH) to get compound 7 (8.4 mg). Fr.1-7 (362.6 mg, eluted by Petroleum ether-EtOAc 80:20) was subjected to an ODS CC with stepwise gradient elution of 55%–100% MeOH–H2O to obtain seven subfractions (Fr.1-7-1 to Fr.1-7-7). Fr.1-7-1 (189.7 mg, eluted by 55% MeOH–H2O) was subjected to semi-preparative RP HPLC with elution of 38% MeOH–H2O (0.1% CH3COOH) to get compounds 11 (95.4 mg), 20 (2.9 mg), and 23 (8.0 mg). Fr.1-7-2 (27.2 mg, eluted with 55% MeOH–H2O) was isolated by semi-preparative RP HPLC with 60% MeOH–H2O (0.1% CH3COOH) to get compound 8 (4.3 mg). The Fr.1-8 (233.6 mg, eluted with Petroleum ether-EtOAc 80:20) was further purified by Sephadex LH-20 eluted with MeOH to obtain two subfractions (Fr.1-8-1 to Fr.1-8-2). Fr.1-8-1 (42.2 mg) was isolated by semi-preparative RP HPLC with 50% MeOH–H2O (0.1% CH3COOH) to get compound 9 (25.3 mg). Fr.1-8-2 (101.9 mg) was subjected to semi-preparative RP HPLC with elution of 70% MeOH–H2O (0.1% CH3COOH) to get compounds 4 (55.7 mg) and 5 (10.0 mg). Fr.1-9 (50.5 mg, eluted with Petroleum ether-EtOAc 80:20) was applied to semi-preparative RP HPLC with 55% CH3CN–H2O (0.1% CH3COOH) to obtain compound 14 (3.4 mg). The Fr.2 (1.07 g, eluted with Petroleum ether-EtOAc 70:30) was chromatographed by an ODS CC with stepwise gradient elution of 45%–100% MeOH–H2O to obtain 10 subfractions (Fr.2-1 to Fr.2-10). Fr.2-2 (125.8 mg, eluted with 55% MeOH–H2O) was isolated by semi-preparative RP HPLC eluted with 55% MeOH–H2O (0.1% CH3COOH) to get compound 10 (14.0 mg). Fr.2-4 (66.0 mg, eluted by 65% MeOH–H2O) was isolated by semi-preparative RP HPLC with 60% MeOH–H2O (0.1% CH3COOH) to acquire compounds 1 (5.1 mg), 2 (2.0 mg), and 3 (6.9 mg). The Fr.3 (8.4 g, eluted with Petroleum ether-EtOAc 60:40) was subjected to an ODS CC with gradient MeOH–H2O (35%–100%) to get 14 subfractions (Fr.3-1 to Fr.3-14). Fr.3-1 (327.1 mg, eluted with 35% MeOH–H2O) was applied to semi-preparative RP HPLC eluted with 15% CH3CN–H2O (0.1% CH3COOH) to obtain compounds 18 (4.5 mg) and 24 (20.9 mg). Fr.3-2 (157.4mg, eluted with 35% MeOH–H2O) was purified by semi-preparative RP HPLC with elution of 20% CH3CN–H2O (0.1% CH3COOH) to obtain compounds 19 (3.9 mg) and 21 (9.8 mg). Fr.3-3 (268.2 mg, eluted with 45% MeOH–H2O) was isolated by semi-preparative RP HPLC eluted with 40% MeOH–H2O (0.1% CH3COOH) to get compounds 15 (7.0 mg) and 17 (8.5 mg). Fr.3-4 (161.7 mg eluted with 45% MeOH-H2O) was subjected to semi-preparative RP HPLC eluted with 45% MeOH–H2O (0.1% CH3COOH) to yield compound 12 (6.9 mg). Fr.3-6 (1.6 g, eluted with 70% MeOH–H2O) was applied to semi-preparative RP HPLC eluted with 45% CH3CN–H2O (0.1% CH3COOH) to get compound 22 (2.2 mg). The Fr.4 (4.3 g, eluted with Petroleum ether-EtOAc 40:60) was isolated by an ODS CC with gradient MeOH–H2O (35%–100%) to get nine subfractions (Fr.4-1 to Fr.4-9). Fr.4-7 (236.3 mg, eluted with 35% MeOH–H2O) was isolated by semi-preparative RP HPLC with elution of 60% MeOH–H2O (0.1% CH3COOH) to yield compound 6 (44.0 mg). The Fr.5 (11.9 g, eluted with Petroleum ether-EtOAc 0:100) was subjected to an ODS CC with stepwise gradient elution of 35%–100% MeOH–H2O to obtain 11 subfractions (Fr.5-1 to Fr.5-11). Fr.5-2 (518.1 mg, eluted with 35% MeOH–H2O) was applied to semi-preparative RP HPLC eluted with 40% MeOH–H2O (0.1% CH3COOH) to get compound 16 (46.2 mg). All structures of isolated compounds are shown in Figure 1.
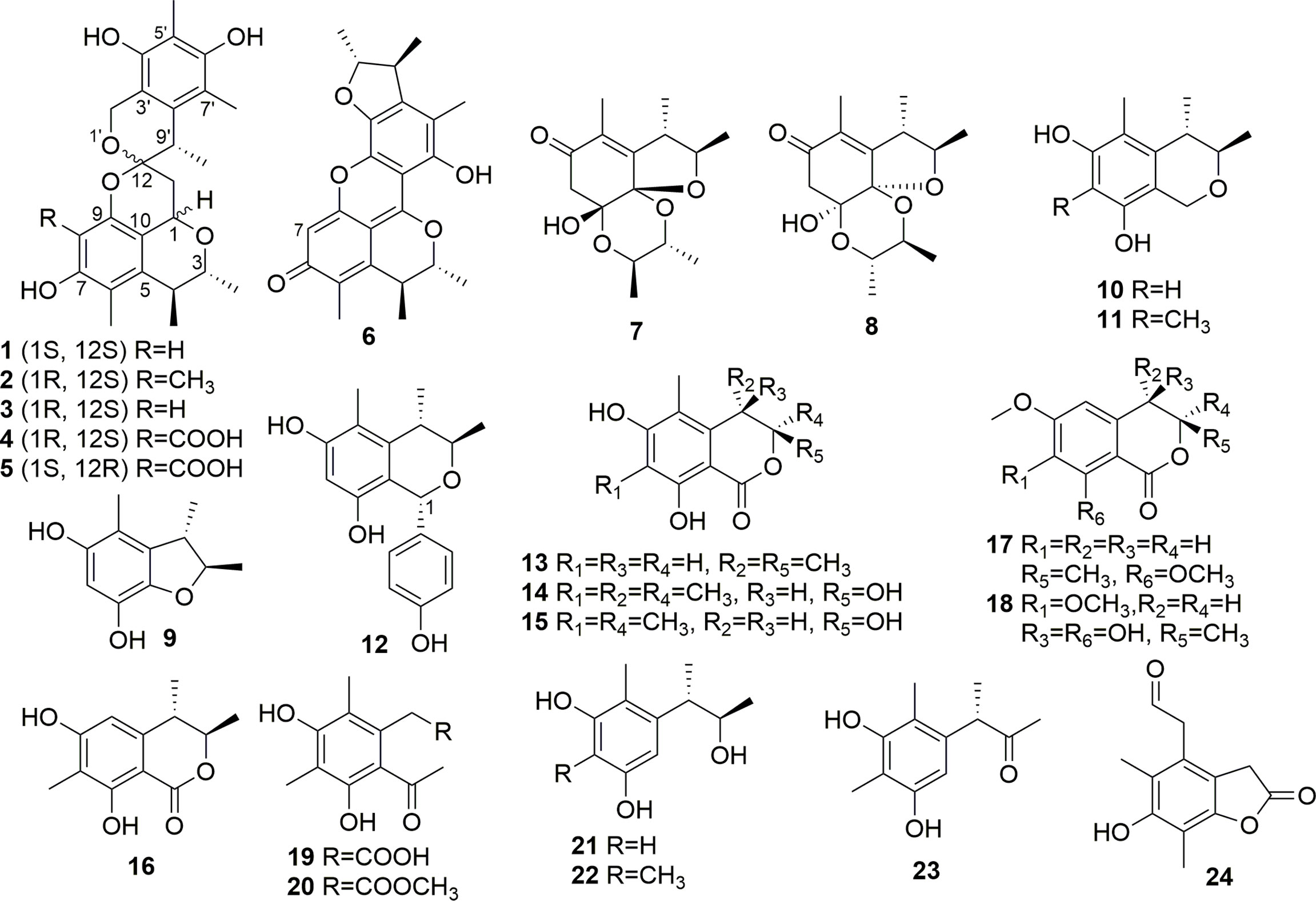
Figure 1 Chemical structures of compounds 1–24.
Xerucitrinin B (1): Pale yellow oil; [α]25D-47.68 (c 0.5, MeOH); UV (MeOH) λmax (log ε), 206 (4.3), 228 (3.6), 284 (3.1); CD (MeOH) (Δε) 209 (−4.00), 225 (−1.48), 230 (−1.57), 291 (+0.08); IR (KBr) νmax, 3,456, 3,356, 2,963, 2,928, 2,870, 1,631, 1,600, 1,555, 1,433, 1,335, 1,116, 874, 842, 777, 714, and 622 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 427.2122 [M + H]+ (calcd for C25H31O6, 427.2121).
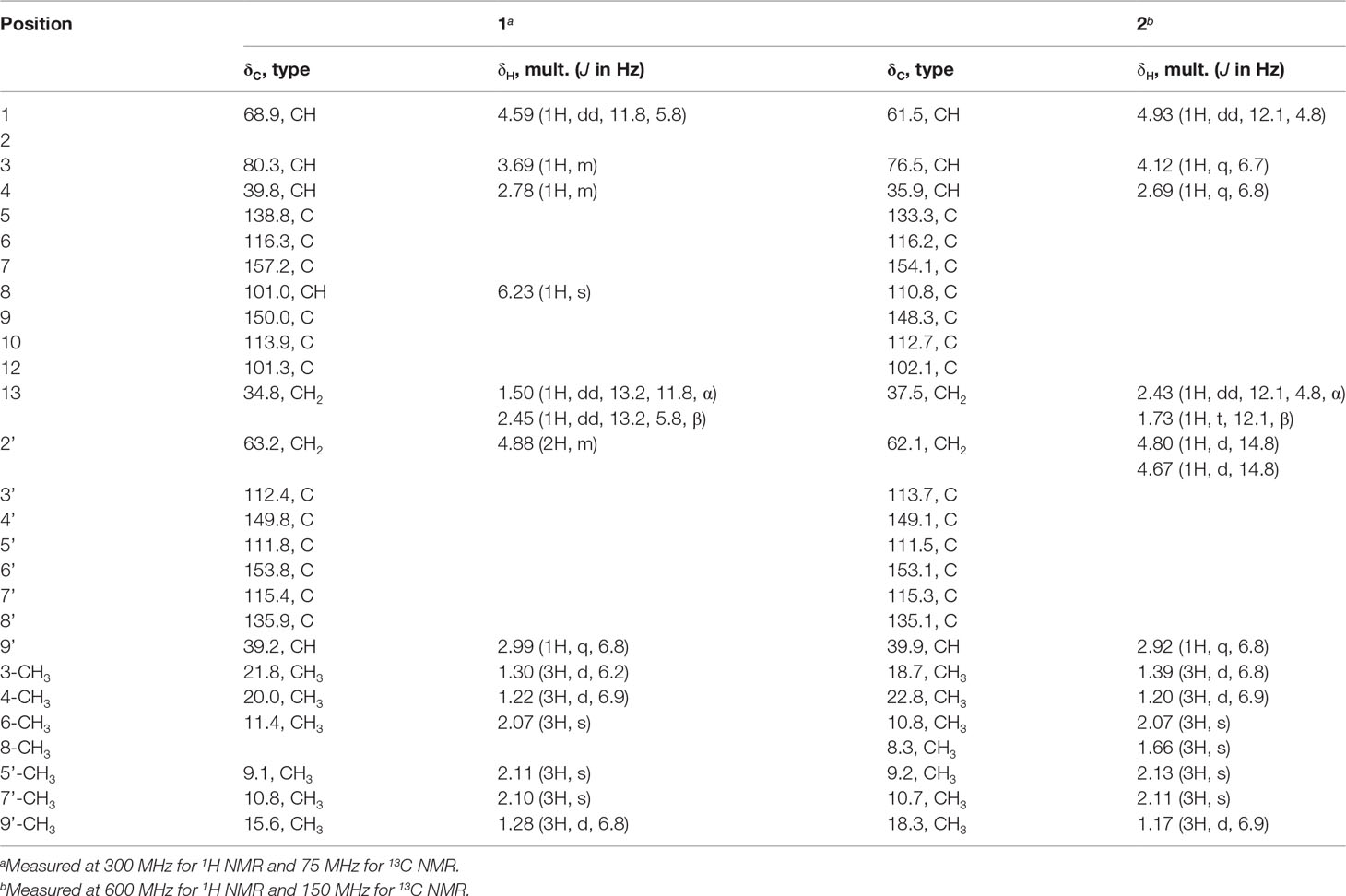
Table 1 1H and 13C NMR spectroscopic data for compounds 1 and 2 in CD3OD.
Xerucitrinin C (2): Pale yellow oil; [α]25D + 39.32 (c 0.5, MeOH); UV (MeOH) λmax (log ε), 206 (4.2), 229 (3.6), 280 (2.8); CD (MeOH) (Δε) 206 (−0.59), 212 (−0.06), 240 (−0.69), 302 (−0.04); IR (KBr) νmax, 3,550, 3,475, 3,411, 2,959, 1,631, 1,556, 1,432, 1,336, 1,156, 878, 780, 714, and 626 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 441.2278 [M + H]+ (calcd for C26H33O6, 441.2277).
Quantum Chemical NMR Calculation of Compound 1
The 3D conformations and subsequent conformer databases of four plausible stereoisomers, (1S*, 3R*, 4S*, 12S*, 9′S*)-1A, (1S*, 3R*, 4S*, 12S*, 9′R*)-1B, (1S*, 3R*, 4S*, 12R*, 9′S*)-1C and (1S*, 3R*, 4S*, 12R*, 9′R*)-1D of compound 1 were acquired using CORINA version 3.4 and CONFLEX version 7.0, respectively. Then, the acceptable conformers were optimized using the HF/6-31G (d) and the wB97XD/6-31G (d) methods in Gaussian 09. From this, 12 stable conformers of (1S*, 3R*, 4S*, 12S*, 9′S*)-1A, 18 stable conformers of (1S*, 3R*, 4S*, 12S*, 9′R*)-1B, 13 stable conformers of (1S*, 3R*, 4S*, 12R*, 9′S*)-1C, and 17 stable conformers of (1S*, 3R*, 4S*, 12R*, 9′R*)-1D were obtained. Then, the optimized stable conformers were used for the NMR calculation at the level of mPW1PW91/6-31+G (d, p) (Frisch et al., 2016). The calculated NMR data were analyzed using the R2, MAE, and DP4+ methods to determine the relative configuration of 1.
Quantum Chemical ECD Calculations of 1 and 2
The conformer databases of (1S, 3R, 4S, 12S, 9′S)-1, (1R, 3S, 4R, 12R, 9′R)-1, (1R, 3R, 4S, 12S, 9′S)-2, and (1S, 3S, 4R, 12R, 9′R)-2 were also generated by CORINA (version 3.4) and CONFLEX (version 7.0). Then, all the acceptable conformers were optimized with HF/6-31G(d) method in Gaussian 09, and subsequent optimization at the APFD/6-31G(d) level for 1 and B3LYP/6-31G(d) level for 2 with methanol led the dihedral angles to be got. After that, stable conformers, 17 for (1S, 3R, 4S, 12S, 9′S)-1, 10 for (1R, 3S, 4R, 12R, 9′R)-1, 14 for (1R, 3R, 4S, 12S, 9′S)-2, and 18 for (1S, 3S, 4R, 12R, 9′R)-2, were acquired. The optimized conformers were subjected to electronic circular dichroism (ECD) calculations, which were performed by Gaussian 09 [APFD/6-311++G(2d,p) for 1, B3LYP/TZVP for 2]. Then, the software SpecDis was used to compare the experimental and calculated spectra (Bruhn et al., 2013), which was also applied in a UV shift to the ECD spectra, Gaussian broadening of the excitations, and Boltzmann weighting of the spectra.
α-Glucosidase Inhibitory Assay
The method of α-glucosidase inhibitory assay followed our previous report (Gou et al., 2021). Briefly, 25 µl samples (1.6 mM) and 50 µl α-glucosidase (0.2 U/ml) were added into 96-well plates. After preincubation at 37°C for 10 min, 25 µl of 5 mM p-NPG was added into each well, and the reaction continued at 37°C for another 5 min. Subsequently, 100 µl of Na2CO3 (0.1 M) was added to stop the reaction. Finally, the optical density was detected using a Synergy HT microplate reader at 405 nm. The α-glucosidase inhibition rate (I%) was calculated by the equation: I%= [(ΔAbscontrol− ΔAbssample)/ΔAbscontrol] × 100 .
Enzyme Kinetics of α-Glucosidase Inhibition Assay
The kinetic study of α-glucosidase inhibition was performed by the Lineweaver–Burk plots. Keeping the enzyme concentration constant (0.2 U/ml), enzymatic reactions were conducted and monitored with different concentrations of tested compounds (0, 100, 200, and 400 μM) and substrate (0.125, 0.25, 0.75, and 1.25 mM). Meanwhile, the inhibition constants (Ki) were calculated using the intersection of Dixon plots (He et al., 2021).
ATP-Citrate Lyase Inhibitory Assay
The ATP-citrate lyase (ACL) inhibitory assay was carried out according to previous reports (Wan et al., 2022; Zhou et al., 2022), in which ADP was quantified using ADP-Glo™ Kinase Assay. First, 2.0 μl of ACL, 2.0 μl of ATP, and 1.0 μl of test compounds were added in a 384-well plate and reacted for 0.5 h under 37°C. Meanwhile, the negative (DMSO) and positive control (BMS-303141) wells were also detected during the kinase assay. Subsequently, the kinase reaction was stopped by adding 2.5 μl of ADP-Glo reagent, and the unconsumed ATP was depleted within 1 h at room temperature. Finally, 5.0 μl of kinase detection reagent was added to convert ADP to ATP. After 1 h of incubation, the luminescence signal was measured using the PerkinElmer EnVision reader.
Molecular Docking Assay
The molecular docking simulations of compounds 1, 3, and 12 with α-glucosidase and compound 4 with ACL were performed following previous report (Wan et al., 2022). First, Chem3D (version 20.0) was used to optimize and acquire the MM2 energy-minimized 3D structures of 1, 3, 4, and 12. Meanwhile, the crystal structures of the receptor proteins, namely, α-glucosidase and ACL, were obtained from Protein Data Bank (www.rcsb.org, α-glucosidase PDB code: 4J5T; ACL PDB code: 6HXH). Then, the ligand and receptor preparation and subsequent docking were performed using AutoDock Vina. Finally, PyMOL version 2.3.4 was used to output visible results.
Statistical Analysis
In this study, GraphPad Prism software version 5 (GraphPad Software, Inc., San Diego, CA, USA) was used to perform the statistical analyses. The final data were expressed as mean ± standard error of mean (SEM), in which each experiment was conducted in triplicate. Multiple comparisons were analyzed using one-way ANOVA and following Tukey’s test. p< 0.05 was considered statistically significant.
Results
Isolation and Structural Elucidation
Compound 1 was obtained as pale yellow oil. The HRESIMS showed a protonated molecular ion at m/z 427.2122 [M + H]+ (calc. for 427.2121), which indicated a molecular formula of C25H30O6 and accounted for 11 degrees of unsaturation. The IR spectrum absorption bands suggested the presence of hydroxyl group (3,456 and 3,356 cm−1), methyl group (2,963, 2,928, and 2,870 cm−1), and aromatic groups (1,631, 1,600, and 1,555 cm−1). The 1H and 13C NMR spectra combined with DEPT 135 and HSQC spectra of 1 displayed the presence of 12 quaternary carbons, five methines, two methylenes, and six methyls. A comparison of the 1H and 13C NMR data with that of xerucitrinin A (3) (Salendra et al., 2021) implied that 1 was a citrinin dimer and they may have the same planar structures, which were confirmed by the analyses of 1H-1H COSY and HMBC correlations (Figure 2 and Supplementary Figures S1–S9).
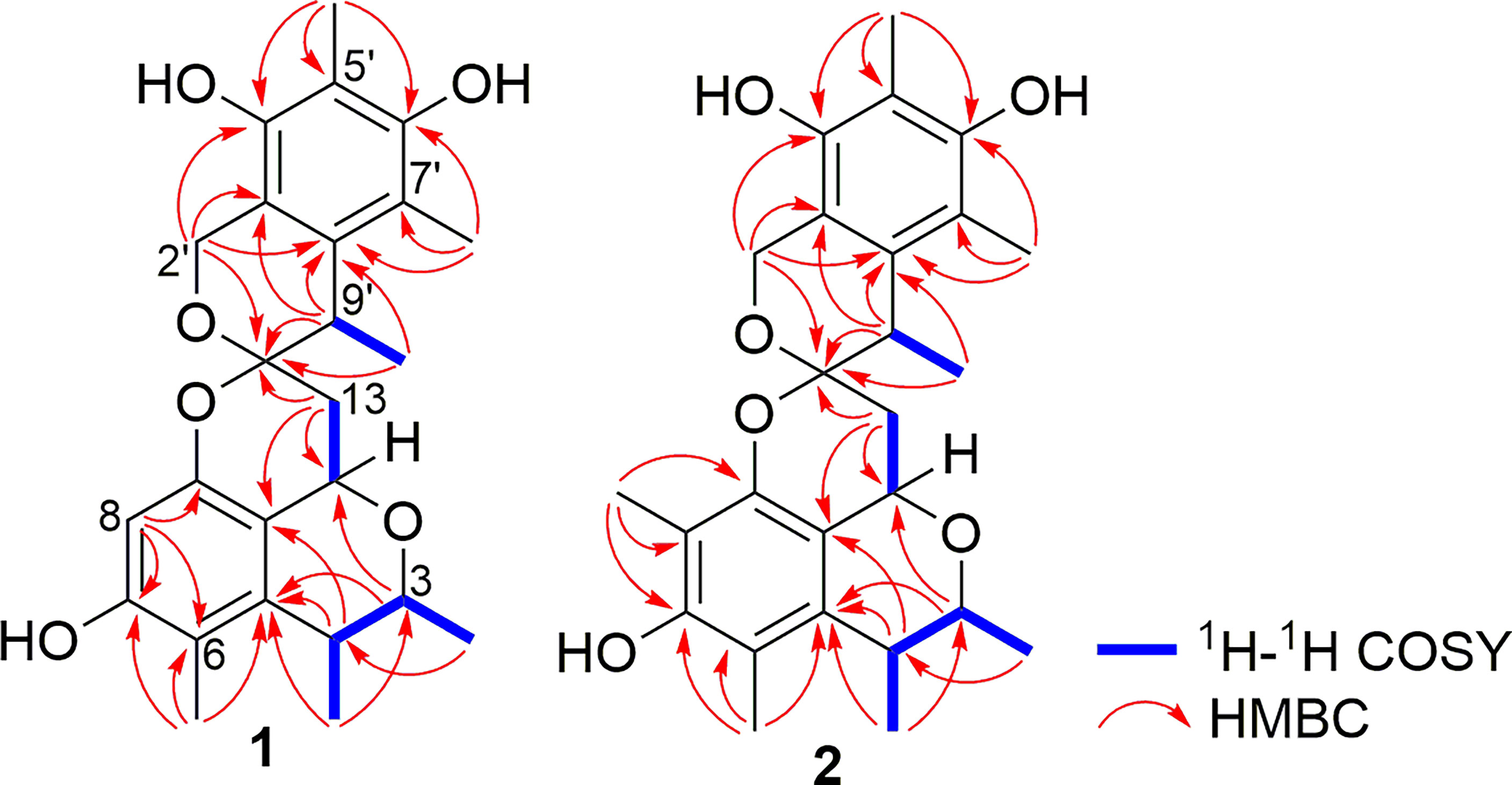
Figure 2 Key 1H-1H COSY and HMBC correlations of compounds 1 and 2.
The relative configuration of 1 was investigated by a NOESY experiment. The key NOESY correlations from H-3 to 4-CH3 and H-1 and from 4-CH3 to H-1 indicated they were on the same face, and the NOESY correlation of 3-CH3 with H-4 suggested that they were on the same face (Figure 3 and Supplementary Figure S10). Thus, the relative configurations of C-1, C-3, and C-4 were identified as 1S*, 3R*, and 4S*. Meanwhile, NOESY correlation of H-1 with H-13β and strong NOESY correlation of H-13α with H-9′ were observed, while no NOESY correlations of 9′-CH3 with H-13α and H-13β and a weak NOESY correlation of H-9’ with H-13β were also observed, making the relative configurations of C-12 and C-9’ ambiguous. To determine the relative configuration of 1, the 13C NMR chemical shift calculations of four possible stereoisomers, (1S*, 3R*, 4S*, 12S*, 9′S*)-1A, (1S*, 3R*, 4S*, 12S*, 9′R*)-1B, (1S*, 3R*, 4S*, 12R*, 9′S*)-1C, and (1S*, 3R*, 4S*, 12R*, 9′R*)-1D (Figure 4A) were performed at the PCM/mPW1PW91/6-31+G(d, p) level using the gauge including atomic orbital (GIAO) method (Supplementary Table S1). The calculation results indicated that 1A showed the highest DP4+ probability (100%, Figure 4B) and linear correlation coefficient (R2 = 0.9982, Figure 4C) and lowest mean absolute error (MAE) value (Figure 4D) among the calculated stereoisomers, suggesting the 1S*, 3R*, 4S*, 12S*, 9′S* relative configuration of 1. Then, the absolute configuration of 1 was established as 1S, 3R, 4S, 12S, 9′S by ECD calculations (Figure 5A). Finally, the structure of compound 1 was determined and named xerucitrinin B.
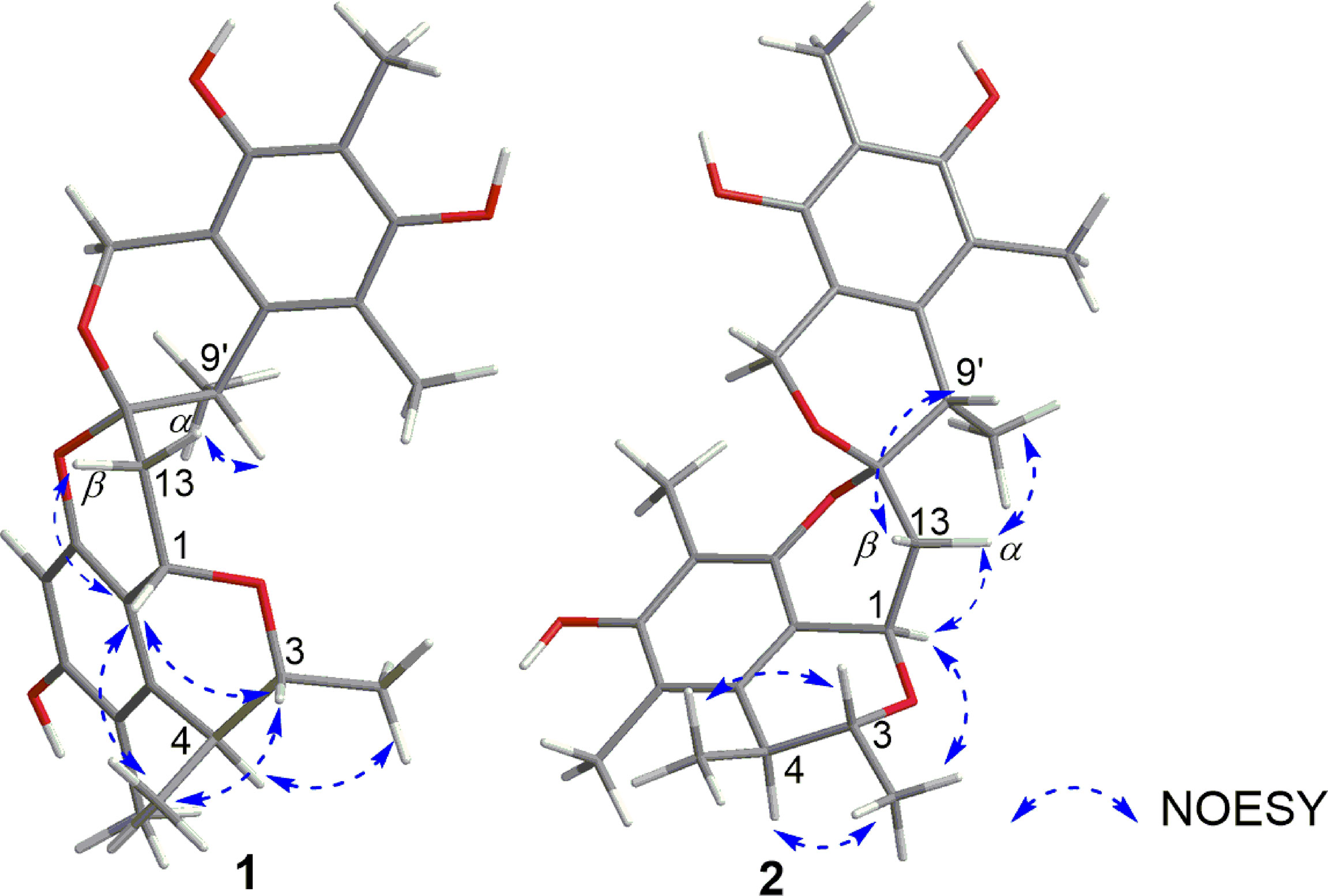
Figure 3 Key NOESY correlations of compounds 1 and 2.
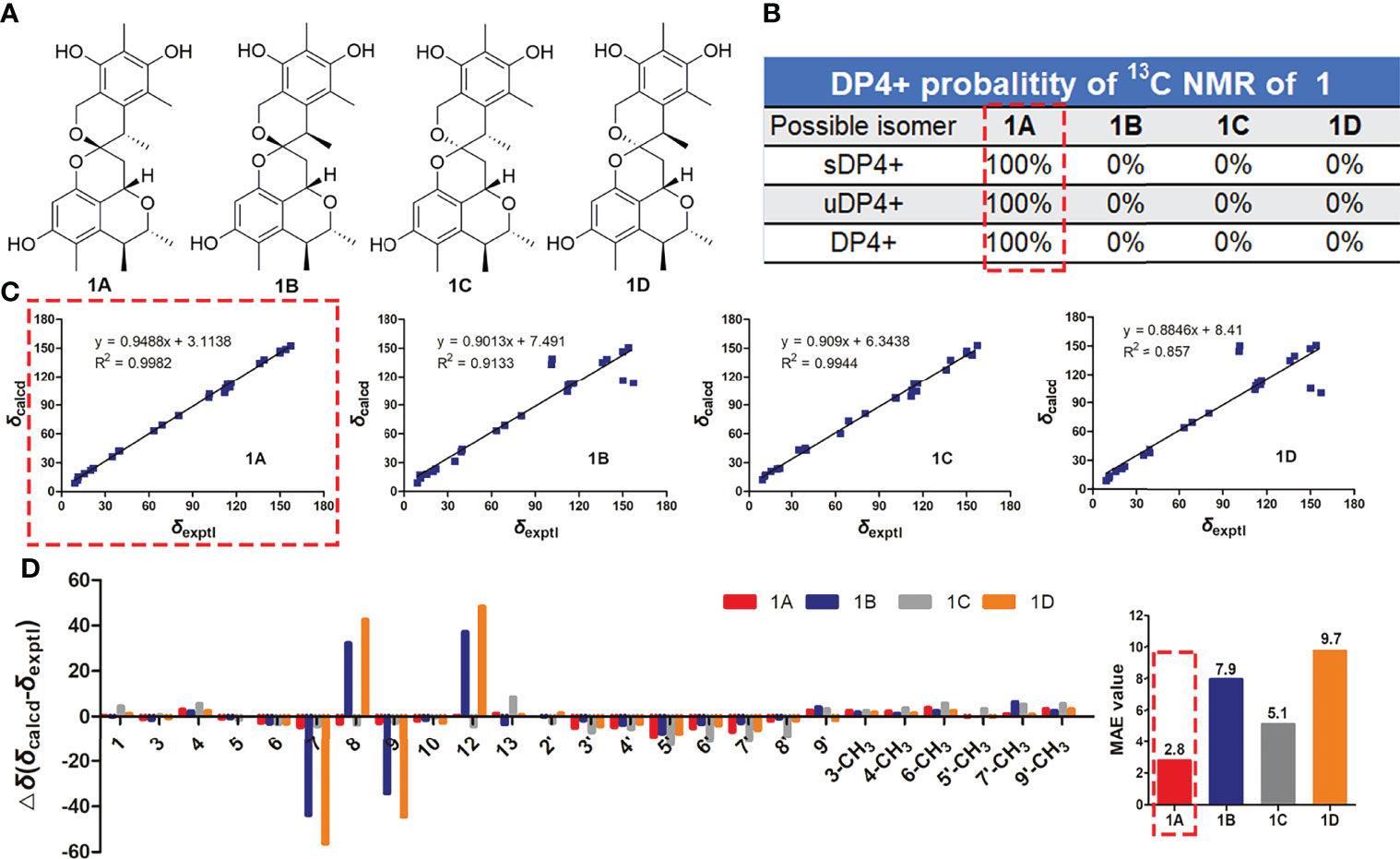
Figure 4 The 13C NMR calculation results of four plausible stereoisomers of 1 at mPW1PW91/6-31+G(d,p) level. (A) The structures of four possible stereoisomers. (B) DP4+ probability analysis. (C) Linear correlation plots of calculated vs. experimental 13C NMR chemical shift values for 1A/1B/1C/1D of 1. (D) Relative errors between the calculated 13C NMR chemical shifts of 1A/1B/1C/1D and the recorded 13C NMR data of 1, and the MAE values analyses.
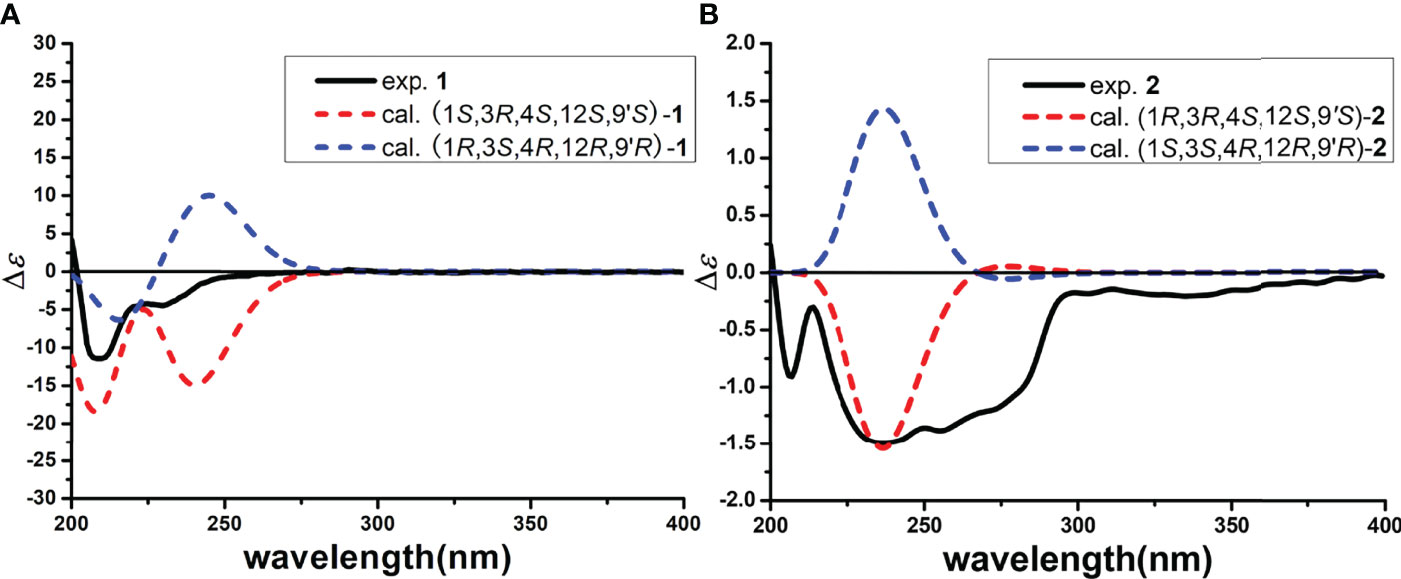
Figure 5 Experimental and calculated ECD spectra of compounds 1 (A) and 2 (B).
Compound 2 was obtained as pale yellow oil. The HRESIMS showed a protonated molecular ion at m/z 441.2278 [M + H]+ (calc. for 441.2277), which indicated a molecular formula of C26H32O6 and accounted for 11 degrees of unsaturation. The IR spectrum absorption bands suggested the presence of hydroxyl group (3,550, 3,475, and 3,411 cm−1), methyl group (2,959 cm−1), and aromatic groups (1,631 and 1,556 cm−1). Comparison of the 1H and 13C NMR data of 2 with that of xerucitrinin A (3) (Salendra et al., 2021) suggested that they had great similarity, and the main difference was the presence of an additional methyl signals at δH 1.66 (3H, s)/δC 8.3 and a quaternary carbon at δC 110.8 (C-8) in 2, which indicated that 2 was a methyl substituted derivative of xerucitrinin A in C-8. The planar structure of 2 was further established based on the analyses of 1H-1H COSY and HMBC data (Figure 2 and Supplementary Figures S11–S19).
The relative configuration of 2 was determined by a NOESY experiment. The NOESY correlations from 3-CH3 to H-1 and H-4 indicated that they were on the same face. The NOESY correlations from H-13α to 9′-CH3 and H-1 and from H-13β to H-9′ indicated that H-1, H-13α, and 9′-CH3 were on the same face. Thus, the relative configuration of 2 was identified as 1R*, 3R*, 4S*, 12S*, 9′S* (Figure 3 and Supplementary Figure S20). Subsequently, the absolute configuration of 2 was determined by ECD calculations. As the calculated ECD spectrum of (1R, 3R, 4S, 12S, 9′S)-2 matched well with the experimental result, the absolute configuration of 2 was identified as 1R, 3R, 4S, 12S, 9′S (Figure 5B). Thus, the structure of 2 was determined and named xerucitrinin C.
The known compounds were identified to be xerucitrinin A (3) (Salendra et al., 2021); xerucitrinic acid A (4) (Sadorn et al., 2016); xerucitrinic acid B (5) (Sadorn et al., 2016); penicitrinone A (6) (Wakana et al., 2006); citrinal A (7) (Wang et al., 2016); citrinal B (8) (Wang et al., 2016), 2, 3, 4-trimethyl-5, 7-dihydroxy-2, 3-dihydrobenzofuran (9) (Chen et al., 2002); decarboxydihydrocitrinin (10) (Wakana et al., 2006); (3R, 4S)-6, 8-dihydroxy-3, 4, 5, 7-tetramethylisochroman (11) (Han et al., 2009); (1S, 3R, 4S)-1-(4-hydroxylphenyl)-3, 4-dihydro-3, 4, 5-trimethyl-1H-2-benzopyran-6, 8-diol (12) (Wu et al., 2015); (3R, 4S)-6, 8-dihydroxy-3, 4, 5-trimethylisocoumarin (13) (Han et al., 2009); (3S, 4S)-sclerotinin A (14) (Nguyen et al., 2017); sclerotinin B (15) (Sassa et al., 1968); (3R, 4S)-6, 8-dihydroxy-3, 4, 7-trimethylisocoumarin (16) (Han et al., 2009); (3R)-3, 4-dihydro-6, 8-dimethoxy-3-methylisocoumarin (17) (Chinworrungsee et al., 2004); 4-hydroxykigelin (18) (Shimada et al., 2004); 4, 6-dimethylcurvulinic acid (19) (Liu et al., 2017); methyl 2-(2-acetyl-3, 5-dihydroxy-4, 6-dimethylphenyl)acetate (20) (Chen et al., 2012); phenol A (21) (Song et al., 2018); 1, 3-benzenediol, 5-[(1R, 2S)-2-hydroxy-1-methylpropyl]-2, 4-dimethyl (22) (Curtis et al., 1968); (S)-3-(3′, 5′-dihydroxy-2′, 4′-methylphenyl)butan-2-one (23) (Han et al., 2009); and 2-(6-hydroxy-5, 7-dimethylbenzofuranone-4-yl) acetaldehyde (24) (Xu et al., 2015), by comparing their MS, NMR, and specific rotation data with those reported. In addition, the CD spectra of 3–5 were provided for the first time (Supplementary Figures S21–S23).
Assessment of α-Glucosidase Inhibitory Activities
The in vitro hypoglycemic assay of compounds 1, 3–19, 21, 23, and 24 was investigated by assessing the inhibitory effects of isolated compounds on α-glucosidase. Acarbose was used as positive control. Results showed that compounds 1, 3, 6, and 12 exerted obvious α-glucosidase inhibitory activities with inhibition rates of 66.3%, 86.9%, 37.4%, and 49.0%, respectively, at a concentration of 400 μM, comparable or even better than that of acarbose (Figure 6A). Furthermore, our study showed that compounds 1, 3, and 12 could inhibit the catalytic activity of α-glucosidase in a dose-dependent manner with IC50 values of 239.8, 176.2, and 424.4 μM, respectively (Figure 6B). Structure–activity relationship analysis showed that the introduction of carboxyl at C-8 of citrinin dimer reduces the α-glucosidase inhibitory activities, since compounds 1 and 3 showed better activities than those of 4 and 5. Additionally, the induction of aromatic ring at C-1 in citrinin monomer increases its α-glucosidase inhibitory activities, since 12 showed better activities than 10, 11, and 13–18.
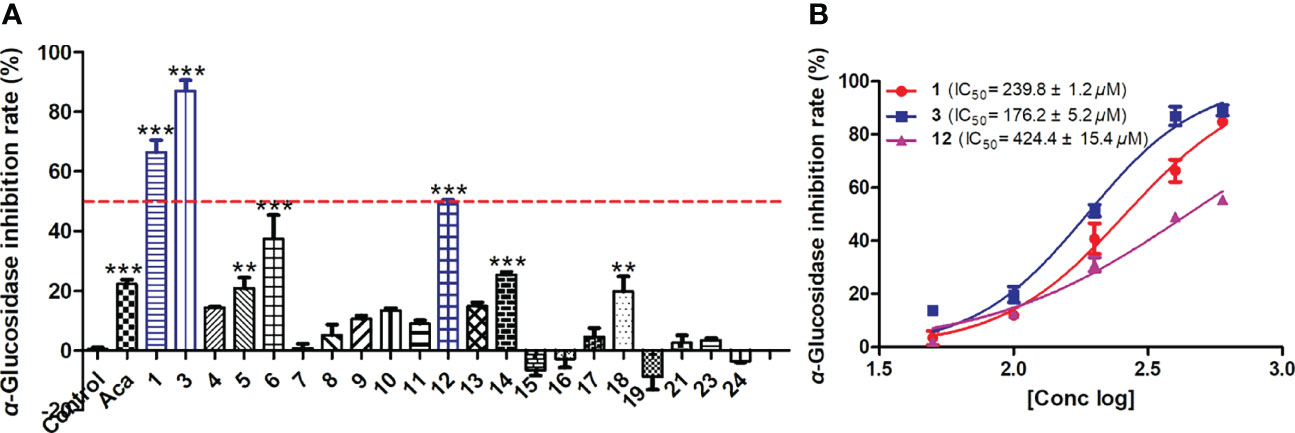
Figure 6 The α-glucosidase inhibitory activities of isolated compounds. (A) The α-glucosidase inhibition of isolated compounds at 400 μM. (B) The IC50 values of compounds 1, 3, and 12. Data were expressed as mean ± SEM (n = 3), **p< 0.01, ***p< 0.001, compared with control.
In order to determine the inhibitory mode of citrinin derivatives on α-glucosidase, the enzyme kinetics of α-glucosidase inhibition for compounds 1 and 3 were investigated using Lineweaver–Burk double reciprocal and Dixon single reciprocal plots. The Lineweaver–Burk plots with the reciprocal of velocity (y-axis) and p-NPG concentrations (x-axis) were drawn to acquire Km and Vmax values, while the Dixon single reciprocal plots with reciprocal of velocity (y-axis) against inhibitory concentrations (x-axis) at varying concentrations of p-NPG were used to elucidate Ki values. In the Lineweaver–Burk plots, the lines of compounds 1 and 3 both intersected on the x-axis, suggesting that the Km values were unchanged, while their Vmax values decreased with the increment of concentration, implying the non-competitive-type inhibitions of 1 and 3 (Figures 7A, C). The Ki values of 1 and 3 were further calculated as 204.3 and 212.7 μM, respectively, by Dixon plots (Figures 7B, D).
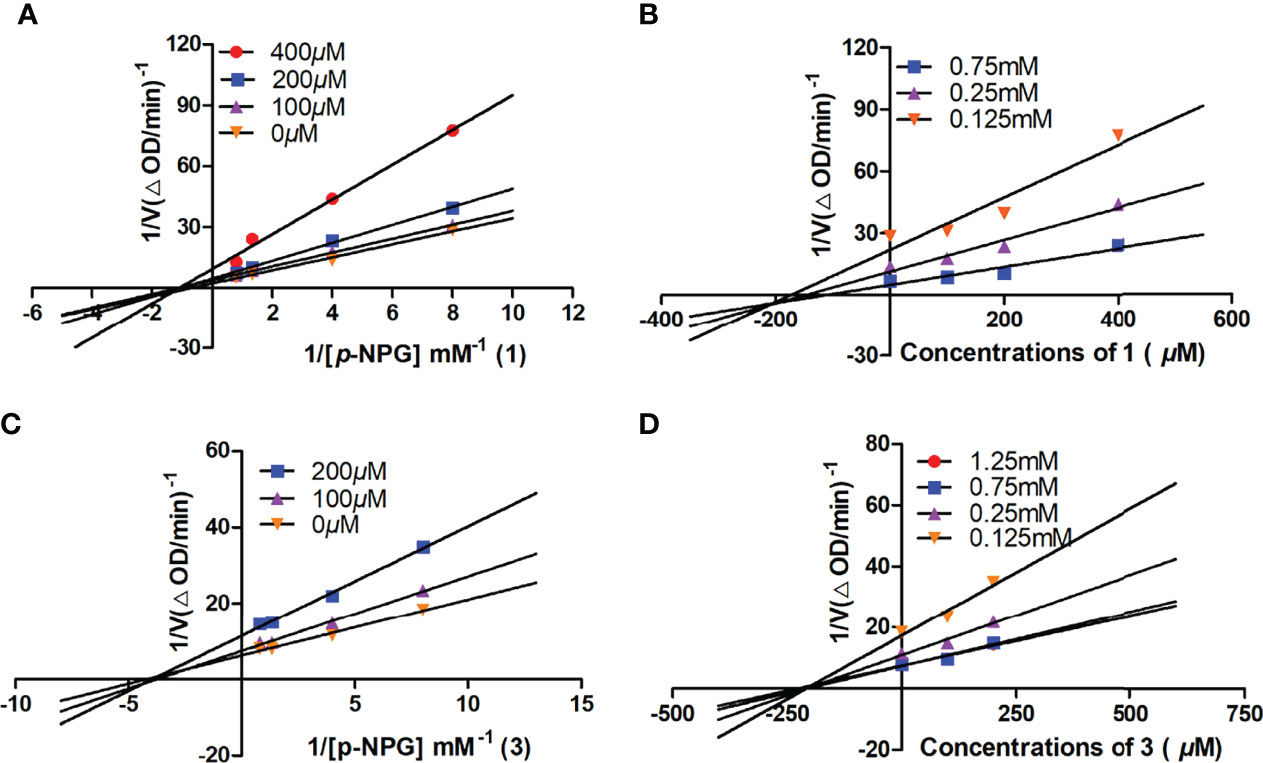
Figure 7 The Lineweaver–Burk and Dixon plots for α-glucosidase inhibition of compounds 1 (A, B) and 3 (C, D).
Furthermore, the molecular docking investigations suggested that compounds 1, 3, and 12 interacted with the catalytic pocket of α-glucosidase. Compound 1 formed two hydrogen bonds with Tyr-29 and His-31 residues (Figure 8A), 3 formed one hydrogen bond with His-31 residue (Figure 8B), and 12 formed four hydrogen bonds with Tyr-29, Arg-28, Asn-9, and Gln-5 residues (Figure 8C). These results were line with the enzymatic experiment and confirmed the hypoglycemic activities of these compounds.
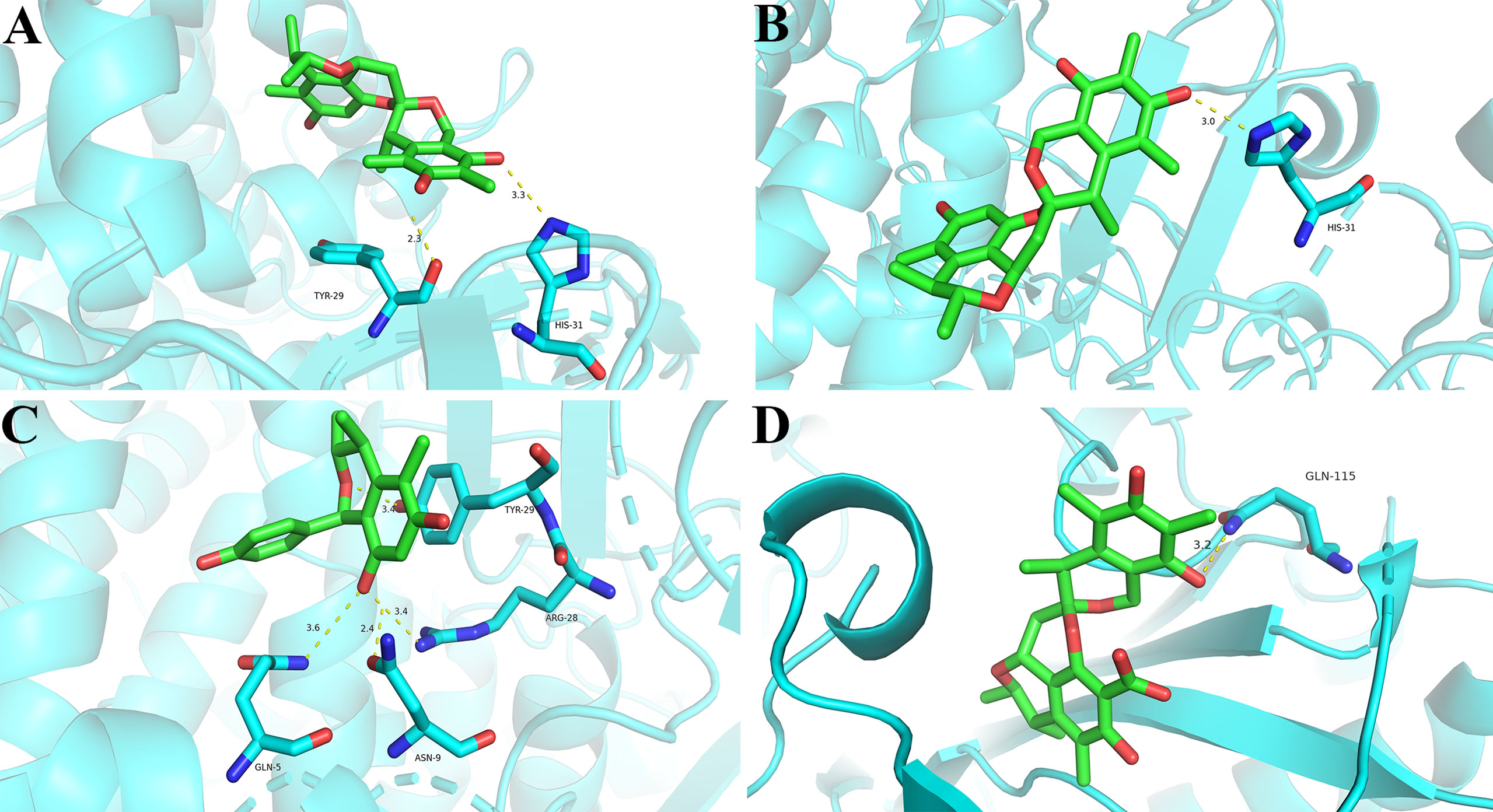
Figure 8 Docking poses and interactions of 1 (A), 3 (B), and 12 (C) with α-glucosidase (PDB code: 4J5T), and 4 (D) with ACL (PDB code: 6HXH). Hydrogen bond was denoted with yellow dash line.
Assessment of ACL Inhibitory Activities
In this study, the ACL inhibitory effects of isolated compounds were assessed by ADP-GloTM Kinase Assay. BMS-303141 was used as positive control (IC50: 0.4 μM). The inhibition rates of compounds 1, 3–21, 23, and 24 were first measured at 20 μM, and only citrinin dimer 4 revealed remarkable inhibitory effect towards ACL with an inhibition rate over 50% (Table 2). Furthermore, dose-dependent experiment showed that compound 4 significantly inhibited the catalytic activity of ACL with an IC50 value of 17.4 μM. To our knowledge, this is the first occurring citrinin with ACL inhibitory activity. The molecular docking assay showed the hydrogen bond interaction of 4 with ACL at Gln-115 residue (Figure 8D), implying 4 interacted with the catalytic pocket of ACL.
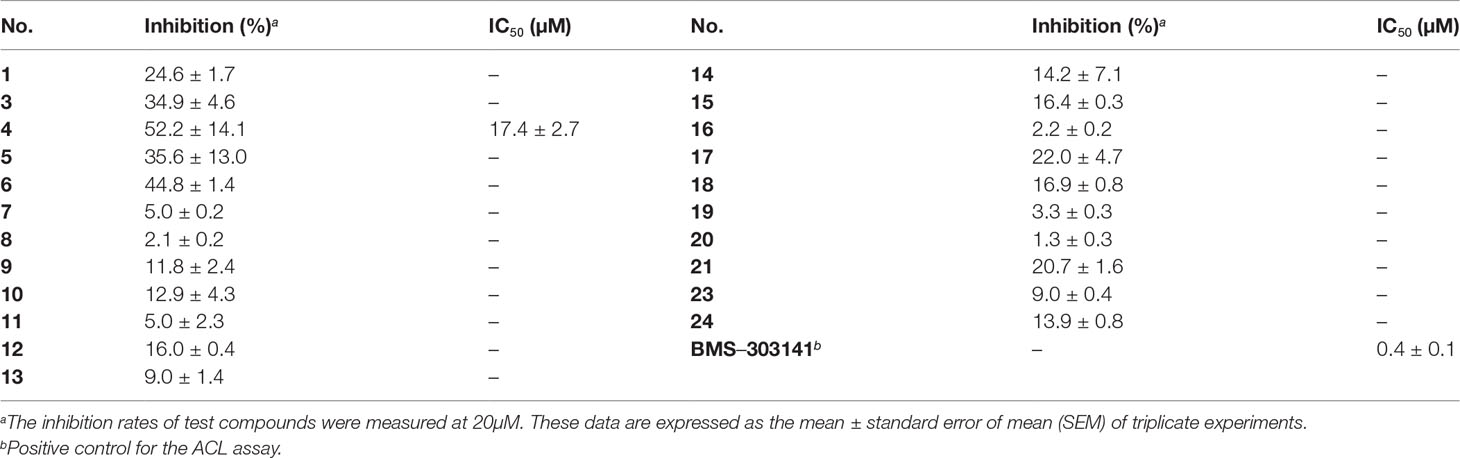
Table 2 Inhibitory activities of compounds 1, 3-21, 23, and 24 against ACL.
Discussion
Compounds 1–5 represent a class of citrinin dimers with unusual 6/6/6/6/6 pentacyclic core scaffold and a 6,6-spiroketal moiety that can be categorized in the azaphilone family having citrinin as a core structure. The difference of their structures lay in the configurations of C-1 and C-12 and the substituents of C-8. The plausible biosynthetic pathway of compounds 4 and 5 has been proposed (Sadorn et al., 2016), which mainly involved a Michael addition of citrinin with its precursor redoxcitrinin, leading to the production of the ketone intermediate. Intramolecular ketal reaction after the aldehyde reduction of the ketone intermediate produced the spiroketal products 1–5. The biosynthetic pathway also explained the possibility of having different configurations in C-1 and C-12 in compounds 1–5.
To our knowledge, the configuration determinations of C-1 and C-12, especially for C-12, were not easy. Here, the configuration of C-1 was identified by 1D and 2D NMR experiments. The configuration of C-12 was identified by extensive spectroscopic analyses and 13C NMR calculation in combination with DP4 +, linear correlation coefficient (R2), and MAE values analyses. In addition, we found that the configuration of C-1 might be identified by the 13C-NMR data, since the lower field 13C resonances of C-1, C-3, C-4, and 3-CH3, especially for C-1 (δC 68.9 and 66.4 in 1 and 5, respectively), were observed in the citrinin dimers with 1S configuration, compared to that with 1R configuration (δC 61.5, 61.0, and 58.9 for C-1 in 2-4, respectively). Additionally, the different configurations of C-12 in compounds 1–5 had less influence on the 1H and 13C NMR data of citrinin dimers. However, significant difference in the Cotton effect was observed in the CD spectra of 1–5. Compounds 1–4 with 12S configuration had negative Cotton effects at 230–300 nm, while compound 5 with 12R configuration had a positive Cotton effect at 230–300 nm (Figure 5 and Supplementary Figures S21–S23). This suggested us that the CD spectrum may be helpful for the configuration determination of C-12. In addition, it was reported that the decarboxylation of C-8 in citrinin will retain its antibiotic activity and reduce its toxicity, suggesting the drug development possibility of citrinin derivatives (Devi et al., 2006).
Conclusion
To sum up, 24 citrinin derivatives, including two new citrinin dimers bearing 6,6-spiroketal moieties (1 and 2), were found from marine-derived fungus P. citrinum Y34. Meanwhile, the α-glucosidase and ACL inhibitory activities of citrinin derivatives were assayed for the first time. Interestingly, citrinin dimers 1 and 3 and citrinin monomer 12 exhibited moderate α-glucosidase inhibitory activities, and citrinin dimer 4 showed potent ACL inhibitory activities. Enzyme kinetics assay indicated the non-competitive-type inhibition of 1 and 3 towards α-glucosidase. Our study enriches the structure diversity of citrinin dimers and provides critical information for their development to be α-glucosidase or ACL inhibitory agents for the treatment of metabolic diseases.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
JW and YM isolated and identified the strain of Penicillium citrinum Y34. XG and MC fermented and extracted the culture of this strain. SC and DT isolated and determined the structures and measured their α-glucosidase inhibitory activities. CL, YS, and JL tested the ACL inhibitory activities. SZ performed the molecular docking assay. BW and JT designed and coordinated the study. JT wrote the paper, while critical revision of the publication was performed by all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by grants from National Key Research and Development Program of China (Grant Numbers: 2018YFC0311002), Guangzhou Basic and Applied Basic Foundation (Grant Numbers: 202201010305), and National Nature Science Foundation of China (No.41876148).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to the support of The High Performance Public Computing Service Platform of Jinan University and that of Ocean college, Zhejiang University.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.961356/full#supplementary-material
References
Anas K., Fauziah F., Bellatasie R. (2021). Potential Bioactive Antibacterial, Cytotoxic, and Antioxidants of Endophytic Fungi Penicillium Citrinum: A Review. World J. Pharm. Pharm. Sci. 10, 1840–1859. doi: 10.20959/wjpps20212-18330
Bakhtra D. D. A., Suryani R., Yuni G. R., Handayani D. (2019). Antimicrobial and Cytotoxic Activities Screening of Symbiotic Fungi Extract Isolated From Marine Sponge Xestospongia Testudinaria DD- 01. J. Chem. Pharm. Sci. 12, 30–34. doi: 10.30558/jchps.20191202001
Bruhn T., Schaumloeffel A., Hemberger Y., Bringmann G. (2013). SpecDics: Quantifying the Comparison of Calculated and Experimental Electronic Circular Dichroism Spectra. Chirality 25, 243–249. doi: 10.1002/chir.22138
Cao F., Sun T. T., Yang J. K., Zhao G. Z., Liu Q. A., Hu L. D., et al. (2019). The Absolute Configuration of Anti-Vibrio Citrinin Dimeric Derivative by VCD, ECD and NMR Methods. Nat. Prod. Res. 33, 2192–2199. doi: 10.1080/14786419.2018.1493590
Chang C. H., Yu F. Y., Wang L. T., Lin Y. S., Liu B. H. (2009). Activation of ERK and JNK Signaling Pathways by Mycotoxin Citrinin in Human Cells. Toxicol. Appl. Pharm. 237 (3), 281–287. doi: 10.1016/j.taap.2009.03.021
Chen L., Huang K., Zhong P., Hu X., Fang Z. X., Wu J. L., et al. (2012). Tumonoic Acids K and L, Novel Metabolites From the Marine-Derived Fungus Penicillium Citrinum. Heterocycles 85, 413–419. doi: 10.3987/COM-11-12380
Chen H. W., Jiang C. X., Li J. Y., Li N., Zang Y., Wu X. Y., et al. (2021). Beshanzoides A-D, Unprecedented Cycloheptanone-Containing Polyketides From Penicillium Commune P-4-1, an Endophytic Fungus of the Endangered Conifer Abies Beshanzuensis. RSC Adv. 11, 39781–39789. doi: 10.1039/d1ra08377e
Chen C. H., Shaw C. Y., Chen C. C., Tsai Y. C. (2002). 2,3,4-Trimethyl-5,7-Dihydroxy-2,3-Dihydrobenzofuran, a Novel Antioxidant, From Penicillium Citrinum F5. J. Nat. Prod. 65, 740–741. doi: 10.1021/np010605o
Chinworrungsee M., Kittakoop P., Isaka M., Maithip P., Supothina S., Thebtaranonth Y. (2004). Isolation and Structure Elucidation of a Novel Antimalarial Macrocyclic Polylactone, Menisporopsin A, From the Fungus Menisporopsis Theobromae. J. Nat. Prod. 67, 689–692. doi: 10.1021/np0304870
Clark B. R., Capon R. J., Lacey E., Tennant S., Gill J. H. (2006). Citrinin Revisited: From Monomers to Dimers and Beyond. Org. Biomol. Chem. 4, 1520–1528. doi: 10.1039/B600960C
Curtis R. F., Hassall C. H., Nazar M. J. (1968). The Biosynthesis of Phenols. Part XV. Some Metabolites of Penicillium Citrinum Related to Citrinin. Chem. Soc Org. 1, 85–93. doi: 10.1039/J39680000085
Delevry D., Gupta E. K. (2021). Bempedoic Acid: Review of a Novel Therapy in Lipid Management. Am. J. Health Syst. Pharm. 78, 95–104. doi: 10.1093/ajhp/zxaa352
Devi P., Naik C. G.,, Rodrigues C. (2006). Biotransformation of Citrinin to Decarboxycitrinin Using an Organic Solvent-Tolerant Marine Bacterium, Moraxella Sp. MB1. Mar. Biotechnol. 8, 129–138. doi: 10.1007/s10126-005-5021-5
El-Neketi M., Ebrahim W., Lin W. H., Gedara S., Badria F., Saad H. E. A., et al. (2013). Alkaloids and Polyketides From Penicillium Citrinum, an Endophyte Isolated From the Moroccan Plant Ceratonia Siliqua. J. Nat. Prod. 76, 1099–1104. doi: 10.1021/np4001366
Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., et al. (2016). Gaussian 16, Revision A.03 (Wallingford CT: Gaussian, Inc.).
Gou X. S., Jia J., Xue Y. X., Ding W. J., Dong Z. T., Tian D. M., et al. (2020). New Pyrones and Their Analogs From the Marine Mangrove-Derived Aspergillus Sp. DM94 With Antibacterial Activity Against Helicobacter Pylori. Appl. Microbiol. Biot. 104, 7971–7978. doi: 10.1007/s00253-020-10792-9
Gou X. S., Tian D. M., Wei J. H., Ma Y. H., Zhang Y. X., Chen M., et al. (2021). New Drimane Sesquiterpenes and Polyketides From Marine-Derived Fungus Penicillium Sp. TW58-16 and Their Anti-Inflammatory and α-Glucosidase Inhibitory Effects. Mar. Drugs 19, 416. doi: 10.3390/md19080416
Han Z., Mei W. L., Zhao Y. X., Deng Y. Y., Dai H. F. (2009). A New Cytotoxic Isocoumarin From Endophytic Fungus Penicillium Sp. 091402 of the Mangrove Plant Bruguiera Sexangula. Chem. Nat. Compd.+ 45, 805–807. doi: 10.1007/s10600-010-9503-y
He X. F., Chen J. J., Huang X. Y., Hu J., Zhang X. K., Guo Y. Q., et al. (2021). The Antidiabetic Potency of Amomum Tsao-Ko and its Active Flavanols, as PTP1B Selective and α-Glucosidase Dual Inhibitors. Ind. Crop Prod. 160, 112908. doi: 10.1016/j.indcrop.2020.112908
Li H. T., Duan R. T., Liu T., Yang R. N., Wang J. P., Liu S. X., et al. (2020). Penctrimertone, a Bioactive Citrinin Dimer From the Endophytic Fungus Penicillium Sp. T2-11. Fitoterapia 146, 104711. doi: 10.1016/j.fitote.2020.104711
Li T. T., Jiang G. X., Qu H. X., Wang Y., Xiong Y. H., Jian Q. J., et al. (2017). Comparative Transcriptome Analysis of Penicillium Citrinum Cultured With Different Carbon Sources Identifies Genes Involved in Citrinin Biosynthesis. Toxins 9, 69. doi: 10.3390/toxins9020069
Liu F., Tian L., Chen G., Zhang L. H., Liu B., Zhang W. Y., et al. (2017). Two New Compounds From a Marine-Derived Penicillium Griseofulvum T21-03. J. Asian Nat. Prod. Res. 19, 678–683. doi: 10.1080/10286020.2016.1231671
Megur A., Daliri E. B. M., Baltriukiene D., Burokas A. (2022). Prebiotics as a Tool for the Prevention and Treatment of Obesity and Diabetes: Classification and Ability to Modulate the Gut Microbiota. Int. J. Mol. Sci. 23 (11), 6097. doi: 10.3390/ijms23116097
Nakajima Y., Iguchi H., Kamisuki S., Sugawara F., Furuichi T., Shinoda Y. (2016). Low Doses of the Mycotoxin Citrinin Protect Cortical Neurons Against Glutamate-Induced Excitotoxicity. Toxicol. Sci. 41, 311–319. doi: 10.2131/jts.41.311
Nguyen T. T. N., Quang T. H., Kim K. W., Kim H. J., Sohn J. H., Kang D. G., et al. (2017). Anti-Inflammatory Effects of Secondary Metabolites Isolated From the Marine-Derived Fungal Strain Penicillium Sp. SF-5629. Arch. Pharm. Res. 40, 328–337. doi: 10.1007/s12272-017-0890-5
Paulose S. K., Chakraborty K. (2022). Ellipyrones A-B, From Oval Bone Cuttlefish Sepia Elliptica: Antihyperglycemic γ-Pyrone Enclosed Macrocyclic Polyketides Attenuate Dipeptidyl Peptidase-4 and Carbolytic Enzymes. Med. Chem. Res. 31, 462–473. doi: 10.1007/s00044-022-02846-6
Sadorn K., Saepua S., Boonyuen N., Laksanacharoen P., Rachtawee P., Pittayakhajonwut P. (2016). Antimicrobial Activity and Cytotoxicity of Polyketides Isolated From the Mushroom Xerula Sp. BCC56836. RSC Adv. 6, 94510–94523. doi: 10.1039/C6RA21898A
Salendra L., Lin X. P., Chen W. H., Pang X. Y., Luo X. W., Long J. Y., et al. (2021). Cytotoxicity of Polyketides and Steroids Isolated From the Sponge-Associated Fungus Penicillium Citrinum SCSIO 41017. Nat. Prod. Res. 35, 900–908. doi: 10.1080/14786419.2019.1610757
Samanthi K. A. U., Wickramaarachchi S., Wijeratne E. M. K., Paranagama P. A. (2015). Two New Antioxidant Active Polyketides From Penicillium Citrinum, an Endolichenic Fungus Isolated From Parmotreama Species in Sri Lanka. J. Natl. Sci. Found. Sri. Lanka 43, 119–126. doi: 10.4038/jnsfsr.v43i2.7939
Sansenya S., Payaka A. (2022). Inhibitory Potential of Phenolic Compounds of Thai Colored Rice (Oryza Sativa L.) Against α- Glucosidase and α- Amylase Through In Vitro and In Silico Studies. J. Sci. Food Agr. doi: 10.1002/jsfa.12039
Sassa T., Aoki H., Namiki M., Munakata K. (1968). Plant Growth- Promoting Metabolites of Sclerotinia Sclerotiorum. I. Isolation and Structures of Sclerotinins A and B. Agric. Biol. Chem. 32, 1432–1439. doi: 10.1080/00021369.1968.10859243
Shimada A., Inokuchi T., Kusano M., Takeuchi S., Inoue R., Tanita M., et al. (2004). 4- Hydroxykigelin and 6- Demethylkigelin, Root Growth Promoters, Produced by Aspergillus Terreus. Z. Naturforsch. C 59, 218–222. doi: 10.1515/znc-2004-3-417
Shi Y. T., Pan C. Q., Cen S. Y., Fu L. L., Cao X., Wang H., et al. (2019). Comparative Metabolomics Reveals Defence- Related Modification of Citrinin by Penicillium Citrinum Within a Synthetic Penicillium- Pseudomonas Community. Environ. Microbiol. 21, 496–510. doi: 10.1111/1462-2920.14482
Song T. F., Chen M. X., Ge Z. W., Chai W. Y., Li X. C., Zhang Z. Z., et al. (2018). Bioactive Penicipyrrodiether A, an Adduct of GKK1032 Analogue and Phenol A Derivative, From a Marine-Sourced Fungus Penicillium Sp. ZZ380. J. Org. Chem. 83, 13395–13401. doi: 10.1021/acs.joc.8b02172
Wakana D., Hosoe T., Itabashi T., Okada K., de Campos Takaki G. M., Yaguchi T., et al. (2006). New Citrinin Derivatives Isolated From Penicillium Citrinum. J. Nat. Med.-Tokyo 60, 279–284. doi: 10.1007/s11418-006-0001-2
Wang W. Y., Liao Y. Y., Zhang B. B., Gao M. L., Ke W. Q., Li F., et al. (2019a). Citrinin Monomer and Dimer Derivatives With Antibacterial and Cytotoxic Activities Isolated From the Deep Sea-Derived Fungus Penicillium Citrinum NLG-S01-P1. Mar. Drugs 17, 46. doi: 10.3390/md17010046
Wang L., Li C. L., Yu G. H., Sun Z. C., Zhang G. J., Gu Q. Q., et al. (2019b). Dicitrinones E and F, Citrinin Dimers From the Marine Derived Fungus Penicillium Citrinum HDN-152-088. Tetrahedron Lett. 60, 151182. doi: 10.1016/j.tetlet.2019.151182
Wang F. Q., Zhu H. C., Ma H. R., Jiang J., Sun W. G., Cheng L., et al. (2016). Citrinal B, a New Secondary Metabolite From Endophytic Fungus Colletotrichum Capsici and Structure Revision of Citrinal A. Tetrahedron Lett. 57, 4250–4253. doi: 10.1016/j.tetlet.2016.08.029
Wan J., Jiang C. X., Tang Y., Ma G. L., Tong Y. P., Jin Z. X., et al. (2022). Structurally Diverse Glycosides of Secoiridoid, Bisiridoid, and Triterpene- Bisiridoid Conjugates From the Flower Buds of Two Caprifoliaceae Plants and Their ATP- Citrate Lyase Inhibitory Activities. Bioorg. Chem. 120, 105630. doi: 10.1016/j.bioorg.2022.105630
Wu Y. Z., Qiao F., Xu G. W., Zhao J., Teng J. F., Li C., et al. (2015). Neuroprotective Metabolites From the Endophytic Fungus Penicillium Citrinum of the Mangrove Bruguiera Gymnorrhiza. Phytochem. Lett. 12, 148–152. doi: 10.1016/j.phytol.2015.03.007
Xu Y., Tian S. S., Zhu H. (2015). A New Lactone Aldehyde Compound Isolated From Secondary Metabolites of Marine Fungus Penicillium Griseofulvum. Nat. Prod. Res. Dev. 27, 559-561. doi: 10.16333/j.1001-6880.2015.04.001
Yang S. Q., Li X. M., Li X., Li H. L., Meng L. H., Wang B. G. (2018). New Citrinin Analogues Produced by Coculture of the Marine Algal-Derived Endophytic Fungal Strains Aspergillus Sydowii EN-534 and Penicillium Citrinum EN-535. Phytochem. Lett. 25, 191–195. doi: 10.1016/j.phytol.2018.04.023
Zhang D. H., Li X. G., Kang J. S., Choi H. D., Jung J. H., Son B. W. J., et al. (2007). Redoxcitrinin, a Biogenetic Precursor of Citrinin From Marine Isolate of Fungus Penicillium Sp. J. Microbiol. Biotechn. 17, 865–867.
Zhong J. Q., Chen Y. C., Liu Z. M., Hu C. Y., Li S. N., Liu H. X., et al. (2022). Bioactive Polyketide Derivatives From the Endophytic Fungus Phaeosphaeriopsis Musa. Phytochem. (Elsevier) 195, 113055. doi: 10.1016/j.phytochem.2021.113055
Keywords: Penicillium citrinum Y34, citrinin dimer, α-Glucosidase inhibitory activity, ACL inhibitory activity, molecular docking
Citation: Chen S, Tian D, Wei J, Li C, Ma Y, Gou X, Shen Y, Chen M, Zhang S, Li J, Wu B and Tang J (2022) Citrinin Derivatives From Penicillium Citrinum Y34 That Inhibit α-Glucosidase and ATP-Citrate Lyase. Front. Mar. Sci. 9:961356. doi: 10.3389/fmars.2022.961356
Received: 04 June 2022; Accepted: 24 June 2022;
Published: 22 July 2022.
Edited by:
Ling Liu, Institute of Microbiology (CAS), ChinaReviewed by:
Youmin Ying, Zhejiang University of Technology, ChinaDewu Zhang, Chinese Academy of Medical Sciences, China
Copyright © 2022 Chen, Tian, Wei, Li, Ma, Gou, Shen, Chen, Zhang, Li, Wu and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Wu, d3ViaW5Aemp1LmVkdS5jbg==; Jinshan Tang, Z3p0YW5namluc2hhbkAxMjYuY29t
†These authors have contributed equally to this work and share first authorship