 Giorgia Palladino1,2
Giorgia Palladino1,2 Simone Rampelli1
Simone Rampelli1 Carles Galià-Camps3,4
Carles Galià-Camps3,4 Daniel Scicchitano1,2
Daniel Scicchitano1,2 Giulia Trapella1
Giulia Trapella1 Enrico Nanetti1
Enrico Nanetti1 Valeria Angelini5
Valeria Angelini5 Daniela Cleo6
Daniela Cleo6 Silvia Turroni1Cinzia Corinaldesi7
Silvia Turroni1Cinzia Corinaldesi7 Marco Candela1,2*
Marco Candela1,2*- 1Unit of Microbiome Science and Biotechnology, Department of Pharmacy and Biotechnology (FaBiT), Alma Mater Studiorum – University of Bologna, Bologna, Italy
- 2Fano Marine Center, The Inter-Institute Center for Research on Marine Biodiversity, Resources and Biotechnologies, Fano, Italy
- 3Departament de Genètica, Microbiologia i Estadística, Universitat de Barcelona, Barcelona, Spain
- 4Institut de Recerca de la Biodiversitat (IRBio), Universitat de Barcelona, Barcelona, Spain
- 5Fondazione Cetacea Onlus, Riccione, Italy
- 6Associazione Blennius, Riccione, Italy
- 7Department of Materials, Environmental Sciences and Urban Planning, Polytechnic University of Marche, Ancona, Italy
Despite their recognized primary importance, marine coastal ecosystems around the globe are currently under threat, being subject to continuous local and global anthropogenic stressors. In this frame, understanding the response of coastal habitat-forming species to multiple stressors and their resilience is fundamental for the sustainable management of coastal ecosystems. In the present study, to provide some glimpses in this direction, we explored the response of the Anemonia viridis-associated microbiota to the combined anthropogenic stressors, which typically affect touristic hotspots at Mediterranean coastal sites. To this aim, two case studies have been carried out, the first in the Riccione coastal site (Italy, Center Mediterranean) and the second at Cap de Creus (Spain, North-western Mediterranean), where the A. viridis microbiota was assessed under the conditions of both high and low anthropogenic pressure. According to our findings, the A. viridis microbiota showed a relevant degree of plasticity in response to combined anthropogenic and environmental stressors, with changes that also mirrored variations in the surrounding seawater, thus indicating a close connection with the environment, from which potential symbiotic partners are selected. However, this potentially adaptive process also has a limitation, as observed in the highly anthropogenic impact site of Cap de Creus, where A. viridis-associated microbiota appeared completely unstructured, as demonstrated by an increased dispersion according to the Anna Karenina principle. This raises the question about the resilience of the A. viridis-associated microbiota under combined climate and anthropogenic threats, as well as of the anthropogenic factors driving the observed dysbiosis changes.
Introduction
Being the connection between land and open sea, the marine coastal environment has a role of primary importance for the health of marine and terrestrial ecosystems, with direct implications on human health and wellbeing (Barbier et al., 2011; Wilkins et al., 2019). Marine coastal ecosystems are known to provide a wide range of ecological and economically important services, including habitat provision, nutrient cycling, coastal protection, fisheries enhancement, and, of course, tourism (Trevathan-Tackett et al., 2019). However, the current and future climate change scenarios are jeopardizing the health of coastal ecosystems, which are already extremely vulnerable, due to the continuous threats from various anthropogenic pressures (Crain et al., 2009; Wambua et al., 2021). In this scenario, it is extremely important to better understand the response of coastal habitat-forming species to local and global stressors, dissecting their inherent limit of adaptability to the anthropogenic pressures, thus allowing the implementation of sustainable practices for the management, preservation, and restoration of coastal ecosystems (Santoro et al., 2021).
Habitat-forming species—such as cnidarians, sponges, seagrasses, and mangroves—represent the foundation of the coastal ecosystems, being at the basis of the coastal trophic webs (Lemieux and Cusson, 2014), where they contribute to primary productivity and create the structural habitat, providing key resources to support the entire ecosystem (Vanwonterghem and Webster, 2020). These marine holobionts live in close association with microbial communities—known as microbiota (Palladino et al., 2021)—representing an integral component of the host biology and providing strategic functionalities, which favor nutrition, protection from diseases, and development (Rohwer et al., 2001; Rosenberg et al., 2007; Morrow et al., 2012; McFall-Ngai et al., 2013; Mcdevitt-Irwin et al., 2017; O’Brien et al., 2018). Being inherently and rapidly adaptable even to extreme environmental conditions, microbiomes are strategic to boost the host’s adaptive performance, complementing the delay in the host genome adaptation (Kolodny and Schulenburg, 2020; Marangon et al., 2021; Rampelli et al., 2021). Therefore, to provide a holistic understanding of the responses of coastal marine organisms to global changes and anthropogenic pressures, the analysis of their associated microbiome is mandatory, assessing its dynamic variations in response to local and global stressors (Trevathan-Tackett et al., 2019).
Among cnidarians, anemones are soft habitat-forming species that have a worldwide distribution in marine coastal environments, with species inhabiting all climate zones and habitat types, providing shelter and food for a wide variety of mobile fauna, including endangered species (Wilkins et al., 2019; Steinberg et al., 2020). Anemones are among the most resistant marine organisms, being able to respond to several stressors, such as shifts in temperature, pH, salinity, and organic and inorganic compounds (Steinberg et al., 2020). It has been shown that the microbiome associated with anemones is characterized by a high taxonomic level core, with local adaptation for what concerns the specific constituents at lower phylogenetic ranks (Brown et al., 2017). Indeed, anemones show local patterns of microbial OTUs (operational taxonomic units), which are not related to latitude or longitude but rather to differences in global and local environmental factors, playing important roles in sorting the compositional structure of the anemone-associated microbiome (Mortzfeld et al., 2016; Morelan et al., 2019). This makes the microbiome of the anemones an extremely fluid partner, possibly complementing the limits of the host physiology and allowing its adaptation or acclimatization to a vast range of environmental conditions, including multiple anthropogenic stressors.
Using A. viridis as a model of soft habitat-forming species (Muller et al., 2016), here, we explored the response of the anemone microbiota to the combined anthropogenic and environmental stressors that typically characterize the touristic hotspots of the Mediterranean coastal sites and that can act synergistically with environmental factors (Danovaro, 2003; Drius et al., 2019). Particularly, the Riviera Romagnola (Italy) and Costa Brava (Spain) represent the important touristic hotspots of the Mediterranean coasts, where the summer tourism peak has been associated with serious threats to the coastal ecosystems (Andolina et al., 2021). Indeed, the close link between growing anthropogenic pressure and increased inputs of nutrients and contaminants in marine coastal systems (including plastics, marine debris, and emerging contaminants such as pharmaceuticals, surfactants, and personal care products like sunscreens), as well as organic enrichment and microbiological pollution induced by sewage discharge, is well recognized (Clark et al., 2001; Craig-Smith et al., 2006; Tovar-Sánchez et al., 2019). In addition, the increased temperature and the consequent lower oxygen levels observed during summer have been reported to exacerbate the negative effects of anthropogenic impacts on marine life and ecosystems.
To highlight the response of the A. viridis microbiota to the combined action of these multiple pressures, we assessed two different case studies. In the former case, we investigated changes in the A. viridis microbiome composition between individuals sampled at the Riccione site (Riviera Romagnola, North Adriatic Sea, Central Mediterranean) in February—with no tourism inflow, low temperature, and high oxygen levels—and in August, with very high tourism inflow, as well as increased temperature and low oxygen levels. Riccione is one of the main coastal tourism hotspots for the Mediterranean Sea (Drius et al., 2019), with a local population increasing from 30,000 people in the winter to 650,000 in August, which is the peak of the touristic summer season (Istat reports 2018-2020). In the latter case, we investigated compositional differences in the A. viridis microbiota from Cap de Creus, Costa Brava (Spain, North-western Mediterranean), where two sites with different impact levels were explored in the same summer season. The first site was considered slightly impacted by tourism and anthropogenic activities, being located near a partial natural reserve where only artisanal fishing and diving activities are allowed, whereas the second site was considered highly impacted by multiple tourism-associated stressors, including wastewater discharge, being located near the urban center Roses (Sardá et al., 2012). Overall, the results obtained from the microbiome analyses in these two case studies provided new insights into the responses of the A. viridis microbiota to the increasing levels of combined anthropogenic and environmental pressures in two different coastal sites and sectors of the Mediterranean Sea.
Materials and methods
Sampling areas and sample collection
Two study areas were considered (Figure 1). The Riccione coastal site is located only 100 km south of the Po delta and receives water discharge from the Po River through the Adriatic Western Coastal Layer (Pettine et al., 1998; Palinkas et al., 2005; Spillman et al., 2007). The Po River, the largest Italian river, accounts for one-third of the total riverine freshwater input in the Adriatic Sea (Spillman et al., 2007). Since the Po valley is one of the most productive agricultural areas in Italy, this river also accounts for approximately 50% of the total nutrients transported into the northern Adriatic Sea (Spillman et al., 2007), also including anthropogenic contaminants collected from the river basin and its tributary (Pettine et al., 1998). Peak flow occurs in the spring and autumn, primarily due to snowmelt and rainfall (Palinkas et al., 2005). Riccione—Riviera Romagnola—is one of the main Italian sites for mass beach tourism since 1960 and, at the peak of the touristic season in August, can reach up to 650,000 presences, while the winter local population is 30,000 (Istat reports 2018-2020). At Cap de Creus (Spain, North-western Mediterranean), two sites were investigated. The first, the low-impact site, was located next to Cadaqués, a small and isolated city with a population of 2,800 inhabitants and no more than 8,000 in summer. Sampling at Cadaqués was performed near a partial natural reserve, where only artisanal fishing and diving activities are allowed. The second, a high-impact site, was located at Roses. Roses has approximately 70,000 inhabitants in the high season, and it is located around 10 km away from Cadaqués, outside the marine protected area, where the beach is commonly highly populated during the touristic season. Moreover, the discharge from wastewater treatment plants is close to the city area and there is a wide recreational port where activity is highly increased in summer due to tourism. Further, several uncontrolled outfalls and associated eutrophication processes were also detected in Roses (Sardá et al., 2012).
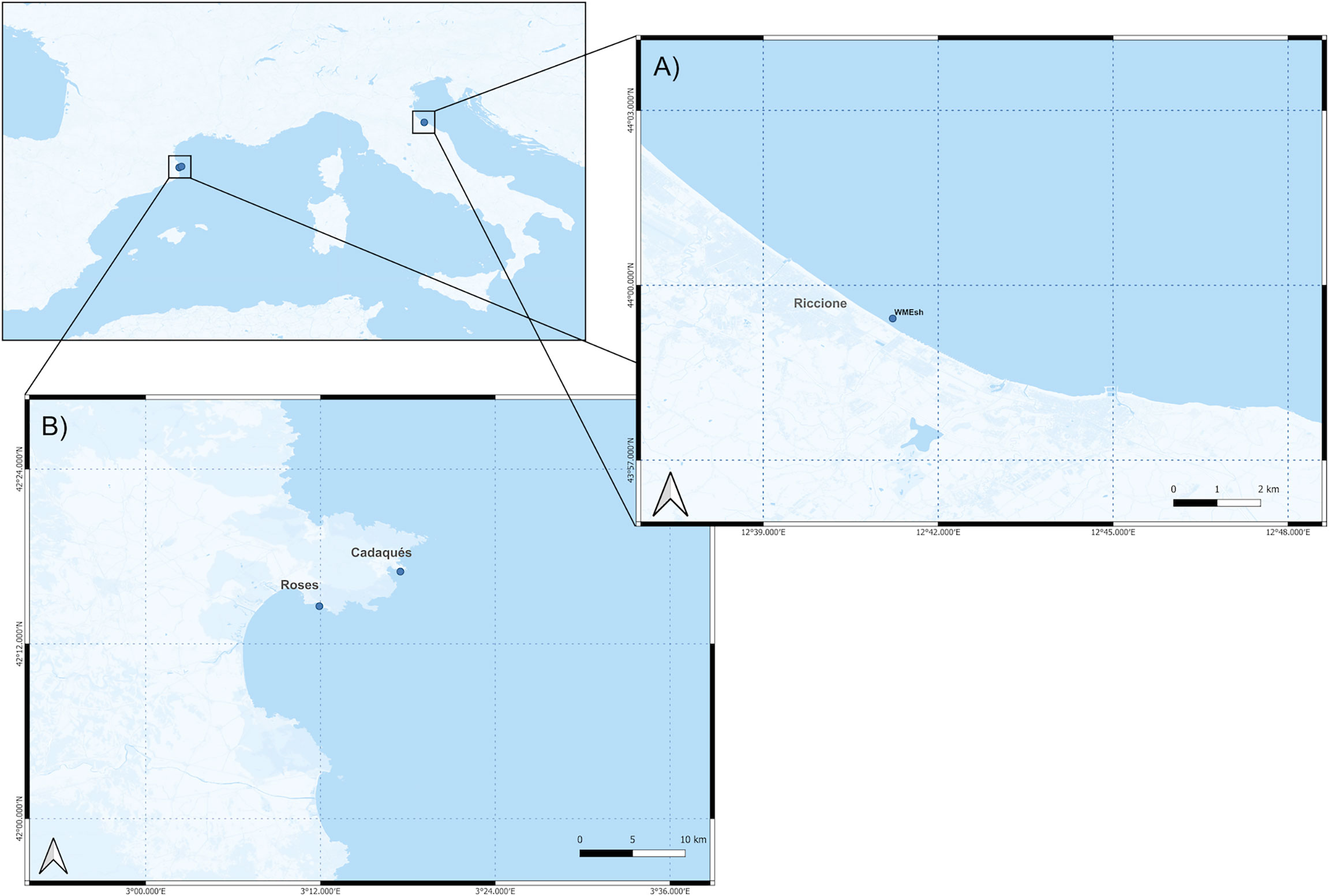
Figure 1 Sampling site description. Maps of the Riccione site (A) at the WMesh location (lat. 43.99049, lon. 12.686993) and of the Cap de Creu site (B) near Cadaqués (lat. 42.282752, lon. 3.291442) and near Roses (lat. 42.243116 lon. 3.198715). Sampling sites are represented as blue dots. All maps were created with the QGIS open-source tool (https://www.qgis.org/it/site/).
The sampling was performed similarly in the two study areas. In the coastal site of Riccione, samples were collected in two different periods (winter, February 2020, and summer, August 2020) along a 100-m-long submerged barrier of reinforced concrete placed approximately 200 m offshore. This framework, referred to as WMesh, was originally designed to prevent coastal erosion, but, thanks to its complex geometry, has provided a hard substrate that ended up hosting a wide variety of marine fauna, including fish, crustaceans, mollusks, and sessile cnidarians (https://www.wmesh.it) (Figure 1A). A total of 20 anemone specimens (10 for each season) were randomly collected by scuba divers and placed into sterile plastic containers. At the same time, five seawater samples (two in winter and three in summer) were collected in sterile 2 L plastic bottles. During the summer sampling, 10 surface sediment samples (0–10 cm) were also collected and placed into sterile Falcon tubes. Environmental parameters at sampling are provided in Supplementary Table S1. In the coastal site of Cap de Creus, a total of 24 A. viridis individuals were collected during the summer period (July 2020) at two different locations: 10 individuals were collected in the protected marine area near Cadaqués, and 14 individuals were collected in the highly touristic site of Roses; the collection was performed in shallow coastal shelves of approximately 50 m2. One seawater sample per site (2 L) was collected alongside anemone individuals (Figure 1B). As described above, anemone samples were placed into sterile plastic containers and seawater samples into sterile 2 L plastic bottles. Environmental parameters at sampling are provided in Supplementary Table S1. Samples collected in Riccione were transported in ice to the laboratory and frozen at -80°C until further processing, whereas samples from Cap de Creus were transported in ice to the laboratory at the University of Barcelona, frozen at -20°C for shipment, then frozen at -80°C until further processing.
Sample processing and DNA extraction
Whole anemones were homogenized using mortar and pestle, and the total bacterial DNA was extracted using the DNeasy PowerBiofilm Kit (Qiagen, Hilden, Germany) (Palladino et al., 2021) following the manufacturer’s protocol with minor adjustments: the homogenization step was performed with a FastPrep instrument (MP Biomedicals, Irvine, CA, USA) at 5.5 movements per second for 1 min, with a 5-min incubation in ice between treatments, and the elution step was preceded by a 5-min incubation at 4°C (Palladino et al., 2021). Each 2 L of the seawater sample was processed via vacuum filtration under sterile conditions using MF-Millipore (Darmstadt, Germany) membrane filters with 0.2-μm pore size, a 47-mm diameter, and a mixed cellulose ester membrane. Each membrane filter was folded using sterilized forceps and placed into a PowerWater DNA Bead Tube and then stored at -80°C until further processing. Seawater microbial DNA extraction was carried out using the DNeasy PowerWater Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. Finally, 0.25–0.35 g of sediment was weighed into PowerBead Tubes, and total bacterial DNA was extracted using the DNeasy PowerSoil Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions with the same minor adjustments described for anemone samples. Total microbial extracted DNA was quantified using NanoDrop ND-1000 (NanoDrop Technologies, Wilmington, DE, United States) and stored at -20°C until further processing.
16S rRNA gene amplification and sequencing
The V3–V4 hypervariable region of the 16S rRNA gene was PCR-amplified in 50-μl final volume containing 25 ng of microbial DNA, 2X KAPA HiFi HotStart ReadyMix (Roche, Basel, Switzerland), and 200 nmol/L of 341F and 785R primers carrying Illumina overhang adapter sequences. For anemone and sediment samples, the thermal cycle consisted of 3 min at 95°C, 25 cycles of 30 s at 95°C, 30 s at 55°C, and 30 s at 72°C, and a final 5-min step at 72°C (Palladino et al., 2021). Seawater samples followed the same PCR amplification protocol with a total of 25 amplification cycles (Musella et al., 2020). PCR products were purified using Agencourt AMPure XP magnetic beads (Beckman Coulter, Brea, CA, United States). Indexed libraries were prepared by limited-cycle PCR with Nextera technology and cleaned up as described above. Libraries were then quantified using a Qubit 3.0 fluorimeter (Invitrogen, Waltham, MA, United States), normalized to 4 nM, and pooled. Finally, the library pool was denatured with 0.2 N NaOH and diluted to 6 pM with a 20% PhiX control. Sequencing was performed on the Illumina MiSeq platform using a 2 × 250-bp paired-end protocol, according to the manufacturer’s instructions (Illumina, San Diego, CA, United States)
Bioinformatics and biostatistics
Raw sequencing outputs for a total of 61 samples were processed with a pipeline combining PANDAseq (Masella et al., 2012) and QIIME 2 (Bolyen et al., 2019). The “fastq filter” function of the Usearch11 algorithm (Edgar, 2010) was applied to retain high-quality reads (min/max length = 350/550 bp). In particular, based on the phred Q score probabilities, reads with an expected error per base E = 0.03 (i.e., three expected errors every 100 bases) were discarded. The retained reads were then binned into amplicon sequence variants (ASVs) using DADA2 (Callahan et al., 2016). A taxonomy assignment was performed using the VSEARCH algorithm (Rognes et al., 2016) and the SILVA database (December 2017 release) (Quast et al., 2012). All the sequences assigned to eukaryotes or unassigned were discarded.
All statistical analyses were performed using the R software (R Core Team; www.r-project.org - last access: March 2021), v. 4.1.2, with the packages “Made4” (Culhane et al., 2005), “vegan” (https://cran.r-project.org/web/packages/vegan/index.html), and MaAsLin2 (Multivariate Association with Linear Models 2, Version 1.8.0) (Mallick et al., 2021). Beta diversity was estimated by computing different metrics, i.e., unweighted UniFrac distance, weighted UniFrac distance, and Bray–Curtis distance. The data separation in the principal coordinate analysis (PCoA) was tested using a permutation test with pseudo-F ratios (function “adonis” in the vegan package). A procrustean randomized test (function “protest” in the vegan package) was performed to highlight significant concordance between different beta-diversity metrics. The Wilcoxon rank-sum test was used to assess significant differences in alpha diversity and taxon relative abundance between groups. P-values were corrected for multiple testing using the Benjamini–Hochberg method, with a false discovery rate (FDR) ≤ 0.05 considered statistically significant. We used MaAsLin2, a modified general linear model for feature-wise multivariate modeling, to identify differentially abundant taxa responsible for possible microbiota-mediated adaptive responses in A. viridis individuals facing multiple and combined anthropic stressors.
Results
Changes in the microbiota of A. viridis with increasing level of multiple impacts: The Riccione case study
The sequencing of the V3–V4 hypervariable region of the 16S rRNA gene from the total microbial DNA resulted in 34 samples producing high-quality output (i.e., 19 anemone, 10 sediment, and 5 seawater samples). The number of high-quality reads in the samples ranged between 1,115 and 14,000 per sample. High-quality reads were subsequently normalized to the lowest number of reads per sample (1,115), resulting in 4,666 ASVs. According to our findings, at the phylum level, the A. viridis microbiota was dominated by Proteobacteria, Bacteroidetes, and Firmicutes, while the corresponding dominant phyla in the surrounding seawater and sediment were, respectively, Proteobacteria, Bacteroidetes, Actinobacteria, Verrucomicrobia, and Cyanobacteria and Proteobacteria, Acidobacteria, and Actinobacteria (Supplementary Figure S1A). At the family level, the dominant members of the A. viridis microbiota were Rhodobacteraceae, Flavobacteriaceae, an unassigned family of the Alphaproteobacteria order, Vibrionaceae, Campylobacteraceae, and Pseudoalteromonadaceae but showing characteristic abundance patterns in the winter and summer samples (Supplementary Figure S2A). Confirming these observations, the PCoAs of the variation of the A. viridis microbiota in the samples set highlighted a significant segregation (permutation test with the pseudo-F ratio, p-value = 0.001) between winter and summer samples, both clustering apart from the corresponding seawater and sediment microbiota samples (Figure 2A). The concordance between A. viridis microbiota variations in different PCoA models was highlighted using a procrustean randomized test (“protest”) (unweighted vs. weighted UniFrac distance and unweighted vs. Bray–Curtis distance resulted in p-value = 0.0001 for both tests, and correlation in a symmetric rotation = 0.8873 and 0.8851, respectively). When we assessed for alpha-diversity patterns, A. viridis microbiota samples were less diverse compared to the corresponding seawater and sediment ecosystems, whereas the diversity of anemone microbiota samples in the different seasons was comparable (Figure 2B). To highlight the compositional differences between the A. viridis microbiota in winter and summer samples, the MaAsLin2 general linear model was applied. According to our findings, several microbiota components were significantly differently represented between winter and summer samples (Supplementary Table S2). The box plots of the relative abundance of the microbial families showing a significantly different distribution between winter and summer samples according to MaAsLin2 are reported in Figure 3. Winter samples were characterized by a higher relative abundance of the families Rhodobacteraceae, Flavobacteriaceae, Lactobacillaceae, Thiotrichaceae, and Xanthomonadaceae, while summer samples were more abundant in Campylobacteraceae, Pseudoalteromonadaceae, Vibrionaceae, Rickettsiaceae and family XII of the order Clostridiales.
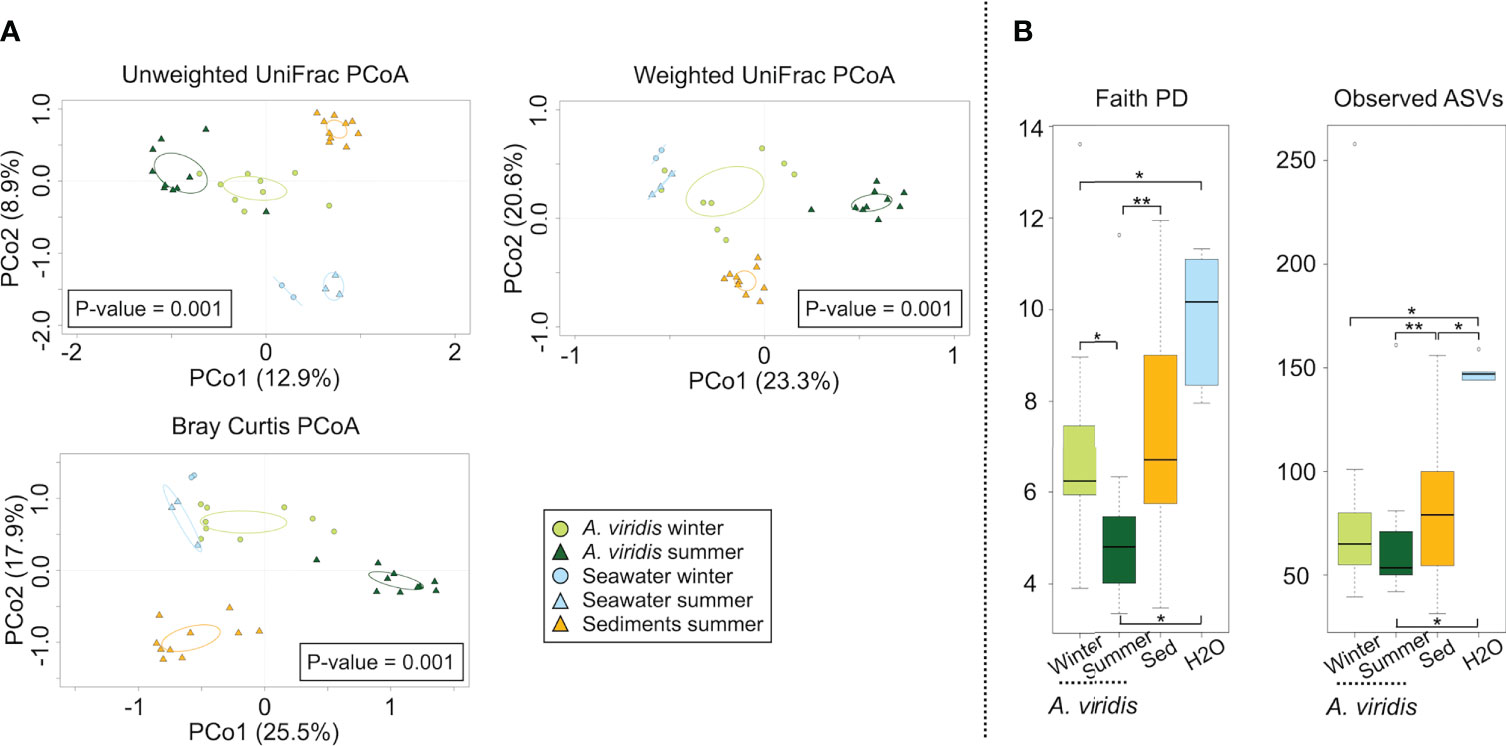
Figure 2 Overall microbiota compositional structure of Anemonia viridis and the surrounding environment in the Riccione site. (A) Principal coordinate analysis (PCoA) based on the unweighted UniFrac distance, weighted UniFrac distance, and Bray–Curtis distance between the microbial profiles of A. viridis and the environmental samples in the Riccione site shows a strong separation between the groups (permutation test with pseudo-F ratio, p-value = 0.001). Concordance between A. viridis microbiota variations in different PCoA models was highlighted using a procrustean randomized test (“protest”) (unweighted vs. weighted UniFrac distance and unweighted vs. Bray–Curtis distance resulted in p-value = 0.0001 for both tests, and correlation in a symmetric rotation = 0.8873 and 0.8851, respectively). The first and second principal components (PCo1 and PCo2) are plotted, and the percentage of variance in the data set explained by each axis is reported. Ellipses include a 95% confidence area based on the standard error of the weighted average of the sample coordinates. A. viridis samples are represented in green (“darkolivegreen1” circles for winter, “darkgreen” triangles for summer), seawater samples are represented in light blue (“lightskyblue1” circles for winter, “lightskyblue1” triangles for summer), and sediment samples are represented in “darkgoldenrod1” triangles. (B) Box-and-whiskers distribution of the alpha diversity metrics Faith’s phylogenetic diversity (PD) and number of observed ASVs. Significant p-values (Wilcoxon rank-sum test controlled for multiple testing using FDR, p-value ≤ 0.05) are reported in the figure with the following symbols: p-value ≤ 0.05 *; p-value ≤ 0.01 **. The color code is the same as in panel A.
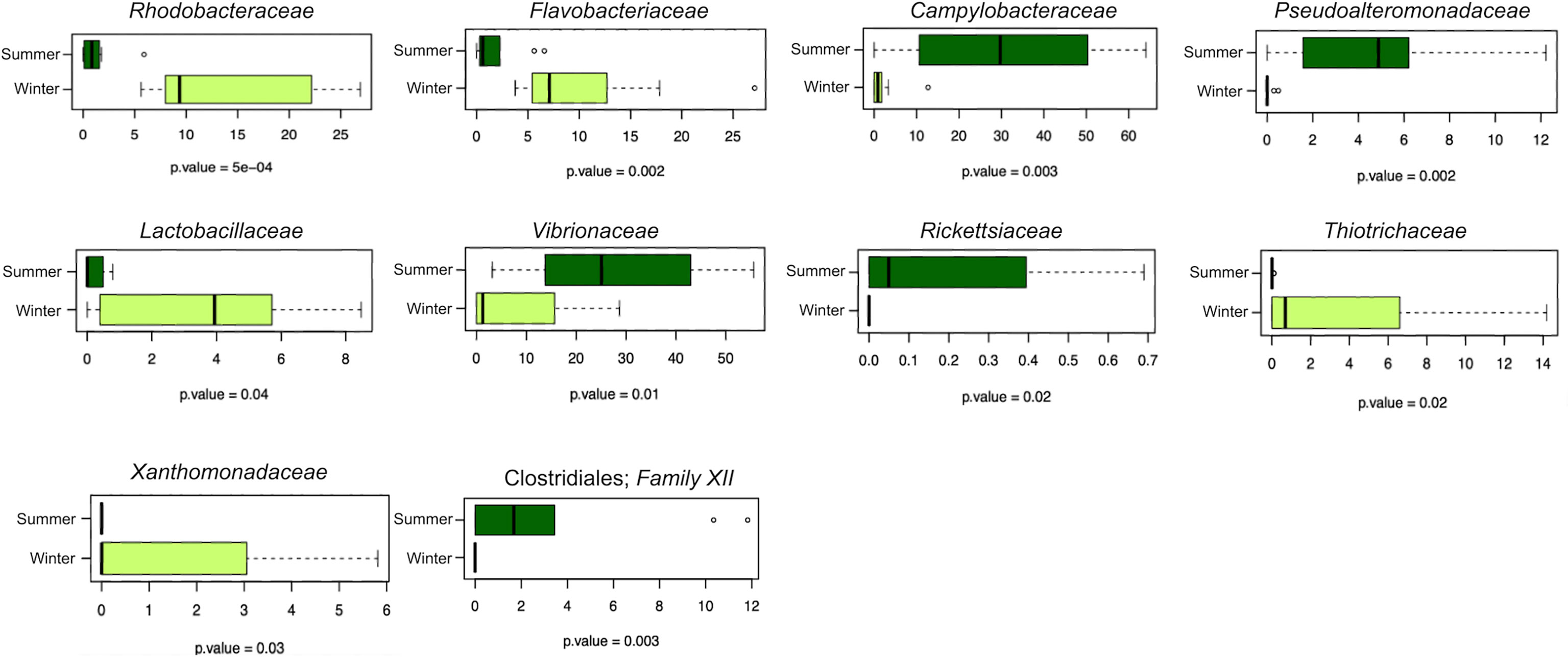
Figure 3 Microbial families responsible for significant compositional differences between the A. viridis microbiota in winter and summer samples collected at the Riccione site. Box-and-whisker plots showing the relative abundance of bacterial families whose differential distribution was significantly different in the A. viridis microbiota in winter (“darkolivegreen1”) and summer (“darkgreen”) samples according to MaAsLin2 (Multivariate Association with Linear Models 2, Version 1.8.0). The central box represents the distance between the 25th and 75th percentiles. The median is marked with a black line. Whiskers identify the 10th and 90th percentiles. P-values are reported below each box plot (Wilcoxon rank-sum test controlled for multiple testing using FDR).
To explore the connections between the A. viridis microbiota and seawater and sediment microbiota, the metacommunity bubble plot was carried out (Figure 4). In the bubble plot, the compositional structure of the winter and summer A. viridis microbiota was compared with the corresponding compositional structure of the surrounding seawater and sediment ecosystems. Interestingly, several bacterial members of the A. viridis microbiota were shared with the microbiota of the surrounding seawater and/or sediments. For instance, an unassigned family of the Gammaproteobacteria order and the bacterial families Campylobacteraceae, Flavobacteriaceae, Halieaceae, Planctomycetaceae, Rhodobacteraceae, Verrucomicrobiaceae, and Vibrionaceae were all shared among the microbiotas of the three systems, while an unassigned family of the Alphaproteobacteria and Rhizobiales orders and the bacterial families Alteromonadaceae, Comamonadaceae, Oceanospirillaceae, Pseudoalteromonadaceae, and Rhodospirillaceae were shared between the microbiotas of the anemones and seawater. Finally, focusing on the A. viridis microbiota components, which were responsible for a different assemblage composition between winter and summer samples, some were shared with the microbiotas of water and/or sediment, such as Flavobacteriaceae, Pseudoalteromonadaceae, Rhodobacteriaceae, Campylobacteraceae, and Vibrionaceae, all showing a winter-to-summer variation in seawater and sediment microbiotas corresponding to the one observed for the A. viridis microbiota. On the contrary, the family XII of Clostridiales, Lactobacillaceae, Thiotrichaceae, and Xantomonadaceae were exclusive to the A. viridis microbiota.
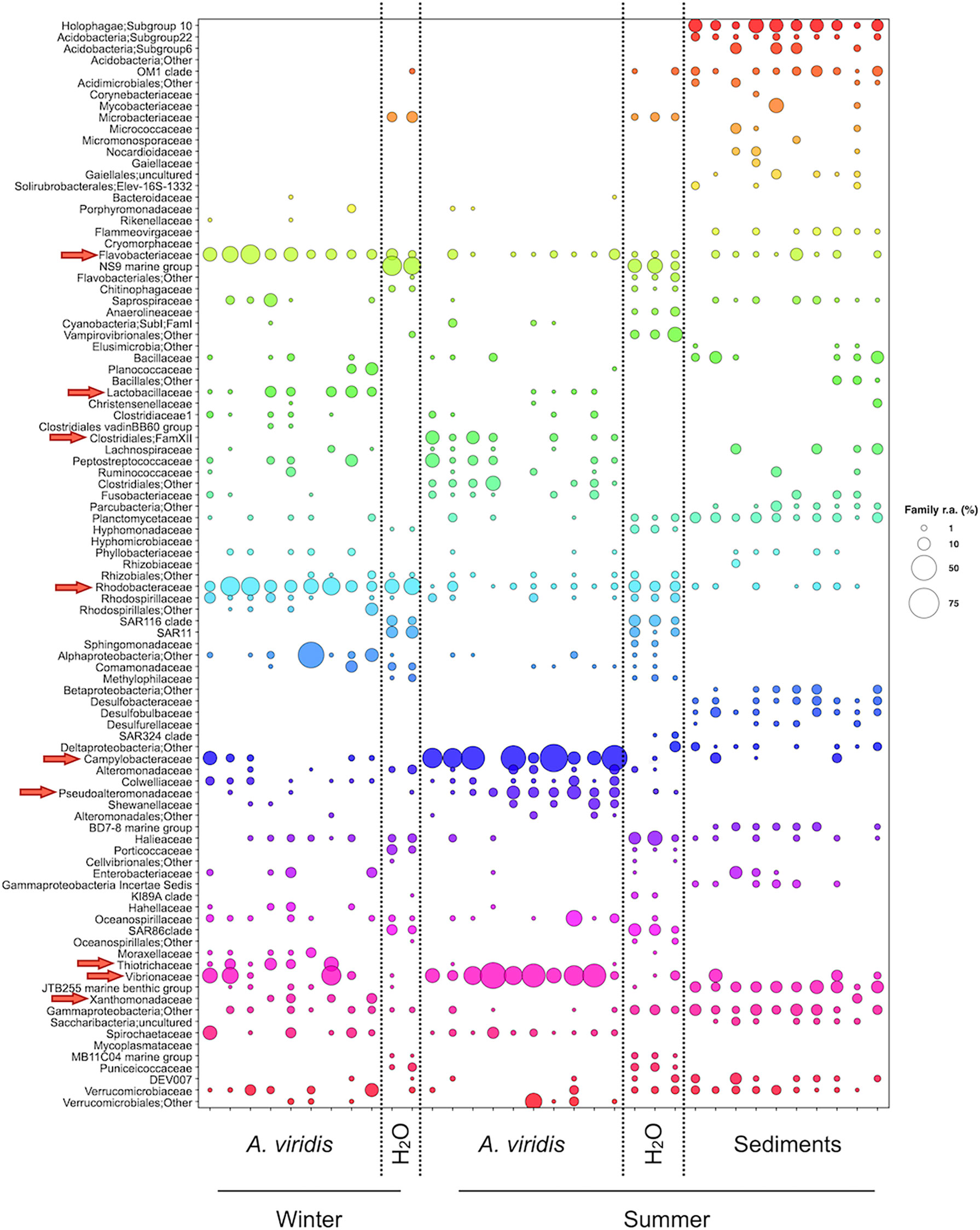
Figure 4 Metacommunity bubble plot at family level in the Riccione site. Bubble plot of the compositional structure of winter and summer A. viridis microbiota samples compared with the surrounding ecosystems (seawater and sediments) at the Riccione site. A. viridis microbiota components showing a differential distribution between winter and summer samples according to the MaAsLin2 model are indicated by red arrows. The size of the bubbles represents the percentage of relative abundance according to the legend on the right.
Changes in A. viridis microbiota in coastal sites with a different level of anthropogenic impact: The Cap de Creus case study
The sequencing of the V3–V4 hypervariable region of the 16S rRNA gene from the total microbial DNA in 24 samples (including 22 anemones and 2 seawater samples) produced a high-quality output. The number of high-quality reads in the samples ranged between 1,358 and 14,090 per sample. High-quality reads were subsequently normalized to the lowest number of reads per sample 1,115, resulting in 4,666 ASVs. As previously observed at the Riccione site, the dominant phyla of the A. viridis microbiota were Proteobacteria, Firmicutes, and Bacteroidetes, while the seawater microbiota was dominated by Proteobacteria, Bacteroidetes, Planctomycetes, Actinobacteria, Verrucomicrobia, and Cyanobacteria (Supplementary Figure S1B). At the family level, the most represented families in the A. viridis microbiota were an unassigned family of the Alphaproteobacteria order and the families Ruminococcaceae, Lachnospiraceae, Moraxellaceae, Pseudoalteromonadaceae, Rhodobacteraceae, Campylobacteraceae, Vibrionaceae, Flavobacteriaceae, and Spirochetaceae, demonstrating an overall family-level structure different from the one observed at the Riccione site (Supplementary Figure S2B). The PCoA of the microbiota compositional structure resulted in a significant segregation (permutation test with pseudo-F ratio, p-value = 0.001) among A. viridis microbiota samples depending on the site (Figure 5A), with the A. viridis microbiota collected at the high-impact site of the Cap de Creus (Roses) showing a higher dispersion in the PCoA plot. A. viridis microbiota variations in different PCoA models using a procrustean randomized test (“protest”) were concordant (unweighted vs. weighted UniFrac distance and Unweighted vs. Bray–Curtis distance resulted in p-value = 0.001 and 0.0001, and correlation in a symmetric rotation = 0.5663 and 0.6665, respectively). For what concerns the alpha diversity, no differences between microbiota at both sites were observed, neither in terms of ASV richness nor Faith’s phylogenetic diversity (PD) index (Figure 5B). To identify the A. viridis microbiota members characteristic from the low- and high-impact sites, the MaAsLin2 test was applied. According to our findings, 11 microbial families were responsible for the significant differences between the microbiota of the two sites (Supplementary Table S3). The corresponding box plot of the relative abundances of bacterial taxa at the low- and high-level impact sites is reported in Figure 6. More specifically, the microbiota of anemones from the low-impact site was characterized by an increased abundance in Clostridiales (vadinBB60 group), Lachnospiraceae, Ruminococcaceae, Campylobacteriaceae, Alteromonadaceae, Pseudoalteromonadaceae, Oceanospirillaceae, and Moraxellaceae compared to the microbiota of the high-impact site. On the contrary, the microbiota of anemones growing at the high-impact site was characterized by a higher abundance of the clade DEV007 belonging to the family Vibrionaceae and Verrucomicrobiaceae.
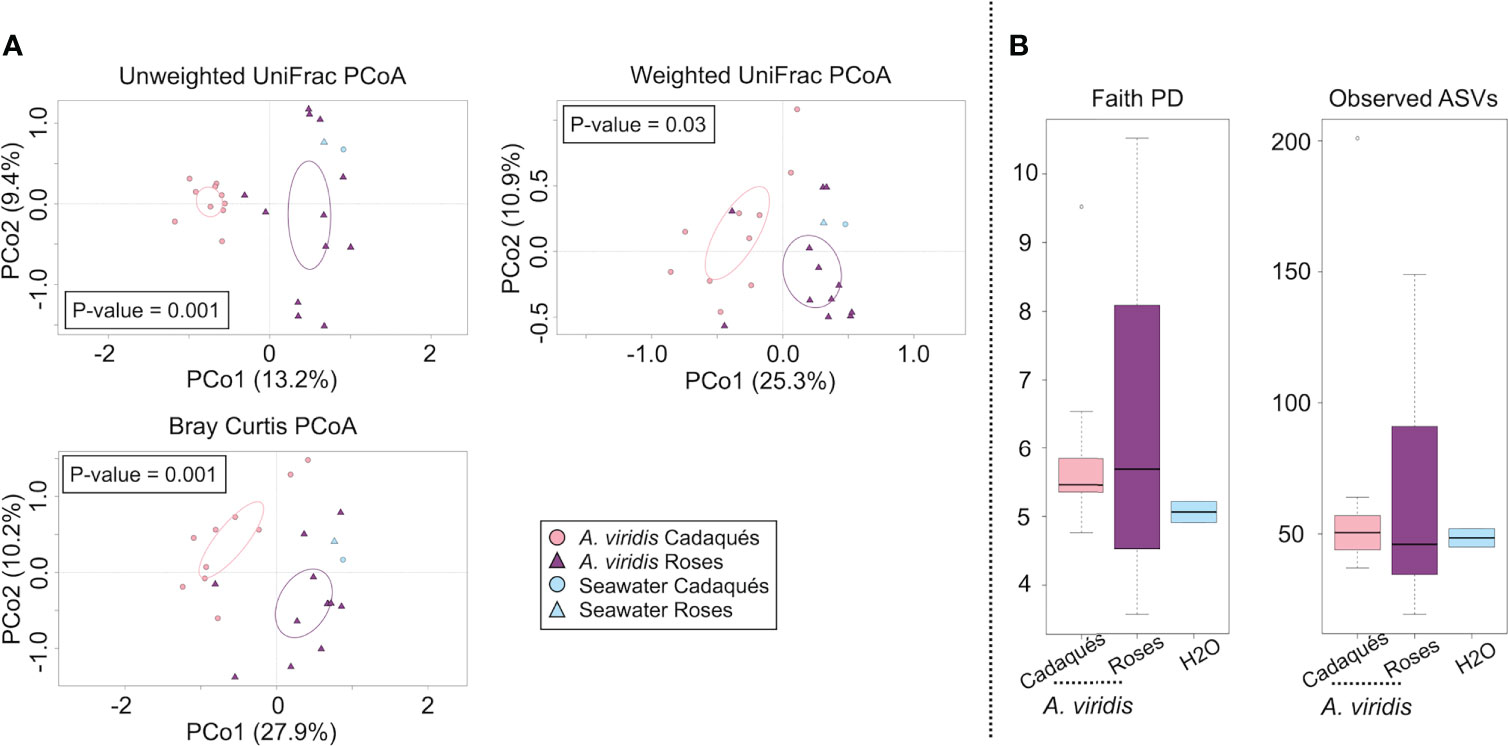
Figure 5 Overall microbiota compositional structure of A. viridis and the surrounding environment in the Cap de Creus site. (A) PCoA based on the unweighted UniFrac distance, weighted UniFrac distance, and Bray–Curtis distance between the microbial profiles of A. viridis and the environmental samples in the Cap de Creus site shows a strong separation between the groups (permutation test with pseudo-F ratio, p-value = 0.001). A. viridis microbiota variations in different PCoA models using a procrustean randomized test (“protest”) were in concordant (unweighted vs. weighted UniFrac distance and unweighted vs. Bray–Curtis distance resulted in p-value = 0.001 and 0.0001 and correlation in a symmetric rotation = 0.5663 and 0.6665, respectively). The first and second principal components (PCo1 and PCo2) are plotted, and the percentage of variance in the data set explained by each axis is reported. Ellipses include a 95% confidence area based on the standard error of the weighted average of the sample coordinate. A. viridis samples are represented in pink shades (“lightpink” circles for Cadaqués, “orchid4” triangles for Roses), and seawater samples are represented in light blue (“lightskyblue1” circles for Cadaqués, “lightskyblue1” triangles for Roses). (B) Box-and-whiskers distribution of the alpha diversity metrics Faith’s PD and number of observed ASVs. No significant p-values were found (Wilcoxon rank-sum test controlled for multiple testing using FDR). The color code is the same as in panel (A).
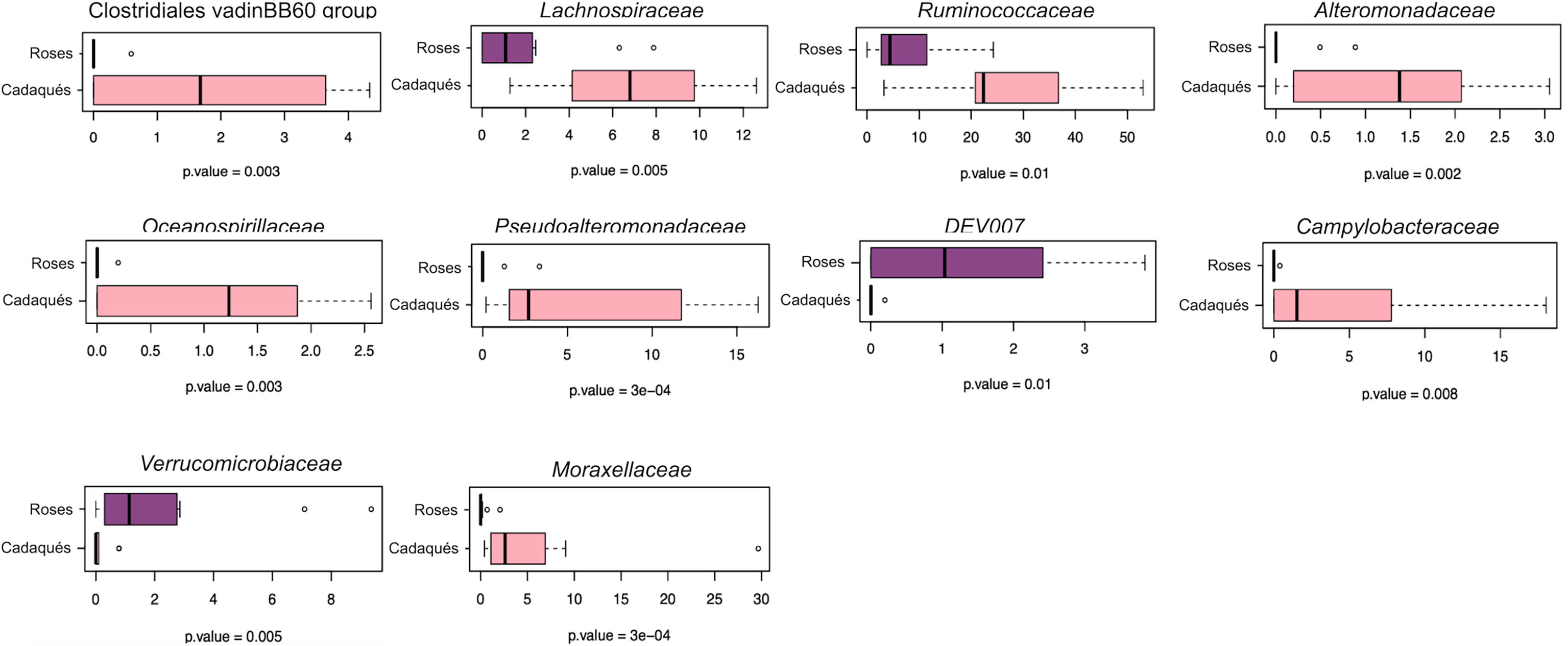
Figure 6 Microbial families responsible for significant compositional differences between the A. viridis microbiota in Cadaqués and Roses samples at the Cap de Creus site. Box-and-whisker plots showing the relative abundance of bacterial families whose differential distribution was significantly different in the A. viridis-associated microbiota in Cadaqués (“lightpink”) and Roses (“orchid4”) sampling sites according to MaAsLin2. The central box represents the distance between the 25th and 75th percentiles. The median is marked with a black line. Whiskers identify the 10th and 90th percentiles. P-values are reported below each box plot (Wilcoxon rank-sum test controlled for multiple testing using FDR).
When exploring the connections between the A. viridis microbiota and the corresponding seawater microbiota by metacommunity bubble plots (Figure 7), we successfully identified several groups shared between the different ecosystems, such as an unassigned family of the Alphaproteobacteria, Gammaproteobacteria, and Rhizobiales orders, the families Alteromonadaceae, Cyanobacteria, Flavobacteriaceae, Halieaceae, the JTB255 marine benthic group (part of the gammaproteobacterial Woeseiaceae), Moraxellaceae, Planctomicetaceae, Rhodobacteraceae, Ruminococcaceae, Vibrionaceae, and Verrucomicrobiaceae, with Alteromonadaceae, Moraxellaceae, and Ruminococcaceae characterizing anemones at the low-impact site and Verrucomicrobiaceae characterizing anemones at the high-impact site.
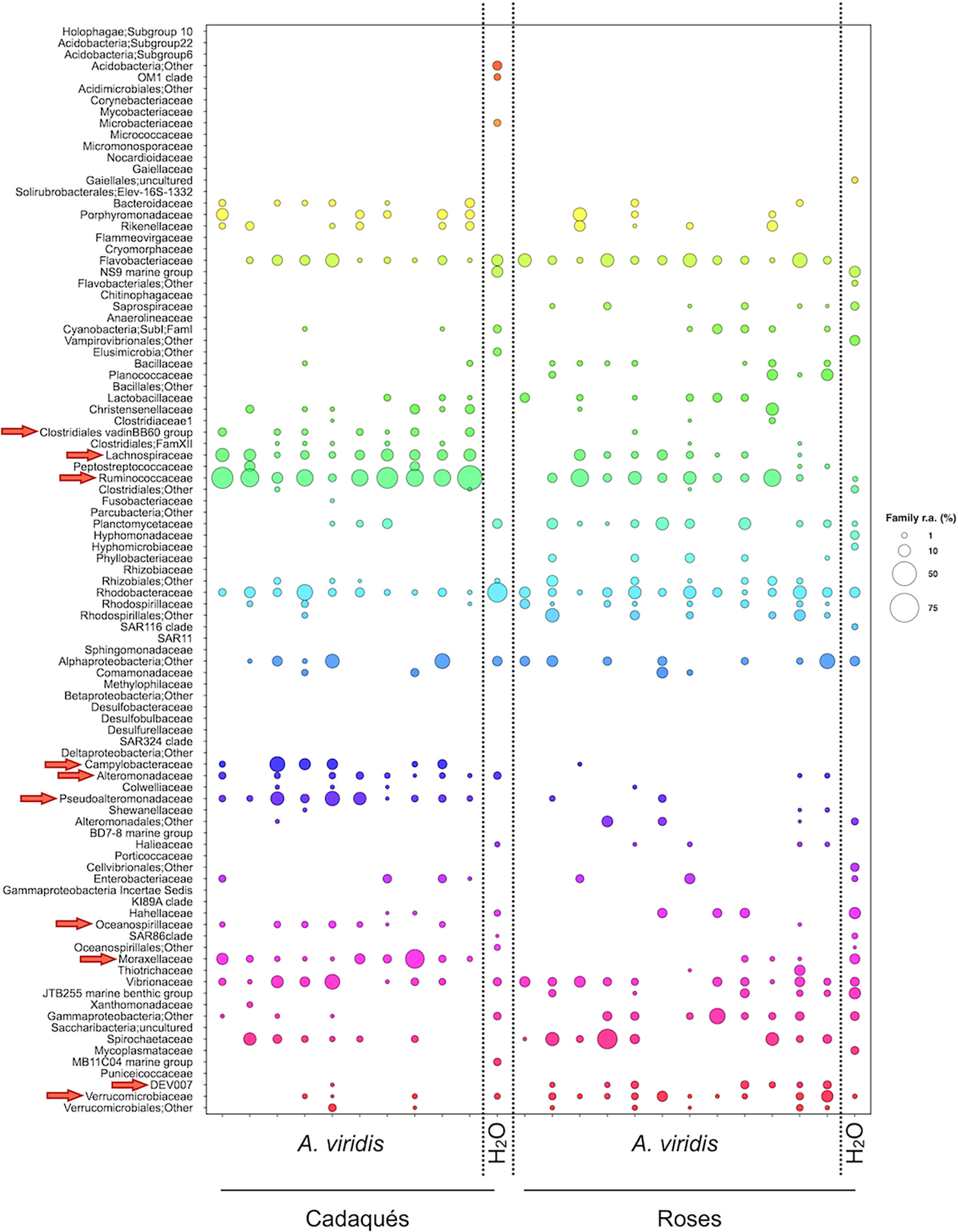
Figure 7 Metacommunity bubble plot at the family level in the Cap de Creus site. Bubble plot of the compositional structure of Cadaqués and Roses A. viridis microbiota samples compared with the respective surrounding seawater at the Cap de Creus site. A. viridis microbiota components showing a differential distribution between Cadaqués and Roses samples according to the MaAsLin2 model are indicated by red arrows. The size of the bubbles represents the percentage of relative abundance according to the legend on the right.
Discussion
In the present study, we found that the A. viridis microbiota was characterized by a conserved compositional structure at high phylogenetic levels while showing a site-specific layout at lower phylogenetic ranks. Indeed, the A. viridis microbiota at Cap de Creus was characterized by several dominant families, such as Lachnospiraceae, Spirochetaceae, Ruminococcaceae, and Moraxellaceae, which were not detected in anemones at the Riccione site. Interestingly, both at the Riccione and the Cap de Creus site, several dominant families of the A. viridis microbiota were shared with the respective surrounding water microbiota. Overall, these findings confirm the local adaptation of the A. viridis microbiota, which remained highly connected with the seawater microbiota, suggesting a strong host filtering, which allows the selection of the symbiont components from a local pool of marine bacteria. Anyway, at both sites, the A. viridis microbiota was characterized by the presence of heterotrophic anaerobes, such as Rhodobacteriaceae, Oceanospirillaceae, Alteromonadaceae, and Pseudoalteromonadaceae, being able to cycle S (sulfur) and N (nitrogen) with possible benefits for the host and the surrounding local ecosystem (Muller et al., 2016).
The A. viridis microbiota showed a relevant degree of plasticity in response to combined anthropogenic and environmental stressors. Indeed, in the Riccione case study, the A. viridis microbiota changed in the same site when subject to increasing multiple impact levels from winter to summer. Interestingly, several changes in the A. viridis microbiota mirrored changes in the microbiota of the surrounding water, such as the increase in Vibrionaceae, Campylobacteraceae, and Pseudoalteromonadaceae, with a parallel decrease in Rohodobacteriaceae and Flavobacteriaceae. The observed increase of Vibrionaceae, Campylobacteraceae, and Pseudoalteromonadaceae in the seawater microbiota (Liu et al., 2011; Luna, 2015; Gutierrez-Barral et al., 2021) might be explained by the synergic action of the organic inputs from the Po River, likely exerting a strong influence on the Adriatic Sea in the summer season, when stratification and eddy circulation make the northern Adriatic a semi-enclosed basin (Spillman et al., 2007), and the enrichment of anthropogenic nutrients in the coastal seawater during the summer tourism peak, as observed by Andolina et al. (2021). This condition may also explain the observed increase in Clostridiales and Rickettsiaceae in the anemone microbiota since these microorganisms have been all previously detected in association with marine holobionts growing in eutrophic conditions (Klinges et al., 2019). Taken together, our findings from the Riccione case study suggest the ability of the A. viridis microbiota to respond to changing environmental conditions and combined anthropogenic and environmental stressors while keeping a close connection with the surrounding environment through the selection of potentially symbiont partners.
In the Cap de Creus case study, the A. viridis microbiota was investigated in two sites with different anthropogenic impact levels (Cadaqués, close to a natural park, and Roses, near an urban center). While the microbiota of A. viridis from Cadaqués was characterized by a compositional structure generally coherent with the one observed for the anemones from the Riccione site, including copiotroph anaerobic Firmicutes (such as Clostridiales, Lachnospiraceae, and Ruminococcaceae), Pseudoalteromonadaceae, and Campylobacteraceae, in Roses, the anemone microbiota appeared to be completely unstructured, as demonstrated by the increased dispersion in the PCoA plot. According to the Anna Karenina principle (Zaneveld et al., 2017), a dysbiotic response of the holobiont microbiome involves its stochastic dispersion, with the microbiome acquiring a random configuration as a result of an impaired host selection. According to other studies (Ahmed et al., 2019), anemones growing under stressed conditions show analogous dysbiotic dispersions. Our data thus indicate that A. viridis growing at the high-impact site in the Cap de Creus case study develops a dysbiotic state, possibly as a result of a maladaptive response to the local environmental conditions.
Although our data need to be confirmed in a wider context (for example, expanding the number of case studies to other geographic sites) and by shotgun functional metagenomics, these suggest the relevant degree of plasticity of the anemone microbiota, which allows the adaptation of the host to different environmental changes and multiple anthropogenic stressors. Microbiome dynamics and strong environmental connections may indeed represent ancestral evolutionary microbiome–host selection systems, based on a continuous and dynamic interplay with changing environmental ecosystems, from which the best available symbionts are selected. In this scenario, it is of great importance to understand the mechanisms involved in this “microbiome gardening” process implemented by the holobiont, as well as the resilience threshold of this ancestral holobiont system, and the possible external factors compromising the host selection processes and resulting in detrimental dysbiotic dispersions, as observed in the Roses site. This information will be instrumental to educate policies to preserve and, eventually, restore coastal holobionts, with cascade benefits for the entire coastal health and ecosystem services.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/ena, PRJEB53200.
Author contributions
MC and CC: conceptualization. GP, CG-C, and GT: methodology. GP and SR: formal analysis. GP, SR, GT, EN, and ST: data curation. GP, CC, and MC: writing–original draft preparation. SR, CG-C, DS, GT, EN, VA, DC, and ST: writing–draft revision. MC: funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the “Controlling Microbiomes Circulations for Better Food Systems” (CIRCLES) project, which was funded by the European Union’s Horizon 2020 research and innovation program under grant agreement no. 818290.
Acknowledgments
We would like to thank the recreational scuba diving association Blennius (Riccione, Italy) for their primary contribution to the sample collection at the Riccione site.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.956899/full#supplementary-material
References
Ahmed H. I., Herrera M., Liew Y. J., Aranda M. (2019). Long-term temperature stress in the coral model aiptasia supports the “Anna karenina principle” for bacterial microbiomes. Front. Microbiol. 10-, 975. doi: 10.3389/fmicb.2019.00975
Andolina C., Signa G., Tomasello A., Mazzola A., Vizzini S. (2021). Environmental effects of tourism and its seasonality on Mediterranean islands: The contribution of the interreg MED BLUEISLANDS project to build up an approach towards sustainable tourism. Environment Dev. Sustainability 23 (6), 8601–8612. doi: 10.1007/s10668-020-00984-8
Barbier E. B., Hacker S. D., Kennedy C., Koch E. W., Stier A. C., Silliman B. R. (2011). The value of estuarine and coastal ecosystem services. Ecol. Monogr. 81 (2)169–193. doi: 10.1890/10-1510.1
Bolyen E., Rideout J. R., Dillon M. R., Bokulich N. A., Abnet C. C., Al-Ghalith G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37 (8), 852–857. doi: 10.1038/s41587-019-0209-9
Brown T., Otero C., Grajales A., Rodriguez E., Rodriguez-Lanetty M. (2017). Worldwide exploration of the microbiome harbored by the cnidarian model, Exaiptasia pallida (Agassiz in verril) indicates a lack of bacterial association specificity at a lower taxonomic rank. PeerJ. 5, e3235. doi: 10.7717/peerj.3235
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., Holmes S. P. (2016). DADA2: High-resolution sample inference from illumina amplicon data. Nat. Methods 13 (7), 581–583. doi: 10.1038/nmeth.3869
Craig-Smith S. J., Tapper R., Font X. (2006). “The coastal and marine environment,” in In tourism and global environmental change (London, UK: Routledge), 121–141.
Crain C. M., Halpern B. S., Beck M. W., Kappel C. V. (2009). Understanding and managing human threats to the coastal marine environment. Ann. N. Y. Acad. Sci. 1162, 39–62. doi: 10.1111/j.1749-6632.2009.04496.x
Culhane A. C., Thioulouse J., Perrière G., Higgins D. G. (2005). MADE4: An r package for multivariate analysis of gene expression data. Bioinformatics 21 (11), 2789–2790. doi: 10.1093/bioinformatics/bti394
Danovaro R. (2003). Pollution threats in the Mediterranean Sea: An overview. Chem. Ecol. 19 (1), 15–32. doi: 10.1080/0275754031000081467
Drius M., Bongiorni L., Depellegrin D., Menegon S., Pugnetti A., Stifter S. (2019). Tackling challenges for Mediterranean sustainable coastal tourism: An ecosystem service perspective. Sci. Total Environ. 652, 1302–1317. doi: 10.1016/j.scitotenv.2018.10.121
Edgar R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26 (19), 2460–2461. doi: 10.1093/bioinformatics/btq461
Gutiérrez-Barral A., Teira E., Hernández-Ruiz M., Fernández E. (2021). Response of prokaryote community composition to riverine and atmospheric nutrients in a coastal embayment: Role of organic matter on vibrionales. Estuarine Coast. Shelf Sci. 251, 107196. doi: 10.1016/j.ecss.2021.107196
Istat Annual report on the tourist movement and hotel and complementary consistency in Emilia romagna. Available at: https://statistica.regione.emilia-romagna.it/documentazione/pubblicazioni/documenti_catalogati/rapporto-annuale-turismo-2020.
Klinges J. G., Rosales S. M., McMinds R., Shaver E. C., Shantz A. A., E.C.and Vega Thurber R. L. (2019). Phylogenetic, genomic, and biogeographic characterization of a novel and ubiquitous marine invertebrate-associated rickettsiales parasite, candidatus aquarickettsia rohweri, gen. nov., sp. nov. ISME J. 13 (12), 2938–2953. doi: 10.1038/s41396-019-0482-0
Kolodny O., Schulenburg H. (2020). Microbiome-mediated plasticity directs host evolution along several distinct time scales. Philos. Trans. R. Soc B 375 (1808), 20190589. doi: 10.1098/rstb.2019.0589
Lemieux J., Cusson M. (2014). Effects of habitat-forming species richness, evenness, identity, and abundance on benthic intertidal community establishment and productivity. PloS One 9 (10), e109261. doi: 10.1371/journal.pone.0109261
Liu M., Dong Y., Zhao Y., Zhang G., Zhang W., Xiao T. (2011). Structures of bacterial communities on the surface of ulva prolifera and in seawaters in an ulva blooming region in jiaozhou bay, China. World J. Microbiol. Biotechnol. 27 (7), 1703–1712. doi: 10.1007/s11274-010-0627-9
Luna G. M. (2015). Diversity of marine microbes in a changing Mediterranean Sea. Rendiconti Lincei 26 (1), 49–58.
Mallick H., Rahnavard A., McIver L. J., Ma S., Zhang Y., Nguyen L. H., et al. (2021). Multivariable association discovery in population-scale meta-omics studies. PloS Comput. 17 (11), e1009442. doi: 10.1371/journal.pcbi.1009442
Marangon E., Laffy P. W., Bourne D. G., Webster N. S. (2021). Microbiome-mediated mechanisms contributing to the environmental tolerance of reef invertebrate species. Mar. Biol. 168 (6), 89. doi: 10.1007/s00227-021-03893-0
Masella A. P., Bartram A. K., Truszkowski J. M., Brown D. G., Neufeld J. D. (2012). PANDAseq: Paired-end assembler for illumina sequences. BMC Bioinf. 13 (1), 31. doi: 10.1186/1471-2105-13-31
McDevitt-Irwin J. M., Baum J. K., Garren M., Vega Thurber R. L. (2017). Responses of coral-associated bacterial communities to local and global stressors. Front. Mar. Sci. 4, 262. doi: 10.3389/fmars.2017.00262
McFall-Ngai M., Hadfield M. G., Bosch T. C., Carey H. V., Domazet-Lošo T., Douglas A. E., et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl Acad. Sci. USA 110 (9), 3229–3236. doi: 10.1073/pnas.1218525110
Morelan I. A., Gaulke C. A., Sharpton T. J., Vega Thurber R., Denver D. R. (2019). Microbiome variation in an intertidal sea anemone across latitudes and symbiotic states. Front. Mar. Sci. 6, 7. doi: 10.3389/fmars.2019.00007
Morrow K. M., Moss A. G., Chadwick N. E., Liles M. R. (2012). Bacterial associates of two Caribbean coral species reveal species-specific distribution and geographic variability. Appl. Environ. Microbiol. 78 (18), 6438–6449. doi: 10.1128/AEM.01162-12
Mortzfeld B. M., Urbanski S., Reitzel A. M., Künzel S., Technau U., Fraune S. (2016). Response of bacterial colonization in nematostella vectensis to development, environment and biogeography. Environ. Microbiol. 18 (6), 1764–1781. doi: 10.1111/1462-2920.12926
Muller E. M., Fine M., Ritchie K. B. (2016). The stable microbiome of inter and sub-tidal anemone species under increasing pCO2. Sci. Rep. 6, 37387. doi: 10.1038/srep37387
Musella M., Wathsala R., Tavella T., Rampelli S., Barone M., Palladino G., et al. (2020). Tissue-scale microbiota of the Mediterranean mussel (Mytilus galloprovincialis) and its relationship with the environment. Sci. Total Environ. 717, 137209. doi: 10.1016/j.scitotenv.2020.137209
O’Brien P. A., Smith H. A., Fallon S., Fabricius K., Willis B. L., Morrow K. M., et al. (2018). Elevated CO2 has little influence on the bacterial communities associated with the pH-tolerant coral, massive porites spp. Front. Microbiol. 9, 2621. doi: 10.3389/fmicb.2018.02621
Palinkas C. M., Nittrouer C. A., Wheatcroft R. A., Langone L. (2005). The use of 7Be to identify event and seasonal sedimentation near the po river delta, Adriatic Sea. Mar. Geol. 222-223, 95–112. doi: 10.1016/j.margeo.2005.06.011
Palladino G., Biagi E., Rampelli S., Musella M., D’Amico F., Turroni S., et al. (2021). Seasonal changes in microbial communities associated with the jewel anemone corynactis viridis. Front. Mar. Sci. 8, 627585. doi: 10.3389/fmars.2021.627585
Pettine M., Patrolecco L., Camusso M., Crescenzio S. (1998). Transport of carbon and nitrogen to the northern Adriatic Sea by the po river. Estuar. Coast. Shelf Sci. 46 (1), 127–142. doi: 10.1006/ecss.1997.0303
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2012). The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41 (D1), D590–D596. doi: 10.1093/nar/gks1219
Rampelli S., Turroni S., Debandi F., Alberdi A., Schnorr S. L., Hofman C. A., et al. (2021). The gut microbiome buffers dietary adaptation in bronze age domesticated dogs. Iscience 24 (8), 102816. doi: 10.1016/j.isci.2021.102816
Rognes T., Flouri T., Nichols B., Quince C., Mahé F. (2016). VSEARCH: A versatile open source tool for metagenomics. PeerJ 4, e2584. doi: 10.7717/peerj.2584
Rohwer F., Breitbart M., Jara J., Azam F., Knowlton N. (2001). Diversity of bacteria associated with the Caribbean coral montastraea franksi. Coral Reefs 20, 85–91. doi: 10.1007/s003380100138
Rosenberg E., Koren O., Reshef L., Efrony R., Zilber-Rosenberg I. (2007). The role of microorganisms in coral health, disease and evolution. Nat. Rev. Microbiol. 5 (5), 355–362. doi: 10.1038/nrmicro1635
Santoro E. P., Borges R. M., Espinoza J. L., Freire M., Messias C. S., Villela H. D., et al. (2021). Coral microbiome manipulation elicits metabolic and genetic restructuring to mitigate heat stress and evade mortality. Sci. Adv. 7 (33), eabg3088. doi: 10.1126/sciadv.abg3088
Sardá R., Rossi S., Martí X., Gili J. M. (2012). Marine benthic cartography of the cap de creus (NE Catalan coast, Mediterranean Sea). Scientia Marina 76 (1), 159–171. doi: 10.3989/scimar.03101.18D
Spillman C. M., Imberger J., Hamilton D. P., Hipsey M. R., Romero J. R. (2007). Modelling the effects of po river discharge, internal nutrient cycling and hydrodynamics on biogeochemistry of the northern Adriatic Sea. J. Mar. Syst. 68 (1-2), 167–200. doi: 10.1016/j.jmarsys.2006.11.006
Steinberg R. K., Dafforn K. A., Ainsworth T., Johnston E. L. (2020). Know thy anemone: A review of threats to octocorals and anemones and opportunities for their restoration. Front. Mar. Sci. 7, 590. doi: 10.3389/fmars.2020.00590
Tovar-Sánchez A., Sánchez-Quiles D., Rodríguez-Romero A. (2019). Massive coastal tourism influx to the Mediterranean Sea: The environmental risk of sunscreens. Sci. Total Environ. 656, 316–321. doi: 10.1016/j.scitotenv.2018.11.399
Trevathan-Tackett S. M., Sherman C. D., Huggett M. J., Campbell A. H., Laverock B., Hurtado-McCormick V., et al. (2019). A horizon scan of priorities for coastal marine microbiome research. Nat. Ecol. Evol. 3 (11), 1509–1520. doi: 10.1038/s41559-019-0999-7
Vanwonterghem I., Webster N. S. (2020). Coral reef microorganisms in a changing climate. Iscience 23 (4), 100972. doi: 10.1016/j.isci.2020.100972
Wambua S., Gourlé H., de Villiers E. P., Karlsson-Lindsjö O., Wambiji N., Macdonald A., et al. (2021). Cross-sectional variations in structure and function of coral reef microbiome with local anthropogenic impacts on the Kenyan coast of the Indian ocean. Front. Microbiol. 12, 673128. doi: 10.3389/fmicb.2021.67312
Wilkins L. G., Leray M., O’Dea A., Yuen B., Peixoto R. S., Pereira T. J., et al. (2019). Host-associated microbiomes drive structure and function of marine ecosystems. PloS Biol. 17 (11), e3000533. doi: 10.1371/journal.pbio.3000533
Keywords: coastal ecosystems, anthropogenic threats, Anemonia viridis, microbiota, Mediterranean Sea
Citation: Palladino G, Rampelli S, Galià-Camps C, Scicchitano D, Trapella G, Nanetti E, Angelini V, Cleo D, Turroni S, Corinaldesi C and Candela M (2022) Plasticity of the Anemonia viridis microbiota in response to different levels of combined anthropogenic and environmental stresses. Front. Mar. Sci. 9:956899. doi: 10.3389/fmars.2022.956899
Received: 30 May 2022; Accepted: 05 September 2022;
Published: 23 September 2022.
Edited by:
Teresa Bottari, Institute for Biological Resources and Marine Biotechnology (CNR), ItalyReviewed by:
Sebastian Fraune, Heinrich Heine University of Düsseldorf, GermanyNar Singh Chauhan, Maharshi Dayanand University, India
Neviaty Zamani, Bogor Agricultural University, Indonesia
Copyright © 2022 Palladino, Rampelli, Galià-Camps, Scicchitano, Trapella, Nanetti, Angelini, Cleo, Turroni, Corinaldesi and Candela. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Candela, bWFyY28uY2FuZGVsYUB1bmliby5pdA==