 Ashok Kumar Jangam1†
Ashok Kumar Jangam1† Vinaya Kumar Katneni1†
Vinaya Kumar Katneni1† Mudagandur S. Shekhar1*
Mudagandur S. Shekhar1* Sudheesh K. Prabhudas1
Sudheesh K. Prabhudas1 Karthic Krishnan1
Karthic Krishnan1 Jani Angel Jesudhas Raymond1
Jani Angel Jesudhas Raymond1 Krishna Sukumaran2Muniyandi Kailasam2
Krishna Sukumaran2Muniyandi Kailasam2 Joykrushna Jena3
Joykrushna Jena3- 1Nutrition Genetics and Biotechnology Division, ICAR-Central Institute of Brackishwater Aquaculture, Chennai, India
- 2Finfish Culture Division, ICAR-Central Institute of Brackishwater Aquaculture, Chennai, India
- 3Fisheries Science Division, Indian Council of Agricultural Research, New Delhi, India
Introduction
Mugil cephalus, commonly known as flathead grey mullet, is an important candidate brackish water food fish species of herbivorous nature positioned at lower trophic levels of the food chain. The biology, genetics, ecology, and fishing aspects of grey mullet were thoroughly reviewed by Whitfield et al. (2012). In the Indian context, though captive broodstock of grey mullet was developed about two decades back (Abraham et al., 2000), large-scale successful induced breeding trials were realized only recently (Sukumaran et al., 2021). Further, the reproductive period of captive grey mullet is reported to be asynchronous between the Eastern and Western Coast stocks of India. The peak reproductive periods are November and June–August for Eastern and Western grey mullet captive stocks, respectively (Sukumaran et al., 2021). A year-round commercial seed production with captive broodstock could not be realized till date due to constraints in captive maturation and seasonal spawning behavior of the species. The availability of genomic and transcriptomic resources would lead to a better understanding of the regulatory pathways involved in captive maturation and breeding of grey mullet. Such information would also facilitate the conduct of functional studies and derive meaningful inferences out of them for the improvement of grey mullet.
Conventionally, transcriptome resources are established by sequencing RNA on an Illumina sequencer wherein the short RNA sequence reads are assembled to generate full-length transcripts. With the development of Pacific Biosciences (PacBio) Single Molecule Real-Time (SMRT) sequencing, the full-length isoform-level transcript sequences could be generated directly without the necessity of assembly. Recently, the PacBio Isoform Sequencing (IsoSeq) based full-length transcript resources were made available for candidate brackish water aquaculture species (Zhang et al., 2019; Katneni et al., 2020; Pootakham et al., 2020) with potential applications to an understanding of economic traits and role in genome annotation. Furthermore, the IsoSeq transcripts can be easily annotated (Zeng et al., 2018) and would help in the discovery of splice sites and structural annotation of the genome.
For grey mullet, transcript information based on short-read sequence datasets is available (Byadgi et al., 2016; Dor et al., 2020), but no full-length transcript resource is available. So far, a full genome is available for only the red lip mullet (Liyanage et al., 2019; Zhao et al., 2021) among fishes in the family Mugilidae. For the first time, we report here a transcriptome resource for grey mullet based on the PacBio IsoSeq reads using nine different adult tissues and five different developmental stages.
Methods
Sample Collection and RNA Extraction
About 9 tissues (muscle, gills, liver, kidney, spleen, heart, stomach, intestine, and gonad) from adult grey mullet and whole specimens from five early developmental stages, including eggs, 3/12/24/55-day old larvae, were collected from the Muttukadu experimental station. The samples were collected following the guidelines approved by the Institutional Animal Ethics Committee of ICAR—Central Institute of Brackishwater Aquaculture (CIBA/IAEC/2021-19). The species identity of the adult specimen is confirmed with the partial sequence of the barcoding gene, cytochrome C oxidase I (GenBank accession: MW584357). All the samples were flash frozen in liquid nitrogen and stored at a temperature of −80°C. The total RNA was extracted using the conventional TRIzol method and its quantity was measured using a Qubit 3.0 fluorometer (Thermofisher #Q33238, Thermofisher Scientific, Massachusetts, USA) using an RNA HS assay kit (Thermofisher #Q32851, Thermofisher Scientific, Massachusetts, USA). The integrity of RNA was evaluated on a 1% agarose gel and an Agilent 2100 Bioanalyzer (Agilent Technologies, California, USA).
Illumina Library Preparation and Sequencing
To generate Illumina compatible RNAseq libraries, 1 ug of total RNA was used for Illumina library construction according to the instructions of the manufacturer using the NEBNext II RNA Library Prep Kit for Illumina® (New England Biolabs Inc., Massachusetts, USA). The libraries were quantified with a Qubit 3.0 fluorometer (Thermofisher Scientific, Massachusetts, USA) using a DNA HS assay kit (Thermofisher Scientific, Massachusetts, USA). The insert size of the libraries was estimated by querying on Tapestation 4150 using high-sensitivity D1000 screentapes (Agilent Technologies, California, USA) following the protocol of the manufacturer. Sequencing of libraries was carried out on an Illumina NovaSeq 6000, S4 Flow Cell (2 × 150 bp read lengths).1, 2
Pacbio IsoSeq Library Preparation and Sequencing
The total RNA of the same 9 tissues and 5 developmental stages was pooled in equimolar concentration and subjected to cDNA synthesis and amplification using the NEBNext® Single Cell/Low Input cDNA Synthesis & Amplification Module (New England Biolabs Inc., Massachusetts, USA) in conjunction with the Iso-Seq Express Oligo Kit (Pacific Biosciences, California, USA). The Pronex beads (Promega, Wisconsin, USA) were used for the purification of the cDNA before amplification and later for size selection of the amplified product. The library was constructed using the SMRTbell Express template Preparation Kit 2.0 (Pacific Biosciences, California, USA) as per the protocol of the manufacturers. The library was purified using Pronex beads (Promega, Wisconsin, USA) and the library size was assessed using a Bioanalyzer (Agilent Technologies, California, USA). About 70 pM of the library was loaded onto 8M SMRTcell and sequenced in the PacBio Sequel II system in CCS/HiFi mode.
RNAseq Based De Novo Transcript Assembly
Good quality RNAseq reads after trimming for poor quality bases and adapters using trimmomatic v0.39 (Bolger et al., 2014), were analyzed in Trinity v2.12.0 (Grabherr et al., 2011) to generate a de novo transcript assembly containing 1,051,143 transcripts. Then the transcripts were processed with TransDecoder v5.5.01 to predict the coding regions in which only one open reading frame (ORF) per transcript that is longer than 100 amino acids was considered further. The selected transcripts were clustered with a similarity threshold of 95% in CD-HIT-EST v4.6 (Li and Godzik, 2006) with default parameters to obtain a final set of 214,899 non-redundant transcripts.
PacBio IsoSeq Based Full-Length Transcripts
Pacbio Isoform sequencing has resulted in total data of about 118.23 Gb, comprising 37.56 million subreads (Table 1). The Iso-Seq3 pipeline from SMRTLink v10.1 was used for data analysis. The Circular Consensus Sequence (CCS) reads were obtained from total data by calling ccs step with parameters as minimum passes = 3 and minimum quality = 0.99, which were further demultiplexed using lima with parameters peek-guess, dump-clips, and dump-removed. The full length non-concatamer sequences (FLNC) were obtained by calling the isoseq3 refine step with parameters require-polya and min-rq = 1. The FLNC reads were further clustered to obtain 87,286 high-quality isoforms with an N50 length of 3,736 bp. Then, Mash v2.2 (Ondov et al., 2019) was used to screen for any possible contaminants with the refseq genomes sketch. Thereafter, the redundant transcript models were collapsed using cDNA_Cupcake v. 28.0.02 (MIN_ALN_COVERAGE = 0.85; MIN_ALN_IDENTITY = 0.95) to obtain 66,538 non-redundant transcripts. While collapsing the isoforms, the differences in 5′ end were ignored. These transcripts were further analyzed in SQANTI 3 v. 4.2 (Tardaguila et al., 2018) to identify 24,102 unique gene models in the M. cephalus transcriptome. The IsoSeq-based transcript assembly was found to be 74.5 and 68.1% complete on assessment using the BUSCO (Seppey et al., 2019) module of OmicsBox v2.0.36 (Omicsbox, 2022) against eukaryote_odb10 (10 September 2020) and actinopterygii_odb10 (5 August 2020) orthologous databases (Kriventseva et al., 2019) respectively (Supplementary Figure 1).

Table 1 Summary of Isoform sequencing and transcriptome of M. cephalus.
PacBio IsoSeq Transcripts Annotation
The full-length coding transcript sequences were subjected to homology-based annotation using blastx (Altschul et al., 1990) against the Actinopterygii (txid7898) cohort of non-redundant protein databases. The protein domain and orthology-based annotation was performed using the Interprosan and EggNOG mapper modules of OmicsBox v2.0.36 (Omicsbox, 2022). These annotations were merged and the final gene ontology based annotation and enzyme code mapping were obtained using OmicsBox v2.0.36 (Omicsbox, 2022). The pathway details of the annotated transcripts was obtained by mapping them against the KEGG database (Kanehisa and Goto, 2000). The blastx hits were obtained for 22,515 (93.42%) transcripts, from which 20,577 (85.38%) were functionally annotated (Table 1, Supplementary Figure 2). Other teleost fishes, Stegastes partitus, Amphiprion ocellaris, Siniperca chuatsi, Lates calcarifer, and Toxotes jaculatrix, were the top-hit species during the blastx search (Supplementary Figure 3). Enzyme codes were obtained for 9,055 (37.57%) of the functionally annotated transcripts. Transferases and hydrolases were the most dominant enzyme classes expressed (Supplementary Figure 4). Gene ontology (Supplementary Figure 5) revealed that the most expressed GO categories were cellular protein metabolic processes (Biological processes), metal ion binding (Molecular function), and intracellular membrane-bound organelles (Cellular components). Based on the InterProScan search, the major domains, families, repeats and sites in the M. cephalus transcriptome were immunoglobulin-like domain, P-loop containing nucleoside triphosphate hydrolase, leucine-rich repeats, and IQ motif, EF-hand binding site, respectively (Supplementary Figures 6–9). For about 69.5% of the transcripts, GO annotations were obtained through EggNOG mapper search while assigning 12.48, 43.95, and 11.51% of COG categories to information storage and processing, cellular processes and signaling, and metabolism, respectively. The SSR analysis of the IsoSeq transcripts using the MISA v1.0 tool identified 47,394 SSRs (Supplementary Figure 10). The monomeric SSRs were most abundant (55.76%), followed by trimeric SSRs (22.92%).
Isoform Diversity
The analysis of full-length transcripts in SQANTI 3 v. 4.2 (Tardaguila et al., 2018) indicated that the 24,102 unique gene models in the M. cephalus transcriptome produce 66,538 unique isoforms of different categories (Figure 1A). Of these, 15.8% (10,525) of isoforms were categorized as Full Splice Match (FSM) as these match with the reference transcript at all the splice junctions while 14.39% (9,576) were classified as Incomplete Splice Match (ISM) as these match with consecutive but not all splice junctions as that of the reference transcripts. About 44.34% of isoforms, were categorized as novel transcripts coded by known genes. Here, a few of them were Novel in Catalog (NIC) isoforms which have either known splice junctions in different combinations or novel splice junctions from known donors and acceptors. A majority of the novel transcripts were categorized as Novel Not in Catalog (NNC), which used alternative donors/acceptors. Another category of novel transcripts that deviated from the splicing pattern of the reference transcripts was intergenic (14.09%), where the entire transcript locus is outside the boundary of a reference gene. Other isoform categories detected were Genic genomic (3.91%; isoforms with partial exon and intron/intergenic overlap of a reference gene) and Fusion (6.38%; isoform spanning over two reference genes). About 60% of the genes were observed to produce only one isoform, and the rest of the genes produced more than one isoform, of which 4.88% (1,176) had more than 10 detected isoforms (Figure 1B). A higher proportion of genes with more isoforms indicated a high degree of transcriptome complexity of the M. cephalus transcriptome. In addition, out of the splice junctions detected in M. cephalus genes, 96.78% (183,204) are canonical with the standard GT-AG (also GC-AG and AT-AC) intron flanking sequence at splice sites while 3.22% (6,088) are non-canonical, having rare intron splice sites (Supplementary Table 1). The splice junctions detected for all of the FSM, ISM, and NIC isoforms were canonical, while 17.5% of NNC isoforms were found to have non-canonical splice junctions (Supplementary Figure 11). We have also verified the splice junctions detected in IsoSeq transcripts with the help of RNAseq reads generated in this study. The splice junctions of more than 99% of FSM, ISM, and NIC isoforms were supported by RNAseq reads, while the splice junctions of only 79.6% of NNC isoforms had support from RNAseq reads (Supplementary Figure 12). The unverified splice junctions could be either false or the result of tissue-specific gene expression. In addition, we have used SUPPA2 (Trincado et al., 2018) with default parameters to estimate the possible alternative splicing (AS) events in the M.cephalus transcriptome. We have identified 25,983 AS events in 5,903 genes, of which 18.51% are A3/A5 (alternative 3′ and 5′ splice sites), 42.64% are AF/AL (alternative first and last exons), 14.73% are SE (skipping exon), 23.09% are RI (retained intron), and 1.04% are MX (mutually exclusive exon) events (Figure 1C).
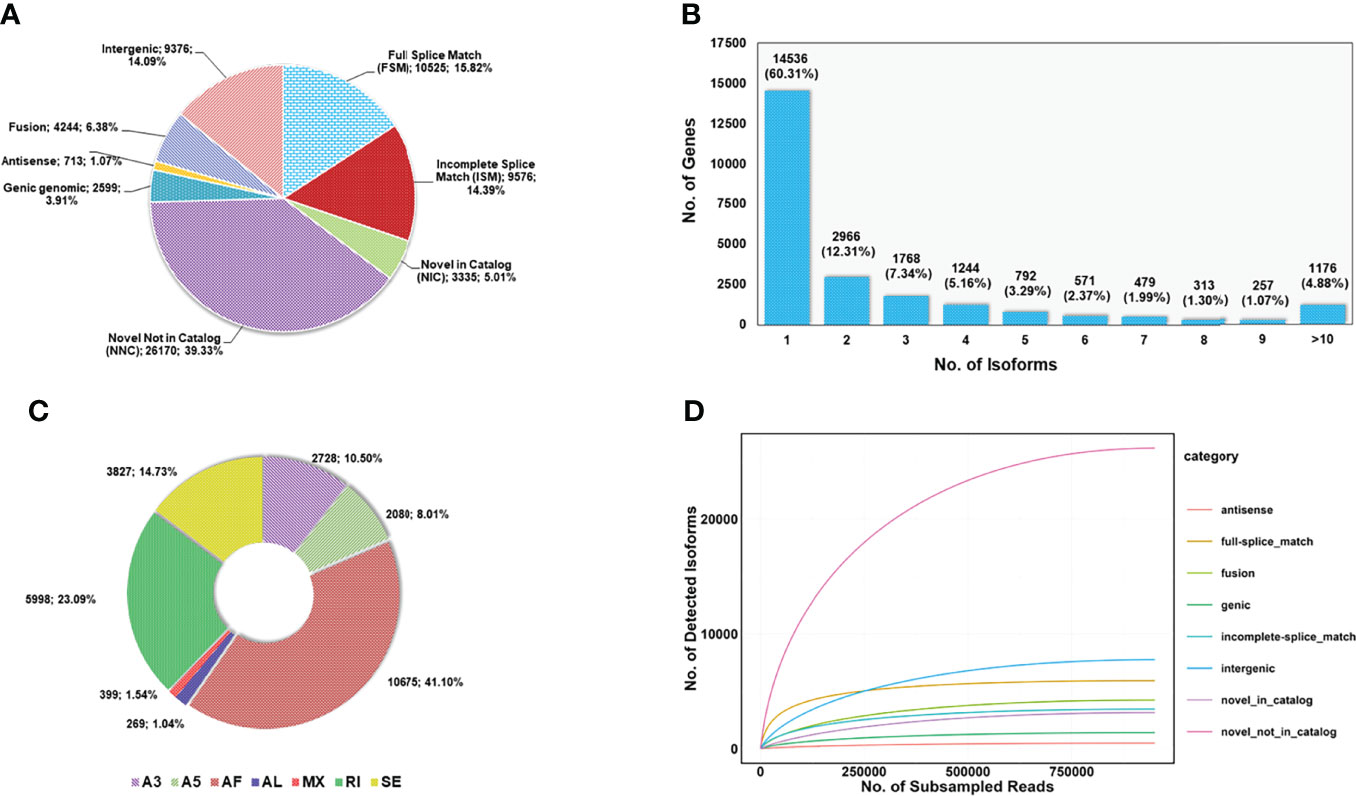
Figure 1 Description of isoform-level transcript features in the M. cephalus transcriptome. (A) Different categories of isoforms identified in M. cephalus transcriptome. (B) Bar plot indicating the number of genes in the M. cephalus transcriptome capable of producing single or multiple isoforms. (C) Various types of alternative splicing events discovered for genes in the M. cephalus transcriptome. (D) Rarefaction curve depicting the sequencing depth required for the discovery of different categories of isoforms.
Rarefaction Curve Analysis
A rarefaction curve (RC) analysis was performed to assess the sufficiency of sequencing toward the discovery of genes and isoforms. The RC analyses were executed with the collapsed transcripts by following the cDNA_Cupcake scripts2, subsample.py (min_fl_count = 2; step = 1,000) and subsample_with_category.py. While the former script was used to subsample at the gene and isoform levels, the latter script was used to sample various isoform-level categories. For M. cephalus IsoSeq data, it was observed that the discovery of isoforms required a higher sequence depth than the discovery of genes to reach saturation (Supplementary Figure 13). Among the various isoform categories identified by Squanti3, the NNC type of isoform needed more sequencing depth to reach saturation (Figure 1D) than any other category of isoform. The tissue-specific expression and low abundance nature of NNC isoforms would explain the necessity of a higher sequence depth to detect them. Overall, the RC analyses indicated the sufficiency of the sequencing data generated in this study toward the detection of genes and isoforms.
Re-Use Potential
For the first time, an isoform-level, full-length transcriptome resource was generated for grey mullet based on the Pacbio Iso-Sequencing using nine different adult tissues and five different developmental stages. The full-length transcripts are of great value for annotation of the M. cephalus genome, the conduct of functional studies, and to support annotations of other mullet fish genomes. The transcript assembly has the potential to contribute to a better understanding of the regulatory pathways involved in the captive maturation and breeding of grey mullet.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA675305, https://figshare.com/, https://doi.org/10.6084/m9.figshare.19634463.v1.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Ethics Committee of ICAR-Central Institute of Brackishwater Aquaculture (CIBA/IAEC/2021-19).
Author Contributions
MS, MK, and JJ conceived and designed the study and experiments. JA and KS collected the samples. SP, KK, AJ, and VK performed bioinformatics analysis. AJ and VK wrote manuscript with inputs from all coauthors. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
The authors acknowledge the funding support from the ICAR-CRP on Genomics project, “Genomic resources for augmentation of economic traits in Indian white shrimp Penaeus indicus and whole genome sequencing of brackishwater aquaculture candidate species”.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the Director of ICAR-CIBA for providing the necessary infrastructure and support for the study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.930017/full#supplementary-material
Footnotes
References
Abraham M., Kailasam M., Kishore Chandra P., Sihranee P., Rajendran K. V., others (2000). Development of Captive Broodstock of the Grey Mullet, Mugil Cephalus (L). Indian J. Fish 47, 91–96.
Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990). Basic Local Alignment Search Tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Byadgi O., Chen Y.-C., Barnes A. C., Tsai M.-A., Wang P.-C., Chen S.-C. (2016). Transcriptome Analysis of Grey Mullet (Mugil Cephalus) After Challenge With Lactococcus Garvieae. Fish \& Shellfish Immunol. 58, 593–603. doi: 10.1016/j.fsi.2016.10.006
Dor L., Shirak A., Curzon A. Y., Rosenfeld H., Ashkenazi I. M., Nixon O., et al. (2020). Preferential Mapping of Sex-Biased Differentially-Expressed Genes of Larvae to the Sex-Determining Region of Flathead Grey Mullet (Mugil Cephalus). Front. Genet. 839. doi: 10.3389/fgene.2020.00839
Grabherr M. G., Haas B. J., Yassour M., Levin J. Z., Thompson D. A., Amit I., et al. (2011). Trinity: Reconstructing a Full-Length Transcriptome Without a Genome From RNA-Seq Data. Nat. Biotechnol. 29, 644. doi: 10.1038/nbt.1883
Kanehisa M., Goto S. (2000). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Katneni V. K., Shekhar M. S., Jangam A. K., Prabhudas S. K., Krishnan K., Kaikkolante N., et al. (2020). Novel Isoform Sequencing Based Full-Length Transcriptome Resource for Indian White Shrimp, Penaeus Indicus. Front. Mar. Sci. 7. doi: 10.3389/fmars.2020.605098
Kriventseva E. V., Kuznetsov D., Tegenfeldt F., Manni M., Dias R., Simão F. A., et al. (2019). OrthoDB V10: Sampling the Diversity of Animal, Plant, Fungal, Protist, Bacterial and Viral Genomes for Evolutionary and Functional Annotations of Orthologs. Nucleic Acids Res. 47, D807–D811. doi: 10.1093/nar/gky1053
Li W., Godzik A. (2006). Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Liyanage D. S., Oh M., Omeka W. K. M., Wan Q., Jin C. N., Shin G.-H., et al. (2019). First Draft Genome Assembly of Redlip Mullet (Liza Haematocheila) From Family Mugilidae. Front. Genet. 10, 1246. doi: 10.3389/fgene.2019.01246
Omicsbox (2022). OmicsBox-Bioinformatics Made Easy (Version 2.0.36). (Valencia, Spain: BioBam Bioinformatics). Available at: https://www.biobam.com/omicsbox/.
Ondov B. D., Starrett G. J., Sappington A., Kostic A., Koren S., Buck C. B., et al. (2019). Mash Screen: High-Throughput Sequence Containment Estimation for Genome Discovery. Genome Biol. 20, 1–13. doi: 10.1186/s13059-019-1841-x
Pootakham W., Uengwetwanit T., Sonthirod C., Sittikankaew K., Karoonuthaisiri N. (2020). A Novel Full-Length Transcriptome Resource for Black Tiger Shrimp (Penaeus Monodon) Developed Using Isoform Sequencing (Iso-Seq). Front. Mar. Sci. 7, 172. doi: 10.3389/fmars.2020.00172
Seppey M., Manni M., Zdobnov E. M. (2019). “BUSCO: Assessing Genome Assembly and Annotation Completeness,” in Gene Prediction. Editor M. Kollmar (Humana, NY: Springer), 227–245. doi: 10.1007/978-1-4939-9173-0_14
Sukumaran K., Thomas D., Rekha M. U., Angel J. R. J., Bera A., Mandal B., et al. (2021). Reproductive Maturation and Induced Breeding of Two Geographical Groups of Grey Mullet, Mugil Cephalus Linnaeu. Aquaculture 536, 736423. doi: 10.1016/j.aquaculture.2021.736423
Tardaguila M., de la Fuente L., Marti C., Pereira C., Pardo-Palacios F. J., Del Risco H., et al. (2018). SQANTI: Extensive Characterization of Long-Read Transcript Sequences for Quality Control in Full-Length Transcriptome Identification and Quantification. Genome Res. 28, 396–411. doi: 10.1101/gr.222976.117
Trincado J. L., Entizne J. C., Hysenaj G., Singh B., Skalic M., Elliott D. J., et al. (2018). SUPPA2: Fast, Accurate, and Uncertainty-Aware Differential Splicing Analysis Across Multiple Conditions. Genome Biol. 19, 1–11. doi: 10.1186/s13059-018-1417-1
Whitfield A. K., Panfili J., Durand J.-D. (2012). A Global Review of the Cosmopolitan Flathead Mullet Mugil Cephalus Linnaeus 1758 (Teleostei: Mugilidae), With Emphasis on the Biology, Genetics, Ecology and Fisheries Aspects of This Apparent Species Complex. Rev. Fish Biol. Fish 22, 641–681. doi: 10.1007/s11160-012-9263-9
Zeng D., Chen X., Peng J., Yang C., Peng M., Zhu W., et al. (2018). Single-Molecule Long-Read Sequencing Facilitates Shrimp Transcriptome Research. Sci. Rep. 8, 1–9. doi: 10.1038/s41598-018-35066-3
Zhang X., Li G., Jiang H., Li L., Ma J., Li H., et al. (2019). Full-Length Transcriptome Analysis of Litopenaeus Vannamei Reveals Transcript Variants Involved in the Innate Immune System. Fish \& Shellfish Immunol. 87, 346–359. doi: 10.1016/j.fsi.2019.01.023
Keywords: isoform sequencing, PacBio, Iso-Seq, RNAseq, flathead grey mullet, Mugil cephalus, transcriptome
Citation: Jangam AK, Katneni VK, Shekhar MS, Prabhudas SK, Krishnan K, Raymond JAJ, Sukumaran K, Kailasam M and Jena J (2022) Isoform Sequencing Based Transcriptome Resource for Flathead Grey Mullet (Mugil cephalus). Front. Mar. Sci. 9:930017. doi: 10.3389/fmars.2022.930017
Received: 27 April 2022; Accepted: 24 May 2022;
Published: 20 June 2022.
Edited by:
Andrew Stanley Mount, Clemson University, United StatesReviewed by:
Francisco Vargas-Albores, Consejo Nacional de Ciencia y Tecnología (CONACYT), MexicoDaniel Garcia-Souto, University of Vigo, Spain
Copyright © 2022 Jangam, Katneni, Shekhar, Prabhudas, Krishnan, Raymond, Sukumaran, Kailasam and Jena. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mudagandur S. Shekhar, TVMuU2hla2hhckBpY2FyLmdvdi5pbg==
†These authors have contributed equally to this work