 Yang Yang
Yang Yang Guohao Wang3
Guohao Wang3 Yaya Li
Yaya Li Jiabao Hu
Jiabao Hu Zhen Tao
Zhen Tao- 1Key Laboratory of Mariculture and Enhancement, Marine Fishery Institute of Zhejiang Province, Zhoushan, China
- 2Marine and Fishery Research Institute, Zhejiang Ocean University, Zhoushan, China
- 3School of Marine Science, Ningbo University, Ningbo, China
- 4School of Fisheries, Zhejiang Ocean University, Zhoushan, China
In teleost, follicle atresia is a common degenerative process that can occur at different stages of ovarian development. In this study, we depicted the cellular morphology of silver pomfret (Pampus argenteus) follicular atresia in detail and divided it into four different stages from Aα to Aδ stages based on the main cellular characteristics. High-throughput RNA sequencing was used to profile follicle atresia from Aα to Aδ stages, and many stage-specific genes were identified. In early atretic ovary, a great number of genes in cytokine-cytokine receptor interaction were obviously downregulated, suggesting that somatic cells might directly induce the follicle atresia by disrupting the normal conservation with germ cells. Meanwhile, the regulatory network of immune cell–related pathways was discovered in the process of atresia. The genes enriched in Th cell differentiation, leukocyte transendothelial migration, cholesterol metabolism, and so on were abundantly expressed, indicating that the immune cells play key roles in the process of in follicle atresia. Moreover, a model was proposed to illustrate how somatic cells mediate the process of follicle atresia. The study provides important insights into the molecular networks underlying follicle atresia in teleost.
Introduction
In teleost, oogenesis is an important biological phenomenon to generate haploid reproductive cells, i.e., eggs (Kagawa, 2013). More recently, emerging research found that fishes have another potential plastic response to unsuitable environmental conditions is to forego egg production until the subsequent spawning (referred to as “skipped spawning”) (Rideout and Tomkiewicz, 2011; Gonzalez-Kother et al., 2020). As a widespread phenomenon among fishes, three general forms of skipped spawning have been identified: retaining, reabsorbing, and resting (Rideout and Tomkiewicz, 2011). The reabsorbing, also known as follicle atresia, is a specific process in which a large number of vitellogenic oocytes were eliminated and reabsorbed prior to ovulation (Morais et al., 2012; Sales et al., 2019). In cultured fishes, follicular atresia may be an important problem when females showing large amounts of vitellogenic follicles failed to mature and ovulate during the spawning season. For example, in Microstomus pacificus, tilapia, and Solea senegalensis, major atresia contributes the females into reproductively “inactive” despite the ovaries still contained some advance yolked oocytes (Tingaud-Sequeira et al., 2009; Rideout and Tomkiewicz, 2011; Sales et al., 2019). In teleost, a plenty of literature have been reported on oogenesis (Kagawa, 2013; Reading et al., 2017; Biran and Levavi-Sivan, 2018). However, the process of follicular atresia remains mysterious and complex, and few quantitative data are available about follicular atresia, including the cellular and molecular aspects of degenerative processes of the oocytes.
Recent studies showed that although teleost fish has substantial diversity of reproductive strategies, the follicular atresia is a conserved dynamic process according to the morphological characteristics that can be divided into three stages: early, advanced, and late. Meanwhile, some main morphological events were observed in this process including the yolk degradation and reabsorption, hypertrophy of the follicular cells, and accumulation of autophagic vacuoles (Santos et al., 2005; Morais et al., 2012; Sales et al., 2019). However, to date, the molecular mechanism in regulating follicular atresia in teleost fish remains unclear. In mammals, apoptosis in follicular cells is termed as the main mechanism involved in mediating the ovarian follicle atresia (Matsuda et al., 2012; Tiwari et al., 2015). Nevertheless, in fish, apoptosis and autophagy of follicular is only observed in late stages of follicle atresia (Miranda et al., 1999; Morais et al., 2012), suggesting that another novel molecular pathway exists in the follicular atresia of fish. More recently, in teleost fishes, some studies also showed that lipid metabolism, oxidative metabolism, and immune cells play pivotal roles in the regulating of follicular atresia. Some related signal pathways and genes were also discovered in Solea senegalensis (Tingaud-Sequeira et al., 2009), Coho salmon (Yamamoto et al., 2016), and Sterlet sturgeon (Akhavan et al., 2016). However, a comprehensive investigation of stage-specific, related gene expression to illustrates that the molecular mechanism of follicular atresia in teleost fishes is currently lacking.
Silver pomfret (Pampus argenteus) is a widely distributed and economically important marine fish species in Japan, China, India, and Malaysia (Gu et al., 2021; Yang et al., 2021a). During spawning season, the fishes in aquaculture have a large number of follicle atresia, and egg fertilization is dramatically reduced, but no precise studies have been performed to analyze this process. In this study, we assessed the follicle atresia in detail based on morphological observation and divided it into four stages. In addition, we further profiled the transcriptome and gene expression dynamics at different stages of follicular atresia, and the results showed that the gonadal somatic cells might play pivotal roles in this process. Overall, this study aimed to uncover new molecular insights into regulating follicular atresia and to reveal the specific characteristics of follicular atresia of fish.
Materials and Methods
Ethics Statement
All experiments described in this study were approved by the Administration of Affairs Concerning Animal Experimentation guidelines from the Science and Technology Bureau of China and were conducted following the Animal Care Committee of Ningbo University. Experimental individuals of silver pomfret were reared indoors in square pools supplied with sand-filtered seawater with a temperature range from 21.3°C to 12.0°C, a dissolved oxygen level of >7.35 ± 0.05 mg/ml and a photoperiod of 12L:12D in the Xiangshan Bay Sci-Tech Co., Ltd., located at Ningbo, Zhejiang Province, China. A suitable food (50.3% crude protein and 16.1% crude lipid) was fed six times daily.
Sample Collection and Isolation
According to the order of gonadal development as described by Yang et al. (2021a), fish samples and mature eggs of silver pomfret were harvested during spawning season (from November to December 2020) to obtain females undergoing the whole atretic follicles regression. The sex of each fish and developmental stages (from oogonia to mature egg) of germ cells were determined by examining gonad tissue slices according to microscopic histological examination. Thirty-five females were selected and were deeply anesthetized with MS-222 (100 mg/L; Sigma-Aldrich, St. Louis, MO, USA) before dissection to minimize suffering. The ovaries were isolated and divided into a half fixed in Bouin’s fluid overnight for histology and a half quickly frozen in liquid nitrogen then stored at −80°C for RNA extraction.
Light Microscopy and Histological Examination
The fixed samples were dehydrated in a graded series of alcohol, cleared in xylene, embedded in paraffin wax, and sectioned into serial slices at 3–5 μm thicknesses. A portion of the sections was stained with hematoxylin and eosin, and the other portion of the samples was stained with Toluidine blue and 4′, 6-diamidino-2-phenylindole (DAPI) for immune cells and nucleus observation. All images were captured by Nikon Ni-E light microscopy equipped with a Nikon DS-Ri2 imaging system and a NIS-Elements BR 4.50.000 software. The number of theca cell and granulosa cell for each developmental stage of germ cell was determined by examining 20 randomly selected samples. Meanwhile, 10 germ cell diameters were measured for each germ cell type analyzed. Only germ cells, theca cells, and granulosa cells with prominent nuclear were measured on the basis of the method described by Yang et al. (2018) to ensure that the determination was more correct.
RNA-Seq and Data Processing
Sixteen samples of ovaries across five developmental stages of atretic follicles regression were harvested, and at least three biological replicates were harvested for each developmental stage. A total of 16 transcriptome profiles were constructed using Illumina TruSeqTM RNA sample preparation Kit and Illumina Novaseq 6000 sequencing platform at Shanghai Majorbio Bio-pharm Biotechnology Co., Ltd. (Shanghai, China). The raw paired end reads were trimmed and quality controlled by SeqPrep and Sickle with default parameters. The clean reads were obtained by filtering out any reads containing adaptor sequences, low-quality reads with an average quality score of less than 20, and reads with unknown base (N). Then, a de novo assembly was run with the clean reads to obtain unigenes using Trinity.
Expression Data Analysis
The unigenes were used for Basic Local Assignment Search Tool (BLAST) searches and annotation against the NCBI's Non-Redundant Proteins (NR), Clusters of Orthologous Groups of Proteins (COG), Gene Ontology (GO), Pfam, Swiss-Prot, and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. The expression level of each transcript was calculated to identify DEGs (differential expression genes) using the transcripts per million reads (TPM) method. RNA-Seq by Expectation Maximization (RSEM) was used to quantify gene abundances. The differential expression analysis was performed using the DESeq2 with Q-value ≤ 0.05 and DEGs with |log2FC| >1 were considered to be significantly different expressed genes. Functional enrichment analysis including GO and KEGG was performed to identify which DEGs were significantly enriched in GO terms and metabolic pathways at Bonferroni-corrected P-value ≤ 0.05. The gene coexpression networks were constructed via the system biology approach weighted gene coexpression network analysis (WGCNA). Before filter, there were 53,982 genes. After background correction and standardization of gene expression, abnormal and small variation genes (mean value of expression quantity <1 and coefficient of variation < 0.1) were filtered. Finally, 11,031 genes were clustered into 11 modules according to gene’s similar expression pattern. The short time-series expression miner (STEM) program with 50 sequential modes, maximum time interval = 1, and P-value ≤ 0.05 was used to analyze differentially expressed genes and identify temporal expression profiles. The protein–protein interaction networks were constructed using STRING database with Danio rerio as reference species. The data including WGCNA, STEM, and protein–protein interaction analysis were performed using the online platform of Majorbio Cloud Platform (www.majorbio.com).
Quantitative Analysis of Gene Expression
The gene expression analysis via quantitative real-time PCR (qRT-PCR) was performed with three biological replicates using the SYBR Premix Ex Taq Kit (Takara, Japan) according to the method in a quantitative thermal cycler (Quantagene, China) as described in our previous study (Gu et al., 2021; Yang et al., 2021a). The 18s and β-actin genes were used as the endogenous reference genes, and all primers were listed in Supplementary Table 1.
Immunofluorescence
The ATPase location analysis was detected using immunofluorescence method as described in our previous study (Yang et al., 2021a). The polyclonal antiserum of ATPase (zebrafish anti-rabbit ATPase, ABmart Biological Company, Hangzhou, China) was diluted to 1:200 and secondary antibody anti-rabbit Immunoglobulin G (IgG) conjugated with Alexa Fluor 488 (Abclonal, Wuhan, China) was diluted to 1:200. Nuclear DNA labeling was performed by 4′, 6-diamidino-2-phenylindole (DAPI), (1:500). The normal goat serum that replaced the primary antibody was conducted as the negative controls, and no signals were detected.
Statistical Analysis
All data in this study were expressed as the mean ± SEM, and the statistical analysis was performed using GraphPad Prism 9 software to detect significant difference with one-way analysis of variance (ANOVA) after Tukey’s HSD (honestly significant difference) test. post hoc test correction. P < 0.05 was considered to indicate a statistically significant difference.
Results
Morphological Hallmarks of Oogenesis and Follicular Atresia
As described in previous studies of most fish, at early follicle formation, oogonia are surrounded by a layer of somatic cells, which called follicle cells, and then developing oocyte become surrounded by a continuous granulosa cell layer and theca cell layer. To uncover the characterization of oogenesis of silver pomfret, we used histological examinations to analyze the morphology of follicular complex and to measure the dynamic changes of diameters of germ cells and numbers of somatic cells. The morphology of different germ cells types (from oogonia to mature egg) was displayed in Figure 1A and was distinguished on the basis of the morphological criteria established in our previous study (Yang et al., 2021a). The diameters of germ cells significantly increased from 10 μm at oogonia to 1,385 μm at mature egg. At late perinucleolar oocyte stage, both the continuous granulosa cells and theca cells appeared, and the numbers of those two types of somatic cells, especially the granulosa cells, exhibited an obvious increase during oogenesis.
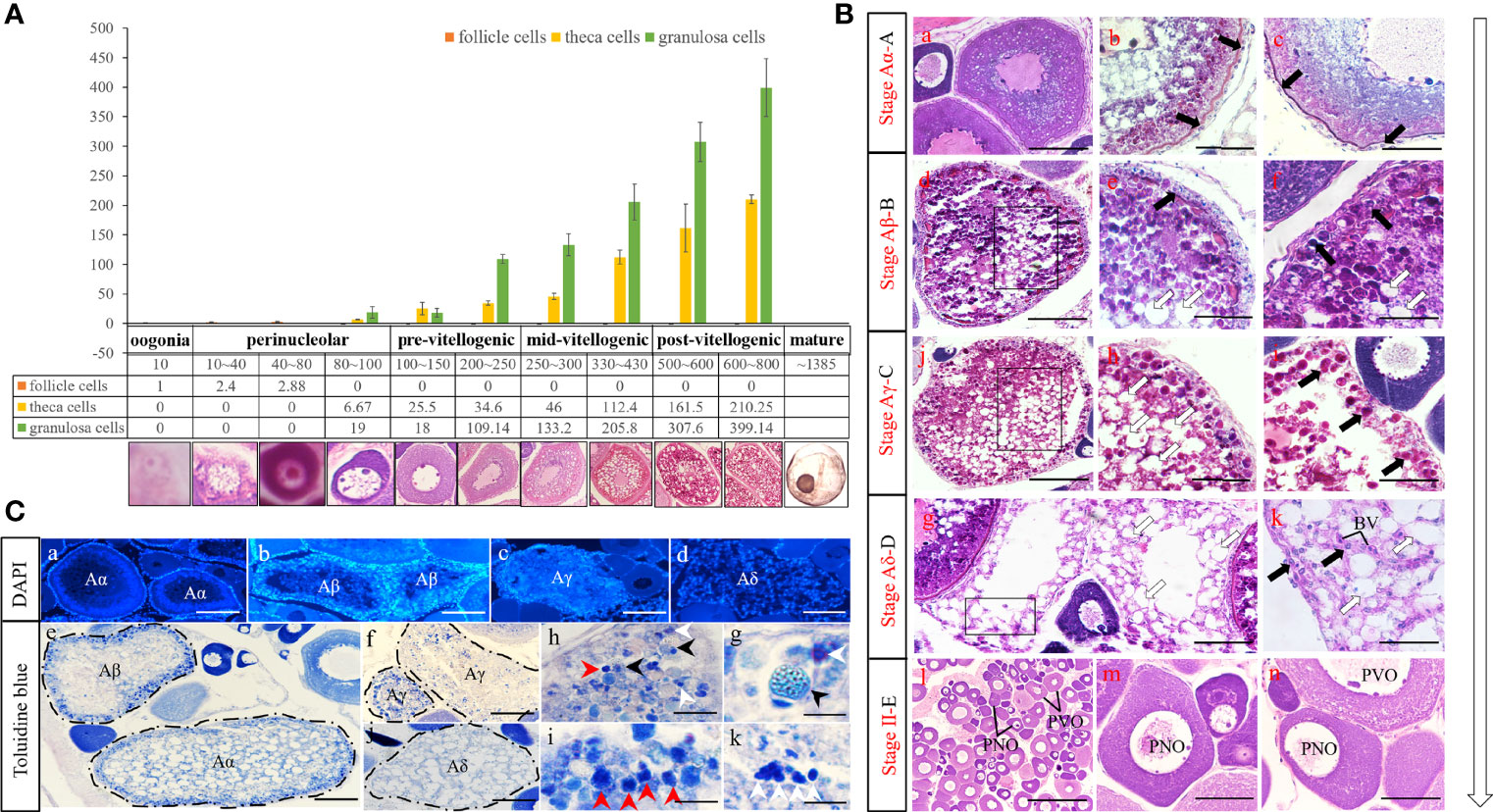
Figure 1 Statistic and morphological analysis of oogenesis and follicle atresia. (A) The number of somatic cells, and morphology and diameter of different germ cell from oogonia to mature egg stages of P. argenteus. (B) Five stages of P. argenteus follicle atresia collected for histological and RNA-seq analysis. White and black arrows indicate phagocytic vacuoles and somatic cells respectively. Scale bar = 200 μm (a, g, n), 150 μm (b, c, e), 250 μm (d, j, m), 100 μm (f, h, i, k), and 600 μm (l). (C) Histological identification of nucleus and immune cells stained by DAPI and Toluidine blue, respectively. White, black, and red arrows indicate mast cells, macrophages, and leucocytes, respectively. Scale bar = 300 μm (a), 200 μm (b, c, d), 400 μm (e, f, j), 35 μm (h), 20 μm (i, k), and 15 μm (g).
To reveal the cellular characteristics of follicular atresia, the gonadal tissue undergoing the whole atretic follicles regression was carefully observed in Figure 1B and divided into four stages, following the main morphological events of the regression of the vitellogenic follicles: Aα (initial atresia), Aβ (intermediate atresia), Aγ (advanced atresia), and Aδ (final atresia) stages. Although there was no statistical data in the present study, the atresia process was seen to be most frequent in vitellogenic follicles and of rare occurrence in previtellogenic follicles. At the Aα stage, the zona pellucida began to shrink and the granulosa cells hypertrophied progressively (Figure 1B, A–C) and still surrounded the oocytes (Figure 1C, A). At the Aβ stage, the yolk globules lost their integrity becoming liquefied (Figure 1B, D), the highly hypertrophied granulosa cells started to proliferate and fold inward the ooplasm (Figures 1B, e, f, c, b) and the zona pellucida initiated to tear and disintegrate (Figures 1B, e). Meanwhile, the phagocytic vacuoles were gradually formed and became numerous in ooplasm (Figures 1B, d–f). At the Aγ stage, the zona pellucida and yolk were almost completely engulfed and resorbed (Figures 1B, i, j), and the granulosa cells completely distributed in the follicular atresia (Figures 1C, c). At the Aδ stage, the granulosa cells were less numerous, and the atretic follicle appeared as a connective tissue scar at the end of the follicular regression process (Figures 1B, g–k, c, d). Completing the whole follicular atresia, the gonads developed into the perinucleolar and pre-vitellogenic stage (stage II, Figures 1B, l–n). In addition, the gonadal tissues were stained by Toluidine blue to reveal the immune cells during the process of follicular atresia. As shown in Figures 1C, e–k, the immune cells, which stained deep blue, gradually increased in Aβ and Aγ stage. Moreover, the macrophages, leucocytes, and mast cells were obviously recognized in follicular atresia (Figures 1C, h–k).
Global Analysis of the Transcriptome of Atretic Ovarian Tissues
To uncover the underlying molecular network that regulates follicular atresia of silver pomfret, we used RNA sequencing (RNA-seq) to analyze the transcriptomes of 16 samples with at least three biological replicates at five developmental stages of atretic follicles regression: Aα (AtresA), Aβ (AtresB), Aγ (AtresC), Aδ (AtresD), and stage II (AtresE). For 16 libraries, the information of raw reads, clean reads, mean Q30, and GC content in each sample was showed in Supplementary Table 2. After transcriptome assembly by Trinity, a total of 53,982 unigenes, which ranged from 201 to 17,632 bp and N50 length of 2,498 bp, were obtained, and the Benchmarking Universal Single-Copy Orthologs (BUSCO) score was 86.8%. The unigenes were annotated by alignment with public protein databases, such as Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Clusters of Orthologous Groups of Proteins (COG), NCBI's Non-Redundant Proteins (NR), Swiss-Prot, and Pfam (Supplementary Table 3). For transcriptome data, we normalized the gene expression levels with TPM method, and a high correlation was observed in the expression profiles of each sample, with the exception of AtresE2A (Figure 2A). Therefore, we deleted the AtresE2A and performed the principal component analysis (PCA) to get insight into the variability in the transcriptomics data. The first two principal components explained 14.6% and 12.2% of the variance among samples, respectively. Overall, PCA distinguished the developmental stages of atretic follicles regression (Figure 2B). However, some replicates within the same stage were not well clustered because the stage assignment of samples was only based on the histological traits. Moreover, the genes from five stages were combined, and a Venn diagram was used to reveal unique and commonly expressed genes among the ovaries (Figure 2C). A total of 13,444 genes were common among all five stages of ovarian tissues. Notably, among them, the AtresE had the least number of specific genes.
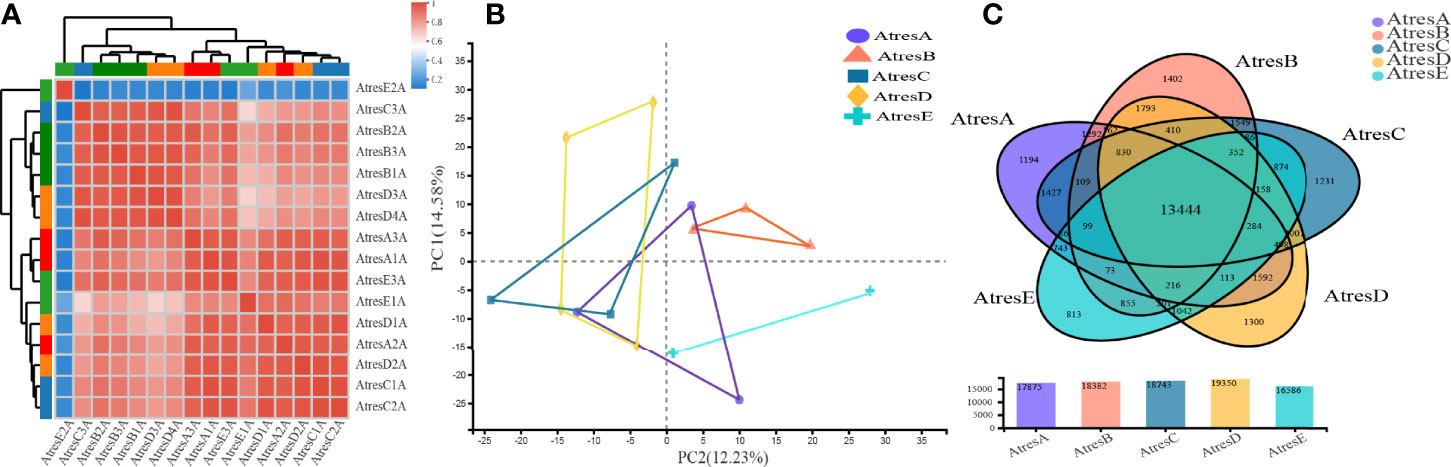
Figure 2 Global analysis of the transcriptomes. (A) Correction analysis of the expression profiles of 16 samples. (B) PCA analysis of 15 samples deleting the AtresE2A. (C) Venn diagram showing the number of shared and uniquely expressed genes among the five stages.
The significant DEGs between every two groups were identified with a padj value less than 0.05 and a log2FC higher than 2 or lower than 2 (Figure 3A). Among them, the largest number of DEGs was identified in AtresD compared with AtresE, of which 630 were downregulated and 79 were upregulated. The least number of DEGs was identified in AtresC compared with AtresD, of which five were downregulated and five were upregulated (Figure 3A). To discover the common and unique DEGs between the process and completion of atresia, a Venn diagram was set up using the DEGs of AtresA, AtresB, AtresC, and AtresD compared with AtresE, respectively (Figure 3B). Only three common DEGs were identified in all four compared groups, but 346 common DEGs were identified in both AtresC vs. AtresE and AtresD vs. AtresE. In addition, a Venn diagram of DEGs of AtresA vs. AtresB, AtresB vs. AtresC, and AtresC vs. AtresD was established to explore the common and unique DEGs in each stage during follicle atresia (Figure 3C). One common DEG was identified in the three compared groups, and the largest number of DEGs was 36 in AtresA vs. AtresB and AtresB vs. AtresC. This analysis indicated that the different regulation networks occur in each stage during follicle atresia, which was also consistent with the obvious morphological difference. However, the least number of DEGs in AtresC compared with AtresD revealed there was not identifiable gene expression divergence between these two stages during follicle atresia.
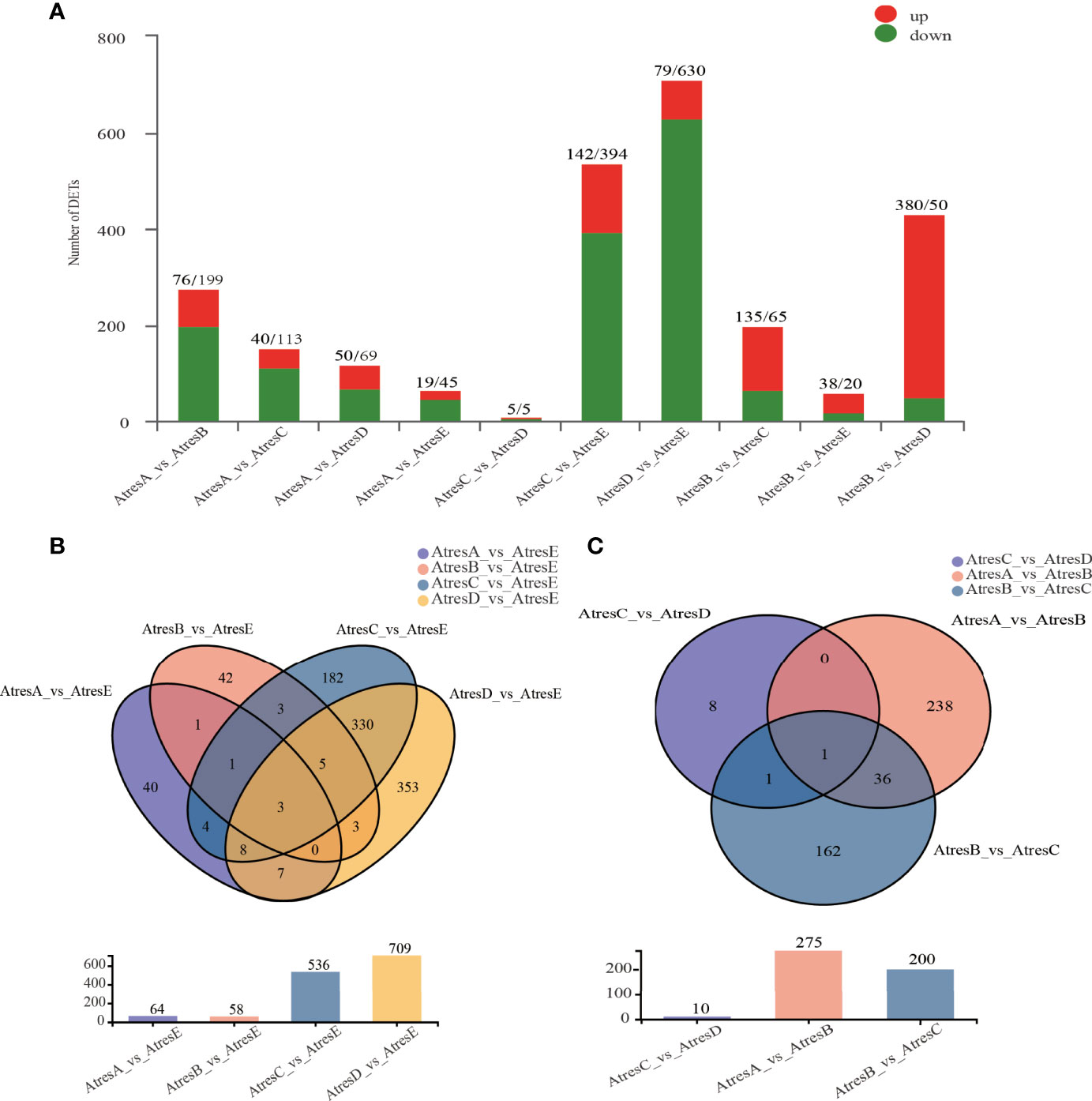
Figure 3 DEGs between five stages. (A) DEGs between every two stages. (B) Venn diagram of DEGs in AtresA, AtresB, AtresC, and AtresD compared with AtresE. (C) Venn diagram of DEGs in AtresA vs. AtresB, AtresB vs. AtresC, and AtresC vs. AtresD.
Coexpression Network Analysis Reveals Distinct Regulatory Programs Among P. argenteus Atretic Ovarian Tissues
To further explore the co-expressed gene networks across the follicle atresia, the WGCNA using 11,031 DEGs was used to identify highly connected gene subnetworks or modules. A total of 11 distinct modules, including one module in gray color reserved for genes outside of all modules, were designed in the WGCNA network (Figure 4 and Supplementary Figure 1). After correlation analysis with phenotypic data, four tissue-specific modules (pink, 109 genes; yellow, 738 genes; magenta, 103 genes; and turquoise, 2448 genes) were further analyzed (Figure 4). The four modules were individually subjected to KEGG enrichment analysis (Supplementary Figure 2). The genes coexpressed in the pink module (AtresA-specific module) were distinctively enriched in “ribosome”, “parkinson disease”, and “thermogenesis” biological processes. In the yellow module (AtresB-specific module), genes were significantly enriched in “choline metabolism in cancer”, “Th17 cell differentiation”, and “chronic myeloid leukemia” pathways. In the magenta module (AtresC-specific module), genes were mainly enriched in “herpes simplex virus 1 infection”, “D-glutamine and D-glutamate metabolism”, and “chagas disease (American trypanosomiasis)” biological processes. The genes in the turquoise module (AtresD- and E-specific modules) were enriched in “amoebiasis”, “AGE-RAGE signaling pathway in diabetic complications”, “pertussis”, “leukocyte transendothelial migration”, and so on biological processes. Furthermore, the most highly regulatory genes in the four specific modules were respectively analyzed to construct and visualize gene networks, in which each node presented a gene and the connecting lines between genes represented coexpression correlations (Figure 4 and Supplementary Table 4). The results revealed the Cox1 (TRINITY_DN1640_c0_g3), TRINITY_DN20436_c0_g1, TRINITY_DN655_c0_g1, and PTRF (TRINITY_DN8507_c0_g2) had the most connections in the network in the pink, yellow, magenta, and turquoise modules, respectively.
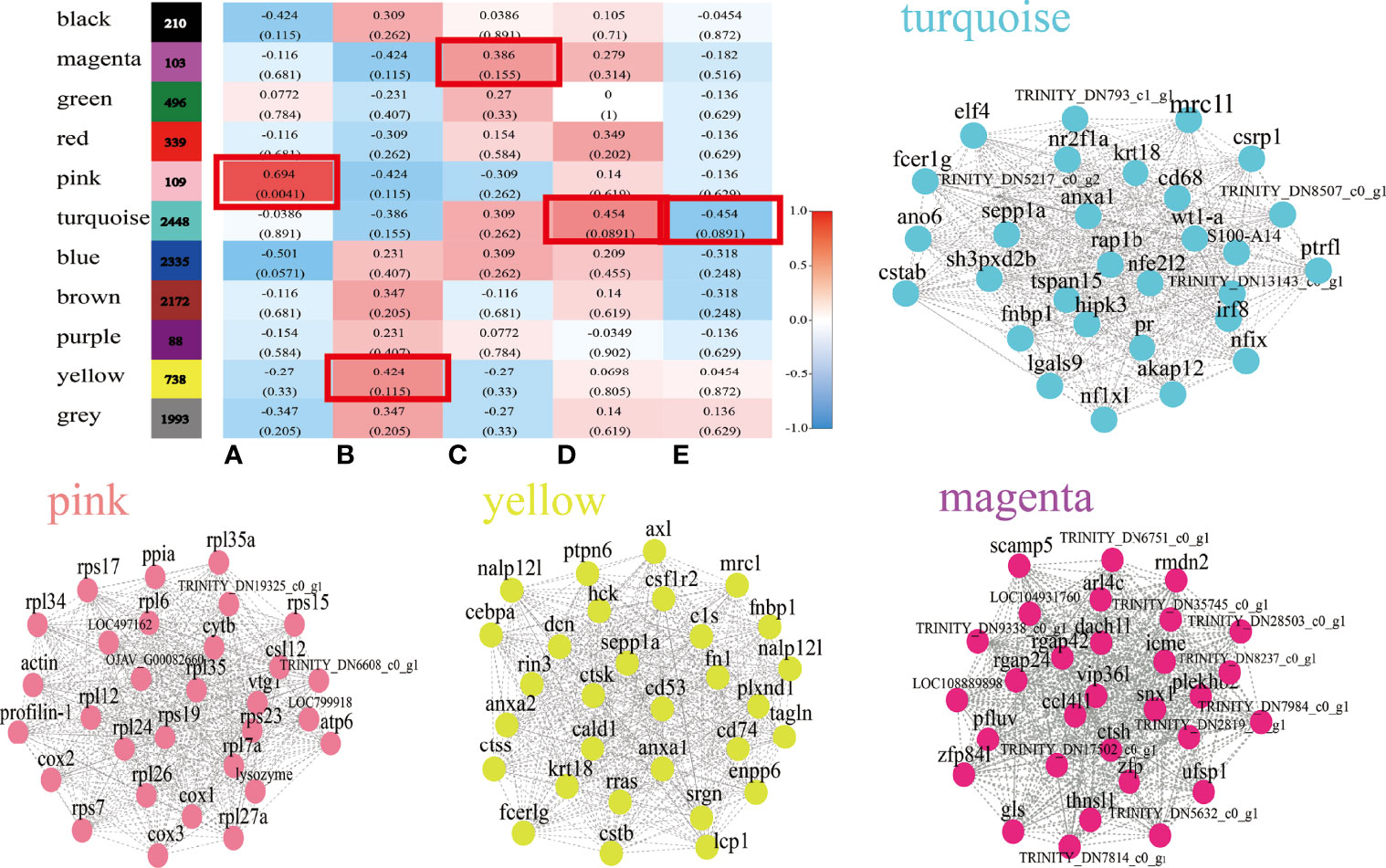
Figure 4 WGCNA analysis of DEGs. A total of 11 distinct modules and Module-phytohormone association in WGCNA. Each row corresponds to a module. Columns correspond to five stages. Visualize correlation networks of 30 DEGs in pink, yellow, magenta, and turquoise modules.
Dynamic Expression of DEGs Related to Follicular Atresia
To identify the key regulator genes between the process and completion of atresia, a total of 982 DEGs in AtresA, AtresB, AtresC, and AtresD compared with AtresE were analyzed for the enrichment of KEGG pathways. As shown in Figure 5A, the 20 most enriched pathways were related to specific biological processes. Among them, 11 most enriched pathways were analyzed for gene expression and protein interaction analysis. When most of the genes in enrichment pathways were the same, they were divided into one class; finally, three classes were obtained. To further identify the key regulator genes related to follicular atresia, the network of protein–protein interactions involved the three groups were constructed, respectively, in which each node represented a gene and the size of node represented the importance degree. The genes, which have the most importance degree in the networks, may be the key regulatory genes. As shown in Figure 5B, in the group composed of “cytokine-cytokine receptor interaction”, “viral protein interaction with cytokine and cytokine receptor”, “cell adhesion molecules (CAMs)”, and “focal adhesion”, of 68 genes, the hub genes included pecam1, fn1a, ptprc, itga5, and cxcr4b. In the group merged by “rheumatoid arthritis”, “staphylococcus aureus infection”, “tuberculosis”, and “phagosome”, of 35 genes, c1qa and cd74a are the central genes (Figure 5C). In the group composed of “transcriptional misregulation in cancer”, “hematopoietic cell lineage”, and “leukocyte transemdothelial migration”, of 43 genes, cxcr4b, pecam1, and mmp9 are the hub gene with the highest importance degree (Figure 5D). Those core genes’ differential expression was analyzed and constructed in hierarchical clusters to show the gene expression profiles during atresia (Figure 5E). In addition, all genes in the 11 enriched pathways differential expression were also display in Supplementary Figure 3. The results revealed that closely all DEGs had a similar expression pattern, with a high expression in AtresA, AtresC, and AtresD and a relative low expression in AtresB and AtresE.
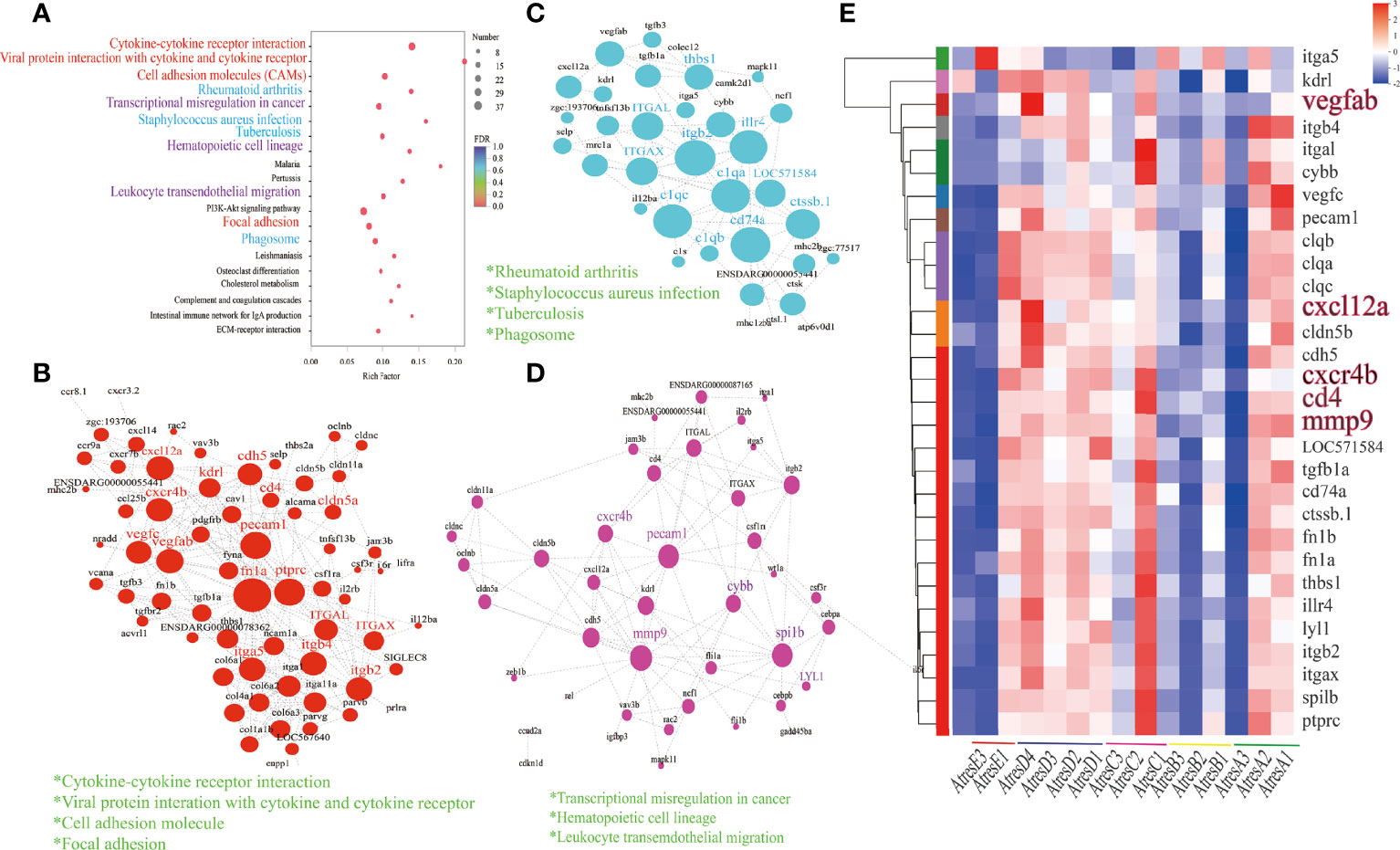
Figure 5 Functions, dynamic changes, and protein–protein interaction networks of DEGs among AtresA, AtresB, AtresC, and AtresD compared with AtresE. (A) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. (B) protein–protein interaction networks of DEGs enriched in “cytokine-cytokine receptor interaction”, “viral protein interaction with cytokine and cytokine receptor”, “cell adhesion molecule”, and “focal adhesion” pathways. (C) Protein–protein interaction networks of DEGs enriched in “rheumatoid arthritis”, “staphylococcus aureus infection”, “tuberculosis”, and “phagosome” pathways. (D) Protein–protein interaction networks of DEGs enriched in “transcriptional misregulation in cancer”, “hematopoietic cell lineage”, and “leukocyte transemdothelial migration” pathways. (E) Hierarchical clustering and expression analysis of core genes.
To categorize different gene expression profiles, the temporal variations in their expression of AtresA, AtresB, AtresC, and AtresD compared with AtresE were, respectively, analyzed to identify the cluster of genes that were most representative of the changes across the follicle atresia. The resulted showed that 14, 27, and 6 statistically significant model profiles (colored profiles) were identified (Figure 6A). The KEGG analysis revealed that “cytokine-cytokine receptor interaction” and “viral protein interaction with cytokine and cytokine receptor” were significantly enriched in profile 14 and 27 and that “parkinson disease”, “oxidative phosphorylation”, and “thermogenesis” were mainly enriched in profile 6 (Figure 6B).
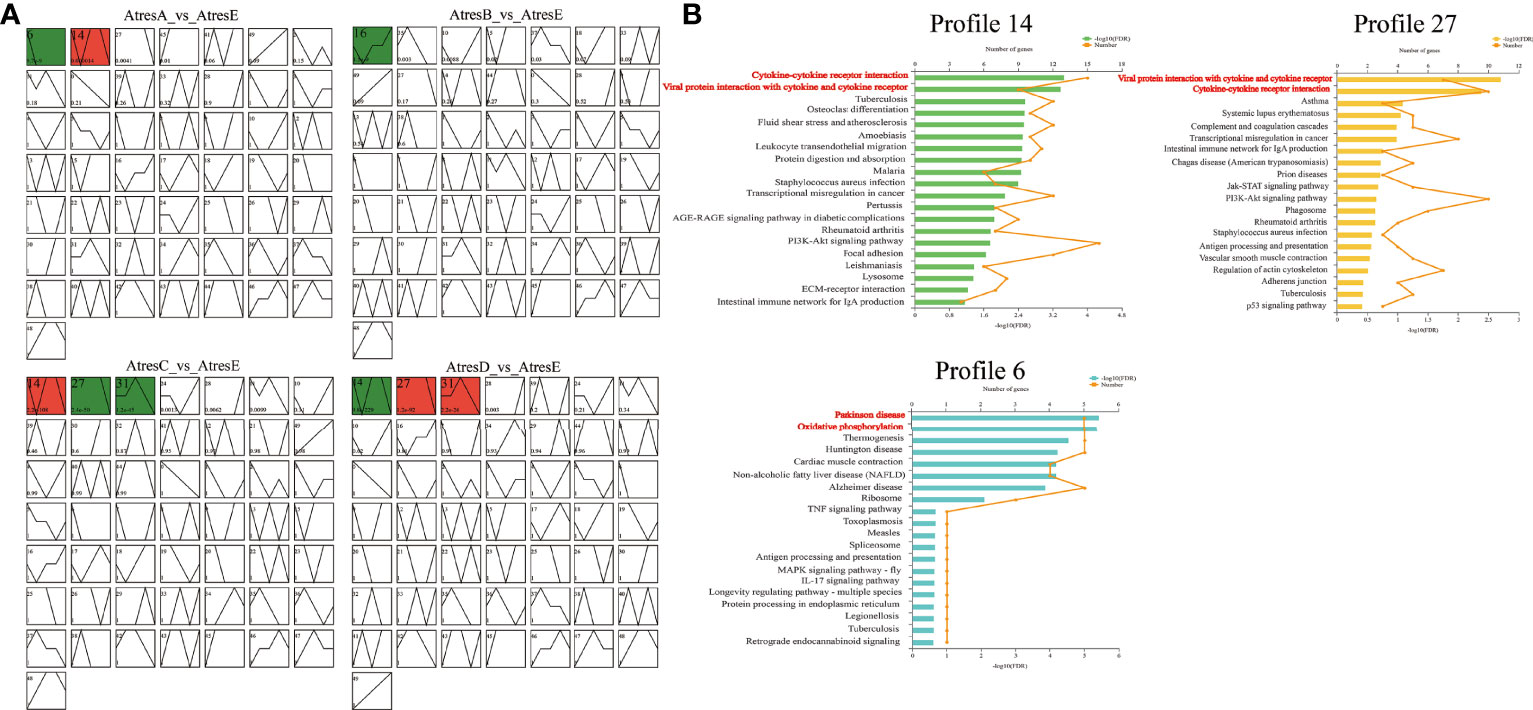
Figure 6 Module profiles and functional enrichment analysis of the genes in five stages of follicle atresia. (A) Module profiles of the genes in AtresA, AtresB, AtresC, and AtresD compared with AtresE, respectively. The STEM analysis shows genes expression trends. The broken line represents the trend of genes expression levels with time, the colored trend graphs are the significant change trends, and the same color represents the same cluster. (B) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis results for the genes clustered in profiles 14, 27, and 6 among the five stages.
Excavation of Key Genes During Follicular Atresia of P. argenteus
To uncover the expression patterns of genes during follicle atresia, the hierarchical clusters of the DEGs of AtresA vs. AtresB, AtresB vs. AtresC, and AtresB vs. AtresD using their differential expression were analyzed and constructed. Because only 10 DEGs existed in AtresC compared with AtresD, the replaced DEGs in AtresB vs. AtresD were maintained in this analysis. Hierarchical clustering of the DEGs revealed two groups of genes based on their expression trends from AtresA to AtresE, in which group 1 genes had the highest expression level in AtresA and little or no expression in other stages, group 2 genes exhibited the highest expression level in AtresC and AtresD, relative low expression in AtresA, and no expression in AtresB (Figure 7A). In addition, the clustering analysis was used to further divide the DEGs into different subclusers with clear and distinct expression profiles (Figure 7B). Genes in cluster 1 were distinctively expressed in AtresA, and genes in cluster 2, 3, and 5 were mainly expressed in AtresA, AtresC, and AtresD (Figure 7B). The KEGG pathways analysis showed that DEGs in group 1 were enriched in “ribosome”, “parkinson disease”, and “IL-17 signaling pathway” biological processes (Figure 7C). In group 2, the DEGs were mainly enriched in “pertussis”, “leukocyte transendothelial migration”, and “complement and coagulation cascades” biological processes (Figure 7D). The network of protein–protein interactions showed that closet all genes were the sub-genes in “ribosome” and “oxidative phosphorylation” pathways (Figure 7E); cxcr4b, cd4, mmp9, and cldn5b were sub-genes in “leukocyte transendothelial migration”, “Th1 and Th2 cell differentiation”, and “Th17 cell differentiation”; and clqa, CD68, clqe, apoc, and apoe were the central genes in “complement and coagulation cascades”, “lysosome”, and “choleterol metabolism” pathways (Figure 7F). Furthermore, the Gene Ontology (GO) analysis of genes in groups 1 and 2 was also formed (Supplementary Figure 4A) and depicted as directed acyclic graphs in Supplementary Figure 4B, which showed the relationships of the GO terms.
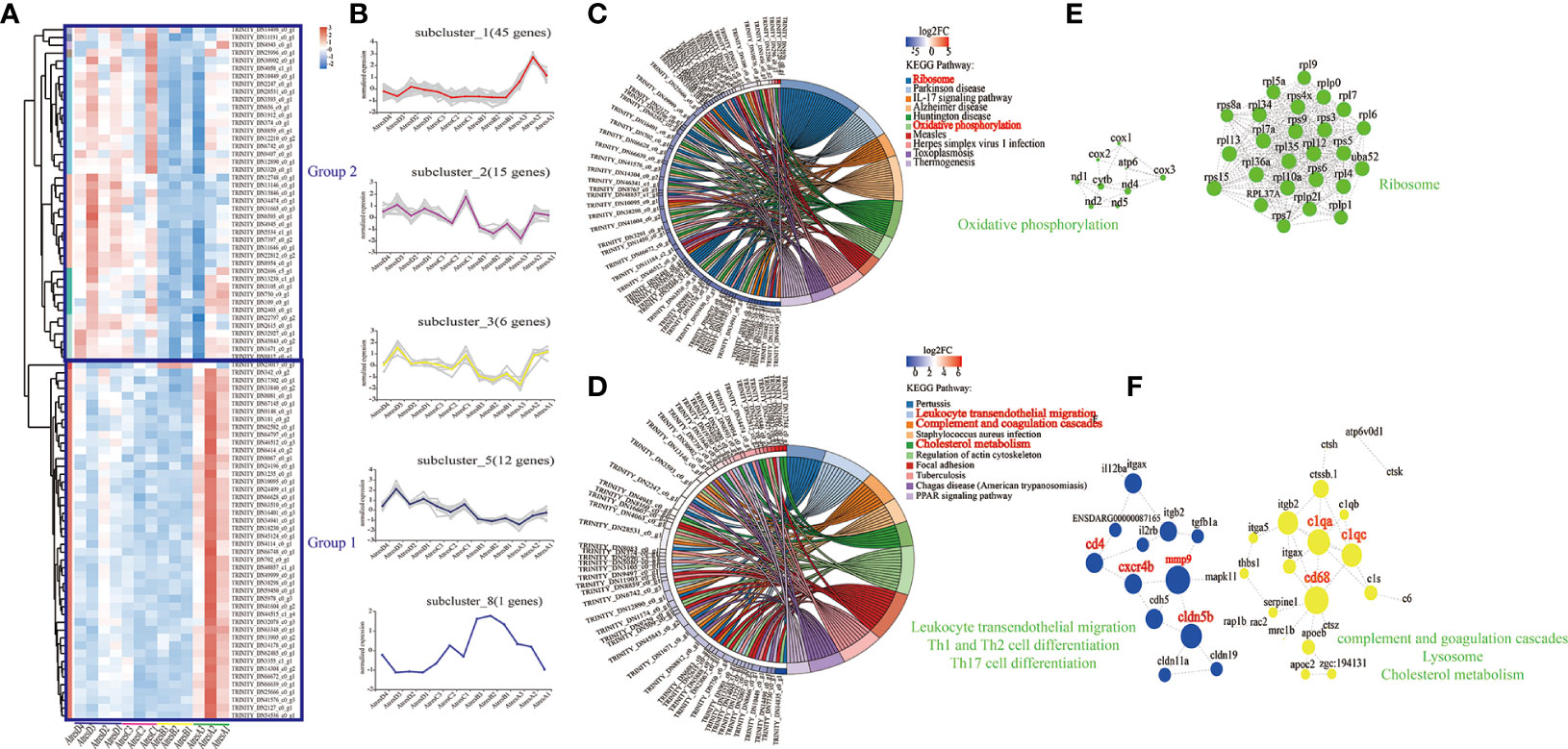
Figure 7 Functions, dynamic changes, and protein–protein interaction networks of DEGs among AtresA vs. AtresB, AtresB vs. AtresC, and AtresB vs. AtresD. (A) Hierarchical clustering showing two groups of the expression levels of DEGs at five stages. (B) Expression trends of genes in five clusters during five stages. (C) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of DEGs in group 1. (D) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of DEGs in group 2. (E) Protein–protein interaction networks of DEGs in group 1 enriched in “oxidative phosphorylation” and “ribosome” pathways. (F) Protein–protein interaction networks of DEGs in group 2 enriched in “leukocyte transendothelial migration”, “Th1 and Th2 cell differentiation”, “Th17 cell differentiation”, “complement and goagulation cascades”, “lysosome”, and “cholesterol metabolism” pathways.
Analysis of the Expression of DEGs at the mRNA Level
To evaluate the quality of the RNA-seq and the reliability of DEGs, we used qRT-PCR to quantify eight DEGs in different pathways from AtresA to AtresE, including cxcr4 and mapk11 in “leukocyte transendothelial migration”, rac2 and il12b in “Th17 cell differentiation”, ctb3 and cox2 in “oxidative phosphorylation”, and rps6 and rps3 in “ribosome”. As shown in Figure 8, the cxcr4, mapk11, rac2, and il12b had the same expression trends, with higher expression levels in AtresC and AtresD. Conversely, the ctb3, cox2, rps6, and rps3 exhibited higher expression levels in AtresA and AtresB. These results were consistent with the RNA-seq data analysis, demonstrating the reliability of RNA-seq data used to analyze differential mRNA expression.
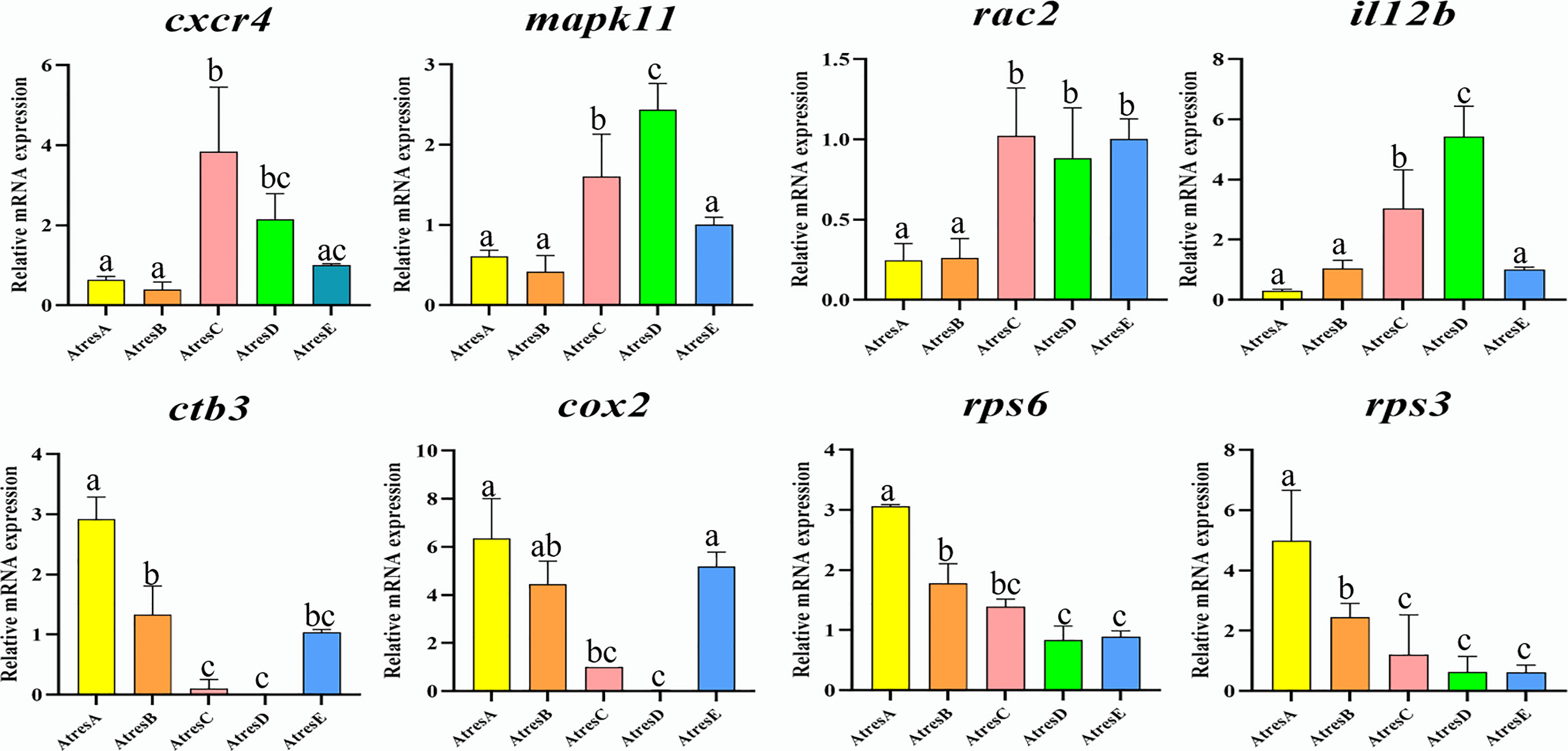
Figure 8 Validation of eight DEGs by qRT-PCR. Three samples in each stage and, finally, 15 samples were used in qRT-PCR analysis. Meanwhile, three replicates in each sample were performed. All data were expressed as the mean ± SEM, and the statistical analysis was performed using GraphPad Prism 9 software to detect significant difference with one-way analysis of variance (ANOVA) after Tukey’s HSD (honestly significant difference) test. post hoc test correction. P < 0.05 was considered to indicate a statistically significant difference.
Subcellular Localization of ATPase
Furthermore, a fluorescence immunohistochemistry analysis was used to characterize the expression pattern of ATPase with distinct expression change in follicle atresia. As shown in Figure 9A, ATPase was mainly located in the cytoplasm around the cell membrane of oocyte in Aα. With the process of atresia, the protein levels of the putative ATPase continuously decreased and nearly disappeared in Aδ. When the atresia was completed, appreciable expression of ATPase distributed in the cytoplasm region around the cell membrane of pre-vitellogenic oocyte (stage II). This expression pattern of ATPase was consistent with the RNA-seq data analysis.
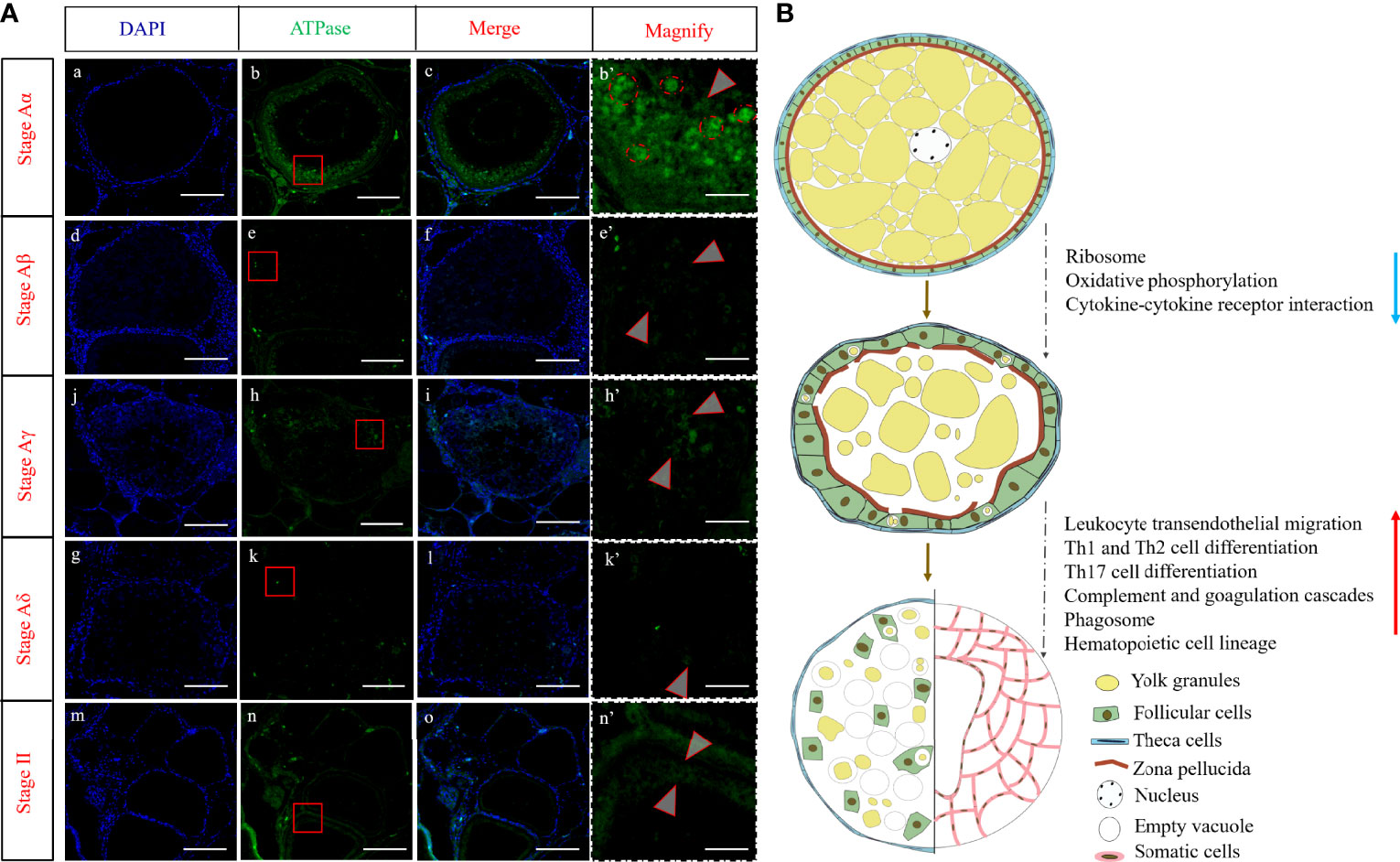
Figure 9 (A)Fluorescence immunohistochemistry analysis of ATPase in five stages of follicular atresia. The b’, e’, h’, k’, and n’ are enlarged images of red boxed regions in b, e, h, k, and n, respectively. Arrows indicate the ATPase signals. The red dashed line circles in b’ represent vitellin with autofluorescence. (B) A schematic model illustrating the molecular changes in follicle atresia. Scale bar = 400 μm (a–o), 70 um (b’–n’).
Discussion
In teleost, follicle atresia is a common degenerative process in ovaries that occurs at different stages of development and is triggered by various exogeneous and endogenous factors (Miranda et al., 1999; Gonzalez-Kother et al., 2020). In this study, we detailly descripted characteristics of the process of follicle atresia and identified many stage-specific genes involving in different stages of follicle atresia by RNA-seq in silver pomfret.
The Phase Transition in Follicle Atresia of Silver Pomfret Is Of Obvious Morphological Difference
In fishes, follicle atresia status is mainly determined using morphological characteristics and is commonly accepted as a process of degeneration of postovulatory follicles or vitellogenic follicles failed to mature and ovulate during the spawning season. A drastic resorption of yolk protein and tissue remodeling were the putative morphological hallmarks for follicular atresia (Santos et al., 2005; Gonzalez-Kother et al., 2020; Sales et al., 2019). More recently, apart from the resorption of yolk protein, more morphological changes of the somatic and germ cells were observed during follicular atresia with exquisite histological examinations, including the hypertrophy and distortion of granulosa cells (Miranda et al., 1999; Morais et al., 2012), presence and formation of immune cells (Mokhtar, 2019; Mokhtar and Hussein, 2020), and autophagy and apoptosis of follicular cells (Morais et al., 2012; Sales et al., 2019). In the present study, we detailly descripted the progress of follicular atresia in silver pomfret (Yang et al., 2021a) and found that it was a continuously dynamic process, which could be divided into four different stages, from Aα to Aδ stage, based on the main cellular characteristics. Similar descriptions for different stages of follicle atresia have been reported in some fishes, such as Leporinus taeniatus, Leporinus obtusidens, Astyanax bimaculatus, Prochilodus argenteus, and Nile tilapia (Miranda et al., 1999; Morais et al., 2012; Sales et al., 2019), suggesting that the process of follicle atresia was a conserved phenomenon in teleost. To our surprise, more stage-specific morphological characteristics of somatic cells were observed in the present study. At Aα stage, granulosa cells that surrounded oocytes became hypertrophied, proliferated, differentiated into immune cells, and gradually fold inward the ooplasm at Aβ stage. Further, those somatic cells completely distributed in the follicular atresia and resorbed yolk protein by phagocytic vacuoles at Aγ stage and gradually disappeared until completing the whole follicular atresia. These stage-specific morphological results strongly indicated that somatic cells might be a modulator in the progress of follicle atresia by proliferation, distortion, differentiation, autophagy, and apoptosis in fish. However, to date, little is known about genes in the ovary related with somatic cells that regulate atresia in fish.
Identification of Key Genes Involved in Early Follicle Atresia of Silver Pomfret
Through analyzing the transcriptomes of five developmental stages of atretic follicles regression, many stage-specific genes were identified, which may contribute to the difference among the process of follicle atresia. Several coexpressed gene sets were identified by the WGCNA, such as pink, yellow, and magenta modules (Figure 4). These stage-specific gene sets were mainly enriched in different biological processes (Supplementary Figure 2). Among them, the genes coexpressed in the pink module were distinctively enriched in “ribosome” and “parkinson disease” and showed Aα-specific expression patterns. In addition, hierarchical clustering of the DEGs only expressed in stage Aα and nearly disappeared in stage Aβ and also showed that the genes were significantly enriched in “ribosome”, “parkinson disease”, and “oxidative phosphorylation” (Figure 7). Furthermore, we used qRT-PCR and fluorescence immunohistochemistry to validate the genes expression levels and protein location of the DEGs including ctb3, cox2, and atpase in “ribosome” and rps3 and rps6 in “parkinson disease” and “oxidative phosphorylation” (Figures 8, 9), and the results indicated that the changes of those genes might be of key roles in the initial stage of follicle atresia. Similarly, previous morphological studies in Astyanax bimaculatus lacustris and Leporinus reinhardti also showed a degradation of organelles including mitochondria in the peripheral ooplasm during the initial stage of follicle atresia (Miranda et al., 1999). Mitochondria and ribosome play important roles in oogenesis, especially in vitellogenesis in oviparous animals. For decades, it has been well known that the oogenesis is a dynamic process harboring accumulation of vitellogenins and process of yolk protein, which is substantially depended on the prodigious generation of protease and energy (Zelazowska and Kilarski, 2009; Reading et al., 2017; Sullivan and Yilmaz, 2018). Thus, ribosome generation and active mitochondrial activities were widely existed in oogenesis of oviparous animals involving the teleost. In previous study, similar with other fishes, such as paddlefish and sturgeon, mitochondria massively increased along with the development of oocytes in silver pomfret (Yang et al., 2021a). Meanwhile, in Senegalese sole, the genes of mitochondrial energy production (cox1, cytb, nd3, nd1, and acca2) were validated to overexpress in oogenesis but downregulate in follicle atresia (Tingaud-Sequeira et al., 2009). In addition, it also has been reported that the genes in ribosome pathway were highly upregulated in oogenesis in mud crabs and mouse (Ihara et al., 2011; Yang et al., 2021b). Therefore, the transcriptome in this study strongly indicated that the downregulation of genes in ribosome and oxidative phosphorylation biological pathways might be the key genes and play critical roles in early follicle atresia of silver pomfret.
Ovarian Somatic Cells Play Various Important Roles in Follicle Atresia of Silver Pomfret
The development and differentiation of female germ cells are controlled by a unique microenvironment created by surrounding somatic cells. In mammals, apoptosis in follicular cells seems to mediate the ovarian follicle atresia (Yu et al., 2004; Matsuda et al., 2012). However, in fish, apoptosis of follicular cells was not relevant at the onset of atresia, although it was also observed during late follicle atresia (Morais et al., 2012; Sales et al., 2019). It is worth mentioning that in this fish species, as in other teleosts (Morais et al., 2012; Sales et al., 2019), the process of ovarian follicle atresia is preceded by the hypertrophy and distortion of the granulosa cells (Figure 1B). In general, follicle development strongly depends on the communication between the germ cells and surrounding somatic cells by cytokine-cytokine receptor interaction such as Kit and KitL (Matzuk et al., 2002; Saatcioglu et al., 2016). In this study, we found a great number of genes in cytokine-cytokine receptor interaction obviously downregulated in early atretic ovary, suggesting that the normal conservation between germ cells and somatic cells was already disrupted. Meanwhile, morphological results in the present study showed the presence of immune cells, such as leukocytes and blood cells, surrounded the early atretic oocytes and invaded in the late follicle atresia (Figures 1B, C). Therefore, it seems that those immune cells were possible transited from the follicular cells. On the other hand, it has been firstly reported in fish that various immune cells including leucocytes, mast cells, lymphocytes, and macrophages were detected in ovarian stroma during ovarian development (Mokhtar, 2019; Mokhtar and Hussein, 2020). It was also possible that some immune cells in atretic follicles were derived from ovarian stroma. Those morphological results suggest a relationship between immune cells and follicular atresia. However, to date, the molecular mechanisms mediating the generation and specific function of immune cells during follicular atresia in fish is still unknown. In the present study, through RNA-seq analysis, we found that DEGs significantly expressed at stages Aγ and Aδ were highly enriched in immune-related biological processes, such as “leukocyte transemdothelial migration”, “hematopietic cell lineage”, and “lysosome” (Figures 5, 7). Meanwhile, some significantly expressed genes, such as cd4, cxcr4, mmp9, cd68, clqc, clqa, and cldn5b, seem to play the key roles in mediating the generation and specific function of immune cells during follicle atresia.
CD4+ T helper (Th) differentiation. In this study, Th cell differentiation–related genes were identified in atretic ovaries, which may contribute to the process of follicle atresia (Figure 7). It is well known that T cells play important roles in immune system, and two general populations of T cells are categorized according to the T-cell receptor (TCR) on the cell surface, CD8+ and CD4+ T cells in vertebrate (Toda et al., 2011; Nakanishi et al., 2015). In the present study, the network of protein–protein interactions showed that cd4 was a key gene (Figures 5, 7). In vertebrate, CD4 is a conserved nonpolymorphic transmembrane glycoprotein molecule, functioned as coreceptor with the T-cell receptor by binding to major histocompatibility complex class II (MHC-II), which is expressed on the surfaces of antigen-presenting cells (APC), dendritic cells, macrophages, and B lymphocytes, and distinctively expressed on the surface of CD4+ helper cells (Nakanishi et al., 2015; Tang et al., 2021). Moreover, CD4+ T cells play a key role in coordinating the immune system through production of various cytokines after activation and differentiation (Ashfaq et al., 2019; Tang et al., 2021). Generally, subsequent to interaction with the MHC-II, the naïve CD4+ T cells get activated and differentiate into specific subtypes, such as Th1, Th2, Th17, Th9, Th22, Th3, and Treg cell lineages, each with a characteristic cytokine profile (Ashfaq et al., 2019; Tang et al., 2021). In this study, we identified a number of Th cell differentiation related genes, such as IL-2, IL-2R, IL-6, IL-12, MHC-II, TGF-β, STAT6, and CD4, which were enriched in “Th1 and Th2 cell differentiation” and “Th17 cell differentiation” biological processes and their expression were positively correlated with the process of follicle atresia (Figure 7). Th17 cells have been reported to be important for the inflammatory responses and autoimmune disorders, due to their ability to serve either as protective/nonpathogenic cells or proinflammatory pathogenic cells (Ashfaq et al., 2019). TGF-β and IL-6 are the major signaling cytokines involved in Th17 cell differentiation from naïve T cells (Wang and Secombes, 2013; Ashfaq et al., 2019; Chatterjee et al., 2020). In addition, IL-6 was able to promote macrophage proliferation locally during inflammatory event. In vertebrate, Th1 cells govern the protective response against the intracellular pathogens by producing lymphocyte proliferation (Ashfaq et al., 2019). Th2 cells stimulate B lymphocytes to produce different antibody isotypes and control extracellular infection. In Th1 and Th2 differentiation, IL-2 plays an important role and promotes naïve CD4+ T cell differentiation into Th1 and Th2 cells via highly combining to their receptors, IL-2R, and also regulates other cells including CD8+ T cells, NK cells, and B cells (Wang et al., 2018). In addition, IL-12 is also the critical cytokine that initiates the downstream signaling cascade to develop Th1 cells (Ashfaq et al., 2019). In a previous study, different cytokines were proved to be of important functions in regulation of the ovarian functions, including IL-6 from granulosa cells, which was detected in ovaries of trout and perch, and had a key role during ovulation (Chatterjee et al., 2020). It is well established that the ovulatory process is similar to the inflammatory responses. In this study, the expression levels of genes involved in CD4+ Th cell differentiation were obviously upregulated during follicle atresia. This result suggested that follicle atresia might also be an inflammatory response, and Th1, Th2, and Th17 cells might play an important role in this process of oocyte degeneration.
Leukocyte transendothelial migration. Gonadal somatic cells migration into oocyte is important and common phenomenon for follicle atresia in fish. In this study, we found that some immune cells fold inward and finally distributed in atretic oocyte from Aβ stage (Figures 1C, 9). Meanwhile, RNA-seq results showed that the genes, such as rac2, itgam, itgb, thy1, cams, cxcr4, cxcl12a, mmp9, and pecam, enriched in “leukocyte transendothelial migration” were significantly expressed during atresic ovaries (Figure 5, 8). This result suggested that those genes in this pathway might participate in the process of follicle atresia and regulate the migration of immune cells into oocyte to act immune responses. The network of protein–protein interactions further showed that the CXCR4, CXCI12A, and MMP9 were the key genes in the leukocyte transendothelial migration during the late stages of follicle atresia (Figure 5). CXCL12, also known as stromal cell–derived factor 1α (SDF-1α), is evolutionarily conserved member of the CXC-type chemokine family (Hara and Tanegashima, 2014). In vertebrate, CXCL12 binds specifically to its receptor CXCR4 to control the migration of various cells including primordial germ cells, hematopoietic stem cells, and inflammation-associated immune cells in development, immune response, and disease (Hara and Tanegashima, 2014; Hersh et al., 2018). In fish, a particularly prominent example is the guidance of primordial germ cells by CXCR4/CXCL12 during the formation of gonads (Schier, 2003; Minina et al., 2007). In this study, CXCR4/CXCL12 were highly expressed in late stages of atretic ovary. Similarly, high expression of chemotaxis (leukocyte cell–derived chemotaxin 2) was also reported in follicle atresia of flatfish (Tingaud-Sequeira et al., 2009). On the basis of the morphology of migration of immune cells into atretic oocytes and RNA-seq, it seems that CXCR4/CXCL12 might play key role in controlling this migration during atresia. MMP9, also known as gelatinase B, is a member of the matrix metalloproteinase (MMP) family, which are important for the remodeling of the extracellular matrix in a number of biological processes including the process of inflammation and tissue remodeling and repair (Kanbe et al., 1999). Meanwhile, MMPs are a family of zinc-containing multi-domain enzymes that regulate the shape of extracellular matrix by degrading and remodeling its components (Yoong et al., 2007). In vertebrate, it has been demonstrated that MMP9 is expressed in a number of immune cells including neutrophils and eosinophils and is of particular interest for angiogenesis and neurogenesis by corrupting basal lamina molecules (Kanbe et al., 1999; Yoong et al., 2007). In addition, in atretic ovary of tilapia, strong immunoreactivity of MMP9 were detected in rodlet cells (Mokhtar and Hussein, 2020). Those results suggested MMP9 were proteolytic enzymes involved in follicular atresia. Although extensive studies of CXCR4/CXCL12 and MMP9 have been conducted in the migration of immune cells in mammals, this is the first study to identify CXCR4/CXCL12 and MMP9 in the process of follicle atresia in teleost.
Macrophages and cholesterol metabolism. Resorption of yolk is a putative morphological hallmark for follicular atresia in teleost. Studies in teleost have shown that there is a massive transfer of the oocyte yolk proteins and lipids into the bloodstream during the process of follicular atresia by the follicular cells (Santos et al., 2005; Yamamoto et al., 2011; Morais et al., 2012). In this study, we found that the expression levels of genes including cd68, lpl, apoe, and apoc, involved in “macrophages” and “cholesterol metabolism”, were upregulated in atretic ovary of Aγ and Aδ stages (Figures 5, 7). The apoe and apoc belong to the apolipoprotein (APO), which play an important role in controlling plasma lipoprotein metabolism by the regulation of several enzymes, such as lipoprotein lipase (LPL). The high apoc, apoe, and lpl transcript levels in atretic ovary of silver pomfret, similar with the RNA-seq results of Senegalese sole (Tingaud-Sequeira et al., 2009), suggested the importance of cholesterol metabolism during follicular atresia in fish, which may facilitate the resorption of energy-rich yolk materials from atretic oocytes. CD68 is a heavily glycosylated glycoprotein, which is highly expressed in macrophages and other mononuclear phagocytes (Chistiakov et al., 2017). Generally, CD68 is exploited as a valuable cytochemical marker to identify macrophages (Chistiakov et al., 2017). In this study, we also found that a number of macrophages existed in atretic ovary by morphological observation (Figure 1). On the basis of the results of the morphology and genes specific expression, we proposed that macrophages had an important role in follicular atresia of teleost.
In summary, we analyzed the histological changes of follicle atresia and divided it into four different stages in silver pomfret. Meanwhile, the transcriptome data provide significant insights during follicle atresia, including the identification of several stage-specific modules, the discovery of important roles of ovarian somatic cells important roles in follicle atresia, including immune-related functions, and the identification of key genes involved in Th cell differentiation, leukocyte transendothelial migration, and cholesterol metabolism during follicle atresia (Figures 9B, 10).
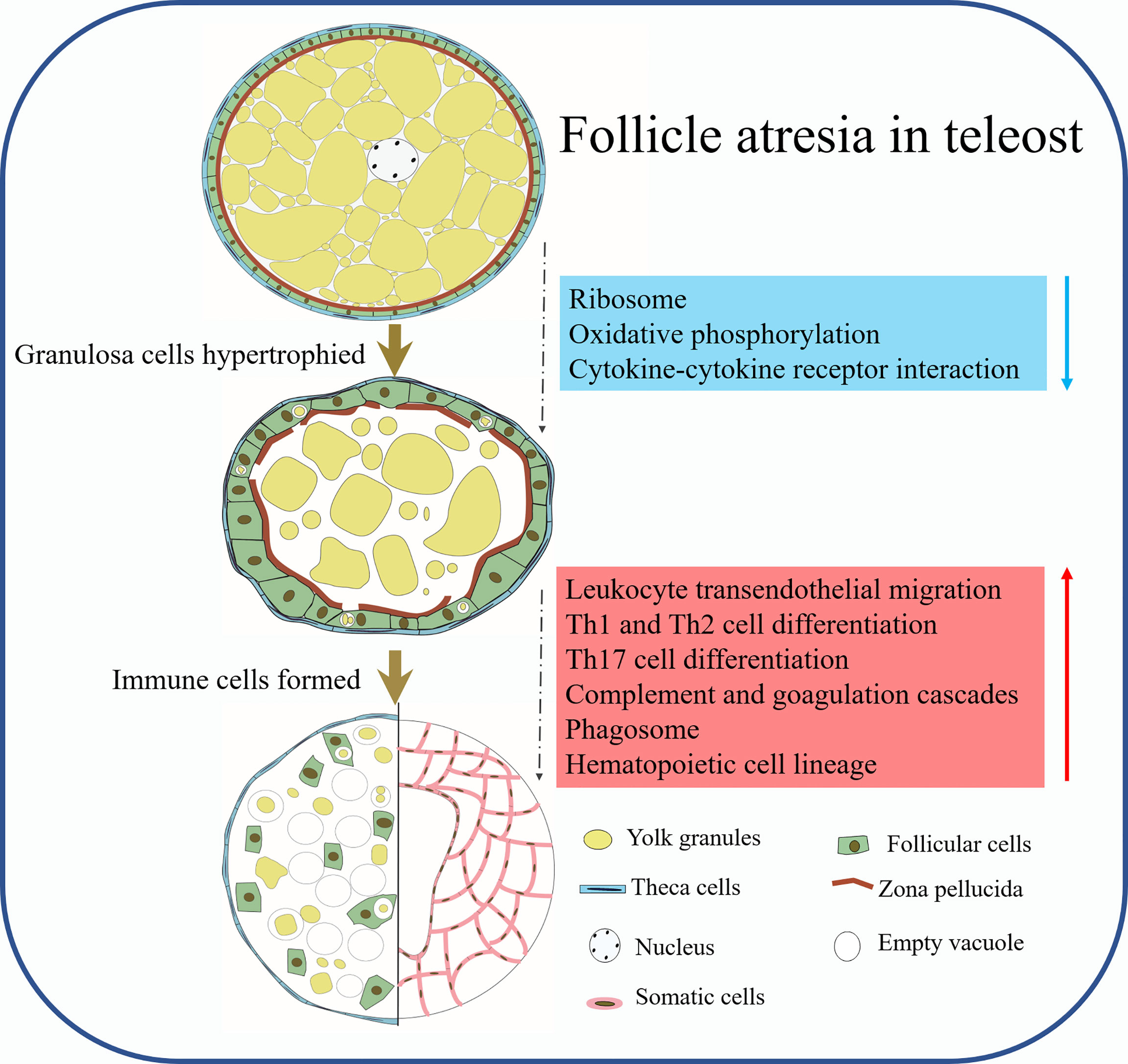
Figure 10 The schematic diagram showing the working hypothesis by which somatic cells mediate the process of follicle atresia in P. argenteus.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA780019.
Ethics Statement
The animal study was reviewed and approved by the Animal Care Committee of Ningbo University.
Author Contributions
YY and ZT conceived and designed the project and wrote the manuscript. GW, YL, and JH collected the samples and carried out the analysis. ZT and YW reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Research Fund of Marine Fishery Institute of Zhejiang Province (2021KF004), Ningbo Public Welfare Science and Technology Planning Project (202002N3035), and the Fundamental Research Funds for Zhejiang Provincial Universities and Research Institutes (2021J012).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.927548/full#supplementary-material
Abbreviations
Aα, AtresA, initial; Aβ, AtresB, intermediate atresia; Aγ, AtresC, advanced atresia; Aδ, AtresD, final atresia; Stage II, AtresE, perinucleolar and pre-vitellogenic stage; DEGs, differential expression genes; WGCNA, weighted gene coexpression network analusis; Th, CD4 + T helper; T-cell receptor; MHC-II major histocompatibility complex class II.
References
Akhavan S. R., Salati A. P., Falahatkar B., Jalali S. A. H. (2016). Changes of Vitellogenin and Lipase in Captive Sterlet Sturgeon Acipenser Ruthenus Females During Previtellogenesis to Early Atresia. Fish. Physiol. Biochem. 42 (3), 967–978. doi: 10.1007/s10695-015-0189-8
Ashfaq H., Soliman H., Saleh M., El-Matbouli M. (2019). CD4: A Vital Player in the Teleost Fish Immune System. Vet. Res. 50 (1), 1. doi: 10.1186/s13567-018-0620-0
Biran J., Levavi-Sivan B. (2018). Endocrine Control of Reproduction, Fish. Encyclopedia Reprod.6, 362–368. doi: 10.1016/B978-0-12-809633-8.20579-7
Chatterjee A., Guchhait R., Maity S., Mukherjee D., Pramanick K. (2020). Functions of Interleukin-6 in Ovulation of Female Climbing Perch, Anabas Testudineus. Anim. Reprod. Sci. 219, 106528. doi: 10.1016/j.anireprosci.2020.106528
Chistiakov D. A., Killingsworth M. C., Myasoedova V. A., Orekhov A. N., Bobryshev Y. V. (2017). CD68/macrosialin: Not Just a Histochemical Marker. Lab. Invest. 97 (1), 4–13. doi: 10.1038/labinvest.2016.116
Gonzalez-Kother P., Oliva M. E., Tanguy A., Moraga D. (2020). A Review of the Potential Genes Implicated in Follicular Atresia in Teleost Fish. Mar. Genomics 50, 100704. doi: 10.1016/j.margen.2019.100704
Gu W., Yang Y., Ning C., Wang Y., Hu J., Zhang M., et al. (2021). Identification and Characteristics of Insulin-Like Growth Factor System in the Brain, Liver, and Gonad During Development of a Seasonal Breeding Teleost, Pampus Argenteus. Gen. Comp. Endocrinol. 300, 113645. doi: 10.1016/j.ygcen.2020.113645
Hara T., Tanegashima K. (2014). CXCL14 Antagonizes the CXCL12-CXCR4 Signaling Axis. Biomolec. Concepts. 5 (2), 167–173. doi: 10.1515/bmc-2014-0007
Hersh T., Dimond A., Ruth B., Lupica N., Bruce J., Kelley J., et al. (2018). A Role for the CXCR4-CXCL12 Axis in the Little Skate, Leucoraja Erinacea. Am. J. Physiol-Regul. Integr. Comp. Physiol. 315, R218–R229. doi: 10.1152/ajpregu.00322.2017
Ihara M., Tseng H., Schultz R. M. (2011). Expression of Variant Ribosomal RNA Genes in Mouse Oocytes and Preimplantation Embryos. Biol. Reprod. 84, 944–946. doi: 10.1095/biolreprod.110.089680
Kagawa H. (2013). Oogenesis in Teleost Fish. Aqua-BioSci. Monogr. 6 (4), 99–127. doi: 10.5047/absm.2013.00604.0099
Kanbe N., Tanaka A., Kanbe M., Itakura A., Kurosawa M., Matsuda H. (1999). Human Mast Cells Produce Matrix Metalloproteinase 9. Eur. J. Immunol. 29 (8), 2645–2649. doi: 10.1002/(SICI)1521-4141(199908)29:08<2645::AID-IMMU2645>3.0.CO;2-1
Matsuda F., Inoue N., Manabe N., Ohkura S. (2012). Follicular Growth and Atresia in Mammalian Ovaries: Regulation by Survival and Death of Granulosa Cells. J. Reprod. Dev. 58 (1), 44–50. doi: 10.1262/jrd.2011-012
Matzuk M. M., Burns K. H., Viveiros M. M., Eppig J. J. (2002). Intercellular Communication in the Mammalian Ovary: Oocytes Carry the Conversation. Science 296 (5576), 2178–2180. doi: 10.1126/science.1071965
Minina S., Reichman-Fried M., Raz E. (2007). Control of Receptor Internalization, Signaling Level, and Precise Arrival at the Target in Guided Cell Migration. Curr. Biol. 17 (13), 1164–1172. doi: 10.1016/j.cub.2007.05.073
Miranda A. C. L., Bazzoli N., Rizzo E., Sato Y. (1999). Ovarian Follicular Atresia in Two Teleost Species: A Histological and Ultrastructural Study. Tissue Cell 31 (5), 480–488. doi: 10.1054/tice.1999.0045
Mokhtar D. M. (2019). Characterization of the Fish Ovarian Stroma During the Spawning Season: Cytochemical, Immunohistochemical and Ultrastructural Studies. Fish. Shellfish. Immunol. 94, 566–579. doi: 10.1016/j.fsi.2019.09.050
Mokhtar D., Hussein M. (2020). Microanalysis of Fish Ovarian Follicular Atresia: A Possible Synergic Action of Somatic and Immune Cells. Microsc. Microanal. 26, 1–10. doi: 10.1017/S1431927620001567
Morais R. D., Thome R. G., Lemos F. S., Bazzoli N., Rizzo E. (2012). Autophagy and Apoptosis Interplay During Follicular Atresia in Fish Ovary: A Morphological and Immunocytochemical Study. Cell Tissue Res. 347 (2), 467–478. doi: 10.1007/s00441-012-1327-6
Nakanishi T., Thome R., Matsuura Y. (2015). T Cells in Fish. Biology 4, 640–663. doi: 10.3390/biology4040640
Reading B. J., Sullivan C. V., Schilling J. (2017). Vitellogenesis in Fishes. Encyclopedia Fish. Physiol., 1–12. doi: 10.1016/B978-0-12-809633-8.03076-4
Rideout R. M., Tomkiewicz J. (2011). Skipped Spawning in Fishes: More Common Than You Might Think. Mar. Coast. Fish. 3 (1), 176–189. doi: 10.1080/19425120.2011.556943
Saatcioglu H. D., Cuevas I., Castrillon D. H. (2016). Control of Oocyte Reawakening by Kit. PLos Genet. 12 (8), e1006215. doi: 10.1371/journal.pgen.1006215
Sales C. F., Melo R. M. C., Pinheiro A. P. B., Luz R. K., Bazzoli N., Rizzo E. (2019). Autophagy and Cathepsin D Mediated Apoptosis Contributing to Ovarian Follicular Atresia in the Nile Tilapia. Mol. Reprod. Dev. 86 (11), 1592–1602. doi: 10.1002/mrd.23245
Santos H. B., Rizzo E., Bazzoli N., Sato Y., Moro L. (2005). Ovarian Regression and Apoptosis in the South American Teleost Leporinus Taeniatus Lütken (Characiformes, Anostomidae) From the São Francisco Basin. J. Fish. Biol. 67 (5), 1446–1459. doi: 10.1111/j.1095-8649.2005.00854.x
Schier A. F. (2003). Chemokine Signaling: Rules of Attraction. Curr. Biol. 13 (5), R192–R194. doi: 10.1016/S0960-9822(03)00122-2
Sullivan C. V., Yilmaz O. (2018). Vitellogenesis and Yolk Proteins, Fish. Encyclopedia Reprod. 6, 266–277. doi: 10.1016/B978-0-12-809633-8.20567-0
Tang H., Jiang X., Zhang J., Pei C., Zhao X., Li L., et al. (2021). Teleost CD4+ Helper T Cells: Molecular Characteristics and Functions and Comparison With Mammalian Counterparts. Vet. Immunol. Immunopathol. 240, 110316. doi: 10.1016/j.vetimm.2021.110316
Tingaud-Sequeira A., Chauvigné F., Lozano J., Agulleiro M. J., Asensio E., Cerdà J. (2009). New Insights Into Molecular Pathways Associated With Flatfish Ovarian Development and Atresia Revealed by Transcriptional Analysis. BMC Genomics 10 (1), 434. doi: 10.1186/1471-2164-10-434
Tiwari M., Prasad S., Tripathi A., Pandey A. N., Ali I., Singh A. K., et al. (2015). Apoptosis in Mammalian Oocytes: A Review. Apoptosis 20 (8), 1019–1025. doi: 10.1007/s10495-015-1136-y
Toda H., Saito Y., Koike T., Takizawa F., Araki K., Yabu T., et al. (2011). Conservation of Characteristics and Functions of CD4 Positive Lymphocytes in a Teleost Fish. Dev. Comp. Immunol. 35 (6), 650–660. doi: 10.1016/j.dci.2011.01.013
Wang T., Hu Y., Wangkahart E., Liu F., Wang A., Zahran E., et al. (2018). Interleukin (IL)-2 Is a Key Regulator of T Helper 1 and T Helper 2 Cytokine Expression in Fish: Functional Characterization of Two Divergent IL2 Paralogs in Salmonids. Front. Immunol. 9, 1683. doi: 10.3389/fimmu.2018.01683
Wang T., Secombes C. J. (2013). The Cytokine Networks of Adaptive Immunity in Fish. Fish. Shellfish. Immunol. 35 (6), 1703–1718. doi: 10.1016/j.fsi.2013.08.030
Yamamoto Y., Luckenbach J. A., Goetz F. W., Young G., Swanson P. (2011). Disruption of the Salmon Reproductive Endocrine Axis Through Prolonged Nutritional Stress: Changes in Circulating Hormone Levels and Transcripts for Ovarian Genes Involved in Steroidogenesis and Apoptosis. Gen. Comp. Endocrinol. 172 (3), 331–343. doi: 10.1016/j.ygcen.2011.03.017
Yamamoto Y., Luckenbach J. A., Young G., Swanson P. (2016). Alterations in Gene Expression During Fasting-Induced Atresia of Early Secondary Ovarian Follicles of Coho Salmon, Oncorhynchus Kisutch. Comp. Biochem. Physiol. Part A: Mol. Integr. Physiol. 201, 1–11. doi: 10.1016/j.cbpa.2016.06.016
Yang Y., Liu Q., Xiao Y., Wang X., An H., Song Z., et al. (2018). Germ Cell Migration, Proliferation and Differentiation During Gonadal Morphogenesis in All-Female Japanese Flounder (Paralichthys Olivaceus). Anat. Rec. 301, 727–741. doi: 10.1002/ar.23698
Yang Y., Ning C., Li Y., Wang Y., Hu J., Liu Y., et al. (2021a). Dynamic Changes in Mitochondrial DNA, Morphology, and Fission During Oogenesis of a Seasonal-Breeding Teleost, Pampus Argenteus. Tissue Cell 72, 101558. doi: 10.1016/j.tice.2021.101558
Yang Y., Zhang Y., Zhuang Y., Zhang C., Bao C., Cui Z. (2021b). Identification of Differentially Abundant mRNA Transcripts and Autocrine/Paracrine Factors in Oocytes and Follicle Cells of Mud Crabs. Anim. Reprod. Sci. 230, 106784. doi: 10.1016/j.anireprosci.2021.106784
Yoong S., O’Connell B., Soanes A., Crowhurst M. O., Lieschke G. J., Ward A. C. (2007). Characterization of the Zebrafish Matrix Metalloproteinase 9 Gene and its Developmental Expression Pattern. Gene Expression Patterns 7 (1), 39–46. doi: 10.1016/j.modgep.2006.05.005
Yu Y. S., Sui H., Bin Z., Li W., Luo M. J., Tan J. H. (2004). Apoptosis in Granulosa Cells During Follicular Atresia: Relationship With Steroids and Insulin-Like Growth Factors. Cell Res. 14 (4), 341–346. doi: 10.1038/sj.cr.7290234
Keywords: follicle atresia, somatic cells, immune, teleost, ovary
Citation: Yang Y, Wang G, Li Y, Hu J, Wang Y and Tao Z (2022) Oocytes Skipped Spawning Through Atresia Is Regulated by Somatic Cells Revealed by Transcriptome Analysis in Pampus argenteus. Front. Mar. Sci. 9:927548. doi: 10.3389/fmars.2022.927548
Received: 24 April 2022; Accepted: 15 June 2022;
Published: 20 July 2022.
Edited by:
Quan Xin Gao, Huzhou University, ChinaReviewed by:
Arnaud Tanguy, Sorbonne Universités, FranceGeng Tian, Shanghai Jiao Tong University, China
Feng Yang, Henan Agricultural University, China
Copyright © 2022 Yang, Wang, Li, Hu, Wang and Tao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhen Tao, dGFvemhlbjEyM0B6am91LmVkdS5jbg==