 Yanhu Ren
Yanhu Ren Zhenhao Luo
Zhenhao Luo Qian Liu
Qian Liu Bin Wei
Bin Wei Yue-Hong Wu
Yue-Hong Wu Wen-Sheng Shu
Wen-Sheng Shu Xue-Wei Xu
Xue-Wei Xu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 15 September 2022
Sec. Aquatic Microbiology
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.923295
Heterotrophic prokaryotes constitute the largest living biomass in the ocean and can be divided into particle-associated (PA) and free-living (FL) fractions. PA and FL prokaryotic communities play critical roles in the biogeochemical cycles of particulate and dissolved organic matter; however, their community assembly processes, biogeographical distribution patterns, and functional properties in oligotrophic surface water remain to be further elucidated. Based on 16S rRNA gene sequencing and shotgun metagenomics, we investigated the assembly mechanisms, biogeography, and functional potential of PA and FL prokaryotes in the surface waters of the West Pacific and Indian Oceans. FL prokaryotic communities were predominantly structured by deterministic processes, whereas their PA counterparts appeared to be shaped by the combined action of deterministic and stochastic processes. PA and FL prokaryotes in the tropical oligotrophic surface ocean exhibit markedly different community structures and functional potentials. Bacterial PA specialists such as Lentimonas, Alteromonas, and Pirellula as well as archaeal PA specialists Marine Group II and Marine Group III were significantly more abundant in PA assemblages, whereas lineages such as Prochlorococcus, SAR11 clade, and Candidatus Actinomarina were significantly more abundant in FL communities. The metabolic potential of the PA community was more abundant in pathways such as polyamine biosynthesis, carbohydrate metabolism, and glycosaminoglycan degradation. In contrast, the FL community was more enriched in functions related to amino acid metabolism, lipid biosynthesis, and aromatic degradation.
As a whole, organic matter in the ocean comprises particulate organic matter (POM) and dissolved organic matter (DOM) (Sharp, 1973). POM, which is formed in the upper ocean layer, is a complex matrix comprising organic and inorganic materials (Boeuf et al., 2019) and can be transported from the surface to the interior of the ocean. This is facilitated by the sinking of POM, which was previously fixed to the biomass of phototrophs on the ocean surface, a process referred to as the biological carbon pump, thereby sequestering atmospheric CO2 in the ocean (De La Rocha & Passow, 2014). Additionally, it has been revealed that marine particles can harbor an anoxic microenvironment, allowing anaerobic metabolism in it (Hollibaugh et al., 2000; Bianchi et al., 2018). In contrast, the DOM fraction constitutes the largest reservoir of reduced carbon in the ocean and is nearly equal to the CO2 pool in the atmosphere (Benner et al., 1992; Zhang et al., 2018). Therefore, the biogeochemical behavior of POM and DOM has important implications for elemental cycling and climate.
Heterotrophic prokaryotes constitute the largest living biomass in the oceans (Morán et al., 2017), playing a key role in global biogeochemical cycles (Arrigo, 2005). Extensive research has shown that marine bacteria have different ecological strategies and can be divided into particle-associated (PA) and free-living (FL) fractions (Crump et al., 1999; Zhang et al., 2007; Ortega-Retuerta et al., 2013). Compared to their FL counterparts, PA bacteria can exhibit higher enzymatic activity rates and specific metabolic activity, which may facilitate the release of nutrients from organic particles to the surrounding water, thereby increasing the DOM bioavailability (Azam & Malfatti, 2007; Ortega-Retuerta et al., 2013). Meanwhile, the FL counterpart often face a massive pool of dissolved organic molecules under oligotrophic conditions (Lauro et al., 2009; Zhang et al., 2018). Moreover, previous studies have indicated that PA and FL archaeal communities are phylogenetically distinct in the ocean, implying that distinct PA and FL niches may also exist in the archaeal domain (Orsi et al., 2015; Tarn et al., 2016).
Due to eutrophication and heterogeneity of physicochemical properties, the community structure and distribution patterns of PA and FL bacteria in the surface waters of rivers, estuaries, and adjacent coastal oceans have been extensively studied over the past few decades (Crump et al., 1999; Ortega-Retuerta et al., 2013; Li et al., 2018; Liu et al., 2020). Furthermore, studies of PA and FL bacteria in other oceanic regions have deepened our understanding of their community structure and diversity (Thiele et al., 2015; Milici et al., 2017; Suzuki et al., 2017; Mestre et al., 2018). Regarding community assembly mechanisms (i.e., the underlying ecological and biochemical drivers that lead to the current community composition), a recent study by Jain et al. (2021) examined and quantified the assembly processes of PA and FL bacterial communities throughout the water column in a high Arctic fjord.
In this context, we investigated community structure, community assembly mechanisms, and biogeographical distribution patterns of PA and FL prokaryotes in the surface waters of the West Pacific and Indian Oceans. Moreover, based on genome-centric metagenomics, we analyzed the functional profile of PA and FL prokaryotes in the surface ocean of the West Pacific and Indian Oceans and further revealed markedly different diversity, microbial compositions, and functional potentials between PA and FL assemblages.
Surface seawater samples from 11 sampling sites (S1–S4 and M1–M7) were collected from the West Pacific Ocean during the DY56 cruise of the R/V Dayang Yi Hao in September and October 2019. S1, S3, and S4 were sampled at a depth of 0–5 m using a rosette of Niskin bottles attached to a CTD profiler (SBE 911 plus, Sea-Bird Inc.), and the other eight samples (S2 and M1–M7) (0–0.5 m) were manually collected using pre-sterilized 10-L Nalgene carboys. Moreover, surface seawater (0–0.5 m) from the other four sampling sites (S5–S8) in the Indian Ocean was manually collected during the DY58 cruise of the R/V Dayang Yi Hao in January 2020 (Figure 1).
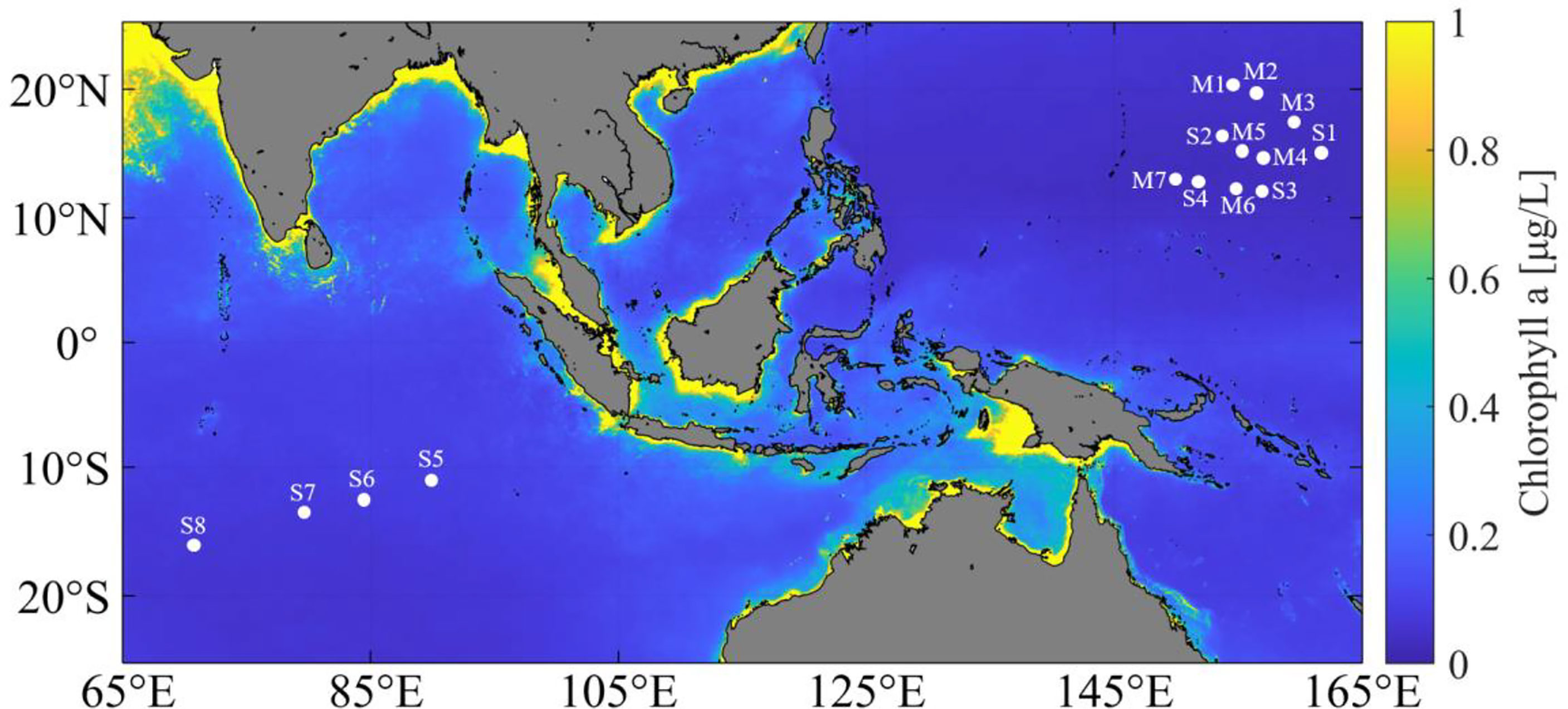
Figure 1 Location of sampling sites. M1-M7 & S1-S4, sampling sites in the West Pacific Ocean. S5-S8, sampling sites in the Indian Ocean. The surface chlorophyll a concentration (mg/L) shown in this map is the annual average value from 2019 to 2020 (obtained from the MODIS-aqua 4 km data set).
Fifty liters of seawater (per sample) was sequentially filtered through 3.0-μm-pore-size and 0.22-μm-pore-size filter membranes (Mixed Cellulose Esters Membrane, 142 mm, Millipore) to collect PA and FL fractions, respectively (Ortega-Retuerta et al., 2013; Suzuki et al., 2017; Liu et al., 2019; Yuan et al., 2021). A peristaltic pump was used to complete the filtration under low pressure, and the membranes were stored at −80°C immediately after filtration for further processing.
The water salinity and temperature of S1, S3, and S4 were obtained in situ using the CTD profiler, whereas the chlorophyll-a on each filter (GF/F, Whatman) was measured with a Trilogy Laboratory Fluorometer after overnight extraction with 90% acetone, and the pH value was measured with a pH meter (Mettle Toledo Inc., Switzerland). The temperature, salinity, and pH of S2 and M1–M7 were measured with the YSI Professional Plus Multi-Parameter Instrument (YSI Inc., USA), and chlorophyll-a concentrations corresponding to their sampling dates were retrieved using NASA’s Aqua satellite (MODIS-Aqua, 2018). The sampling sites and their corresponding metadata are listed in Supplemental Table S1.
Total DNA was extracted from the PA and FL fraction membranes using an ALFA-SEQ Advanced Water DNA Kit (Findrop Biosafety Technology, Guangzhou, China), following the manufacturer’s instructions. The DNA concentration was measured using NanoDrop One (Thermo Fisher Scientific, Waltham, USA). The V4 region of the prokaryotic 16S rRNA gene was amplified using polymerase chain reaction (PCR) with the universal primers 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) (Caporaso et al., 2011). PCR amplification was conducted with a 50-μl mixture containing 1× PCR buffer, 1.5 mM MgCl2, 0.2 mM dNTP mix, 0.5 μM of each primer, 2 U of Invitrogen Platinum Taq DNA polymerase (Life Technologies), and 2.5 μl of DNA extracts from each sample as templates. The PCR program consisted of an initial denaturation at 98°C for 30 s, 30 cycles of denaturation at 94°C for 30 s, annealing at 52°C for 30 s, elongation at 72°C for 60 s, and a final elongation step at 72°C for 5 min. Mixed PCR products were purified using the E.Z.N.A. Gel Extraction Kit (Omega, USA). Sequencing libraries were generated using the NEB Next® Ultra™ II DNA Library Prep Kit for Illumina® (New England Biolabs, MA, USA) following the manufacturer’s recommendations, and index codes were added. The libraries were then sequenced on an Illumina Nova6000 platform and 250-bp paired-end reads were generated. To obtain metagenomic data, the total community DNA was used for library preparation with the NEB Next® Ultra™ DNA Library Prep Kit for Illumina® (New England Biolabs, MA, USA) following the manufacturer’s recommendations. Library quality was assessed on the Qubit 3.0 Fluorometer (Life Technologies, Grand Island, NY) and Agilent 4200 (Agilent, Santa Clara, CA) system. The library was sequenced on an Illumina NovaSeq platform (150-bp paired-end reads) to obtain approximately 20 Gbp of metagenomic data for the sequenced samples.
Data of 16S rRNA genes (S1–S8 and M1–M7) were processed and analyzed using the QIIME 2 (version 2020.6) workflow (Bolyen et al., 2019). Briefly, raw sequence data were demultiplexed and quality-filtered using the q2-demux plugin, followed by denoising with DADA2 (Callahan et al., 2016) to obtain amplicon sequence variants (ASVs). ASVs were firstly aligned with MAFFT (Katoh et al., 2002), and then the resulting masked alignment was used to construct a phylogenetic tree with fasttree2 (Price et al., 2010) by using the command of “qiime phylogeny align-to-tree-mafft-fasttree”. To make samples comparable, alpha and beta diversity metrics were calculated after samples were rarefied to 47,000 sequences per sample by using the “qiime diversity core-metrics-phylogenetic” command. The Naïve Bayes classifier against 99% OTUs reference sequences of Silva_138_release (Quast et al., 2013) was trained and used to assign taxonomic annotations to ASVs using QIIME 2 feature-classifier plugin (Bokulich et al., 2018).
A previously proposed conceptual framework explained that community structure and dynamics are governed by four main processes: selection, dispersal, ecological drift, and speciation (Vellend, 2016; Logares et al., 2020). To quantify the contribution of various ecological processes to PA and FL prokaryotic community assembly, the community composition and phylogenetic tree generated by QIIME 2 were put into the iCAMP (version 1.5.4) (Ning et al., 2020) workflow with parameters set as “ds = 0.2, bin.size.limit = 12” and others were set to default values.
Raw metagenomic reads of S1–S8 were quality-filtered and trimmed using Trimmomatic (version 0.39) (Bolger et al., 2014) with the following parameters: LEADING:3 TRAILING:3 SLIDINGWINDOW:5:20 MINLEN:50. Then, quality-controlled reads of each sample were de novo assembled using MEGAHIT (version 1.2.9) (Li et al., 2015) with the following parameters: k-min 35, k-max 95, k-step 20, and min-contig 500; during this process, scaffolds with a length of <500 bp in each assembly were removed for further analysis. Gene prediction was carried out using Prodigal (version 2.6.3) with the parameters set as -f gff, -g 11, -p meta, -q, and -c (Hyatt et al., 2010). To evaluate the dynamics of individual genes, quality-controlled sequencing reads of each sample were mapped back to the predicted genes to calculate coverage information using BBMap (Version 38.86; https://sourceforge.net/projects/bbmap/) with the following parameters: minid = 0.90, build = 1, trimq =10, and local = f. Moreover, the coverage information of the genes in each sample was calculated and normalized using the TPM method (Knight et al., 2018) to obtain the relative abundance of predicted genes. To obtain KEGG Orthology (KO), cluster of orthologous group (COG), and archaeal COG (arCOG), blastp was used to assign predicted proteins to the KEGG database (Kanehisa et al., 2021) and eggNOG database (version 5.0.0) (Huerta-Cepas et al., 2019) with the parameters set as -k 1, -f 6, and -e 10−5.
To carry out genome-centric analysis of PA and FL prokaryotic communities, quality-controlled reads of PA/FL fractions from the same oceanic region were co-assembled using MEGAHIT (version 1.2.9) (Li et al., 2015) with the following parameters: k-min 31, k-max 111, k-step 20, and min-contig 1000. The assemblies were then put into the MetaWRAP pipeline (version 1.2) (Uritskiy et al., 2018) to perform genomic binning with default parameters. Briefly, initial binning was conducted using CONCOCT (version 1.1.0) (Alneberg et al., 2014), MaxBin2 (version 2.2.5) (Wu et al., 2015), and metaBAT2 (version 2.12.1) (Kang et al., 2019) in the BINNING module, and then the metagenome-assembled genomes (MAGs) generated above were consolidated and refined using Binning_refiner (version 1.2) (Song & Thomas, 2017) in the BIN_REFINEMENT module. Completeness and contamination of MAGs were evaluated using CheckM (version 1.1.3) (Parks et al., 2015), and only MAGs with completeness > 50% and contamination < 5% were retained for subsequent analysis. After de-replicating MAGs at 95% identity using dRep (version 3.0.0) (Olm et al., 2017), quality-controlled reads of each sample were mapped back to MAGs to calculate their relative abundance, and taxonomy was assigned to MAGs based on the Genome Taxonomy Database (GTDB) (version r207) using GTDB-Tk (version 2.0.0) (Chaumeil et al., 2019). Finally, de-replicated MAGs were annotated by EnrichM v0.6.4 (https://github.com/geronimp/enrichM) using the KO database to compare their metabolic pathways.
All statistical analyses were performed using several packages in the statistical programming language R (version 4.0.3). Barplots generated by ggplot2 (v3.3.5) were used to visualize the relative abundance of taxa in each sample. The Bray–Curtis dissimilarity index was used to construct dissimilarity matrices for the community structure and function profiles of PA and FL prokaryotes. Principal coordinate analysis (PCoA) was used to further reveal the clustering patterns of the community structure and function profiles of PA and FL assemblages (vegan, v2.5-4). Variance partitioning analysis was conducted using the “varpart” function of the vegan package. Significant differences (p < 0.05) in community structure and function profiles between the PA and FL assemblages were tested using permutation multivariate analysis of variance (Adonis, 999 permutations). The non-parametric Wilcoxon rank-sum test was used to distinguish statistically significant differences in the normalized abundance of a given taxon or gene between classified groups of samples, and the P-values for multiple comparisons were adjusted by the false discovery rate (FDR).
The phylum-level taxonomic compositions of PA and FL assemblages in the West Pacific and Indian Oceans are shown in Figure S1. The sequences were mainly assigned to Pseudomonadota, Cyanobacteria, Bacteroidota, Actinomycetota, Candidatus Thermoplasmatota, Planctomycetota, Verrucomicrobiota, Candidatus Marinimicrobia (SAR406 clade), and SAR324 clade (Marine Group B). Actinomycetota and Candidatus Marinimicrobia (SAR406 clade) were more abundant in the FL assemblages, whereas Candidatus Thermoplasmatota, Planctomycetota, Verrucomicrobiota, and SAR324 clade (Marine Group B) were more enriched in the PA assemblages. Moreover, PCoA based on the Bray–Curtis distance significantly (p < 0.05) divided the community structure into four clusters, which represent PA and FL fractions in the West Pacific and Indian Oceans, respectively (Figure 2A).
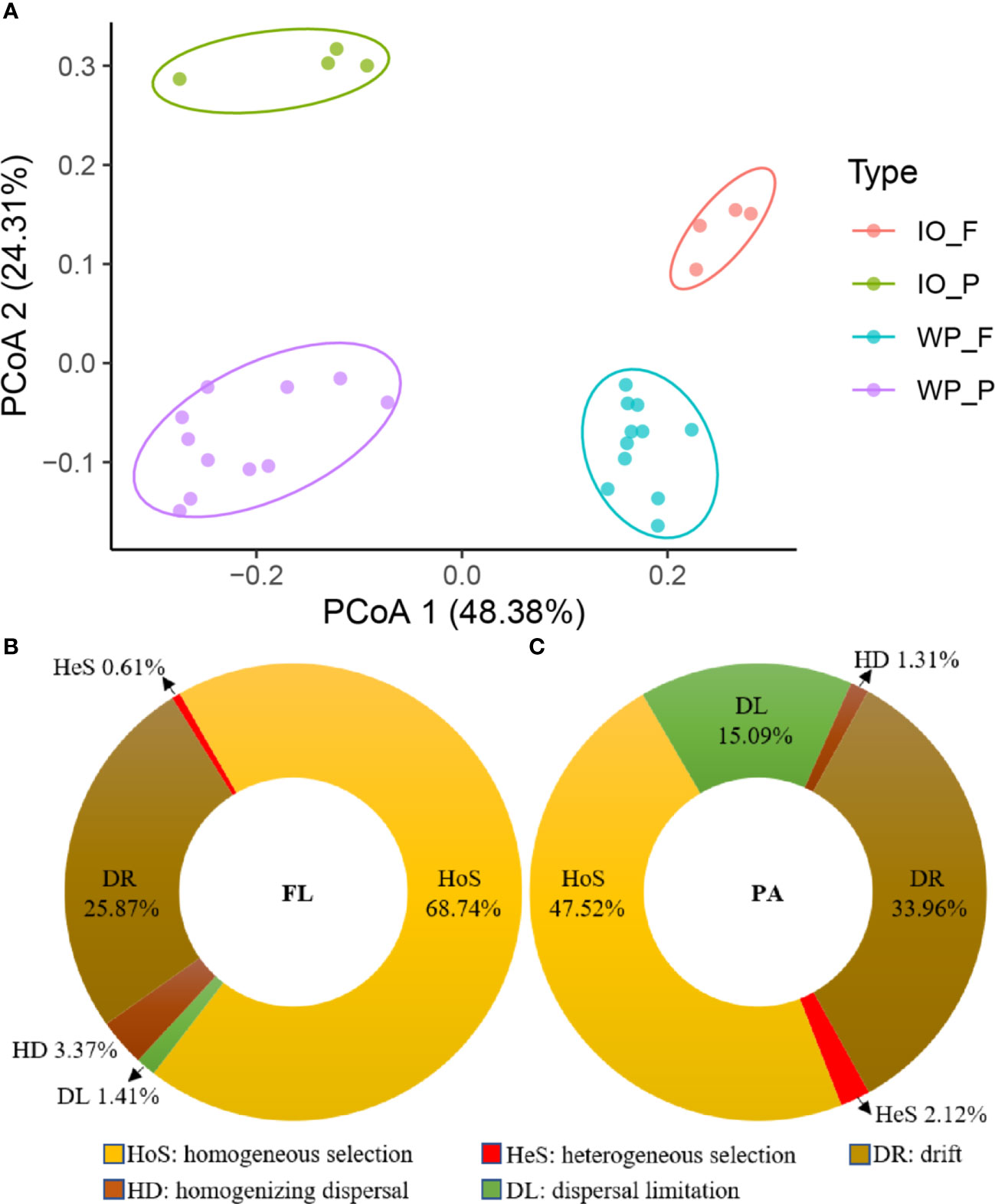
Figure 2 (A) Prokaryotic community structure (genus level) of PA and FL assemblages in the Indian Ocean (IO) and West Pacific Ocean (WP) revealed by 16S rRNA gene amplicon sequencing. (B, C) Relative influences of assembly processes governing turnover of FL (B) and PA (C) communities in the Indian and West Pacific Oceans.
Based on the results of the community assembly mechanism analysis, homogeneous selection (HoS) and drift (DR) were found to be the main mechanisms structuring the FL prokaryotic community, with relative importance values of 68.74% and 25.87% (Figure 2B), respectively. In contrast, heterogeneous selection (HeS), dispersal limitation (DL), and homogenizing dispersal (HD) represent only a small percentage of the variation in FL prokaryotic communities. Moreover, as shown in Figure 2C, HoS, DR, and DL were the major processes shaping PA prokaryotic communities, with relative importance of 47.52%, 33.96%, and 15.09%, respectively. In addition, HeS and HD were found to have limited explanations for the variation in PA prokaryotic communities.
The surface waters of the West Pacific Ocean (S1–S4) and Indian Ocean (S5–S8) were selected to further explore the distribution pattern and metabolic potential of PA and FL prokaryotes on large geographic scales. PCoA based on the Bray–Curtis distance indicated that the community structures of PA and FL assemblages were divided into four clusters. Furthermore, the first and second axes of PCoA explained 48.48% and 29.62% of the total variance of the community structure, respectively (Figure 3A). After calculating the normalized coverage of genes for the KEGG and eggNOG functional annotations, PCoA based on the Bray–Curtis distance once again revealed that functional profiles of PA and FL assemblages have significantly different clustering patterns (p < 0.05). The first and second axes of the KEGG PCoA plot (Figure S2) explained 85.65% and 6.162% of the total variance of functional profiles, respectively. Similarly, the first and second axes of the eggNOG PCoA plot explained 85.19% and 7.369% of the total variance of functional profiles, respectively (Figure 3B).
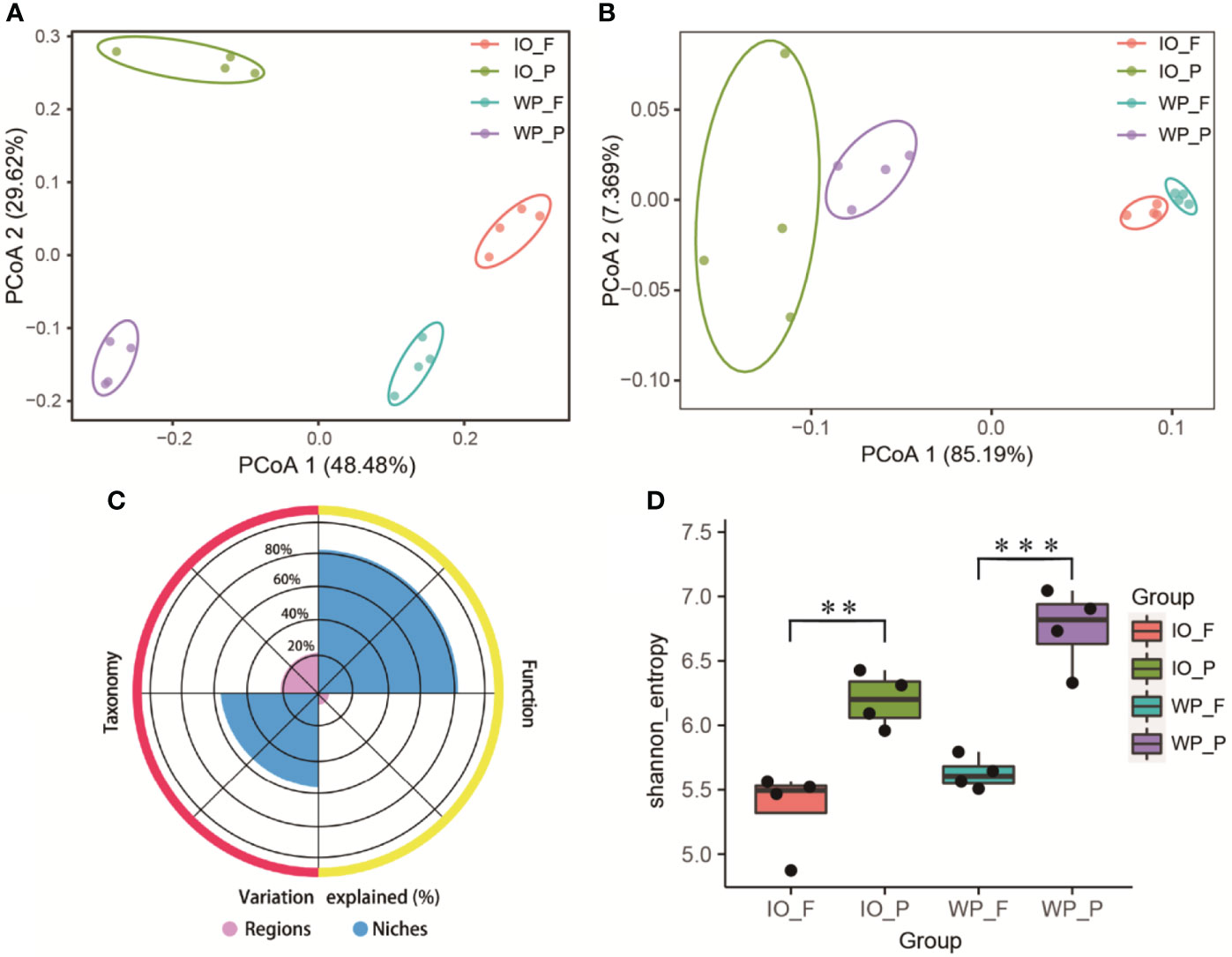
Figure 3 (A) Prokaryotic community structure (genus level) and (B) functional profile (based on eggNOG annotation) differences of PA and FL assemblages revealed by 16S rRNA gene amplicon sequencing and metagenomics, respectively. (C) Variances of community structure and functional profile explained by oceanic regions and PA and FL niches. (D) Differences in Shannon index between PA and FL communities. IO, Indian Ocean. WP, West Pacific Ocean. Asterisks represent the following statistical significance: **0.001 ≤ P < 0.01; ***P < 0.001.
Variance partitioning analysis was applied to assess the influence of oceanic regions and lifestyle on PA and FL prokaryotes. According to the variance partitioning analysis results (Figure 3C), oceanic regions explained 22% and 6% of the taxonomic and functional variation of the prokaryotic community, respectively. In contrast, PA and FL niches accounted for 54% and 81% of the variation of the community structure and functional profile, respectively.
The Shannon index, observed features, and Pielou’s evenness were analyzed to estimate the diversity, species richness, and evenness of PA and FL assemblages. The Shannon index (Figure 3D) and observed features (Figure S3A) indices in PA assemblages were significantly higher than those in their FL counterparts, demonstrating that PA prokaryotic communities exhibit higher diversity and richness than FL communities. The Pielou’s evenness index for the PA assemblages was higher than their FL counterparts; however, a significant difference was only found in the West Pacific Ocean (Figure S3B).
Based on the normalized relative abundances of PA and FL prokaryotes at genus level, the top 20 taxa with significant differences (p < 0.05) between PA and FL assemblages, as well as the affiliation between phylum/class and finer levels, were analyzed to better understand the differences in community structure (Figure S4 and Supplemental Table S2). For instance, PA specialists including Lentimonas, Alteromonas, and Pirellula were more abundant in PA assemblages, whereas taxa such as Prochlorococcus, SAR11 clade, and Candidatus Actinomarina were more enriched in FL fractions (Figure 4).
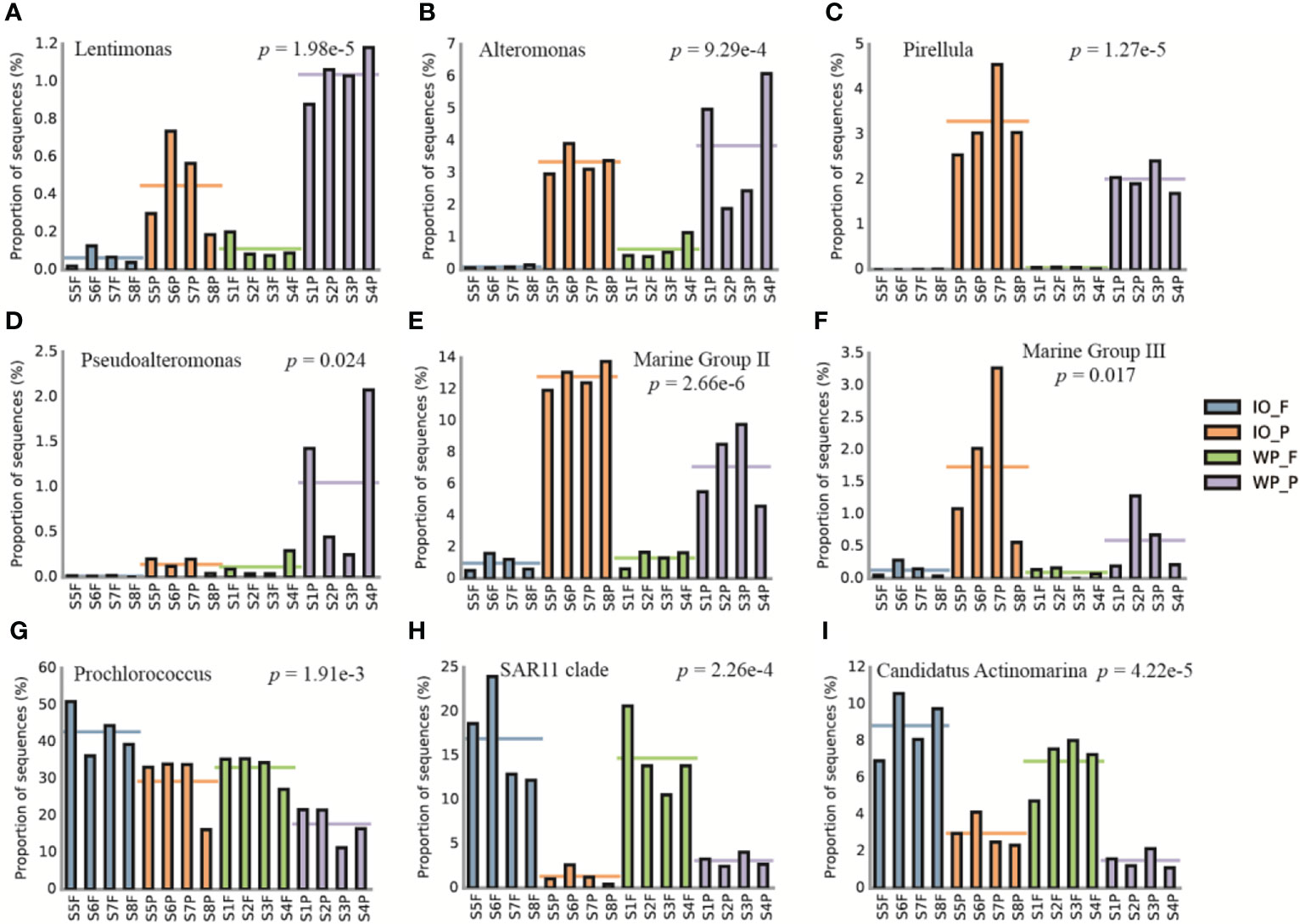
Figure 4 Examples of taxa with significant differences between PA and FL prokaryotes revealed by 16S rRNA gene amplicon sequencing. Relative abundance of (A) Lentimonas, (B) Alteromonas, (C) Pirellula, (D) Pseudoalteromonas, (E) Marine Group II, (F) Marine Group III, (G) Prochlorococcus, (H) SAR11 clade, and (I) Candidatus Actinomarina. The horizontal lines represent the mean values of the relative abundance of PA and FL assemblages. IO, Indian Ocean. WP, West Pacific Ocean. F, free-living. P, particle-associated.
The relative abundance of 71 MAGs was found to be significantly different between PA and FL fractions, which may represent the genomes of prokaryotes with potential PA and FL niche differences. The GTDB classification and genome quality information for the 71 MAGs were presented in Figure 5 and Supplemental Table S3. The taxonomic composition of these genomes was similar to that of the amplicon analysis and was mainly affiliated with Pseudomonadota, Bacteroidota, Myxococcota, Verrucomicrobiota, Candidatus Thermoplasmatota, Planctomycetota, Cyanobacteria, Actinomycetota, Desulfobacterota, and SAR324 clade.
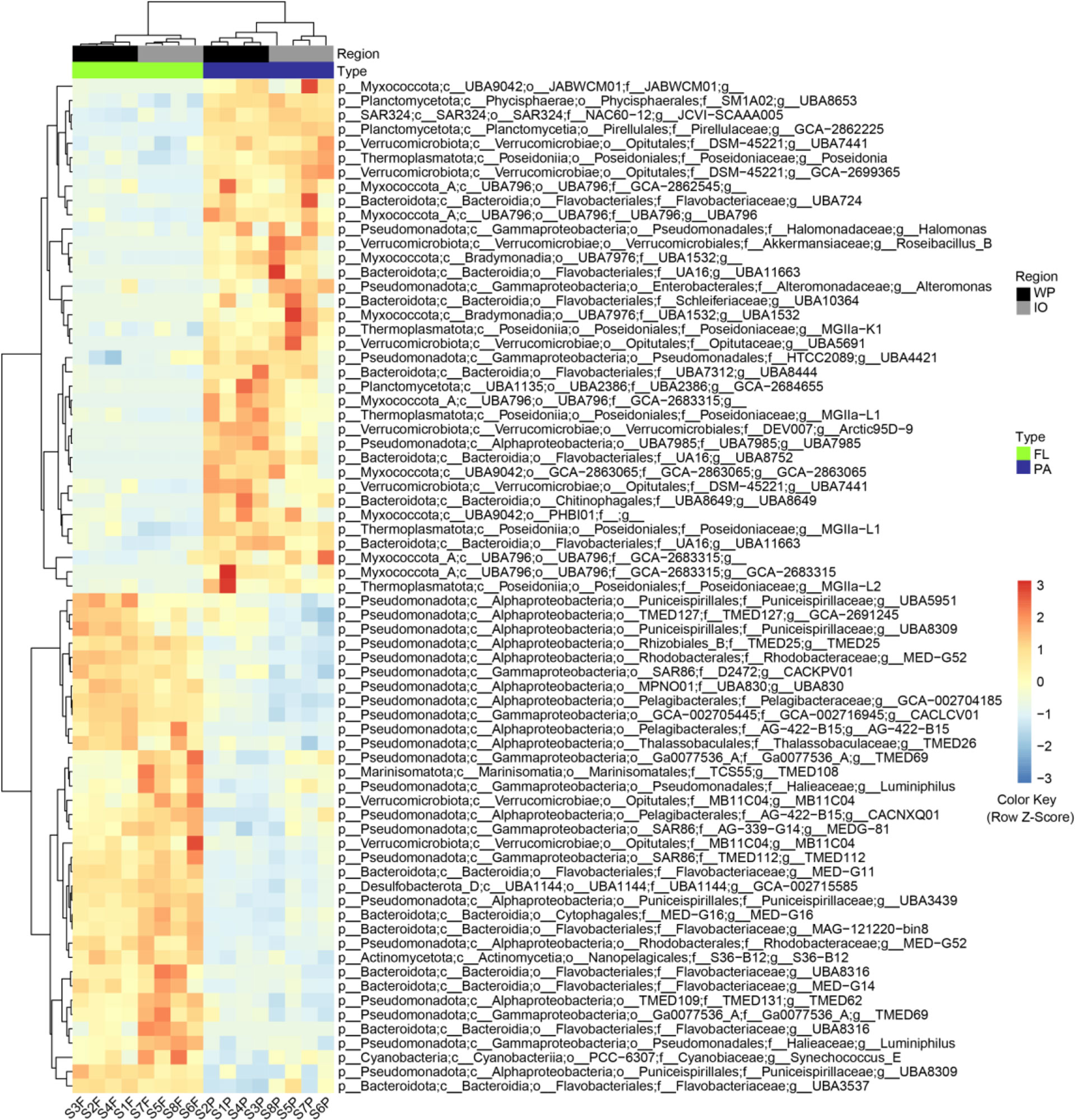
Figure 5 Metagenomic analysis revealed 71 MAGs with significant differences (p < 0.05) in relative abundance between PA and FL assemblages. WP, West Pacific Ocean. IO, Indian Ocean. FL, free-living. PA, particle-associated; p, phylum; c, class; o, order. f, family; g, genus.
Genome-centric metagenomics revealed significant differences (p < 0.05) in metabolic pathways between the PA and FL assemblages (Figure 6). The FL fractions were enriched in most amino acid metabolism pathways, whereas PA fractions were more abundant in pathways related to serine, threonine, and polyamine biosynthesis. Carbohydrate metabolism, nitrogen metabolism, and sulfur metabolism were found more abundant in PA assemblages, whereas photosynthesis, ATP synthesis, and aromatic degradation were more enriched in the FL fractions. Moreover, in contrast to fatty acid, sterol, glycan, and lipopolysaccharide biosynthesis pathways, which were more enriched in FL assemblages, glycosaminoglycan degradation was more enriched in the PA fractions.
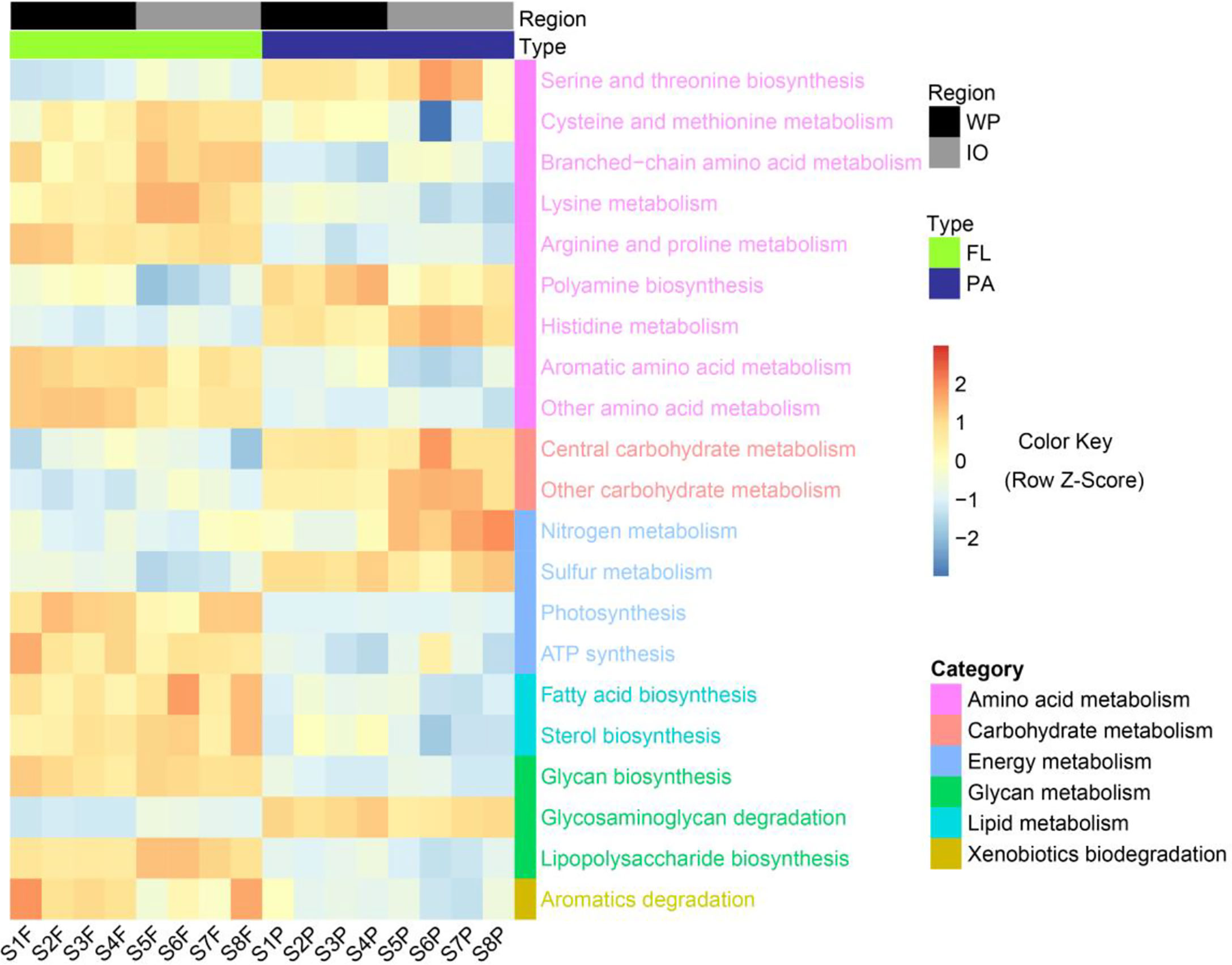
Figure 6 Metabolic pathways with significant differences (p < 0.05) in relative abundance between PA and FL assemblages revealed by genome-centric metagenomics. WP, West Pacific Ocean. IO, Indian Ocean. FL, free-living. PA, particle-associated. KEGG pathways were colored according to their functional categories.
Our results indicated that HoS was the dominant process structuring PA and FL prokaryotic communities (Figures 2B, C), which is in line with the relatively stable physicochemical parameters we obtained (Supplemental Table S1). This suggests that homogeneous environmental pressures drive the similarity of PA or FL prokaryotic communities in the same oceanic region (Figure 2A). Moreover, our study supports previous observations (Jain et al., 2021) that the relative influence of HoS is higher in FL assemblages than in their PA counterparts. Particularly, previous studies have indicated that individual particles highly differed in colonizing communities (Bižić-Ionescu et al., 2018; Szabo et al., 2022), whereas HeS can poorly explain the community turnover of PA communities in our study. This result may be explained by the fact that the PA assemblages in this study were collected from the upper oligotrophic oceans, where changes in PA communities related to carbon availability may be much slower due to constant supply of labile, easily degradable organic matter (Bižić-Ionescu et al., 2018). It is apparent that DL showed higher influence on PA fractions compared to their FL counterparts. A possible explanation for this is that the colonization of PA prokaryotes on particles may reduce their dispersal rate. Furthermore, DL, HD, and DR are largely considered to be stochastic processes (Zhou & Ning, 2017; Ning et al., 2020), and their relatively high influence on PA assemblages suggests that stochastic processes play an important role in the community assembly of PA prokaryotes. Collectively, the FL prokaryotic communities were predominantly structured by deterministic processes, whereas their PA counterparts appeared to be shaped by the combined action of deterministic and stochastic processes.
Previous studies have concluded that PA bacteria are more diverse than their FL counterparts in most marine environments (Bižić-Ionescu et al., 2015). Our research is consistent with this and showed that the Shannon index of PA prokaryotes was significantly higher than those of their FL counterparts (Figure 3D). In addition, as HoS is an ecological factor that drives the similarity of community structure, it can lead to an observed reduction in species richness (Crespo et al., 2013; Yuan et al., 2021). Therefore, the higher relative importance of HoS in the FL community compared to the PA community is in accordance with the results of the observed features (Figure S3A).
The biogeography and functional profile of PA and FL prokaryotes in the West Pacific and Indian Oceans were revealed by PCoA and variance partitioning analysis (Figures 3A–C). These results indicate that niche differentiation was the dominant factor shaping the community structure and functional profile of PA and FL prokaryotes in the tropical oligotrophic surface ocean. Furthermore, consistent with earlier studies suggesting that the PA community may be largely influenced by factors such as particle composition and quality (Yung et al., 2016; Bižić-Ionescu et al., 2018), PA assemblages in the West Pacific and Indian Oceans demonstrated more distinct biogeographic distribution patterns and higher variability than their FL counterparts.
The community composition and structure of the PA and FL prokaryotes in this study were very similar to previous results (Figure 4; Figure S1; Figure S4). Taxa such as Lentimonas, Alteromonas, and Marine Group II were significantly more abundant in PA assemblages, which is consistent with previous findings suggesting that they are potential PA specialists and may play an important role in POM degradation (Ganesh et al., 2014; Simon et al., 2014; Bižić-Ionescu et al., 2015; Orsi et al., 2015; Sichert et al., 2020). Conversely, lineages such as Prochlorococcus, SAR11 clade, AEGEAN-169 marine group, and Candidatus Actinomarina were significantly more enriched in FL assemblages, which have been proven to be FL and play a major role in the ocean carbon cycle (Partensky et al., 1999; Grote et al., 2012; Ulloa et al., 2021).
Genome-centric metagenomics revealed significant differences in the functional potential between PA and FL prokaryotic communities (Figure 6). Most amino acid metabolism pathways were found to be more enriched in the FL fractions, suggesting that FL prokaryotes may have great metabolic potential in the metabolism of low-molecular-weight amino acids. Conversely, polyamine biosynthesis was more abundant in the PA assemblages. As polyamines have been shown necessary for bacterial biofilm formation (Michael, 2018), their enrichment in PA assemblages suggests that polyamines may play an active role in the particle attachment of PA bacteria. Furthermore, it has been previously reported that polysaccharides constitute a large fraction of the organic matter produced and degraded in the ocean, and carbohydrate-active enzymes are important tools used by heterotrophic microbes to initiate the degradation of marine polysaccharides (Arnosti et al., 2021). In this study, carbohydrate metabolism and glycosaminoglycan degradation pathways were significantly enriched in the PA assemblages, implying that PA prokaryotic communities may play a key role in mediating the degradation of polysaccharide particles.
Our research provided a systematic and detailed genetic investigation of PA and FL prokaryotes in tropical surface oceans. Consistent with relatively stable oligotrophic oceanic environments, HoS dominated the PA and FL prokaryotic community assembly. The PA prokaryotic community exhibited higher diversity and possessed a significantly higher number of particle specialists compared to the FL fraction, whereas taxa characterized by FL were significantly more enriched in the FL prokaryotic community. Genome-centric metagenomics further indicated that the PA prokaryotic community showed great metabolic potential for polysaccharide degradation, whereas the FL counterparts might play a vital role in the biogeochemical cycling of amino acids. Being limited to samples collected from the ocean surface, this study was unable to decipher the dynamics and functional characteristics of PA and FL prokaryotic communities in deep seawater. Comprehensive investigations of PA and FL prokaryotes in the total water column would be appropriate for future studies to fully understand their roles in the ocean carbon cycle.
Raw reads of 16S rRNA gene amplicons and metagenomic sequencing are available for download from the Short Reads Archive with NCBI BioProject accession no. PRJNA764824 and PRJNA824599.
YR, W-SS, and X-WX contributed to conception and design of the study. Y-HW, W-SS, and X-WX implemented sampling, DNA extraction, and sequencing. YR did data analysis and wrote the manuscript with help from ZL, QL, and BW. All authors contributed to manuscript revision, read, and approved the submitted version.
This work was supported by China Ocean Mineral Resources R & D Association (COMRA) Special Foundation (DY135-B2-10), National Natural Science Foundation of China (No. 91851114, 31770004 and 81903534), and Scientific Research Fund of the Second Institute of Oceanography, MNR (grant No. HYGG1901).
We would like to thank senior engineer Xianming Deng, Dr. Dong Sun, Dr. Hong Cheng, and Dr. Li-Li Guo for their help in the sample collection during the DY56 and DY58 cruise of the R/V Dayang Yi Hao. We also thank Xinyang Wang for providing diagram of Figure 1, as well as the Guangdong Magigene Biotechnology Co., Ltd. China for the assistance in data analysis.
Guangdong Magigene Biotechnology Co., Ltd. had the following involvement with the study: Data analysis. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.923295/full#supplementary-material
Alneberg J., Bjarnason B. S., de Bruijn I., Schirmer M., Quick J., Ijaz U. Z., et al. (2014). Binning metagenomic contigs by coverage and composition. Nat. Methods 11 (11), 1144–1146. doi: 10.1038/nmeth.3103
Arnosti C., Wietz M., Brinkhoff T., Hehemann J. H., Probandt D., Zeugner L., et al. (2021). The biogeochemistry of marine polysaccharides: Sources, inventories, and bacterial drivers of the carbohydrate cycle. Annu. Rev. Mar. Sci. 13, 81–108. doi: 10.1146/annurev-marine-032020-012810
Arrigo K. R. (2005). Marine microorganisms and global nutrient cycles. Nature 437 (7057), 349–355. doi: 10.1038/nature04159
Azam F., Malfatti F. (2007). Microbial structuring of marine ecosystems. Nat. Rev. Microbiol. 5 (10), 782–791. doi: 10.1038/nrmicro1747
Benner R., Pakulski J. D., McCarthy M., Hedges J. I., Hatcher P. G. (1992). Bulk chemical characteristics of dissolved organic matter in the ocean. Science 255 (5051), 1561–1564. doi: 10.1126/science.255.5051.1561
Bianchi D., Weber T. S., Kiko R., Deutsch C. (2018). Global niche of marine anaerobic metabolisms expanded by particle microenvironments. Nat. Geosci. 11 (4), 263–268. doi: 10.1038/s41561-018-0081-0
Bižić-Ionescu M., Ionescu D., Grossart H.-P. (2018). Organic particles: Heterogeneous hubs for microbial interactions in aquatic ecosystems. Front. Microbiol. 9, 2569. doi: 10.3389/fmicb.2018.02569
Bižić-Ionescu M., Zeder M., Ionescu D., Orlić S., Fuchs B. M., Grossart H.-P., et al. (2015). Comparison of bacterial communities on limnic versus coastal marine particles reveals profound differences in colonization. Environmental Microbiology 17 (10), 3500–3514. doi: 10.1111/1462-2920.12466
Boeuf D., Edwards B. R., Eppley J. M., Hu S. K., Poff K. E., Romano A. E., et al. (2019). Biological composition and microbial dynamics of sinking particulate organic matter at abyssal depths in the oligotrophic open ocean. Proc. Natl. Acad. Sci. United States America 116 (24), 11824–11832. doi: 10.1073/pnas.1903080116
Bokulich N. A., Kaehler B. D., Rideout J. R., Dillon M., Bolyen E., Knight R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6 (1), 90. doi: 10.1186/s40168-018-0470-z
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Bolyen E., Rideout J. R., Dillon M. R., Bokulich N. A., Abnet C. C., Al-Ghalith G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37 (9), 1091–1091. doi: 10.1038/s41587-019-0252-6
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., Holmes S. P. (2016). DADA2: High-resolution sample inference from illumina amplicon data. Nat. Methods 13 (7), 581–58+. doi: 10.1038/Nmeth.3869
Caporaso J. G., Lauber C. L., Walters W. A., Berg-Lyons D., Lozupone C. A., Turnbaugh P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. United States America 108, 4516–4522. doi: 10.1073/pnas.1000080107
Chaumeil P.-A., Mussig A. J., Hugenholtz P., Parks D. H. (2019). GTDB-tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36 (6), 1925–1927. doi: 10.1093/bioinformatics/btz848%JBioinformatics
Crespo B. G., Pommier T., Fernández-Gómez B., Pedrós-Alió C. (2013). Taxonomic composition of the particle-attached and free-living bacterial assemblages in the Northwest Mediterranean Sea analyzed by pyrosequencing of the 16S rRNA. MicrobiologyOpen 2 (4), 541–552. doi: 10.1002/mbo3.92
Crump B. C., Armbrust E. V., Baross J. A. (1999). Phylogenetic analysis of particle-attached and free-living bacterial communities in the Columbia river, its estuary, and the adjacent coastal ocean. Appl. Environ. Microbiol. 65 (7), 3192–3204. doi: 10.1128/AEM.65.7.3192-3204.1999
De La Rocha C. L., Passow U. (2014). “The biological pump,” in Holland H. D., Turekian K. K. (Eds.), Treatise on geochemistry (Second edition) (Oxford: Elsevier) 93–122. doi: 10.1016/B978-0-08-095975-7.00604-5
Ganesh S., Parris D. J., DeLong E. F., Stewart F. J. (2014). Metagenomic analysis of size-fractionated picoplankton in a marine oxygen minimum zone. ISME J. 8 (1), 187–211. doi: 10.1038/ismej.2013.144
Grote J., Thrash J. C., Huggett M. J., Landry Z. C., Carini P., Giovannoni S. J., et al. (2012). Streamlining and core genome conservation among highly divergent members of the SAR11 clade. Mbio 3 (5), e00252-00212. doi: 10.1128/mBio.00252-12
Hollibaugh J., Wong P., Murrell M. (2000). Similarity of particle-associated and free-living bacterial communities in northern San Francisco bay, California. Aquat. Microbial Ecol. 21, 103–114. doi: 10.3354/ame021103
Huerta-Cepas J., Szklarczyk D., Heller D., Hernandez-Plaza A., Forslund S. K., Cook H., et al. (2019). eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47 (D1), D309–D314. doi: 10.1093/nar/gky1085
Hyatt D., Chen G. L., LoCascio P. F., Land M. L., Larimer F. W., Hauser L. J. (2010). Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11, 119. doi: 10.1186/1471-2105-11-119
Jain A., Balmonte J. P., Singh R., Bhaskar P. V., Krishnan K. P. (2021). Spatially resolved assembly, connectivity and structure of particle-associated and free-living bacterial communities in a high Arctic fjord. FEMS Microbiol. Ecol. 97 (11). doi: 10.1093/femsec/fiab139
Kanehisa M., Furumichi M., Sato Y., Ishiguro-Watanabe M., Tanabe M. (2021). KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 49 (D1), D545–D551. doi: 10.1093/nar/gkaa970
Kang D. D., Li F., Kirton E., Thomas A., Egan R., An H., et al. (2019). MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359. doi: 10.7717/peerj.7359
Katoh K., Misawa K., Kuma K., Miyata T. (2002). MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30 (14), 3059–3066. doi: 10.1093/nar/gkf436
Knight R., Vrbanac A., Taylor B. C., Aksenov A., Callewaert C., Debelius J., et al. (2018). Best practices for analysing microbiomes. Nat. Rev. Microbiol. 16 (7), 410–422. doi: 10.1038/s41579-018-0029-9
Lauro F. M., McDougald D., Thomas T., Williams T. J., Egan S., Rice S., et al. (2009). The genomic basis of trophic strategy in marine bacteria. Proc. Natl. Acad. Sci. U. S. A. 106 (37), 15527–15533. doi: 10.1073/pnas.0903507106
Li D. H., Liu C. M., Luo R. B., Sadakane K., Lam T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de bruijn graph. Bioinformatics 31 (10), 1674–1676. doi: 10.1093/bioinformatics/btv033
Li J. L., Salam N., Wang P. D., Chen L. X., Jiao J. Y., Li X., et al. (2018). Discordance between resident and active bacterioplankton in free-living and particle-associated communities in estuary ecosystem. Microb. Ecol. 76 (3), 637–647. doi: 10.1007/s00248-018-1174-4
Liu Y., Lin Q., Feng J., Yang F., Du H., Hu Z., et al. (2020). Differences in metabolic potential between particle-associated and free-living bacteria along pearl river estuary. Sci. Total Environ. 728, 138856. doi: 10.1016/j.scitotenv.2020.138856
Liu J., Zheng Y., Lin H., Wang X., Li M., Liu Y., et al. (2019). Proliferation of hydrocarbon-degrading microbes at the bottom of the Mariana trench. Microbiome 7 (1), 47. doi: 10.1186/s40168-019-0652-3
Logares R., Deutschmann I. M., Junger P. C., Giner C. R., Krabberød A. K., Schmidt T. S. B., et al. (2020). Disentangling the mechanisms shaping the surface ocean microbiota. Microbiome 8 (1), 55. doi: 10.1186/s40168-020-00827-8
Mestre M., Ruiz-González C., Logares R., Duarte C. M., Gasol J. M., Sala M. M. (2018). Sinking particles promote vertical connectivity in the ocean microbiome. Proc. Natl. Acad. Sci. U. S. A. 115 (29), E6799–E6807. doi: 10.1073/pnas.1802470115
Michael A. J. (2018). Polyamine function in archaea and bacteria. J. Biol. Chem. 293 (48), 18693–18701. doi: 10.1074/jbc.TM118.005670
Milici M., Vital M., Tomasch J., Badewien T. H., Giebel H. A., Plumeier I., et al. (2017). Diversity and community composition of particle-associated and free-living bacteria in mesopelagic and bathypelagic southern ocean water masses: Evidence of dispersal limitation in the bransfield strait. Limnology Oceanography 62 (3), 1080–1095. doi: 10.1002/lno.10487
MODIS-Aqua (2018) Moderate-resolution imaging spectroradiometer (MODIS) aqua chlorophyll concentration data (Greenbelt, MD, USA: NASA OB.DAAC). Available at: https://oceancolor.gsfc.nasa.gov/l3/order/ (Accessed 03/14/2022). NASA Goddard Space Flight Center, Ocean Ecology Laboratory, Ocean Biology Processing Group.
Morán X. A. G., Gasol J. M., Pernice M. C., Mangot J. F., Massana R., Lara E., et al. (2017). Temperature regulation of marine heterotrophic prokaryotes increases latitudinally as a breach between bottom-up and top-down controls. Glob Chang Biol. 23 (9), 3956–3964. doi: 10.1111/gcb.13730
Ning D., Yuan M., Wu L., Zhang Y., Guo X., Zhou X., et al. (2020). A quantitative framework reveals ecological drivers of grassland microbial community assembly in response to warming. Nat. Commun. 11 (1), 4717. doi: 10.1038/s41467-020-18560-z
Olm M. R., Brown C. T., Brooks B., Banfield J. F. (2017). dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11 (12), 2864–2868. doi: 10.1038/ismej.2017.126
Orsi W. D., Smith J. M., Wilcox H. M., Swalwell J. E., Carini P., Worden A. Z., et al. (2015). Ecophysiology of uncultivated marine euryarchaea is linked to particulate organic matter. Isme J. 9 (8), 1747–1763. doi: 10.1038/ismej.2014.260
Ortega-Retuerta E., Joux F., Jeffrey W. H., Ghiglione J. F. (2013). Spatial variability of particle-attached and free-living bacterial diversity in surface waters from the Mackenzie river to the Beaufort Sea (Canadian Arctic). Biogeosciences 10 (4), 2747–2759. doi: 10.5194/bg-10-2747-2013
Parks D. H., Imelfort M., Skennerton C. T., Hugenholtz P., Tyson G. W. (2015). CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25 (7), 1043–1055. doi: 10.1101/gr.186072.114
Partensky F., Hess W. R., Vaulot D. (1999). Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiol. Mol. Biol. Rev. 63 (1), 106–10+. doi: 10.1128/Mmbr.63.1.106-127.1999
Price M. N., Dehal P. S., Arkin A. P. (2010). FastTree 2-approximately maximum-likelihood trees for Large alignments. PloS One 5 (3), e9490. doi: 10.1371/journal.pone.0009490
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41 (D1), D590–D596. doi: 10.1093/nar/gks1219
Sharp J. H. (1973). Size classes of organic carbon in seawater. Limnology Oceanography 18 (3), 441–447. Available at: https://aslopubs.onlinelibrary.wiley.com/doi/pdf/10.4319/lo.1973.18.3.0441.
Sichert A., Corzett C. H., Schechter M. S., Unfried F., Markert S., Becher D., et al. (2020). Verrucomicrobia use hundreds of enzymes to digest the algal polysaccharide fucoidan. Nat. Microbiol. 5 (8), 1026–1039. doi: 10.1038/s41564-020-0720-2
Simon H. M., Smith M. W., Herfort L. (2014). Metagenomic insights into particles and their associated microbiota in a coastal margin ecosystem. Front. Microbiol 5 466. doi: 10.3389/fmicb.2014.00466
Song W.-Z., Thomas T. (2017). Binning_refiner: Improving genome bins through the combination of different binning programs. Bioinformatics 33 (12), 1873–1875. doi: 10.1093/bioinformatics/btx086%J.Bioinformatics
Suzuki S., Kaneko R., Kodama T., Hashihama F., Suwa S., Tanita I., et al. (2017). Comparison of community structures between particle-associated and free-living prokaryotes in tropical and subtropical pacific ocean surface waters. J. Oceanography 73 (3), 383–395. doi: 10.1007/s10872-016-0410-0
Szabo R. E., Pontrelli S., Grilli J., Schwartzman J. A., Pollak S., Sauer U., et al. (2022). Historical contingencies and phage induction diversify bacterioplankton communities at the microscale. Proc. Natl. Acad. Sci. U. S. A. 119 (30), e2117748119. doi: 10.1101/2021.09.27.461956%JbioRxiv. 2027.461956.
Tarn J., Peoples L. M., Hardy K., Cameron J., Bartlett D. H. (2016). Identification of free-living and particle-associated microbial communities present in hadal regions of the Mariana trench. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00665
Thiele S., Fuchs B. M., Amann R., Iversen M. H. (2015). Colonization in the photic zone and subsequent changes during sinking determine bacterial community composition in marine snow. Appl. Environ. Microbiol. 81 (4), 1463–1471. doi: 10.1128/AEM.02570-14
Ulloa O., Henriquez-Castillo C., Ramirez-Flandes S., Plominsky A. M., Murillo A. A., Morgan-Lang C., et al. (2021). The cyanobacterium prochlorococcus has divergent light-harvesting antennae and may have evolved in a low-oxygen ocean. Proc. Natl. Acad. Sci. United States America 118 (11). doi: 10.1073/pnas.2025638118
Uritskiy G. V., DiRuggiero J., Taylor J. (2018). MetaWRAP–a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6 (1), 158. doi: 10.1186/s40168-018-0541-1
Wu Y.-W., Simmons B. A., Singer S. W. (2015). MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32 (4), 605–607. doi: 10.1093/bioinformatics/btv638%J.Bioinformatics
Yuan H., Li T., Li H., Wang C., Li L., Lin X., et al. (2021). Diversity distribution, driving factors and assembly mechanisms of free-living and particle-associated bacterial communities at a subtropical marginal Sea. Microorganisms 9 (12). doi: 10.3390/microorganisms9122445
Yung C. M., Ward C. S., Davis K. M., Johnson Z. I., Hunt D. E. (2016). Insensitivity of diverse and temporally variable particle-associated microbial communities to bulk seawater environmental parameters. Appl. Environ. Microbiol. 82 (11), 3431–3437. doi: 10.1128/aem.00395-16
Zhang C., Dang H., Azam F., Benner R., Legendre L., Passow U., et al. (2018). Evolving paradigms in biological carbon cycling in the ocean. Natl. Sci. Rev. 5 (4), 481–499. doi: 10.1093/nsr/nwy074%JNationalScienceReview
Zhang R., Liu B., Lau S. C., Ki J. S., Qian P. Y. (2007). Particle-attached and free-living bacterial communities in a contrasting marine environment: Victoria harbor, Hong Kong. FEMS Microbiol. Ecol. 61 (3), 496–508. doi: 10.1111/j.1574-6941.2007.00353.x
Keywords: oligotrophic ocean, prokaryotes, particle-associate, free-living, community assembly mechanisms, biogeography, metabolic potential
Citation: Ren Y, Luo Z, Liu Q, Wei B, Wu Y-H, Shu W-S and Xu X-W (2022) Insights into community assembly mechanisms, biogeography, and metabolic potential of particle-associated and free-living prokaryotes in tropical oligotrophic surface oceans. Front. Mar. Sci. 9:923295. doi: 10.3389/fmars.2022.923295
Received: 19 April 2022; Accepted: 22 August 2022;
Published: 15 September 2022.
Edited by:
Mar Benavides, Institut de Recherche Pour le Développement (IRD), FranceReviewed by:
Mina Bizic, Leibniz-Institute of Freshwater Ecology and Inland Fisheries (IGB), GermanyCopyright © 2022 Ren, Luo, Liu, Wei, Wu, Shu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xue-Wei Xu, eHV4d0BzaW8ub3JnLmNu; Wen-Sheng Shu, c2h1d2Vuc2hlbmdAbS5zY251LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.