 Gaoyou Yao
Gaoyou Yao Hua Zhang1,3,4
Hua Zhang1,3,4 Maoxian He
Maoxian He
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 22 July 2022
Sec. Deep-Sea Environments and Ecology
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.920327
Knowledge about community structure and genetic diversity can help assess the potential for change in the loss and restoration of biodiversity, thereby facilitating effective management and ecosystem protection. Macroinvertebrate communities are an important biotic component of deep sea cold seep ecosystems. As Haima cold seep is increasingly being assessed for its potential gas hydrate mineral wealth, knowledge of community characteristics and genetic diversity of macrobenthos is needed to anticipate the potential impacts on biodiversity. In this study, we examined species diversity and community structure at five sites in the Haima cold seep using a remote-operated vehicle (ROV) for in situ surveying. The results identified 12 macrobenthic species from 5 phyla and 12 families. The macrobenthos community could be divided into two communities (H1 and H2=mussel bed community, and H3 and H4= vesicomyid clams community) based on CLUSTER and NMDS analyses. Gigantidas haimaensis (Mollusca), Branchipolynoe pettiboneae (Annelida), and Histampica haimaensis (Echinodermata) were most dominant within their respective phyla, with values of the dominance of 0.160, 0.021 and 0.114, respectively. The genetic diversity of these three typical macrobenthic species in the Haima cold seep was evaluated using the mitochondrial cytochrome c oxidase subunit I (COI) gene, haplotype, and nucleotide diversity values were 0.651 to 0.912 and 0.00148 to 0.00812, respectively, representing high haplotype diversity but low nucleotide diversity. Finally, mitochondrial concatenated dataset (MCD) sequences from three mitochondrial genes (ATP6, COI, and NAD4) and 294,734 genome-wide single nucleotide polymorphisms (SNPs) from restriction site-associated DNA-sequencing (RAD-seq) data were obtained from 60 individuals from two sites (H1 and H2), providing deep insight into the genetic diversity and structure of G. haimaensis, the engineer species in Haima cold seep. No significant genetic differentiation between G. haimaensis in H1 and H2 was detected based on MCD sequences. Nevertheless, when using SNP datasets, a small but clear genetic subdivision between G. haimaensis in the two sites as revealed by STRUCTURE and principal component analysis (PCA). The results comprehensively illuminate macrobenthos biodiversity in the Haima cold seep ecosystem and provide a baseline against which population dynamics may be assessed in the future.
Cold seeps previously believed to support minimal life are generally situated in the deep sea, with extremely high levels of toxic compounds and exceptionally high pressure (Yang et al., 2012). Unlike shallow-water ecosystems, which are based on photosynthesis, cold seep ecosystems rely on chemosynthetic production, in which chemotrophic bacteria perform oxidative reduction reactions to generate energy for carbon fixation (Feng et al., 2018). The high chemosynthetic primary production enables these unique ecosystems to develop high biomass in deep-water ecosystems (Ramirez-Llodra et al., 2010). Knowledge about these unique deep-sea chemotrophic ecosystems could expand our understanding of Earth’s biodiversity (Vrijenhoek, 2010; Baco et al., 2016).
Macroinvertebrate communities are important biotic components of aquatic ecosystems, and a sound understanding of the genetic diversity and community structure of macroinvertebrates can provide a basis for assessing community changes following anthropogenic or natural disturbances (Hunter and Halanych, 2008). In particular, genetic diversity is associated with population fitness and is central to many conservation challenges such as species response to environmental changes, ecosystem recovery, and the viability of recently endangered populations (Frankham, 2010; Romiguier et al., 2014). Loss of genetic diversity can result in inbreeding depression, which can trigger a vortex of extinction, even in seemingly healthy populations, especially in populations with low effective sizes that are prone to genetic drift (Tanaka, 2000). Although maintenance of genetic diversity has been recognised as three essential components in conservation biology, genetic diversity is greatly ignored in management and policy (Laikre et al., 2010). In deep-sea ecosystems, patchy distribution of low-quality food, low faunal abundance, and low temperatures slow biological processes (McClain et al., 2012; Jones et al., 2017), making deep-sea species vulnerable to environmental change (Miyazaki et al., 2013; Breusing et al., 2015). By monitoring community structure and genetic diversity in deep-sea ecosystems, we can assess the potential for change in the loss and restoration of diversity and ecosystem resilience. However, investigating these remote ecosystems is challenging due to technical and financial limitations. As a result, information about the community structure of macrobenthos and genetic diversity in deep-sea ecosystems remains scarce (Breusing et al., 2015; Thaler et al., 2017).
Haima cold seep, discovered in the South China Sea in 2015 (Liang et al., 2017), covers an area of 618 km2, including an active cold seep area of 350 km2(Zhao et al., 2020). However, huge amounts of gas hydrates providing a potentially vital energy resource have been detected in the shallow sediments of the Haima cold seep (Fang et al., 2019), making the area a potential target of gas hydrates extractions, which threatens deep-sea ecosystems. Therefore, there is an urgent need for efficient biodiversity surveys to inform conservation. Studies on biodiversity of some deep-sea species in Haima cold seep have been reported (Sun et al., 2018; Dong et al., 2021; Ke et al., 2022); however, knowledge on community characteristics and genetic diversity of macrobenthos in Haima cold seep remains limited. As Haima cold seep is increasingly being assessed for its gas hydrate mineral wealth, baseline data on the biodiversity of benthic invertebrates is needed for effective management and ecosystem protection (Everett and Park, 2018). In the present work, remote operated vehicle (ROV) in situ surveying, mitochondrial DNA fragment analysis of dehydrogenase subunit 4 (NAD4), ATP synthase F0 subunit 6 (ATP6), and cytochrome c oxidase submit I (COI), and restriction site-associated DNA sequencing (RAD-seq) were used to comprehensively characterise genetic diversity and composition of the macrobenthos communities in Haima cold seep. The results provide a valuable baseline biodiversity assessment prior to gas hydrates extractions.
Five sites were selected in Haima cold seep (Figure 1). During cruise HYDZ6-202005 of the Haiyang 6 research vessel of the Guangzhou Marine Geological Survey, specimens were collected using the ROV Haima on September 1-6, 2020. Specimens were sampled using a 30 cm diameter, and 50 cm length sampling net manipulated by mechanical arms and photographed and videotaped using high-definition underwater cameras during the dive. The sampling net was held perpendicular to the sea floor when sampling and moved in parallel until the macrobenthos reached the bottom of the sampler. Megabenthos (e.g., Neolithodes brodiei) that a sampler cannot capture was directly grabbed using the mechanical arms or qualitatively analysed based on video records. The collected species were photographed immediately after being transported to the deck, then placed in liquid nitrogen and stored at -80°C. Morphological characteristics were used to preliminarily identify species for collected organisms according to relevant references (Zhao et al., 2020; Dong et al., 2021), and DNA barcodes were used for further species identification. The description of the species identified was given in supplementary electronic material.
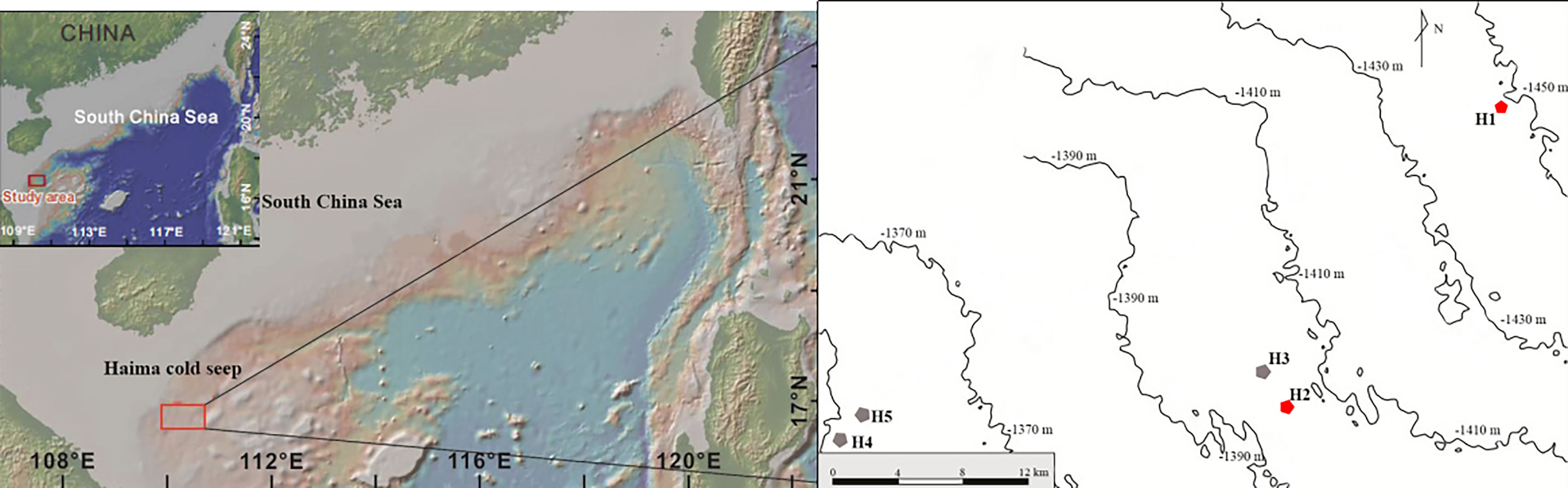
Figure 1 Locations for sample collection of G. haimaensis. Five investigating sampling sites were set, only H1 and H2 that were marked by red hexagon were found to have G. haimaensis populations. H3, H4 and H5 in which no mussel population were marked by grey hexagon.
Species richness was estimated based on species presence/absence data (Marshall and Stepien, 2020). The percentage of species richness detected was calculated as a number of species collected at a given locality divided by the total number of species collected at the Haima cold seep. The relative abundances of species were calculated from species count data. Shannon-wiener (Shannon, 1948), Pielou (Pielou, 1966), and Margalef (Margalef, 1968) indices were used to assess biodiversity. The dominant value (Y) was used to calculate species dominance in Haima cold seep using the followed formula:
Y =fi´pi,pi= ni/N (1)
“N” is the total number of samples collected, “pi” is the proportion of species “i” within the total number of samples collected, and “fi” is the frequency of occurrence of species “i” at sampling points. Nonmetric multidimensional scaling (NMDS) and CLUSTER analyses were used to visualise the community structure of macrobenthos patterns using Primer 7 (https://www.primer-e.com/). Site H5 was removed from the analysis since this station has no fauna.
The type species from each phylum in Haima cold seep were chosen by the dominance values for analysis of population genetic diversity, which were G. haimaensis (Mollusca), Branchipolynoe pettiboneae (Annelida) and Histampica haimaensis (Echinodermata) Representative Cnidaria and Arthropoda species were absent due to insufficient samples. A total of 60 specimens (30 each for H1 and H2) of G. haimaensis, 14 specimens of B. pettiboneae, and 16 specimens of H. haimaensis (including 32 pre-existing H. haimaensis COI sequences downloaded from GenBank) were used for subsequent genetic diversity analysis based on COI.
Primers HCO2198 and LCO1490 (Vrijenhoek, 1994) were used to amplify COI. Total genomic DNA was extracted using an EasyPure Marine Animal Genomic DNA Kit (Transgen, Beijing, China) following the manufacturer’s instructions. PCR amplification was carried out in a volume of 30 μL containing 1 µL of DNA template (100-200 ng/µL), 0.5 μL of each primer (10 μM), 13 µL of ddH2O, and 15 μL of SuperMix (Transgen). Thermal cycling included an initial denaturation for 5 min at 94°C, followed by 34 cycles of denaturation for 30 s at 94°C, annealing for 30 s at 52°C, and extending for 30 s at 72°C, followed by a final extension at 72°C for 10 min. PCR products were separated by 1% agarose gel electrophoresis, stained with ethidium bromide, then sequenced directionally (BGI, Shenzhen, China). Raw sequences were verified by visualising sequence peaks and assembled using the NASTAR Lasergene package. Sequence alignment was performed using MEGA v6.0 with default settings for sequence variation analysis (complete deletion), including parsimony informative sites and segregating sites (Tamura et al., 2011).
G. haimaensis is the most important, iconic, and engineer species in the Haima cold seep ecosystem (Liang et al., 2017; Xu et al., 2019). Dense mussel beds generate a highly complex habitat for many deep-sea species to inhabit. Thus, genetic diversity and structure were further assessed using three mitochondrial genes (COI, ATP6, and NAD4). Sixty (60) specimens of G. haimaensis from H1 (30 individuals) and H2 (30 individuals) were sampled. ATP6 (Xu et al., 2019) and NAD4 (Bielawski and Gold, 1996) were amplified using primers as previously described. The procedure for amplification was the same as that described above. Aligned sequences were removed and trimmed simultaneously. Finally, manual assembly yielded a mitochondrial concatenated dataset (MCD, ATP6+COI+NAD4) which was used for subsequent analysis.
The above samples were subjected to RAD-seq, and the library was created according to an established method (Etter et al., 2012) using 0.1−1 μg of genomic DNA from each sample. DNA was digested using the restriction enzyme EcoRI, Solexa P1 Adapter, and Solexa P2 Adapter were ligated to the digested products, and libraries were sequenced on an Illumina platform to obtain 150 bp paired-end reads. To ensure that reads were reliable and without artificial bias in subsequent analyses, raw data (raw reads) in FASTQ format were first processed through a series of quality-control (QC) procedures. QC standards were as follows: (1) reads with ≥10% unidentified nucleotides (Ns) were removed; (2) reads with >50% of bases having a Phred quality<5 were removed; (3) reads with >10 nt aligned to the adapter were removed, allowing ≤10% mismatches. Finally, we obtained high-quality reads (average Q20 = 94.52% and average Q30 = 87.5%). The error rate reads were calculated using in-house Perl scripts made by Novogene Co., Ltd (Beijing, China;Setting:-r 150 -N 0.1 -q 33 -L 5 -p 0.5). Next, RAD tags were extracted from the draft genome (Sun et al., 2017), which served as a reference for downstream alignment and SNP discovery. Burrows-Wheeler Aligner (BWA) software was used to align clean reads from each individual against the reference genome (settings = mem–t − 4–k 32–M) (Li & Durbin, 2009). Alignment files were converted to bamfiles using SAMtools software (settings = rmdup). After alignment, SNP calling was performed using a conservative Bayesian approach as implemented in SAMtools (Li et al., 2009). To improve the accuracy of SNP genotyping, additional filters were applied using (1) coverage depth >3 and (2) global minor allele frequency (MAF) ≥0.05.
Genetic diversity, including haplotype diversity (Hd), the number of haplotypes (H), and nucleotide diversity (π), were estimated using DNasp5.10 (Librado and Rozas, 2009). Population demography was examined using two different approaches based on COI sequences. Demographic history was investigated by mismatch distributions using DnaSP5.10 (Librado and Rozas, 2009), and Tajima’s D (Tajima, 1989) and Fu’s Fs (Fu, 1997) tests were conducted to examine the neutrality of sequences with Arlequin 3.5 (Shen et al., 2016).
The genetic diversity index of G. haimaensis based on MCD was calculated using the method described above, including Hd, H and π. Expected heterozygosity (He), the effective number of alleles (Ne), and observed heterozygosity (Ho) were calculated using genome-wide SNPs. To examine the genealogical relationships among mitochondrial DNA (mtDNA) haplotypes, haplotype network analysis based on COI, NAD4, and ATP6 was performed using Network 4.6 (Bandelt et al., 1999). Genetic divergence (Fst) of G. haimaensis between H1 and H2 was examined based on MCD sequences using Arlequin 3.5 (Excoffier and Lischer, 2010). The population structure of G. haimaensis for H1 and H2 was examined using the Bayesian algorithm implemented in STRUCTURE 2.3.4 (Pritchard et al., 2000) based on the SNP dataset. The optimal value of K was estimated using both Puechmaille’s method (Puechmaille, 2016) and Evanno’s ΔK method (Evanno et al., 2005) as implemented in STRUCTURE SELECTOR (Li and Liu, 2018). FST values between the two populations of G.haimaensis based on SNPs were calculated using Arlequin, with 10,000 permutations to determine significance. Principle component analysis (PCA) was performed to explore population division.
Fst vs heterozygosity analysis was implemented to identify the outliers loci derived from habitat selection. A locus with heterozygosity and an FST value above the 99.95th percentile of the related distribution is considered an outlier (Bovo et al., 2020). FST and heterozygosity of H1 and H2 were calculated with vcftools v 0.1.14 and the script (Setting:fst-window-size: 10,000; fst-window-step: 5,000). The top 0.05 Hp and top 0.05 Fst of the two sites of G. haimaensis were intersected, and then the intersection of the two populations was combined for deduplicating. Finally, the outliers loci were filtered using VCFtools.
Twelve macrobenthic species from five phyla and 12 families were identified. Two of 12 macrofauna species were identified for the first time in Haima cold seep (Table S1). Regarding relative abundance, Mollusca (41.95%), Annelida (32.33%) and Echinodermata (15.36%) were the most represented phyla. The most abundant species was G. haimaensis (30.09%), followed by B. pettiboneae (27.22%) and Histampica haimaensis (14.23%; Figure 2). According to the values of the dominance of macrobenthos in Haima cold seep, G. haimaensis (0.160), B. pettiboneae (0.145) and Histampica haimaensis (0.114) were the most represented members of Mollusca, Annelida and Echinodermata, respectively (Table S2).
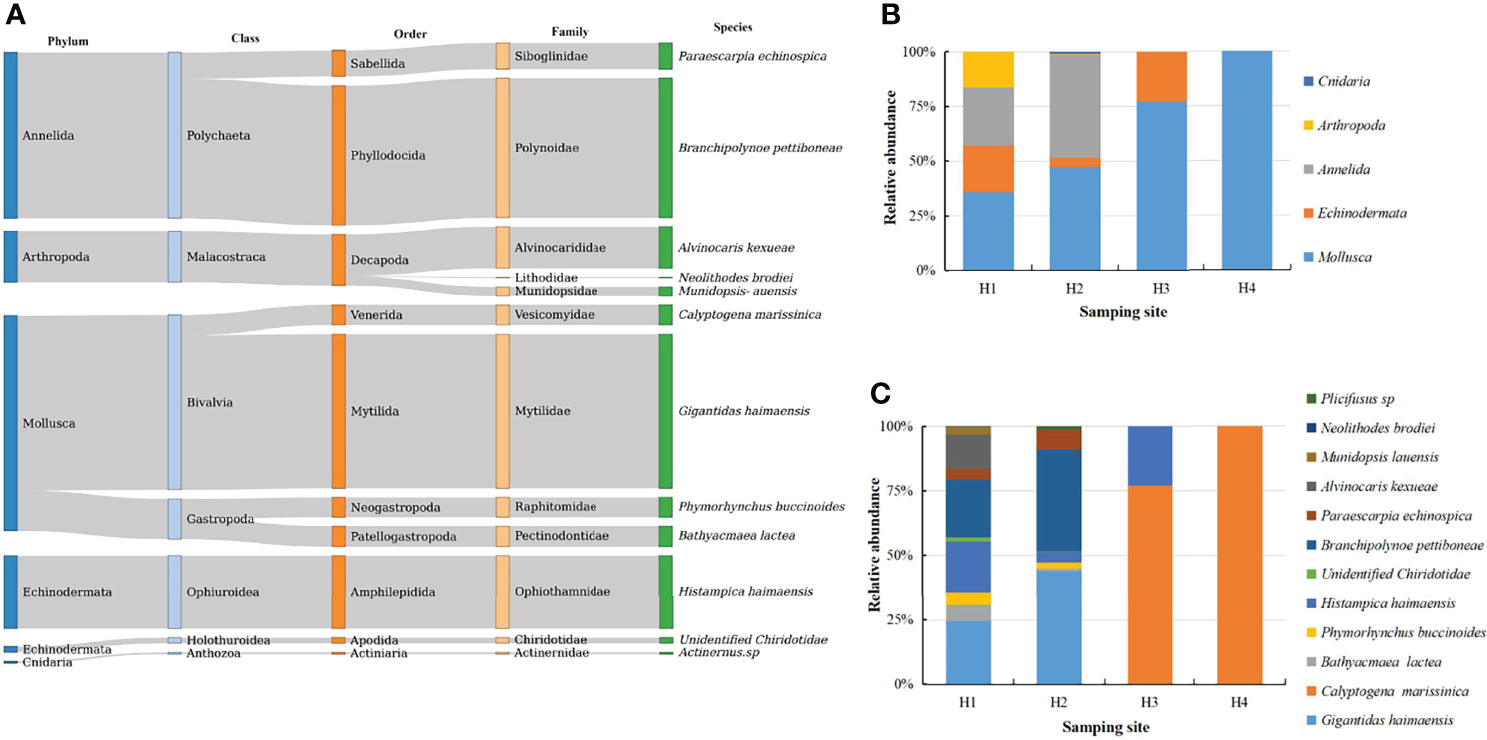
Figure 2 Relative abundance of epibenthic macrofauna in Haima cold seep at each taxonomic level (A), at the phylum level (B) and species level (C) in every sampling site.
Regarding community structure, 10, 9, 2 and 1 species were identified at sites H1, H2, H3 and H4, respectively (Table S1). The highest species richness, species abundance and biomass were observed in site H1 (S = 10, D = 1.45, H = 1.906) and H2 (S = 9, D = 1.421, H = 1.267) (Table 1). The macrobenthos community could be divided into two communities (H1 and H2=mussel bed community, and H3 and H4= vesicomyid clams community) based on CLUSTER and NMDS analyses (Figure S1).
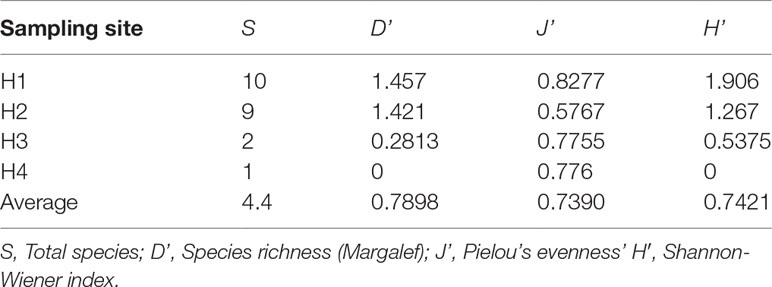
Table 1 Indexes of the biodiversity analysis of the macrobenthos community in Haima cold seep.
Based on COI sequences, the haplotype diversity, the number of haplotypes, and nucleotide diversity of the three representative macrobenthic species are presented in Table 2. For G. haimaensis, 55 COI sequences (633 bp) were successfully amplified, along with 13 segregating sites (2.05%), four of which were parsimony informative, and 16 haplotypes were detected. The nucleotide and haplotype diversity values of G. haimaensis were 0.651 and 0.00148, respectively. For B. pettiboneae, COI sequences (624 bp) were successfully amplified, along with 25 segregating sites (4.00%), eight of which were parsimony informative, and eight haplotypes were detected. The nucleotide diversity and haplotype diversity values of B. pettiboneae was 0.00812 and 0.912, respectively. For H. haimaensis, 48 COI sequences (528 bp) were obtained, along with 11 segregating sites (2.08%), six of which were parsimony informative, and 11 haplotypes were detected. The nucleotide diversity and haplotype diversity values of B. pettiboneae were 0.00214 and 0.748, respectively.
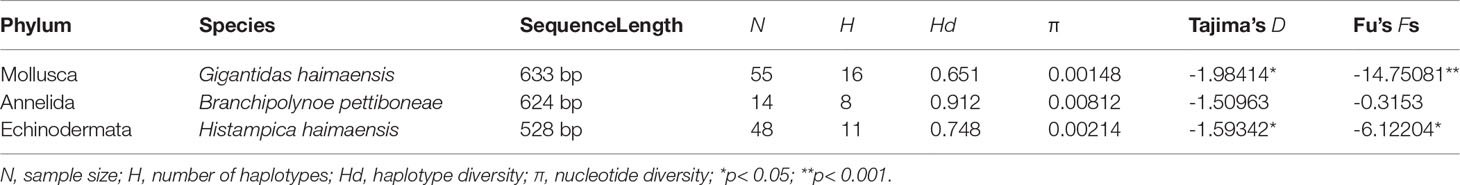
Table 2 DNA polymorphism and neutrality tests values for the three representative macrobenthos species in Haima cold seep based on COI sequences.
To provide insight into the demographic history of the three representative macrobenthos species, Fu’s Fs and mismatch distribution were inferred. Both Fu’s Fs and Tajima’s D values were found to be negative and significant for G. haimaensis and H. haimaensis populations in Haima cold seep (p<0.05), and all mismatch distribution shapes were clearly unimodal, apart from the B. pettiboneae population in Haima cold seep (Figure 3).
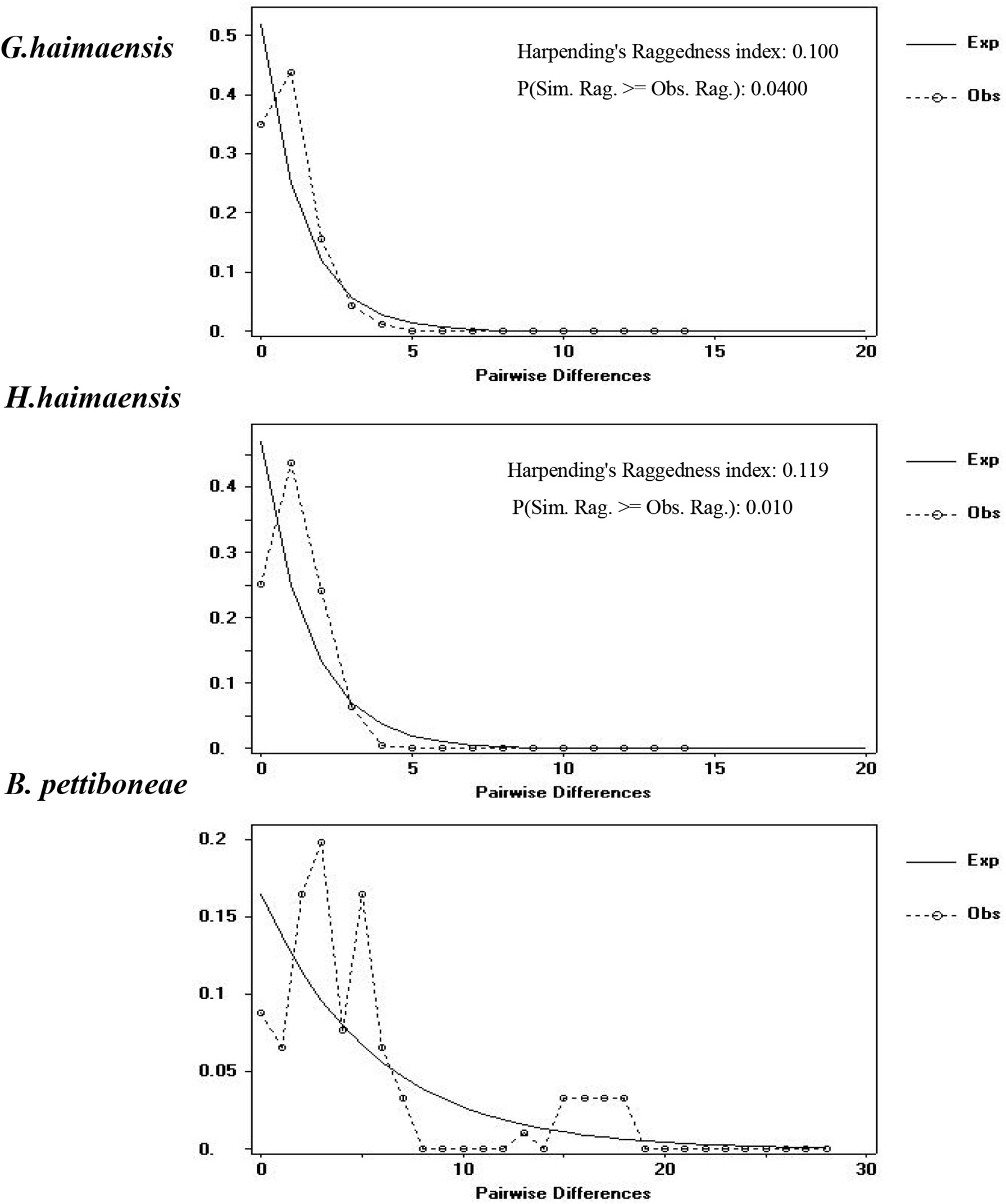
Figure 3 Observed (broken lines) and expected (solid lines) mismatch distribution for three representative macrobenthos species basing on COI sequence. X-axis = pairwise differences and Y-axis = frequency.
Based on MCD sequences, the haplotype diversity, the number of haplotypes, and nucleotide diversity for the two groups of G. haimaensis are presented in Table 3. A total of 41 haplotypes were identified from 51 G. haimaensis individuals (Table S3). The Hd value for H1 and H2 was 0.988 and 0.970, respectively. The π value was 0.00191 and 0.00179. Genetic diversity analysis of G. haimaensis detected a similar level of nucleotide and haplotype diversity between the two sites. The network exhibited a star-like shape, with the most frequent haplotypes shared between the two groups in the centre, surrounded by private and low-frequency haplotypes. No clade of haplotypes was revealed in haplotype networks constructed using a median-joining network (MJN) approach (Figure 4). The Fst between the two sites was 0.00745 (p >0.05), indicating no significant genetic differentiation between the two sites for G. haimaensis (Table 4).

Table 3 Summary genetic statistics of each local group of G. haimaensis based on MCD sequences.
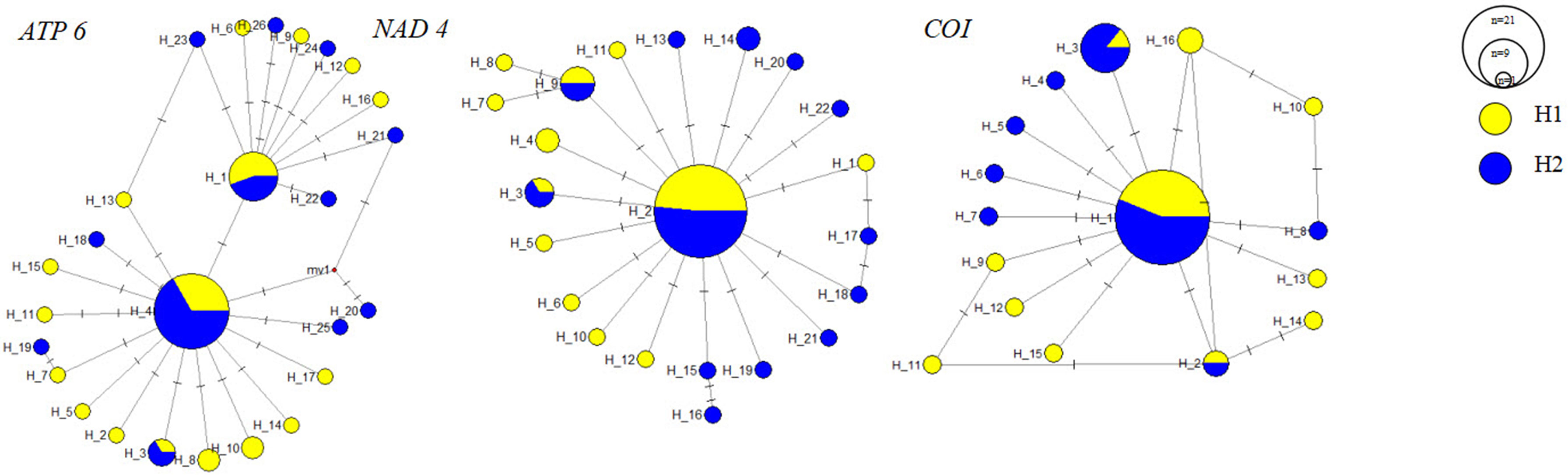
Figure 4 Statistical parsimony network for haplotypes from G. haimaensis samples collected at Haima cold seep. Large circles represent a single individual. H1 represented by yellow circles. H2 represented by blue circles.
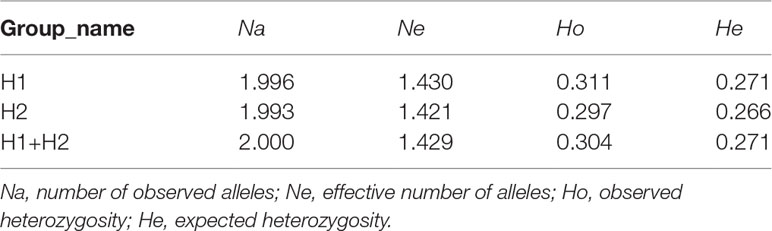
Table 4 Genetic diversity of G. haimaensis among the two sites based on SNPs.
RAD-seq yielded ~953 million reads in total, with a mean of 15.88 million reads per individual. The average mapping rate for each individual was 95.53%, with an average depth per individual of 14.00 (Table S4). The high mapping rate and sequencing depth indicated that the raw data could be used for subsequent SNP analysis. After genotyping and strict filtering, 294,734 SNPs were identified. Genetic diversity analysis of G. haimaensis based on SNPs detected a similar level between H1 and H2, with Na, Ne, Ho and He values of 2.000, 1.429, 0.304 and 0.271, respectively (Table 4). The pairwise Fst value calculated based on the entire set of 294,734 SNPs was 0.0106 (p<0.05). STRUCTURE analyses (i.e., optimal K = 2) revealed a small but clear differentiation between the two sites (Figure 5; Figure S3). The PCA results also detected this minor genetic subdivision of population genetic structure (the two minor separated clusters in the Figure 6). To test whether the genetic differentiation is not driven by a few outlier loci derived from habitat selection, Fst vs heterozygosity analyses were implemented. A total of 475 SNPs in 50 regions were eliminated, and the result showed that, with a few exceptions, individuals from two groups (H1 and H2) form distinct two clades (Figure S4). In addition, PCA at different allele coverage depths (dp=3 vs. dp=5) was also compared to determine whether the allele coverage depth might have affected the results. The result agrees with our findings, with identical results (Figure S5). The new results (Fst vs heterozygosity and dp3 vs. dp5) showed that the genetic differentiation is not driven by a few outlier loci and is affected by allele coverage depth, which is in close agreement with our conclusion (the genetic differentiation between the two mussel beds of G. haimaensis).
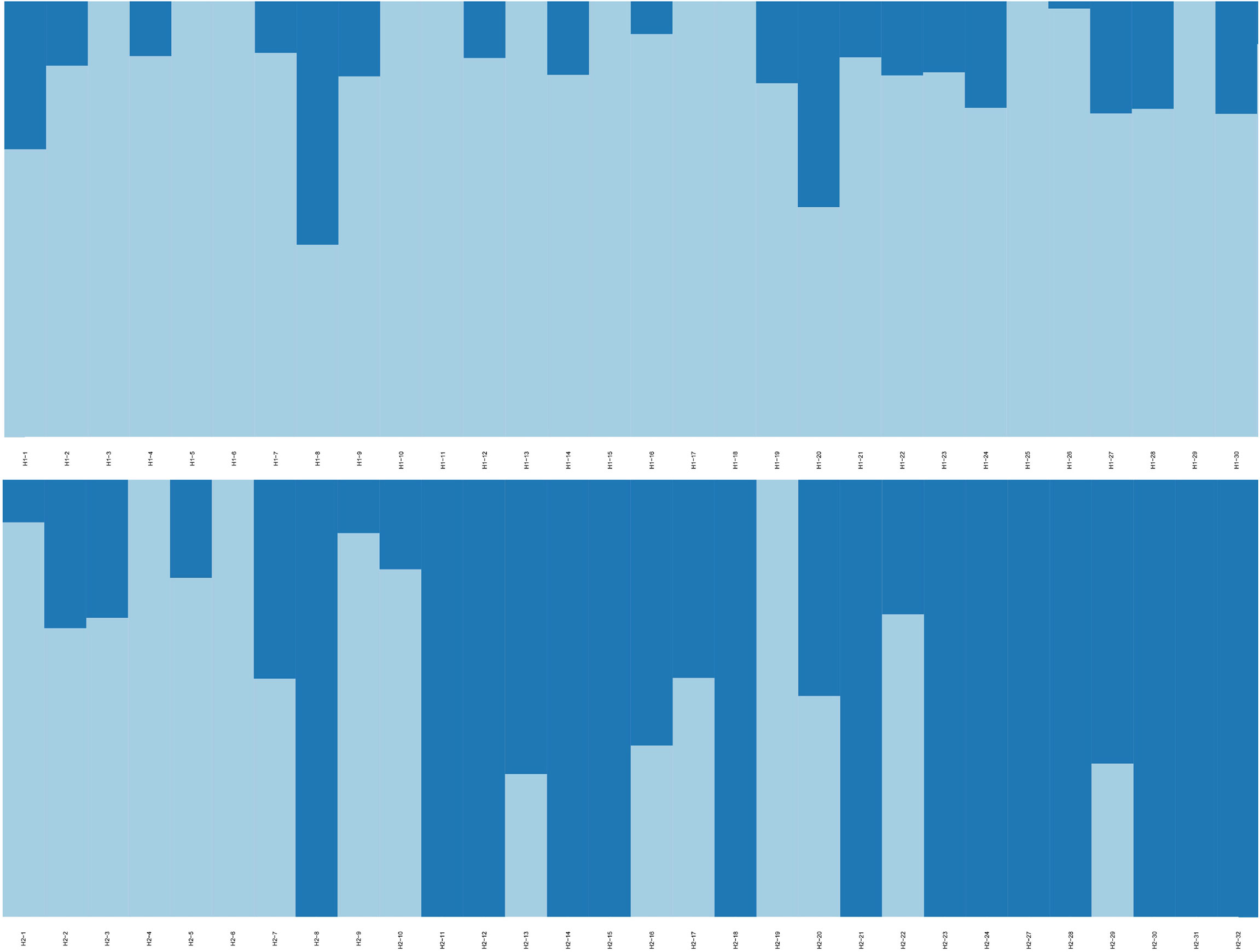
Figure 5 Population genetic structure of G. haimaensis at H1 and H2 sites based on the entire set of 294,734 SNPs using STRUCTURE analyses (K=2). Each individual is represented by a single bar, with different colors showing membership fractions of each inferred cluster. H1 is shown above and H2 is shown below.
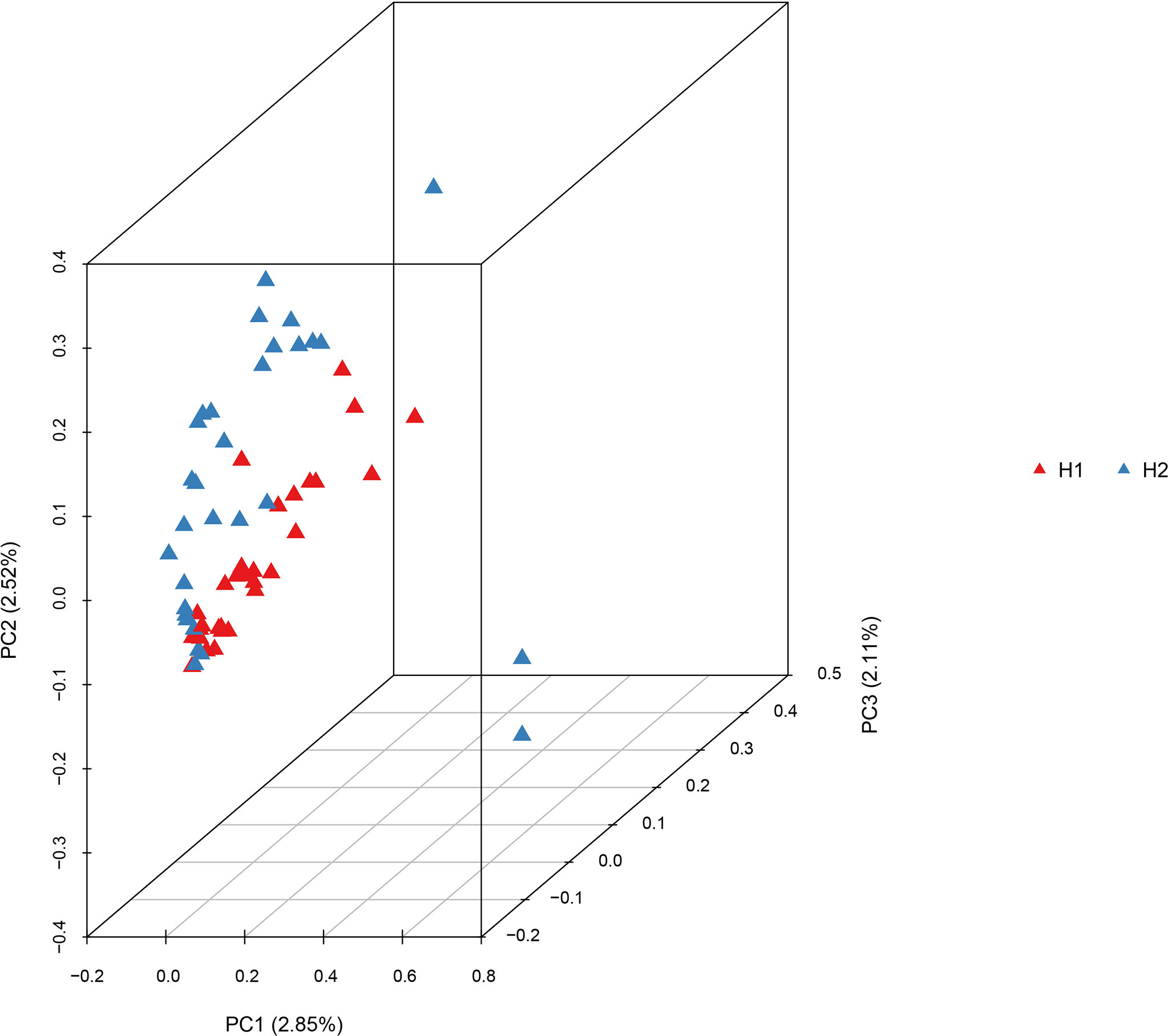
Figure 6 Principal component analysis (PCA) plot of G. haimaensis at two sites based on the 294,734 SNPs.
Knowledge about species diversity is fundamental for biodiversity conservation (Yang et al., 2004). In the present study, we identified 12 macrobenthic species from five phyla, and 12 families in the Haima cold seep surveys. By comparison, Dong et al. (2021) detected 24 macrofauna species in Haima cold seep. This apparent difference in species richness may be explained by differences in sampling effort (Nakajima et al., 2014). Data used in the present study was based on a single dive at each site, while multiple dives were implemented in Dong et al. (2021). Species richness at Haima cold seep is higher than 42 previously reported chemosynthesis-based ecosystems (range 2−38, mean 9.8) in the northwest Pacific (Nakajima et al., 2014). Biodiversity patterns are shaped by energy availability in deep-sea ecosystems (Woolley et al., 2016). The high species richness at Haima cold seep may be attributed to high active seep activities, the foundation for chemotrophic communities. Intensive investigation in recent years could be another reason for the apparent high species richness at Haima cold seep. In addition, our study identified two new macrofauna species for the first time in the Haima cold seep ecosystem, and the new records can expand our understanding of the geographic distributions of some species. For instance, A. kexueae (Figure S6) was previously only described from hydrothermal vents in the Manus Basin in the Southwest Pacific (WANG and SHA, 2017). However, its presence in Haima cold seep indicates that A. kexueae is not an endemic species because it inhabits both vents and cold seeps.
Two clear different kinds of communities (deep sea mussels and vesicomyid clams) were found in Haima cold seep, and the mussel community is much more diversified than the vesicomyid community. The dense mussel beds generate a highly complex habitat for many deep-sea species to inhabit (Vrijenhoek, 2010). Moreover, this may also be related to the fact that seepages are less vigorous for the clam beds, Calyptogena being able to collect sulfides in the sediment with their foot, leaving less chance for the epibenthic fauna to feed and survive. The community structure at Haima cold seep is similar to one of the three currently known active cold seeps (site F) in the South China Sea (SCS) (Feng et al., 2018). Several species are shared between Haima cold seep and site F, including B. pettiboneae, Shinkaia crosnieri and B. lactea, indicating possible community connectivity between the two sites. However, no study has confirmed this hypothesis. Future research should therefore determine whether gene exchange between benthic species. Although some species are shared between Haima cold seep and site F, substantial differences were observed. For example, the engineer species at Haima cold seep is G. haimaensis, whereas G. platifrons is dominant at site F. Apart from the position of umbones (G. haimaensis = subterminal and G. platifrons = terminal), the two species are similar in morphology (Xu et al., 2019) and genetics based on the mitochondrial genome (Zhang et al., 2021). However, the difference is that G. platifrons inhabiting both vents and cold seeps is widely distributed in the Northwest Pacific, while G. haimaensis is endemic to SCS in Northwest Pacific. In addition, Calyptogena marissinica, Ophiophthalmus sp and Phymorhynchus buccinoides are common species at Haima cold seep, but none were found at site F. Characterised by patchy distributions and high heterogeneity, deep-sea cold seeps reportedly harbour highly specialised communities (Teixeira et al., 2013), which might partially explain the finding that macrobenthos in Haima cold seep had a distinct and discernible community structure.
Genetic diversity is closely related to the survivability of populations and long-term adaptability, especially in drastically and suddenly changing marine environments (Barrett and Schluter, 2008). The present work examined the genetic diversity of three representative macrobenthic species (G. haimaensis, H. haimaensis and B. pettiboneae) in Haima cold seep using mtDNA markers for the first time. Genetic diversity analysis of G. haimaensis, H. haimaensis and B. pettiboneae populations in Haima cold seep detected a similar level of genetic diversity compared with other closely related species populations (Hurtado et al., 2004; Shen et al., 2016). For instance, haplotype diversity and nucleotide diversity of G. haimaensis populations were 0.980 and 0.00186, respectively, similar to most other deep-sea mussel species such as G. platifrons, for which Hd was 0.965 to 0.970 and π was 0.0015 to 0.0021 (Xu et al., 2018). Based on the haplotype and nucleotide diversity results, these three species exhibited low nucleotide diversity but high haplotype diversity (Hd >0.5 and π<0.05) (Grant and Bowen, 1998). This trend in genetic diversity was also observed for other deep-sea macrobenthic species populations, and it seems to be shared among macrobenthic communities in deep-sea ecosystems, especially deep-sea mussel communities (Table S5). These types of genetic diversity are also common in coastal mussel (eg. Perna viridis, Mytella charruana and Mytilus galloprovincialis. G. haimaensis population in Haima cold seep exhibits high haplotype diversity, but low nucleotide diversity, which may be explained by the high mutational rate of the mitochondrial gene. Haplotype diversity can increase rapidly in a short period of time by accumulating single nucleotide variations, while nucleotide polymorphism cannot co-occur (Avise et al., 1984). In addition, when a pioneer population with a low effective population size colonizes nascent vents or seeps, the founder effect might also account for the low genetic diversity of the macrobenthos in the Haima cold seep.
A total of 294,734 SNPs and three mitochondrial genes (ATP6, COI, and NAD4) were analysed to understand the genetic structure of G. haimaensis in the Haima cold seep. There was no significant genetic differentiation between H1 and H2 sites using the three concatenated mitochondrial genes (MCD). Furthermore, no apparent genetic structure was revealed in haplotype networks or Fst values. These results align with a previous study (Xu et al., 2018) based on mitochondrial genes, suggesting high connectivity and lack of population differentiation between deep-sea mussel populations. However, the results of PCA based on 294,734 SNPs detected a genetic subdivision, and analysis by STRUCTURE provided further evidence of genetic differentiation between the H1 and H2 populations of G. haimaensis. There may be a lack of demographic connectivity between the H1 and H2 mussel beds. Due to the different types of genetic markers and the number of genetic markers used, there may be discrepancies when assessing SNPs and mitochondrial genes. Analyses of only a few genetic markers can lead to relatively larger confidence intervals for minimal values of Fst, which may result in non-statistical significance between populations (Blanco Bercial and Bucklin, 2016). Discrepancies between results obtained from SNPs and mitochondrial markers have also been reported in genetic structure studies of G. platifrons and S. crosnieri (Sun et al., 2018; Cheng et al., 2020). Selective sweeps of mitochondrial genes might be more common for organisms living in chemosynthesis-based ecosystems in the deep ocean compared with those in other marine environments (Roterman et al., 2016). The significantly negative Fu’s FS and Tajima’s D values for G. haimaensis indicate a signature of selective sweeps of mitochondrial genes (Fu, 1997) (Tajima, 1989).
In general, geographic distances have a negative impact on gene exchange between populations (Taguchi et al., 2014). However, in this study, within a geographic distance of only ~18 km, a small but clear genetic subdivision was observed for G. haimaensis between the two sites based on STRUCTURE and PCA using the entire dataset of 294,734 SNPs. In contrast, no significant genetic divergence was found between populations of close deep sea mussel G. platifrons in the Northwest Pacific, despite a geographic distance of >2,000 km. This scenario is usually referred to as chaotic genetic patchiness (CGP) that patterns of genetic structure that cannot be explained by any clear geographic trend. The driving mechanisms of chaotic genetic patchiness are recognized to be selection, sweepstakes reproductive success, temporal shifts in local population dynamics and collective dispersal (Iannucci et al., 2020). In our study, collective dispersal and sweepstakes reproductive success may be the driving factor for this chaotic genetic patchiness in G. haimaensis populations. Collective dispersal indicates any process leading to gene flow by groups of individuals (Yearsley et al., 2013). This type of dispersal may arise from individual dispersal strategies, such as collective larval dispersal by ocean currents. For deep sea mussel larva, only a few larvae can colonize new vents or seeps (Vrijenhoek, 2010). Free-swimming planktotrophic deep sea mussle larvae are expected to have long-distance dispersal potential. Thus, there has been a focus on understanding the genetic connectivity between populations separated by greater distances while ignoring those geographically close populations. Our results indicate that chaotic genetic patchiness is present within deep-sea mussels populations that are geographically close. This scenario may also exist in other deep-sea benthic communities.
Assessment of species and genetic diversity can reveal adaptability to environmental disturbance (Vrijenhoek, 2010). Compared with other healthy aquatic ecosystems, the macrobiotic species population in Haima cold seep is characterised by lower nucleotide diversity, suggesting that these populations may be vulnerable to environmental changes when gas hydrates extractions. The possibility of regime changes poses challenges for mitigation programs and environmental management. Therefore, it is necessary to understand whether such a regime change will have a significant adverse impact, which should influence management. Managers need to understand the magnitude of local changes in populations and community structures in order to anticipate the cumulative effects and environmental impacts of post-disruption regime changes (Thaler et al., 2017).
Using ROV in situ surveys, mitochondrial DNA fragments, and RAD-seq approaches, the species diversity and genetic diversity of macrobenthos in the Haima cold seep were estimated, and 12 macrobenthic species from five phyla and 12 families were identified. The macrobenthos community can be divided into three groups (H1 and H2=community type 1 and H3 and H4=community type 2) based on CLUSTER and NMDS analyses. G. haimaensis (Mollusca), Branchipolynoe pettiboneae (Annelida) and H. haimaensis (Echinodermata) had the highest values of dominance (0.160, 0.021 and 0.114, respectively) among members in their respective phyla. All three typical macrobenthic species presented high haplotype diversity but low nucleotide diversity. Mismatch distribution and neutrality analyses suggested that G. haimaensis and H. haimaensis populations underwent recent population expansions. When using three mitochondrial genes, no significant genetic differentiation was detected between sites H1 and H2 for G. haimaensis. Nevertheless, genetic subdivision between the two populations of G. haimaensis was evident when using SNP datasets. The results comprehensively illuminate macrobenthos biodiversity in the Haima cold seep ecosystem, and provide a baseline against which population dynamics may be assessed in the future.
The original contributions presented in the study are publicly available. The data for the mitochondrial gene sequences can be found here: NCBI, MW384429 to MW384487 for nad4, MW408123 to MW408176for COI, and MW384488 to MW384545 for atp6.The RAD-Seq data can be found here: NCBI, PRJNA837114.
GY performed most of the experiments, analysed data, and wrote the manuscript. MH planned and designed the research. YS, HZ, PX and HJ assisted in the experiment. All authors contributed to the article and approved the submitted version.
This project was supported by the Key Special Project for Introduced Talents Team of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (GML2019ZD0401), the Major Project of Basic and Applied Basic Research of Guangdong Province (2019B030302004), and the Science and Technology Planning Project of Guangdong Province (2020B1212060058).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Data and samples were collected on board of research vessel (R/V) Haiyang 6 of Guangzhou Marine Geological Survey (China), we thank the crew for their help during the cruise. We also want to thank Dr Eric for editing this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.920327/full#supplementary-material
Avise J. C., Neigel J. E., Arnold J. (1984). Demographic Influences on Mitochondrial DNA Lineage Survivorship in Animal Populations. J. Mol. Evol. 20 (2), 99–105. doi: 10.1007/BF02257369
Baco A. R., Etter R. J., Ribeiro P. A., Von der Heyden S., Beerli P., Kinlan B. P. (2016). A Synthesis of Genetic Connectivity in Deep-Sea Fauna and Implications for Marine Reserve Design. Mol. Ecol. 25 (14), 3276–3298. doi: 10.1111/mec.13689
Bandelt H., Forster P., Röhl A. (1999). Median-Joining Networks for Inferring Intraspecific Phylogenies. Mol. Biol. Evol. 16 (1), 37–48. doi: 10.1093/oxfordjournals.molbev.a026036
Barrett R. D., Schluter D. (2008). Adaptation From Standing Genetic Variation. Trends Ecol. Evol. 23 (1), 38–44. doi: 10.1016/j.tree.2007.09.008
Bielawski J. P., Gold J. R. (1996). Unequal Synonymous Substitution Rates Within and Between Two Protein-Coding Mitochondrial Genes. Mol. Biol. Evol. 13 (6), 889–892. doi: 10.1093/oxfordjournals.molbev.a025649
Blanco Bercial L., Bucklin A. (2016). New View of Population Genetics of Zooplankton: RAD-Seq Analysis Reveals Population Structure of the North Atlantic Planktonic Copepod Centropages Typicus. Mol. Ecol. 25 (7), 1566–1580. doi: 10.1111/mec.13581
Bovo S., Ribani A., Muñoz M., Alves E., Araujo J. P., Bozzi R., et al. (2020). Whole-Genome Sequencing of European Autochthonous and Commercial Pig Breeds Allows the Detection of Signatures of Selection for Adaptation of Genetic Resources to Different Breeding and Production Systems. Genet. Sel. Evol. 52 (1), 1–19. doi: 10.1186/s12711-020-00553-7
Breusing C., Johnson S. B., Tunnicliffe V., Vrijenhoek R. C. (2015). Population Structure and Connectivity in Indo-Pacific Deep-Sea Mussels of the Bathymodiolus Septemdierum Complex. Conserv. Genet. 16 (6), 1415–1430. doi: 10.1007/s10592-015-0750-0
Cheng J., Hui M., Li Y., Sha Z. (2020). Genomic Evidence of Population Genetic Differentiation in Deep-Sea Squat Lobster Shinkaia Crosnieri (Crustacea: Decapoda: Anomura) From Northwestern Pacific Hydrothermal Vent and Cold Seep. Deep Sea Res. Part I: Oceanographic Res. Papers. 156, 103188. doi: 10.1016/j.dsr.2019.103188
Dong D., Li X., Yang M., Gong L., Li Y., Sui J., et al. (2021). Report of Epibenthic Macrofauna Found From Haima Cold Seeps and Adjacent Deep-Sea Habitats, South China Sea. Mar. Life Sci. Tech. 3 (1), 1–12. doi: 10.1007/s42995-020-00073-9
Etter P. D., Bassham S., Hohenlohe P. A., Johnson E. A., Cresko W. A. (2012). SNP Discovery and Genotyping for Evolutionary Genetics Using RAD Sequencing (Clifton: Humana Press).
Evanno G., Regnaut S., Goudet J. (2005). Detecting the Number of Clusters of Individuals Using the Software STRUCTURE: A Simulation Study. Mol. Ecol. 14 (8), 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Everett M. V., Park L. K. (2018). Exploring Deep-Water Coral Communities Using Environmental DNA. Deep Sea Res. Part II: Topical Stud. Oceanogr. 150, 229–241. doi: 10.1016/j.dsr2.2017.09.008
Excoffier L., Lischer H. E. (2010). Arlequin Suite Ver 3.5: A New Series of Programs to Perform Population Genetics Analyses Under Linux and Windows. Mol. Ecol. Resour. 10 (3), 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Fang Y., Wei J., Lu H., Liang J., Lu J., Fu J., et al. (2019). Chemical and Structural Characteristics of Gas Hydrates From the Haima Cold Seeps in the Qiongdongnan Basin of the South China Sea. J. Asian Earth Sci. 182, 103924. doi: 10.1016/j.jseaes.2019.103924
Feng D., Qiu J., Hu Y., Peckmann J., Guan H., Tong H., et al. (2018). Cold Seep Systems in the South China Sea: An Overview. J. Asian Earth Sci. 168, 3–16. doi: 10.1016/j.jseaes.2018.09.021
Frankham R. (2010). Challenges and Opportunities of Genetic Approaches to Biological Conservation. Biol. Conserv. 143 (9), 1919–1927. doi: 10.1016/j.biocon.2010.05.011
Fu Y. (1997). Statistical Tests of Neutrality of Mutations Against Population Growth, Hitchhiking and Background Selection. Genetics. 147 (2), 915–925. doi: 10.1017/S0016672397002966
Grant W., Bowen B. W. (1998). Shallow Population Histories in Deep Evolutionary Lineages of Marine Fishes: Insights From Sardines and Anchovies and Lessons for Conservation. J. Hered. 89 (5), 415–426. doi: 10.1093/jhered/89.5.415
Hunter R. L., Halanych K. M. (2008). Evaluating Connectivity in the Brooding Brittle Star Astrotoma Agassizii Across the Drake Passage in the Southern Ocean. J. Hered. 99 (2), 137–148. doi: 10.1093/jhered/esm119
Hurtado L. A., Lutz R. A., Vrijenhoek R. C. (2004). Distinct Patterns of Genetic Differentiation Among Annelids of Eastern Pacific Hydrothermal Vents. Mol. Ecol. 13 (9), 2603–2615. doi: 10.1111/j.1365-294X.2004.02287.x
Iannucci A., Cannicci S., Caliani I., Baratti M., Pretti C., Fratini S. (2020). Investigation of Mechanisms Underlying Chaotic Genetic Patchiness in the Intertidal Marbled Crab Pachygrapsus Marmoratus (Brachyura: Grapsidae) Across the Ligurian Sea. BMC Evol. Biol. 20 (1), 1–13. doi: 10.1186/s12862-020-01672-x
Jones D. O. B., Kaiser S., Sweetman A. K., Smith C. R., Menot L., Vink A., et al. (2017). Biological Responses to Disturbance From Simulated Deep-Sea Polymetallic Nodule Mining. PLoS One 12 (2), e171750. doi: 10.1371/journal.pone.0171750
Ke Z., Li R., Chen Y., Chen D., Chen Z., Lian X., et al. (2022). A Preliminary Study of Macrofaunal Communities and Their Carbon and Nitrogen Stable Isotopes in the Haima Cold Seeps, South China Sea. Deep Sea Res. Part I: Oceanographic Res. Papers. 184, 103774. doi: 10.1016/j.dsr.2022.103774
Laikre L., Allendorf F. W., Aroner L. C., Baker C. S., Gregovich D. P., Hansen M. M., et al. (2010). Neglect of Genetic Diversity in Implementation of the Convention on Biological Diversity. Conserv. Biol. 24 (1), 86–88. doi: 10.1111/j.1523-1739.2009.01425.x
Liang Q., Hu Y., Feng D., Peckmann J., Chen L., Yang S., et al. (2017). Authigenic Carbonates From Newly Discovered Active Cold Seeps on the Northwestern Slope of the South China Sea: Constraints on Fluid Sources, Formation Environments, and Seepage Dynamics. Deep Sea Res. Part I: Oceanographic Res. Papers. 124, 31–41. doi: 10.1016/j.dsr.2017.04.015
Librado P., Rozas J. (2009). DnaSP V5: A Software for Comprehensive Analysis of DNA Polymorphism Data. Bioinformatics. 25 (11), 1451–1452. doi: 10.1093/bioinformatics/btp187
Li H., Durbin R. (2009). Fast and Accurate Short Read Alignment With Burrows–Wheeler Transform. Bioinformatics. 25 (14), 1754–1760. doi: 10.1093/bioinformatics/btp324
Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al. (2009). The Sequence Alignment/Map Format and SAMtools. Bioinformatics. 25 (16), 2078–2079. doi: 10.1093/bioinformatics/btp352
Li Y. L., Liu J. X. (2018). StructureSelector: A Web-Based Software to Select and Visualize the Optimal Number of Clusters Using Multiple Methods. Mol. Ecol. Resour. 18 (1), 176–177. doi: 10.1111/1755-0998.12719
Marshall N. T., Stepien C. A. (2020). Macroinvertebrate Community Diversity and Habitat Quality Relationships Along a Large River From Targeted eDNA Metabarcode Assays. Environ. DNA. 2 (4), 572–586. doi: 10.1002/edn3.90
McClain C. R., Allen A. P., Tittensor D. P., Rex M. A. (2012). Energetics of Life on the Deep Seafloor. Proc. Natl. Acad. Sci. 109 (38), 15366–15371. doi: 10.1073/pnas.1208976109
Miyazaki J., Beppu S., Kajio S., Dobashi A., Kawato M., Fujiwara Y., et al. (2013). Dispersal Ability and Environmental Adaptability of Deep-Sea Mussels ;Bathymodiolus (Mytilidae: Bathymodiolinae). Open J. Mar. Science. 03 (01), 31–39. doi: 10.4236/ojms.2013.31003
Nakajima R., Yamakita T., Watanabe H., Fujikura K., Tanaka K., Yamamoto H., et al. (2014). Species Richness and Community Structure of Benthic Macrofauna and Megafauna in the Deep-Sea Chemosynthetic Ecosystems Around the Japanese Archipelago: An Attempt to Identify Priority Areas for Conservation. Divers. Distrib. 20 (10), 1160–1172. doi: 10.1111/ddi.12204
Pielou E. C. (1966). The Measurement of Diversity in Different Types of Biological Collections. J. Theor. Biol. 13, 131–144. doi: 10.1016/0022-5193(66)90013-0
Pritchard J. K., Stephens M., Donnelly P. (2000). Inference of Population Structure Using Multilocus Genotype Data. Genetics. 155 (2), 945–959. doi: 10.1093/genetics/155.2.945
Puechmaille S. J. (2016). The Program Structure Does Not Reliably Recover the Correct Population Structure When Sampling is Uneven: Subsampling and New Estimators Alleviate the Problem. Mol. Ecol. Resour. 16 (3), 608–627. doi: 10.1111/1755-0998.12512
Ramirez-Llodra E., Brandt A., Danovaro R., De Mol B., Escobar E., German C. R., et al. (2010). Deep, Diverse and Definitely Different: Unique Attributes of the World’s Largest Ecosystem. Biogeosciences. 7 (9), 2851–2899. doi: 10.5194/bg-7-2851-2010
Romiguier J., Gayral P., Ballenghien M., Bernard A., Cahais V., Chenuil A., et al. (2014). Comparative Population Genomics in Animals Uncovers the Determinants of Genetic Diversity. Nature. 515 (7526), 261–263. doi: 10.1038/nature13685
Roterman C. N., Copley J. T., Linse K. T., Tyler P. A., Rogers A. D. (2016). Connectivity in the Cold: The Comparative Population Genetics of Vent-Endemic Fauna in the Scotia Sea, Southern Ocean. Mol. Ecol. 25 (5), 1073–1088. doi: 10.1111/mec.13541
Shannon C. E. (1948). A Mathematical Theory of Communication. Bell system Tech. J. 27 (3), 379–423. doi: 10.1002/j.1538-7305.1948.tb01338.x
Shen Y., Kou Q., Chen W., He S., Yang M., Li X., et al. (2016). Comparative Population Structure of Two Dominant Species, Shinkaia Crosnieri (Munidopsidae: Shinkaia) and Bathymodiolus Platifrons (Mytilidae: Bathymodiolus), Inhabiting Both Deep-Sea Vent and Cold Seep Inferred From Mitochondrial Multi-Genes. Ecol. Evolution. 6 (11), 3571–3582. doi: 10.1002/ece3.2132
Sun Y., Liang Q., Sun J., Yang Y., Tao J., Liang J., et al. (2018). The Mitochondrial Genome of the Deep-Sea Tubeworm Paraescarpia Echinospica (Siboglinidae, Annelida) and its Phylogenetic Implications. Mitochondrial DNA Part B. 3 (1), 131–132. doi: 10.1080/24701394.2017.1334773
Sun P., Tang B., Yin F. (2018). Population Genetic Structure and Genetic Diversity of Chinese Pomfret at the Coast of the East China Sea and the South China Sea. [Journal Article; Res. Support Non-U.S. Gov’t]. Mitochondrial DNA A DNA Mapp Seq Anal. 29 (4), 643–649. doi: 10.1080/24701394.2017.1334773
Sun J., Zhang Y., Xu T., Zhang Y., Mu H., Zhang Y., et al. (2017). Adaptation to Deep-Sea Chemosynthetic Environments as Revealed by Mussel Genomes. Nat. Ecol. Evol. 1 (5), 1–7. doi: 10.1038/s41559-017-0121
Taguchi M., King J. R., Wetklo M., Withler R. E., Yokawa K. (2014). Population Genetic Structure and Demographic History of Pacific Blue Sharks (Prionace Glauca) Inferred From Mitochondrial DNA Analysis. Mar. Freshw. Res. 66 (3), 267–275. doi: 10.1071/MF14075
Tajima F. (1989). Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics. 123 (3), 585–595. doi: 10.1101/gad.3.11.1801
Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. (2011). MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 28 (10), 2731–2739. doi: 10.1093/molbev/msr121
Tanaka Y. (2000). Extinction of Populations by Inbreeding Depression Under Stochastic Environments. Popul. Ecol. 42 (1), 55–62. doi: 10.1007/s101440050009
Teixeira S., Olu K., Decker C., Cunha R. L., Fuchs S., Hourdez S., et al. (2013). High Connectivity Across the Fragmented Chemosynthetic Ecosystems of the Deep Atlantic Equatorial Belt: Efficient Dispersal Mechanisms or Questionable Endemism? Mol. Ecol. 22 (18), 4663–4680. doi: 10.1111/mec.12419
Thaler A. D., Saleu W., Carlsson J., Schultz T. F., Van Dover C. L. (2017). Population Structure Ofbathymodiolus Manusensis ,a Deep-Sea Hydrothermal Vent-Dependent Mussel From Manus Basin Papua New Guinea. PeerJ. 5, e3655. doi: 10.7717/peerj.3655
Vrijenhoek R. (1994). DNA Primers for Amplification of Mitochondrial Cytochrome C Oxidase Subunit I From Diverse Metazoan Invertebrates. Mol. Mar. Biol. Biotechnol. 3 (5), 294–299.
Vrijenhoek R. C. (2010). Genetic Diversity and Connectivity of Deep-Sea Hydrothermal Vent Metapopulations. Mol. Ecol. 19 (20), 4391–4411. doi: 10.1111/j.1365-294X.2010.04789.x
WANG Y., SHA Z. (2017). A New Species of the Genus Alvinocaris Williams and Chace 1982 (Crustacea: Decapoda: Caridea: Alvinocarididae) From the Manus Basin Hydrothermal Vents, Southwest Pacific. Zootaxa. 4226 (1), 126–136. doi: 10.11646/zootaxa.4226.1.7
Woolley S. N. C., Tittensor D. P., Dunstan P. K., Guillera-Arroita G., Lahoz-Monfort J. J., Wintle B. A., et al. (2016). Deep-Sea Diversity Patterns are Shaped by Energy Availability. Nature. 533 (7603), 393–396. doi: 10.1038/nature17937
Xu T., Feng D., Tao J., Qiu J. (2019). A New Species of Deep-Sea Mussel (Bivalvia: Mytilidae: Gigantidas) From the South China Sea: Morphology, Phylogenetic Position, and Gill-Associated Microbes. Deep Sea Res. Part I: Oceanographic Res. Papers. 146, 79–90. doi: 10.1016/j.dsr.2019.03.001
Xu T., Sun J., Watanabe H. K., Chen C., Nakamura M., Ji R., et al. (2018). Population Genetic Structure of the Deep-Sea Mussel Bathymodiolus Platifrons (Bivalvia: Mytilidae) in the Northwest Pacific. Evol. Appl. 11 (10), 1915–1930. doi: 10.1111/eva.12696
Yang J., Lu B., Chen D., Yu Y., Yang F., Nagasawa H., et al. (2012). When did Decapods Invade Hydrothermal Vents? Clues From the Western Pacific and Indian Oceans. Mol. Biol. Evol. 30 (2), 305–309. doi: 10.1093/molbev/mss224
Yang Y., Tian K., Hao J., Pei S., Yang Y. (2004). Biodiversity and Biodiversity Conservation in Yunnan, China. Biodiversity Conserv. 13 (4), 813–826. doi: 10.1023/B:BIOC.0000011728.46362.3c
Yearsley J. M., Viard F., Broquet T. (2013). The Effect of Collective Dispersal on the Genetic Structure of a Subdivided Population. Evolution. 67 (6), 1649–1659. doi: 10.1111/evo.12111
Zhang K., Sun J., Xu T., Qiu J., Qian P. (2021). Phylogenetic Relationships and Adaptation in Deep-Sea Mussels: Insights From Mitochondrial Genomes. Int. J. Mol. Sci. 22 (4), 1900. doi: 10.3390/ijms22041900
Keywords: macrobenthos, Gigantidas haimaensis, genetic diversity, mitochondrial gene, RAD-seq, Haima cold seep, deep sea, chaotic genetic patchiness
Citation: Yao G, Zhang H, Xiong P, Jia H, Shi Y and He M (2022) Community Characteristics and Genetic Diversity of Macrobenthos in Haima Cold Seep. Front. Mar. Sci. 9:920327. doi: 10.3389/fmars.2022.920327
Received: 14 April 2022; Accepted: 23 June 2022;
Published: 22 July 2022.
Edited by:
Chiara Romano, Anton Dohrn Zoological Station, ItalyReviewed by:
Chunsheng Wang, Ministry of Natural Resources, ChinaCopyright © 2022 Yao, Zhang, Xiong, Jia, Shi and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maoxian He, aG14MkBzY3Npby5hYy5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.