 Yu-Qi Ye
Yu-Qi Ye Zhi-Peng Hao1
Zhi-Peng Hao1 Meng-Qi Ye
Meng-Qi Ye Zong-Jun Du
Zong-Jun Du
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 20 June 2022
Sec. Aquatic Microbiology
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.919253
This article is part of the Research TopicMicrobes from Marine Distinctive EnvironmentsView all 19 articles
Two novel rod-shaped and Gram-negative bacterial strains, designated A6E486T and 5E331T, were isolated from a coastal sediment sample taken from Xiaoshi Island, Weihai, China, and a fresh kelp sample collected from a kelp culture area, Rongcheng, China, respectively. Growth of strain A6E486T occurred at 20°C–43°C (optimum, 33°C–35°C) at pH 5.5–7.5 (optimum, 6.5) and in the presence of 1.0%–5.5% (w/v) NaCl (optimum, 2.5%–3.0%). Strain 5E331T grew with 1.5%–5.0% (w/v) NaCl (optimum, 3.0%) at 15°C–40°C (optimum, 33°) and pH 6.0–8.5 (optimum, 7.0). The similarity of 16S rRNA gene sequence between the two strains was 95.2%. The phylogenetic analysis based on 16S rRNA gene sequence showed that strains A6E486T and 5E331T belong to the genus Kordiimonas, sharing the highest similarity to the genus Kordiimonas (94.6%–96.8%, 94.9%–96.1%, respectively). Strains A6E486T and 5E331T had percentage of conserved protein (POCP) values of 56.0%–67.3% and average nucleotide identity (ANI) values of 68.8%–73.1% to members of the genus Kordiimonas. The major polar lipids detected in the two strains were phosphatidylethanolamine (PE), phosphatidylglycerol (PG), diphosphatidylglycerol (DPG), and unidentified glycolipids, aminolipids, and lipids. The predominant respiratory quinone of the two strains was Q-10. Based upon the results presented in this study, strains A6E486T and 5E331T represent two novel species of the genus Kordiimonas, for which the names Kordiimonas marina and Kordiimonas laminariae are proposed with the type strains A6E486T (= KCTC 82758T = MCCC 1H00470T) and 5E331T (= KCTC 92199T = MCCC 1H00515T), respectively. Comparative genomic analysis showed that seven species of the genus Kordiimonas shared 1,258 core genes and had differences in carbohydrate metabolism, energy metabolism, and cofactor and vitamin metabolism. The pan-genome of the genus Kordiimonas was open. The prediction of secondary metabolites showed that most strains of the genus Kordiimonas had the ability to produce homoserine lactones, one of the most important signal molecules in the quorum-sensing system of Gram-negative bacteria. Additionally, numerous genes involved in bacterial defense, motility and chemotaxis, cold adaptation, and environment stress response were found in the genus Kordiimonas, indicating the marine-adapted lifestyle of members of the genus Kordiimonas.
The genus Kordiimonas was firstly proposed as a member of the order Kordiimonadales in the class Alphaproteobacteria by Kwon et al. (2005), with Kordiimonas gwangyangensis being the type species. At the time of writing, the genus Kordiimonas consists of seven validly published species according to the List of Prokaryotic names with Standing in Nomenclature1, including K. gwangyangensis, Kordiimonas lacus, Kordiimonas aestuarii, Kordiimonas aquimaris, Kordiimonas lipolytica, Kordiimonas sediminis, and Kordiimonas pumila. All members of the genus Kordiimonas are aerobic, oxidase- and catalase-positive, except K. sediminis (facultatively anaerobic, oxidase- and catalase- negative) (Zhang et al., 2016). All of the type strains of the genus Kordiimonas are isolated from marine environments and habitats, such as marine sediment (Kwon et al., 2005; Xu X. et al., 2011; Math et al., 2012; Zhao et al., 2018), seawater (Yang et al., 2013; Wu et al., 2016), and sea cucumber culture pond sediments (Zhang et al., 2016), and have Q-10 as the predominant respiratory quinone. In this study, we describe two bacterial strains, A6E486T isolated from a coastal sediment sample and 5E331T isolated from a fresh kelp sample. Polyphasic investigations showed that the two strains represent two novel species affiliated with the genus Kordiimonas. Comparative genomic analyses of these two strains and the related species are contributed to understand the marine-adapted lifestyle of the genus Kordiimonas.1
Strain A6E486T was isolated from an intertidal sediment sample collected from Xiaoshi Island (37°32’30”N, 122°6’20”E), one of the largest continuous ecosystems in Weihai, China. The sediment sample was treated with an enrichment culture technique at 33°C for 30 days (Mu et al., 2018), and 100-μl enrichment cultures were spread on marine agar 2216 (MA; BD) by using the standard 10-fold dilution plating technique. After incubation at 33°C for 7 days, a beige colony designated A6E486T was picked and pure cultures were obtained using purification procedures. Strain 5E331T was obtained from fresh kelp samples, which were taken from a kelp culture area (37°15’96”N, 122°36’49”E) in Rongcheng, China. Here, 10 g of fresh kelps were cut into small squares with sides of 1.5 cm under sterile conditions. Then, treated samples were placed in a conical flask containing glass beads and 100 ml seawater and shaken at 300 rpm for 1.5 h. Subsequently, the kelp treatment fluid was serially diluted to 10−3 with sterile seawater, and 100 μl aliquot was spread on MA. The incubation was performed at 28°C for 2 weeks. Strains A6E486T and 5E331T were stored at –80°C in sterile 1% (w/v) saline supplemented with 15% (v/v) glycerol. The type strains K. gwangyangensis JCM 12864T and K. lipolytica JCM 30877T obtained from the Japan Collection of Microorganisms were used as experiment control strains and cultured under optimum conditions.
The morphological and physiological features of strains A6E486T and 5E331T were examined after incubation at 33°C for 3 days on MA medium. The Gram staining reaction was determined according to the method described previously (Dong and Cai, 2001). Cell morphology and size were examined employing optical microscopy (E600, Nikon), transmission electron microscopy (JEM-1200, Jeol), and scanning electron microscopy (model Nova NanoSEM450, FEI). Growth ranges and optima of temperature were determined on MA at 0°C, 4°C, 15°C, 20°C, 25°C, 28°C, 30°C, 33°C, 35°C, 37°C, 40°C, 43°C, 45°C. The pH range for growth was tested in marine broth 2216 (MB; BD) by adding the appropriate buffers (20 mM), including 2-Morpholinoethanesulfonic acid (MES) (pH 5.5−6.0), 1,4-Piperazinediethanesulfonic acid (PIPES) (pH 6.5−7.0), N-(2-Hydroxyethyl) piperazine-N'-2-ethanesulfonic acid (HEPES) (pH 7.5−8.0), Tricine (pH 8.5), and 3-(Cyclohexylamino)-2-hydroxy-1-propanesulfonic acid (CAPSO) (pH 9.0−9.5). The optimal condition with NaCl for growth was tested in the following medium (0.1% yeast extract, 0.5% peptone), prepared with artificial seawater (per liter: 3.2 g MgSO4, 2.2 g MgCl2, 1.2 g CaCl2, 0.7 g KCl, 0.2 g NaHCO3) containing NaCl at concentrations from 0% to 10% (w/v, in 0.5% intervals). The strains were incubated at optimum temperature for 7 days. Growth experiments with leucine as sole carbon source were tested in the following liquid media, artificial seawater with inorganic carbon (NaHCO3) removed was supplemented with 0.1% NH4Cl as a nitrogen source with or without leucine. Leucine solution was sterilized with 0.22-μm filters. The final concentration of leucine in the medium was 1 g/L. The strains were incubated at optimum temperature for 11 days. Bacterial growth was monitored using a spectrophotometer at 600 nm.
Catalase activity was tested by adding 3% (v/v) H2O2 solution to the plate with fresh colonies. Oxidase activity was determined using an oxidase test reagent (bioMérieux). Anaerobic growth was determined by incubating strains on MA with or without 0.1% (w/v) KNO3 in an AnaeroPack for 14 days. Hydrolysis tests of Tweens (20, 40, 60, and 80), alginate, casein, CM-cellulose, DNA, and starch were carried out according to methods described previously (Dong and Cai, 2001). The API 50CH and API 20E kits (bioMérieux) were employed to investigate acid production and additional biochemical characteristics, respectively. Additional enzyme activities were checked using API ZYM kit (bioMérieux), and carbon source oxidation was tested employing BIOLOG GEN III MicroPlates. All of the API and BIOLOG tests were carried out according to the manufacturer’s instructions, except for the modification of adjusting salinity to 3% (w/v). Antibiotic susceptibility was determined on MA at 33°C for 7 days using the disc diffusion method (Jorgensen and Turnidge, 2015).
Strains A6E486T and 5E331T and experiment control strains were cultured in MB under optimum conditions, and the cells at the late stage of exponential growth phase were harvested and freeze-dried in order to investigate chemotaxonomic characterizations. Fatty acids were extracted from freeze-dried cell biomasses (10 mg) and then analyzed using a gas chromatograph (model 6890N, Agilent) and the Microbial Identification System (MIDI database: TSBA40) (Sasser, 1990). Isoprenoid quinones were obtained from freeze-dried cell biomasses (300 mg) and identified by high-performance liquid chromatography (HPLC), as described previously (Minnikin et al., 1984). Total lipids were extracted with chloroform/methanol/water system (2.5:5:2, v/v/v) and analyzed by two-dimensional TLC according to the modified method described previously (Komagata and Suzuki, 1987).
The 16S rRNA gene was amplified employing PCR technology with the primer pair 27F and 1492R (Lane, 1991) and was sequenced by RuiBiotech (Qingdao, China). To obtain nearly complete 16S rRNA genes, purified PCR products were ligated into the pMD18-T vector (Takara), and ligation products were transferred into Escherichia coli DH5a receptor cells according to the manufacturer’s instructions. For comparative analysis, the 16S rRNA gene sequences of strains A6E486T and 5E331T were submitted to the National Center for Biotechnology Information (NCBI) database2 and the EzBioCloud server.3
The 16S rRNA gene sequences of strains A6E486T, 5E331T, and relatives were aligned using MUSCLE (Edgar, 2004), and then the trimmed sequences were used to reconstruct phylogenetic trees employing the neighbor-joining (NJ) and maximum-parsimony (MP) algorithm implemented in the software MEGA X (Felsenstein, 1981; Saitou and Nei, 1987; Kumar et al., 2018). The maximum-likelihood (ML) tree was reconstructed using the best-fit substitution model GTR+G+I. Bootstrap analysis was performed with 1,000 replications to provide confidence estimates for tree topologies.
Genomic DNA of strains A6E486T and 5E331T was extracted using a bacteria genomic DNA kit (Takara), and the draft genomes were determined by Beijing Novogene Bioinformatics Technology Co. Ltd. (Beijing, China). Sequencing was performed on the Illumina NovaSeq 6000 platform (Illumina Inc., San Diego, CA, USA) using the paired-end 150-bp sequencing protocol. Raw sequencing data were generated by Illumina base-calling software CASAVA v1.8.24, and the sequenced reads were assembled using SOAPdenovo software (Li et al., 2009). Other relevant genomes in this study were obtained from the NCBI Prokaryotic reference genome database.
The completeness values of the used genomes were estimated based on the method of lineage-specific CheckM v1.1.3 (Parks et al., 2015). Gene content was annotated using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP). The average nucleotide identity (ANI) was calculated using the online ANI calculator5. The percentage of conserved protein (POCP) value was calculated as described by Qin et al. (2014).6 The ribosomal protein phylogenetic tree was reconstructed by FastTree (Price et al., 2009) using JTT+ CAT parameters and IQ-TREE (Nguyen et al., 2015; Chernomor et al., 2016) using the LG+F+I+G4 model. The 15 ribosomal proteins (L2, L3, L4, L5, L6, L14, L15, L16, L22, L24, S3, S8, S10, S17, and S19) were obtained and aligned using Python script6 with the methods described by Wiegand et al. (2020). All genomes were predicted using Prodigal Server for uniformity (Hyatt et al., 2010). The analysis of core and unique genes was performed using the Bacterial Pan Genome Analysis (BPGA) tool, an ultrafast pan-genome analysis pipeline, with a threshold of 50% amino acid sequence identity (Chaudhari et al., 2016). Metabolic pathways were analyzed in detail by using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa et al., 2016), and the presence of gene clusters encoding secondary metabolites was predicted by antiSMASH server7. Carbohydrate-active enzymes of the genus Kordiimonas were annotated employing the online dbCAN28, a meta server for automated carbohydrate-active enzyme annotation (Zhang et al., 2018). The analysis of marine adaptation metabolic features was performed by Rapid Annotation using Subsystem Technology (RAST) version 2.0 (Aziz et al., 2008) and the KEGG database (Kanehisa et al., 2016).
Morphological observations by transmission electron microscopy and scanning electron microscopy showed that cells of strains A6E486T (with widths of 0.5–0.7 μm and lengths of 1.0–2.5 μm) and 5E331T (with widths of 0.2–0.5 μm and lengths of 0.8–2.7 μm) were both short rod-shaped (Supplementary Figure S1). For strain A6E486T, the oxidations of the sole carbon source were positive for D-maltose, D-trehalose, D-fructose, glycyl-L-proline, L-glutamic acid, L-galactonic acid lactone, and D-glucuronic acid, which was consistent with K. gwangyangensis JCM 12864T, while that of strain 5E331T was negative as K. lipolytica JCM 30877T (Supplementary Table S1). Strains A6E486T and 5E331T exhibited some similarities to K. gwangyangensis and K. lipolytica JCM 30877T, including their abilities to hydrolyze Tweens (20, 40, and 60). The differential characteristics of strains A6E486T and 5E331T and experiment control strains were summarized in Table 1 and Supplementary Table S1. Both strains A6E486T and 5E331T were susceptible to (μg per disc) penicillin (10), chloramphenicol (30), norfloxacin (30), cefotaxime sodium (30), gentamycin (10), ofloxacin (5), clarithromycin (15), carbenicillin (100), and ceftriaxone (30) but resistant to tetracycline (30) and kanamycin (30).
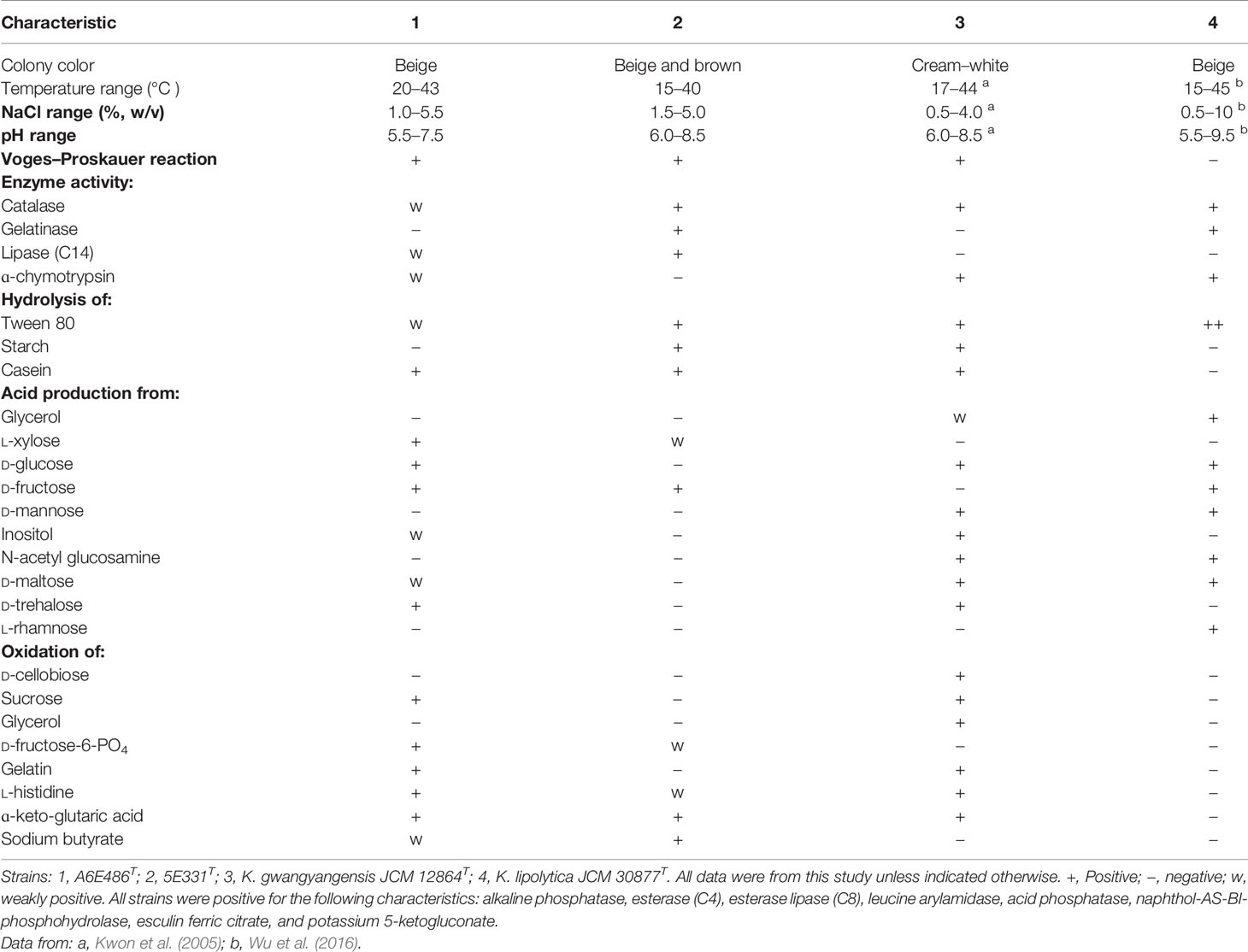
Table 1 Differential characteristics between strains A6E486T and 5E331T and experiment control strains.
The major cellular fatty acids (>10%) of strain A6E486T consisted of iso-C15:0, iso-C17:0, iso-C17:1 ω9c, and summed feature 3 (comprising C16:1 ω6c and/or C16:1 ω7c) while those of strain 5E331T were iso-C15:0, iso-C17:1 ω9c, and summed feature 3 (Table 2). The fatty acid compositions of the two strains were similar to experiment control strains but with differences in the proportions of some fatty acids. For example, iso-C17:0 was one of the major fatty acids in strains A6E486T and K. gwangyangensis JCM 12864T but not in strains 5E331T and K. lipolytica JCM 30877T. Strains A6E486T and 5E331T had Q-10 as the predominant respiratory quinone, which was consistent with the quinone type of the genus Kordiimonas. The major polar lipids detected in the two strains were phosphatidylethanolamine (PE), phosphatidylglycerol (PG), diphosphatidylglycerol (DPG), and unidentified glycolipids, aminolipids, and lipids. PE, PG, DPG, and unidentified glycolipids were detected in all tested Kordiimonas species (Supplementary Figure S2).
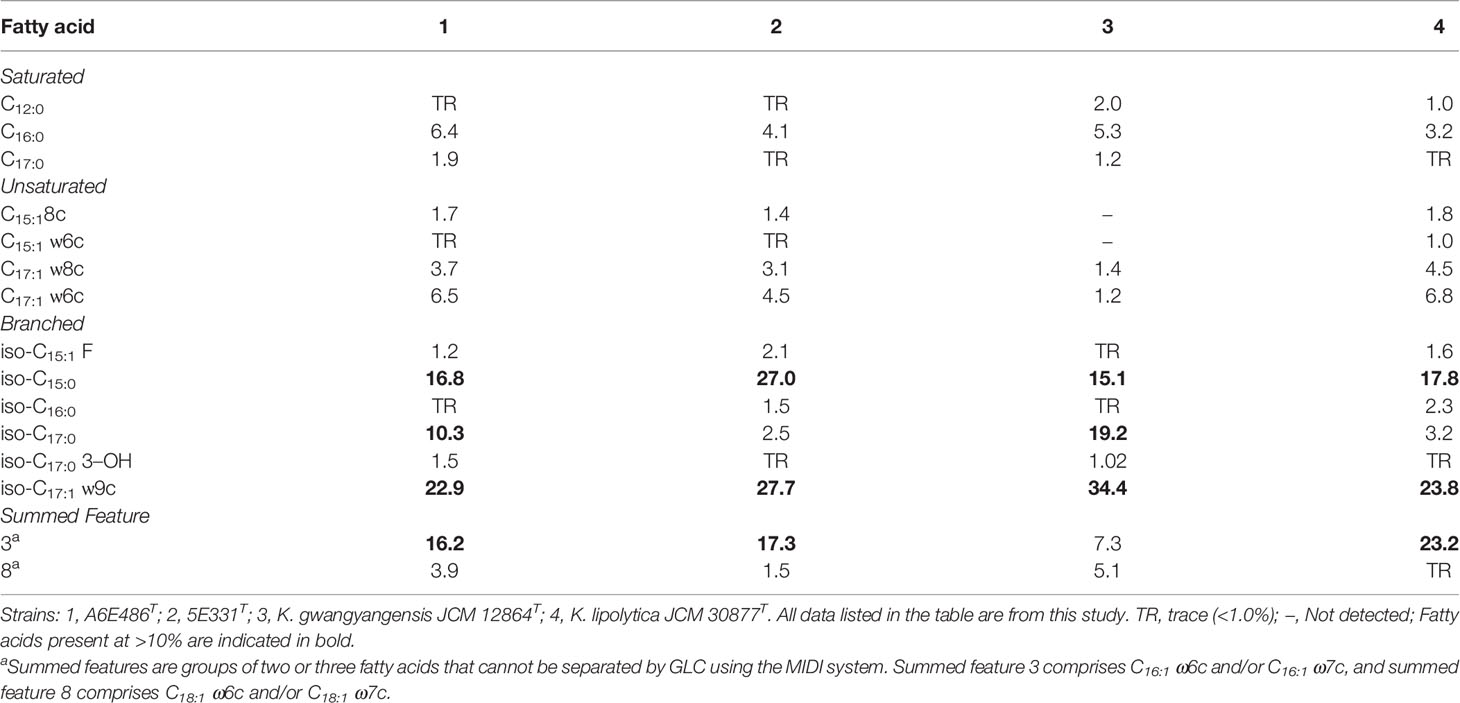
Table 2 Cellular fatty acid composition (%) of strains A6E486T and 5E331T and experiment control strains.
Nearly complete 16S rRNA gene sequences of strains A6E486T (1,461 bp; GenBank accession number: MZ901372) and 5E331T (1,462 bp; GenBank accession number: OM663707) were obtained according to the methods described in the Materials and Methods section. Comparative analysis results using EzBioCloud databases showed that strains A6E486T and 5E331T shared the highest 16S rRNA gene sequence similarity values of 96.8% and 96.1%, respectively, with K. lipolytica JCM 30877T. NJ phylogenetic tree based on 16S rRNA gene indicated that strains A6E486T and 5E331T were affiliated with the genus Kordiimonas (Figure 1). Similar tree topologies were also obtained with the ML and MP algorithms.
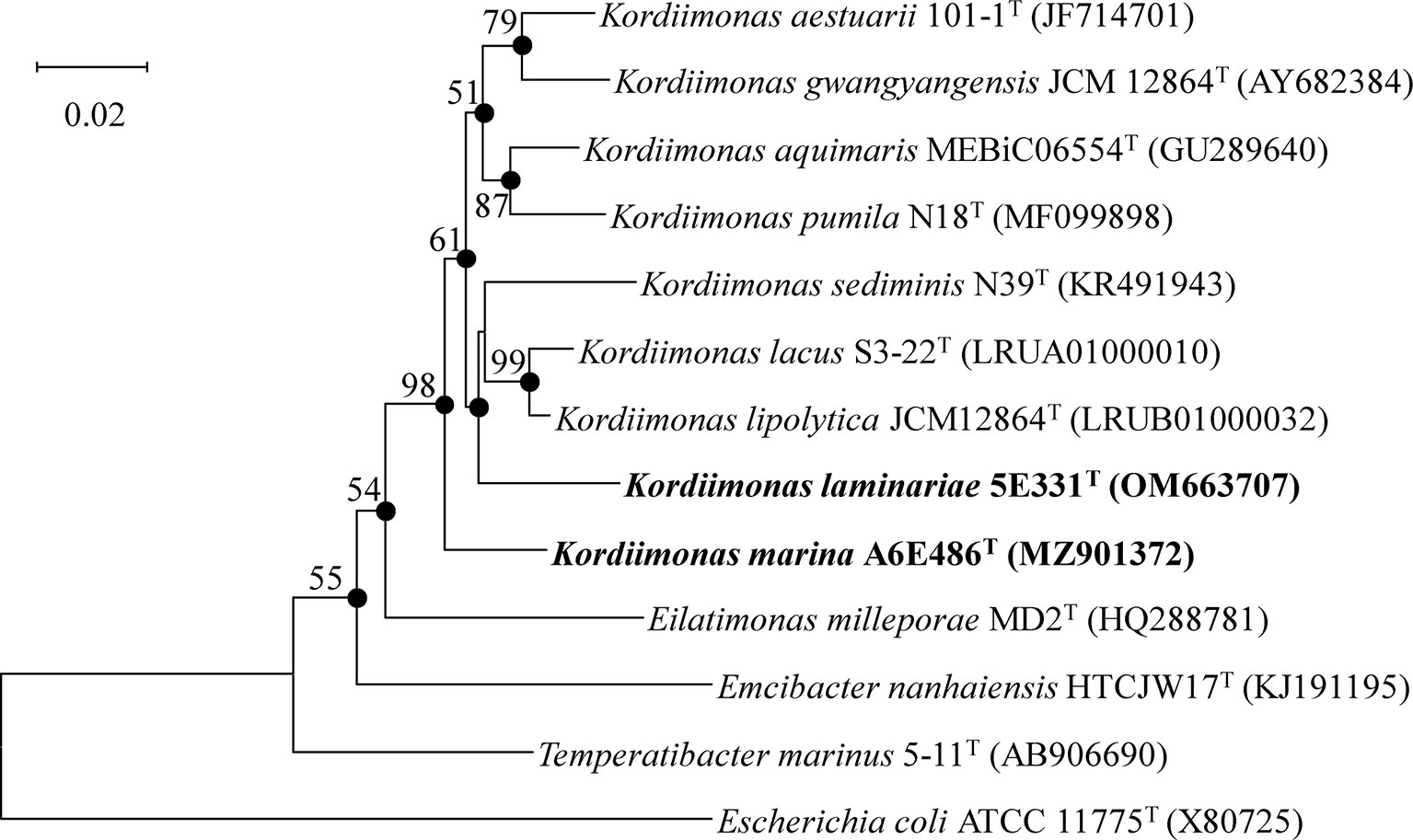
Figure 1 Neighbor-joining phylogenetic tree based on 16S rRNA gene sequences of strains A6E486T and 5E331T and other closely related species. Filled circles indicate branches that were recovered with all three methods (neighbor-joining, maximum-likelihood, and maximum-parsimony). Percentage bootstrap values above 50% (1,000 replicates) are shown at branch nodes. Escherichia coli ATCC 11775T was used as the out-group. Bar, 0.02 substitutions per nucleotide position.
The genome lengths of the seven species of the genus Kordiimonas (strains A6E486T, 5E331T, K. gwangyangensis JCM 30877T, K. lipolytica JCM 12864T, K. lacus CGMCC 1.9109T, K. pumila N18T, and K. sediminis KCTC 42590T) were 3,243,597–4,557,767 bp with a mean size of 3,894,463 bp. The completeness values of these genomes were 99.6%–100.0%. Among them, strain 5E331T had the minimum DNA G+C content (46.2%), while strain A6E486T possessed the maximum content (59.9%). The NCBI PGAP results showed that a total of 3,436 genes were predicted for strain A6E486T, including 3,381 protein-coding genes and 53 RNA genes (46 tRNA genes, 3 rRNA genes, and 4 ncRNA genes), while strain 5E331T had 3,505 predicted genes. The detailed PGAP results of the seven species were shown in Table 3.
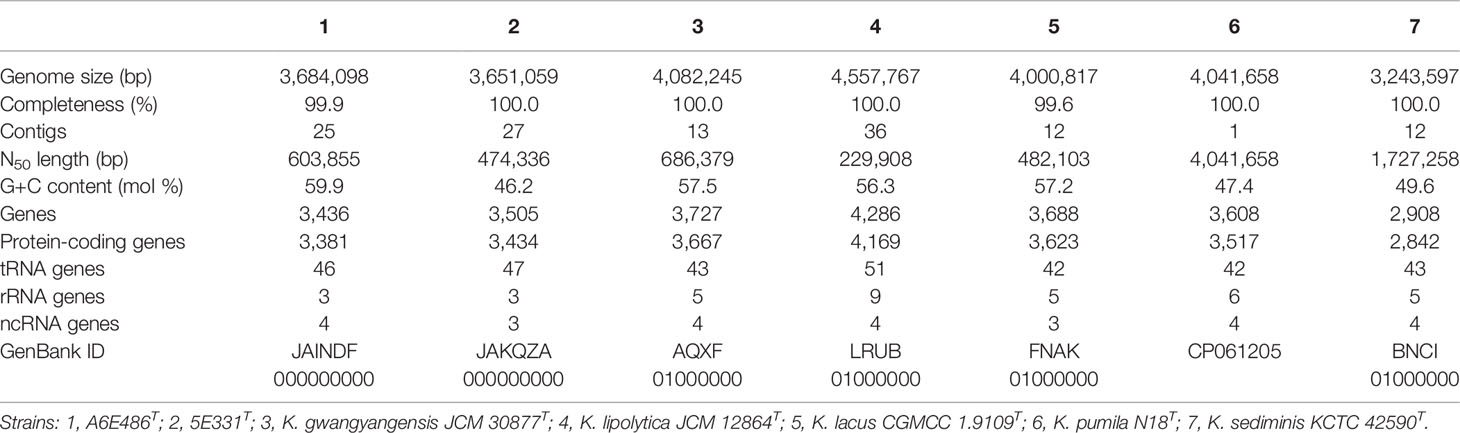
Table 3 Genome statistics of strains A6E486T and 5E331T and five members of the genus Kordiimonas.
The POCP and ANI values were calculated to identify the genomic similarities of strains A6E486T and 5E331T with some members of the genus Kordiimonas (Figure 2). The POCP and ANI values between strain A6E486T (strain 5E331T) and some species of the genus Kordiimonas were 56.3%–67.3% (56.0%–61.5%) and 68.6%–73.1% (68.8%–69.9%), respectively. The POCP values were higher than the genus delineation threshold (50%) (Qin et al., 2014a), and the ANI values were lower than the species delineation threshold (95%) (Yoon et al., 2017), which indicated that strains A6E486T and 5E331T represented two novel species of the genus Kordiimonas. The ribosomal protein phylogenetic tree showed the evolutionary relationships of some members of genus Kordiimonas (Figure 2).
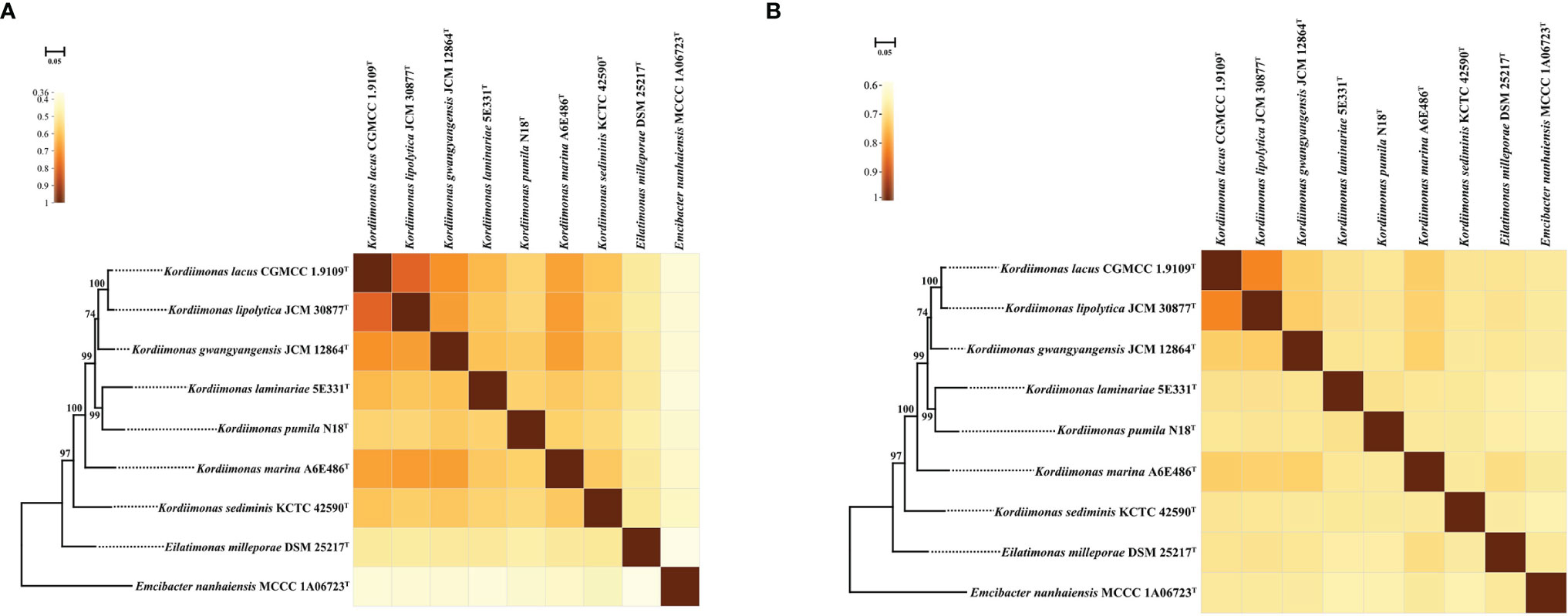
Figure 2 Heat maps showing POCP and ANI values among A6E486T, 5E331T, and members of the family Temperatibacteraceae. (A) Heat map based on POCP values. (B) Heat map based on ANI values. IQ-TREE based on ribosomal protein sequences shows the phylogenetic relationships among these strains. Percentage bootstrap values (1,000 replicates) are shown at branch nodes. Emcibacter nanhaiensis MCCC 1A06723T was used as the out-group. Bar, 0.05 substitutions per nucleotide position. Same tree topology was also obtained using FastTree. ANI, average nucleotide identity; POCP, percentage of conserved proteins.
Pan-genome analysis using BPGA was performed to identify orthologous groups among strains A6E486T, 5E331T, K. gwangyangensis JCM 30877T, K. lipolytica JCM 12864T, K. lacus CGMCC 1.9109T, K. pumila N18T, and K. sediminis KCTC 42590T. Comparative analysis based on orthologous groups of proteins revealed that 1,258 core genes were shared by the seven species (Figure 3A). The percentages of core genes in each genome ranged from 34.3% to 44.1% (Figure 3B), and compared with unique genes and accessory genes, core genes contributed more to energy metabolism, nucleotide metabolism, replication and repair, and translation (Supplementary Figure S3). The proportion of accessory genes and unique genes in the seven genomes varied greatly (Figure 3B). Accessory genes and unique genes were more distributed than core genes in amino acid metabolism, carbohydrate metabolism, signal transduction, and xenobiotic biodegradation and metabolism (Supplementary Figure S3), which indicated that these genes conferred metabolic diversity and marine environment adaptability to the members of the genus Kordiimonas (Innamorati et al., 2020). Results of the core pan-genome prediction model showed that the pan-genome increased with the addition of new strains and was far from saturation (Supplementary Figure S4), which indicated that the pan-genome of the genus Kordiimonas was open. Compared with a previous study (Geng et al., 2022), the complement of strains A6E486T and 5E331T in this study reduced the number of shared core genes of the genus Kordiimonas, showing that core genes decreased with the increase of species (Supplementary Figure S4).
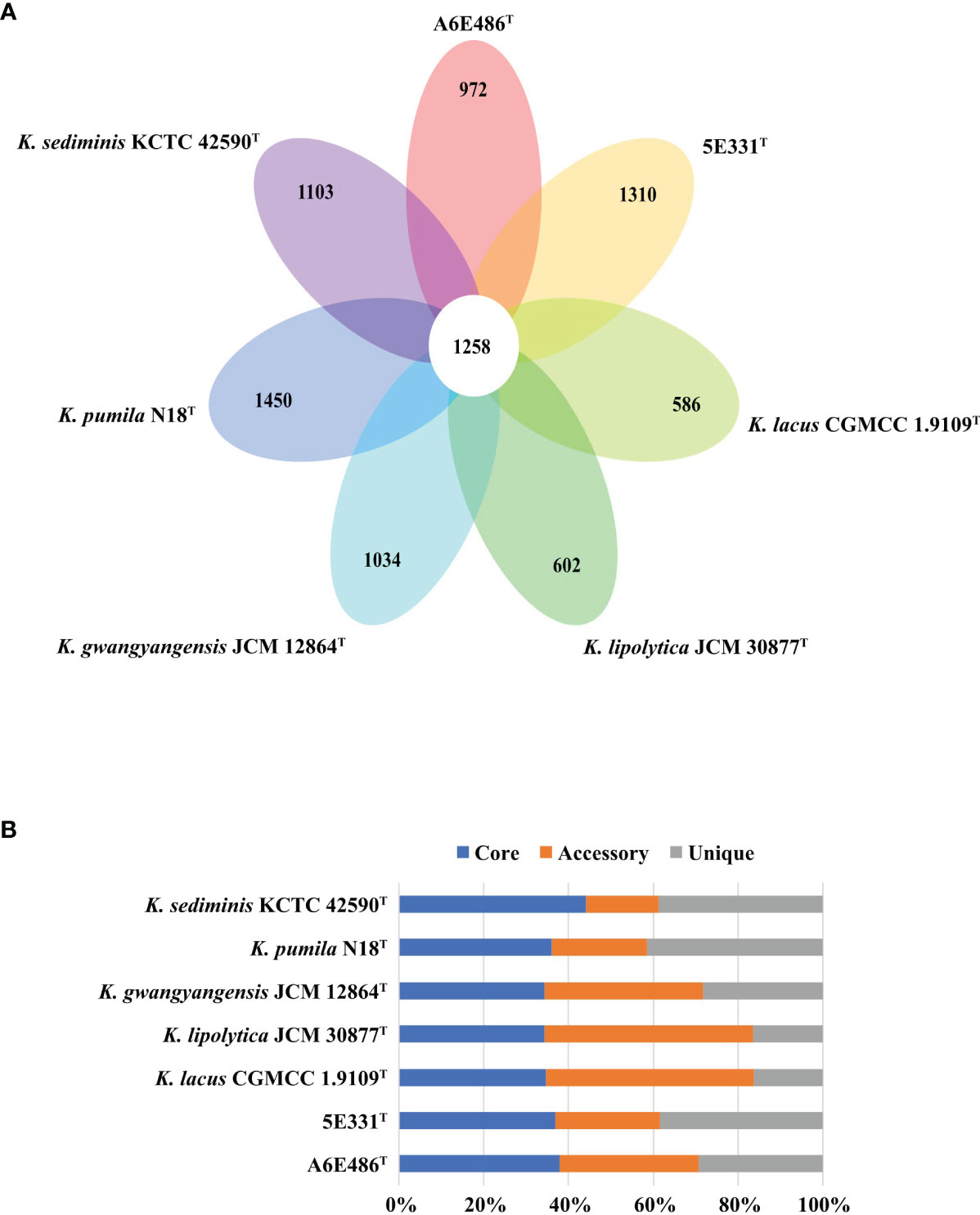
Figure 3 Comparisons of Kordiimonas orthologous protein groups in the seven Kordiimonas genomes. (A) Venn diagram displaying the numbers of core gene families and unique genes for each of the seven Kordiimonas strains. (B) Percentage of core, accessory, and unique genes in each of the seven genomes.
Metabolic pathways were analyzed using KEGG’s BlastKOALA service. The results showed that the seven species of the genus Kordiimonas had the most genes in gene information processing, followed by signaling and cellular processes and carbohydrate metabolism (Supplementary Figure S5). In addition, the metabolic pathways of the seven species were compared and analyzed, including carbohydrate metabolism, energy metabolism, lipid metabolism, amino acid metabolism, and metabolism of cofactors and vitamins. Among the seven Kordiimonas species, all strains have complete pathways of gluconeogenesis (M00003), pyruvate oxidation (M00307), citrate cycle (M00009), pentose phosphate pathway (M00007), and glyoxylate cycle (M00012), whereas only strains A6E486T and K. lipolytica JCM 30877T possess a complete glycolysis pathway (M00001) (Figure 4). The seven species also have similarities and differences in other metabolic pathways. For example, a complete PE biosynthesis pathway (M00093) was found in all strains, which was consistent with the polar lipid results of the genus Kordiimonas. The initiation pathways of fatty acid synthesis (M00082) and cobalamin biosynthesis (M00122) are complete only in strain 5E331T, while the cytochrome o ubiquinol oxidase (M00417) and betaine biosynthesis (M00555) pathways are complete only in strain A6E486T (Figure 4). Considering that the incomplete glycolysis pathway in most Kordiimonas strains leads to pyruvate deficiency and thus to the decrease of acetyl-CoA from the pyruvate oxidation pathway, the leucine degradation pathway is important due to the production of acetyl-CoA (Figure 5A). It was proven that strains A6E486T, 5E331T, K. gwangyangensis JCM 30877T, and K. lipolytica JCM 12864T could grow slowly in the medium with leucine as the solo carbon source (Supplementary Figure S6). In addition, dTDP-L-rhamnose biosynthesis pathway (M00793) was completely annotated in the genomes of the seven Kordiimonas species (Figure 5B).
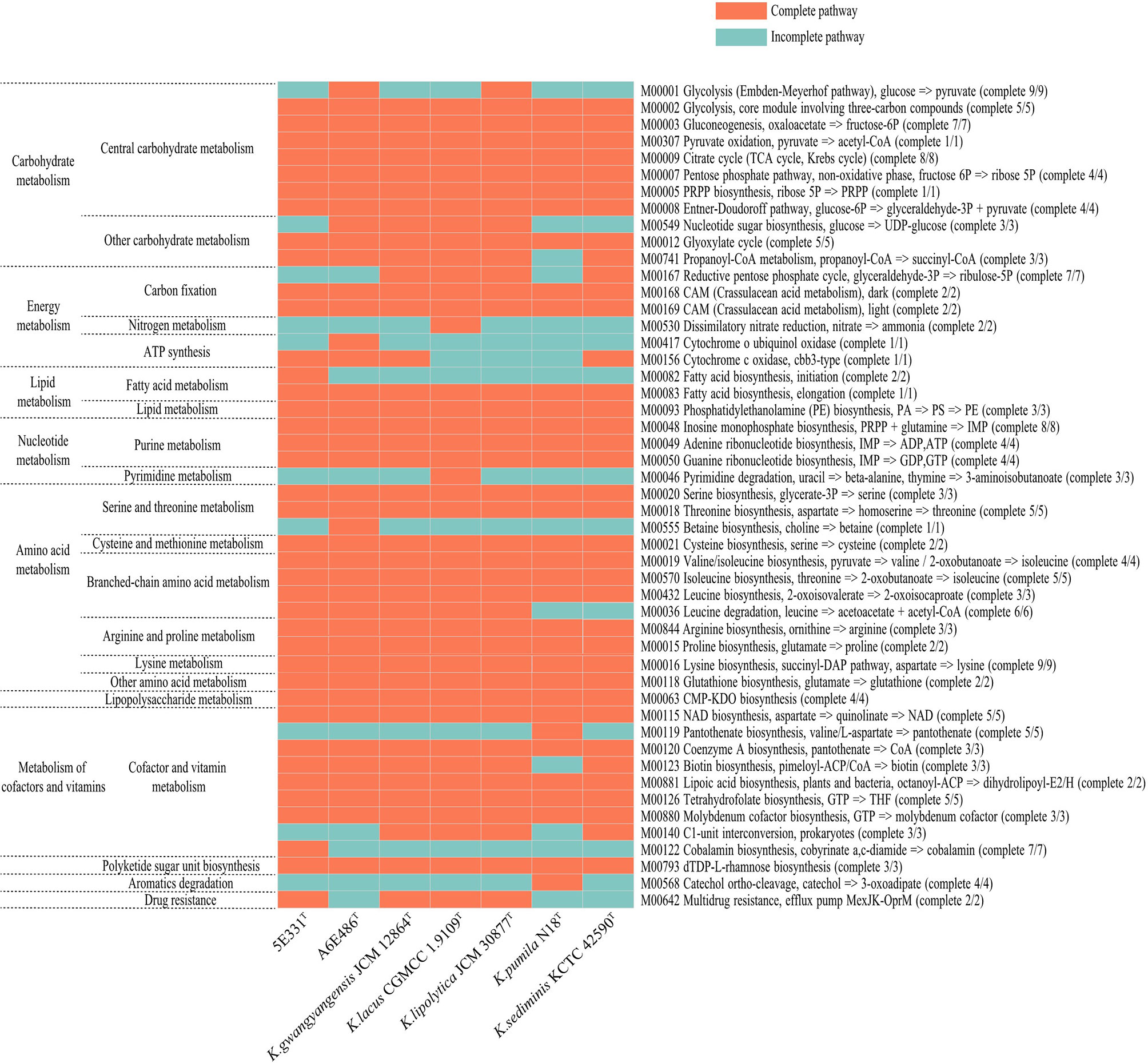
Figure 4 Heat maps of complete and incomplete metabolic pathways in the genomes of strains A6E486T, 5E331T, K. gwangyangensis JCM 30877T, K. lipolytica JCM 12864T, K. lacus CGMCC 1.9109T, K. pumila N18T, and K. sediminis KCTC 42590T.
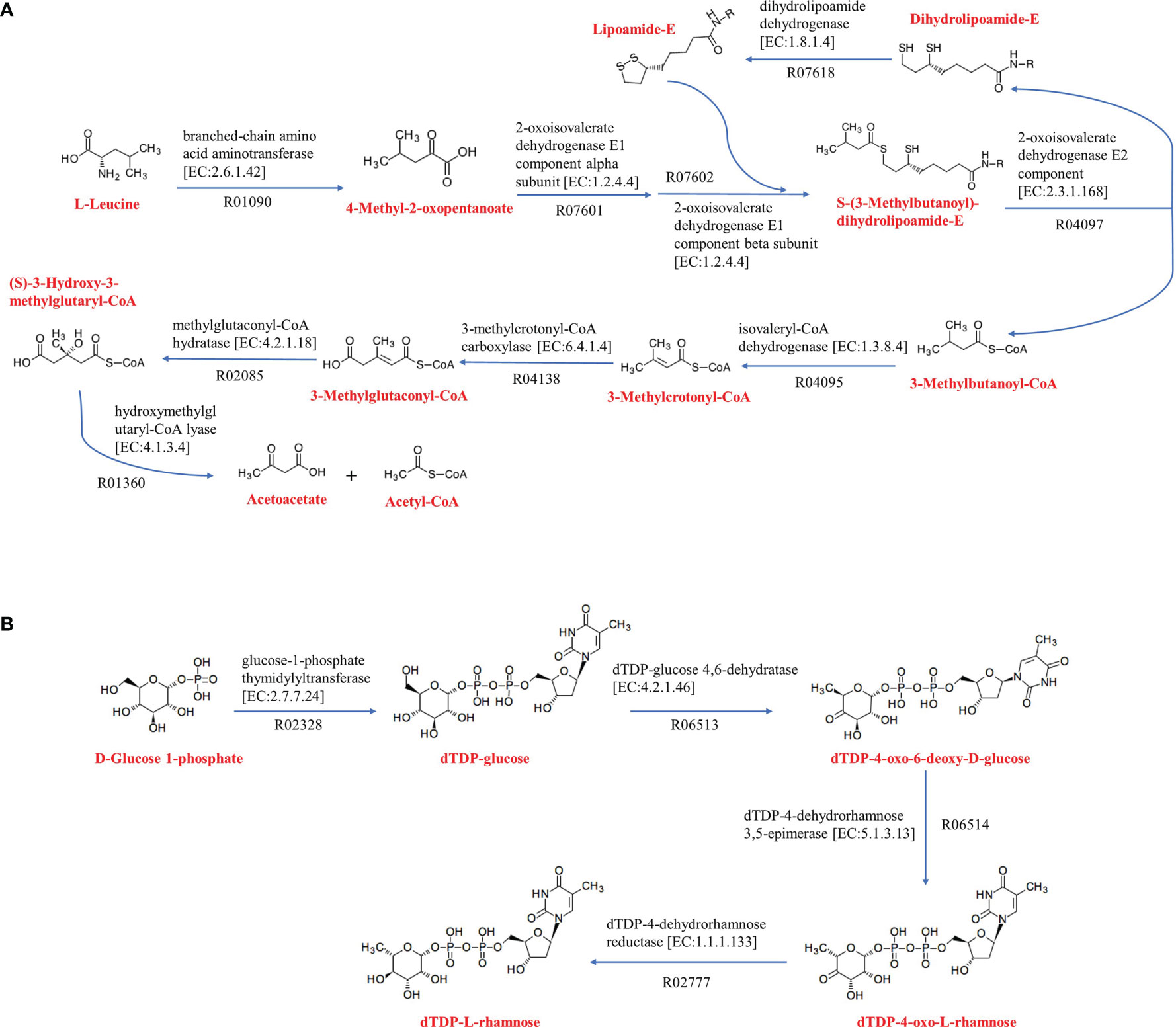
Figure 5 Amino acid metabolism and polyketide sugar unit biosynthesis of Kordiimonas strains. (A) Leucine degradation. (B) dTDP-L-rhamnose biosynthesis. The metabolic pathway maps are based on the KEGG analysis. Other reactants and products have been omitted.
AntiSMASH server was used to predict the secondary metabolites of the seven species. Results showed that all of the seven species contained gene clusters encoding RiPP-like (Other unspecified ribosomally synthesized and posttranslationally modified peptide product). Except for K. gwangyangensis JCM 12864T and K. pumila N18T, the genomes of the remaining five strains contained terpene gene clusters. The gene clusters encoding hserlactone (homoserine lactone) were found in genomes of the six strains except K. pumila N18T, which had β-lactone (β-lactone containing protease inhibitor) gene clusters (Supplementary Table S2). Homoserine lactones, as one of the most important signal molecules in the quorum-sensing system of Gram-negative bacteria, regulate the expression of many physiological characteristics (Parsek and Greenberg, 2000; Qin et al., 2020). Cluster Pfam (Protein Families Database) analysis and Pfam-based GO (Gene Ontology) term annotation results showed that the core biosynthetic genes in all of the hserlactone gene clusters encoded autoinducer synthase, and there were additional biosynthetic genes encoding luxR family regulatory proteins upstream and downstream of the core genes (Supplementary Figure S7). The luxR regulators are important components of the quorum-sensing system, which are distributed to sense and respond to N-acyl homoserine lactone by autoinducer-binding domain (Subramoni and Venturi, 2009; Santos et al., 2012).
Considering that the strains had differences in acid production and oxidation with different carbohydrates as substrates (Table 1; Supplementary Table S1), carbohydrate-active enzymes of the genus Kordiimonas were annotated and analyzed. The results showed that K. sediminis KCTC 42590T had the least carbohydrate-active enzymes (86), while K. gwangyangensis JCM 30877T had the most (137). Among these carbohydrate-active enzymes, most enzymes in the genomes of the seven species were assigned to the glycosyltransferase (GT) family and glycoside hydrolase (GH) family, which was consistent with a previous study (Geng et al., 2022); however, polysaccharide lyases (PLs) were found in parts of the Kordiimonas species (Figure 6). Strain A6E486T had 53 GTs and 50 GHs, which were more than those in strain 5E331T, K. lipolytica JCM 12864T, K. lacus CGMCC 1.9109T, K. pumila N18T, and K. sediminis KCTC 42590T. Strain 5E331T harbored 107 carbohydrate-active enzymes, including 42 GTs, 36 GHs, 16 carbohydrate esterases (CEs), 6 carbohydrate-binding modules (CBMs), and 7 auxiliary activities (AAs). Detailed comparison results were shown in Figure 6.
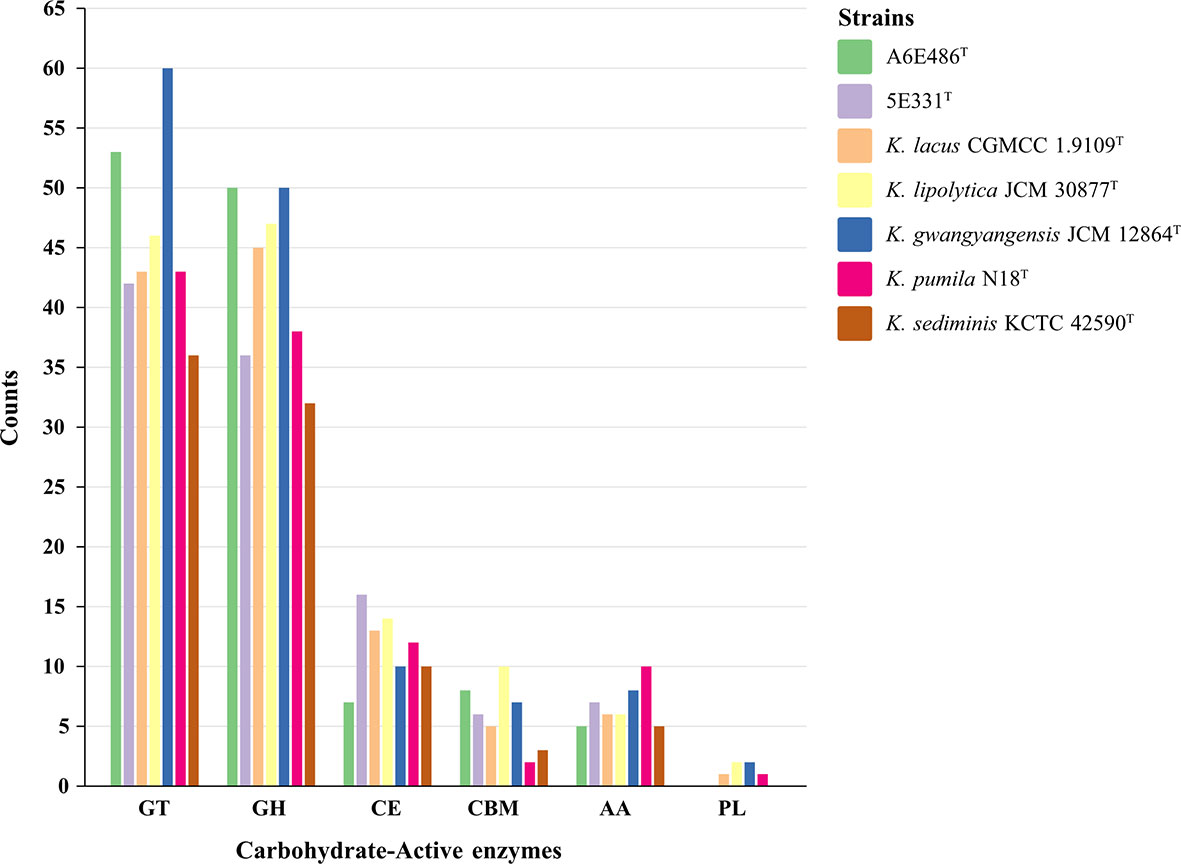
Figure 6 Histogram of predicted carbohydrate-active enzymes in strains A6E486T, 5E331T, K. gwangyangensis JCM 30877T, K. lipolytica JCM 12864T, K. lacus CGMCC 1.9109T, K. pumila N18T, and K. sediminis KCTC 42590T. Numbers of each enzyme detected in the genomes were shown in the map. GT, glycosyltransferase; GH, glycoside hydrolase; CE, carbohydrate esterase; CBM, carbohydrate-binding module; AA, auxiliary activity; PL, polysaccharide lyase.
Considering that members of the genus Kordiimonas were all isolated from marine environments and habitats, metabolic features related to functional categories were analyzed to investigate the marine-adapted lifestyle of the genus Kordiimonas (Table 4). In the first place, 26–51 genes associated with “Virulence, Disease and Defense” were found in the genomes of members of the genus Kordiimonas, which may be crucial for bacteria to resist marine environment pollutants including heavy-metal ions, antibiotics, and other toxic compounds. Secondly, flagellar motility and bacterial chemotaxis genes were found, and there were differences in the number of the related genes. Most of the marine environments are oligotrophic; flagellar motility and bacterial chemotaxis genes, which assist marine bacteria in the acquisition of nutrients, may play an important role in the marine sediment-adapted lifestyle (Qin et al., 2011). Thirdly, there were different numbers of genes related to capsular and extracellular polysaccharides and polysaccharide export proteins in the genomes. Exopolysaccharides (EPSs) are essential for the survival of bacteria in marine environments, as EPSs assist them to endure extremes of temperature, salinity, and nutrient availability (Nichols et al., 2005). The existence of genes related to EPS synthesis and export suggested that members of the genus Kordiimonas may be able to resist low temperature through EPS secretion. Additionally, cold-shock proteins and tRNA-dihydrouridine synthase play roles in the cold-adapted lifestyle of marine bacteria by helping protein folding and increasing tRNA flexibility at low temperatures, respectively (Qin et al., 2014b). Fourthly, all members of the genus Kordiimonas had numerous genes involved in the “stress response,” which may give these species the ability to cope with pressures such as oxygen and temperature in marine environments.
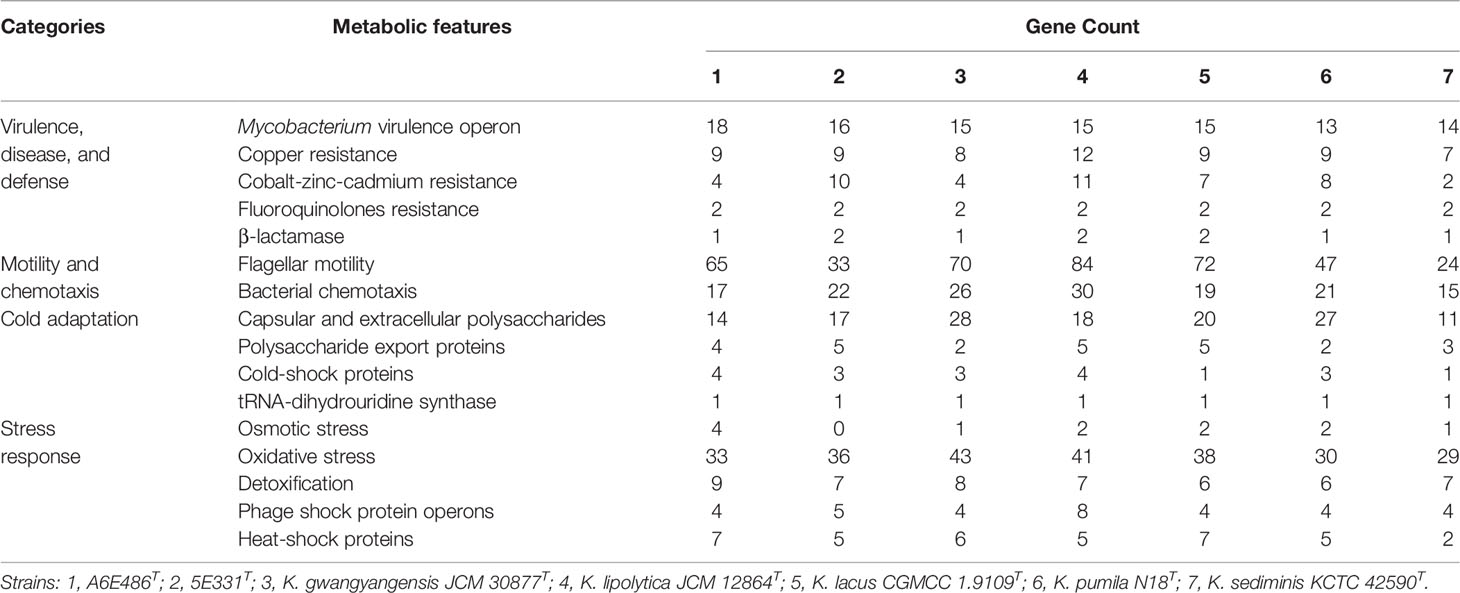
Table 4 Metabolic features and selected genes related to the adaptation to marine environments.
Considering the high osmotic pressure of marine environments and the protective effect of compatible solutes on microorganisms under high osmotic condition (Da et al., 1998; Wood et al., 2001), compatible solute synthesis and transport of the genus Kordiimonas were analyzed using the KEGG database. The results showed that only strain A4E486T had a complete betaine synthesis pathway (M00555) among the seven species of the genus Kordiimonas (Figure 4). Strains 5E331T, K. gwangyangensis JCM 30877T, K. lipolytica JCM 12864T, K. lacus CGMCC 1.9109T, K. pumila N18T, and K. sediminis KCTC 42590T encode choline dehydrogenase (CHDH), which converts choline to betaine aldehyde (Zou et al., 2016). The genes encoding glycine betaine transporter were found only in the genome of strain A4E486T. In the hypertonic environment, the accumulation of amino acids in cells, such as proline (Hoffmann et al., 2012) and arginine (Xu S. et al., 2011), also plays a positive role in the anti-osmotic ability of microorganisms (Da et al., 1998; Roesser and Muller, 2001). The seven genomes had the proline biosynthesis pathway (M00015) and arginine biosynthesis pathway (M00844) identified by the KEGG database (Figure 4). Numerous complete amino acid synthesis pathways in members of the genus Kordiimonas may help to resist cell damage caused by high osmotic pressure in marine environments. Finally, the antioxidant systems used to scavenge free radicals were analyzed. The antioxidant system mainly includes superoxide dismutase, catalase, glutathione, and cytochrome oxidase (Pomposiello and Demple, 2002). The analysis showed that all seven species of the genus Kordiimonas encoded superoxide dismutase (Fe-Mn family) and catalase-peroxidase. Strains 5E331T and K. pumila N18T had genes that encoded catalase. The genes encoding cytochrome c peroxidase were found in strains 5E331T, K. gwangyangensis JCM 30877T, K. lipolytica JCM 12864T, K. lacus CGMCC 1.9109T, and K. sediminis KCTC 42590T. Additionally, all seven members of the genus Kordiimonas had a complete glutathione synthesis pathway (M00118) (Figure 4).
The analysis of metabolic features and related genes revealed the genetic basis of the genus Kordiimonas to adapt to the marine environment.
Kordiimonas marina (ma.ri’na. L. fem. adj. marina, pertaining to the isolation of the type strain from the marine environment).
Cells are Gram-negative and facultatively anaerobic with rod shape. Growth is observed at pH 5.5–7.5 (optimum, 6.5) and temperatures of 20°C–43°C (optimum, 33°C–35°C) and with 1.0%–5.5% (w/v, optimum, 2.5%–3.0%) NaCl. Growth occurs under anaerobic conditions, and nitrate is reduced to nitrite. The activities of trypsin and oxidase are positive, and the activities of ɑ-chymotrypsin, ɑ-galactosidase, ɑ-glucosidase, ɑ-mannosidase, and ɑ-fucosidase are weakly positive. Acids are produced from D-arabinose, D-ribose, D-xylose, L-sorbose, D-tagatose, and potassium 5-ketogluconate. The major cellular fatty acids (>10%) are iso-C15:0, iso-C17:0, iso-C17:1 ω9c, and summed feature 3 (comprising C16:1 ω6c and/or C16:1 ω7c). The predominant respiratory quinone is Q-10. The major polar lipids consist of PE, PG, DPG, and unidentified glycolipids, aminolipids, and lipids. The DNA G+C content of type strain is 59.9 mol%.
The type strain is A6E486T (= KCTC 82758T = MCCC 1H00470T), which was isolated from coastal sediments collected off the coast of Weihai, China.
Kordiimonas laminariae (la.mi.na’ri.ae. N.L. gen. fem. n. laminariae, pertaining to the kelp Laminaria, from which the type strain was isolated).
Cells are Gram-negative and aerobic with rod shape. Growth is observed at pH 6.0–8.5 (optimum,7.0) and temperatures of 15°C–40°C (optimum, 33°C) and with 1.5%–5.0% (w/v, optimum, 3.0%) NaCl. Growth does not occur under anaerobic condition, and nitrate is not reduced to nitrite. The activities of trypsin and oxidase are positive, but the activities of ɑ-chymotrypsin, ɑ-galactosidase, ɑ-glucosidase, ɑ-mannosidase, and ɑ-fucosidase are negative. Acids are produced from D-ribose, L-sorbose, D-turanose, D-lyxose, D-tagatose, and potassium 5-ketogluconate. The major cellular fatty acids (>10%) are iso-C15:0, iso-C17:1 ω9c, and summed feature 3 (comprising C16:1 ω6c and/or C16:1 ω7c). The predominant respiratory quinone is Q-10. The major polar lipids consist of PE, PG, DPG, and unidentified glycolipids, aminolipids, and lipids. The DNA G+C content of type strain is 46.2 mol%.
The type strain is 5E331T (= KCTC 92199T = MCCC 1H00515T), which was isolated from fresh kelps collected from a kelp culture area, Rongcheng, China.
The data presented in this study are deposited in the GenBank. The accession number for the 16S rRNA gene sequence of strains A6E486T and 5E331T are MZ901372 and OM663707, respectively. The accession number for the whole genome shotgun project of strains A6E486T and 5E331T are JAINDF000000000 and JAKQZA000000000, respectively.
Y-QY and Z-PH isolated the strain A6E486T and performed material preparation, experimental operation, and data collection and analysis. Y-YY helped process cell samples and photograph cell morphology. LM isolated the strain 5E331T, and Y-QY finished the experiment and article. Z-JD and M-QY offered experiment guidance and critical revision of the article. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (32070002) and the National Science and Technology Fundamental Resources Investigation Program of China (2019FY100700).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The implementation of scanning electron microscope was supported by the Physical-Chemical Materials Analytical and Testing Center of Shandong University at Weihai.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.919253/full#supplementary-material
ANI, average nucleotide identity; POCP, percentage of conserved proteins; KEGG, Kyoto Encyclopedia of Genes and Genomes; MCCC, Marine Culture Collection of China; KCTC, Korean Collection for Type Cultures; JCM, Japan Collection of Microorganisms; NCBI, National Center for Biotechnology Information; PGAP, Prokaryotic Genome Annotation Pipeline; HPLC, high-performance liquid chromatography; NJ, neighbor-joining; ML, maximum-likelihood; MP, maximum-parsimony; Pfam, Protein Families Database; GO, Gene Ontology; RAST, Rapid Annotation using Subsystem Technology; BPGA, Bacterial Pan Genome Analysis.
Aziz R. K., Bartels D., Best A. A., Dejongh M., Disz T., Edwards R. A., et al. (2008). The RAST Server: Rapid Annotations Using Subsystems Technology. BMC Genomics 9, 75. doi: 10.1186/1471-2164-9-75
Chaudhari N. M., Gupta V. K., Dutta C. (2016). BPGA- an Ultra-Fast Pan-Genome Analysis Pipeline. Sci. Rep. 6, 24373. doi: 10.1038/srep24373
Chernomor O., von Haeseler A., Minh B. Q. (2016). Terrace Aware Data Structure for Phylogenomic Inference From Supermatrices. Syst. Biol. 65 (6), 997–1008. doi: 10.1093/sysbio/syw037
Da C. M., Santos H., Galinski E. A. (1998). An Overview of the Role and Diversity of Compatible Solutes in Bacteria and Archaea. Adv. Biochem. Eng. Biotechnol. 61, 117–153. doi: 10.1007/BFb0102291
Dong X. Z., Cai M. Y. (2001). “"Determination of Biochemical Characteristics,",” in Manual for the Systematic Identification of General Bacteria. Eds. Dong X. Z., Cai M. Y. (Beijing: Science Press), 370–398.
Edgar R. C. (2004). MUSCLE: Multiple Sequence Alignment With High Accuracy and High Throughput. Nucleic Acids Res. 32 (5), 1792–1797. doi: 10.1093/nar/gkh340
Felsenstein J. (1981). Evolutionary Trees From DNA Sequences: A Maximum Likelihood Approach. J. Mol. Evol. 17 (6), 368–376. doi: 10.1007/BF01734359
Geng N., Yang D., Hua J., Huang L. J., Dong H., Sun C., et al. (2022). Complete Genome Sequence of Kordiimonas Pumila N18T Sheds Light on Biogeochemical Roles of the Genus Kordiimonas. Mar. Genomics 62, 100930. doi: 10.1016/j.margen.2022.100930
Hoffmann T., von Blohn C., Stanek A., Moses S., Barzantny H., Bremer E. (2012). Synthesis, Release, and Recapture of Compatible Solute Proline by Osmotically Stressed Bacillus Subtilis Cells. Appl. Environ. Microbiol. 78 (16), 5753–5762. doi: 10.1128/AEM.01040-12
Hyatt D., Chen G. L., Locascio P. F., Land M. L., Larimer F. W., Hauser L. J. (2010). Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinf. 11, 119. doi: 10.1186/1471-2105-11-119
Innamorati K. A., Earl J. P., Aggarwal S. D., Ehrlich G. D., Hiller N. L. (2020). “"The Bacterial Guide to Designing a Diversified Gene Portfolio,",” in The Pangenome: Diversity, Dynamics and Evolution of Genomes. Eds. Tettelin H., Medini D. (Cham, CH: Springer), 51–87.
Jorgensen J. H., Turnidge J. D. (2015). “"Susceptibility Test Methods: Dilution and Disk Diffusion Methods,",” in Manual of Clinical Microbiology. Eds. Jorgensen J. H., Carroll K. C., Funke G., Pfaller M. A., Landry M. L., Richter S. S., Warnock D. W. (New York, NY: Wiley), 1253–1273.
Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M. (2016). KEGG as a Reference Resource for Gene and Protein Annotation. Nucleic Acids Res. 44 (D1), 457–462. doi: 10.1093/nar/gkv1070
Komagata K., Suzuki K. (1987). Lipid and Cell-Wall Analysis in Bacterial Systematics. Methods Microbiol. 19, 161–207. doi: 10.1016/S0580-9517(08)70410-0
Kumar S., Stecher G., Li M., Knyaz C., Tamura K. (2018). MEGA X: Molecular Evolutionary Genetics Analysis Across Computing Platforms. Mol. Biol. Evol. 35 (6), 1547–1549. doi: 10.1093/molbev/msy096
Kwon K. K., Lee H. S., Yang S. H., Kim S. J. (2005). Kordiimonas Gwangyangensis Gen. Nov., Sp. Nov., a Marine Bacterium Isolated From Marine Sediments That Forms a Distinct Phyletic Lineage (Kordiimonadales Ord. Nov.) in the 'Alphaproteobacteria'. Int. J. Syst. Evol. Microbiol. 55 (5), 2033–2037. doi: 10.1099/ijs.0.63684-0
Lane D. J. (1991). “"16s/23s rRNA Sequencing,",” in Nucleic Acid Techniques in Bacterial Systematics. Eds. Stackebrandt E., Goodfellow M. (New York, NY: Wiley), 115–175.
Li R. Q., Yu C., Li Y. R., Lam T., Yiu S. M., Kristiansen K., et al. (2009). SOAP2: An Improved Ultrafast Tool for Short Read Alignment. Bioinformatics 25 (15), 1966–1967. doi: 10.1093/bioinformatics/btp336
Math R. K., Jeong S. H., Jin H. M., Park M. S., Kim J. M., Jeon C. O. (2012). Kordiimonas Aestuarii Sp. Nov., a Marine Bacterium Isolated From a Tidal Flat. Int. J. Syst. Evol. Microbiol. 62 (Pt 12), 3049–3054. doi: 10.1099/ijs.0.038943-0
Minnikin D. E., O'Donnell A. G., Goodfellow M., Alderson G., Athalye M., Schaal A., et al. (1984). An Integrated Procedure for the Extraction of Bacterial Isoprenoid Quinones and Polar Lipids. J. Microbiol. Meth. 2 (5), 233–241. doi: 10.1016/0167-7012(84)90018-6
Mu D. S., Liang Q. Y., Wang X. M., Lu D. C., Shi M. J., Chen G. J., et al. (2018). Metatranscriptomic and Comparative Genomic Insights Into Resuscitation Mechanisms During Enrichment Culturing. Microbiome 6 (1), 230. doi: 10.1186/s40168-018-0613-2
Nguyen L. T., Schmidt H. A., von Haeseler A., Minh B. Q. (2015). IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi: 10.1093/molbev/msu300
Nichols C. A., Guezennec J., Bowman J. P. (2005). Bacterial Exopolysaccharides From Extreme Marine Environments With Special Consideration of the Southern Ocean, Sea Ice, and Deep-Sea Hydrothermal Vents: A Review. Mar. Biotechnol. (NY). 7 (4), 253–271. doi: 10.1007/s10126-004-5118-2
Parks D. H., Imelfort M., Skennerton C. T., Hugenholtz P., Tyson G. W. (2015). CheckM: Assessing the Quality of Microbial Genomes Recovered From Isolates, Single Cells, and Metagenomes. Genome Res. 25 (7), 1043–1055. doi: 10.1101/gr.186072.114
Parsek M. R., Greenberg E. P. (2000). Acyl-Homoserine Lactone Quorum Sensing in Gram-Negative Bacteria: A Signaling Mechanism Involved in Associations With Higher Organisms. Proc. Natl. Acad. Sci. U. S. A. 97 (16), 8789–8793. doi: 10.1073/pnas.97.16.8789
Pomposiello P. J., Demple B. (2002). Global Adjustment of Microbial Physiology During Free Radical Stress. Adv. Microb. Physiol. 46, 319–341. doi: 10.1016/s0065-2911(02)46007-9
Price M. N., Dehal P. S., Arkin A. P. (2009). FastTree: Computing Large Minimum Evolution Trees With Profiles Instead of a Distance Matrix. Mol. Biol. Evol. 26 (7), 1641–1650. doi: 10.1093/molbev/msp077
Qin Q. L., Li Y., Zhang Y. J., Zhou Z. M., Zhang W. X., Chen X. L., et al. (2011). Comparative Genomics Reveals a Deep-Sea Sediment-Adapted Life Style of Pseudoalteromonas Sp. SM9913. ISME J. 5 (2), 274–284. doi: 10.1038/ismej.2010.103
Qin X., Thota G. K., Singh R., Balamurugan R., Goycoolea F. M. (2020). Synthetic Homoserine Lactone Analogues as Antagonists of Bacterial Quorum Sensing. Bioorg Chem. 98, 103698. doi: 10.1016/j.bioorg.2020.103698
Qin Q. L., Xie B. B., Yu Y., Shu Y. L., Rong J. C., Zhang Y. J., et al. (2014b). Comparative Genomics of the Marine Bacterial Genus Glaciecola Reveals the High Degree of Genomic Diversity and Genomic Characteristic for Cold Adaptation. Environ. Microbiol. 16 (6), 1642–1653. doi: 10.1111/1462-2920.12318
Qin Q. L., Xie B. B., Zhang X. Y., Chen X. L., Zhou B. C., Zhou J., et al. (2014a). A Proposed Genus Boundary for the Prokaryotes Based on Genomic Insights. J. Bacteriol. 196 (12), 2210–2215. doi: 10.1128/JB.01688-14
Roesser M., Muller V. (2001). Osmoadaptation in Bacteria and Archaea: Common Principles and Differences. Environ. Microbiol. 3 (12), 743–754. doi: 10.1046/j.1462-2920.2001.00252.x
Saitou N., Nei M. (1987). The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 4 (4), 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Santos C. L., Correia-Neves M., Moradas-Ferreira P., Mendes M. V. (2012). A Walk Into the LuxR Regulators of Actinobacteria: Phylogenomic Distribution and Functional Diversity. PLos One 7 (10), e46758. doi: 10.1371/journal.pone.0046758
Sasser M. (1990). “Identification of Bacteria by Gas Chromatography of Cellular Fatty Acids,” in MIDI Technical Note 101 (Newark, DE: MIDI Inc).
Subramoni S., Venturi V. (2009). LuxR-Family 'Solos': Bachelor Sensors/Regulators of Signalling Molecules. Microbiol. (Reading England). 155 (5), 1377–1385. doi: 10.1099/mic.0.026849-0
Wiegand S., Jogler M., Boedeker C., Pinto D., Vollmers J., Rivas-Marin E., et al. (2020). Cultivation and Functional Characterization of 79 Planctomycetes Uncovers Their Unique Biology. Nat. Microbiol. 5 (1), 126–140. doi: 10.1038/s41564-019-0588-1
Wood J. M., Bremer E., Csonka L. N., Kraemer R., Poolman B., van der Heide T., et al. (2001). Osmosensing and Osmoregulatory Compatible Solute Accumulation by Bacteria. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 130 (3), 437–460. doi: 10.1016/s1095-6433(01)00442-1
Wu Y. H., Jian S. L., Meng F. X., Maripatay, Tohty D., Wang C. S., et al. (2016). Kordiimonas Lipolytica Sp. Nov., Isolated From Seawater. Int. J. Syst. Evol. Microbiol. 66 (6), 2198–2204. doi: 10.1099/ijsem.0.001007
Xu X. W., Huo Y. Y., Bai X. D., Wang C. S., Oren A., Li S. Y., et al. (2011). Kordiimonas Lacus Sp. Nov., Isolated From a Ballast Water Tank, and Emended Description of the Genus Kordiimonas. Int. J. Syst. Evol. Microbiol. 61 (Pt 2), 422–426. doi: 10.1099/ijs.0.018200-0
Xu S., Zhou J. W., Liu L. M., Chen J. (2011). Arginine: A Novel Compatible Solute to Protect Candida Glabrata Against Hyperosmotic Stress. Process Biochem. 46 (6), 1230–1235. doi: 10.1016/j.procbio.2011.01.026
Yang S. H., Kim M. R., Seo H. S., Lee S. H., Lee J. H., Kim S. J., et al. (2013). Description of Kordiimonas Aquimaris Sp. Nov., Isolated From Seawater, and Emended Descriptions of the Genus Kordiimonas Kwon Et Al. 2005 Emend. Xu Et Al. 2011 and of its Existing Species. Int. J. Syst. Evol. Microbiol. 63 (Pt 1), 298–302. doi: 10.1099/ijs.0.038893-0
Yoon S. H., Ha S. M., Lim J., Kwon S., Chun J. (2017). A Large-Scale Evaluation of Algorithms to Calculate Average Nucleotide Identity. Antonie Van Leeuwenhoek. 110 (10), 1281–1286. doi: 10.1007/s10482-017-0844-4
Zhang H., Yohe T., Huang L., Entwistle S., Wu P., Yang Z., et al. (2018). Dbcan2: A Meta Server for Automated Carbohydrate-Active Enzyme Annotation. Nucleic Acids Res. 46 (W1), W95–W101. doi: 10.1093/nar/gky418
Zhang H. X., Zhao J. X., Chen G. J., Du Z. J. (2016). Kordiimonas Sediminis Sp. Nov., Isolated From a Sea Cucumber Culture Pond. Antonie Van Leeuwenhoek. 109 (5), 705–711. doi: 10.1007/s10482-016-0671-z
Zhao J., Zhang R., Hou X. J., Han S. B., Li Y., Cong S., et al. (2018). Kordiimonas Pumila Sp. Nov., Isolated From Coastal Sediment. Int. J. Syst. Evol. Microbiol. 68 (5), 1743–1748. doi: 10.1099/ijsem.0.002740
Keywords: Kordiimonas, polyphasic taxonomy, comparative genomic analysis, metabolic pathways, homoserine lactone, marine-adapted lifestyle
Citation: Ye Y-Q, Hao Z-P, Yue Y-Y, Ma L, Ye M-Q and Du Z-J (2022) Characterization of Kordiimonas marina sp. nov. and Kordiimonas laminariae sp. nov. and Comparative Genomic Analysis of the Genus Kordiimonas, A Marine-Adapted Taxon. Front. Mar. Sci. 9:919253. doi: 10.3389/fmars.2022.919253
Received: 13 April 2022; Accepted: 19 May 2022;
Published: 20 June 2022.
Edited by:
Shan He, Ningbo University, ChinaReviewed by:
Jian He, Nanjing Agricultural University, ChinaCopyright © 2022 Ye, Hao, Yue, Ma, Ye and Du. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meng-Qi Ye, eWVtZW5ncWlAMTI2LmNvbQ==; Zong-Jun Du, ZHV6b25nanVuQHNkdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.