 Lusheng Xin1,2,3*†
Lusheng Xin1,2,3*† Jiangnan Yu1,2,3†Bowen Huang1Chen Li1,3Changming Bai1,3*
Jiangnan Yu1,2,3†Bowen Huang1Chen Li1,3Changming Bai1,3* Qin Liu4*
Qin Liu4* Chongming Wang1,3
Chongming Wang1,3- 1Key Laboratory of Environment Controlled Aquaculture Ministry of Education, Dalian Ocean University, Dalian, China
- 2Function Laboratory for Marine Fisheries Science and Food Production Processes, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
- 3Qingdao Key Laboratory of Mariculture Epidemiology and Biosecurity, Key Laboratory of Maricultural Organism Disease Control, Ministry of Agriculture, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao, China
- 4Guangxi Key Laboratory for Agricultural Resources Chemistry and Biotechnology, College of Biology and Pharmacy, Yulin Normal University, Yulin, China
Background
Vibrios are widely distributed in the marine environment. Several Vibrio species, including V. lentus, V. kanaloae, V. pomeroyi, and V. chagasii, could hardly be distinguished phenotypically from V. splendidus and were known as splendidus-clade strains (Gay et al., 2004b; Roux and Austin, 2006). The classification of these splendidus-clade strains had been characterized at a molecular level (gyrase B subunit, 16S rRNA sequences and amplified fragment length polymorphism) (Le Roux et al., 2004; Rojas et al., 2015), whereas there have been few systematic studies on the characterization of these splendidus-clade strains at the genomic level. These splendidus-clade strains had been found to be associated with the outbreak events of mortality among molluscs (Le Roux et al., 2002; Rojas et al., 2015). These various strains might act in an additive/synergistic manner, resulting in higher Crassostrea gigas mortality rates (Gay et al., 2004a; Gómez-León et al., 2005), or few strains displayed an individual pathogenicity as V. splendidus strain JZ6 causing obvious lesions on the adductor muscle of Yesso scallop (Liu et al., 2013).
The key virulence factors of bacteria influence their pathogenicity. However, little is known about the pathophysiology and virulence factors of these splendidus-clade strains. (Zhang and Li, 2021). Few studies have revealed several virulence factors of these splendidus-clade strains. Extracellular products, such as metalloproteases and siderophores as virulence factors competing for host available iron (Wyckoff et al., 2015; Song et al., 2018), flagellum protein FliC and dihydrolipoic acid dehydrogenase (DLDH) as adhesion factors (Dai et al., 2020), and the quorum sensing system as regulatory factor, have been verified to contribute to the pathogenesis of these splendidus-clade strains (Henke and Bassler, 2004). Iron chelators had been shown to reduce the virulence of V. splendidus, suggesting that the iron uptake mechanism may influence its virulence. (Zhang et al., 2016).
A recent study by us showed that V. kanaloae strain KH-4 could lead to acute hepatopancreas necrosis, and final death of ark clams (Huang et al., 2021). However, the pathogenic molecular mechanism of KH-4 is largely unknown, and the lack of the complete genome of V. kanaloae isolated from ark clams limits the advance of relevant research. The genomic sequence of V. kanaloae strain KH-4, isolated from the hepatopancreas of diseased ark clams in December 2020, was drawn in this work. Research on the identification of virulence factors in KH-4 was carried out to reveal its pathogenesis. Meanwhile, genomic comparative analysis of KH-4 and 19 selected splendidus-clade related strains was performed for the characterization of the differentiation and evolution among these splendidus-clade strains.
Data Description
Genome Sequencing and Assembly
The genomic DNA of V. kanaloae strain KH-4 was extracted from monoclonal pure culture that was isolated in our previous study (Huang et al., 2021) according to the manufacturer’s instructions of HiPure Bacterial DNA Kits (Magen, Guangzhou, China). Qubit (Thermo Fisher Scientific, Waltham, MA, USA) and Nanodrop (Thermo Fisher Scientific, Waltham, MA, USA) were used to determine the DNA quantity. A combination of PacBio III and Illumina II technologies were used to sequence the bacterial genome. Genomic DNA was fragmented and end-repaired using G-tubes (Covaris, Woburn, MA, USA) for PacBio sequencing, and then processed using the SMRTbell Template Prep Kit v.1.0 (with a fragment size >10 Kb, as determined by the blue Pippin system) according to the manufacturer’s instructions. (PacBio, Menlo Park, CA, USA). A Qubit 2.0 Flurometer (Life Technologies, CA, USA) was used to quantify the library DNA, and a Bioanalyzer 2100 was used to estimate average fragment size (Agilent, Santa Clara, CA, USA). Standard protocols were followed for Single-molecule real-time (SMRT) sequencing on the Pacific Biosciences Sequel (PacBio, Menlo Park, CA, USA). Continuous long reads were attained from PacBio sequencing and were used for de novo assembly using Falcon v0.3.0 (Chin et al., 2013). The PacBio data included 817,423 polymerase reads with an N50 of 8548 bp and a total of 6.21Gb (Table S1).
For Illumina sequencing, genomic DNA was randomly sonicated, then end-repaired, adenine nucleotide-tailed, and adaptor ligated using NEBNext® ΜLtra™ DNA Library Prep Kit (NEB, USA) according to the preparation protocol. 300-400 bp DNA fragments were enriched by PCR. Finally, the enriched amplicons were purified using AMPure XP system (Beckman Coulter, Brea, CA, USA), their size distribution was analyzed by Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA) and quantified using real-time PCR. Genome was sequenced on the Illumina Novaseq 6000 sequencer by the pair-end method (PE 150). Raw data from Illumina sequencing was filtered using FASTP v0.20.0 (Chen et al., 2018). After filtering, the Illumina data consisted of 7.05 million paired-end 2 × 150 bp reads corresponding to 1.06 Gbp (Table S1). Clean reads of Illumina data were used to correct the draft genome sequences to improve the quality of the assembly and determine the final genome sequences using Pilon v1.23 (Walker et al., 2014). The whole genome sequencing of KH-4 were finally assembled into two circular chromosomes of 1,516,539 bp (Figure 1A) and 3,166,113 bp (Figure 1B) with GC content of 43.6%-44.1%. Their circularity was checked and verified by mapping reads across the linkage of the head and the tail of the chromosomes without breakpoint (Figure S1).
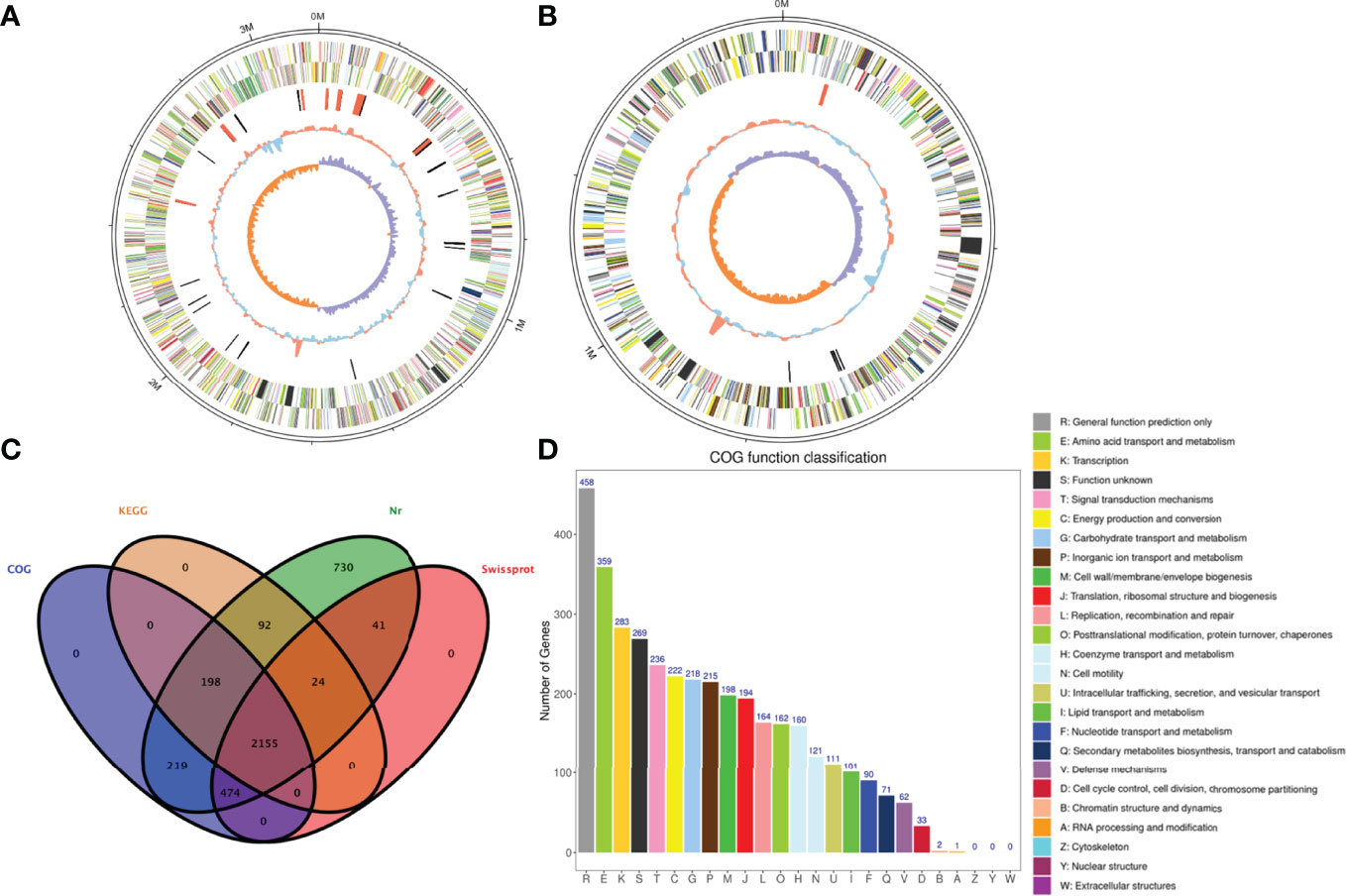
Figure 1 The circular genome maps of the strain KH-4. (A), Chromosome 1. (B) Chromosome 2. The tracks from outside to inside represent the identity of the genome size, CDS (+), CDS (−), rRNA(red), tRNA(black), GC contents, and GC skews. On both strands of CDS, all annotated ORFs were colored differently according to the COG annotation. (C) Annotation summary of unigenes. (D) Functional categorization of KH-4 based on the COG database.
Genomic Component Prediction
The coding sequences were predicted using Prokka v1.1.2 (Seemann, 2014) and NCBI prokaryotic genome annotation pipeline (Tatusova et al., 2016). rRNAs were identified by rRNAmmer v1.2 (Lagesen et al., 2007) and tRNAs by tRNAscan-SE v2.0 (Chan et al., 2021). Results showed that Chr1 coded 2673 proteins, 40 rRNA genes, and 116 tRNA genes, while Chr2 coded 1261 proteins, 3 rRNA genes, and 16 tRNA genes. Interspersed repeat elements were identified by RepeatMasker v4.1.0 (Chen, 2004). A total of 61 interspersed repeats (Table S2) were found in the genome of KH-4, including 39 short interspersed repeated sequences (SINEs), 14 long interspersed repeated sequences (LINEs), one long terminal repeat sequence (LTR), and seven DNA elements. Tandem repeat elements were identified by TRF v4.0.9 (Benson, 1999). Transposons were predicted using TransposonPSI. A total of 0.23% (10,821 bases) as TEs were found to scatter throughout the genome of KH-4, with 11 of class I retrotransposons (0.20% of assembled genome) and three of class II DNA transposons (0.03% of assembled genome), respectively (Table S3).
Function Annotations
Gene annotation was performed by alignment with the various protein databases including NCBI non-redundant protein sequence (Nr) database, UniProt/Swiss-Prot (Swissport), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Cluster of Orthologous Groups of proteins (COG). Overall, Nr, Swissport, COG, and KEGG annotations were obtained respectively for 3933, 2694, 3046, and 2469 unigenes (Figure 1C). There were 22 COG categories of the predicted open reading frames (ORFs) in KH-4 (Figure 1D), with the top three as the following: R (458 ORFs), E (359 ORFs), K (283 ORFs). About 1361 of the 3933 annotated unigenes were linked to 129 KEGG pathways. Major KEGG pathway representations included metabolic pathways (54.67%), biosynthesis of secondary metabolites (24.85%), microbial metabolism in diverse environments (15.65%), and ABC transporters (10.95%).
Blast was used to explore potential virulence factors in KH-4 respectively against Pathogen Host Interactions (PHI) and Virulence Factors of Pathogenic Bacteria (VFDB). Blast against PHI revealed several homologous virulence factors in KH-4 referring to intestinal diseases (Table S4), including RNA-binding protein Hfq (locus_tag LWM38_12790), protease LonA (locus_tag LWM38_04625), quorum-sensing regulator luxO (locus_tag LWM38_09410), et al. One hundred seventy-two homologous virulence genes were finally identified in KH-4 against VFDB (Table S5), including ABC transporter components, hemolysin, heme transport protein, and flagellins.
Comparative Genomic Analysis
A comparative genomic investigation was carried out among the strains of V. splendidus (strain: 10N.286.52.F10, BST398, CECT 8714, and OU02), V. lentus (strain: 40M4T, BSW13, 5F79 and 10N.286.45.C8), V. kanaloae (strain: KH-4, R17, S12 and CCUG 56968), V. chagasii (strain: ECSMB14107, LC2-408, LMG 21353, and CCUG 48643), and V. tasmaniensis (strain: SM1924, 10N.222.45.A2, 1F-187, and ZS-17). The genomes of above strains were downloaded from NCBI database and analyzed. The software Diamond and OrthoMCL were used to identify orthologous unigenes among species by default. Briefly, all the unigenes were blasted with each other, E-value<1e-5 and query coverage>30% were thought as orthologous genes. Following that, OrthoMCL classified orthologous genes from different species into one protein family, as well as other species-specific genes into different families. One thousand five hundred thirty orthologous unigenes were identified (Table S6). The phylogenetic tree was constructed according to protein sequence alignment of orthologous unigenes with 1000 bootstrap replicates. The phylogenetic tree showed that KH-4 clustered much more closely with the other selected V. kanaloae strains (Figure 2). However, several splendidus-clade strains (OU02, SM1924, ZS-17) could not be well clustered according to their previous identities. These strains were named and classified based on several housekeeping genes, while their accurate classification status should be reconsidered. The pangenome analysis was carried out with the default parameters of the bacterial pan genome analysis tool (BPGA). Twenty genome sequences of these splendidus-clade strains had a pangenome of 76,703 genes, including a core-genome of 44,282 genes (shared by all genomes), an accessory genome of 31,795 genes (partially shared by genomes), and a unique-genome of 626 genes in KH-4 (Table S7).
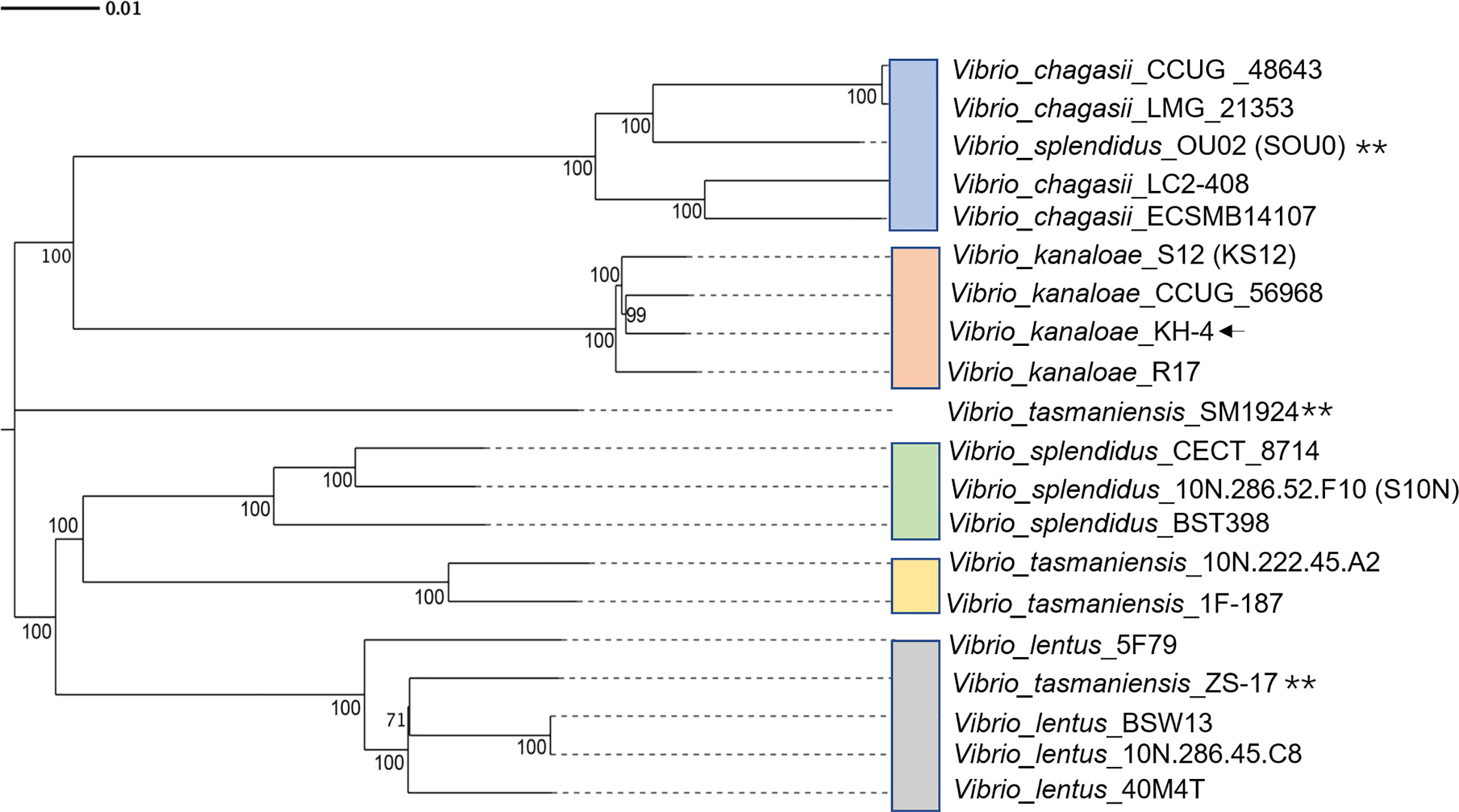
Figure 2 Phylogenetic tree based on concatenated orthologous unigenes of selected splendidus-clade strains. Clusters of V. splendidus, V. kanaloae, V. chagasii, and V. tasmaniensis are colored. Asterisks show the strains that are not well clustered according to their previous identities. ** The strains that were not well clustered.
Conclusions
The 4.68 Mbp complete genome sequence of V. kanaloae strain KH-4 was released here. The genome of KH-4 was compared to the earlier described 19 genomes, resulting in a comprehensive recognition of splendidus-clade strains. The results revealed 1530 orthologous unigenes among selected splendidus-clade strains and 626 specific genes in V. kanaloae strain KH-4 compared with others. In all, our data provide a valuable resource for better cognition and differentiation of these splendidus-clade strains for future research.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: BioProject, accession number PRJNA791127.
Author Contributions
LX, CB, and QL conceived the idea and collected the animal samples. LX, JY, BH, and CL performed the experiments and the bioinformatics analysis. LX, JY, CB, and CW prepared the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was financially supported by the National Natural Science Foundation of China (31902400), China Agriculture Research System of MOF and MARA.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to all the laboratory members for their technical support and helpful suggestions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.902660/full#supplementary-material
References
Benson G. (1999). Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Chan P. P., Lin B. Y., Mak A. J., Lowe T. M. (2021). Trnascan-SE 2.0: Improved Detection and Functional Classification of Transfer RNA Genes. Nucleic Acids Res. 49, 9077–9096. doi: 10.1093/nar/gkab688
Chen N. (2004). Using Repeat Masker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinf. 5, 4.10. doi: 10.1002/0471250953.bi0410s05
Chen S., Zhou Y., Chen Y., Gu J. (2018). Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Chin C.-S., Alexander D. H., Marks P., Klammer A. A., Drake J., Heiner C., et al. (2013). Nonhybrid, Finished Microbial Genome Assemblies From Long-Read SMRT Sequencing Data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Dai F., Li Y., Shao Y., Li C., Zhang W. (2020). FliC of Vibrio Splendidus-Related Strain Involved in Adhesion to Apostichopus Japonicus. Microbial. Pathog. 149, 104503. doi: 10.1016/j.micpath.2020.104503
Gay M., Berthe F. C., Le Roux F. (2004a). Screening of Vibrio Isolates to Develop an Experimental Infection Model in the Pacific Oyster Crassostrea Gigas. Dis. Aquat. Org. 59, 49–56. doi: 10.3354/dao059049
Gay M., Renault T., Pons A.-M., Le Roux F. (2004b). Two Vibrio Splendidus Related Strains Collaborate to Kill Crassostrea Gigas: Taxonomy and Host Alterations. Dis. Aquat. Org. 62, 65–74. doi: 10.3354/dao062065
Gómez-León J., Villamil L., Lemos M., Novoa B., Figueras A. (2005). Isolation of Vibrio Alginolyticus and Vibrio Splendidus From Aquacultured Carpet Shell Clam (Ruditapes Decussatus) Larvae Associated With Mass Mortalities. Appl. Environ. Microbiol. 71, 98–104. doi: 10.1128/AEM.71.1.98-104.2005
Henke J. M., Bassler B. L. (2004). Three Parallel Quorum-Sensing Systems Regulate Gene Expression in Vibrio Harveyi. J. Bacteriol. 186, 6902–6914. doi: 10.1128/JB.186.20.6902-6914.2004
Huang B., Zhang X., Wang C., Bai C., Li C., Li C., et al. (2021). Isolation and Characterization of Vibrio Kanaloae as a Major Pathogen Associated With Mass Mortalities of Ark Clam, Scapharca Broughtonii, in Cold Season. Microorganisms 9, 2161. doi: 10.3390/microorganisms9102161
Lagesen K., Hallin P., Rødland E. A., Stærfeldt H.-H., Rognes T., Ussery D. W. (2007). RNAmmer: Consistent and Rapid Annotation of Ribosomal RNA Genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Le Roux F., Gay M., Lambert C., Nicolas J. L., Gouy M., Berthe F. (2004). Phylogenetic Study and Identification of Vibrio Splendidus-Related Strains Based on gyrB Gene Sequences. Dis. Aquat. Org. 58, 143–150. doi: 10.3354/dao058143
Le Roux F., Gay M., Lambert C., Waechter M., Poubalanne S., Chollet B., et al. (2002). Comparative Analysis of Vibrio Splendidus-Related Strains Isolated During Crassostrea Gigas Mortality Events. Aquat. Living Resour. 15, 251–258. doi: 10.1016/S0990-7440(02)01176-2
Liu R., Qiu L., Yu Z., Zi J., Yue F., Wang L., et al. (2013). Identification and Characterisation of Pathogenic Vibrio Splendidus From Yesso Scallop (Patinopecten Yessoensis) Cultured in a Low Temperature Environment. J. Invertebr. Pathol. 114, 144–150. doi: 10.1016/j.jip.2013.07.005
Rojas R., Miranda C. D., Opazo R., Romero J. (2015). Characterization and Pathogenicity of Vibrio Splendidus Strains Associated With Massive Mortalities of Commercial Hatchery-Reared Larvae of Scallop Argopecten Purpuratus (Lamarck 1819). J. Invertebr. Pathol. 124, 61–69. doi: 10.1016/j.jip.2014.10.009
Roux F. L., Austin B. (2006). Vibrio Splendidus. Biol. Vibrios, 285–296. doi: 10.1128/9781555815714.ch21
Seemann T. (2014). Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Song T., Liu H., Lv T., Zhao X., Shao Y., Han Q., et al. (2018). Characteristics of the Iron Uptake-Related Process of a Pathogenic Vibrio Splendidus Strain Associated With Massive Mortalities of the Sea Cucumber Apostichopus Japonicus. J. Invertebr. Pathol. 155, 25–31. doi: 10.1016/j.jip.2018.05.001
Tatusova T., Dicuccio M., Badretdin A., Chetvernin V., Nawrocki E. P., Zaslavsky L., et al. (2016). NCBI Prokaryotic Genome Annotation Pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Walker B. J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., et al. (2014). Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PloS One 9, e112963. doi: 10.1371/journal.pone.0112963
Wyckoff E. E., Allred B. E., Raymond K. N., Payne S. M. (2015). Catechol Siderophore Transport by Vibrio cholerae. J. Bacteriol. 197, 2840–2849. doi: 10.1128/JB.00417-15
Zhang W., Li C. (2021). Virulence Mechanisms of Vibrios Belonging to the Splendidus Clade as Aquaculture Pathogens, From Case Studies and Genome Data. Rev. Aquac. 13, 2004–2026. doi: 10.1111/raq.12555
Keywords: ark clam, Vibrio, Vibrio kanaloae, genome, comparative
Citation: Xin L, Yu J, Huang B, Li C, Bai C, Liu Q and Wang C (2022) Complete Genome Sequence of Vibrio kanaloae Strain KH-4 From Ark Clams, Scapharca broughtonii. Front. Mar. Sci. 9:902660. doi: 10.3389/fmars.2022.902660
Received: 23 March 2022; Accepted: 28 April 2022;
Published: 26 May 2022.
Edited by:
Hao Chen, Institute of Oceanology (CAS), ChinaReviewed by:
Yi Lan, Hong Kong University of Science and Technology, Hong Kong SAR, ChinaArun Prasanna, King Abdullah University of Science and Technology, Saudi Arabia
Copyright © 2022 Xin, Yu, Huang, Li, Bai, Liu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lusheng Xin, eGlubHNAeXNmcmkuYWMuY24=; Changming Bai, YmFpY21AeXNmcmkuYWMuY24=; Qin Liu, bGl1cWluQHlsdS5lZHUuY24=
†These authors have contributed equally to this work