 Fan Ling1,2
Fan Ling1,2 Suhelen Egan
Suhelen Egan Yingrui Zhuang
Yingrui Zhuang Luyang Xiao
Luyang Xiao Qin Yang
Qin Yang Gaoge Wang
Gaoge Wang- 1College of Marine Life Sciences, Ocean University of China, Qingdao, China
- 2Institute of Evolution and Marine Biodiversity, Ocean University of China, Qingdao, China
- 3Centre for Marine Science and Innovation and School of Biological, Earth and Environmental Sciences, The University of New South Wales, Sydney, NSW, Australia
- 4Weihai Changqing Ocean Science and Technology Co., Ltd, Rongcheng, China
Recent bacterial induced bleaching disease events of the commercially farmed brown seaweed Saccharina japonica has resulted in significant reduction in healthy sporeling supply. However, to date the host associated epimicrobial community shifts with the disease progression have not been characterized. We investigated the dynamic shifts in the composition of the epibacterial communities of S. japonica with disease progression using 16S rRNA gene amplicon sequencing. We found that the alpha diversity was significantly higher in the control group than in the infected group over disease progression (P < 0.01). There was a significant shift in the composition and predicted functions of the epibacterial communities in both control and infected groups. Indicator species, belonging to Stenotrophomonas, Pseudomonas and Sphingomona were significantly higher in the control group than infected group, suggesting that these taxa are associated with healthy S. japonica. In contrast, Vibrio, Pseudoalteromonas and Colwellia were keystone taxa in the infected group at 24 h, and thus maybe considered to be the secondary opportunistic pathogens. Our study describes the changes of epibacterial communities associated with the progression of bleaching disease in S. japonica. This new information not only extends our baseline knowledge of the S. japonica epimicrobiome, but also paves the way for developing measures to mitigate disease outbreaks for the sustainable aquaculture of S. japonica.
Introduction
Macroalgae (seaweeds) associated bacteria are critical for the normal development and health of their host (Barott et al., 2011; Egan et al., 2013; Singh and Reddy, 2014; Saha and Weinberger, 2019). For example, they are thought to provide vital nutrients for the healthy growth of the host (Croft et al., 2006) and produce antimicrobial compounds for fouling defence (Penesyan et al., 2009; Egan et al., 2013) as well as produce chemical signals that can induce the release and settlement of algal spores (Joint et al., 2007; Weinberger et al., 2007). However, seaweed associated microbes are not always beneficial and as in other systems [e.g. corals (Damjanovic et al., 2019), humans (Hajishengallis and Lamont, 2016) and shrimp (Lu et al., 2020)], disruption of the hosts natural microbiota has been correlated with the occurrence of various disease in seaweeds (Egan and Gardiner, 2016; Kumar et al., 2016; Longford et al., 2019; Egan et al., 2020; Li et al., 2022).
Microbiome shifts have been found in bleaching diseased red seaweed Delisea pulchra. Healthy and diseased D. pulchra harbor distinct microbial communities (Campbell et al., 2011; Case et al., 2011; Fernandes et al., 2011), with the abundance of known and putative pathogens (e.g. Aquimarina sp. (healthy: 27%, diseased: 55%), Phaeobacter gallaeciensis) increased double roughly during the disease progression (Zozaya-Valdes et al., 2015; Kumar et al., 2016; Zozaya-Valdes et al., 2017). Further, it has been suggested that beneficial bacteria can impede this bacterial shift and thus reduce bleaching rates (Longford et al., 2019; Li et al., 2021). Similar correlation of microbiome change and bleaching disease was also found in the habitat forming kelp Ecklonia radiata (Marzinelli et al., 2015). These studies indicate that bleaching disease in seaweeds is generally accompanied by host-associated microbiome shifts.
Microbial communities are often characterized by keystone taxa, which drive community composition and function irrespective of their abundance (Banerjee et al., 2018). Microbial co-occurrence network analysis is a powerful analytical tool for identifying the keystone taxa (i.e. highly connected taxa within the community) and has been used for human (Shetty et al., 2017; Banerjee et al., 2018), plant (Agler et al., 2016; Weiss et al., 2016) and shrimp (Dai et al., 2020) studies. For example keystone taxa found in the gut of diseased shrimp have been identified as candidate pathogens across multiple studies (Banerjee et al., 2018; Dai et al., 2018; Dai et al., 2020). Similar approaches have also been applied to identify keystone taxa associated with the gut microbiota of healthy shrimp with the potential to be developed into novel probiotics (Dai et al., 2018; Xiong, 2018). However, to date, whether there exist keystone taxa and how they change with the disease progression in seaweeds has not been investigated.
Saccharina japonica is one of the most important commercially farmed seaweeds worldwide. Like land crops, S. japonica has continued to be under threat from various diseases at both nursery and field cultivation stages since the successful implement of scale-cultivation in 1958 in China (Gachon et al., 2010; Wang et al., 2014). In some severe cases, diseases can cause a 20 - 50% reduction in yield volume (Gachon et al., 2010). In addition to previously characterized diseases (such as rotten hole disease (Wang et al., 2008; Zhang R. et al., 2020) and green rotten disease (Li et al., 2020), a new bleaching disease has recently been described to occur during the late nursery stage of S. japonica (Zhang et al., 2022a). Previous studies have observed differences in the microbial communities associated with healthy and diseased individuals (Li et al., 2020; Zhang R. et al., 2020). Moreover, laboratory assays have shown that this disease is in part due to an infection with the bacterium Pseudoalteromonas piscicida X-8 (PpX-8) (Zhang et al., 2022a). However, nothing is known about the interaction of the pathogen with the natural epimicrobiome of S. japonica or if and how the host epimicrobiome changes during an infection. Given the importance of the microbiome in maintaining the health status in a range of host systems, the main objectives of this study were to use 16S rRNA gene amplicon sequencing to (a) assess the dynamic changes in the S. japonica epimicrobiome during disease progression after infection with PpX-8 and (b) identify potential health associated bacteria and putatively pathogenic bacteria by analysis of indicator species and keystone taxa associated with farmed S. japonica. Our results will serve as a basis for developing disease diagnosis and therapies for the nursery farms of S. japonica.
Materials and Methods
Sample Collection
Healthy juvenile sporophytes (10 - 15 cm in length) of Saccharina japonica were freshly collected from Weihai Changqing Ocean Science & Technology Co., Ltd, located in Rongcheng, Shandong province, China (37°19’ 41″ N, 122°17′ 21″ E), on 24th December 2020. Samples were shipped in sterile sealed bag with ice packs within 4 h to the laboratory. After the samples arrived at the laboratory, juvenile sporophytes were immediately washed with sterile seawater and then were cultured in the incubator (10 ± 1°C, 80 μE m-2 s-1, light: dark= 12 h: 12 h).
Infection Assay and Epibacterial Collection
The pathogen Pseudoalteromonas piscicida X-8 (PpX-8) (Zhang et al., 2022a) was routinely cultured on the Zobell 2216E marine agar plates at 25°C. For infection assays, a single colony was inoculated into 100 mL Zobell 2216E medium and incubated shaking for 8 h at 25°C. Thereafter 1mL was subcultured into 100 mL of fresh medium until the optical density (OD600) reached 0.5 (bacterial density: 1.0 × 108 CFU/mL). PpX-8 cultures were centrifuged at 2000 g for 15 min and the cell pellet was rinsed at least twice with sterilized seawater and were resuspended in an equal volume of sterilized seawater.
Forty eight healthy juvenile sporophytes were washed with sterilized seawater at least three times. Six small tissue pieces (3.0 × 3.0 cm) were cut from each sporophyte with a sterile surgical blade resulting in a total of 288 tissue pieces. Each independent biological replicate consisted of six tissue pieces randomly assigned into each well of a 6 - well cell culture plates (USA). There were 8 replicates (each replicate included six tissue pieces) for both control and infected groups at each time point, resulting in a total of 48 replicate samples (i.e. 8 biological replicates × 2 treatments × 3 time points, Figure S1). Individual wells were then inoculated with either 1 mL of the PpX-8 suspension (described above) or 1 mL of sterile seawater as a control (C). The plates were incubated at 10 ± 1°C, 80 μE m-2 s-1 (light: dark= 12 h:12 h) for 4 h, 14 h and 24 h, respectively. Tissue pieces were rinsed with sterilized seawater at least three times to remove PpX-8 before collecting epiphytic bacteria. Epiphytic bacteria were sampled by swabbing tissue surface with sterile cotton swab. Three sampling time points (4 h, 14 h and 24 h) were designed for both control (C) and PpX-8 infected groups (X-8). The collected epiphytic bacteria were kept frozen at -80°C until DNA extraction. Microscopic images were taken at each sampling time to record the disease symptoms under microscope (Nikon, Japan).
DNA Extraction, Amplification, and Sequencing of the 16S rRNA Gene Amplicon Sequencing
DNA of epiphytic bacteria was extracted using the HiPure Soil DNA Kits (Magen, Guangzhou, China) according to manufacturer’s protocols. The V3-V4 hypervariable regions of the bacterial 16S rRNA gene were amplified with the primers 341F (5’ -CCTACGGGNGGCWGCAG-3’) and 806R (5’ -GGACTACHVGGGTATCTAAT-3’). PCR reactions were performed in 50 μL mixture contained 100 ng template DNA, 10 × Buffer KOD (5 μL), dNTPs (5 μL, 2 mM), primers 341F and 806R (1.5 μL of each, 10 μM), KOD DNA Polymerase (Toyobo, Japan). PCR cycling conditions were as follows: 94°C for 2 minutes, 30 cycles of 98°C for 10 seconds, 62°C for 30 seconds, 68°C for 30 seconds and final extension for 5 minutes at 68°C. Amplicons were extracted from 2% agarose gels using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, U.S.) according to the manufacturer’s instructions and quantified using ABI StepOnePlus Real-Time PCR System (Life Technologies, Foster City, USA). Addition of sequencing adapters, the qualified amplicons was then sequenced on the Illumina Novaseq6000 platform to generate 2×250 bp paired-end reads by Guangzhou Genedenovo Biotechnology (Guangzhou, China).
Analyses of Illumina Sequencing Data
The raw reads were first filtered to remove low-quality reads (containing more than 10% of unknown nucleotides) using FASTP. The paired end clean reads were merged as raw tags using FLSAH 1.2.11 with a minimum overlap of 10 bps and mismatch error rates were less than 2%. After raw tags filtering, the clean tags were clustered into operational taxonomic units (OTUs) of ≥ 97% similarity using UPARSE (Edgar and Robert, 2013). The tag sequence with highest abundance was selected as representative sequence within each cluster. The representative OTU sequences were classified into organisms by a naive Bayesian model using RDP classifier 2.2 (Wang et al., 2007) based on SILVA database (v132) (Pruesse et al., 2007) with the confidence threshold values ranging from 0.8 to 1. On the basis of this classification, sequences of chloroplast, mitochondria and archaea origin were removed from the dataset. The raw reads were deposited into the NCBI Sequence Read Archive (SRA) database (Accession Number PRJNA775653).
Indicator Species Analysis
To identify the bacterial taxa that are indicative of the epibacterial communities associated with either control and infected groups, calculation of IndVal and associated tests was performed using the labdsv package (version2.0-1) in R project (Roberts, 2016). The indicator species were selected at genus level with p<0.01 and IndVal>0.95 (Lu et al., 2020).
Association Network Analysis
Interspecies interactions in epimicrobial communities were assessed by the association network analysis. The association network was acquired using Pearson correlation coefficient from the OTUs abundance. The co-occurrence OTU pairs was selected if the Pearson correlation coefficient |r| > 0.7 and statistically significant at P < 0.05 using R psych package (Banerjee et al., 2018). Each network (three networks for control and three networks for the infected group) was constructed using all 8 replicates. For each group, putative keystone OTUs were defined according to previous studies (Banerjee et al., 2018; Gao et al., 2021) as those with degree centrality higher than 11 and closeness centrality greater than 0.6.
Shift of the Predicted Function of Epibacterial Communities Over Disease Progression
The KEGG (Kyoto Encyclopedia of Genes and Genomes) Pathway was performed using Tax4Fun software to predict the function of epibacterial communities based on the OTU abundance (Aßhauer et al., 2015). Cluster analysis was conducted on the abundance of multiple KEGG functions, and the sample functional distribution was visually displayed with the heatmap (Pheatmap Package in R Project Version 1.0.12).
Statistical Analysis
The stacked bar plot of the community composition was visualized with ggplot2 package 2.2.1. Chao1 and Shannon index were calculated in QIIME and the alpha diversity comparison between groups was calculated by Welch’s t-test. Canonical correspondence analysis (CCA) and permutational multivariate analysis of variance (PERMANOVA) were performed to evaluate the differences among sampling time points and infected treatment in the epibacteria based on Bray-Curtis metric using Vegan package 2.5.3.
Results
Distribution of Epiphytic Bacterial Communities of Control and Infected Juvenile Sporophytes
Morphological images of both healthy juvenile sporophytes and tissue pieces of S. japonica were showed in Figures 1A, B. No bleaching symptoms were observed in the control group at 4 h (Figure 1C), 14 h (Figure 1D) and 24 h (Figure 1E). There was no significantly different in tissue color between control (Figure 1C) and PpX-8 infected tissues (Figure 1F) at 4 h. While, compared to the corresponding control at 14 h and 24 h, PpX-8 infected group showed moderate bleaching symptoms at 14 h (Figure 1G) and high levels of bleaching were observed at 24 h (Figure 1H).

Figure 1 The microscopic images of healthy juvenile sporophytes of S. japonica and the morphological images of tissue pieces in both control and PpX-8 infected groups. Control and infected tissue pieces were cultured under condition at 10 ± 1°C with the light intensity of 80 µE m2 s–1 and the photoperiod of 12 h: 12 h (L: D). (A) healthy juvenile sporophytes; (B), tissue of healthy juvenile sporophytes; (C–E) control group at 4, 14 and 24 h, respectively; (F) infected group at 4 h; (G) infected group at 14 h with moderate bleaching symptom; (H) infected group at 24 h with high level of bleaching. Bars, a: 1.2 cm; b-h: 400 μm.
A total of 6,088,736 reads, with an average of 126,849 ± 5574 (mean ± standard deviation) reads per sample, were obtained from the 48 samples. Overall, 11,397 OTUs were assigned across all the samples based on the SILVA database at the cut-off level of 97%. Control group harbored a higher number of OTUs than the PpX-8 infected group. Rarefaction analysis indicated that the sequencing effort reached saturation all samples (see Figure S2) and this further supported Good’s coverage estimates of greater than 98% for all samples. The obtained OTUs were classified into 30 phyla, 66 classes, 131 orders, 203 families, 330 genera.
Regarding alpha diversity, Shannon indices were significantly higher in the control group than in the infected group at 4 h, 14 h and 24 h (t-test, p < 0.001). Species richness estimates (Chao 1) were significantly higher in the control group than in infected group at 14 h and 24 h (t-test, p < 0.01). In addition, diversity (Shannon index) gradually increased from 4 h to 24 h, whereas the richness (Chao 1) decreased gradually across the time courses in the infected group (Table 1).
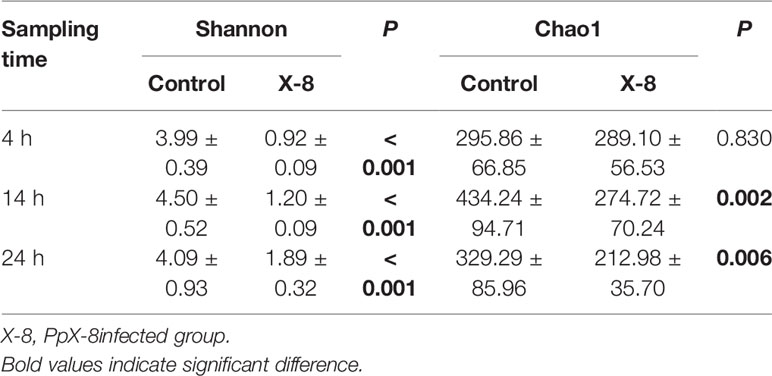
Table 1 Alpha diversity of the epibacterial communities associated with control and the infected groups at each time point.
The most dominant phylum was Proteobacteria in both control (87.14%) and infected groups (99.70%). Other major phyla including Verrucomicrobia (4.79%), Planctomycetes (2.94%) and Bacteroidetes (2.72%) in control group (Figure 2A). Pseudoalteromonas and Vibrio were the most abundant genera in both control and infected groups. In particular, the relative abundance of Pseudoalteromonas increased gradually in both control (ranging from 19.94% to 35.88%) and infected groups (ranging from 2.45% to 13.22%) over the time. This included sequences belonging to Pseudoalteromonas strain PpX-8, which represented 0.8% and 0.2% of the community in the control and infected groups, respectively. In contrast, the relative abundance of Vibrio showed the high but slightly decreasing overtime (from 95.34% to 81.68%) in the infected group (Figure 2B).
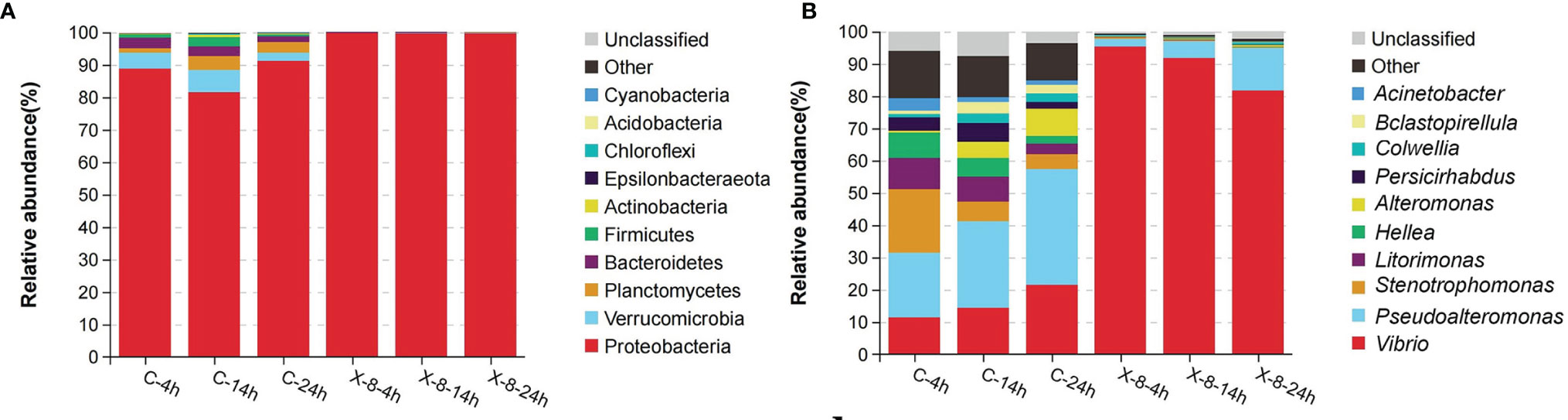
Figure 2 Epibacterial communities associated with control and the PpX-8 infected S. japonica. C, control group; X-8, PpX-8 infected group. 4 h, 14 h and 24 h were sampling time points. Relative abundance of OTUs (top 10) representing in (A) phylum, (B) genus.
The Shifts of Epibacterial Community Structure Associated With the Juvenile Sporophytes of S. japonica Infected by PpX-8
A canonical correspondence analysis (CCA) biplot showed that there was no distinct separation over time in control and infected group, whereas infected treatment significantly affected the structures of the epibacterial communities (Figure 3). Further, a PERMANOVA revealed that sampling time, infected treatment and their interaction contributed 0.2328, 0.6164 and 0.716 variation in the epibacterial communities of S. japonica, respectively (Table 2). By pairwise comparisons, the distance of epibacterial community structure between control group and infected group was decreased gradually over disease progression (Table 3).
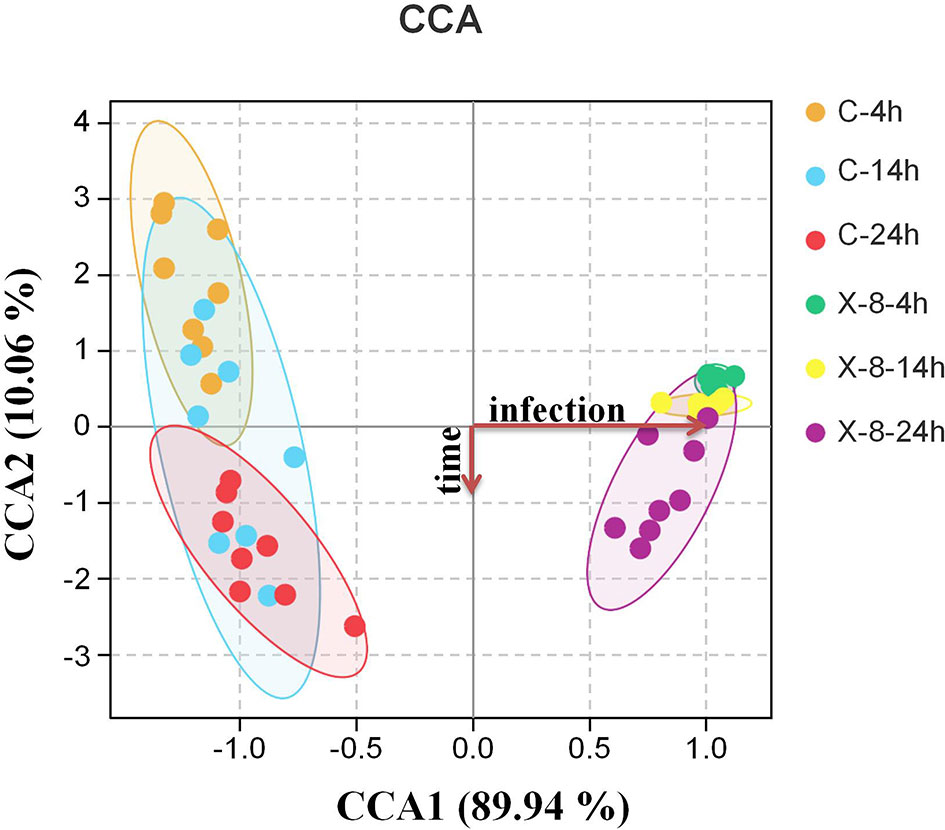
Figure 3 Canonical correspondence analysis (CCA) biplot of epibacterial communities associated with control and infected groups at each time point based on Bray-Curtis distance. C, control group; X-8, PpX-8 infected group. 4 h, 14 h and 24 h were sampling time points.
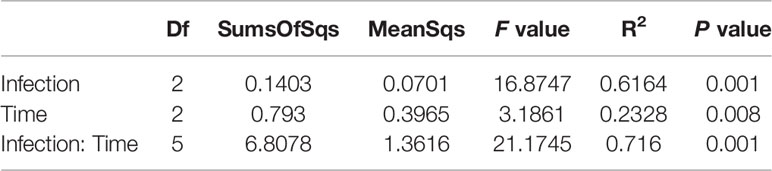
Table 2 The effects of sampling time and PpX-8 on variation in community composition using permutational multivariate analysis of variance (PERMANOVA) with adonis based on Bray–Curtis.
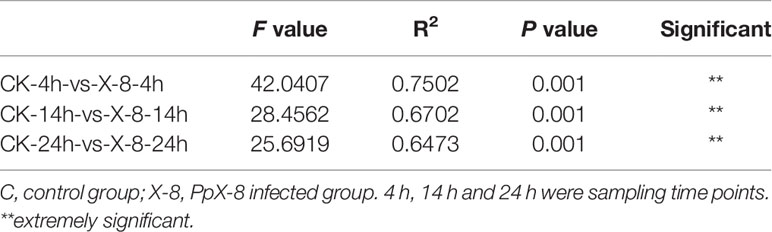
Table 3 Pairwise comparison using permutational multivariate analysis of variance (PERMANOVA) with adonis based on Bray–Curtis.
Indicator analysis showed indicator species at each time point to reflect the impact of PpX-8 on epibacterial dynamics during the disease progression (Figure 4). With respected to infected group, Stenotrophomonas, Persicirhabdus, Acinetobacter, Pseudomonas and Sphingomonas were indicator species at 4 h, 14 h and 24 h at control group. Specially, the relative abundance of Stenotrophomonas, Pseudomonas and Sphingomonas was significantly higher in control group (t-test, p < 0.05) than those in infected group (the average relative abundance was between 0.03% and 0.3%). These three genera are thought to be positively associated with healthy S. japonica (Figure S3). While, for the PpX-8 infected group, Vibrio was the indicator species at 4 h, 14 h and 24 h.
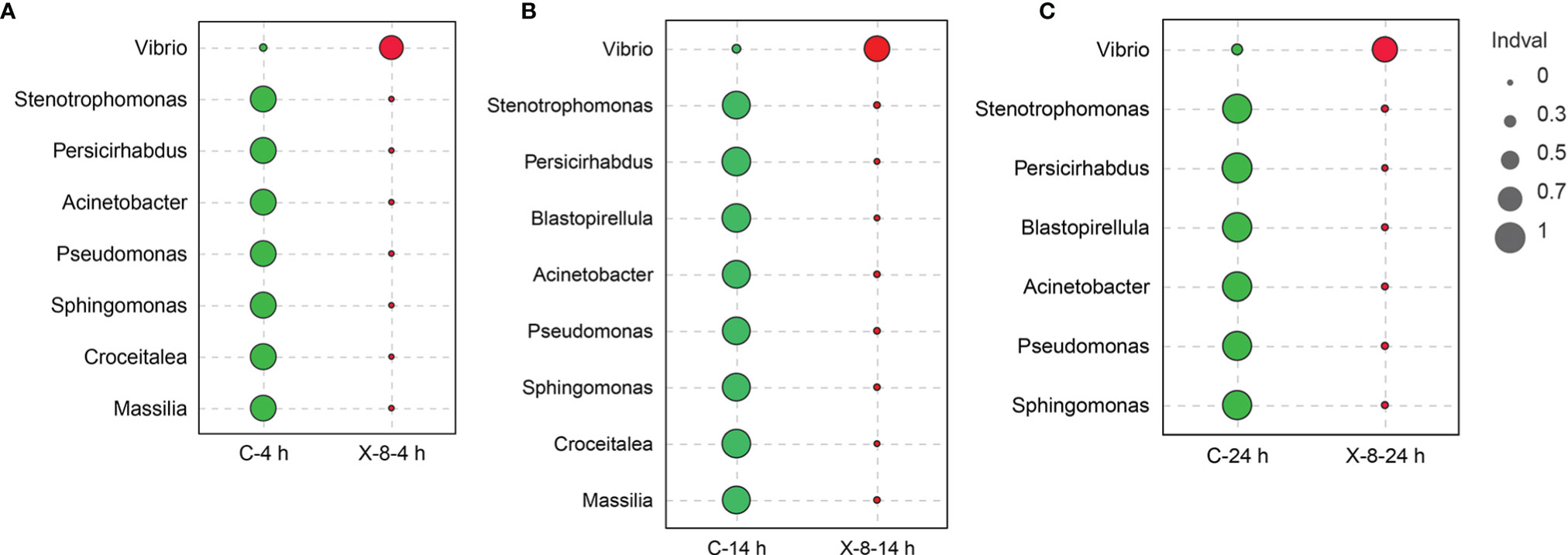
Figure 4 The succession of indicator species at (A) 4 h; (B) 14 h; (C) 24 h. The sizes of the circle represented Indval; green represented control group, red represented PpX-8 infected group.
Association Network of Epibacterial Communities
To determine the effects of PpX-8 infection on the interaction between epiphytic bacterial members, we performed an association network analysis and found distinct networks according to sample type (Figure 5). The number of edges in the networks increased over time in both control and infected groups (Figure 5), with less total number of edges in the infected group. Whereas, the number of negatively correlated edges in the infected group (28-91) was all more than those in the control group (12-71) at each sampling time points. The average clustering coefficient increased with time, suggesting that the more complexity and higher connectivity of network (Table S1).
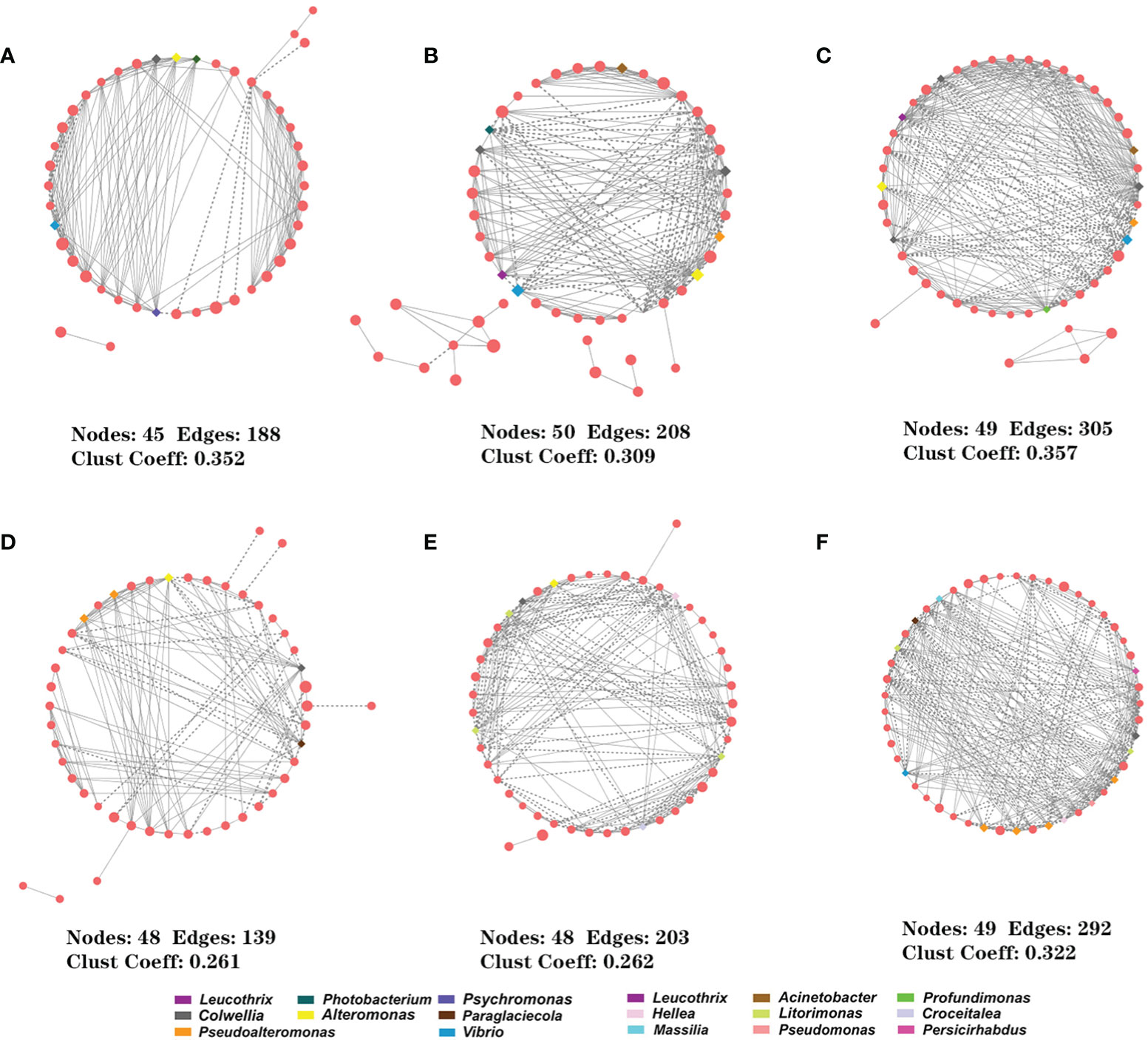
Figure 5 Interspecies interactions in both control and infected groups. (A–C) control group at 4 h, 14 h and 24 h; (D–F) PpX-8 infected group at 4 h, 14 h and 24 h. The solid (or dashed) represents the positive (or negative). The squares represent keystone taxa.
Putative keystone taxa were identified based on the highest degree and highest closeness centrality scores (Banerjee et al., 2018; Dai et al., 2019; Gao et al., 2021). For each network, OTUs with degree > 11, closeness centrality > 0.6 were selected as the putative keystone taxa. Firstly, there were a total of 24 and 25 keystone taxa in the control and in the infected networks, respectively (Table S2). There were some changes in putative keystone taxa with time in both control and infected groups, suggesting that PpX-8 infection greatly affected the structure of epibacterial community. The putative keystone taxa in the control group included 11 genera, which mainly belonged to family Alteromonadaceae and accounted for 62.5%. In contrast, predicted keystone taxa of the infected group contained 10 genera, among which belonged to family Alteromonadaceae and Hyphomonadaceae, each accounting for 28% respectively, while, Pseudoalteromonadaceae accounted for 24%. Interestingly, Vibrio, Colwellia and Pseudoalteromonas were the common putative keystone taxa at 24 h in both control and infected groups (Table S2), they were also indicator species for the infected group at 24 h (Figure 4). In addition, Hellea and Litorimonas became keystone taxa at 14 h and 24 h in the infected group, whereas were not found in the control group (Table S2). Therefore, these two taxa may play a role during the progression of bleaching disease.
To assess the importance of these predicted keystone taxa, we performed a co-occurrence network analysis in which these were removed. The results demonstrate that the absence of keystone taxa leads to fragmentation of the network (Figure S4), which indicated that these keystone taxa were important for maintaining the balance of bacterial communities of S. japonica.
The Predicted Gene Functions of Epibacterial Communities After PpX-8 Infection
The functional changes of epibacterial communities in both control and infected groups were predicted using Tax4Fun in Figure 6. Among them, the abundance of 9 pathways in infected group was significantly higher than in control group (t-test, p < 0.05), including metabolism (carbohydrate metabolism, lipid metabolism, glycan biosynthesis and metabolism, metabolism of terpenoids and polyketides), environmental information processing (membrane transport, signal transduction and molecules and interaction), infectious diseases and cell communication. Twelve pathways were significantly enriched in the control group (t-test, p < 0.05), and included metabolism (energy metabolism, amino acid metabolism, metabolism of cofactors and vitamins and biosynthesis of other secondary metabolites), genetic information processing (translation, replication and repair, folding, sorting and degradation, transcription), cellular processes (cell motility, growth and death) and environmental adaption (Figure 6).

Figure 6 Functional prediction of epibacterial communities associated with S. japonica. C, control group; X-8, PpX-8infected group. 4 h, 14 h and 24 h were sampling times.
Discussion
In this paper, we assessed the diversity of epibacterial communities during bleaching disease progression in farmed S. japonica after inoculation with the pathogenic bacterium - PpX-8. We observed a significant temporal shift in epibacterial communities specific to both the control and infected S. japonica groups. In addition, the healthy associated microbes in the control group – Stenotrophomonas, Pseudomonas and Sphingomona and potentially opportunistic pathogens in the infected group - Vibrio, Pseudoalteromonas and Colwellia all suggest that the bleaching disease of S. japonica may be linked to an imbalance of the epibacterial communities. The imbalance could cause the bleaching disease symptoms and provide the opportunity for one or multiple secondary colonizers to exacerbate disease symptoms. To our knowledge, this is the first characterization of the epibacterial shifts during the bleaching disease progression in farmed seaweeds.
Usually, diverse microbial communities function as highly stable and resilient for the health of the host (Rungrassamee et al., 2016; Longford et al., 2019) and can provide colonization resistance against the invading pathogens and perturbation (Loreau et al., 2001; Girvan et al., 2005; De Schryver and Vadstein, 2014). Compared to the control group, we found that the richness and diversity of epibacterial communities was significantly decreased after treatment with the algal pathogen PpX-8. This decrease in diversity was likely due to the sharp increase in the relative abundance of the most two dominant genera Vibrio and Pseudoalteromonas in the infected group (Figure 2B). Similar decreases in bacterial diversity after infection with a pathogen and/or in diseased hosts compared to healthy counterparts have been seen in both seaweeds (Zozaya-Valdes et al., 2015; Kumar et al., 2016; Longford et al., 2019), corals (Damjanovic et al., 2019) and shrimp (Huang et al., 2016; Rungrassamee et al., 2016; Xiong, 2018; Dai et al., 2019) and maybe an early indicator of poor host health.
We identified specific bacterial taxa that were indicative of both control and infected epibacterial communities. These indicator species included taxa belonging to Stenotrophomonas, Pseudomonas and Sphingomona, which were in high abundance and decreased over the time in the control group. Therefore, these three genera are thought to be associated with healthy S. japonica. Stenotrophomonas is ubiquitously present in the environment, plants and soil etc. (Ryan et al., 2009) and engages in beneficial interactions with plants (Vilchis-Carmona et al., 2021). For example, Stenotrophomonas can produce factors which promote plant growth, and secrete antibacterial compounds protecting plants from damage (Ryan et al., 2009; Elufisan et al., 2020). Pseudomonas, especially Pseudomonas putida, can protect plants against phytopathogens because of their type VI secretion systems (T6SS) (Bernal et al., 2017). Sphingomonas are environmentally friendly bacteria and can degrade various hazardous organic compounds (Zhang et al., 2021) and enhance the growth of microalgal strain Oocystis sp. KNUA044 (Na et al., 2021). In addition, Sphingomonas are often found in soils and have been identified as beneficial microbes in crops, such as rice and tomato, degrading toxic substances and directly reducing pathogen population (Chen et al., 2021; Cheng et al., 2021; Choi et al., 2021; Feng et al., 2021). Based on the beneficial functions of the above three taxa, we speculate that these taxa may play a similarly beneficial role for maintaining the health of farmed S. japonica. Moreover, the results in this study are in part similar to our previous studies, in which Xanthomonadaceae (Stenotrophomonas) were enriched in healthy S. japonica (Zhang et al., 2020; Han et al., 2021). However, the specific beneficial effects of these three taxa on S. japonica need to be further elucidated.
Compared to the control group, the shifts in microbiome composition occurred as early as at 4 h after PpX-8 infection in the infected group. Both Vibrio and Pseudoalteromonas became the most dominant two genera and their relative abundance reached more than 90% (Figure 2B). Vibrio has been identified as the pathogenic bacteria for shrimp white feces syndrome (Lu et al., 2020), coral bleaching (Ben-Haim and Rosenberg, 2002; Rosenberg and Falkovitz, 2004), mariculture fish (Zhang X-H. et al., 2020), rotten hole disease of Laminaria japonica (synonymous of Saccharina japonica) (Wang et al., 2008) and yellow spot disease of Pyropia (Yang et al., 2020; Zhu et al., 2021). Therefore, it seemed that Vibrio was one of the secondary opportunistic pathogens in this study.
Pseudoalteromonas and Colwellia were keystone species at 24 h in the infected group. Moreover, the relative abundance of Pseudoalteromonas and Colwellia increased during the infection period. Previous studies have shown that Pseudoalteromonas and Colwellia seaweed isolates are capable of agar degradation (Sánchez Hinojosa et al., 2018), with some Pseudoalteromonas producing cold-adapted hydrolytic enzymes and exhibiting high agar-hydrolyzing activity, even at low temperatures (Hollants et al., 2013; Tropeano et al., 2013; Ivanova et al., 2014; Borchert et al., 2017; Sánchez Hinojosa et al., 2018). Other Pseudoalteromonas isolates have been identified as causative agents of disease of Laminaria japonica (Sawabe et al., 1998) and S. japonica (Zhang et al., 2022). Similarly, isolates of Colwellia have the ability to digest agar and other algal phycocolloids (Martin et al., 2015; Xu et al., 2017; Christiansen et al., 2018). Higher abundance of Colwellia in the infected group is also consistent with that of bleaching diseased D. pulchra (Egan et al., 2013). Given the high relative abundance of these Vibrio, Pseudoalteromonas and Colwellia in the infected group, together with previous reports of virulence traits associated with related strains, it is possible that these taxa represent secondary opportunistic pathogens which may act to hasten disease development in S. japonica. In addition, the Litorimonas and Hellea were keystone taxa in infected group at 14 h and 24 h. The genus Litorimonas, created by Jung et al. (2011), was considered as opportunistic pathogen of green rotten disease in S. japonica because of their relative abundance in diseased samples increased 7.5 times (Li et al., 2020). Hellea was generally found in seaweed, and the relative abundance in diseased S. japonica and D. pulchra was significantly higher than healthy ones (Zozaya-Valdes et al., 2017; Zhang et al., 2020). Thus, we speculated that Litorimonas and Hellea may play an important role in the progression of disease. However, further work including isolation and virulence characterization of these abundant strains will be required to determine their importance in the progression of bleaching disease in farmed S. japonica. Compared with the control group, predicted functions of epibacteria in infected group changed significantly as early as 4 h after PpX-8 infection. During the course of 24 h infection, immune system of the epibacterial community in the control group was more abundant than that in the infected group. Immune system has been found to be associated with the gut microbiome of healthy shrimp (Lu et al., 2020) and may prevent the invasion of opportunistic pathogens. Therefore, we speculate that KEGG pathways of immune system of epimicrobiome in control group may also maintain the health of S. japonica. While, KEGG pathways of signaling molecules interaction and cell communication were significantly abundant in the infected group. These pathways have been found to be closely related to disease, particularly cell communication can increase the virulence of opportunistic bacteria (Chadha et al., 2021) and were reported to be present in algal pathogens of Roseobacters (Wagner-Dobler et al., 2005; Case et al., 2011). The potential enrichment of these pathways within the epibacterial communities of the infected group may facilitate the progression of bleaching disease after Pp-X-8 infection. In addition, infectious diseases were significantly more abundant in the infected group. The KEGG pathway of infectious diseases has been reported in white feces syndrome shrimp (Lu et al., 2020). An increase in this pathway may contribute to promote invasiveness of other opportunistic pathogens. Moreover, the enriched pathways associated with carbohydrate metabolism in the infected group may be due to the potentially secondary opportunistic pathogen (such as Pseudoalteromonas and Colwellia) with activity of digesting agar and other algal phycocolloids.
Conclusions
Considering the frequency and severity of the disease outbreaks at the nursery stage in farmed S. japonica, it is important and urgent to understand the mechanisms of disease pathogenesis and resistance. Our results add to the growing body of evidence that pathogenic bacteria-induced epibacterial shifts during the disease progression. Bacterial taxa associated with healthy seaweed and the secondary opportunistic pathogens identified in this study enrich our baseline knowledge of the biology of farmed seaweeds and pave the way for the application of beneficial microbiota to protect against disease in seaweed farming industries.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
FL and GW conceived and designed the experiments and wrote the manuscript. FL finished the experiments. SE participated sequencing analysis and English writing. YZ helped the analysis of sequencing data. LC and LX provided the samples of Saccharina japonica. QY involved in designing the experiments. All authors contributed to the article and approved the submitted version.
Funding
This study was sponsored by National Key R & D Program of China (2018YFD0900305), the National Natural Science Foundation of China (42076106; 41576158), Sino-German Center for Research Promotion (GZ1357) and the Qingdao postdoctoral foundation for application research.
Conflict of Interest
Authors LC and LX are employed by Weihai Changqing Ocean Science and Technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Guangzhou Genedenovo Biotechnology Co. Ltd for their kindly technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.865224/full#supplementary-material
References
Aßhauer K. P., Wemheuer B., Daniel R., Meinicke P. (2015). Tax4Fun: Predicting Functional Profiles From Metagenomic 16S rRNA Data. Bioinformatics 31, 2882–2884. doi: 10.1093/bioinformatics/btv287
Agler M. T., Ruhe J., Kroll S., Morhenn C., Kim S. T., Weigel D., et al. (2016). Microbial Hub Taxa Link Host and Abiotic Factors to Plant Microbiome Variation. PloS Biol. 14, 1–31. doi: 10.1371/journal.pbio.1002352
Banerjee S., Schlaeppi K., van der Heijden M. G. A. (2018). Keystone Taxa as Drivers of Microbiome Structure and Functioning. Natl. Rev. Microbiol. 16, 567–576. doi: 10.1038/s41579-018-0024-1
Barott K. L., Rodriguez-Brito B., Janouškovec J., Marhaver K. L., Smith J. E., Keeling P., et al. (2011). Microbial Diversity Associated With Four Functional Groups of Benthic Reef Algae and the Reef-Building Coral Montastraea Annularis. Environ. Microbiol. 13, 1192–1204. doi: 10.1111/j.1462-2920.2010.02419.x
Ben-Haim Y., Rosenberg E. (2002). A Novel Vibrio Sp. Pathogen of the Coral Pocillopora Damicornis. Mar. Biol. 141, 47–55. doi: 10.1007/s00227-002-0797-6
Bernal P., Allsopp L., Filloux A., Llamas M. A. (2017). The Pseudomonas Putida T6SS Is a Plant Warden Against Phytopathogens. ISME J. 11, 972–987. doi: 10.1038/ismej.2016.169
Borchert E., Knobloch S., Dwyer E., Flynn S., Jackson S. A., Jóhannsson R., et al. (2017). Biotechnological Potential of Cold Adapted Pseudoalteromonas Spp. Isolated From ‘Deep Sea’ Sponges. Mar. Drugs 15, 184. doi 10.3390/md15060184
Campbell A. H., Harder T., Nielsen S., Kjelleberg S., Steinberg P. D. (2011). Climate Change and Disease: Bleaching of a Chemically Defended Seaweed. Global Change Biol. 17, 2958–2970. doi: 10.1111/j.1365-2486.2011.02456.x
Case R. J., Longford S. R., Campbell A. H., Low A., Tujula N., Steinberg P. D., et al. (2011). Temperature Induced Bacterial Virulence and Bleaching Disease in a Chemically Defended Marine Macroalga. Environ. Microbiol. 13, 529–537. doi: 10.1111/j.1462-2920.2010.02356.x
Chadha J., Harjai K., Chhibber S. (2021). Revisiting the Virulence Hallmarks of Pseudomonas Aeruginosa: A Chronicle Through the Perspective of Quorum Sensing. Environ. Microbiol. doi: 10.1111/1462-2920.15784
Cheng H., Zhang D., Ren L., Song Z., Li Q., Wu J., et al. (2021). Bio-Activation of Soil With Beneficial Microbes After Soil Fumigation Reduces Soil-Borne Pathogens and Increases Tomato Yield. Environ. Pollut. 283, 117160. doi: 10.1016/j.envpol.2021.117160
Chen Z., Lin L., Li Y., Chen Y., Zhang H., Han H., et al. (2021). Shifts in Rhizosphere Bacterial Community Structure, Co-Occurrence Network, and Function of Miscanthus Following Cadmium Exposure. Huanjing Kexue 42, 3997–4004. doi 10.13227/j.hjkx.202011198
Choi O., Lee Y., Park J., Kang B., Chun H. J., Kim M. C., et al. (2021). A Novel Toxoflavin-Quenching Regulation in Bacteria and Its Application to Resistance Cultivars. Microbial. Biotechnol. 14, 1657–1670. doi: 10.1111/1751-7915.13831
Christiansen L., Bech P. K., Schultz-Johansen M., Martens H. J., Stougaard P. (2018). Colwellia Echini Sp. Nov., an Agar-and Carrageenan-Solubilizing Bacterium Isolated From Sea Urchin. Int. J. Syst. Evol. Microbiol. 68, 687–691. doi: 10.1099/ijsem.0.002568
Croft M. T., Warren M. J., Smith A. G. (2006). Algae Need Their Vitamins. Eukaryot. Cell 5, 1175–1183. doi: 10.1128/EC.00097-06
Dai W., Qiu Q., Chen J., Xiong J. (2019). Gut Eukaryotic Disease-Discriminatory Taxa Are Indicative of Pacific White Shrimp (Litopenaeus Vannamei) White Feces Syndrome. Aquaculture 506, 154–160. doi: 10.1016/j.aquaculture.2019.03.034
Dai W., Sheng Z., Chen J., Xiong J. (2020). Shrimp Disease Progression Increases the Gut Bacterial Network Complexity and Abundances of Keystone Taxa. Aquaculture 517, 734802. doi: 10.1016/j.aquaculture.2019.734802
Dai W., Yu W., Xuan L., Tao Z., Xiong J. (2018). Integrating Molecular and Ecological Approaches to Identify Potential Polymicrobial Pathogens Over a Shrimp Disease Progression. Appl. Microbiol. Biotechnol. 102, 3755–3764. doi: 10.1007/s00253-018-8891-y
Damjanovic K., van Oppen M. J. H., Menéndez P., Blackall L. L. (2019). Experimental Inoculation of Coral Recruits With Marine Bacteria Indicates Scope for Microbiome Manipulation in Acropora Tenuis and Platygyra Daedalea. Front. Microbiol. 10, 1702. doi: 10.3389/fmicb.2019.01702
De Schryver P., Vadstein O. (2014). Ecological Theory as a Foundation to Control Pathogenic Invasion in Aquaculture. ISME J. 8, 2360–2368. doi: 10.1038/ismej.2014.84
Edgar R. C. (2013). UPARSE: Highly Accurate OTU Sequences From Microbial Amplicon Reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Egan S., Fukatsu, Takema, Francino M. P. (2020). Opportunities and Challenges to Microbial Symbiosis Research in the Microbiome Era. Front. Microbiol. 11, 1150. doi: 10.3389/fmicb.2020.01150
Egan S., Gardiner M. (2016). Microbial Dysbiosis: Rethinking Disease in Marine Ecosystems. Front. Microbiol. 7, 991. doi: 10.3389/fmicb.2016.00991
Egan S., Harder T., Burke C., Steinberg P., Kjelleberg S., Thomas T. (2013). The Seaweed Holobiont: Understanding Seaweed-Bacteria Interactions. FEMS Microbiol. Rev. 37, 462–476. doi: 10.1111/1574-6976.12011
Elufisan T. O., Rodríguez-Luna I. C., Oyedara O. O., Sánchez-Varela1 A., Hernández-Mendoza A., Gonzalez E. D., et al. (2020). The Polycyclic Aromatic Hydrocarbon (PAH) Degradation Activities and Genome Analysis of a Novel Strain Stenotrophomonas Sp. Pemsol Isolated From Mexico. Microbiology 8, e8102. doi doi: 10.7717/peerj.8102
Feng F., Zhan H., Wan Q., Wang Y., Li Y., Ge J., et al. (2021). Rice Recruits Sphingomonas Strain HJY-Rfp via Root Exudate Regulation to Increase Chlorpyrifos Tolerance and Boost Residual Catabolism. J. Exp. Bot. 72, 5673–5686. doi: 10.1093/jxb/erab210
Fernandes N., Case R. J., Longford S. R., Seyedsayamdost M. R., Steinberg P. D., Kjelleberg S., et al. (2011). Genomes and Virulence Factors of Novel Bacterial Pathogens Causing Bleaching Disease in the Marine Red Alga Delisea Pulchra. PloS One 6, e27387. doi: 10.1371/journal.pone.0027387
Gachon C. M. M., Sime-Ngando T., Strittmatter M., Chambouvet A., Kim G. H. (2010). Algal Diseases: Spotlight on a Black Box. Trends Plant Sci. 15, 633–640. doi: 10.1016/j.tplants.2010.08.005
Gao M., Xiong C., Gao C., Tsui C. K. M., Zhou X., Wang M., et al. (2021). Disease-Induced Changes in Plant Microbiome Assembly and Functional Adaptation. Microbiome 9, 187. doi: 10.1186/s40168-021-01138-2
Girvan M. S., Campbell C. D., Killham K., Prosser J. I., Glover L. A. (2005). Bacterial Diversity Promotes Community Stability and Functional Resilience After Perturbation. Environ. Microbiol. 7, 301–313. doi: 10.1111/j.1462-2920.2005.00695.x
Hajishengallis G., Lamont R. J. (2016). Dancing With the Stars: How Choreographed Bacterial Interactions Dictate Nososymbiocity and Give Rise to Keystone Pathogens, Accessory Pathogens, and Pathobionts. Trends Microbiol. 24, 477–489. doi: 10.1016/j.tim.2016.02.010
Han Q., Zhang X., Chang L., Xiao L., Ahmad R., Saha M., et al. (2021). Dynamic Shift of the Epibacterial Communities on Commercially Cultivated Saccharina Japonica From Mature Sporophytes to Sporelings and Juvenile Sporophytes. J. Appl. Phycol. 33, 1171–1179. doi: 10.1007/s10811-020-02329-4
Hollants J., Leliaert F., Clerck O. D., Willems A. (2013). What We Can Learn From Sushi: A Review on Seaweed-Bacterial Associations. FEMS Microbiol. Ecol. 8, 1–16. doi: 10.1111/j.1574-6941.2012.01446.x
Huang Z., Li X., Wang L., Shao Z. (2016). Changes in the Intestinal Bacterial Community During the Growth of White Shrimp, Litopenaeus Vannamei. Aquac. Res. 47, 1737–1746. doi: 10.1111/are.12628
Ivanova E. P., Ng H. J., Webb H. K. (2014). The Family Pseudoalteromonadaceae. Prokaryotes 28, 575–582. doi: 10.1007/978-3-642-38922-1_229
Joint I., Tait K., Wheeler G. (2007). Cross-Kingdom Signalling: Exploitation of Bacterial Quorum Sensing Molecules by the Green Seaweed Ulva. Philos. Trans. - R. Soc. Biol. Sci. 362, 1223–1233. doi: 10.1098/rstb.2007.2047
Jung J., Kim J. M., Jin H. M., Kim S. Y., Park W., Jeon C. O. (2011). Litorimonas taeanensis gen. nov., sp. nov., Isolated from a Sandy Beach. Int. J. Syst. Evol. Microbiol. 61, 1534–1538. doi: 10.1099/ijs.0.022129-0
Kumar V., Zozaya Valdes E., Kjelleberg S., Thomas T., Egan S. (2016). Multiple Opportunistic Pathogens Can Cause a Bleaching Disease in the Red Seaweed Delisea Pulchra. Environ. Microbiol. 18, 3962–3975. doi: 10.1111/1462-2920.13403
Li J., Majzoub M. E., Marzinelli E. M., Dai Z., Thomas T., Egan S. (2022). Bacterial Controlled Mitigation of Dysbiosis in a Seaweed Disease. ISME J. 16, 378–387. doi: 10.1038/s41396-021-01070-1
Li J., Pang S., Shan T., Su L. (2020). Changes of Microbial Community Structures Associated With Seedlings of Saccharina Japonica at Early Stage of Outbreak of Green Rotten Disease. J. Appl. Phycol. 32, 1323–1327. doi: 10.1007/s10811-019-01975-7
Longford S. R., Campbell1 A. H., Nielsen S., Case R. J., Kjelleberg S., Steinberg P. D. (2019). Interactions Within the Microbiome Alter Microbial Interactions With Host Chemical Defences and Affect Disease in a Marine Holobiont. Sci. Rep. 9, 1363. doi: 10.1038/s41598-018-37062-z
Loreau M., Naeem S., Inchausti P., Bengtsson J., Grime J. P., Hector A., et al. (2001). Biodiversity and Ecosystem Functioning: Current Knowledge and Future Challenges. Science 294, 804–808. doi: 10.1126/science.1064088
Lu J., Zhang X., Qiu Q., Chen J., Xiong J. (2020). Identifying Potential Polymicrobial Pathogens: Moving Beyond Differential Abundance to Driver Taxa. Microbial. Ecol. 80, 447–458. doi: 10.1007/s00248-020-01511-y
Martin M., Barbeyron T., Martin R., Portetelle D., Michel G., Vandenbol M. (2015). The Cultivable Surface Microbiota of the Brown Alga Ascophyllum Nodosum Is Enriched in Macroalgal-Polysaccharide-Degrading Bacteria. Front. Microbiol. 6, 1487. doi: 10.3389/fmicb.2015.01487
Marzinelli E. M., Campbell A. H., Zozaya Valdes E., Vergés A., Nielsen S., Wernberg T., et al. (2015). Continental-Scale Variation in Seaweed Host Associated Bacterial Communities Is a Function of Host Condition, Not Geography. Environ. Microbiol. 17, 4078–4088. doi: 10.1111/1462-2920.12972
Na H., Jo S. W., Do J. M., Kim I. S., Yoon H. S. (2021). Production of Algal Biomass and High-Value Compounds Mediated by Interaction of Microalgal Oocystis Sp. KNUA044 and Bacterium Sphingomonas Knu100. J. Microbiol. Biotechnol. 31, 387–397. doi: 10.4014/jmb.2009.09055
Penesyan A., Marshall-Jones Z., Holmstrom C., Kjelleberg S., Egan S. (2009). Antimicrobial Activity Observed Among Cultured Marine Epiphytic Bacteria Reflects Their Potential as a Source of New Drugs. FEMS Microbiol. Ecol. 69, 113–124. doi: 10.1111/j.1574-6941.2009.00688.x
Pruesse E., Quast C., Knittel K., Fuchs B. M., Ludwig W., Peplies J., et al. (2007). SILVA: A Comprehensive Online Resource for Quality Checked and Aligned Ribosomal RNA Sequence Data Compatible With ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Roberts D. W. (2016). Labdsv: Ordination and Multivariate Analysis for Ecology. Available at: https://CRAN.R-project.org/package=labdsv
Rosenberg E., Falkovitz L. (2004). The Vibrio Shiloi/Oculina Patagonica Model System of Coral Bleaching. Annu. Rev. Microbiol. 58, 143–159. doi: 10.1146/annurev.micro.58.030603.123610
Rungrassamee W., Klanchui A., Maibunkaew S., Karoonuthaisiri N. (2016). Bacterial Dynamics in Intestines of the Black Tiger Shrimp and the Pacific White Shrimp During Vibrio Harveyi Exposure. J. Invertebr Pathol. 133, 12–19. doi: 10.1016/j.jip.2015.11.004
Ryan R., Monchy S., Cardinale M., Taghavi S., Crossman L., Avison M. B., et al. (2009). The Versatility and Adaptation of Bacteria From the Genus Stenotrophomonas. Nat. Rev. Microbiol. 7, 514–525. doi: 10.1038/nrmicro2163
Saha M., Weinberger F. (2019). Microbial “Gardening” by a Seaweed Holobiont: Surface Metabolites Attract Protective and Deter Pathogenic Epibacterial Settlement. J. Ecol. 107, 2255–2265. doi: 10.1111/1365-2745.13193
Sánchez Hinojosa V., Asenjo J., Leiva S. (2018). Agarolytic Culturable Bacteria Associated With Three Antarctic Subtidal Macroalgae. World J. Microbiol. Biotechnol. 34, 73. doi: 10.1007/s11274-018-2456-1
Sawabe T., Makino H., Tatsumi M., Nakano K., Tajima K., Iqbal M. M., et al. (1998). Pseudoalteromonas Bacteriolytica Sp. Nov., A Marine Bacterium That Is the Causative Agent of Red Spot Disease of Laminaria Japonica. Int. J. Syst. Evol. Microbiol. 48, 769–774. doi: 10.1099/00207713-48-3-769
Shetty S. A., Hugenholtz F., Lahti L., Smidt H., de Vos W. M. (2017). Intestinal Microbiome Landscaping: Insight in Community Assemblage and Implications for Microbial Modulation Strategies. FEMS Microbiol. Rev. 41, 182–199. doi: 10.1093/femsre/fuw045
Singh R. P., Reddy C. (2014). Seaweed-Microbial Interactions: Key Functions of Seaweed-Associated Bacteria. FEMS Microbiol. Ecol. 88, 213–230. doi: 10.1111/1574-6941.12297
Tropeano M., Vázquez S., Coria S., Turjanski A., Cicero D., Bercovich A., et al. (2013). Extracellular Hydrolytic Enzyme Production by Proteolytic Bacteria From the Antarctic. Polish Polar Res. 34, 253–267. doi: 10.2478/popore-2013-0014
Vilchis-Carmona J. A., Rodríguez-Luna I. C., Elufisan T. O., Sánchez−Varela A., BibbinsMartínez M., Rivera G., et al. (2021). The Decolorization and Degradation of Azo Dyes by Two Stenotrophomonas Strains Isolated From Textile Effluent (Tepetitla, Mexico). Braz. J. Microbiol. doi: 10.1007/s42770-021-00542-y
Wagner-Dobler I., Thiel V., Eberl L., Allgaier M., Bodor A., Meyer S., et al (2005). Discovery of Complex Mixtures of Novel Long-Chain Quorum Sensing Signals in Free-Living and Host-Associated Marine Alphaproteobacteria. Chem. Biochem. 6, 2195–2206. doi: 10.1002/cbic.200500189
Wang Q., Garrity G. M., Tiedje J. M., Cole J. R. (2007). Naive Bayesian Classifier for Rapid Assignment of rRNA Sequences Into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wang G., Lu B., Shuai L., Li D., Zhang R. (2014). Microbial Diseases of Nursery and Field-Cultivated Saccharina Japonica (Phaeophyta) in China. Algol. Stud. 2, 39–51. doi: 10.1127/1864-1318/2014/0167
Wang G., Shuai L., Li Y., Lin W., Zhao X., Duan D. (2008). Phylogenetic Analysis of Epiphytic Marine Bacteria on Hole-Rotten Diseased Sporophytes of Laminaria Japonica. J. Appl. Phycol. 20, 403–409. doi: 10.1007/s10811-007-9274-4
Weinberger F., Beltran J., Correa J. A., Lion U., Pohnert G., Kumar N., et al. (2007). Spore Release in Acrochaetium Sp. (Rhodophyta) Is Bacterially Controlled. J. Phycol. 43, 235–241. doi: 10.1111/j.1529-8817.2007.00329.x
Weiss S., Van Treuren W., Lozupone C., Faust K., Friedman J., Deng Y., et al. (2016). Correlation Detection Strategies in Microbial Data Sets Vary Widely in Sensitivity and Precision. ISME J. 10, 1–13. doi: 10.1038/ismej.2015.235
Xiong J. (2018). Progress in the Gut Microbiota in Exploring Shrimp Disease Pathogenesis and Incidence. Appl. Microbiol. Biotechnol. 102, 7343–7350. doi: 10.1007/s00253-018-9199-7
Xu Z. X., Zhang H. X., Han J. R., Dunlap C. A., Rooney A. P., Mu D. S., et al. (2017). Colwellia Agarivorans Sp. Nov., an Agar-Digesting Marine Bacterium Isolated From Coastal Seawater. Int. J. Syst. Evol. Microbiol. 67, 1969–1974. doi: 10.1099/ijsem.0.001897
Yang R., Liu Q., He Y., Tao Z., Xu M., Luo Q., et al. (2020). Isolation and Identification of Vibrio Mediterranei 117-T6 as a Pathogen Associated With Yellow Spot Disease of Pyropia (Bangiales, Rhodophyta). Aquaculture 526, 735372. doi: 10.1016/j.aquaculture.2020.735372
Zhang R., Chang L., Xiao L., Zhang X., Han Q., Li N., et al. (2020). Diversity of the Epiphytic Bacterial Communities Associated With Commercially Cultivated Healthy and Diseased Saccharina Japonica During the Harvest Season. J. Appl. Phycol. 32, 2071–2080. doi: 10.1007/s10811-019-02025-y
Zhang X., Chen Y., Saha M., Zhuang Y., Chang L., Xiao L., et al. (2022). Pseudoalteromonas Piscicida X-8 Causes Bleaching Disease in Farmed Saccharina Japonica. Aquaculture 546, 737354. doi: 10.1016/j.aquaculture.2021.737354
Zhang X.-H., He X., Austin B. (2020). Vibrio Harveyi: A Serious Pathogen of Fish and Invertebrates in Mariculture. Mar. Life Sci. Technol. 2, 231–245. doi: 10.1007/s42995-020-00037-z
Zhang X., Xia X., Dai M., Cen J., Zhou L., Xie J. (2021b). Microplastic Pollution and Its Relationship With the Bacterial Community in Coastal Sediments Near Guangdong Province, South China. Sci. Total Environ. 760, 1–9. doi: 10.1016/j.scitotenv.2020.144091
Zhu Z., Xu M., Liu Q., Li D., Yang R., Chen H. (2021). Bacteriophage Therapy on the Conchocelis of Pyropia Haitanensis (Rhodophyta) Infected by Vibrio Mediterranei 117-T6. Aquaculture 531, 735853. doi: 10.1016/j.aquaculture.2020.735853
Zozaya-Valdes E., Egan S., Thomas T. (2015). A Comprehensive Analysis of the Microbial Communities of Healthy and Diseased Marine Macroalgae and the Detection of Known and Potential Bacterial Pathogens. Front. Microbiol. 6, 146. doi: 10.3389/fmicb.2015.00146
Keywords: bleaching disease, epimicrobiome shifts, pathogenic bacteria, 16S rRNA gene amplicon sequencing, Saccharina japonica
Citation: Ling F, Egan S, Zhuang Y, Chang L, Xiao L, Yang Q and Wang G (2022) Epimicrobiome Shifts With Bleaching Disease Progression in the Brown Seaweed Saccharina japonica. Front. Mar. Sci. 9:865224. doi: 10.3389/fmars.2022.865224
Received: 29 January 2022; Accepted: 21 March 2022;
Published: 26 April 2022.
Edited by:
Wei Huang, Ministry of Natural Resources, ChinaReviewed by:
Jinbo Xiong, Ningbo University, ChinaNaihao Ye, Chinese Academy of Fishery Sciences (CAFS), China
Copyright © 2022 Ling, Egan, Zhuang, Chang, Xiao, Yang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qin Yang, eWFuZ3FpbkBvdWMuZWR1LmNu; Gaoge Wang, d2dhb2dlQG91Yy5lZHUuY24=