 Yongxue Li
Yongxue Li Wenjin Hao
Wenjin Hao Saijun Peng
Saijun Peng Tingting Sun
Tingting Sun Jianmin Zhao
Jianmin Zhao Zhijun Dong
Zhijun Dong
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 11 May 2022
Sec. Microbial Symbioses
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.864872
This article is part of the Research Topic Interaction between Marine Invertebrates and Symbiotic Microbes in a Changing Environment: Community Structure and Ecological Functions View all 10 articles
Jellyfish and their associated microbes form an ecological unit called the holobiont. Changes in the composition of dominant microbial assemblages may influence the environmental resilience and function of the holobionts. Differentiating the microbial communities from diverse jellyfish is essential for characterizing the functional contributions of microorganisms but has not been fully explored. In this study, based on 16S rRNA gene sequencing, we investigated the composition of microbial communities associated with two Aurelia polyp species (Aurelia coerulea and Aurelia solida) obtained from seven locations, which were maintained under the same environmental conditions. Sequence analysis showed that the genera Sphingomonas, Phyllobacterium, and Ralstonia were the most abundant members of the Aurelia-associated microbial communities and dominated the core microbiome of the Aurelia polyps in this study. Functional prediction revealed that chemoheterotrophy and aerobic chemoheterotrophy, based on the FAPROTAX dataset, were the primary functions of the associated microbes of Aurelia polyps. In addition, the comparison of microbial communities from different Aurelia polyp populations revealed interspecific instead of intraspecific variation, indicating a correlation between the composition of the symbiotic microbiota and genetic background of Aurelia polyps.
Interactions between organisms are factors determining the coexistence of species and maintenance of biodiversity. It is well established that animals act as hosts for multilineage consortia of microbial communities (i.e., bacteria, archaea, eukaryotes, and viruses) (McFall-Ngai et al., 2013), and microbial assemblages are an important factor in the regulation of host biology. Host-associated microbial communities exert nonnegligible forces on physiological metabolism regulation, immune function, and complex host behaviors (Rook et al., 2017). For instance, the gut microbial communities of humans and insects assist in inhibiting the invasion and colonization of pathogenic bacteria and provide essential amino acids and vitamins for the physiological metabolism of hosts (Esser et al., 2019). The intestinal microbial communities of several insects significantly improve the environmental adaptability of hosts (Zhang and Leadbetter, 2012). Therefore, understanding the composition and structure of host-associated microbial communities is important in enabling insights into the functional contributions of microbes to hosts.
The colonization of a symbiotic consortium is influenced by many factors, such as the transmission of microbes from the external environment to hosts (Martinson et al., 2017), among-microbe interactions (Martinson et al., 2017), and ecological drift (Costello et al., 2012). Studies have found obvious dissimilarities between the microbiomes associated with aquatic animals (Stevens and Olson, 2015), amphibians (Walke et al., 2014), and terrestrial animals (Ren et al., 2017) from different regions. Additionally, the hosts act as ecological filters to selectively assemble specific species from the regional species pool based on their requirements and resources (Adair et al., 2020), causing the microbiomes of different species living in the same environment to be significantly different. Likewise, hosts of the same species often retain similarities in their microbial communities across various environmental conditions (Cheng et al., 2020). This retained microbiome, termed the “core microbiome”, may play an important role in supporting the basic physio-chemical metabolism of hosts (Dietz et al., 2020).
As bacterial life had already existed for approximately three billion years when animals first evolved, microbe-animal interactions likely coevolved with the hosts over millions of years (Knoll, 2015). Based on their early appearance on the evolutionary scene, Cnidarians were likely among the first animals to establish associations with microorganisms (Bosch, 2013). The moon jellyfish Aurelia belongs to the phylum Cnidaria (class: Scyphozoa) and is the most common scyphozoan jellyfish in global coastal waters (Dong et al., 2018). The diphasic life cycle of Aurelia alternates between free-living pelagic medusa and sessile benthic polyp phases (Lucas, 2001). The composition and structure of microbial communities associated with the jellyfish Aurelia have been well investigated. Weiland-Braüuer et al. found that the composition of the microbiota associated with Aurelia aurita changed with the compartments of the adult medusae (mucus versus gastric cavity) and the life stages, particularly during the transition from the benthic to the pelagic stages (Weiland-Bräuer et al., 2015). Kramar et al. reported that the composition of Aurelia solida-associated microbial communities changed in relation to the period of the bloom (Kramar et al., 2019). Moreover, the presence of potentially pathogenic bacteria (i.e., Vibrio and Mycoplasmataceae) in the Aurelia-associated microbiome regarded Aurelia as a vector of pathogens (Tinta et al., 2019; Peng et al., 2021).
In the present study, we focused on the polyps of two moon jellyfish species, Aurelia coerulea and A. solida, which were incubated in the same environmental conditions with the same food source for 6 months to explore the relationship between polyps and symbiotic microbes. Aurelia polyp is a suitable model organism for the interaction between microbes and animals because of its simple body construction (basic immune and nervous systems), easy culture in the laboratory, high regeneration output, and short asexual reproduction cycle (Chiaverano and Graham, 2017). Based on the genetic and evolutionary relationships of the species, we hypothesized that the microbial community structure and functional characteristics of the species would vary under the same environmental conditions. In addition, the potential ecological functions of bacterial communities associated with Aurelia polyps were also discussed in the present study.
The polyps of A. coerulea and A. solida were originally obtained from the laboratory of Agustin Schiariti (Instituto Nacional de Investigación y Desarrollo Pesquero). A. coerulea were collected from the USA, China, Japan, Spain, and France and abbreviated as ACUSA, ACCHI, ACJAP, ACSPA and ACFRA, respectively. A. solida were collected from Israel and Slovenia and abbreviated as ASISR and ASSLO, respectively, as detailed in Table 1. The polyps of the different Aurelia populations were kept separately in tanks. All polyps were cultured at 15°C in ambient fresh seawater with a salinity of 30 practical salinity units and kept on a day:night lighting rhythm of 12:12 h for 6 months of incubation time. Freshly hatched Artemia salnia were fed to the Aurelia polyps as their sole food source once every two days during the incubation period. Homogenization of cultivation conditions was performed to ensure stability and monovariability (i.e., host genotypes) of the symbiotic microbial communities of polyps from different Aurelia species and populations. The polyps were incubated in sterile seawater (0.22-μm filtered) baths for 1 day to clear the digestive system before DNA extraction. Species were identified based on 16S mtRNA pairwise sequence alignment technology.
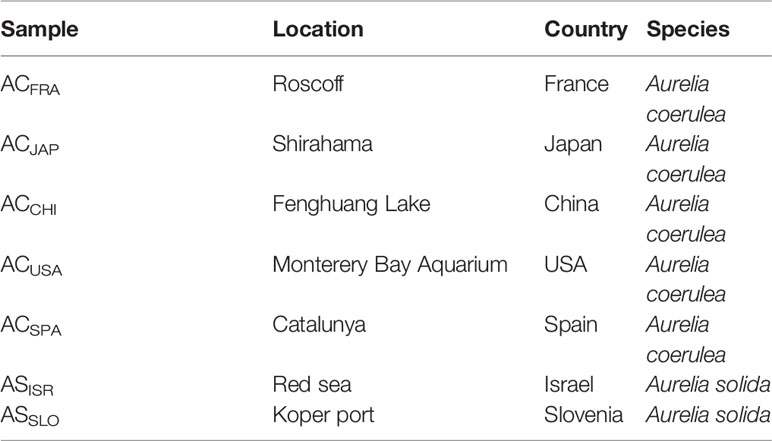
Table 1 The native locations of Aurelia polyps.
Bacterial DNA of Aurelia polyps from 7 populations was isolated using a Wizard genomic DNA purification kit (Promega, Madison, WI, USA) according to the manufacturer’s protocols. Each Aurelia population encompassed 5 replicates (10 polyps each) to yield read libraries sufficient for analysis. PCR amplification of the V4 hypervariable region of the 16S rRNA gene was performed with the primers 515F (5’-GTGCCAGCMGCCGCGGTAA-3’) and 806R (5’-GGACTACHVGGGTWTCTAAT-3’) (Hugerth et al., 2014). All PCRs were carried out in 30 µL reactions with 15 µL of Phusion® High-Fidelity PCR Master Mix (New England Biolabs, USA), 0.2 µM forward and reverse primers, and approximately 10 ng of template DNA. Thermal cycling consisted of initial denaturation at 98°C for 1 min, followed by 30 cycles of denaturation at 98°C for 10 s, annealing at 50°C for 30 s and elongation at 72°C for 30 s, with a final elongation at 72°C for 5 min. PCR products were detected by electrophoresis in a 2% (w/v) agarose gel. PCR amplicons of each sample with bright bands were mixed in equal-density ratios and purified with a GeneJET™ Gel Extraction Kit (Thermo Scientific, USA). Sequencing libraries were generated using an Ion Plus Fragment Library Kit 48 Rxns (Thermo Scientific) following the manufacturer’s recommendations. Then, the library concentration was assessed with a Qubit@ 2.0 Fluorometer (Thermo Scientific). The amplicon libraries were sequenced on the Ion S5™ XL platform at Novogene Bioinformatics Technology Co., Ltd. (Beijing, China).
Single-end reads were assigned to samples based on their unique barcode and truncated by cutting off the barcode and primer sequence. Quality filtering of the raw reads was performed under specific filtering conditions to obtain high-quality clean reads according to the Cutadapt (v1.9.1, http://cutadapt.readthedocs.io/en/stable/) quality control process (Kechin et al., 2017). The reads were compared with the reference database (Silva 132 database, https://www.arb-silva.de/) (Quast et al., 2013) using the UCHIME algorithm (UCHIME algorithm, http://www.drive5.com/usearch/manual/uchime_algo.html) (Edgar et al., 2011) to detect chimera sequences and then the chimera sequences were removed to obtain clean reads (Haas et al., 2011). The singleton and non-target sequences were removed from the analysis.
Sequence analyses were performed using UPARSE software (UPARSE v7.0.100 http://drive5.com/uparse/) (Edgar, 2013). First, sequences with ≥ 97% similarity were assigned to the same operational taxonomic unit (OTU), and a representative sequence for each OTU was screened for further annotation. Then, the Silva 132 database (https://www.arb-silva.de/) (Quast et al., 2013) was used to annotate each representative sequence with taxonomic information based on the Mothur algorithm, and a representative sequence for each OTU was assigned to a taxonomic level using the RDP classifier (Edgar, 2013). Furthermore, multiple sequence alignments were conducted using MUSCLE software (version 3.8.31, http://www.drive5.com/muscle/) (Edgar, 2004) to study the phylogenetic relationships among different OTUs and the divergence in the dominant species among different samples (groups).
Microbial communities normally consist of a few abundant and many rare species (Easson et al., 2020). In this study, the thresholds for rare, conditionally rare, and abundant taxa were defined based on relative sequence abundance cutoffs, with reference to recent publications (Liu et al., 2017). “Rare taxa” were defined as OTUs with a relative sequence abundance < 0.01% in all samples. “Conditionally rare taxa” were defined as OTUs that were rare (relative sequence abundance < 0.01%) in some but not all samples and were never abundant (relative sequence abundance ≥ 1%). “Abundant taxa” were defined as the OTUs that did not fall in either the rare or conditionally rare categories. “Core taxa” was defined as the OTUs present in all Aurelia polyps in this study.
The OTU abundance data were normalized corresponding to the sample with the fewest sequences for further analysis of alpha diversity and beta diversity. The Shannon and CHAO1 diversity indices were calculated based on the normalized OTU matrix with QIIME (version 1.7.0) and visualized with the ggplot2 package of R software (version 2.15.3). Nonparametric Kruskal–Wallis tests, nonparametric Mann–Whitney U tests, and t tests were performed to identify significant differences in functional and relative bacterial abundance between different samples. The beta diversity of samples was calculated based on Bray–Curtis dissimilarity at the OTU level and used to perform principal coordinate analysis (PCoA), which was visualized with the ggplot2 and vegan packages of R software (version 2.15.3). In addition, Wilcoxon match-pair tests and permutational analysis of molecular variance (PERMANOVA) were constructed to test for significant differences in microbial alpha and beta diversity between Aurelia polyp species using the vegan package of R software (version 2.15.3). Furthermore, the relative sequence abundance of samples at diverse classification levels was visualized by the ggplot2 and reshape2 packages of R software (version 2.15.3). The relative sequence abundance of functional bacterial communities was calculated based on the FAPROTAX database and visualized by the pheatmap package of R software (version 2.15.3).
After filtering the raw data, a total of 2,456,681 clean reads from 35 Aurelia polyp samples were obtained, with an average sequence length of 372 bp. A total of 1,213 OTUs were present in the Aurelia polyp samples, clustered into 36 phyla, 49 classes, 107 orders, 190 families, and 400 genera.
A total of 159 OTUs were present in all A. coerulea polyp groups, accounting for 82.06% (ACSPA)–98.58% (ACCHI) of the total relative sequence abundance (Figure 1). In the A. coerulea polyps, 36 bacterial phyla, 48 classes, 98 orders, 179 families, and 366 genera were detected. The microbiomes of A. solida polyps contained 25 phyla, 40 classes, 84 orders, 145 families, and 257 genera. A total of 334 OTUs were shared between both groups of A. solida polyps and accounted for 96.64% and 98.97% of the total relative sequence abundance in ASISR and ASSLO, respectively (Figure 1). Each Aurelia polyp group contained unique OTUs, which all had low relative sequence abundance (< 1%, Figure 1).
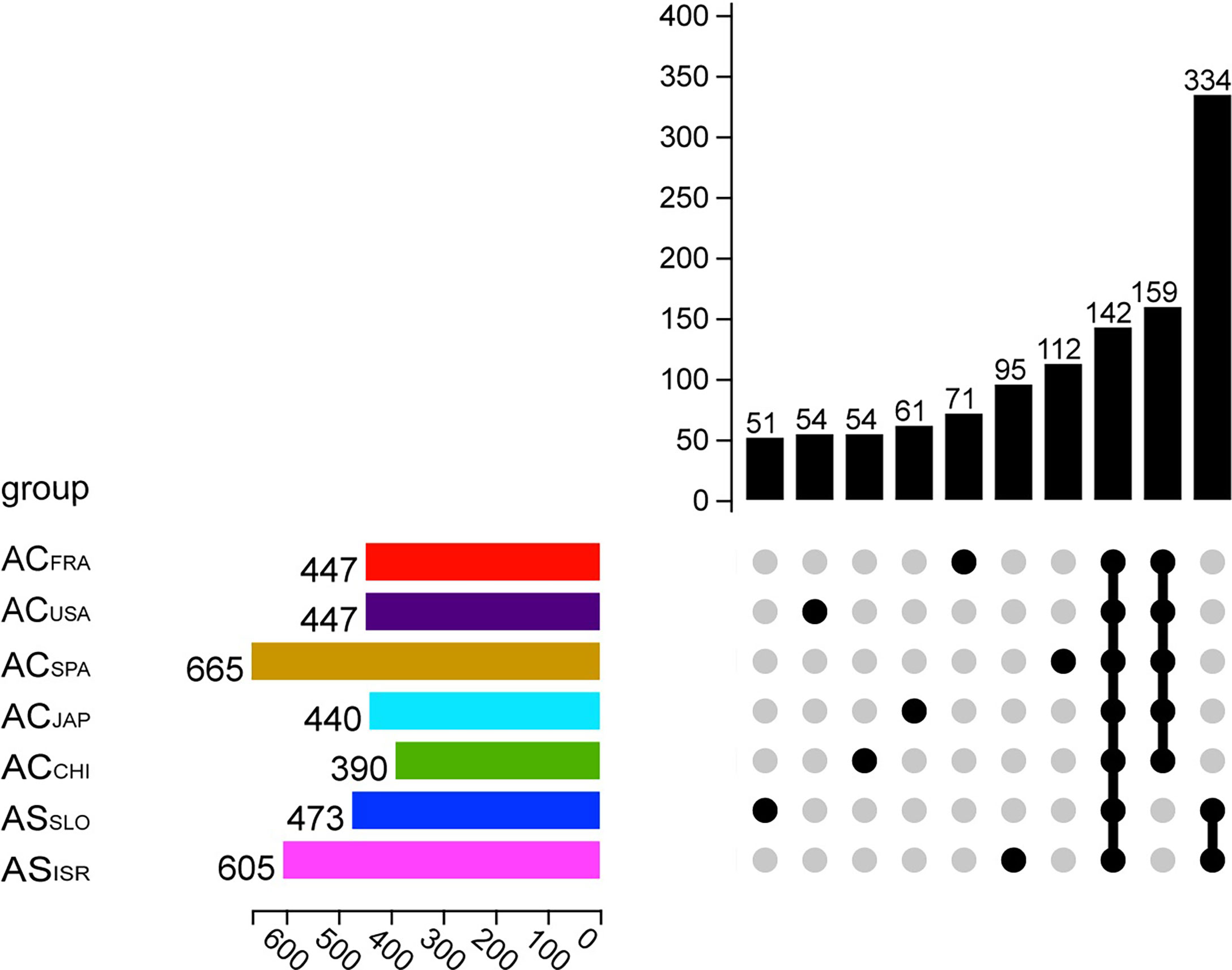
Figure 1 Upset plot showing OTU information of different Aurelia polyp groups. The horizontal column on the left represents the average number of OTUs in the microbiome of each group (n = 5). Each single point in the matrix below indicates that the vertical column above it showd the number of OTUs unique to the corresponding Aurelia polyp group, while the points on the matrix grouped by a line indicate that the vertical column shows the number of OTUs common to those groups.
At the phylum and class levels, bacterial communities associated with both Aurelia polyp species were dominated by Proteobacteria (mainly classes Alphaproteobacteria and Gammaproteobacteria), Firmicutes (mainly class Clostridia), Bacteroidetes, Acidobacteira and Actinobacteria, together comprising 97.01% (ACSPA) – 99.71% (ACJAP) of the total richness (Figure 2). The relative abundances of the other 31 phyla in each Aurelia polyp population were <1%, together comprising 0.29% (ACJAP) – 2.99% (ACSPA) of the total richness (Figure 2). At the family and genus levels, the polyp microbiomes were dominated by Sphingomonadaceae (mainly the genera Sphingomonas and Sphingobacterium), Rhizobiaceae (mainly the genus Phyllobacterium) and Burkholderiaceae (mainly the genus Ralstonia), comprising 53.2% (ACSPA)–95.5% (ACCHI) of the total bacterial abundance associated with A. coerulea and A. solida (Figure 2).
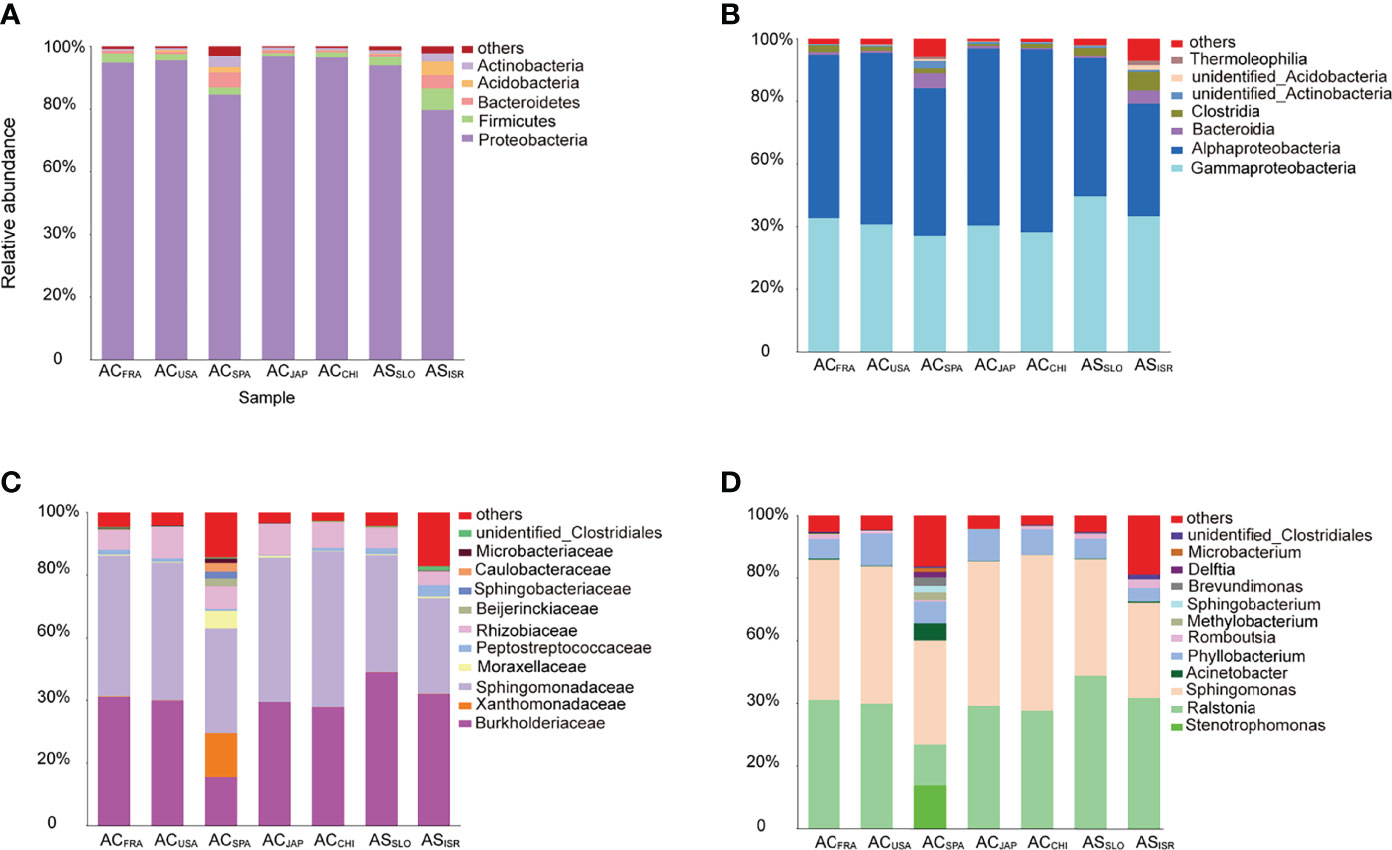
Figure 2 Stacked bar plots of bacterial taxa with relative sequence abundance higher than 1% at the (A) phylum level, (B) class level, (C) family level, and (D) genus level in the Aurelia-associated bacterial communities. The less abundant taxa are grouped under “others”.
At the family level, unidentified Clostridiales (Mann–Whitney U test, p = 0.039) were significantly more abundant in A. solida than in A. coerulea, while Sphingomonadaceae (t test, p = 0.038) and Rhizobiaceae (t test, p = 0.024) were more abundant in A. coerulea. At the genus level, the A. coerulea polyps had significantly higher relative sequence abundance of the genera Sphingomonas (t test, p = 0.043) and Phyllobacterium (t test, p = 0.023) than A. solida polyps.
To further reveal the inter- and intraspecific differences in the microbiomes of Aurelia polyps, the microbes of the two Aurelia polyp species were compared based on microbial alpha diversity and beta diversity, as detailed in Figure 3.
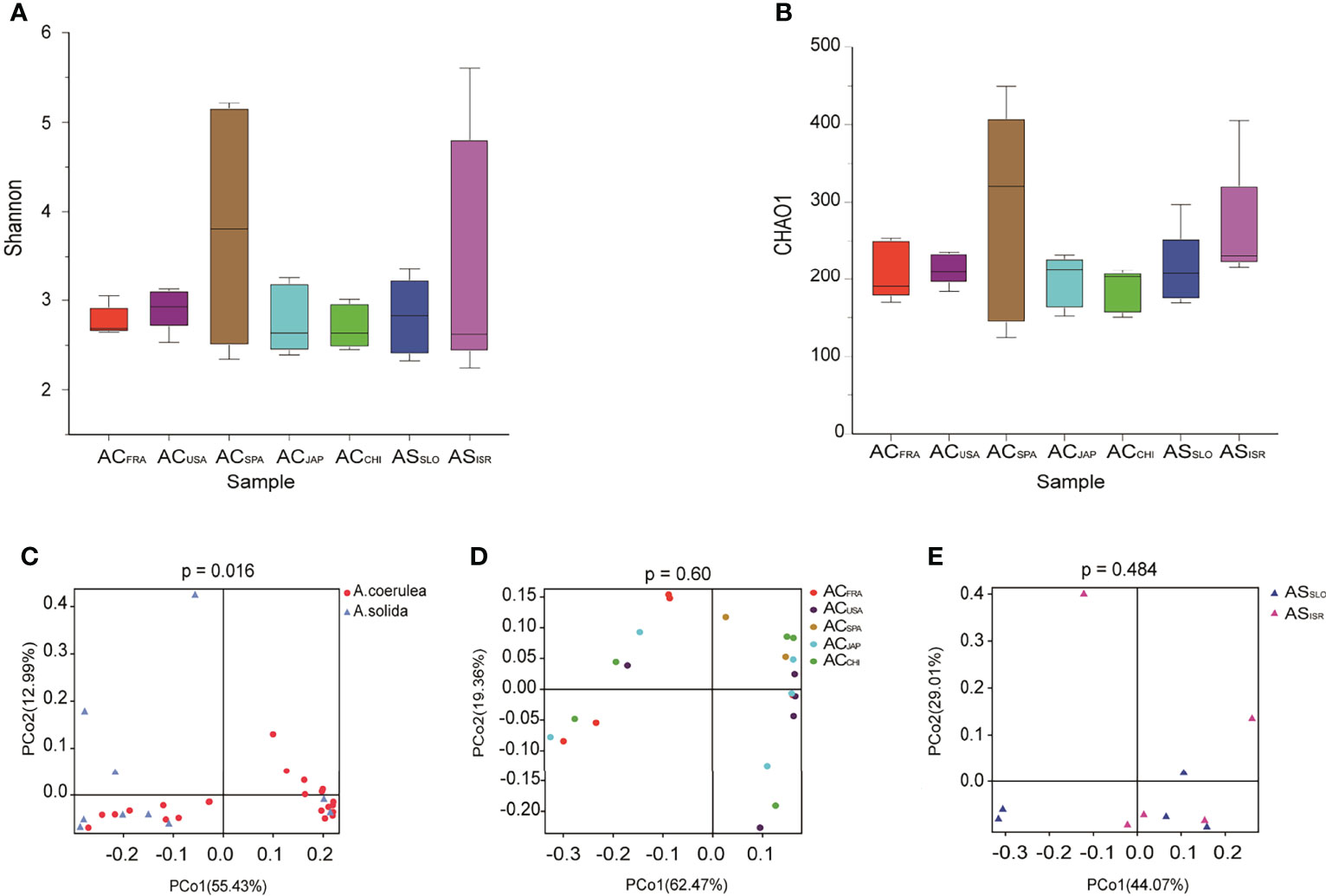
Figure 3 Variation in microbial diversity of different polyp groups. Alpha diversity and richness are represented by Shannon indices (A) and CHAO1 (B) of Aurelia polyp microbiomes. PCoA (Principal coordinates analysis) visualization of bacterial beta - diversity of (C) all samples grouped by different types without outline, (D) A. coerulea polyps samples grouped by location without Outliers, and (E) A. solida polyp samples grouped by location. PERMANOVA tests were used to analyze significant differences between microbial diversity in different groups.
Regarding interspecific differences in microbial alpha diversity, A. coerulea polyps had a lower average Shannon index (2.998) than A. solida polyps (3.119), as well as a lower average CHAO1 index (219 and 237 in A. coerulea and A. solida polyps, respectively, Figure 3). However, no significant difference between the A. coerulea and A. solida groups was detected in either the Shannon indices or CHAO1 indices (Wilcoxon match-pairs test, p = 0.5566, Figure 3). Regarding intraspecific differences, ACCHI had the highest average Shannon indices of the A. coerulea groups (3.367), followed by ACSPA, ACFRA, ACUSA, and ACJAPhad the lowest (2.635). The highest average CHAO1 index of the A. coerulea polyps was found in ACSPA (285), followed by ACUSA, ACFRA and ACJAP, and the lowest was found in ACCHI (186). Within the A. solida groups, ASISR had a higher average CHAO1 index (263) than ASSLO (212) but had a lower mean Shannon index (2.449 in ASISR and 2.831 in ASSLO). Nonetheless, no significant difference was found between the Shannon and CHAO1 indices of polyps in the A. coerulea and A. solida groups (Wilcoxon match-pairs test, p > 0.05, Figure 3).
In terms of the interspecific comparison, PCoA based on Bray–Curtis distance demonstrated a significant difference in microbial beta diversity between the microbiomes of the A. coerulea and A. solida polyps (PERMANOVA test, p = 0.016, Figure 3). Furthermore, significant differences were identified between the abundant and core microbial taxa associated with A. coerulea polyps and A. solida polyps (PERMANOVA test, p = 0.044 and p = 0.03, respectively), but there was no significant difference between the rare microbial taxa associated with the two species (PERMANOVA test, p = 0.403, Table 2). Similarity percentage analysis (Simper) illustrated that the genera Stenotrophomonas, Ralstonia, and Sphingobacterium were the main contributors to the difference in the microbial community between the two species, which contributed 26.8%, 26.1%, and 10.6% to the difference value, respectively (Figure 4). In terms of the intraspecific comparison, PCoA showed no significant intraspecific variation among the groups of A. coerulea polyps (PERMANOVA test, p = 0.60, Figure 3) or A. solida polyps (PERMANOVA test, p = 0.484, Figure 3). Furthermore, no significant difference was detected in the rare, conditionally rare, abundant taxa, or core taxa of the intraspecific groups (PERMANOVA test, p > 0.05, Table 2).
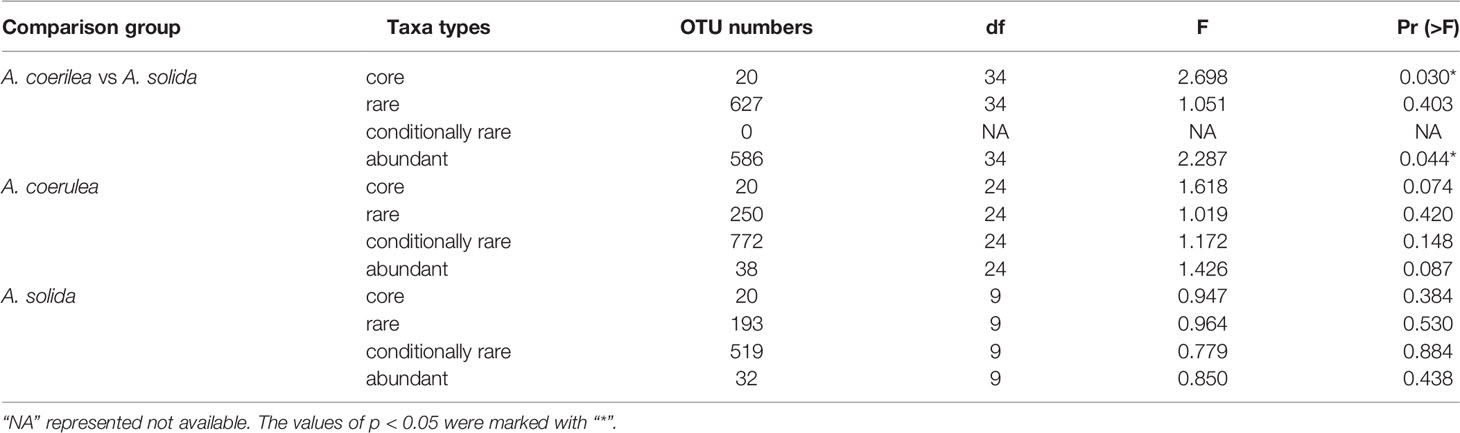
Table 2 PERMANOVA tests of bacterial community structure of Aurelia polyps based on Bray–Curtis dissimilarities of OTU abundance.
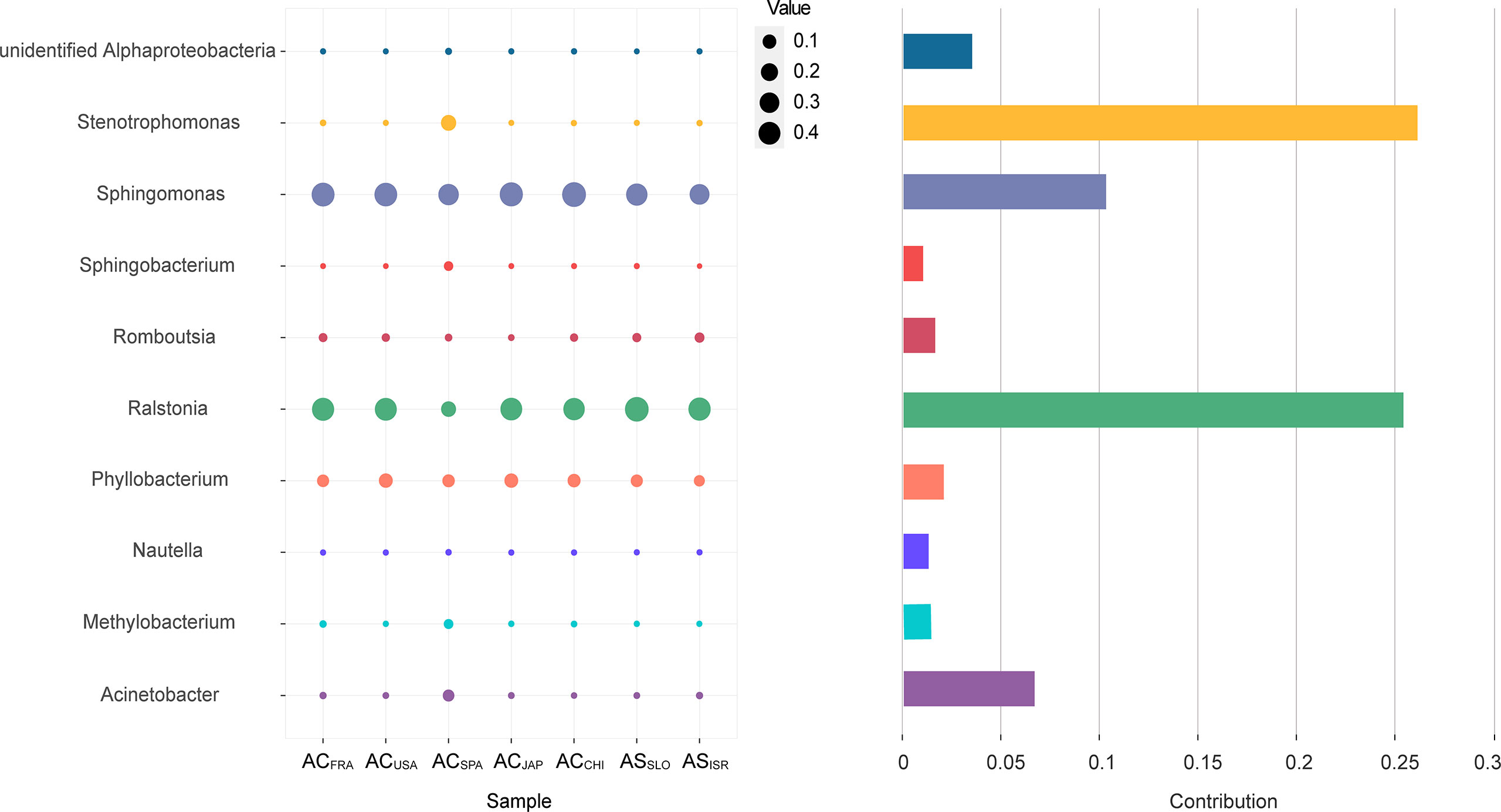
Figure 4 The top 10 genera contributing to the interspecific difference in the microbial community. The vertical axis represents the species. The horizontal axis represents the samples. The bubble size represents the relative sequence abundance of the species. Contribution represents the contribution of the genus in the difference between the A. coerulea and A. solida.
In total, 20 OTUs were found in all Aurelia polyp samples. These 20 OTUs were defined as the “core microbiome” of Aurelia polyps, accounting for 57.19% (ACSPA)–96.86% (ACCHI) of the total microbial richness (Figure 5). The genera Sphingomonas, Ralstonia, and Phyllobacterium were prominent in the core microbiome of the Aurelia polyps, accounting for 79.76% (ASISR)–92.64% (ASSLO) and 57.19% (ACSPA)–96.13% (ACCHI) of the total core microbial richness in A. coerulea polyps and A. solida polyps, respectively (Figure 5). Notably, 6 OTU members of the core microbiome had significantly different abundances between A. coerulea and A. solida polyps: OTU_2 (Mann–Whitney U test, p = 0.034), OTU_1043 (Mann–Whitney U test, p = 0.026), and OTU_1244 (Mann–Whitney U test, p = 0.037) all belonging to the genus Ralstonia, were significantly more abundant in A. solida polyps than in A. coerulea polyps. However, OTU_3 (genus Phyllobacterium, t test, p = 0.026), OTU_1100 (genus Sphingomonas, t test, p = 0.026), and OTU_1287 (genus Sphingomonas, Mann–Whitney U test, p = 0.011) were significantly more abundant in A. coerulea polyps than in A. solida polyps.
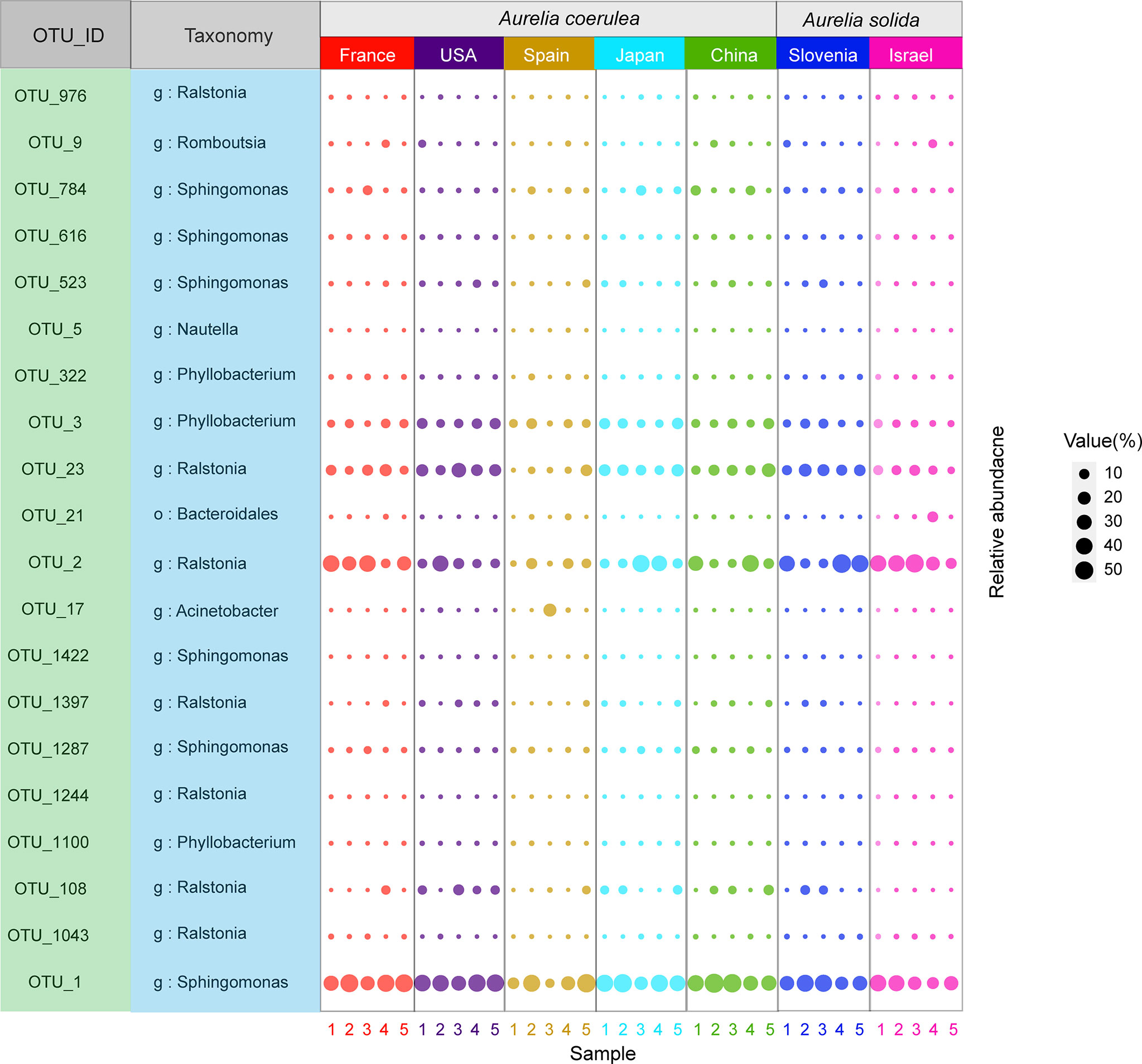
Figure 5 Bubble plot of the core microbiome of Aurelia polyps, with bubble size indicating the relative sequence abundance of bacterial taxa.
Ecological functional annotation of polyp-associated microbial communities was conducted based on the FAPROTAX database. A total of 1,247 functional assignments for 8, 326 OTUs were obtained. OTUs without any functional annotation were excluded from the analysis. Functional group abundances in each sample were calculated as the cumulative relative sequence abundance of OTUs assigned to each functional group after normalizing by the cumulative abundances of OTUs associated with at least one function. Generally, chemoheterotrophy (42.7~70.0% of total OTUs) and anaerobic chemoheterotrophy (36.8~65.5% of total OTUs) were the primary functions in both A. coerulea and A. solida polyps, followed by functions related to the N cycle, such as nitrate reduction, nitrogen respiration, and denitrification (Figure 6). Furthermore, microbial communities associated with A. coerulea polyps had significantly higher abundances of chemoheterotrophic bacterial taxa than those associated with A. solida polyps (Mann–Whitney U test, p = 0.004).
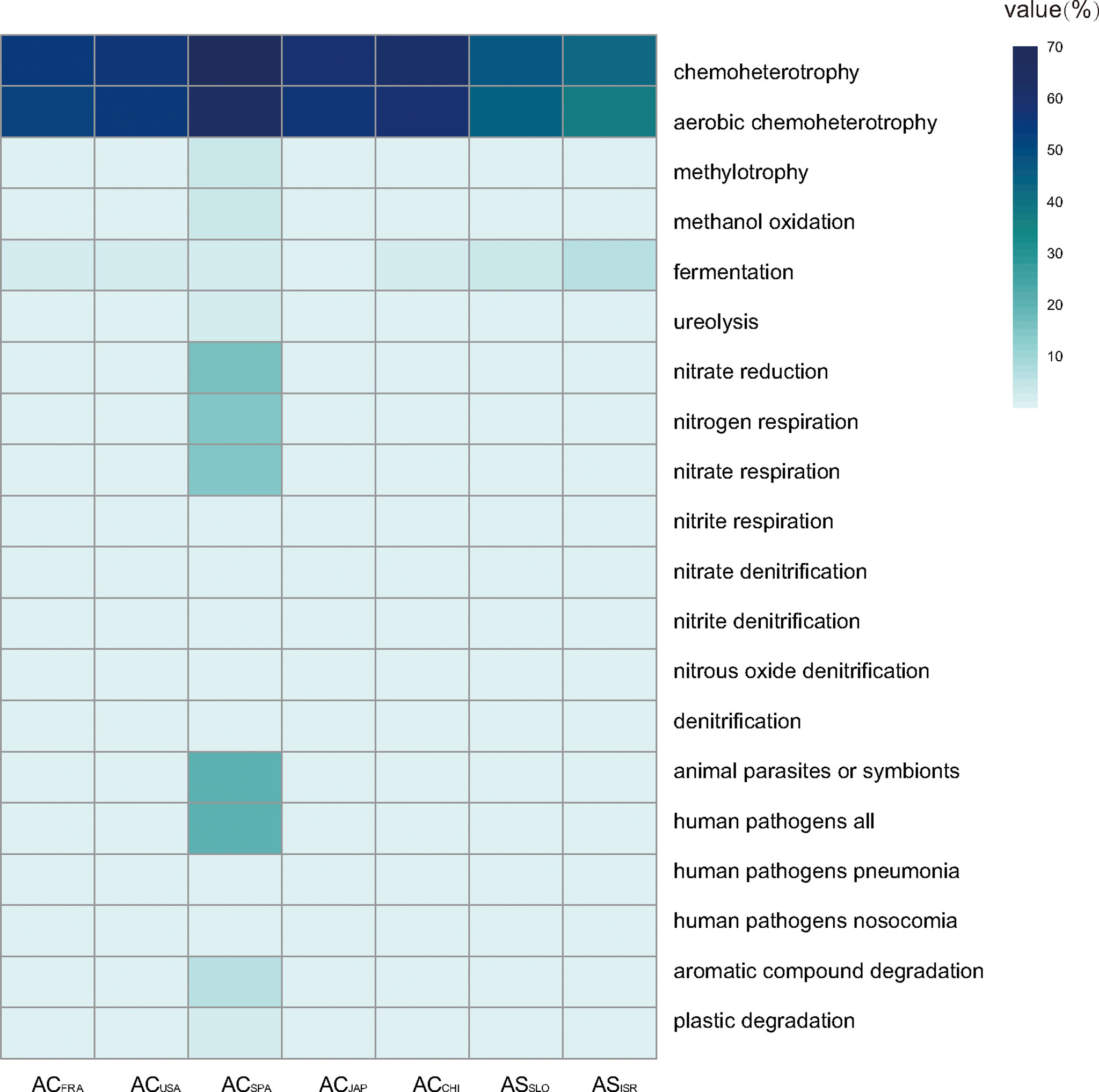
Figure 6 Heatmap matrix on the top 20 functional groups with the highest cumulative OTU relative sequence abundance in Aurelia polyp samples based on analysis of FAPROTAX.
Bacterial communities associated with hosts are shaped by host selection and environmental conditions (Gould et al., 2018). Previous researchers have highlighted environmental factors shaping the structure of microbial communities in hosts such as coral (Zhang et al., 2014; Osman et al., 2020), sponge (Zhang et al., 2014; Easson et al., 2020), and sea anemone (Mortzfeld et al., 2016; Morelan et al., 2019). In this study, despite long-term cultivation in the same environment and feed, we found interspecific variation in beta diversity among Aurelia polyps and intraspecific similarity among Aurelia polyps. Therefore, our results suggest that the genotype of Aurelia polyps is an important factor in the structure of the symbiotic bacterial communities. Weiland-Bräuer et al. concluded that there were large differences in the microbial compositions of Aurelia polyps between North Sea/Roscoff and Baltic Sea subpopulations, similar to the present study (Weiland-Bräuer et al., 2015).
Animal hosts are known to be able to modulate their associated microbiome in terms of genotype regulation, such as via genetic expression or innate immune response activation (Borges, 2017). For example, in research using hydras as model animals, the variation in antimicrobial peptide genes in different species led to variation in the microbiomes among those species (Bosch, 2013). As part of the evolutionarily ancient marine phylum Cnidaria, Aurelia jellyfish may influence the structure of their microbiomes by producing various proteins or antimicrobial peptides via gene expression, which interferes with bacterial quorum sensing (selecting microbes from the environment or promoting the colonization and enrichment of targeted microbes) and inhibits bacterial colonization (Weiland-Bräuer et al., 2019). In addition, vertical transmission of bacterial communities from parents to offspring could be another reason for the differences between the bacterial communities of A. coerulea and A. solida polyps.
Hosts can conditionally shape the structure of microbial communities by interacting to maintain an overall stable state, especially cnidarians (Ainsworth et al., 2015). In the present study, the families Sphingomonadaceae (i.e., genera Sphingomonas), Rhizobiaceae (i.e., genera Phyllobacterium), and Burkholderiaceae (i.e., genera Ralstonia) were detected in all Aurelia polyp samples with high relative sequence abundances. Similarly, Weiland-Bräuer et al. proposed that the relative sequence abundance of Sphingomonadaceae was high in Aurelia collected in Roscoff, the North Sea, and the Baltic Sea throughout the life stages and decreased with the transition of Aurelia polyps to strobila to ephyra to juvenile medusae (Weiland-Bräuer et al., 2015). Peng et al. also detected a high abundance of Sphingomonadaceae and Burkholderiaceae in the associated bacterial community of wild Aurelia medusae (Peng et al., 2021). Several studies have suggested that cnidarian-associated bacterial communities are potentially involved in functional interactions and play a positive role in host-environment adaptation (Ziegler et al., 2017; Podell et al., 2020; Roach et al., 2020; Tong et al., 2020). Hence, the family Sphingomonadaceae may be a member of the core microbial communities associated with Aurelia populations worldwide. Further studies on the function of Sphingomonadaceae would help to gain insight into the impact of microorganisms on hosts.
Bacteria are functional components of most marine multicellular organisms, especially cnidarians (Morelan et al., 2019). Previous studies have agreed that the core microbiome was closely related to the health, growth, environmental adaptability, and production of the hosts (Bosch, 2013; Ainsworth et al., 2015; Brown et al., 2017; Weigel and Erwin, 2017; Weiland-Bräuer et al., 2020). In our study, a total of 20 core OTUs were identified in the bacterial communities of the scyphozoan body parts, predominantly the genera Sphingomonas, Ralstonia, and Phyllobacterium. The genus Sphingomonas (family Sphingomonadaceae), a dibenzofuran- and dibenzodioxin-degrading bacterium with potentially interesting properties for bioaugmentation of contaminated sites (Roggo et al., 2013), was the most abundant taxon in the microbiomes of all Aurelia polyps. Furthermore, Feng et al. (2017) identified that certain Sphingomonas spp. were potentially able to degrade chlorpyrifos, indicating that Sphingomonas could enhance the survival rate of Aurelia polyps in contaminated environments. The genus Phyllobacterium, which can produce exopolysaccharide (Li et al., 2017), was also dominant in Aurelia polyps. The exopolysaccharide could form protective barriers between cells and the environment, regulate cell growth and senescence, and affect cell division and differentiation (Flores-Felix et al., 2018), suggesting that the genus Phyllobacterium may be closely related to the transformation process between Aurelia life stages and host-environment adaptation.
Functional prediction is the first step in determining how microbiome biochemical processes affect the ecological functions of hosts (Ross et al., 2018). FAPROTAX is a promising tool for predicting ecologically relevant functions of bacterial and archaeal taxa derived from 16S rRNA amplicon sequencing (Louca et al., 2016). In this study, according to the FAPROTAX database, aerobic chemoheterotrophy in relation to C cycling was the primary function of Aurelia polyp-associated bacteria associated with numerous bacteria, such as Sphingomonadaceae and Microbacteriaceae. This demonstrated that Aurelia polyps were the main foundation of essential nutrients to support the microbial growth of the associated bacterial communities. Ross et al. (2018) reported that Aurelia medusae are potential bacterial vectors and may harm aquaculture activities, as their microbiomes harbor potential fish pathogens. Similarly, in this study, some potential animal pathogens (i.e., Coxiellaceae) or parasites (i.e., Bdellovibrionaceae and Haliangiaceae) were present in both A. coerulea and A. solida polyps. Moreover, nitrification and denitrification, two functions related to the N cycle that have been reported to be involved in waste removal in host coral (Beman et al., 2007), were potential functions that were abundant in both Aurelia polyp species. Hence, these bacteria may have similar N cycle-related functions in Aurelia polyps, assisting with the adaptation of polyps to nitrate stress in the ambient environment.
Each intraspecific group had unique bacterial taxa, which accounted for less than 1% of the relative sequence abundance. These unique bacteria are often overlooked because of their low abundance but may be critical to the functional maintenance of hosts (Shade et al., 2014). For example, Actinobacteria sp. and Ralstonia sp., located in zooxanthellae and coral intestinal epithelial cells, have low abundance and perform a vital role in the metabolism of the coral Acropora granulosa (Ainsworth et al., 2015). Unlike the relative stability of the core microbiome in Aurelia polyps (Figure 1; Table 2), the unique microbes were less controlled and more sensitive to environmental variation. We speculated that unique microbes were preserved by the hosts from the native environment and potentially contributed to the environmental resilience of Aurelia polyps. Further study on unique microbes could comprehensively elucidate the mechanism of host selection for microbes in the environment.
In this study, we investigated the bacterial communities associated with two moon jellyfish species (A. coerulea and A. solida) obtained from seven locations and incubated under the same environmental conditions. We found that the genera Sphingomonas, Phyllobacterium, and Ralstonia dominated the core microbial communities of the Aurelia polyps. These Aurelia-associated microbes may potentially play an important role in host’s fitness and host’s environmental adaptation by promoting nutrient uptake. Furthermore, the comparison of microbial communities from different Aurelia polyp populations revealed interspecific variation, indicating a correlation between the composition of the associated bacterial community and genetic background of Aurelia polyps.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/ PRJNA799918.
YL: Data analysis, writing - original draft, writing - review & editing. WH: writing - review & editing. SP: Data analysis. TS: Sample process. JZ: Conceptualization, funding acquisition. ZD: Conceptualization, investigation, data analysis, funding acquisition, writing - review & editing. All authors contributed to the article and approved the submitted version.
This research was supported by grants from the NSFC-Shandong Joint Fund (No. U2106208), National Natural Science Foundation of China (No. 41876138), the National Key Research and Development Program of China (2018YFC1406501), and the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDA23050301).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We wish to thank Marine Biology Station Piran, National Institute of Biology, for making the collection possible, and to Dr. Agustin Schiariti from the Instituto Nacional de Investigación y Desarrollo Pesquero for keeping the cultures and providing us with Aurelia polyps.
Adair K. L., Bost A., Bueno E., Kaunisto S., Kortet R., Peters-Schulze G., et al. (2020). Host Determinants of Among-Species Variation in Microbiome Composition in Drosophilid Flies. Isme J. 14, 217–229. doi: 10.1038/s41396-019-0532-7
Ainsworth T. D., Krause L., Bridge T., Torda G., Raina J. B., Zakrzewski M., et al. (2015). The Coral Core Microbiome Identifies Rare Bacterial Taxa as Ubiquitous Endosymbionts. Isme J. 9, 2261–2274. doi: 10.1038/ismej.2015.39
Beman J. M., Roberts K. J., Wegley L., Rohwer F., Francis C. A. (2007). Distribution and Diversity of Archaeal Ammonia Monooxygenase Genes Associated With Corals. Appl. Environ. Microb. 73, 5642–5647. doi: 10.1128/Aem.00461-07
Borges R. M. (2017). Co-Niche Construction Between Hosts and Symbionts: Ideas and Evidence. J. Genet. 96, 483–489. doi: 10.1007/s12041-017-0792-9
Bosch T. C. G. (2013). Cnidarian-Microbe Interactions and the Origin of Innate Immunity in Metazoans. Annu. Rev. Microbiol. 67, 499–518. doi: 10.1146/annurev-micro-092412-155626
Brown T., Otero C., Grajales A., Rodriguez E., Rodriguez-Lanetty M. (2017). Worldwide Exploration of the Microbiome Harbored by the Cnidarian Model, Exaiptasia Pallida (Agassiz in Verrill 1864) Indicates a Lack of Bacterial Association Specificity at a Lower Taxonomic Rank. PeerJ 5, e3235. doi: 10.7717/peerj.3235
Cheng Z. Q., Lei S. N., Li Y., Huang W., Ma R. Q., Xiong J., et al. (2020). Revealing the Variation and Stability of Bacterial Communities in Tomato Rhizosphere Microbiota. Microorganisms 8, 170. doi: 10.3390/microorganisms8020170
Chiaverano L. M., Graham W. M. (2017). Morphological Plasticity in Aurelia Polyps, With Subsequent Effects on Asexual Fecundity and Morphology of Young Medusae. Mar. Ecol. Prog. Ser. 582, 79–92. doi: 10.3354/meps12314
Costello E. K., Stagaman K., Dethlefsen L., Bohannan B. J. M., Relman D. A. (2012). The Application of Ecological Theory Toward an Understanding of the Human Microbiome. Science 336, 1255–1262. doi: 10.1126/science.1224203
Dietz M. W., Salles J. F., Hsu B. Y., Groothuis T. G. G., van der Velde M., Verkuil Y. I., et al. (2020). Prenatal Transfer of Gut Bacteria in Rock Pigeon. Microorganisms 8, 61. doi: 10.3390/microorganisms8010061
Dong Z. J., Wang L., Sun T. T., Liu Q. Q., Sun Y. F. (2018). Artificial Reefs for Sea Cucumber Aquaculture Confirmed as Settlement Substrates of the Moon Jellyfish Aurelia Coerulea. Hydrobiologia 818, 223–234. doi: 10.1007/s10750-018-3615-y
Easson C. G., Chaves-Fonnegra A., Thacker R. W., Lopez J. V. (2020). Host Population Genetics and Biogeography Structure the Microbiome of the Sponge Cliona Delitrix. Ecol. Evol. 10, 2007–2020. doi: 10.1002/ece3.6033
Edgar R. C. (2004). MUSCLE: Multiple Sequence Alignment With High Accuracy and High Throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Edgar R. C. (2013). UPARSE: Highly Accurate OTU Sequences From Microbial Amplicon Reads. Nat. Methods 10, 996. doi: 10.1038/Nmeth.2604
Edgar R. C., Haas B. J., Clemente J. C., Quince C., Knight R. (2011). UCHIME Improves Sensitivity and Speed of Chimera Detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Esser D., Lange J., Marinos G., Sieber M., Best L., Prasse D., et al. (2019). Functions of the Microbiota for the Physiology of Animal Metaorganisms. J. Innate Immun. 11, 393–404. doi: 10.1159/000495115
Feng F. Y., Ge J., Li Y. S., Cheng J. J., Zhong J. F., Yu X. Y. (2017). Isolation, Colonization, and Chlorpyrifos Degradation Mediation of the Endophytic Bacterium Sphingomonas Strain HJY in Chinese Chives (Allium Tuberosum). J. Agr. Food. Chem. 65, 1131–1138. doi: 10.1021/acs.jafc.6b05283
Flores-Felix J. D., Velazquez E., Garcia-Fraile P., Gonzalez-Andres F., Silva L. R., Rivas R. (2018). Rhizobium and Phyllobacterium Bacterial Inoculants Increase Bioactive Compounds and Quality of Strawberries Cultivated in Field Conditions. Food Res. Int. 111, 416–422. doi: 10.1016/j.foodres.2018.05.059
Gould A. L., Zhang V. V., Lamberti L., Jones E. W., Obadia B., Korasidis N., et al. (2018). Microbiome Interactions Shape Host Fitness. P. Natl. Acad. Sci. U. S. A. 115, E11951–E11960. doi: 10.1073/pnas.1809349115
Haas B. J., Gevers D., Earl A. M., Feldgarden M., Ward D. V., Giannoukos G., et al. (2011). Chimeric 16s rRNA Sequence Formation and Detection in Sanger and 454-Pyrosequenced PCR Amplicons. Genome Res. 21, 494–504. doi: 10.1101/gr.112730.110
Hugerth L. W., Wefer H. A., Lundin S., Jakobsson H. E., Lindberg M., Rodin S., et al. (2014). DegePrime, a Program for Degenerate Primer Design for Broad-Taxonomic-Range PCR in Microbial Ecology Studies. Appl. Environ. Microb. 80, 5116–5123. doi: 10.1128/Aem.01403-14
Kechin A., Boyarskikh U., Kel A., Filipenko M. (2017). CutPrimers: A New Tool for Accurate Cutting of Primers From Reads of Targeted Next Generation Sequencing. J. Comput. Biol. 24, 1138–1143. doi: 10.1089/cmb.2017.0096
Knoll A. H. (2015). Life on a Young Planet: The First Three Billion Years of Evolution on Earth (Princeton, NJ: Princeton University Press). doi: 10.1515/9781400866045
Kramar M. K., Tinta T., Lucic D., Malej A., Turk V. (2019). Bacteria Associated With Moon Jellyfish During Bloom and Post-Bloom Periods in the Gulf of Trieste (Northern Adriatic). PloS One 14, e0198056. doi: 10.1371/journal.pone.0198056
Liu M., Yu Z., Yu X. Q., Xue Y. Y., Huang B. Q., Yang J. (2017). Invasion by Cordgrass Increases Microbial Diversity and Alters Community Composition in a Mangrove Nature Reserve. Front. Microbiol. 8, 2503. doi: 10.3389/fmicb.2017.02503
Li Y. P., Zhang G. L., Du C. Y., Mou H. J., Cui J. F., Guan H. S., et al. (2017). Characterization of High Yield Exopolysaccharide Produced by Phyllobacterium Sp 921F Exhibiting Moisture Preserving Properties. Int. J. Biol. Macromol. 101, 562–568. doi: 10.1016/j.ijbiomac.2017.03.089
Louca S., Parfrey L. W., Doebeli M. (2016). Decoupling Function and Taxonomy in the Global Ocean Microbiome. Science 353, 1272–1277. doi: 10.1126/science.aaf4507
Lucas C. H. (2001). Reproduction and Life History Strategies of the Common Jellyfish, Aurelia Aurita, in Relation to Its Ambient Environment. Hydrobiologia 451, 229–246. doi: 10.1023/A:1011836326717
Martinson V. G., Douglas A. E., Jaenike J. (2017). Community Structure of the Gut Microbiota in Sympatric Species of Wild Drosophila. Ecol. Lett. 20, 629–639. doi: 10.1111/ele.12761
McFall-Ngai M., Hadfield M. G., Bosch T. C. G., Carey H. V., Domazet-Loso T., Douglas A. E., et al. (2013). Animals in a Bacterial World, a New Imperative for the Life Sciences. P. Natl. Acad. Sci. U. S. A. 110, 3229–3236. doi: 10.1073/pnas.1218525110
Morelan I. A., Gaulke C. A., Sharpton T. J., Thurber R. V., Denver D. R. (2019). Microbiome Variation in an Intertidal Sea Anemone Across Latitudes and Symbiotic States. Front. Mar. Sci. 6, 7. doi: 10.3389/fmars.2019.00007
Mortzfeld B. M., Urbanski S., Reitzel A. M., Kunzel S., Technau U., Fraune S. (2016). Response of Bacterial Colonization in Nematostella Vectensis to Development, Environment and Biogeography. Environ. Microbiol. 18, 1764–1781. doi: 10.1111/1462-2920.12926
Osman E. O., Suggett D. J., Voolstra C. R., Pettay D. T., Clark D. R., Pogoreutz C., et al. (2020). Coral Microbiome Composition Along the Northern Red Sea Suggests High Plasticity of Bacterial and Specificity of Endosymbiotic Dinoflagellate Communities. Microbiome 8, 8. doi: 10.1186/s40168-019-0776-5
Peng S. J., Hao W. J., Li Y. X., Wang L., Sun T. T., Zhao J. M., et al. (2021). Bacterial Communities Associated With Four Blooming Scyphozoan Jellyfish: Potential Species-Specific Consequences for Marine Organisms and Humans Health. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.647089
Podell S., Blanton J. M., Oliver A., Schorn M. A., Agarwal V., Biggs J. S., et al. (2020). A Genomic View of Trophic and Metabolic Diversity in Clade-Specific Lamellodysidea Sponge Microbiomes. Microbiome 8, 97. doi: 10.1186/s40168-020-00877-y
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Ren T. T., Boutin S., Humphries M. M., Dantzer B., Gorrell J. C., Coltman D. W., et al. (2017). Seasonal, Spatial, and Maternal Effects on Gut Microbiome in Wild Red Squirrels. Microbiome 5, 163. doi: 10.1186/s40168-017-0382-3
Roach T. N. F., Little M., Arts M. G. I., Huckeba J., Haas A. F., George E. E. (2020). A Multiomic Analysis of in Situ Coral-Turf Algal Interactions. P. Natl. Acad. Sci. U. S. A. 117, 13588–13595. doi: 10.1073/pnas.1915455117
Roggo C., Coronado E., Moreno-Forero S. K., Harshman K., Weber J., van der Meer J. R. (2013). Genome-Wide Transposon Insertion Scanning of Environmental Survival Functions in the Polycyclic Aromatic Hydrocarbon Degrading Bacterium Sphingomonas Wittichii RW1. Environ. Microbiol. 15, 2681–2695. doi: 10.1111/1462-2920.12125
Rook G., Bakhed F., Levin B. R., Mcfall-Ngai M. J., Mclean A. R. (2017). Evolution, Human-Microbe Interactions, and Life History Plasticity. Lancet 390, 521–530. doi: 10.1016/S0140-6736(17)30566-4
Ross A. A., Muller K. M., Weese J. S., Neufeld J. D. (2018). Comprehensive Skin Microbiome Analysis Reveals the Uniqueness of Human Skin and Evidence for Phylosymbiosis Within the Class Mammalia. P. Natl. Acad. Sci. U. S. A. 115, E5786–E5795. doi: 10.1073/pnas.1801302115
Shade A., Jones S. E., Caporaso J. G., Handelsman J., Knight R., Fierer N., et al. (2014). Conditionally Rare Taxa Disproportionately Contribute to Temporal Changes in Microbial Diversity. Mbio 5, e01371-14. doi: 10.1128/mBio.01371-14
Stevens J. L., Olson J. B. (2015). Bacterial Communities Associated With Lionfish in Their Native and Invaded Ranges. Mar. Ecol. Prog. Ser. 531, 253–262. doi: 10.3354/meps11323
Tinta T., Kogovsek T., Klun K., Malej A., Herndl G. J., Turk V. (2019). Jellyfish-Associated Microbiome in the Marine Environment: Exploring its Biotechnological Potential. Mar. Drugs 17, 94. doi: 10.3390/md17020094
Tong H. Y., Cai L., Zhou G. W., Zhang W. P., Huang H., Qian P. Y. (2020). Correlations Between Prokaryotic Microbes and Stress-Resistant Algae in Different Corals Subjected to Environmental Stress in Hong Kong. Front. Microbiol. 11, 686. doi: 10.3389/fmicb.2020.00686
Walke J. B., Becker M. H., Loftus S. C., House L. L., Cormier G., Jensen R. V., et al. (2014). Amphibian Skin may Select for Rare Environmental Microbes. Isme J. 8, 2207–2217. doi: 10.1038/ismej.2014.77
Weigel B. L., Erwin P. M. (2017). Effects of Reciprocal Transplantation on the Microbiome and Putative Nitrogen Cycling Functions of the Intertidal Sponge, Hymeniacidon Heliophila. Sci. Rep.-Uk 7, 43247. doi: 10.1038/srep43247
Weiland-Bräuer N., Fischer M. A., Pinnow N., Schmitz R. A. (2019). Potential Role of Host-Derived Quorum Quenching in Modulating Bacterial Colonization in the Moon Jellyfish Aurelia Aurita. Sci. Rep.-Uk 9, 34. doi: 10.1038/s41598-018-37321-z
Weiland-Bräuer N., Neulinger S. C., Pinnow N., Kunzel S., Baines J. F., Schmitz R. A. (2015). Composition of Bacterial Communities Associated With Aurelia Aurita Changes With Compartment, Life Stage, and Population. Appl. Environ. Microb. 81, 6038–6052. doi: 10.1128/Aem.01601-15
Weiland-Bräuer N., Pinnow N., Langfeldt D., Roik A., Gullert S., Chibani C. M., et al. (2020). The Native Microbiome is Crucial for Offspring Generation and Fitness of Aurelia Aurita. Mbio 11, e02336-20. doi: 10.1128/mBio.02336-20
Zhang X. N., Leadbetter J. R. (2012). Evidence for Cascades of Perturbation and Adaptation in the Metabolic Genes of Higher Termite Gut Symbionts. Mbio 3, e00223-12. doi: 10.1128/mBio.00223-12
Zhang F., Vicente J., Hill R. T. (2014). Temporal Changes in the Diazotrophic Bacterial Communities Associated With Caribbean Sponges Ircinia Stroblina and Mycale Laxissima. Front. Microbiol. 5, 561. doi: 10.3389/fmicb.2014.00561
Keywords: microbiome, Aurelia, host genotypes, high-throughput sequencing, jellyfish blooms
Citation: Li Y, Hao W, Peng S, Sun T, Zhao J and Dong Z (2022) Composition and Potential Functions of Bacterial Communities Associated With Aurelia Polyps. Front. Mar. Sci. 9:864872. doi: 10.3389/fmars.2022.864872
Received: 28 January 2022; Accepted: 14 April 2022;
Published: 11 May 2022.
Edited by:
Jie Li, Chinese Academy of Sciences, ChinaReviewed by:
Tinkara Tinta, National Institute of Biology, SloveniaCopyright © 2022 Li, Hao, Peng, Sun, Zhao and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhijun Dong, empkb25nQHlpYy5hYy5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.