 Feng Liu
Feng Liu James T. Melton III
James T. Melton III Hongshu Wang
Hongshu Wang Jing Wang
Jing Wang Juan M. Lopez-Bautista
Juan M. Lopez-Bautista- 1CAS Key Laboratory of Marine Ecology and Environmental Sciences, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China
- 2Marine Ecology and Environmental Science Laboratory, Pilot National Laboratory for Marine Science and Technology (Qingdao), Qingdao, China
- 3Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao, China
- 4Spelman College, Atlanta, GA, United States
- 5Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL, United States
To gain more insights into the evolution of mitochondrial genomes (mitogenomes or mtDNAs) in the green macroalgal genus Ulva (Ulvophyceae, Chlorophyta), we sequenced seven Ulva mitogenomes from six species as well as one Percursaria mitogenome as outgroup, and compared them with the available Ulva mtDNA data. Our comparative analyses unveiled many novel findings. First, the Ulva mitogenomes shared a total of 62 core genes including 29 protein-coding genes (PCGs), three ribosomal RNA genes (rRNAs), 26 transfer RNA genes (tRNAs), three conserved free-standing open reading frames (orfs), and one putative RNA subunit of RNase P (rnpB). The rrn5 gene previously unrecognized is present in all sequenced ulvalean mitogenomes, which is situated between trnG(ucc) and trnW(cca). Second, the evolution of tRNAs in Ulva mitogenomes is related to different processes, including duplication, transposition, remolding, degeneration, loss and recruitment of tRNAs. The duplication of three tRNAs, i.e., trnT1(ugu), trnI1(gau), and trnM2(cau), was observed in Ulva mitogenomes. Third, the DNA-directed RNA polymerases (rpos), belonging to single-subunit DNA-dependent RNA polymerase (ssRNAP) family, are common in ulvalean mitogenomes. A total of three full-length and 55 split rpos have been detected in these 33 ulvalean mitogenomes. Fourth, six types of group I/II introns are detected at 29 insertion sites which are related to seven host genes (atp1, cox1, cox2, nad3, nad5, rnl, and rns) in these ulvalean mitogenomes. One group IB intron, i.e., intron cox1-214 which carried a GIY-YIG homing endonuclease (GHE), was observed for the first time in Ulva organelle genomes. Finally, phylogenomic analyses based on mitogenome dataset showed that the Ulva was split into two sister clades, representing Ulva lineage I and II, which was consistent to the results based on plastid genome dataset. Our study provides more important findings to better understand the evolution of mitochondrial genome in green algae.
Introduction
The green macroalgal genus Ulva Linnaeus 1753 (Ulvophyceae, Chlorophyta) is the most speciose genus in the order Ulvales, and harbors at least 86 species accepted taxonomically all over the world (Guiry and Guiry, 2021). Ulva species are widely distributed in marine and estuarine environments, and some species could live in fresh water (Mareš et al., 2011; Rybak, 2016). Ulva species are known for their rapid, proliferous growth in eutrophic conditions, forming harmful macroalgal blooms known as green tides (Wang et al., 2019). For example, the green tides caused by Ulva prolifera have broken out continuously in the Yellow Sea of China for 15 years from 2007 to 2021 (Liu et al., 2013; Wang et al., 2021), which had serious negative impacts on the local economy and ecosystem (Ye et al., 2011; Wang et al., 2015). Morphological characteristics of Ulva species are highly variable at the intraspecific level, due to different environmental conditions (Blomster et al., 2002; Gao et al., 2016; Liu F. et al., 2020). Phylogenetic methods based on common DNA markers, e.g., the nuclear internal transcribed spacer (ITS) region (ITS1-5.8S-ITS2) and the chloroplast RUBISCO LSU (rbcL) gene, are more reliable for species identifications in Ulva (Hayden and Waaland, 2004; Hughey et al., 2019).
Recently, organelle genomes (mtDNAs and cpDNAs) as potential molecular markers have been proved to be a valuable tool as for phylogenetic and evolutionary studies to understand the molecular species concepts and species boundaries in the genus Ulva (Fort et al., 2020; Liu F. et al., 2020; Liu and Melton, 2021). Mitochondria, which evolved from a single endosymbiotic event involving an α-proteobacterium-like ancestor (Gray et al., 2001; Martin et al., 2015), apparently remained more faithful to their eukaryotic host compared with plastids which were acquired via primary or secondary or higher-order symbiotic events (Keeling, 2010). Compared with the common DNA markers, the mitochondrial genome (mitogenome or mtDNA) with rich genetic information can accurately reflect the evolutionary history of nuclear genome (Burger and Nedelcu, 2012), and systematically describe intraspecific and interspecific evolutionary relationships at various taxonomic levels (e.g., Joardar et al., 2012; Park et al., 2015).
Mitogenome data are currently growing at an accelerated pace using faster high-throughput sequencing technologies. Thus far, a total of 25 mitogenomes from 15 Ulva species have been sequenced and deposited in the GenBank database (Table 1). On the whole, the sequenced Ulva mitogenomes were highly conserved in content of core genes, gene order, and genome architecture at the intragenus level (e.g., Liu and Pang, 2016; Melton and Lopez-Bautista, 2016; Zhou et al., 2016a,b; Liu M. et al., 2020). Some important findings have been unveiled previously in the sequenced Ulva mitogenomes. The content of group I/II introns varied greatly at the intraspecific level (e.g., Ulva australis and Ulva compressa), due to the inconsistent dispersal or invasion of introns as self-splicing and mobile genetic elements (Liu et al., 2017; Liu F. et al., 2020). The frequent integration and rapid turnover of foreign DNA fragments which usually contain specific open reading frames (orfs) and transfer RNA genes (tRNAs) caused the variations in gene content at both intraspecific and interspecific levels (Liu F. et al., 2020). Many repeat sequences are scattered in Ulva mitogenomes and change rapidly even at the intraspecific level (Hanyuda et al., 2016; Cai et al., 2018a,b). Genome rearrangement causes changes in the distribution of core genes, from coding on one strand to two strands (Liu F. et al., 2020).
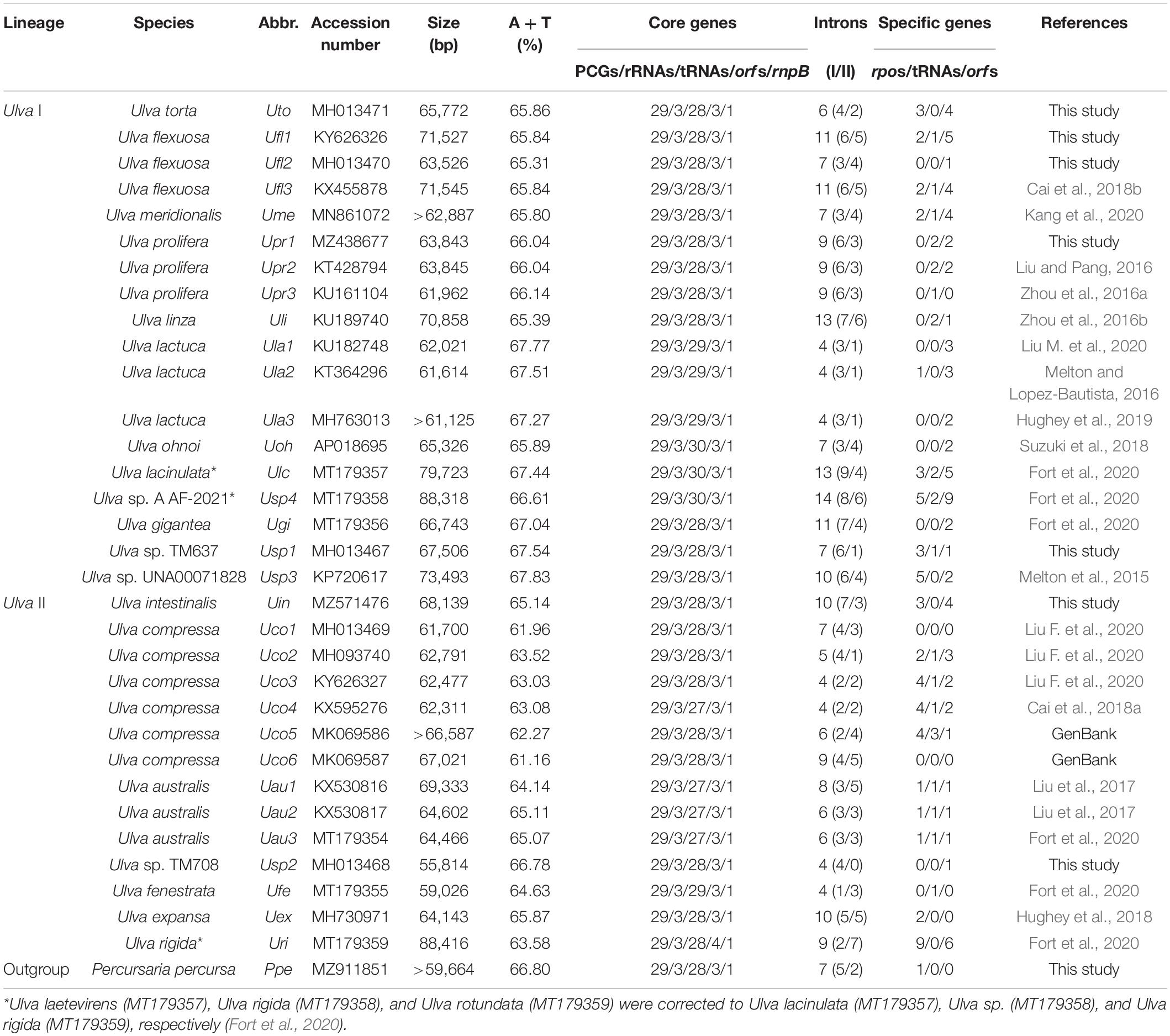
Table 1. General features of 33 ulvalean mitochondrial genomes from 19 Ulva species and one Percursaria species for comparative analysis.
With the accumulation of Ulva mitochondrial genome data, intraspecific and interspecific comparisons could be carried out more systematically. In the present study, we sequenced seven Ulva mitogenomes from six distinct species including Ulva flexuosa (Ufl1 and Ufl2), Ulva torta (Uto), Ulva prolifera (Upr1), Ulva intestinalis (Uin), Ulva sp. TM637 (Usp1), and Ulva sp. TM708 (Usp2), as well as one Percursaria mitogenome as an outgroup. These newly sequenced mitogenomes as well as the available Ulva mtDNA data from the GenBank database were comparatively analyzed to reveal more details of mitochondrial genome evolution.
Materials and Methods
Sample Collection and DNA Extraction
Three algal samples including Ulva flexuosa (Ufl1), Ulva prolifera (Upr1), and Ulva intestinalis (Uin) collected in China were transported to laboratory in coolers (5–8°C) after sampling (Supplementary Table 1). Fresh tissue from one individual thallus was used for DNA extraction using a Plant Genomic DNA Kit (Tiangen Biotech, Beijing, China) according to the manufacturer’s instructions. Four algal samples including Ulva torta (Uto), U. flexuosa (Ufl2), Ulva sp. TM637 (Usp1), and Ulva sp. TM708 (Usp2) from the United States were preserved in silica gel after collection and as herbarium vouchers, which were submitted to the University of Alabama Herbarium (UNA) (Supplementary Table 1). The algal thallus (UTEX LB 1423) of Percursaria percursa (Ppe) was from the UTEX Culture Collection of Algae1. Dried tissue from one individual thallus for United States samples was used to extract DNA with a Qiagen Plant DNA Extraction Kit (QIAGEN, Valencia, CA, United States). Species identification was performed based on phylogenetic analyses of two common marker datasets (the nuclear ITS region and the chloroplast rbcL gene) (Liu et al., 2013).
DNA Sequencing and Mitogenome Assembly
The concentration and quality of isolated DNA were measured with a NanoDrop ND-1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States). For the samples from China, the purified DNA was fragmented into 350 bp and used to construct short-insert libraries. The short fragments were sequenced using an Illumina HiSeq 4000 sequencing platform. For the samples from the United States, paired end reads (150 bp) were sequenced at Cold Spring Harbor Laboratory on an Illumina MiSeq platform. Poor quality sequences and sequencing adapters were removed using Trim Galore! v0.3.7. Eight ulvalean mitogenomes were constructed using a combination of de novo and reference-guided assemblies. The mitogenome of U. prolifera (KT428794) was used as the reference genome for assembly. Mitogenome assembly was done with both A5 (Tritt et al., 2012) and Geneious R7 (Kearse et al., 2012). The mtDNA assembly was examined using the MEM algorithm of BWA v0.7.17 (Li and Durbin, 2010), and the mutation sites were verified using VarScan v2.3.9 (Koboldt et al., 2009). Incomplete genomes were closed by iteratively mapping the trimmed reads on to the contig with the Geneious 7.1 software (Biomatters2).
Genome Annotation
Protein-coding genes (PCGs) were annotated by Open Reading Frame (ORF) Finder at the National Center for Biotechnology Information (NCBI) website3, DOGMA (Wyman et al., 2004) and ORF finder in the Geneious 7.1 software. Transfer RNA genes (tRNAs) were searched for by reconstructing their cloverleaf structures using the tRNA scan-SE 1.21 software with default parameters (Chan et al., 2021). The large and small subunit ribosomal RNA genes (rRNAs) were identified by comparing newly sequenced ulvalean mtDNAs with homologous rRNA genes from the known Ulva mtDNAs deposited in the GenBank database, respectively (Table 1). The 5S rRNA gene (rrn5) was found by the RNAweasel Tool4 and RNA Folding (Zuker, 2003). Intron insertion-sites were identified manually based on the alignments of nucleotide (nt) sequences for homologous genes with or without introns from these 33 ulvalean mitogenomes. The corresponding genes (atp1, cox1, cox2, nad3, nad5, rnl, and rns) in the U. compressa (KY626327) (Uco3) mitogenome were used as a reference (Liu F. et al., 2020). Intron name was defined as host gene plus insertion site. The free-standing and intronic orfs greater than 300 bp were found by Open Reading Frame Finder at the NCBI website. The class and core structure of all these introns were determined using the software RNAweasel and RNA Folding. We observed that some annotations were incomplete or incorrect in the Ulva mitogenome data deposited in the GenBank database. In order to ensure the accuracy of our comparative analysis, we re-annotated all of the deposited Ulva mitogenomes with the same method.
Phylogenetic Analysis of the Core RNA Polymerase Domains
The aa sequences of DNA-dependent RNA polymerase genes (rpos) were searched based on three full-length rpo genes found in Ulva mitogenomes as a query dataset through the BLAST program at the NCBI website5. A total of 45 closest Rpo proteins were obtained from the GenBank database for phylogenetic analysis. The core Rpo domains in these 48 Rpo proteins were determined by significant Pfam-A matches (Punta et al., 2012). Multiple sequence alignments of core Rpo domains were conducted using ClustalX 1.83 with the default settings (Thompson et al., 1997). To avoid phylogenetic artifacts caused by convergent base composition induced by synonymous substitutions (Cox et al., 2014), the ML phylogenetic tree was constructed based on aa sequences of core Rpo domains with 1,000 bootstrap replicates using MEGA 7.0 (Kumar et al., 2016). The phylogenetic relationships were inferred based on the Jones et al. w/freq. model (Jones et al., 1992). Initial tree(s) for the heuristic search were obtained by applying the Neighbor-Joining method to a matrix of pairwise distances estimated using a JTT model. There was a total of 902 positions in the final dataset of Rpos.
Phylogenetic Analysis of the Reverse Transcriptase Domains in Group IIA/IIB Introns
In group IIA/IIB introns of Ulva organelle genomes, the intron-encoded protein (IEP or intronic orf) was one reverse transcriptase/maturase (RTM). Three novel group IIA introns including intron atp1-1095, cox1-312, and nad5-1057 have been found for the first time in Ulva mitogenomes. To further understand the relationships between the new RTMs and those previously reported (Liu F. et al., 2020; Liu and Melton, 2021), we performed the phylogenetic analysis of the conserved reverse transcriptase (RT) domains which were determined by significant Pfam-A matches (Punta et al., 2012). The amino acid (aa) sequences of 94 RT domains (54 in mtDNAs and 40 in cpDNAs) from group IIA/IIB introns were subjected to concatenated alignments using ClustalX 1.83 with the default settings (Thompson et al., 1997). Maximum Likelihood (ML) phylogenetic tree was constructed for the RT dataset based on the Jones et al. w/freq. model (Jones et al., 1992) with 1,000 bootstrap replicates using MEGA 7.0 (Kumar et al., 2016). Initial tree(s) for the heuristic search were obtained by applying the Neighbor-Joining method to a matrix of pairwise distances estimated using a JTT model. There was a total of 330 positions in the final dataset of RT domains.
Comparative Genomic and Phylogenomic Analyses
Base composition of these 33 ulvalean mtDNAs was determined using MEGA 7.0 (Kumar et al., 2016). The nt sequences of 61 genes (including 29 PCGs, three rRNAs, 26 tRNAs, and three conserved orfs) and the aa sequences of 32 genes (including 29 PCGs and three orfs) were subjected to concatenated alignments using ClustalX 1.83 with the default settings, respectively (Thompson et al., 1997). For the nt sequence dataset of the 61 genes, the evolutionary history was inferred by using the ML method based on the Tamura-Nei model (Tamura and Nei, 1993). Initial tree(s) for the heuristic search were obtained by applying the Neighbor-Joining method to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach. For the aa sequence dataset of the 32 genes, the evolutionary history was inferred by using the ML method based on the Jones et al. w/freq. model (Jones et al., 1992). Initial tree(s) for the heuristic search were obtained by applying the Neighbor-Joining method to a matrix of pairwise distances estimated using a JTT model. There was a total of 39,070 and 10,491 positions in the final nt and aa datasets, respectively. Phylogenomic analysis was conducted with 1,000 bootstrap replicates using MEGA 7.0 (Kumar et al., 2016).
Results and Discussion
Mitogenome Features and Gene Content
Eight newly sequenced ulvalean mitochondrial genomes (seven Ulva mtDNAs and one Percursaria mtDNA) were acquired in this study and compared with 25 known Ulva mitogenomes to understand the evolution of Ulva mtDNAs. The Ulva mitogenomes ranged in size from the smallest one, 55,814 bp in Ulva sp. TM708 (Usp2), to the largest one, 88,416 bp in U. rigida (Uri) (Table 1). The Usp2 mtDNA is the second smallest mitochondrial genome in Ulvophyceae to date, which lies between the 45,971-bp mitogenome of Codium fragile (Bryopsidales) and the 56,761-bp mitogenome of Oltmannsiellopsis viridis (Oltmansiellopsidales) (Pombert et al., 2004, 2006). All ulvalean mitogenomes were biased toward A + T nucleotides and the A + T content ranged from 61.16% in U. compressa (Uco6) to 67.83% in Ulva sp. (Usp3).
These 33 ulvalean mitogenomes shared a total of 62 core genes including 29 PCGs, three rRNAs (rnl, rns and rrn5), 26 tRNAs, three conserved free-standing orfs, and one putative RNA subunit of RNase P (rnpB) (Table 2). The homologs of these three conserved orfs share identical positions in all sequenced ulvalean mtDNAs, but based on blastp and Pfam, we can not determine their function. In mitogenome of U. rigida (Uri), the conserved orf located between rps2 and trnL1(uaa) was split into two orfs, orf326 and orf199 (Figure 1). All these core genes are coded on the same strand, and have the identical gene order in these ulvalean mitogenomes with the exception of one U. compressa mitogenome (Uco1), in which a collinear block of eight genes (rps11-rps19-rps4-rpl16-trnR3-trnQ-trnE-trnS3) has been inverted (Liu F. et al., 2020).
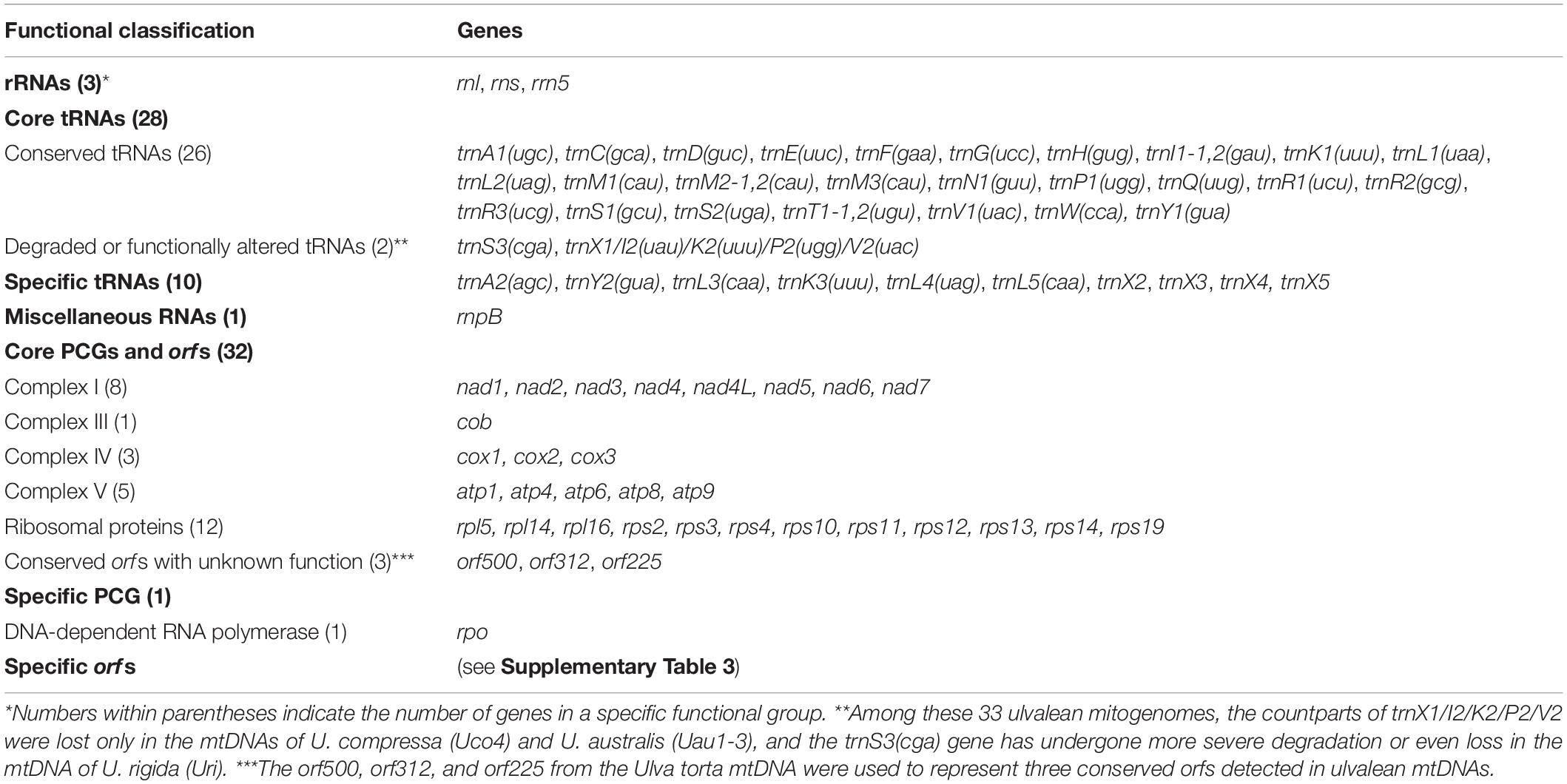
Table 2. Functional classification of genes (including orfs) identified among these 33 ulvalean mitochondrial genomes.

Figure 1. Comparison of genome organization and gene order of these 33 sequenced ulvalean mitochondrial genomes (A,B). The arrows indicated the direction of gene transcription. Different gene types were shown as filled boxes in different colors.
It is worth noting that the mitochondrial 5S rRNA gene (rrn5) previously unrecognized in Ulva mitogenomes was situated between trnG(ucc) and trnW(cca) in all sequenced ulvalean mitogenomes, while it was flanked by rnl and trnI(gau) in ulotrichalean mtDNAs. The sequences of domain β were conserved among the mitochondrial 5S rRNAs in Ulvales, but were different from the counterparts in Ulotrichales. Similar to that in mtDNAs of brown algae and some Ochrophyta lineages, the mitochondrial rrn5 gene in Ulvales was folded into a secondary structure by adopting a permuted triskelion shape (Valach et al., 2014; Figure 2A). We observed that the 5S rRNAs in ulvalean mtDNAs displayed a much larger structural variability than these in other lineages. The interior loop B in stem C is very asymmetric (Figure 2B), which makes it difficult to recognize the rrn5 genes. The sequences (approximately 35 bp) of domain γ including helix V, loop E, helix IV and loop D were highly conserved among rrns genes in not only Ulvales but also Ulotrichales. However, the homologous sequence was not detected in the mitogenomes of Bryopsidales and Oltmannsiellopsidales, as might be due to the sequence divergence, compositional bias and/or structural deviation of mitochondrial 5S rRNA genes (Valach et al., 2014).
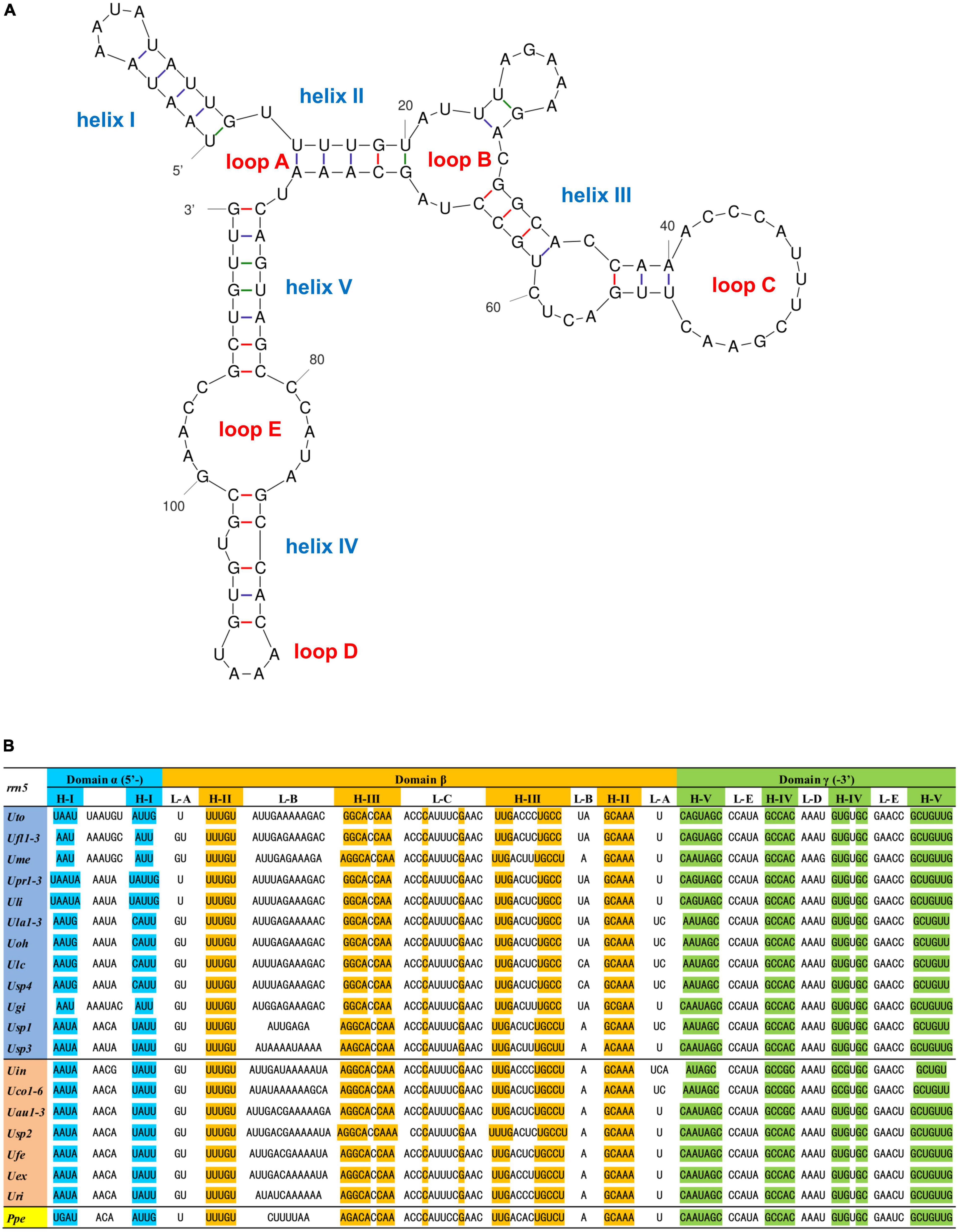
Figure 2. The permuted 5S rRNA genes (rrn5) detected in ulvalean mitochondrial genomes (A,B). (A) Potential secondary structure of the mitochondrial 5S rRNA gene in Ulva prolifera (Upr1-3). (B) Alignment of the mitochondrial 5S rRNA sequences in Ulvales. Shaded nucleotides indicated that bases could be paired.
The putative RNA subunit of mitochondrial RNase P (rnpB) could be detected in ulvalean mitogenomes using RNAweasel, which was situated in the latter part of the conserved orf (e.g., orf500 in U. torta) between rps2 and trnL1(uaa). The rnpB gene is encoding the RNA component of RNase P which participates in the generation of mature 5′-ends of tRNAs by cleaving the 5′-leader elements of tRNA precursors (Daoud et al., 2012; Shaukat et al., 2021). The highly variable sequence of rnpB makes it difficult to be found in mtDNAs. Thus far, the mitochondrial rnpB gene was found only in mtDNAs of the prasinophyte green alga Nephroselmis olivacea (Turmel et al., 1999), some ascomycete fungi (Seif et al., 2005) and jakobid protists (Lang et al., 1997; Burger et al., 2013).
Evolution of Transfer RNA Genes in Ulva Mitogenomes
The number of tRNA genes in Ulva mitogenomes showed slight differences at the interspecific level, ranging from 27 to 30 (Table 2). A total of 26 core tRNA genes are highly conserved in terms of both composition and structure, and shared by all sequenced ulvalean mitogenomes. The secondary structure of most tRNAs showed typical clover structures with the exception of the trnS3(cga) gene located between trnE(uuc) and nad5. It seems that trnS3(cga) has undergone insertion or deletion mutations in Ulva mtDNAs, causing the change of their secondary structures, or even severe degradation in the DHU arm and DHU loop, which led to the loss of this gene in the mtDNA of U. rigida (Uri). The trnX1 flanked by cox3 and rpl14 displayed variation in the anticodon loop sequences, which caused a marked change in function among different Ulva mtDNAs (Liu F. et al., 2020). This tRNA gene has been remolded into trnK2(uuu) in U. flexuosa (Ufl1-3) and U. meridionalis (Ume); or trnP2(ugg) in U. prolifera (Upr1-3) and U. linza (Uli); or trnI2(uau) in U. compressa (Uco1-3, 5, and 6); or trnV2(uac) in Ulva sp. TM708 (Usp2), U. fenestrata (Ufe), U. expansa (Uex), and U. rigida (Uri). This gene was lost in the mtDNAs of U. compressa (Uco4) and U. australis (Uau1-3) (Liu et al., 2017; Liu F. et al., 2020). Loss and structural changes of tRNAs at interspecific and intraspecific level reflected their rapid evolution in Ulva mtDNAs (Noutahi et al., 2019). The functions of the lost tRNA genes could be replaced by those from the nucleus or other organelles (Adams and Palmer, 2003; Pino et al., 2010).
We found that the tRNA duplication events occurred frequently in the evolution of Ulva mitogenomes. The mitogenomes of U. ohnoi (Uoh), U. lacinulata (Ulc), Ulva sp. (Usp4), and U. rigida (Uri) have two perfect copies of trnT1(ugu), both of which are located between rnl and cob (Figure 3A). Considering that Uoh, Ulc, and Usp4 have a relatively distant relationship with Uri, it seems that the duplication of trnT1(ugu) happened at least twice independently in Ulva mitogenomes. The trnI1-1(gau) located between trnK1(uuu) and trnA1(ugc) was duplicated in U. fenestrata (Ufe), and the trnI1-2(gau) has been translocated at the upstream of trnV2 (Figure 3B). Our previous study found that trnI1(gau) in U. australis (Uau1-3) was translocated from the intergenic region of trnK1-trnA1 to that of cox3-rpl14 (Liu et al., 2017), which was similar to that in Ulva sp. TM708 (Usp2). Based on their phylogenetic relationships, it is more reasonable that the transposition of trnI1(gau) occurred in common ancestor of the U. australis-Ulva sp.-U. fenestrata clade after its duplication, and then the previous trnI1-1(gau) was subsequently lost in mtDNAs of Uau1-3 and Usp2. In the U. lactuca-U. ohnoi-U. lacinulata-Ulva sp. clade, there are two copies of trnM2(cau) situated in different locations (Figure 3C), i.e., the intergenic regions of rns-trnY1 and cox3-trnX1, respectively, indicating that trnM2(cau) experienced the process of duplication and transposition in this clade.
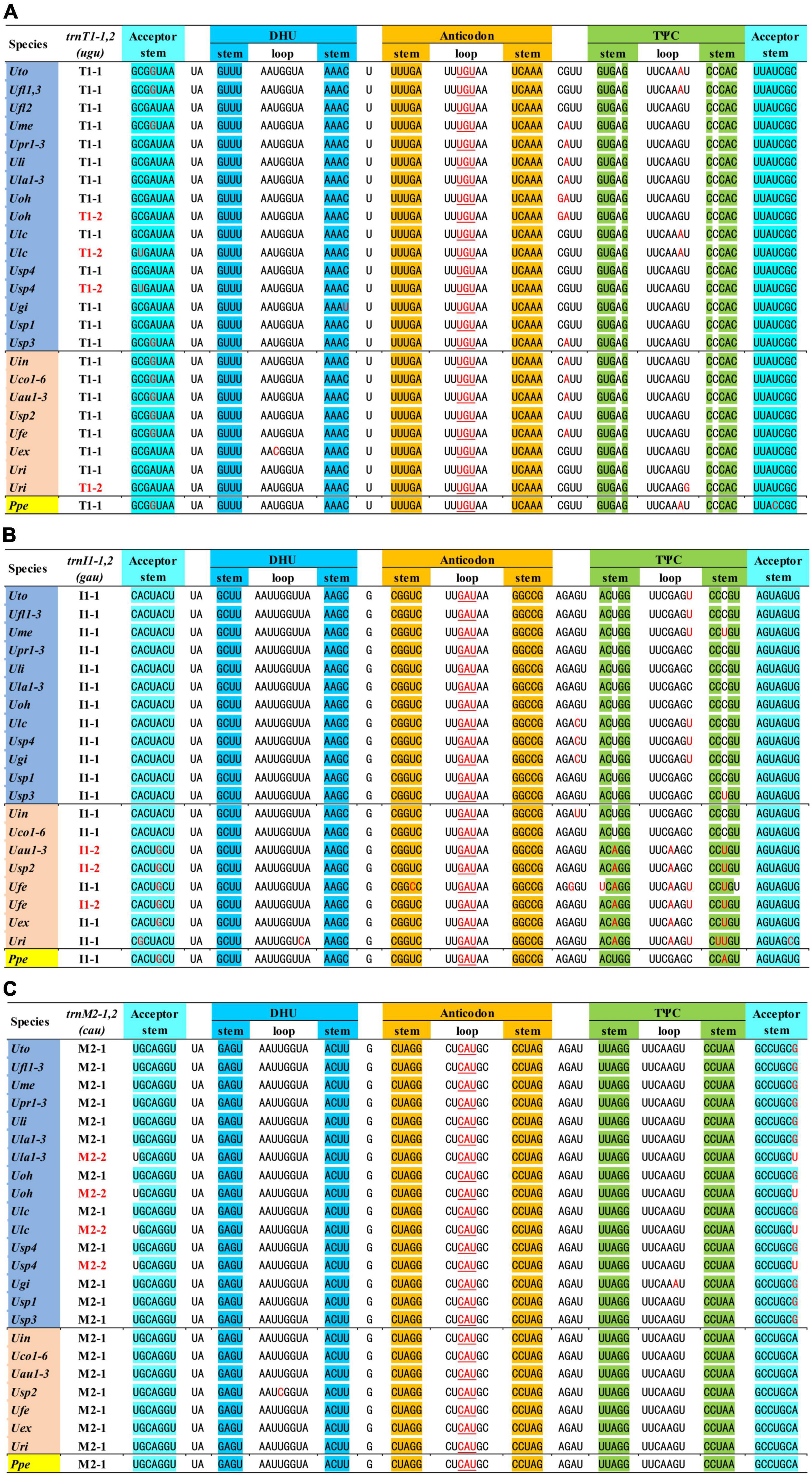
Figure 3. The aligned sequences of three tRNA genes with duplication mutation in Ulva mitogenomes. (A) trnT1(ugu). (B) trnI1(gau). (C) trnM2(cau). Shaded nucleotides indicated that bases could be paired.
The frequency of some specific tRNAs differed significantly at the interspecific level (Table 2). For example, trnY2(gua) was observed to exist only in the mtDNAs of U. prolifera (Upr1-3) and U. linza (Uli), and trnX2 was present only in mtDNAs of U. lacinulata (Ulc) and Ulva sp. (Usp4). These specific tRNAs are most likely recruited into the mitogenomes through the integration of exogenous DNA fragments (e.g., mitochondrial plasmids) (Handa, 2008). Overall, the evolution of tRNAs in Ulva mitogenomes is similar to that in other eukaryotic lineages (e.g., insects, fungi and higher plants), which is related to different evolutionary processes, including duplication, transposition, remolding, degeneration, loss and recruitment of tRNAs (e.g., Lang, 2014; Zhang et al., 2018; Li et al., 2021).
DNA-Dependent RNA Polymerase Genes and Specific Free-Standing Open Reading Frames
Among these specific free-standing orfs, a total of 58 orfs were annotated as the full-length or split DNA-dependent RNA polymerase genes (rpos) in these 33 ulvalean mitogenomes. Only three rpos were complete, including orf903 in U. rigida (Uri), orf956 in Ulva sp. TM637 (Usp1) and orf973 in Ulva sp. (Usp4), and the left 55 orfs ranging in size from 98 to 371 aa seem to be the different remnants after the degradation of the intact rpos (Figure 4). Obviously, the rapid mutation accumulation (e.g., insertion and deletion) led to the frameshift and destruction of rpos in these ulvalean mtDNAs. These rpos were detected to be located in nine specific intergenic regions including nad6-trnS1, trnV1-trnS2, trnM2-1-trnY1, trnY1-trnG, rnl-trnT1-1, trnK1-trnI1-1, trnA1-trnR1, cox3-trnX1, and trnE-trnS3, either in a forward or reverse order (Figure 4).
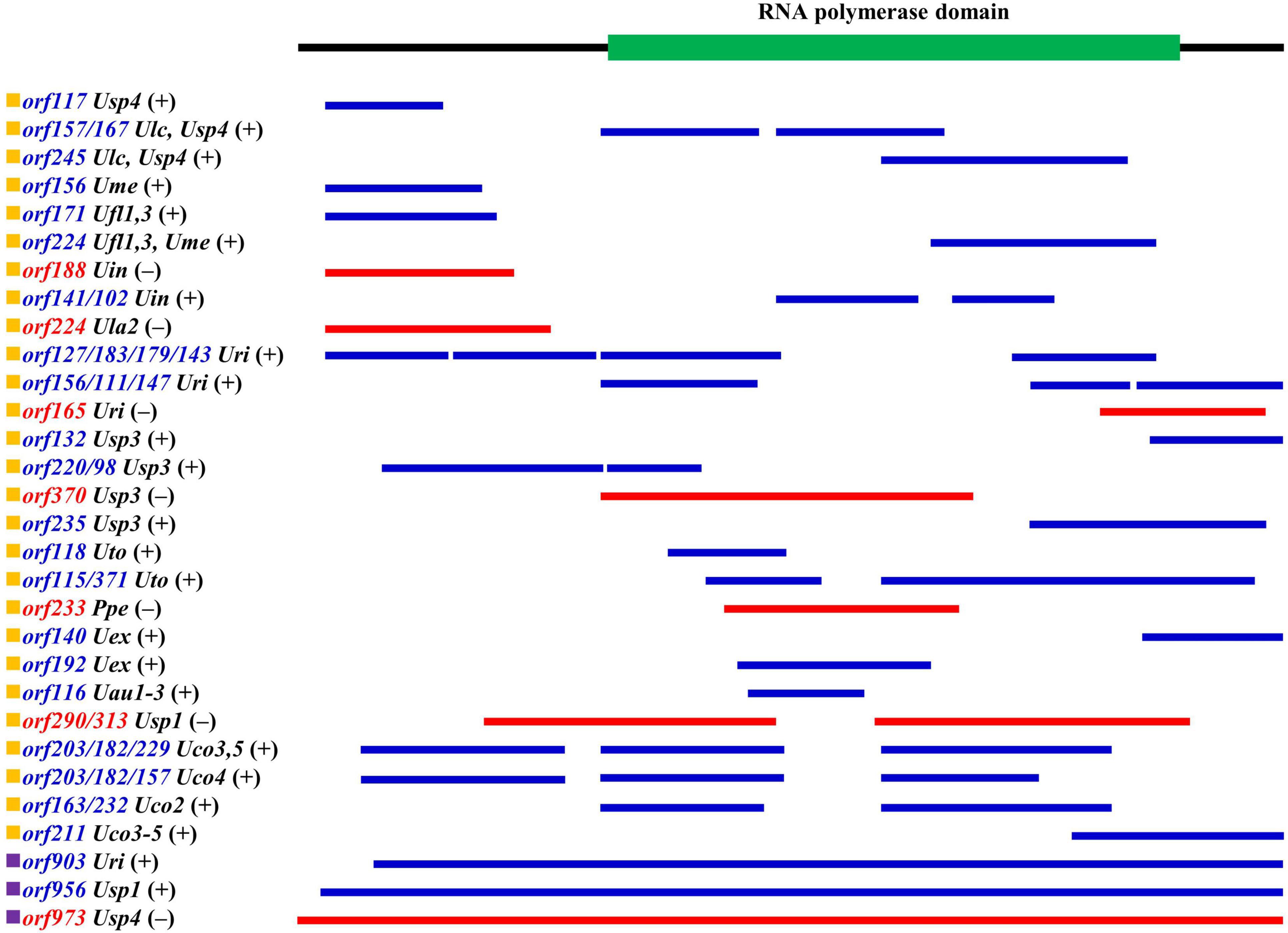
Figure 4. The schematic alignment of three full-length and 55 split RNA polymerase (Rpo) proteins in ulvalean mitogenomes. Proteins shown in blue are transcribed on the same chain as the core genes, and proteins shown in red are transcribed on the minus strand. The core Rpo domains which was identified in Rpo proteins by Pfam are displayed above the alignment.
The Ulva mitochondrial Rpos belong to T7-phage-type RNA polymerase which was a group of single-subunit Rpo (ss-Rpo) family. The ss-Rpo family is structurally and evolutionarily distinct from the multi-subunit family of Rpos (including bacterial and eukaryotic sub-families) (Cermakian et al., 1997). Ulva mitogenomes lack the ancestral a-proteobacterial polymerase genes, as was the same as almost all eukaryotic mitogenomes except jacobids (Yin et al., 2010; Peralta-Castro et al., 2020). Transcription of mitochondrial proteins involves nuclear or mitochondrial encoded single subunit T7-phage-type Rpos which probably has replaced the a-proteobacterial polymerase. In this regard, the mitogenomes in Ulva are completely different from chloroplast genomes which still retain the core subunits (rpoA, rpoB, rpoC1, and rpoC2) of the plastid-encoded Rpo derived from their cyanobacterial ancestor (Liu and Melton, 2021).
Based on the blastp searches, the entire T7-phage-type rpo genes closely related to those in Ulva mtDNAs could be found in the nuclear or mitochondrial genomes of eukaryotes (higher plants, brown algae and fungi), as well as genomes of prokaryotes (bacteria and viruses). A total of 45 Rpo proteins from the GenBank database were finally used for phylogenetic analysis. The ML tree inferring from the analysis of core Rpo domains in the 48 rpo genes showed that three Ulva full-length Rpos clustered together, representing a Chlorophyta-specific Rpo lineage (Figure 5). As found in higher plants and fungi (Clark-Walker, 1992; Handa, 2008; Warren et al., 2016), the observed Rpo genes in Ulva mtDNAs most likely come from mitochondrial linear plasmids which carry genes usually coding for an RNA/DNA polymerase or more often for both, as well as other ORFs (Cermakian et al., 1996). These linear plasmids were observed to be sometimes entirely integrated into the mitogenome. The mitogenome of brown alga Pylaiella littoralis contains a putative completely integrated linear plasmid which harbors an entire T7-phage-type Rpo gene (Rousvoal et al., 1998; Oudot-Le Secq et al., 2001).
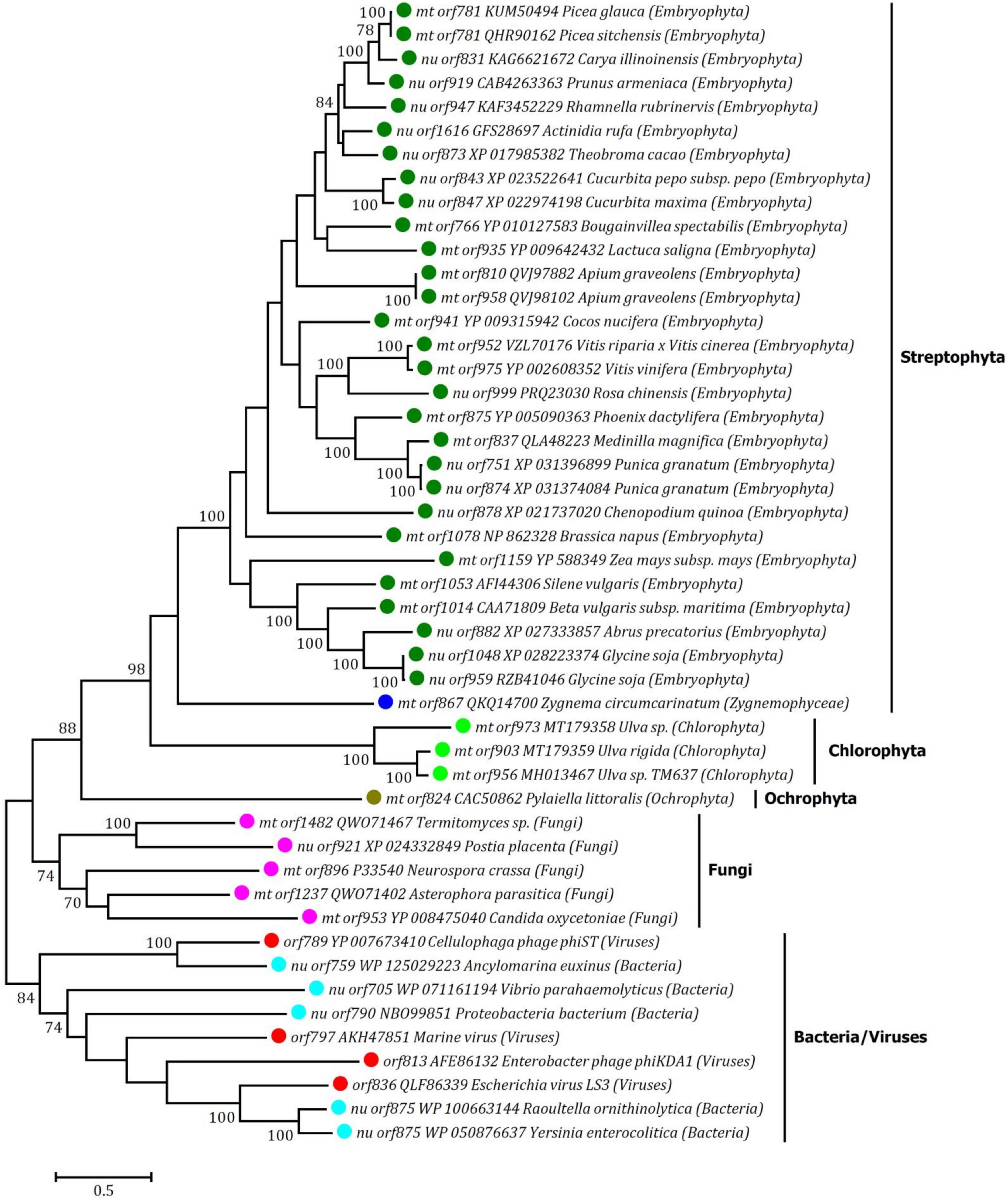
Figure 5. Phylogenetic tree based on Maximum Likelihood (ML) analysis of amino acid (aa) sequences of 48 RNA polymerase (Rpo) domains from Rpo proteins. The ML analysis was conducted with 1,000 bootstrap replicates using MEGA 7.0. The bootstrap support values greater than 70% were displayed at branches. Branch lengths were proportional to the amount of sequence change, which were indicated by the scale bar below the trees. The tree was rooted with Rpo proteins from bacteria and viruses as outgroups.
At the interspecific level, Ulvalean mitogenomes sometimes shared some specific homologous orfs which were located in different positions in these genomes (Supplementary Table 3). For example, among the 17 homologous orfs in group 1, which were from four intergenic regions (Supplementary Table 3), 13 orfs showed sequence similarity to the putative bacterial transposase with alignment scores of approximately 50%, and the left four orfs are more like remnants of their degradation. The orf124 which was only found in mtDNA of Ulva sp. TM708 (Usp2) contained the transferase hexapeptide (six repeats) of putative bacterial origin. The left specific orfs had little sequence similarity to any PCGs in the GenBank database based on the search of blastp.
All sequenced Ulva mitogenomes shared the same set of core genes, but they showed great variations in the content of specific genes including rpo genes, specific orfs (more than 100 codons) and recruited tRNAs (Tables 1, 2). These variations are mainly caused by different acquisitions of foreign DNA fragments from diverse sources including nucleus, plastids, bacteria, viruses, and mitochondrial plasmids (e.g., Wang et al., 2007; Gandini and Sanchez-Puerta, 2017; Liu F. et al., 2020). The integrations of foreign fragments in Ulva mtDNAs lead to the generation of large specific intergenic regions. Nine intergenic regions flanked by core genes vary greatly in size and sequence even at intraspecific levels, indicating that these hot spot regions are undergoing drastic dynamic changes which involve the recent capture of exogenous DNA fragments (Liu and Melton, 2021) and the frequent mutation or loss of intergenic regions which may occur through recombination-excision processes or by slipped-strand mispairing for small regions (Clark-Walker, 1992). Based on these hot spot regions, specific DNA markers could be designed and developed to specifically identify species or populations in the genus Ulva.
Diversity and Evolution of Ulva Mitochondrial Introns
Obviously, these sequenced ulvalean mitogenomes show great changes in intron content from four introns in Ulva sp. TM708 (Usp2), U. lactuca (Ula1-3), U. compressa (Uco3 and 4) and U. fenestrata (Ufe) to 14 in Ulva sp. (Usp4) (Table 3). Introns account for 8.77% of mitogenome in Usp2 to 27.80% in U. linza (Uli) in size. The reported ulvophycean mitogenomes contain a certain number of introns (Pombert et al., 2004, 2006; Melton et al., 2015; Turmel et al., 2016), which are not only ribozymes that catalyze their own splicing, but also retroelements that usually harbor an intron-encoded protein (IEP) and can insert themselves into new locations. Most of introns encoded a homing endonuclease or a reverse transcriptase/maturase (RTM) in Ulva mtDNAs, while IEPs in some introns degenerated and even completely lost (Liu et al., 2017; Liu F. et al., 2020).

Table 3. The updated information on insertion site, size and group of introns detected in these 33 ulvalean~mitogenomes.
From a comparative analysis of these intron locations, a total of 29 intron insertion sites were detected at seven mitochondrial genes (atp1, cox1, cox2, nad3, nad5, rnl, and rns) (Table 3). Three types of group I introns including group IB (complete, LHE), group IB (complete, GHE) and group ID, and three of group II introns including group IIA, group IIB and group II (LHE) were detected in these ulvalean mtDNAs. Five intron insertion sites were found for the first time in Ulva mitogenomes, two introns (intron cox1-214 and cox1-900) belonging to group IB and three (intron atp1-1095, cox1-312, and nad5-1057) belonging to group IIA. Intron DNA sequences at the same insertion site were homologous among these ulvalean mtDNAs, and these cognate introns shared the highly conserved RNA secondary structures of ribozyme components. Similar to that in chloroplast genomes, group IB intron was the most prevalent in ulvalean mitogenomes, and was found at ten insertion sites. The LHEs from different families of group IB introns displayed great genetic diversity (Liu and Melton, 2021), but their ribozyme components showed similarity in RNA secondary structures. One group ID intron (intron cox1-709) formerly detected to be present only in the U. prolifera-U. linza-U. flexuosa-Ulva sp. clade (Liu F. et al., 2020), were found to be scattered in mitogenomes of two Ulva lineages (I and II), indicating this group ID intron might be frequently homing or jumping.
In Ulva chloroplast genomes, intron-encoded homing endonucleases from three distinct families (LAGLIDADG, GIY-YIG, and H-N-H) have been found in group I introns (Wang et al., 2021), while all ORF-containing group I introns previously known in Ulva mitogenomes encode a LAGLIDADG homing endonuclease (LHE). In this study, we found that the IEPs from nine group IB (complete) introns were LAGLIDADG homing endonucleases (LHEs), while intron cox1-214 carried a GIY-YIG homing endonuclease (GHE). Group IB (complete,
GHE) intron was observed to be present only in the mtDNAs of two closely related species, U. lacinulata (Ulc) and Ulva sp. (Usp4), indicating that it is most probably the result of an independent insertion event occurring in their common progenitor. In addition, the GHE was also observed to be encoded in group I (derived, A) intron (e.g., intron psbB-489, psbB-772, psbC-882, and psbD-740) in Ulva chloroplast genomes.
Group IIA/IIB introns were present at eight and five insertion sites in these ulvalean mitogenomes, respectively, and both of them usually encoded an RTM (Dai et al., 2003; Liu F. et al., 2020). All of three newly discovered group II intron, i.e., intron atp1-1095, cox1-312, and nad5-1057, belonged to group IIA intron, indicating that group IIA intron is frequently involved in recent invasion or homing in Ulva mitogenomes. Based on phylogenetic analysis of reverse transcriptase (RT) domains in RTMs from Ulva mtDNAs and cpDNAs, RTs can be clearly clustered in two clades, representing group IIA and IIB lineages, respectively (Figure 6). RT family encoded in intron nad5-1057 had a close relationship with that in intron cox1-760, while both of RT families encoded in intron atp1-1095 and cox1-312 showed novel sequence characteristics, respectively, and represented two new intron families. The conserved YXRYADDXXXGXXG catalytic motif in RT segment 5 was shared by RT domains from group IIA introns (Supplementary Figure 1), which was much longer than those (RYADD) from group IIB introns (Supplementary Figure 2; Bonen and Vogel, 2001). All of group IIA introns (100%) contained an intact tripartite RTM gene in these ulvalean mitogenomes, while the RTMs in some group IIB introns have obviously degenerated or been completely lost, e.g., intron nad3-216 and rnl-1963 in U. gigantea (Ugi), and intron rns-780 in U. ohnoi (Uoh). Similar phenomena have been found in P. littoralis (Ikuta et al., 2008), but the mechanism by which the integrity of RTMs in group IIA introns is maintained successfully remains an open question. Group IIB introns with incomplete or missing RTMs should retain splicing competence to ensure that housekeeping genes function properly.
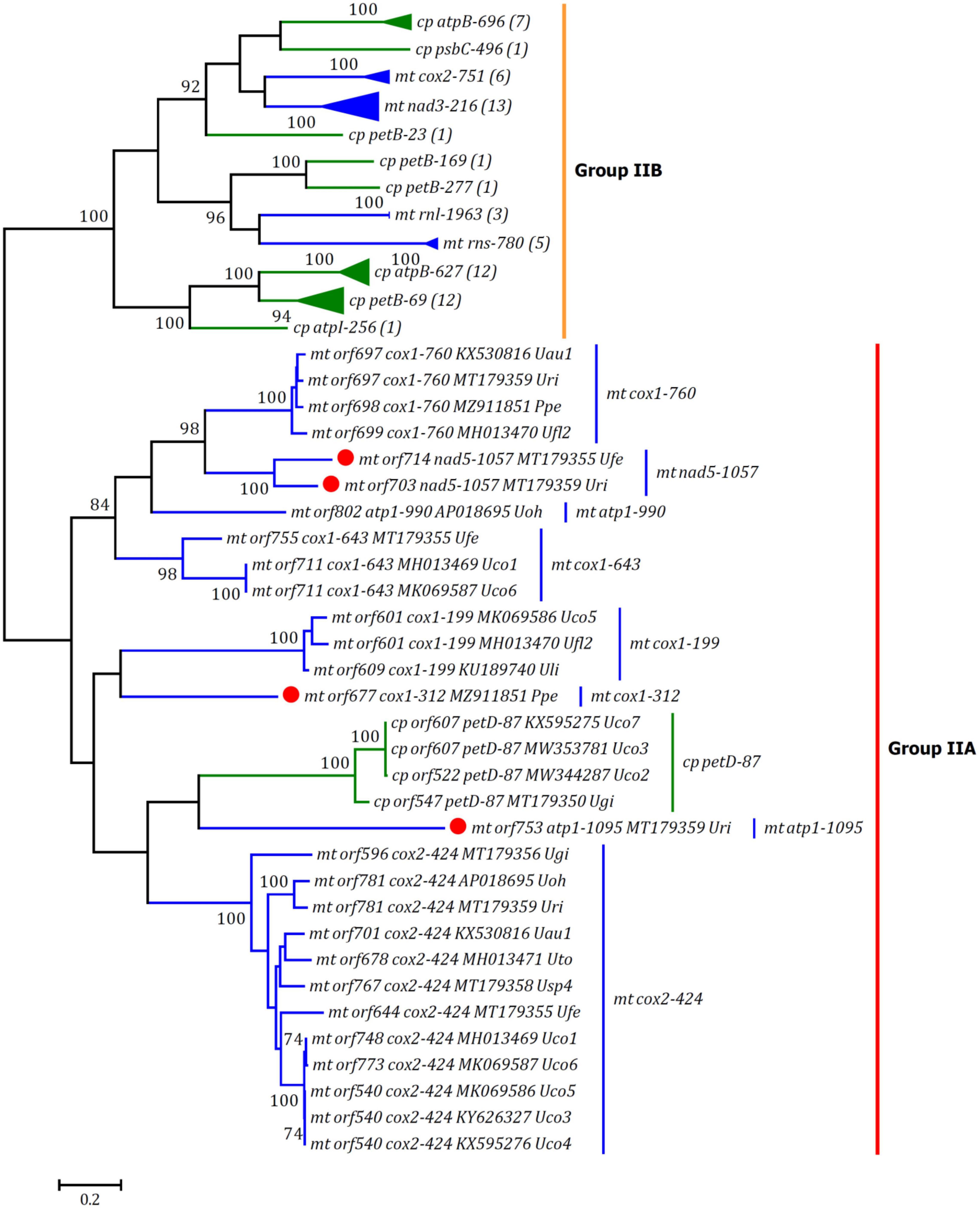
Figure 6. Unrooted phylogenetic tree based on Maximum Likelihood (ML) analysis of amino acid (aa) sequences of 94 reverse transcriptase (RT) domains (54 in mtDNAs and 40 in cpDNAs) from group IIA/IIB introns. RT domains from mtDNAs were shown in blue and RT domain from cpDNAs in green. Numbers within parentheses in group IIB clade indicated the number of RT domains found in each cognate intron family. The ML analysis was conducted with 1,000 bootstrap replicates using MEGA 7.0. The bootstrap support values greater than 70% were displayed at branches. Branch lengths were proportional to the amount of sequence change, which were indicated by the scale bar below the trees.
Interestingly, some group II introns in Ulva mitogenomes did not encode an RTM, but instead encoded an LHE. These mitochondrial LHEs in group II introns have close relationships with that in group IB introns (Liu and Melton, 2021). For a long time, it is considered that there is no genetic evolution relationship between group I and group II introns (Kelchner, 2002; Haugen et al., 2005). These findings indicated that there was a certain genetic relationship between group I and group II introns, at least between group IB (LHE) and group II (LHE) introns. Considering the great differences in ribozyme structure and splicing mechanism between group IB (LHE) and group II (LHE) introns (Seetharaman et al., 2006; Stoddard, 2011), the close genetic relationships of their IEPs are likely related to the evolution processes for both of introns. It appears that group I and II introns might recruit or employ the same IEP components (e.g., LHE) in their evolution, but these processes are still unclear.
Phylogenomic Analysis
Single DNA markers (e.g., ITS, rbcL, and 18S rDNA) or combined marker sequences contain limited phylogenetic signals to understand genetic relationships in Ulva species. Trees inferred from phylogenetic analysis based on these datasets have important implications for the phylogeny and classification of Ulva species (e.g., Hayden et al., 2003; Hayden and Waaland, 2004), but it is still restricted by the number of genetic difference signals in these DNA markers, especially in the differentiation of closely related species (e.g., U. linza vs. U. prolifera) or intraspecific relationships (Shimada et al., 2010; Liu et al., 2013). Phylogenomic trees based on mitogenome data and/or plastid genome data as well as organelle genome structure information could provide a more comprehensive understanding of evolutionary systematics and molecular species concepts in this morphologically simple group of macroalgae, due to their rich genetic information at the genome level (Liu F. et al., 2020; Liu and Melton, 2021).
The Ulva mitogenomes sequenced thus far represent a large portion of the genetic diversity at the intragenus level as they were sampled from each of the major clades in this genus. Phylogenomic analysis using maximum likelihood (ML) method based on the mitochondrial nt and aa sequence datasets showed that the 19 Ulva species were divided into two sister clades with strong support values (100%), representing Ulva lineage I and II, respectively (Figure 7). Many mutation sites in mtDNAs are completely consistent within each of these two lineages, but there are significant differences between these two lineages. For example, a three-base insertion occurred at the 5′ end of rps14 in all mtDNAs of Ulva lineage II, not in the mtDNAs of lineage I and P. percursa (Ppe) (Supplementary Figure 3), and a 15-base insertion was detected in rps2 of Ulva lineage II, not in that of lineage I (Supplementary Figure 4). In addition, some genomic features such as gene content, tRNA duplication, distribution of some introns, gene order, and genome rearrangement could partially support the current evolutionary relationships in lineage I and II. The trnY2(gua) was present only in the U. linza-U. prolifera (LP) clade, two copies of trnM2(cau) were present only in the U. lactuca-U. ohnoi-U. lacinulata-Ulva sp. clade, and the GHE-containing group IB introns were found only in the U. lacinulata-Ulva sp. clade.
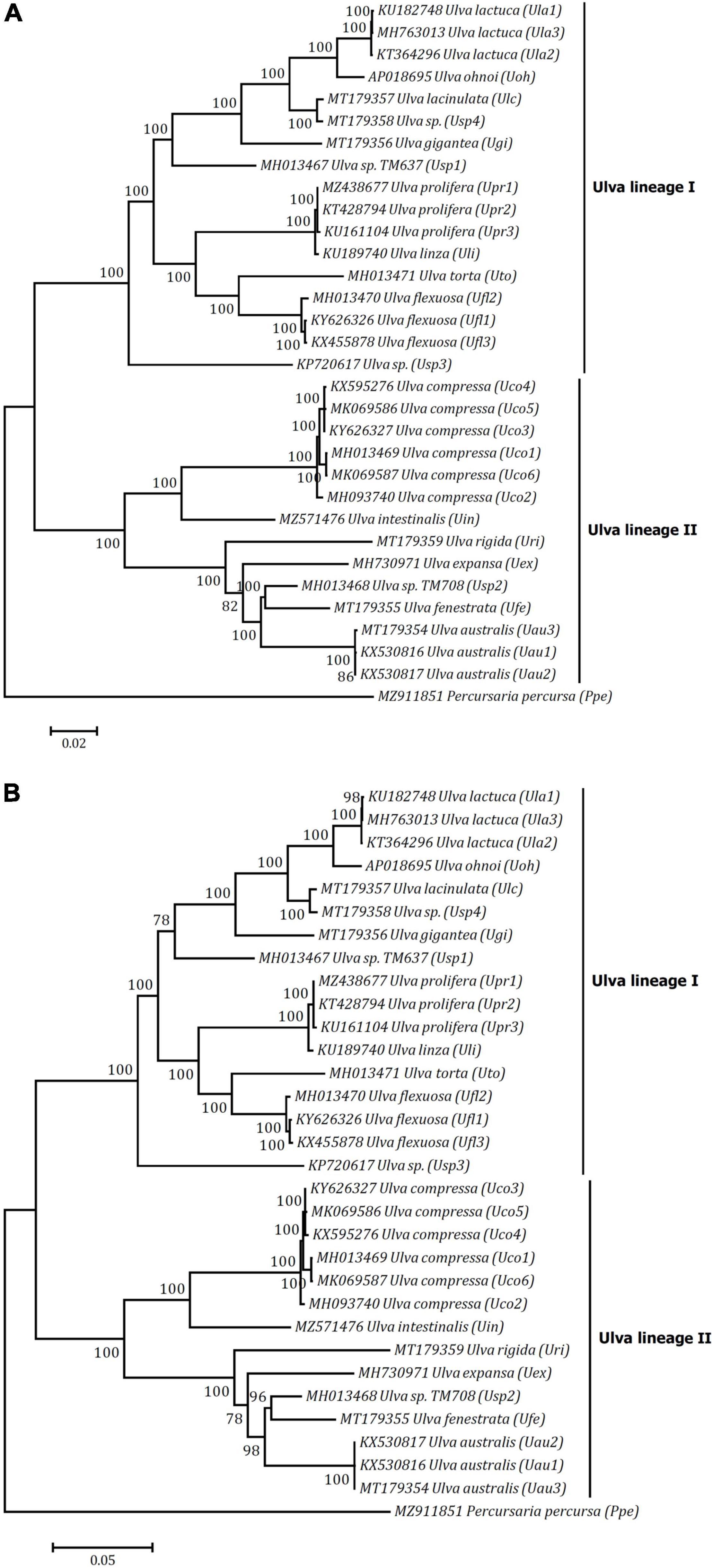
Figure 7. Phylogenomic trees based on Maximum Likelihood (ML) analysis of the nucleotide (nt) sequences of 61 genes (A) and the amino acid (aa) sequences of 32 genes (B) in 32 ulvalean mitogenomes. The mitogenome of Ulva meridionalis (Ume) was not included due to its incomplete cox1 gene. The ML analysis was conducted with 1,000 bootstrap replicates using MEGA 7.0. The bootstrap support values greater than 70% were displayed at branches. Branch lengths were proportional to the amount of sequence change, which were indicated by the scale bar below the trees. The tree was rooted with Percursaria percursa (Ppe) as an outgroup.
Relationships of taxa in the present tree are generally congruent with those based on plastid genome dataset (Liu and Melton, 2021; Wang et al., 2021). Minor topological differences between trees are most likely due to the different evolutionary rates between two gene datasets from mitogenomes and plastid genomes. Although two monophyletic clades in Ulva, namely lineage I and II, were well supported by genomic data (Figure 7), few morphological synapomorphies for these two clades were identified in this group of green macroalgae, due to their high degree of phenotypic plasticity caused by environment conditions (Hayden et al., 2003). Both lineage I and II consist of green seaweeds with multiple morphotypes including tubular and blade morphologies. Considering that the genus Ulva contains more than 80 known species worldwide as well as many cryptic species, further phylogenetics studies including more taxa is needed to clarify the infrageneric taxonomy and to understand the evolutionary relationships in Ulva.
Conclusion
Size variations of Ulva mitogenomes caused by integration of foreign DNA fragments, gain or loss of group I/II introns, and abundance of repetitive sequences have been well shown at the interspecific and intraspecific level in our previous studies (Liu et al., 2017; Liu F. et al., 2020). This study uncovered many novel important findings in the evolution of Ulva mitogenomes. These ulvalean mitogenomes shared a total of 62 core genes including 29 PCGs, three rRNAs, 26 tRNAs, three conserved orfs, and one putative rnpB. The rrn5 gene previously unrecognized is present in all ulvalean mitogenomes, and this gene is folded into a secondary structure by adopting a permuted triskelion shape. The evolution of tRNAs in Ulva mitogenomes is related to duplication, transposition, remolding, degeneration, loss or recruitment. The DNA-directed RNA polymerases (rpos) are common in ulvalean mitogenomes and a total of three full-length and 55 split rpos have been detected in these 33 ulvalean mitogenomes. The GHE-containing group IB introns were found for the first time in Ulva mtDNAs, which expand our understanding of intron diversity in Ulva mitogenomes. All of three newly discovered group II intron belonged to group IIA intron, indicating that group IIA intron is frequently involved in recent invasion or homing in Ulva mitogenomes. Phylogenomic analyses based on mitogenome dataset showed that the Ulva was split into two sister clades, representing Ulva lineage I and II. This study provides new insights on the genetics, systematics, and evolution of Ulva species. The comparative analysis of these ulvalean mitogenomes enriches our understanding of the mitogenome evolution in Ulvophyceae.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI (accessions: MH013471, KY626326, MH013470, MZ438677, KP720617, MZ571476, MH013468, and MZ911851).
Author Contributions
FL and JM designed the study and contributed equally to this work. FL, JM, HW, JW, and JL-B performed the experiments. FL performed the analysis and wrote the manuscript. All authors have read and approved the final version of the manuscript.
Funding
This work was financially supported by the Strategic Priority Research Program of Chinese Academy of Sciences (No. XDA23050302/XDA23050403), the Science and Technology Basic Resources Investigation Program of China (No. 2018FY100200), the Key Research Program of Frontier Sciences, Chinese Academy of Sciences (No. QYZDB-SSW-DQC023), the Major Scientific and Technological Innovation Project of Shandong Province (No. 2019JZZY020706), the National Natural Science Foundation of China (No. 41876165), the National Science Foundation (NSF) HBCU-UP (No. 1436759) for postdoctoral fellowship, and NSF through Assembling the Tree of Life for Green Algae GRAToL (DEB 1036495).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors wish to thank Wei Luan, Manman Liu, Yu Wang, and Nansheng Chen for their assistance in algal collection and data analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.850710/full#supplementary-material
Footnotes
- ^ https://utex.org/products/utex-lb-1423
- ^ http://www.geneious.com
- ^ https://www.ncbi.nlm.nih.gov/orffinder
- ^ https://megasun.bch.umontreal.ca/cgi-bin/RNAweasel/RNAweaselInterface.pl
- ^ http://www.ncbi.nlm.nih.gov/blast
References
Adams, K. L., and Palmer, J. D. (2003). Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol. Phylogenet. Evol. 29, 380–395. doi: 10.1016/s1055-7903(03)00194-5
Blomster, J., Bäck, S., Fewer, D. P., Kiirikki, M., Lehvo, A., Maggs, C. A., et al. (2002). Novel morphology in Enteromorpha (Ulvophyceae) forming green tides. Am. J. Bot. 89, 1756–1763. doi: 10.3732/ajb.89.11.1756
Bonen, L., and Vogel, J. (2001). The ins and outs of group II introns. Trends Genet. 17, 322–331. doi: 10.1016/s0168-9525(01)02324-1
Burger, G., Gray, M. W., Forget, L., and Lang, B. F. (2013). Strikingly bacteria-like and gene-rich mitochondrial genomes throughout jakobid protists. Genome Biol. Evol. 5, 418–438. doi: 10.1093/gbe/evt008
Burger, G., and Nedelcu, A. M. (2012). “Mitochondrial genomes of algae,” in Advances in Photosynthesis and Respiration Including Bioenergy and Related Processes: Genomics of Chloroplasts and Mitochondria, eds R. Bock and V. Knoop (Dordrecht: Springer), 125–157. doi: 10.1089/omi.2011.0074
Cai, C., Liu, F., Jiang, T., Wang, L., Jia, R., Zhou, L., et al. (2018a). Comparative study on mitogenomes of green tide algae. Genetica 146, 529–540. doi: 10.1007/s10709-018-0046-7
Cai, C., Wang, L., Jiang, T., Zhou, L., He, P., and Jiao, B. (2018b). The complete mitochondrial genomes of green tide algae Ulva flexuosa (Ulvophyceae, Chlorophyta). Conserv. Genet. Resour. 10, 415–418. doi: 10.1007/s12686-017-0838-6
Cermakian, N., Ikeda, T. M., Cedergren, R., and Gray, M. W. (1996). Sequences homologous to yeast mitochondrial and bacteriophage T3 and T7 RNA polymerases are widespread throughout the eukaryotic lineage. Nucleic Acids Res. 24, 648–654. doi: 10.1093/nar/24.4.648
Cermakian, N., Ikeda, T. M., Miramontes, P., Lang, B. F., Gray, M. W., and Cedergren, R. (1997). On the evolution of the single-subunit RNA polymerases. J. Mol. Evol. 45, 671–681. doi: 10.1007/pl00006271
Chan, P. P., Lin, B. Y., Mak, A. J., and Lowe, T. M. (2021). tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096. doi: 10.1093/nar/gkab688
Clark-Walker, G. D. (1992). Evolution of mitochondrial genomes in fungi. Int. Rev. Cytol. 141, 89–127. doi: 10.1016/s0074-7696(08)62064-1
Cox, C. J., Li, B., Foster, P. G., Embley, T. M., and Civán, P. (2014). Conflicting phylogenies for early land plants are caused by composition biases among synonymous substitutions. Syst. Biol. 63, 272–279. doi: 10.1093/sysbio/syt109
Dai, L., Toor, N., Olson, R., Keeping, A., and Zimmerly, S. (2003). Database for mobile group II introns. Nucleic Acids Res. 31, 424–426. doi: 10.1093/nar/gkg049
Daoud, R., Forget, L., and Lang, B. F. (2012). Yeast mitochondrial RNase P, RNase Z and the RNA degradosome are part of a stable supercomplex. Nucleic Acids Res. 40, 1728–1736. doi: 10.1093/nar/gkr941
Fort, A., McHale, M., Cascella, K., Potin, P., Usadel, B., Guiry, M. D., et al. (2020). Foliose Ulva species show considerable inter-specific genetic diversity, low intra-specific genetic variation, and the rare occurrence of inter-specific hybrids in the wild. J. Phycol. 57, 219–233. doi: 10.1111/jpy.13079
Gandini, C. L., and Sanchez-Puerta, M. V. (2017). Foreign plastid sequences in plant mitochondria are frequently acquired via mitochondrion-to-mitochondrion horizontal transfer. Sci. Rep. 7:43402. doi: 10.1038/srep43402
Gao, G., Zhong, Z. H., Zhou, X. H., and Xu, J. T. (2016). Changes in morphological plasticity of Ulva prolifera under different environmental conditions: a laboratory experiment. Harmful Algae 59, 51–58. doi: 10.1016/j.hal.2016.09.004
Gray, M. W., Burger, G., and Lang, B. F. (2001). The origin and early evolution of mitochondria. Genome Biol. 2:reviews1018.1–reviews1018.5. doi: 10.1186/gb-2001-2-6-reviews1018
Guiry, M. D., and Guiry, G. M. (2021). AlgaeBase. Available online at: http://www.algaebase.org (Accessed September 25, 2021)
Handa, H. (2008). Linear plasmids in plant mitochondria: peaceful coexistences or malicious invasions?. Mitochondrion 8, 15–25. doi: 10.1016/j.mito.2007.10.002
Hanyuda, T., Heesch, S., Nelson, W., Sutherland, J., Arai, S., Boo, S. M., et al. (2016). Genetic diversity and biogeography of native and introduced populations of Ulva pertusa (Ulvales. Chlorophyta). Phycol. Res. 64, 102–109. doi: 10.1111/pre.12123
Haugen, P., Simon, D. M., and Bhattacharya, D. (2005). The natural history of group I introns. Trends Genet. 21, 111–119. doi: 10.1016/j.tig.2004.12.007
Hayden, H. S., Blomster, J., Maggs, C. A., Silva, P. C., Stanhope, M. J., and Waaland, J. R. (2003). Linnaeus was right all along: Ulva and Enteromorpha are not distinct genera. Eur. J. Phycol. 38, 277–294.
Hayden, H. S., and Waaland, J. R. (2004). A molecular systematic study of Ulva (Ulvaceae, Ulvales) from the northeast Pacific. Phycologia 43, 364–382.
Hughey, J. R., Maggs, C. A., Mineur, F., Jarvis, C., Miller, K. A., Shabaka, S. H., et al. (2019). Genetic analysis of the Linnaean Ulva lactuca (Ulvales, Chlorophyta) holotype and related type specimens reveals name misapplications, unexpected origins, and new synonymies. J. Phycol. 55, 503–508. doi: 10.1111/jpy.12860
Hughey, J. R., Miller, K. A., and Gabrielson, P. W. (2018). Mitogenome analysis of a green tide forming Ulva from California, USA confirms its identity as Ulva expansa (Ulvaceae, Chlorophyta). Mitochondrial DNA B Resour. 3, 1302–1303. doi: 10.1080/23802359.2018.1535859
Ikuta, K., Kawai, H., Müller, D. G., and Ohama, T. (2008). Recurrent invasion of mitochondrial group II introns in specimens of Pylaiella littoralis (brown alga), collected worldwide. Curr. Genet. 53, 207–216. doi: 10.1007/s00294-008-0178-x
Joardar, V., Abrams, N. F., Hostetler, J., Paukstelis, P. J., Pakala, S., Pakala, S. B., et al. (2012). Sequencing of mitochondrial genomes of nine Aspergillus and Penicillium species identifies mobile introns and accessory genes as main sources of genome size variability. BMC Genomics 13:698. doi: 10.1186/1471-2164-13-698
Jones, D. T., Taylor, W. R., and Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282. doi: 10.1093/bioinformatics/8.3.275
Kang, X., Liu, J., Yang, X., Cui, J., Zhao, L., Wen, Q., et al. (2020). The complete mitochondrial genome of a green macroalgae species: Ulva meridionalis (Ulvales: Ulvaceae). Mitochondrial DNA B Resour. 5, 760–761. doi: 10.1080/23802359.2020.1715856
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Keeling, P. J. (2010). The endosymbiotic origin, diversification and fate of plastids. Philos. Trans. R. Soc. B. 365, 729–748. doi: 10.1098/rstb.2009.0103
Kelchner, S. A. (2002). Group II introns as phylogenetic tools: structure, function, and evolutionary constraints. Am. J. Bot. 89, 1651–1669. doi: 10.3732/ajb.89.10.1651
Koboldt, D. C., Chen, K., Wylie, T., Larson, D. E., McLellan, M. D., Mardis, E. R., et al. (2009). VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25, 2283–2285. doi: 10.1093/bioinformatics/btp373
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lang, B. F. (2014). “Mitochondrial genomes in fungi,” in Molecular Life Sciences, eds R. D. Wells, J. S. Bond, J. Klinman, B. S. S. Masters, and E. Bell (New York: Springer), 1–7.
Lang, B. F., Burger, G., O’Kelly, C. J., Cedergren, R., Golding, G. B., Lemieux, C., et al. (1997). An ancestral mitochondrial DNA resembling a eubacterial genome in miniature. Nature 387, 493–497. doi: 10.1038/387493a0
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, R., Lei, Z., Li, W., Zhang, W., and Zhou, C. (2021). Comparative mitogenomic analysis of Heptageniid mayflies (Insecta: Ephemeroptera): conserved intergenic spacer and tRNA gene duplication. Insects 12:170. doi: 10.3390/insects12020170
Liu, F., and Melton, J. T. (2021). Chloroplast genomes of the green-tide forming alga Ulva compressa: comparative chloroplast genomics in the genus Ulva (Ulvophyceae, Chlorophyta). Front. Mar. Sci. 8:668542. doi: 10.3389/fmars.2021.668542
Liu, F., Melton, J. T., and Bi, Y. (2017). Mitochondrial genomes of the green macroalga Ulva pertusa (Ulvophyceae, Chlorophyta): novel insights into the evolution of mitogenomes in the Ulvophyceae. J. Phycol. 53, 1010–1019. doi: 10.1111/jpy.12561
Liu, F., Melton, J. T., Lopez-Bautista, J. M., and Chen, N. (2020). Multiple intraspecific variations of mitochondrial genomes in the green-tide forming alga, Ulva compressa Linnaeus (Ulvophyceae, Chlorophyta). Front. Mar. Sci. 7:714. doi: 10.3389/fmars.2020.00714
Liu, F., and Pang, S. (2016). The mitochondrial genome of the bloom-forming green alga Ulva prolifera. Mitochondrial DNA A DNA Mapp. Seq. Anal. 27, 4530–4531. doi: 10.3109/19401736.2015.1101548
Liu, F., Pang, S., Chopin, T., Gao, S., Shan, T., Zhao, X., et al. (2013). Understanding the recurrent large-scale green tide in the Yellow Sea: temporal and spatial correlations between multiple geographical, aquacultural and biological factors. Mar. Environ. Res. 83, 38–47. doi: 10.1016/j.marenvres.2012.10.007
Liu, M., Liu, F., Chen, N., Melton, J. T., and Luo, M. (2020). Mitochondrial genomes and phylogenomic analysis of Ulva lactuca Linnaeus (Ulvophyceae, Chlorophyta). Mitochondrial DNA B. 5, 1638–1639. doi: 10.1080/23802359.2020.1745712
Mareš, J., Leskinen, E., Sitkowska, M., Skácelová, O., and Blomster, J. (2011). True identity of the European freshwater Ulva (Chlorophyta, Ulvophyceae) revealed by a combined molecular and morphological approach. J. Phycol. 47, 1177–1192. doi: 10.1111/j.1529-8817.2011.01048.x
Martin, W. F., Garg, S., and Zimorski, V. (2015). Endosymbiotic theories for eukaryote origin. Philos. Trans. R. Soc. B. 370:20140330. doi: 10.1098/rstb.2014.0330
Melton, J. T., Leliaert, F., Tronholm, A., and Lopez-Bautista, J. M. (2015). The complete chloroplast and mitochondrial genomes of the green macroalga Ulva sp. UNA00071828 (Ulvophyceae, Chlorophyta). PLoS One 10:e0121020. doi: 10.1371/journal.pone.0121020
Melton, J. T., and Lopez-Bautista, J. M. (2016). De novo assembly of the mitochondrial genome of Ulva fasciata Delile (Ulvophyceae, Chlorophyta), a distromatic blade-forming green macroalga. Mitochondrial DNA A DNA Mapp. Seq. Anal. 27, 3817–3819. doi: 10.3109/19401736.2015.1082095
Noutahi, E., Calderon, V., Blanchette, M., El-Mabrouk, N., and Lang, B. F. (2019). Rapid Genetic Code Evolution in Green Algal Mitochondrial Genomes. Mol. Biol. Evol. 36, 766–783. doi: 10.1093/molbev/msz016
Oudot-Le Secq, M. P., Fontaine, J. M., Rousvoal, S., Kloareg, B., and Loiseaux-De Goër, S. (2001). The complete sequence of a brown algal mitochondrial genome, the Ectocarpale Pylaiella littoralis (L.) Kjellm. J. Mol. Evol. 53, 80–88. doi: 10.1007/s002390010196
Park, S., Grewe, F., Zhu, A., Ruhlman, T. A., Sabir, J., Mower, J. P., et al. (2015). Dynamic evolution of Geranium mitochondrial genomes through multiple horizontal and intracellular gene transfers. New Phytol. 208, 570–583. doi: 10.1111/nph.13467
Peralta-Castro, A., García-Medel, P. L., Baruch-Torres, N., Trasviña-Arenas, C. H., Juarez-Quintero, V., Morales-Vazquez, C. M., et al. (2020). Plant Organellar DNA Polymerases Evolved Multifunctionality through the Acquisition of Novel Amino Acid Insertions. Genes 11:1370. doi: 10.3390/genes11111370
Pino, P., Aeby, E., Foth, B. J., Sheiner, L., Soldati, T., Schneider, A., et al. (2010). Mitochondrial translation in absence of local tRNA aminoacylation and methionyl tRNA Met formylation in Apicomplexa. Mol. Microbiol. 76, 706–718. doi: 10.1111/j.1365-2958.2010.07128.x
Pombert, J.-F., Beauchamp, P., Otis, C., Lemieux, C., and Turmel, M. (2006). The complete mitochondrial DNA sequence of the green alga Oltmannsiellopsis viridis: evolutionary trends of the mitochondrial genome in the Ulvophyceae. Curr. Genet. 50, 137–147. doi: 10.1007/s00294-006-0076-z
Pombert, J.-F., Otis, C., Lemieux, C., and Turmel, M. (2004). The complete mitochondrial DNA sequence of the green alga Pseudendoclonium akinetum (Ulvophyceae) highlights distinctive evolutionary trends in the Chlorophyta and suggests a sister-group relationship between the Ulvophyceae and Chlorophyceae. Mol. Biol. Evol. 21, 922–935. doi: 10.1093/molbev/msh099
Punta, M., Coggill, P. C., and Eberhardt, R. Y. (2012). The Pfam protein families database. Nucleic Acids Res. 40, D290–D301.
Rousvoal, S., Oudot, M.-P., Fontaine, J.-M., Kloareg, B., and Loiseaux-de Goër, S. (1998). Witnessing the evolution of transcription in mitochondria: the mitochondrial genome of the primitive brown alga Pylaiella littoralis (L.) Kjellm. encodes a T7-like RNA polymerase. J. Mol. Biol. 277, 1047–1057. doi: 10.1006/jmbi.1998.1679
Rybak, A. S. (2016). Ecological preferences of freshwater Ulva flexuosa (Ulvales; Ulvophyceae): development of macroalgal mats in a Tulce fishpond (Wielkopolska Region, Poland). Oceanol. Hydrobiol. Stud. 45, 100–111. doi: 10.1515/ohs-2016-0010
Seetharaman, M., Eldho, N. V., Padgett, R. A., and Dayie, K. T. (2006). Structure of a self-splicing group II intron catalytic effect or domain 5: parallels with spliceosomal U6 RNA. RNA 12, 235–247. doi: 10.1261/rna.2237806
Seif, E., Leigh, J., Liu, Y., Roewer, I., Forget, L., and Lang, B. F. (2005). Comparative mitochondrial genomics in zygomycetes: bacteria-like RNase P RNAs, mobile elements and a close source of the group I intron invasion in angiosperms. Nucleic Acids Res. 33, 734–744. doi: 10.1093/nar/gki199
Shaukat, A.-N., Kaliatsi, E. G., Skeparnias, I., and Stathopoulos, C. (2021). The Dynamic Network of RNP RNase P Subunits. Int. J. Mol. Sci. 22:10307. doi: 10.3390/ijms221910307
Shimada, S., Nagano, M., Hiraoka, M., Ichihara, K., Mineur, F., and Zhu, W. (2010). Phylogeographic analysis of the genus Ulva (Ulvales, Chlorophyta), including bloom sample in Qingdao, China. Coast. Mar. Sci. 34, 117–122.
Stoddard, B. L. (2011). Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure 19, 7–15. doi: 10.1016/j.str.2010.12.003
Suzuki, S., Yamaguchi, H., Hiraoka, M., and Kawachi, M. (2018). Mitochondrial and chloroplast genome sequences of Ulva ohnoi, a green-tide forming macroalga in the Southern coastal regions of Japan. Mitochondrial DNA B Resour. 3, 765–767. doi: 10.1080/23802359.2018.1483778
Tamura, K., and Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526. doi: 10.1093/oxfordjournals.molbev.a040023
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997). The ClustalX windows interface flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. doi: 10.1093/nar/25.24.4876
Tritt, A., Eisen, J. A., Facciotti, M. T., and Darling, A. E. (2012). An integrated pipeline for de novo assembly of microbial genomes. PLoS One 7:e42304. doi: 10.1371/journal.pone.0042304
Turmel, M., Lemieux, C., Burger, G., Lang, B. F., Otis, C., Plante, I., et al. (1999). The complete mitochondrial DNAsequences of Nephroselmis olivacea and Pedinomonas minor: two radically different evolutionary patterns within green algae. Plant Cell 11, 1717–1730. doi: 10.1105/tpc.11.9.1717
Turmel, M., Otis, C., and Lemieux, C. (2016). Mitochondrion-to-chloroplast DNA transfers and intragenomic proliferation of chloroplast group II introns in Gloeotilopsis green algae (Ulotrichales, Ulvophyceae). Genome Biol. Evol. 8, 2789–2805. doi: 10.1093/gbe/evw190
Valach, M., Burger, G., Gray, M. W., and Lang, B. F. (2014). Widespread occurrence of organelle genome-encoded 5S rRNAs including permuted molecules. Nucleic Acids Res. 42, 13764–13777. doi: 10.1093/nar/gku1266
Wang, D., Wu, Y. W., Shih, A. C. C., Wu, C. S., Wang, Y. N., and Chaw, S. M. (2007). Transfer of chloroplast genomic DNA to mitochondrial genome occurred at least 300 MYA. Mol. Biol. Evol. 24, 2040–2048. doi: 10.1093/molbev/msm133
Wang, H., Liu, F., Wang, J., and Chen, N. (2021). Phylogenomic analysis of the chloroplast genome of the green-tide forming macroalga Ulva intestinalis Linnaeus (Ulvophyceae, Chlorophyta). Mitochondrial DNA B 6, 3052–3054. doi: 10.1080/23802359.2021.1978889
Wang, Y., Liu, F., Liu, X., Shi, S., Bi, Y., and Moejes, F. W. (2019). Comparative transcriptome analysis of four co-occurring Ulva species for understanding the dominance of Ulva prolifera in the Yellow Sea green tides. J. Appl. Phycol. 31, 3303–3316.
Wang, Z., Xiao, J., Fan, S., Li, Y., Liu, X., and Liu, D. (2015). Who made the world’s largest green tide in China?—an integrated study on the initiation and early development of the green tide in Yellow Sea. Limnol. Oceanogr. 60, 1105–1117.
Warren, J. M., Simmons, M. P., Wu, Z., and Sloan, D. B. (2016). Linear Plasmids and the Rate of Sequence Evolution in Plant Mitochondrial Genomes. Genome Biol. Evol. 8, 364–374. doi: 10.1093/gbe/evw003
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Ye, N., Zhang, X., Mao, Y., Liang, C., Xu, D., Zou, J., et al. (2011). ‘Green tides’ are overwhelming the coastline of our blue planet: taking the world’s largest example. Ecol. Res. 26, 477–485. doi: 10.1007/s11284-011-0821-8
Yin, C., Richter, U., Börner, T., and Weihe, A. (2010). Evolution of plant phage-type RNA polymerases: the genome of the basal angiosperm Nuphar advena encodes two mitochondrial and one plastid phage-type RNA polymerases. BMC Evol. Biol. 10:379. doi: 10.1186/1471-2148-10-379
Zhang, L., Yu, D., Storey, K. B., Cheng, H., and Zhang, J. (2018). Higher tRNA gene duplication in mitogenomes of praying mantises (Dictyoptera, Mantodea) and the phylogeny within Mantodea. Int. J. Biol. Macromol. 111, 787–795. doi: 10.1016/j.ijbiomac.2018.01.016
Zhou, L., Wang, L., Zhang, J., Cai, C., and He, P. (2016a). Complete mitochondrial genome of Ulva linza, one of the causal species of green macroalgal blooms in Yellow Sea, China. Mitochondrial DNA B 1, 31–33. doi: 10.1080/23802359.2015.1137806
Zhou, L., Wang, L., Zhang, J., Cai, C., and He, P. (2016b). Complete mitochondrial genome of Ulva prolifera, the dominant species of green macroalgal blooms in Yellow Sea, China. Mitochondrial DNA B. 1, 76–78. doi: 10.1080/23802359.2015.1137831
Keywords: mitochondrial genome, Ulvophyceae, green algae, group I/II intron, DNA-directed RNA polymerase, mitochondrial 5S rRNA
Citation: Liu F, Melton JT III, Wang H, Wang J and Lopez-Bautista JM (2022) Understanding the Evolution of Mitochondrial Genomes in the Green Macroalgal Genus Ulva (Ulvophyceae, Chlorophyta). Front. Mar. Sci. 9:850710. doi: 10.3389/fmars.2022.850710
Received: 08 January 2022; Accepted: 09 February 2022;
Published: 16 March 2022.
Edited by:
Jin Liu, Peking University, ChinaCopyright © 2022 Liu, Melton, Wang, Wang and Lopez-Bautista. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Liu, bGl1ZmVuZ0BxZGlvLmFjLmNu; cHJjbGl1ZmVuZ0BzaW5hLmNu