 Chunqi Jiang
Chunqi Jiang Sayaka Mino
Sayaka Mino Tomoo Sawabe
Tomoo Sawabe
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 23 March 2022
Sec. Microbial Symbioses
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.844983
This article is part of the Research Topic Interaction between Marine Invertebrates and Symbiotic Microbes in a Changing Environment: Community Structure and Ecological Functions View all 10 articles
Vibrionaceae is one of the most diverse bacterial families and is currently classified into over 50 clades, some members of which play an important role in the symbiotic relationships with humans and animals. Halioticoli clade, which currently consists of 10 species: 8 species associated with the gut of abalone (symbiotic), 1 species (V. breoganii) from bivalves, and 1 species (V. ishigakensis) from subtropical seawater (planktonic). To accelerate studies in the evolution, ecogenomics, and biotechnology of Halioticoli clade species, the genomic backbones and pangenome analyses based on complete genome sequences are needed. Genome sizes of Halioticoli clade species ranged from 3.5 Mb to 4.8 Mb, with V. ishigakensis the biggest. The evolutionary relationships using multilocus sequence analysis based on eight housekeeping genes and 125 single-copy core genes revealed a division of five sub-clades in this clade; 1) V. breoganii, V. comitans, V. inusitatus and V. superstes, 2) V. ezurae, V. neonatus, and V. halioticoli, 3) V. rarus, 4) V. gallicus, and 5) V. ishigakensis. The pan-genomic analysis combined with function and metabolism estimations showed that the planktonic group (sub-clade 5) contained the greatest number of specific genes, and more genes responsible for carbohydrate metabolisms, especially the genes encoding D-galactonate degradation. These results demonstrated that the genome expanded by acquiring more abilities for utilizing various carbohydrates during the evolution from symbiotic to a planktonic lifestyle. Moreover, according to Carbohydrate-Active enZYmes (CAZy) profiling, genes encoding alginate degrading enzymes (aly), classified into PL6, PL7, PL15, and PL17 were common in the ten genomes, but sub-clade 1 had the most. Meanwhile, sub-clade 1and 5 also possessed abundant genes related to macroalgae substrates degradation (GHs), which are also responsible for the genome expansion of sub-clade 1 and 5.
Currently, over 190 species consisting of 9 genera have been accurately described in the family Vibrionaceae (Parte et al., 2020), which is one of the most diverse bacterial families, play an important role in geochemistry, pathogenicity, ecology, and systematics (Thompson et al., 2004; Gomez-Gil et al., 2014; Lee and Raghunath, 2018). Vibrios were further classified into 51 clades by multilocus sequence analysis (MLSA) in the most recent description of “Vibrio clade 3.0” (Jiang et al., 2022). Members of these clades have continued to be of great interest because of their symbiotic relationships with humans and animals, which can be referred to as parasitism, mutualism or commensalism (Moya et al., 2008). Representatively, Vibrio cholerae, the causative agent of the potent diarrheal disease cholera, is one of the most notorious human pathogens (Orata et al., 2015). The pathogenicity of members in the Cholerae clade has also been discovered although they were first described as non-pathogenic environmental strains (Kirchberger et al., 2014; Guardiola-Avila et al., 2021). On the other hand, the mutualism symbiosis between Hawaiian bobtail squid Euprymna scolopes and the bioluminescent bacteria Vibrio fischeri has been studied for decades because this model is uniquely suited to the investigation of symbiosis from both host and bacterial perspectives, putting the Vibrio-squid symbiosis at the forefront of host-microbe interactions (Nyholm and McFall-Ngai, 2004; Septer, 2019). Moreover, the recent rapid expansion in bacterial genome data has provided insights into the adaptive, diversifying and reductive evolutionary processes that occur in host-microbe interactions (Toft and Andersson, 2010). It has been reported about the general feature of genome-size reduction and AT content increase in endosymbiont genomes than free-living relatives, and the degree of them was related to the age of association (Wernegreen, 2002; Moya et al., 2008).
V. halioticoli was originally isolated from the gut of abalone Haliotis discus hannai as a non-motile alginolytic vibrio in 1998 (Sawabe et al., 1998). The dominance in the gut of Japanese abalone and the acetic acid production by V. halioticoli via fermentation of alginate, which is major components of ingested kelps, suggested a mutual relationship between V. halioticoli and abalones (Tanaka et al., 2002; Sawabe, 2006). The Halioticoli clade was first proposed in 2007 (Sawabe et al., 2007b), and most of the species have been discovered associated with abalone, particular in the guts (Table 1). Currently, ten species, V. breoganii, V. comitans, V. ezurae, V. gallicus, V. halioticoli, V. inusitatus, V. ishigakensis, V. neonatus, V. rarus, and V. superstes, have been described. Halioticoli clade species were discovered not only in Japanese abalone but also major abalone species outside Japan; V. neonatus (H. discus discus), V. ezurae (H. diversicolor diversicolor), V. comitans (H. gigantea), V. rarus (H. madaka) and V. inusitatus (H. rufescens) were isolated from the gut of Japanese abalone (Sawabe et al., 2004a; Sawabe et al., 2007a). V. superstes was isolated from the gut of Australian abalone H. laevigata and H. rubra (Hayashi et al., 2003), and V. gallicus was isolated from the gut of the French abalone H. tuberculate (Sawabe et al., 2004b). In 2009, the first non-abalone associated Halioticoli clade species, V. breoganii, was discovered from Spanish clams Ruditapes philippinarum and Ruditapes decussatus (Beaz Hidalgo et al., 2009). Nevertheless, draft genomes of V. halioticoli, V. superstes, were reported in 2014, but not much genome characterization has been completed yet. Recently, a genome of a reference strain of V. breoganii was completed, and the genome was rich in genes responsible for degrading macroalgal carbohydrates, which is likely to be characterized as a vegetarian vibrio compared to V. halioticoli genome (Corzett et al., 2018).
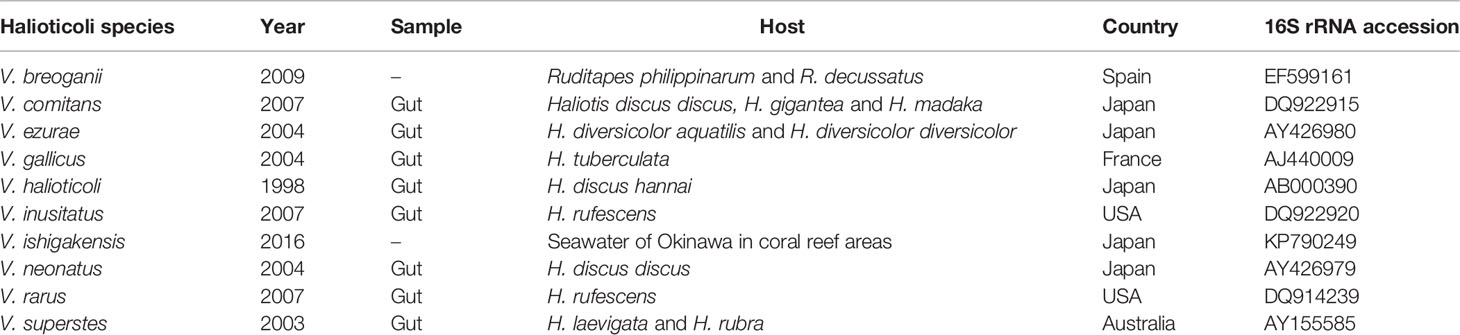
Table 1 Isolation information of Halioticoli clade species.
More interestingly, a first planktonic Halioticoli clade species, V. ishigakensis, was isolated from seawater taken in the Okinawa coral reef area, Japan (Gao et al., 2016). Rather different phenotypes (Table S1) of the non-motile Halioticoli species could be a key reference species to elucidate evolutionary processes from symbiotic to planktonic or vice versa in the Halioticoli clade species. However, the lack of genome sequences limits our knowledge of this clade. Here, we present the complete genome sequences of type strains of all current Halioticoli clade species and performed the first genomic analyses for this clade to evaluate their ecogenomics, evolutionary history, and possible biotechnology applications.
DNA extraction was performed using Wizard genomic DNA purification kit (Promega, USA) following the manufacturer’s instructions. The Nanopore sequencing library was prepared using the Rapid Barcoding Kit (SQK-RBK004) and sequenced using MinION device (Oxford Nanopore Technologies, Oxford, UK). Raw reads were basecalled using Guppy 1.1. The Illumina DNA library was prepared using Nextera XT DNA Library Preparation Kit (Illumina) and sequenced with the Illumina MiSeq platform. Then, the complete genome sequences of Halioticoli clade type strains were assembled by means of the hybrid assembly approach using both Nanopore and Illumina reads by Unicycler 0.4.7 (Tanaka et al., 2018; Jiang et al., 2022). Finally, the genome sequences were annotated using DDBJ Fast Annotation and Submission Tool (DFAST) (Tanizawa et al., 2018) and deposited in the DDBJ/GenBank/ENA under BioProject PRJDB11924 with accession numbers as Table 2.
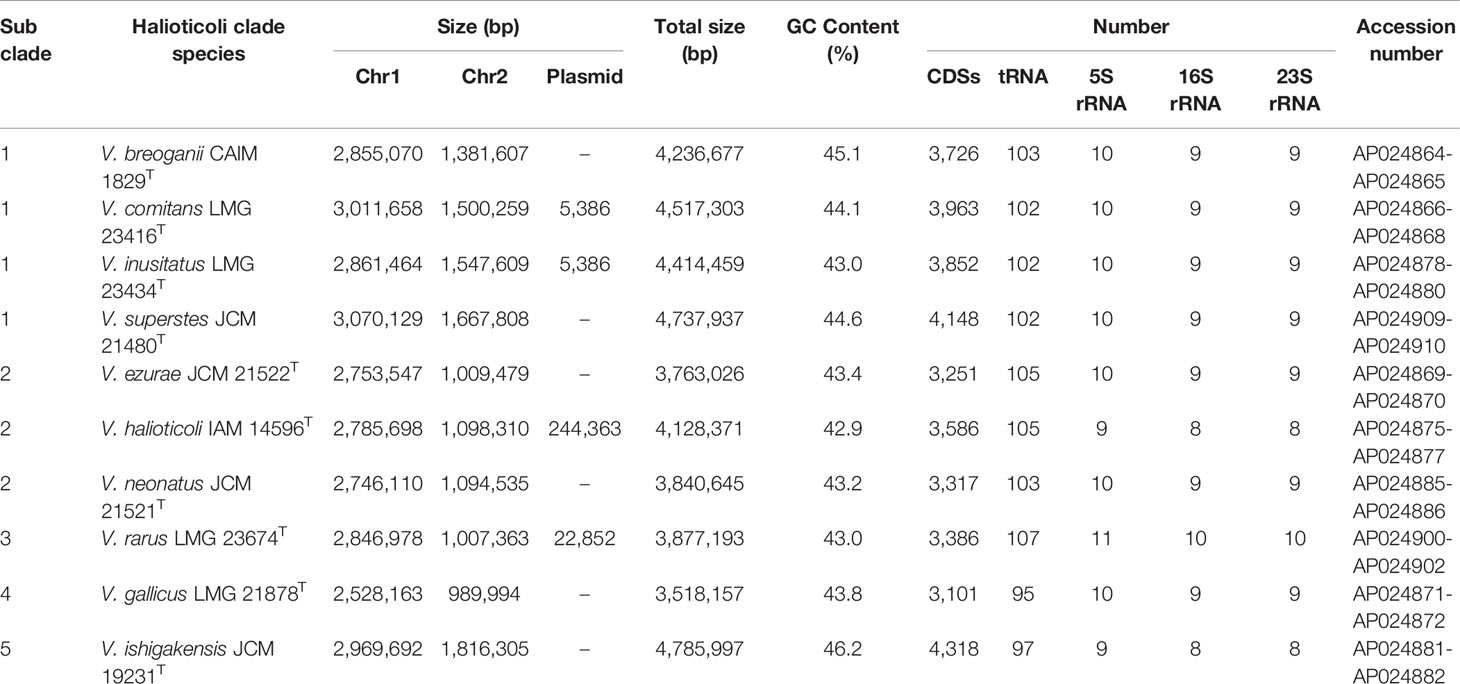
Table 2 General genomic characteristics of Halioticoli clade species.
MLSA was performed according to the previous description (Sawabe et al., 2013). Briefly, the entire nucleotide sequences of the eight housekeeping genes (ftsZ, gapA, gyrB, mreB, pyrH, recA, rpoA, and topA) were obtained after genome annotation. The sequences were aligned using MUSCLE (Edgar, 2004). Split decomposition analysis using the concatenated sequence was performed using SplitsTree 4.14.8 with a neighbor net drawing and a Jukes-Cantor correction. Phylogenetic analysis using the same concatenated sequence was constructed using Maximum Likelihood (ML), Neighbor-Joining (NJ), and Minimum-Evolution (ME) methods with 500 bootstraps by MEGA-X v10.1.8 (Kumar et al., 2018).
Genomic comparisons were performed based on chromosomes. The ten genomes from the Halioticoli clade were compared with the genome of V. ishigakensis JCM 19231T by BLASTn and visualized using BRIG v.0.95 (Alikhan et al., 2011). Synteny between the same genomes was analyzed using the Artemis Comparison Tool (ACT) v.18.1.0 (Carver et al., 2005).
Pan-genome analysis was performed using Halioticoli clade genomes by the Anvi’o program ver. 7 (Eren et al., 2015). Firstly, each genome sequence file was converted to an anvi’o contigs database (anvi-gen-contigs-database) using Prodigal (Hyatt et al., 2010), these contigs databases were decorated with hits from HMM models (anvi-run-hmms). An anvi’o genome storage was generated (anvi-gen-genomes-storage) using prepared contigs databases, and then, the pan-genome was analyzed (anvi-pan-genome) using NCBI’s blastp for amino acid sequence similarity search and the MCL algorithm (Van Dongen and Abreu-Goodger, 2012) for cluster identification in amino acid sequence similarity search results. In addition, Average Nucleotide Identity (ANI) values were calculated using the PyANI with ANIb method (anvi-compute-genome-similarity) (Pritchard et al., 2016). Finally, it was visualized and decorated (anvi-display-pan). Core genes were filtered (anvi-get-sequences-for-gene-clusters) and extracted in fasta files (anvi-get-sequences-for-gene-clusters) for further analysis.
Gene annotation was performed using Clusters of Orthologous Groups 2020 (COG20) (Galperin et al., 2021) for function estimation (anvi-run-ncbi-cogs), and Kyoto Encyclopedia of Genes and Genomes (KEGG) (Aramaki et al., 2020) for metabolism estimation (anvi-estimate-metabolism) (Muto et al., 2013). In addition, the enrichment scores of function/metabolism in different groups were identified using an R script developed by Amy Willis (anvi-compute-functional-enrichment) (Shaiber et al., 2020).
Carbohydrate-Active enZYmes (CAZy) were annotated using the dbCAN2 meta server (HMMdb v9) with HMMER, DIAMOND, and eCAMI tools (Zhang et al., 2018), and domains supported by more than two tools were used in this study. Genomic islands (GEIs) predictions were calculated by IslandViewer4 with IslandPick, IslandPath-DIMOB, and SIGI-HMM methods (Bertelli et al., 2017) using the GenBank files after DFAST annotation, predictions supported by at least one method were used in this study. Results were visualized using ggplot2.
Genomes of all species consisted of two chromosomes and four of them (V. comitans LMG 23416T, V. halioticoli IAM 14596T, V. inusitatus LMG 23434T, and V. rarus LMG 23674T) had one plasmid (Table 2). The genome sizes of Chromosome 1 (Chr. 1) ranged from 2,528,163 to 3,070,129 bp, and those of Chromosome 2 (Chr. 2) ranged from 989,994 to 1,816,305 bp. These genomes showed 42.9-46.2% GC content, identified 3,101-4,318 CDS, 25-31 rRNA, and 95-107 tRNA. V. ishigakensis JCM 19231T had the biggest genome with the highest GC content, the largest numbers of CDS, rRNA, and tRNA. In contrast, V. gallicus LMG 21878T had the smallest genome with the lowest GC content and the smallest numbers of CDS and tRNA. Compared to the genome size and GC content among 189 other Vibrionaceae species, Halioticoli clade species showed a relatively narrow GC content range but variable genome size (Figure 1).
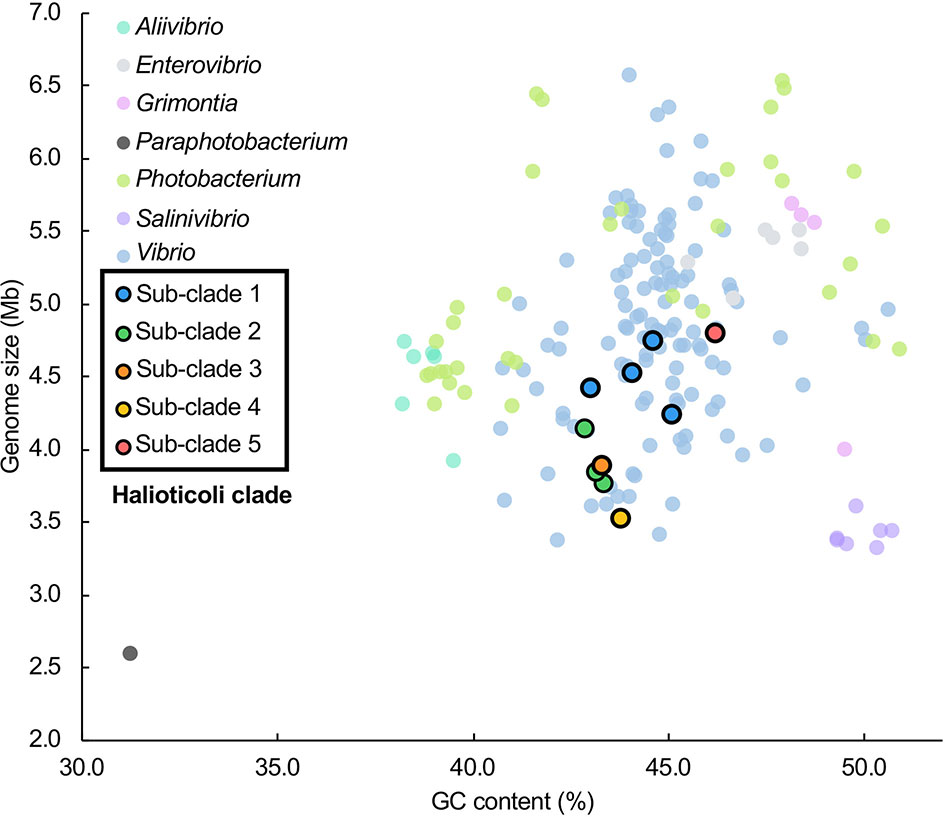
Figure 1 Genome size and GC content relationship for Halioticoli clade species in 189 Vibrionaceae species (7 genera). Data were obtained from Jiang et al. (2022). Compared to the genome size and GC content among 189 other Vibrionaceae species, Halioticoli clade species showed a relatively narrow GC content range but variable genome size. In addition, the clustering of sub-clades and the differentiation between different sub-clades species suggest that genome expansions may occur during evolution.
To explore the evolutionary history of Halioticoli clade species, the MLSA network and phylogenetic tree using concatenated eight house-keeping genes with V. cholerae ATCC 14035T and E. coli K-12 MG1655 as outgroups were constructed (Figures 2, S1). Both methods indicated five evolutionary directions in the Halioticoli clade: sub-clade 1) V. breoganii, V. comitans, V. inusitatus, and V. superstes, sub-clade 2) V. ezurae, V. neonatus, and V. halioticoli, sub-clade 3) V. rarus, sub-clade 4) V. gallicus, and sub-clade 5) V. ishigakensis. In which, sub-clade 1 to 4 consist of symbiotic species, and sub-clade 5 consists of the only one planktonic species. In addition, genome size and GC content relationship also showed the clustering of sub-clades and the differentiation between symbiotic and planktonic species (Figure 1), suggesting that genome expansions may occur during evolution.
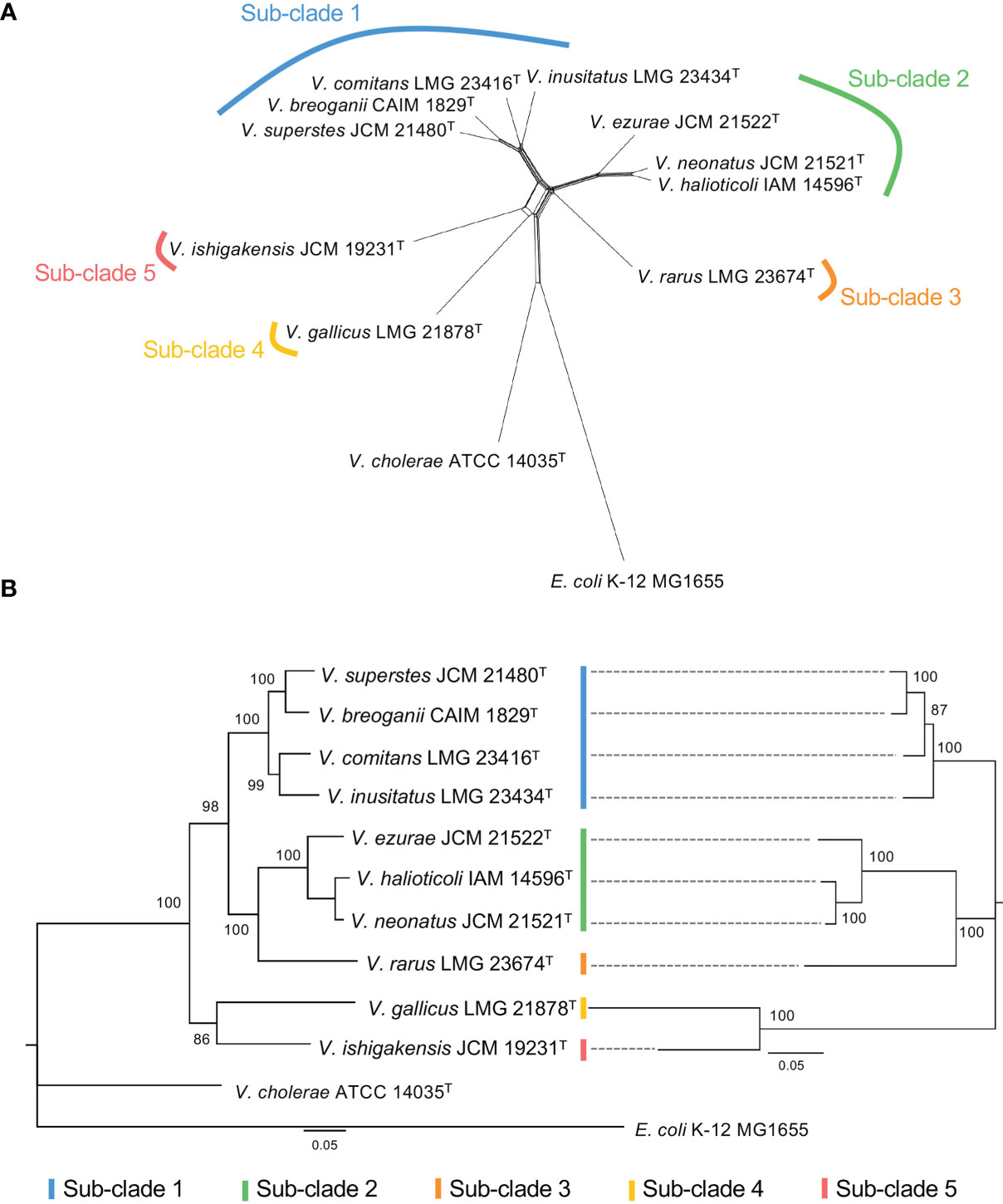
Figure 2 (A) Split network of Halioticoli clade based on concatenated sequences of eight protein coding genes (ftsZ, gapA, gyrB, mreB, pyrH, recA, rpoA, and topA). (B) Phylogenetic analysis of Halioticoli clade based on 8 eight protein coding genes (Left) and 125 single copy core genes (Right) using Maximum Likelihood (ML) method and General Time Reversible model with 500 bootstraps. Bootstrap values are shown at the branch points. All branches were both recovered in Neighbor-Joining (NJ) and Minimum-Evolution (ME) trees.
Analyses of homologous gene conservation and gene order across two or more genomes of different species play a vital role in comparative genomics since they can provide further insights into evolutionary processes that contribute to diversity, chromosomal dynamics, and interspecies rearrangement rates (Bhutkar et al., 2006; Lee et al., 2018; de la Haba et al., 2019). The nucleotide identity comparison using BLASTn of both chromosomes (Chr. 1 and Chr. 2) for Halioticoli clade species was performed using V. ishigakensis JCM 19231T as the reference genome (Figure 3A). The analysis revealed a higher nucleotide similarity in Chr. 1 but a lower similarity in Chr. 2, which indicates the genomes of Chr. 1 were highly conserved but those of Chr. 2 was relatively varied. Intra-sub-clade and inter-sub-clade genome rearrangement mappings of both chromosomes demonstrated that 1) similar gene arrangements amongst genomes of intra-subclade species in sub-clades 1 and 2, and 2) less similar of those arrangements among those of inter-subclade species (Figures 3B, S2).
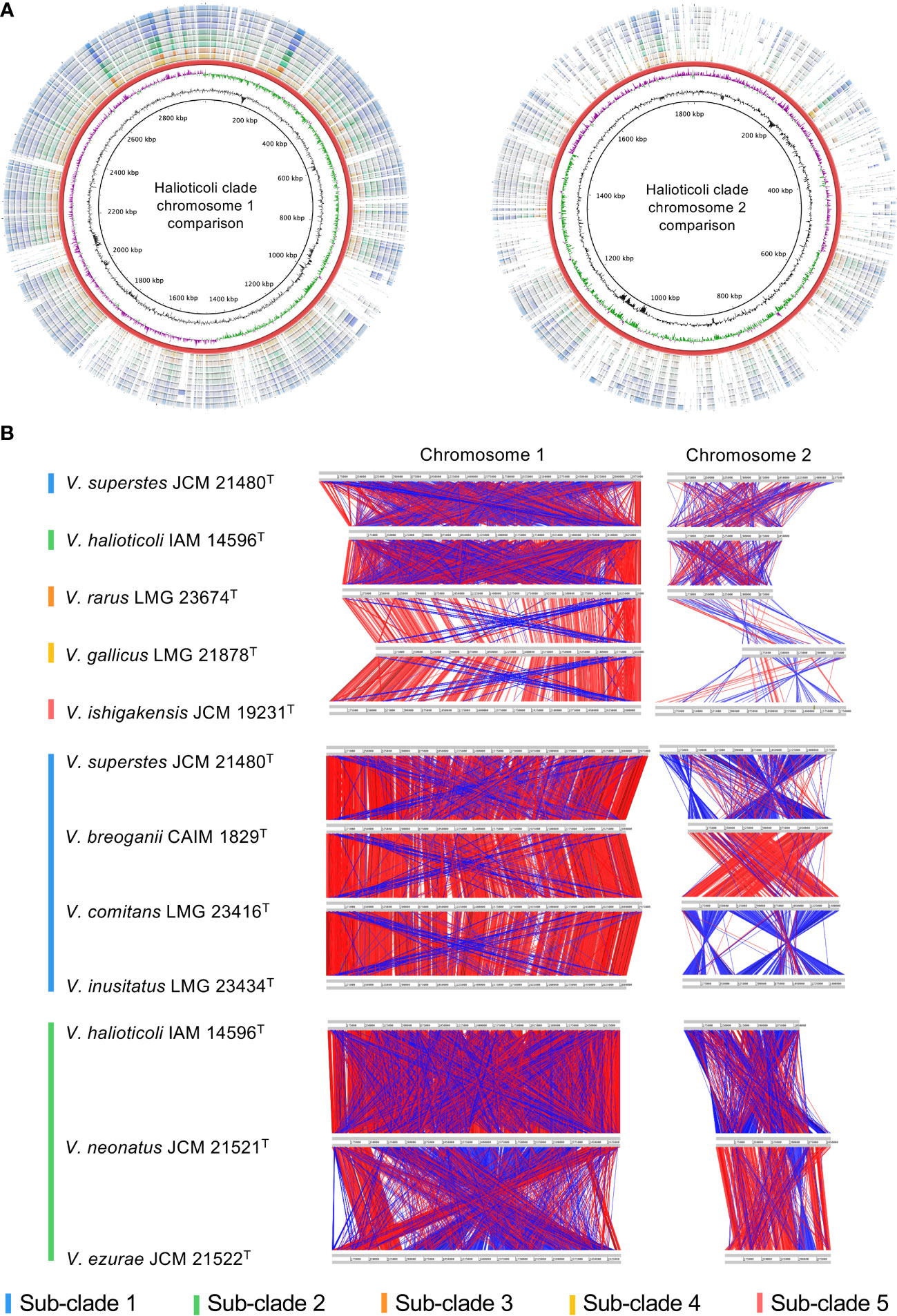
Figure 3 Genomic comparison in Halioticoli clade. (A) Circular map designed to compare the nucleotide identity of all genomes against V. ishigakensis JCM 19231T. The genomes were compared by BLASTn, and the percent identity between them was determined by the intensity of color in each ring. The rings from inner to outer are presented as follows: the GC content and CG skew of V. ishigakensis JCM 19231T, the genomes of V. ishigakensis JCM 19231T, V. gallicus LMG 21878T, V. rarus LMG 23674T, V. halioticoli IAM 14596T, V. neonatus JCM 21521T, V. ezurae JCM 21522T, V. inusitatus LMG 23434T, V. comitans LMG 23416T, V. breoganii CAIM 1829T, and V. superstes JCM 21480T, respectively. (B) Genomic synteny plots were analyzed using Artemis Comparison Tool. Gray lines indicate each genome size. Red bars indicate the conserved genomic regions, and blue bars indicate genomic inversions. A bigger and clearer plot is available in Figure S1.
Relatively more GEIs and transposase/integrase were predicted in sub-clades 2 and 3, which were likely to be shared common ancestry (Figures 2, S1, S3). The lowest number of GEIs and transposase/integrase were found in V. gallicus (sub-clade 4) and V. comitans (sub-clade 1), respectively. Interestingly, V. ishigakensis showed opposite results that much higher GEIs numbers but much lower transposase/integrase numbers.
Pan-genomics is capable of investigating the relationships between a given group of genomes by means of characterizing the core and accessory genes, providing a unique insight in the phylogeny and taxonomy analysis (Eren et al., 2015; Delmont and Eren, 2018). The ten genomes of Halioticoli clade species were used for pan-genome analysis using Anvi’o v7. In the Halioticoli clade pan-genome (Figure 4), a total of 8,062 gene clusters (GCs) with 36,612 genes were defined, in which 2,130 GCs with 22,123 genes (60%) were recognized in the core-genome (1,973 GCs with 19,730 genes were recognized as the single-copy core-genome), and 2,456 GCs with 10,881 genes (30%) were recognized in the accessory-genome. The remaining genes were recognized as species-specific genes as in Figure 4, among which, V. ishigakensis (sub-clade 5) possessed the most specific genes (922), 2-5 times more than other species. In addition, gene cluster analysis also showed that sub-clade 5 gained the highest number of GCs (4134), followed by sub-clade 1 (3,732 in average), sub-clade 2 (3,250 in average), sub-clade 3 (3193), and sub-clade 4 (2987). These results showed positive relationships with genome size.
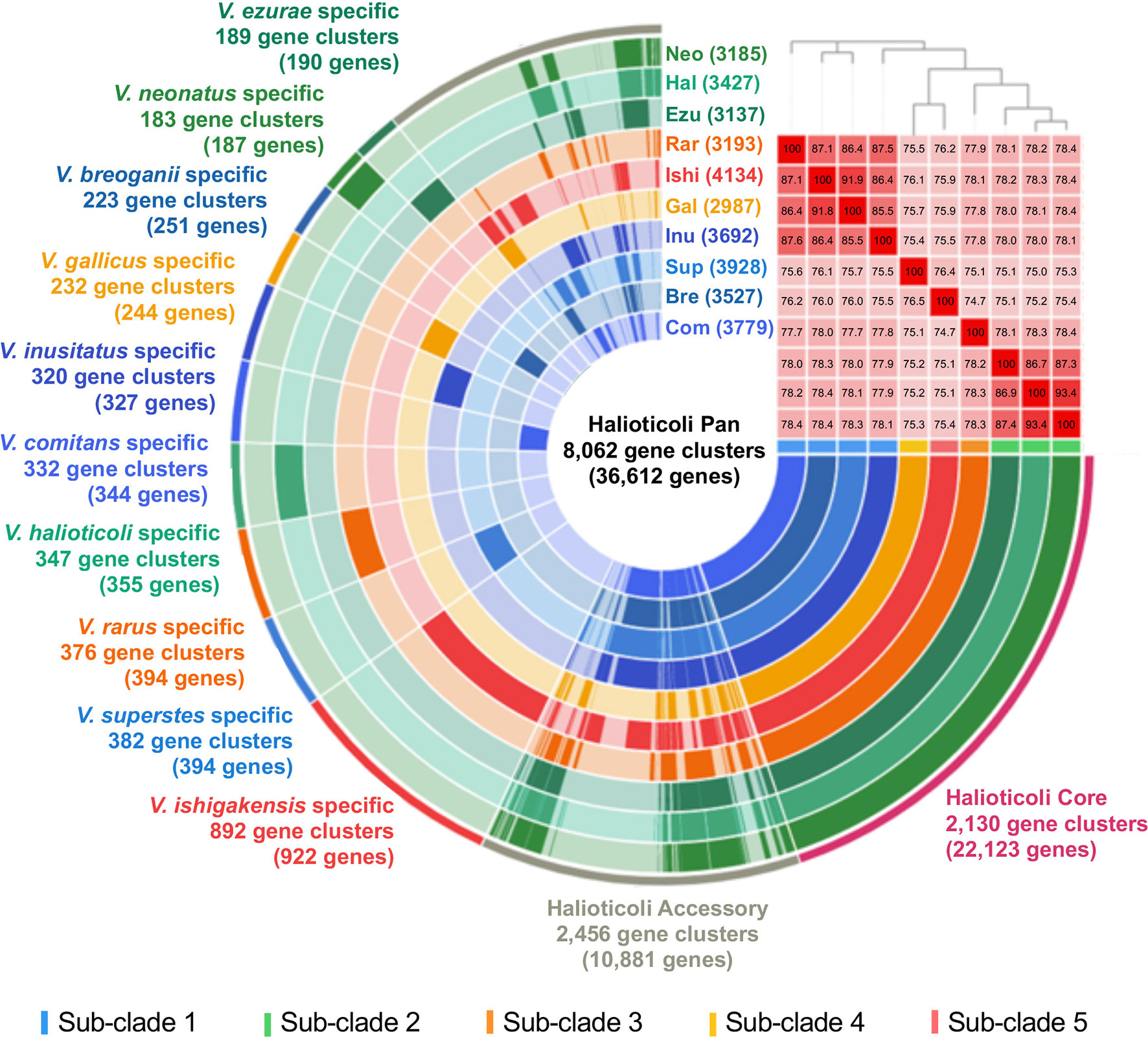
Figure 4 The pan-genome analysis of the Halioticoli clade species. Circle bars represent the occurrence of gene clusters in each genome. Gene cluster represents a group of homologues identified based on the amino acid sequence similarity. Heatmap in the upper right corner represents ANI calculation between these genomes (the barrier for species identification is 95%), and the above phylogenetic tree was constructed using amino acid sequences of 125 better single-copy genes by embedded FastTree tool. Abbreviations for the Halioticoli clade species are represented as Bre, V. breoganii CAIM 1829T; Com, V. comitans LMG 23416T; Ezu, V. ezurae JCM 21522T; Gal, V. gallicus LMG 21878T; Hal, V. halioticoli IAM 14596T; Inu, V. inusitatus LMG 23434T; Ish, V. ishigakensis JCM 19231T; Neo, V. neonatus JCM 21521T; Rar, V. rarus LMG 23674T; and Sup, V. superstes JCM 21480T. Numbers in parentheses after abbreviations represent the total numbers of gene clusters in each species.
Due to the difficulty to use a large number of single-copy genes (SCGs), a set of 125 better-SCGs was filtered using a custom setting (–min-geometric-homogeneity-index 1, –max-functional-homogeneity-index 0.9). The concatenated amino acid sequence of the 125 better-SCGs was used for constructing a more accurate phylogenetic tree, and the result showed that the topology was congruent with the one constructed by eight house-keeping genes (Figures 2B, 4), the five sub-clades could be identified as well. Moreover, the five sub-clades could also be illustrated by clustering in the ANI matrix, and sub-clade species showed at least 85.5% intra ANI similarity.
The Clusters of Orthologous Genes (COGs) database has been a popular tool for functional and comparative genomics of bacteria and archaea in recent decades with the newest update of COG20 (Galperin et al., 2021). COG20 function estimation for each genome of Halioticoli clade species showed that the same function structure was shared among the species, as well as in the core-genome (Figure S4A). However, the result in the species-specific genomes was diverse in some aspects (Figure 5A). Despite the poorly characterized and unknown functions, the planktonic species, V. ishigakensis-specific genome had the most diverse and abundant gene sets in each function category. Among them, V. ishigakensis showed more abundant in the metabolism functions, in particular in the “Carbohydrate transport and metabolism” function. In addition, the functions of “Transcription”, “Signal transduction mechanisms”, and “Cell wall membrane envelope biogenesis” were also abundant. It is likely that these functions were an adaptation for survival in planktonic environments. The symbiotic species-specific genomes showed poorer ability in metabolizing, with the V. superstes-specific one being the most powerful. To further investigate the metabolism functions in the Halioticoli clade, metabolism pathway modules were reconstructed using the KEGG database. As in the function structure, the Halioticoli clade species shared the common metabolism structure in complete genomes (Figure S4B), but different structures in species-specific genomes (Figure 5B). A wide range of genes responsible for diverse metabolism pathways was only detected in the V. ishigakensis-specific genome, in particular carbohydrate metabolism genes were highly detected. On the other hand, other species-specific genomes, demonstrated their advantages, such as a remarkable detection for “cofactor and vitamin metabolism” in the V. rarus-specific genome. In addition, the enrichment analyses of metabolism pathway modules indicated that the V. ishigakensis shared almost all enriched modules with other species with the exception of D-galactonate degradation (M00552), which is exclusively and most enriched in itself (Figure 6).

Figure 5 Heatmap representation based on the number of hits in specific genes for each Halioticoli clade species, (A) COG20 function prediction, and (B) KEGG metabolism prediction. The left and right axis ticks represent different subcategories and categories, respectively. Abbreviations for the Halioticoli clade species are represented as Figure 4.
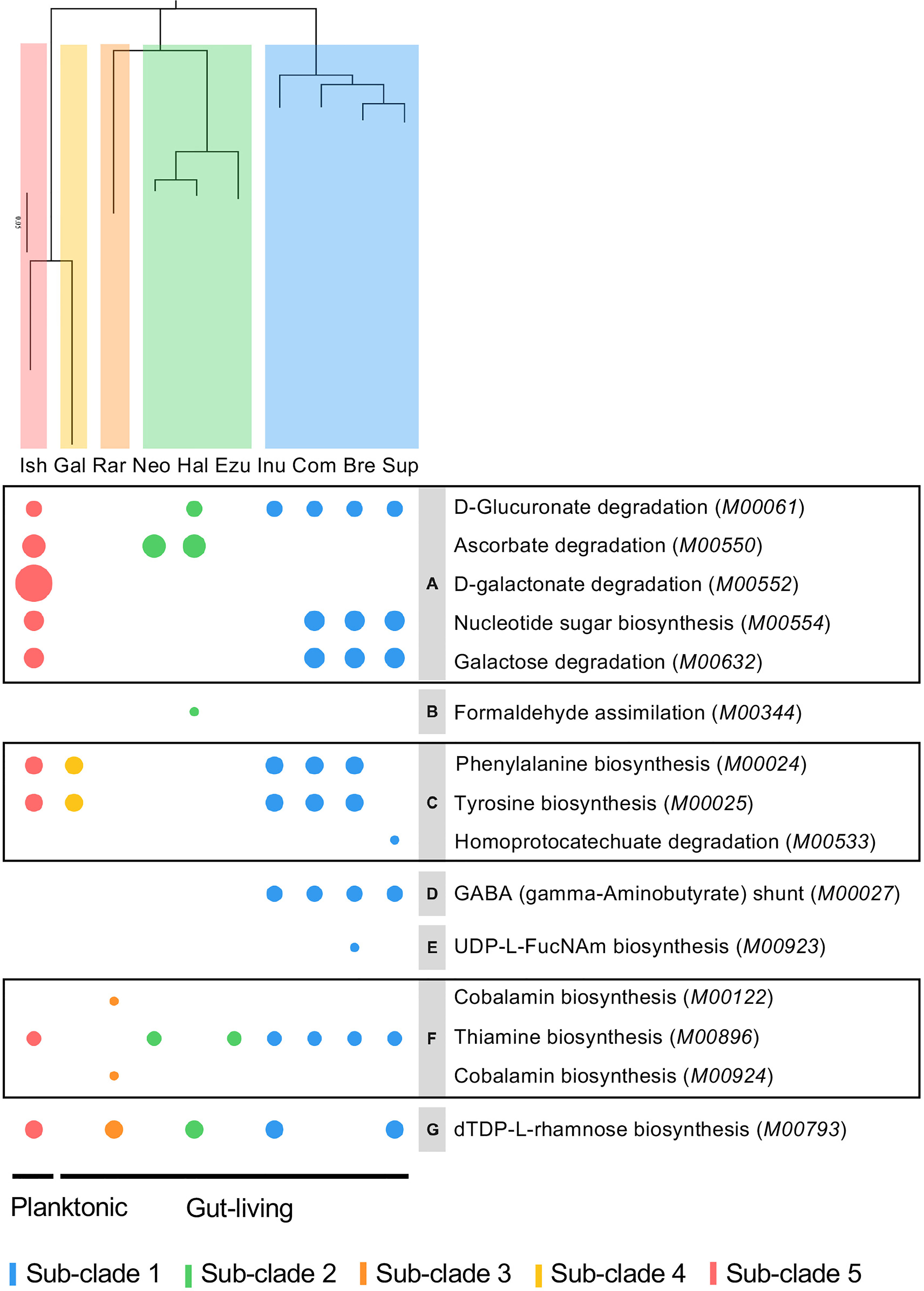
Figure 6 Bubble plot based on enrichment of KEGG pathway modules in Halioticoli clade. Bubble size represents the enrichment score, colour represents different groups. Phylogenetic tree was constructed accordingly to Figure 2B. The italic contents of parentheses represent the module accession numbers in the KEGG database. Gray parts indicate the secondary category of the KEGG module as follows, (A) Other carbohydrate metabolism, (B) Methane metabolism, (C) Aromatic amino acid metabolism, (D) Other amino acid metabolism, (E) Lipopolysaccharide metabolism, (F) Cofactor and vitamin metabolism, (G) Polyketide sugar unit biosynthesis. Related genes were listed in Table S2.
Furthermore, the COG20 and KEGG annotations were also performed for gene clusters (GCs) of each Halioticoli clade species. Same COG20 functions were shared among the clade but with different abundance. V. ishigakensis, which has the biggest genome size, gained the highest number of GCs with a wide range of COG functions, in particular GCs classified into “Transcription (K)”, “Cell wall/membrane/envelope biogenesis (M)”, and “Carbohydrate transport and metabolism (G)”. In more detail, numbers of GCs were 1.2, 1.6, 1.7, and 1.7 folds in K, 1.1, 1.3, 1.3, and 1.4 folds in M, and 1.4, 1.9, 2.3, and 1.9 folds in G, compared with those numbers of sub-clades 1, 2, 3 and 4, respectively. On the contrary, the lowest number of GCs were observed in V. gallicus, numbers of GCs classified to “Cell cycle control, cell division, chromosome partitioning (D)”, “Posttranslational modification, protein turnover, chaperones (O)”, and “Defense mechanisms (V)” were reduced (Figure 7A). KEGG annotation of GCs which were lost or gained showed “Polyamine biosynthesis” (E6, in Figures 7B, S5) were gained in sub-clades 1, 3, and 5 but lost in sub-clades 2 and 4, and GCs encoded trans-2,3-dihydro-3-hydroxyanthranilate isomerase [EC:5.3.3.17] in “Biosynthesis of other bacterial compounds (I5)” were gained in sub-clades 1 and 5 but lost in other sub-clades. In more details, glycine/D-amino acid oxidase (deaminating) (dadA) (PDB:3AWI) and acyl-CoA reductase or other NAD-dependent aldehyde dehydrogenase (adhE) (PDB:1A4S), both of which can use putrescine to produce GABA (M00136), were gained in the sub-clades 1 and 3; and genes (rfbB, rfbC, rfbD, and rmlA1) involved in the biosynthesis of the dTDP-L-rhamnose (M00793) were lost in the sub-clade4 (Figure S6).
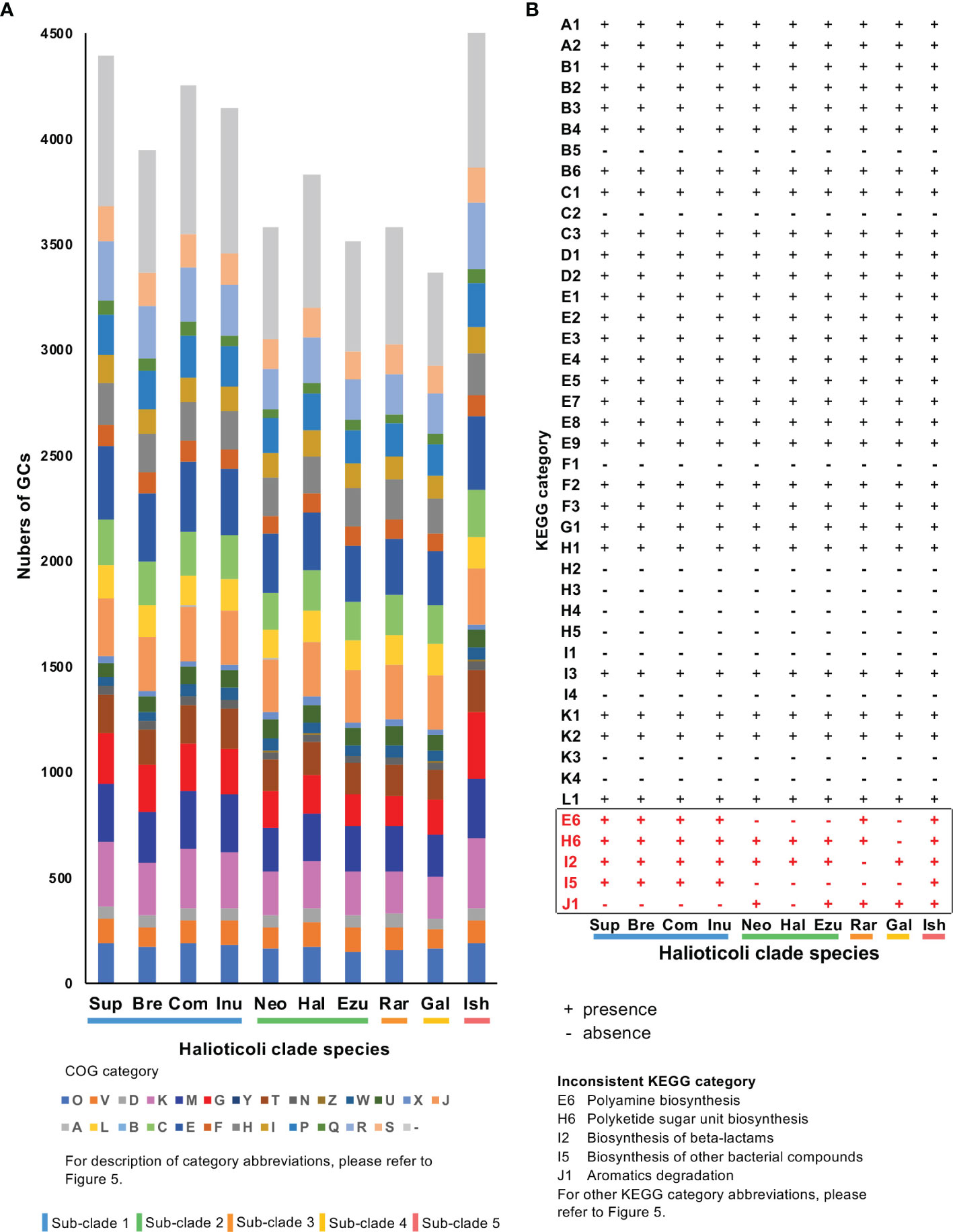
Figure 7 COG and KEGG annotation of gene clusters (GCs) for each Halioticoli clade species. (A) Numbers of GCs for each COG functional category. (B) Presence and absence of each KEGG category. Numbers of GCs for each KEGG category please refer to Figure S5. Abbreviations for COG and KEGG categories are represented as Figure 5. The Abbreviations for the Halioticoli clade species are represented as Figure 4.
Carbohydrate-Active enZYmes (CAZy) were predicted for each genome to describe the catalytic modules (enzymes) encoded in these genomes. Generally, each genome of the Halioticoli clade species contained 4 main enzymes classes: Carbohydrate Esterases (CEs), Glycoside Hydrolases (GHs), GlycosylTransferases (GTs), and Polysaccharide Lyases (PLs); and one associated module: Carbohydrate-Binding Modules (CBMs); only some of them contained few Auxiliary Activities (AAs) class enzymes (Figure 8E). Among them, Sub-clade 5 (V. ishigakensis) had the most CAZy (120), followed by sub-clade 1 (95-109), as a result of the abundance of GHs in these sub-clades (Figure 8A). Meanwhile, kinds of polysaccharide lyases (PL6, PL7, PL15, and P17) involved in alginate degradation were found enriched in the Halioticoli clade species, while V. ishigakensis had fewer numbers (15) than most symbiotic species (19-21) (Figure 8C). Compared to the planktonic sub-clade, symbiotic sub-clades showed higher ability on alginate degradation but with different advantages, which is a higher capacity of intracellular degradation (PL15 and PL17) in sub-clade 1, but a higher capacity of extracellular degradation (PL6 and PL7) in sub-clade 2 and 3.These finds indicate that V. ishigakensis obtained a powerful ability for degrading diverse glycosidic bonds but became weaker in degrading polysaccharides during the evolution from the gut environment to the planktonic environment.
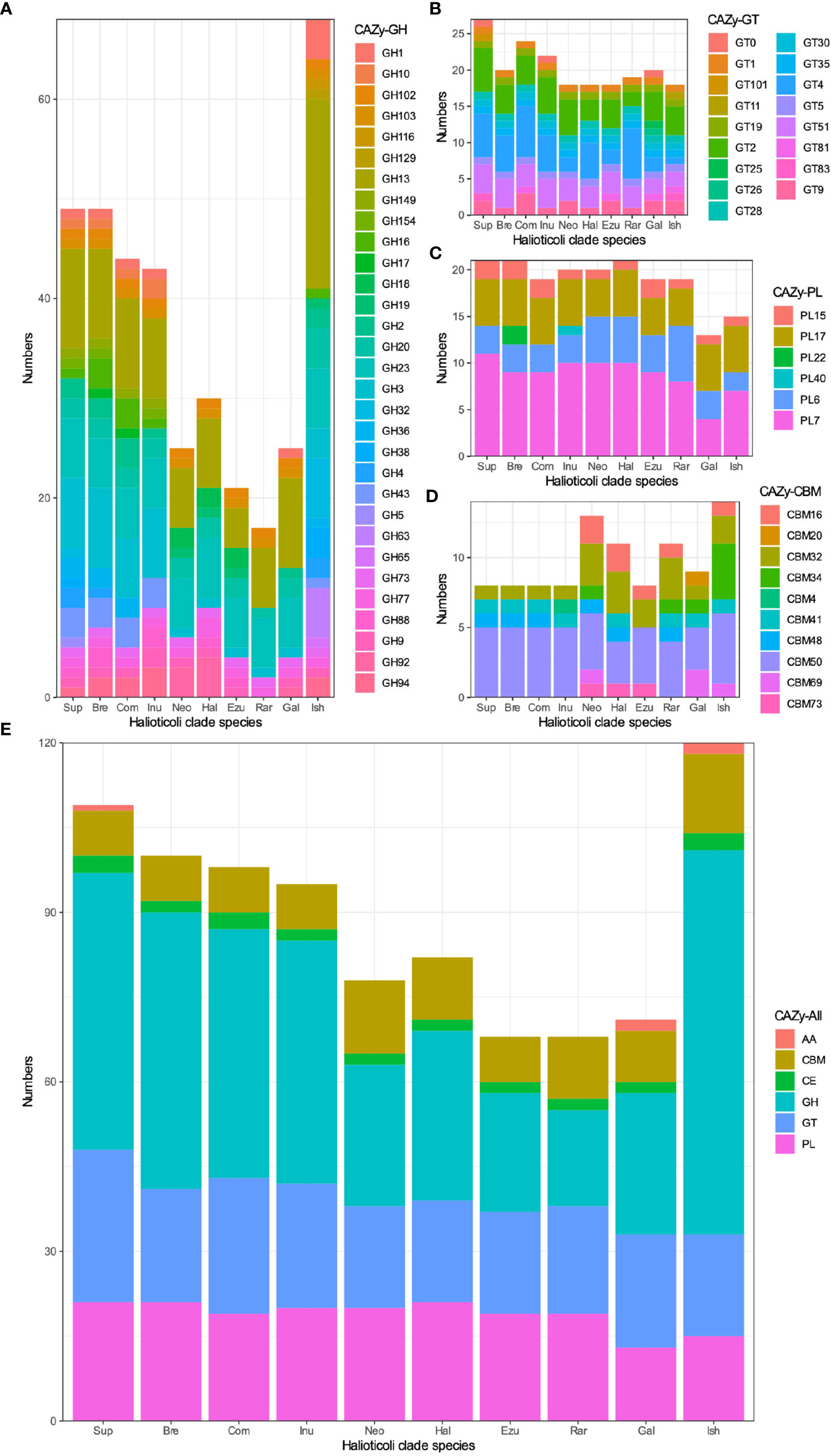
Figure 8 The numbers of Carbohydrate-active enzyme (CAZy) predicted in each Halioticoli clade species, annotated by dbCAN2 meta server. (A) Glycoside Hydrolases (GH), (B) GlycosylTransferases (GT), (C) Polysaccharide Lyases (PL), (D) Carbohydrate-Binding Modules (CBM), and (E) The total CAZy numbers. Abbreviations for the Halioticoli clade species are represented as Figure 4.
Ten species in the Halioticoli clade, including the first described species of V. halioticoli (Sawabe et al., 1998) and the most recently described one of V. ishigakensis (Gao et al., 2016), have been found to date, making the clade robust in the family Vibrionaceae (Jiang et al., 2022). However, the evolutionary history of them remains unknown due to the lack of complete genome sequence comparisons. We succeeded in getting all complete genomes, and it showed that all of them were composed of two chromosomes while part of them with one additional plasmid, in which, the planktonic species, V. ishigakensis had the biggest genome size and highest GC content (Table 2). On the basis of the genome sequences, phylogenetic analyses using three methods all clearly showed the five evolutionary directions (Figures 2, S1). V. gallicus were deeply branched, followed to V. ishigakensis in this clade.
Comparative genomic analyses have been widely employed to explore the diversity, evolution, and chromosomal dynamics between given genomes, such as Methylophilaceae (Jimenez-Infante et al., 2016), Salinivibrio (de la Haba et al., 2019), and Erysipelothrix (Grazziotin et al., 2021). Here, we utilized the complete genomes of Halioticoli clade species to construct a circular map of nucleotide identity comparison and a linear synteny comparison for both chromosomes (Chr. 1 and Chr. 2). As with previous studies (Okada et al., 2005; Kirkup et al., 2010), the results showed that the structure of Chr. 1 was more stable and conserved than Chr. 2 during the evolution (more gaps could be found), and it was more evident in the intra-subclades species than inter-subclades species (Figure 3). This may be explained by the mutations, rearrangements or horizontal gene transfer (HGT) of genomes, in which, HGT is an important mechanism for the evolution of microbial genomes, enabling the bacteria to adapt to the environment (Dobrindt et al., 2004). A significant part of the HGT has been facilitated by genomic islands (GEIs), which plays an important role in promoting the adaptive evolution of commensal, symbiotic and environmental bacteria (Dobrindt et al., 2004; Juhas et al., 2009). Results showed that V. gallicus contained the least number of GEIs. The numbers of GEIs predicted on Chr. 2 were higher than those on Chr. 1 in most species while the opposite occurred in the sub-clade 2 species (Figure S3A). Besides, although the HGT of GEIs was predicted from accessory genes, some genes/proteins could be found shared in the same sub-clade species, such as pectin degradation protein KdgF (kdgF) in sub-clade 1 and DNA methyltransferase (hsdM) in sub-clade 2. We also searched transposase and integrase as presence of HGT events, transposase related genes were significantly abundant (Figure S3B). In which, except of the one integrase/recombinase xerC and xerD were found shared on the Chr. 1 of core-genomes, the other transposase and integrase related genes were distributed among the accessory and specific genomes.
In this study, we also estimated the COG function and reconstructed the metabolism pathway for each species genome, core/accessory genomes, and species-specific genomes. The function and metabolism structures were shared in the whole genome of each halioticoli clade species and their core-genome (Figure S4), but diverged in the accessory-genome and species-specific genomes (Figure 5). According to the reconstruction of metabolism pathways using KEGG database, most of the genes were involved in the carbohydrate and amino acid metabolisms. Moreover, there was a significant detection in the cofactor and vitamin metabolism by V. rarus-specific genomes, due to the related genes of cobalamin/B12 biosynthesis. Furthermore, the enrichment scores of metabolism pathway modules between different groups showed that almost all enriched pathway modules in the planktonic group (sub-clade 5, V. ishigakensis) were shared with other groups, except the D-galactonate degradation (M00552) was enriched exclusively in itself and was the most enriched module (Figure 6). This module has been reported involved in the catabolism of carrageenan, which is one of main components of red algal cell walls (Gobet et al., 2018; Schultz-Johansen et al., 2018). For the symbiotic group, most enriched modules were detected in the sub-clade 1, and subclade-specific enriched modules could be found as well, for example, GABA (γ-aminobutyrate) shunt (M00027) in sub-clade 1 and cobalamin biosynthesis (M00122 and M00924) in sub-clade 3 (V. rarus). The γ-aminobutyrate (GABA) shunt is a metabolic pathway that bypasses two steps of the tricarboxylic-acid (TCA) cycle to produce succinate, as an alternative route in plants and mammals, while it has not been extensively studied in bacteria but is thought to play a role in glutamate metabolism, anaplerosis, and antioxidant defense (Bouche et al., 2003; Feehily et al., 2013). In addition, GABA has also been found abundant in many algae species (Belghit et al., 2017), the enrichment of related modules could be caused by algae associations in the gut of algae-eating animals.
Prediction of Carbohydrate-Active enZYmes (CAZy) showed a subclade-based grouping as well. V. ishigakensis contained the most CAZy, which was due to the abundance of GHs (Figure 8). The significant presence (19) of GH13, which is a main α-amylase family (van der Maarel et al., 2002; Janeček and Zámocká, 2020), was likely responsible for the diversity and abundance of carbohydrate metabolisms detected above for V. ishigakensis. Sub-clade 1 and 5 species are likely capable of utilizing a variety of β-glucans, which are an important group of glucose-based polysaccharides composed of β-glycosidic bonds found primarily in algal cell walls (Corzett et al., 2018), due to the possesses of enzymes identified in GH3 and GH16. We also found other enzymes involved in algal carbohydrates (Mann et al., 2013) exclusively in these two sub-clades, including GH 36 and GH43, which is related to breaking down carrageenans/carbohydrates and cell wall-degrading, respectively (Tang et al., 2017). Furthermore, genes encoding alginate degrading enzymes (aly), classified into PL6, PL7, PL15, and PL17 were commonly found in the genomes of Halioticoli clade species, but the number in V. ishigakensis was relatively smaller. A similar signature of GH and PL CAZy has been described in V. breoganii, indicating the evolution of specialization for macroalgal substrates (Corzett et al., 2018). As a result of the above results, it appears that all species of sub-clade 1 have evolved to specialize in macroalgae, which could function as alternative sources for bioenergy production using macroalgae. In addition, chitin utilization is a conservative function in the family Vibrionaceae except for V. breoganii (Hunt et al., 2008; Corzett et al., 2018), but it has not been mentioned in other Halioticoli clade species. In our study, same as V. breoganii, members of sub-clade 1, sub-clade 3, and sub-clade 4 lacked any domain of GH18, GH19, GH116, and GH129, or motif of CBM5, CBM14, and CBM73, which are implicated in chitin utilization (Figure S7). However, two chitinase (ChiA, GH18 family), one chitodextrinase (GH19) and one motif (CBM73) were found distributed among the members of sub-clade 2. These results indicate that species of sub-clade 2 were likely to be able to utilize chitin while that of sub-clade 1, sub-clade 3, and sub-clade 4 were not.
In this study, the first pan-genomic analysis of the Halioticoli clade was completed thanks to the complete genomes of the type strains of this clade. The results obtained regarding the phylogenetic analysis and pan-genome analysis, as well as function and metabolism estimation, will help us to elucidate the evolutionary processes of these species from symbiotic to planktonic lifestyle. It appears that genome expansion encoding more carbohydrate metabolism occurred during symbiotic as a gut-living to free-living environments, planktonic species acquired more abilities to utilize a variety of carbohydrates for surviving in the environment while symbiotic species were evolved to specialize in macroalgae utilization. These generic backbones could contribute to developing bioenergy potential using macroalgae as biocatalysts.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: DDBJ [accession: PRJDB11924].
CJ conceived, designed and performed the experiments, analyzed the data, visualized the data, and drafted and reviewed the manuscript. SM analyzed the data and reviewed the manuscript. TS conceived and designed the experiments and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This study was partly supported by MEXT KAKEN 19H03041.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.844983/full#supplementary-material
Supplementary Figure 1 | The concatenated split network based on nucleotide sequences of eight housekeeping genes retrieved from 191 Vibrionaceae species. The ftsZ, gapA, gyrB, mreB, pyrH, recA, rpoA, and topA gene sequences were concatenated and the tree was reconstructed using the SplitsTree4 ver. 4.14.8. Sequence data was obtained from Jiang et al., 2022.
Supplementary Figure 2 | Genomic synteny plots were analyzed using Artemis Comparison Tool. Gray lines indicate each genome size. Red bars indicate the conserved genomic regions, and blue bars indicate genomic inversions.
Supplementary Figure 3 | (A) The numbers of Genomic island (GEI) predicted of both chromosomes in each Halioticoli clade species, annotated by IslandViewer 4. (B) The numbers of transposase and integrase related genes predicted in each Halioticoli clade species. Abbreviations for the Halioticoli clade species are represented as Bre, V. breoganii CAIM 1829T; Com, V. comitans LMG 23416T; Ezu, V. ezurae JCM 21522T; Gal, V. gallicus LMG 21878T; Hal, V. halioticoli IAM 14596T; Inu, V. inusitatus LMG 23434T; Ish, V. ishigakensis JCM 19231T; Neo, V. neonatus JCM 21521T; Rar, V. rarus LMG 23674T; and Sup, V. superstes JCM 21480T.
Supplementary Figure 4 | Distribution of (A) COG20 function and (B) KEGG metabolism predication across the accessory-genome, core-genome, and each complete genome of Halioticoli clade species. Abbreviations for the Halioticoli clade species are represented as Bre, V. breoganii CAIM 1829T; Com, V. comitans LMG 23416T; Ezu, V. ezurae JCM 21522T; Gal, V. gallicus LMG 21878T; Hal, V. halioticoli IAM 14596T; Inu, V. inusitatus LMG 23434T; Ish, V. ishigakensis JCM 19231T; Neo, V. neonatus JCM 21521T; Rar, V. rarus LMG 23674T; and Sup, V. superstes JCM 21480T.
Supplementary Figure 5 | Numbers of gene clusters (GCs) for each KEGG category. The Abbreviations for the Halioticoli clade species are represented as Bre, V. breoganii CAIM 1829T; Com, V. comitans LMG 23416T; Ezu, V. ezurae JCM 21522T; Gal, V. gallicus LMG 21878T; Hal, V. halioticoli IAM 14596T; Inu, V. inusitatus LMG 23434T; Ish, V. ishigakensis JCM 19231T; Neo, V. neonatus JCM 21521T; Rar, V. rarus LMG 23674T; and Sup, V. superstes JCM 21480T.
Supplementary Figure 6 | Gene loci mapping of Halioticoli clade. (A) Circular map of CDS comparison between all genomes against V. ishigakensis JCM 19231T, performed by CGView Server (cgview.ca). (B) Gene loci of gained or lost gene clusters.
Supplementary Figure 7 | Prediction of chitin utilization related genes in each Halioticoli clade species. Abbreviations for the Halioticoli clade species are represented as Bre, V. breoganii CAIM 1829T; Com, V. comitans LMG 23416T; Ezu, V. ezurae JCM 21522T; Gal, V. gallicus LMG 21878T; Hal, V. halioticoli IAM 14596T; Inu, V. inusitatus LMG 23434T; Ish, V. ishigakensis JCM 19231T; Neo, V. neonatus JCM 21521T; Rar, V. rarus LMG 23674T; and Sup, V. superstes JCM 21480T. Members of sub-clade 1, sub-clade 3, and sub-clade 4 lacked any domain of GH18, GH19, GH116, and GH129, or motif of CBM5, CBM14, and CBM73, which are implicated in chitin utilization.
Alikhan N. F., Petty N. K., Ben Zakour N. L., Beatson S. A. (2011). BLAST Ring Image Generator (BRIG): Simple Prokaryote Genome Comparisons. BMC Genomics 12, 402. doi: 10.1186/1471-2164-12-402
Aramaki T., Blanc-Mathieu R., Endo H., Ohkubo K., Kanehisa M., Goto S., et al. (2020). KofamKOALA: KEGG Ortholog Assignment Based on Profile HMM and Adaptive Score Threshold. Bioinformatics 36, 2251–2252. doi: 10.1093/bioinformatics/btz859
Beaz Hidalgo R., Cleenwerck I., Balboa S., Prado S., De Vos P., Romalde J. L. (2009). Vibrio Breoganii Sp. Nov., a Non-Motile, Alginolytic, Marine Bacterium Within the Vibrio Halioticoli Clade. Int. J. Syst. Evol. Microbiol. 59, 1589–1594. doi: 10.1099/ijs.0.003434-0
Belghit I., Rasinger J. D., Heesch S., Biancarosa I., Liland N., Torstensen B., et al. (2017). In-Depth Metabolic Profiling of Marine Macroalgae Confirms Strong Biochemical Differences Between Brown, Red and Green Algae. Algal. Res. 26, 240–249. doi: 10.1016/j.algal.2017.08.001
Bertelli C., Laird M. R., Williams K. P., Lau B. Y., Hoad G., Winsor G. L., et al. (2017). IslandViewer 4: Expanded Prediction of Genomic Islands for Larger-Scale Datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Bhutkar A., Russo S., Smith T. F., Gelbart W. M. (2006). Techniques for Multi-Genome Synteny Analysis to Overcome Assembly Limitations. Genome Inform. 17, 152–161. doi: 10.11234/gi1990.17.2_152
Bouche N., Fait A., Bouchez D., Moller S. G., Fromm H. (2003). Mitochondrial Succinic-Semialdehyde Dehydrogenase of the γ-Aminobutyrate Shunt is Required to Restrict Levels of Reactive Oxygen Intermediates in Plants. Proc. Natl. Acad. Sci. 100, 6843–6848. doi: 10.1073/pnas.1037532100
Carver T. J., Rutherford K. M., Berriman M., Rajandream M. A., Barrell B. G., Parkhill J. (2005). ACT: The Artemis Comparison Tool. Bioinformatics 21, 3422–3423. doi: 10.1093/bioinformatics/bti553
Corzett C. H., Elsherbini J., Chien D. M., Hehemann J. H., Henschel A., Preheim S. P., et al. (2018). Evolution of a Vegetarian Vibrio: Metabolic Specialization of Vibrio Breoganii to Macroalgal Substrates. J. Bacteriol. 200, e00020–18. doi: 10.1128/JB.00020-18
De la Haba R. R., López-Hermoso C., Sánchez-Porro C., Konstantinidis K. T., Ventosa A. (2019). Comparative Genomics and Phylogenomic Analysis of the Genus Salinivibrio. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.02104
Delmont T. O., Eren A. M. (2018). Linking Pangenomes and Metagenomes: The Prochlorococcus Metapangenome. PeerJ 6, e4320. doi: 10.7717/peerj.4320
Dobrindt U., Hochhut B., Hentschel U., Hacker J. (2004). Genomic Islands in Pathogenic and Environmental Microorganisms. Nat. Rev. Microbiol. 2, 414–424. doi: 10.1038/nrmicro884
Edgar R. C. (2004). MUSCLE: Multiple Sequence Alignment With High Accuracy and High Throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eren A. M., Esen Ö.C., Quince C., Vineis J. H., Morrison H. G., Sogin M. L., et al. (2015). Anvi’o: An Advanced Analysis and Visualization Platform for ‘Omics Data. PeerJ 3, e1319. doi: 10.7717/peerj.1319
Feehily C., O’Byrne C. P., Karatzas K. A. G. (2013). Functional γ-Aminobutyrate Shunt in Listeria Monocytogenes: Role in Acid Tolerance and Succinate Biosynthesis. Appl. Environ. Microbiol. 79, 74–80. doi: 10.1128/AEM.02184-12
Galperin M. Y., Wolf Y. I., Makarova K. S., Vera Alvarez R., Landsman D., Koonin E. V. (2021). COG Database Update: Focus on Microbial Diversity, Model Organisms, and Widespread Pathogens. Nucleic Acids Res. 49, D274–D281. doi: 10.1093/nar/gkaa1018
Gao F., Al-saari N., Rohul Amin A. K. M., Sato K., Mino S., Suda W., et al. (2016). Vibrio Ishigakensis Sp. Nov., in Halioticoli Clade Isolated From Seawater in Okinawa Coral Reef Area, Japan. Syst. Appl. Microbiol. 39, 330–335. doi: 10.1016/j.syapm.2016.04.002
Gobet A., Barbeyron T., Matard-Mann M., Magdelenat G., Vallenet D., Duchaud E., et al. (2018). Evolutionary Evidence of Algal Polysaccharide Degradation Acquisition by Pseudoalteromonas Carrageenovora 9T to Adapt to Macroalgal Niches. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.02740
Gomez-Gil B., Thompson C. C., Matsumura Y., Sawabe T., Iida T., Christen R., et al. (2014). “The Famlily Vibrionaceae,” in The Prokaryotes (Berlin, Heidelberg: Springer Berlin Heidelberg), 659–747. doi: 10.1007/978-3-642-38922-1_225
Grazziotin A. L., Vidal N. M., Hoepers P. G., Reis T. F. M., Mesa D., Caron L. F., et al. (2021). Comparative Genomics of a Novel Clade Shed Light on the Evolution of the Genus Erysipelothrix and Characterise an Emerging Species. Sci. Rep. 11, 3383. doi: 10.1038/s41598-021-82959-x
Guardiola-Avila I., Sánchez-Busó L., Acedo-Félix E., Gomez-Gil B., Zúñiga-Cabrera M., González-Candelas F., et al. (2021). Core and Accessory Genome Analysis of Vibrio Mimicus. Microorganisms 9, 191. doi: 10.3390/microorganisms9010191
Hayashi K., Sawabe T., Thompson F. L., Swings J., Gudkovs N., Christen R., et al. (2003). Vibrio Superstes Sp. Nov., Isolated From the Gut of Australian Abalones Haliotis Laevigata and Haliotis Rubra. Int. J. Syst. Evol. Microbiol. 53, 1813–1817. doi: 10.1099/ijs.0.02625-0
Hunt D. E., Gevers D., Vahora N. M., Polz M. F. (2008). Conservation of the Chitin Utilization Pathway in the Vibrionaceae. Appl. Environ. Microbiol. 74, 44–51. doi: 10.1128/AEM.01412-07
Hyatt D., Chen G. L., LoCascio P. F., Land M. L., Larimer F. W., Hauser L. J. (2010). Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinf. 11, 119. doi: 10.1186/1471-2105-11-119
Janeček Š., Zámocká B. (2020). A New GH13 Subfamily Represented by the α-Amylase From the Halophilic Archaeon Haloarcula Hispanica. Extremophiles 24, 207–217. doi: 10.1007/s00792-019-01147-y
Jiang C., Tanaka M., Nishikawa S., Mino S., Romalde J. L., Thompson F. L., et al. (2022). Vibrio Clade 3.0: New Vibrionaceae Evolutionary Units Using Genome-Based Approach. Curr. Microbiol. 79, 1–15. doi: 10.1007/s00284-021-02725-0
Jimenez-Infante F., Ngugi D. K., Vinu M., Alam I., Kamau A. A., Blom J., et al. (2016). Comprehensive Genomic Analyses of the OM43 Clade, Including a Novel Species From the Red Sea, Indicate Ecotype Differentiation Among Marine Methylotrophs. Appl. Environ. Microbiol. 82, 1215–1226. doi: 10.1128/AEM.02852-15
Juhas M., van der Meer J. R., Gaillard M., Harding R. M., Hood D. W., Crook D. W. (2009). Genomic Islands: Tools of Bacterial Horizontal Gene Transfer and Evolution. FEMS Microbiol. Rev. 33, 376–393. doi: 10.1111/j.1574-6976.2008.00136.x
Kirchberger P. C., Turnsek M., Hunt D. E., Haley B. J., Colwell R. R., Polz M. F., et al. (2014). Vibrio Metoecus Sp. Nov., a Close Relative of Vibrio Cholerae Isolated From Coastal Brackish Ponds and Clinical Specimens. Int. J. Syst. Evol. Microbiol. 64, 3208–3214. doi: 10.1099/ijs.0.060145-0
Kirkup B. C., Chang L., Chang S., Gevers D., Polz M. F. (2010). Vibrio Chromosomes Share Common History. BMC Microbiol. 10, 137. doi: 10.1186/1471-2180-10-137
Kumar S., Stecher G., Li M., Knyaz C., Tamura K. (2018). MEGA X: Molecular Evolutionary Genetics Analysis Across Computing Platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lee J., Lee D., Sim M., Kwon D., Kim J., Ko Y., et al. (2018). Mysyntenyportal: An Application Package to Construct Websites for Synteny Block Analysis. BMC Bioinf. 19, 216. doi: 10.1186/s12859-018-2219-x
Lee L. H., Raghunath P. (2018). Editorial: Vibrionaceae Diversity, Multidrug Resistance and Management. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.00563
Mann A. J., Hahnke R. L., Huang S., Werner J., Xing P., Barbeyron T., et al. (2013). The Genome of the Alga-Associated Marine Flavobacterium Formosa Agariphila KMM 3901T Reveals a Broad Potential for Degradation of Algal Polysaccharides. Appl. Environ. Microbiol. 79, 6813–6822. doi: 10.1128/AEM.01937-13
Moya A., Peretó J., Gil R., Latorre A. (2008). Learning How to Live Together: Genomic Insights Into Prokaryote–Animal Symbioses. Nat. Rev. Genet. 9, 218–229. doi: 10.1038/nrg2319
Muto A., Kotera M., Tokimatsu T., Nakagawa Z., Goto S., Kanehisa M. (2013). Modular Architecture of Metabolic Pathways Revealed by Conserved Sequences of Reactions. J. Chem. Inf. Model. 53, 613–622. doi: 10.1021/ci3005379
Nyholm S. V., McFall-Ngai M. J. (2004). The Winnowing: Establishing the Squid - Vibrios Symbiosis. Nat. Rev. Microbiol. 2, 632–642. doi: 10.1038/nrmicro957
Okada K., Iida T., Kita-Tsukamoto K., Honda T. (2005). Vibrios Commonly Possess Two Chromosomes. J. Bacteriol. 187, 752–757. doi: 10.1128/JB.187.2.752-757.2005
Orata F. D., Kirchberger P. C., Méheust R., Barlow E. J., Tarr C. L., Boucher Y. (2015). The Dynamics of Genetic Interactions Between Vibrio Metoecus and Vibrio Cholerae, Two Close Relatives Co-Occurring in the Environment. Genome Biol. Evol. 7, 2941–2954. doi: 10.1093/gbe/evv193
Parte A. C., Sardà Carbasse J., Meier-Kolthoff J. P., Reimer L. C., Göker M. (2020). List of Prokaryotic Names With Standing in Nomenclature (LPSN) Moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 70, 5607–5612. doi: 10.1099/ijsem.0.004332
Pritchard L., Glover R. H., Humphris S., Elphinstone J. G., Toth I. K. (2016). Genomics and Taxonomy in Diagnostics for Food Security: Soft-Rotting Enterobacterial Plant Pathogens. Anal. Methods 8, 12–24. doi: 10.1039/C5AY02550H
Sawabe T. (2006). “The Mutual Partnership Between Vibrio Halioticoli and Abalones”, in The Biology of Vibrios. Eds. Thompson F. L., Austin B., Swings J.. (Washington, D.C: ASM Press), 219–230. Available at: http://hdl.handle.net/2115/48579.
Sawabe T., Fujimura Y., Niwa K., Aono H. (2007a). Vibrio Comitans Sp. Nov., Vibrio Rarus Sp. Nov. And Vibrio Inusitatus Sp. Nov., From the Gut of the Abalones Haliotis Discus Discus, H. Gigantea, H. Madaka and H. Rufescens. Int. J. Syst. Evol. Microbiol. 57, 916–922. doi: 10.1099/ijs.0.64789-0
Sawabe T., Hayashi K., Moriwaki J., Fukui Y., Thompson F. L., Swings J., et al. (2004a). Vibrio Neonatus Sp. Nov. And Vibrio Ezurae Sp. Nov. Isolated From the Gut of Japanese Abalones. Syst. Appl. Microbiol. 27, 527–534. doi: 10.1078/0723202041748154
Sawabe T., Hayashi K., Moriwaki J., Thompson F. L., Swings J., Potin P., et al. (2004b). Vibrio Gallicus Sp. Nov., Isolated From the Gut of the French Abalone Haliotis Tuberculata. Int. J. Syst. Evol. Microbiol. 54, 843–846. doi: 10.1099/ijs.0.02804-0
Sawabe T., Kita-Tsukamoto K., Thompson F. L. (2007b). Inferring the Evolutionary History of Vibrios by Means of Multilocus Sequence Analysis. J. Bacteriol. 189, 7932–7936. doi: 10.1128/JB.00693-07
Sawabe T., Ogura Y., Matsumura Y., Feng G., Amin A. R., Mino S., et al. (2013). Updating the Vibrio Clades Defined by Multilocus Sequence Phylogeny: Proposal of Eight New Clades, and the Description of Vibrio Tritonius Sp. Nov. Front. Microbiol. 4. doi: 10.3389/fmicb.2013.00414
Sawabe T., Sugimura I., Ohtsuka M., Nakano K., Tajima K., Ezura Y., et al. (1998). Vibrio Halioticoli Sp. Nov., a Non-Motile Alginolytic Marine Bacterium Isolated From the Gut of the Abalone Haliotis Discus Hannai. Int. J. Syst. Bacteriol. 48, 573–580. doi: 10.1099/00207713-48-2-573
Schultz-Johansen M., Bech P. K., Hennessy R. C., Glaring M. A., Barbeyron T., Czjzek M., et al. (2018). A Novel Enzyme Portfolio for Red Algal Polysaccharide Degradation in the Marine Bacterium Paraglaciecola Hydrolytica S66T Encoded in a Sizeable Polysaccharide Utilization Locus. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.00839
Septer A. N. (2019). The Vibrio-Squid Symbiosis as a Model for Studying Interbacterial Competition. mSystems 4, e00180–19. doi: 10.1128/mSystems.00108-19
Shaiber A., Willis A. D., Delmont T. O., Roux S., Chen L. X., Schmid A. C., et al. (2020). Functional and Genetic Markers of Niche Partitioning Among Enigmatic Members of the Human Oral Microbiome. Genome Biol. 21, 292. doi: 10.1186/s13059-020-02195-w
Tanaka M., Mino S., Ogura Y., Hayashi T., Sawabe T. (2018). Availability of Nanopore Sequences in the Genome Taxonomy for Vibrionaceae Systematics: Rumoiensis Clade Species as a Test Case. PeerJ 6, e5018. doi: 10.7717/peerj.5018
Tanaka R., Sawabe T., Yoshimizu M., Ezura Y. (2002). Distribution of Vibrio Halioticoli Around an Abalone-Farming Center in Japan. Microbes Environ. 17, 6–9. doi: 10.1264/jsme2.2002.6
Tang K., Lin Y., Han Y., Jiao N. (2017). Characterization of Potential Polysaccharide Utilization Systems in the Marine Bacteroidetes Gramella Flava JLT2011 Using a Multi-Omics Approach. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00220
Tanizawa Y., Fujisawa T., Nakamura Y. (2018). DFAST: A Flexible Prokaryotic Genome Annotation Pipeline for Faster Genome Publication. Bioinformatics 34, 1037–1039. doi: 10.1093/bioinformatics/btx713
Thompson F. L., Iida T., Swings J. (2004). Biodiversity of Vibrios. Microbiol. Mol. Biol. Rev. 68, 403–431. doi: 10.1128/MMBR.68.3.403-431.2004
Toft C., Andersson S. G. E. (2010). Evolutionary Microbial Genomics: Insights Into Bacterial Host Adaptation. Nat. Rev. Genet. 11, 465–475. doi: 10.1038/nrg2798
van der Maarel M. J. E., van der Veen B., Uitdehaag J. C., Leemhuis H., Dijkhuizen L. (2002). Properties and Applications of Starch-Converting Enzymes of the α-Amylase Family. J. Biotechnol. 94, 137–155. doi: 10.1016/S0168-1656(01)00407-2
Wernegreen J. J. (2002). Genome Evolution in Bacterial Endosymbionts of Insects. Nat. Rev. Genet. 3, 850–861. doi: 10.1038/nrg931
Keywords: vibrionaceae, marine invertebrate, symbiosis, planktonic, halioticoli clade, complete genome
Citation: Jiang C, Mino S and Sawabe T (2022) Genomic Analyses of Halioticoli Clade Species in Vibrionaceae Reveal Genome Expansion With More Carbohydrate Metabolism Genes During Symbiotic to Planktonic Lifestyle Transition. Front. Mar. Sci. 9:844983. doi: 10.3389/fmars.2022.844983
Received: 29 December 2021; Accepted: 07 March 2022;
Published: 23 March 2022.
Edited by:
Fabiano Thompson, Federal University of Rio de Janeiro, BrazilReviewed by:
Xiao-Hua Zhang, Ocean University of China, ChinaCopyright © 2022 Jiang, Mino and Sawabe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomoo Sawabe, c2F3YWJlQGZpc2guaG9rdWRhaS5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.