
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 06 April 2022
Sec. Marine Evolutionary Biology, Biogeography and Species Diversity
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.839225
 Chaowei Zhou1,2†
Chaowei Zhou1,2† Yan Li1,2†Yan Zhou1,2†Yu Zou3†
Yan Li1,2†Yan Zhou1,2†Yu Zou3† Dengyue Yuan1,2Xingxing Deng4Luo Lei5Jian Su6Chengke Zhu1,2Hua Ye1,2Hui Luo1,2Guangjun Lv1,2Xinghua Zhou1,2Gangqiao Kuang1,2Chuang Zhang7Jun Wu6Zonglin Zheng1,2
Dengyue Yuan1,2Xingxing Deng4Luo Lei5Jian Su6Chengke Zhu1,2Hua Ye1,2Hui Luo1,2Guangjun Lv1,2Xinghua Zhou1,2Gangqiao Kuang1,2Chuang Zhang7Jun Wu6Zonglin Zheng1,2 Shijun Xiao3*Minghui Li2*
Shijun Xiao3*Minghui Li2*Northern snakehead, Channa argus (C. argus), is an important economic and ecological fish species. The wild population of the species was sharply declined in the last decade. A high-quality reference genome could lay a solid foundation for the genetic and conservation studies for C. argus. In this work, we report a chromosomal genome assembly with PacBio and Hi-C technology using the albino northern snakehead, a color variety of C. argus. A 644.1-Mb genome with 24 chromosomes was obtained with a contig and scaffold N50 of 11.78 and 27.8 Mb, respectively. We inferred that C. argus diverged from A. testudineus around 85.6 million years ago. 514 expanded gene families and 214 positively selected genes were identified in the C. argus genome. The chromosome-level genome provides a valuable high-quality genomic resource for population, as well as genetic and evolutionary studies for C. argus and other species in Channidae.
Northern snakehead fish, Channa argus (C. argus), belonging to Osteichthyes, Perciformes, and Channidae, is an important ecological and economic fish species in tropical and subtropical Asia and Africa (Cheng and Zheng, 1987; Courtenay and Williams, 2004). C. argus possesses many excellent characteristics for its roles in aquaculture including strong fecundity, fast growth, anti-hypoxia, delicious taste, exquisite meat quality, less bone spurs, and high nutritional value (Glass et al., 1986; Chen and Yang, 2013). In addition, C. argus has been reported to have medicinal values such as removing blood stasis, generating muscle and blood, nourishing, and conditioning and was usually used as a primary food of daily tonic and wound healing (Yuan et al., 2005; Qin and Jiang, 2010). Moreover, C. argus has excellent hypoxia tolerance due to the upper gill organ, enabling long-distance transportation for the species (Xiao et al., 2019). C. argus has become a widely farmed aquatic species in China, leading to a rapid development in its breeding industry in recent years (Li et al., 2007; Xiao et al., 2015).
The albino northern snakehead (C. argus var.) is a member of the Channidae family of the Perciformes and is distributed mainly in the middle-lower reaches of the Jialing River Basin in Sichuan Province, China (Shi et al., 1980). According to records, C. argus var. was originally considered to be a subspecies of the Northern snakehead C. argus (Wang et al., 1992). Subsequently, a large amount of molecular biological evidence proved that C. argus var. was not a subspecies of C. argus but an albino population (Li et al., 2016; Zhou et al., 2017). Meanwhile, because of its freshness, high nutritional value (e.g., high polyunsaturated fatty acid omega-6 levels), and potential ornamental value (e.g., all white of body) compared to those of the C. argus (Figure 1), it is a valuable economic and ornamental fish in China (Zou et al., 2017; Zhou et al., 2018). The average market price of C. argus var. is about 3–4 times higher than that of C. argus (Zhou et al., 2018). However, due to the environmental deterioration and overfishing, the wild populations of the species were declining in the last decade (Zhou et al., 2018). Additionally, the low survival rate and aberration rate in larval breeding seriously limited the development of intensive aquaculture of C. argus var.

Figure 1 A picture of the albino northern snakehead C. argus var. for the reference genome construction.
Genome is one of the most important genetic resources in the ecological and breeding studies for species, especially for research aiming at improving economic traits for farming animals. In recent years, the genomes of many fish species have been successfully reported, including Takifugu rubripes (Aparicio et al., 2002), Oryzias latipes (Kasahara et al., 2007), Danio rerio (Howe et al., 2013), Cyprinus carpio (Xu et al., 2014), Litopenaeus vannamei (Zhang et al., 2019), Oxygymnocypris stewartii (Liu et al., 2019), Datnioides undecimradiatus (Sun et al., 2020), and Platycephalus sp.1 (Xu et al., 2021). A previous study has shown that the genomic application of a reference genome largely depends on the continuity and completeness quality of genome sequences (Xiao et al., 2020). The genome of C. argus has been reported in 2017 by Jian Xu (Xu et al., 2017), which provides basic genomic data for studies of the species. However, the public genome of C. argus was assembled using short reads from next-generation sequencing technologies and was highly fragmented with the contig N50 length of 81.4 kb (Xu et al., 2017). More importantly, the genome was not assembled into the chromosomal level, making the genome not being able to provide sufficient genomic information for the following chromosome evolution and fine mapping of functional genes for important economic traits (Dan et al., 2018). There is a great demand for a chromosome-level high-quality reference genome of C. argus to facilitate and prompt evolutionary and conservation studies and functional gene mapping of the critical economic traits for the species.
Here, for the first time, we presented a high-quality chromosomal genome assembly for the albino northern snakehead (C. argus var.) with a combined strategy of Illumina, PacBio, and Hi-C technology. The contig and scaffold N50 length reached 11.78 and 27.8 Mb, respectively. More than 95.8% of the sequence reads were assembled into 24 chromosomes, demonstrating the outstanding completeness and sequence continuity of the reference genome. 22,593 protein-coding genes were predicted in the assembled genome, and more than 91.9% of those genes were successfully functionally annotated. We believe that the high-quality chromosomal genome would provide a valuable reference not only for the genomic dissection of the phenotypic variation in the species but also for the evolutionary investigation of Channidae family among teleosts.
A female individual of C. argus var. was reared in Neijiang Fish Farm (Neijiang City, Sichuan Province, China) and was used for the genome sequencing and assembly (Figure 1). A total of 12 tissues, including white muscle, skin, spleen, liver, intestinal, ovary, swim bladder, kidney, heart, brain, eye, and gill, were collected and then quickly frozen and stored in liquid nitrogen for 6 h. Of these tissues, the white muscle was used for DNA sequencing for genome assembly and all tissues were used for transcriptome sequencing.
To construct a DNA sequencing library, we extracted the genomic DNA from the muscle tissue of a female individual using the standard phenol/chloroform extraction method. The quality of genomic DNA molecules was checked, and we required that the main band of extracted DNA was around 20 kb in the agarose gel electrophoresis experiment, and the DNA spectrophotometer ratio (SP) 260/280 was larger than 1.8. Subsequently, short-read (insert size: 250 bp) and long-read (insert size: 20 kb) DNA sequencing libraries were created according to the protocols of manufacturers, the former one for the whole genome sequencing based on the Illumina HiSeq X Ten platform and the latter for the PacBio Sequel platform, respectively.
RNA-seq data can be used to improve the quality of the genome annotation. To include as many tissue-specific expressed transcripts for the analysis as possible, RNAs of all the 12 collected tissues were extracted using the TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). The purified RNA quantity and quality for each tissue were assessed, and we required that the absorbance be larger than 1.7 at 260 nm/280 nm based on the NanoDrop ND-1000 spectrophotometer (LabTech, Hopkinton, MA, USA) and the RIN value was larger than 8.5 on the basis of the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), respectively. Then, RNA molecules from all the 12 collected tissues were equally mixed before the subsequent transcriptome sequencing with the Illumina HiSeq X Ten platform according to the manufacturer’s protocol. Briefly, 3 µg of RNA molecules was used for library construction. After the library construction including purification, fragmentation, cDNA synthesis, adaptor ligation, and fragment selection, the transcripts were sequenced with Illumina HiSeq X Ten (Illumina Inc., San Diego, CA, USA) system using the paired-end 150-bp mode.
The short sequencing data of C. argus var. from the Illumina platform were used for the genome character evaluation using the Kmer-based method (Liu et al., 2013). The sequencing data were firstly quality checked and filtered before the analysis. HTQC (Yang et al., 2013) was used for the low-quality base/read filtering, and FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was applied for the quality control. All adapter sequences that reside in reads were removed, and paired-end reads with more than 10% ambiguous bases or with more than 50% low-quality bases (Phred score <5) were filtered. The 17mers were generated from the sequencing data using the Jellyfish package (Marçais and Kingsford, 2011), and the frequency of all 17mers was plotted to illuminate the genome characters.
Long reads from the whole-genome PacBio sequencing were used for the de novo genome assembly. Falcon 2.1.4 (Chin et al., 2016) was used for the genome assembly with the parameters listed in Supplementary Table S1. To eliminate ineluctable base errors in the assembly, the PacBio long reads and Illumina short reads were used again for the base correction. Firstly, the PacBio long reads were mapped upon the preliminary genome using blasr software (Chaisson and Tesler, 2012) and the alignment results were used for the sequence polish using arrow utility (ARROW in GCpp v1.9.0) (Chin et al., 2013) with the minCoverage of 15. Secondly, two rounds of sequence polish using Illumina short reads were performed using BWA (Abuín et al., 2015) for read alignment and Pilon 1.23 (Walker et al., 2014) for the base correction.
The muscle tissue was used for the Hi-C library construction and sequencing. Two micrograms of tissue from the same individual used for the genome assembly was collected. The chromatin cross-linking, lysis, digestion, marking with biotin, ligation, chromatin cross-linking reversal, and DNA fragment collection for Hi-C library construction were performed using the identical experimental process in the previous study (Xu et al., 2018b). The DNA molecules were used for the library construction and sequencing as traditional genome sequencing project using the Illumina HiSeq X Ten platform (Illumina, San Diego, CA, USA).
The interaction frequencies among contigs were estimated from the sequencing data; however, the data analysis was different from that of a traditional whole-genome sequencing project, since abundant chimeric reads could be observed in the Hi-C library sequencing. We first applied an iterative alignment strategy to align the reads to the assembled contigs using Bowtie (Langmead, 2010) with a single end mode. Only read pairs where both ends were uniquely aligned to the contigs were selected for the following study. Then, the interaction frequencies among contigs were evaluated using the hiclib python library (Imakaev et al., 2012). At last, contigs were clustered, ordered, and orientated to restore their relative locations along chromosomes using an agglomerative hierarchical clustering method implemented in Lachesis (no version) (Burton et al., 2013).
Before the protein-coding gene annotation in the C. argus var. genome, both tandem and interspersed repeats were predicted and masked. Tandem Repeat Finder (TRF 4.09) (Benson, 1999) was used to detect tandem repeats in the genome. RepeatMasker 4.1.2 and RepeatProteinMask were used to predict interspersed repeats based on the Repbase database (Jurka et al., 2005). The de novo prediction was applied with RepeatMasker 4.1.2 using the combined library from RepeatModeler 2.0.2a and LTR-FINDER 1.0.7 (Xu and Wang, 2007). All repeat types were merged to eliminate the redundancy.
To predict protein-coding genes in the C. argus var. genome, de novo and homolog- and RNA-seq-based methods were used. De novo prediction was performed with Augustus 3.4.0 (Stanke et al., 2006). For the homolog-based method, protein sequences of Anabas testudineus, Danio rerio, Oryzias latipes, Tetraodon nigroviridis, and Xiphophorus maculatus were downloaded from the Ensembl database release 96 (Flicek et al., 2007) and aligned to the C. argus var. genome with TBLASTN utility (Altschul, 2012), which was processed with GeneWise 2.4.1 (Birney et al., 2004) to obtain the gene models. TopHat 2.1.1 was used to map RNA-seq data upon the C. argus var. genome (Trapnell and Pachter Lsalzberg, 2009), and Cufflinks 2.2.1 was then used to assemble transcripts (Ghosh and Chan, 2016). Packages were used to map RNA-seq data upon the C. argus var. genome and extract gene information. The MAKER 3.01.02 (Cantarel et al., 2008) package was used to merge all gene models from de novo and homolog- and RNA-seq-based methods.
All the final protein-coding genes were searched against NR, TrEMBL, Swissport, and COG databases using BLAST 2.11.0+ utility (Altschul, 2012) with a maximal e-value of 1e-5. Blast2GO 5.2.5 (Conesa et al., 2005) software was used for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation.
Coding sequences and corresponding protein sequences for 11 fish species, including Callorhinchus milii, Lepisosteus oculatus, D. rerio, Gadus morhua, X. maculates, O. latipes, Anabas testudineus, Gasterosteus aculeatus, Larimichthys crocea, T. nigroviridis, and Takifugu rubripes, were downloaded from the Ensembl database. The longest transcript and encoded protein sequence for each gene locus was selected, and the OrthoMCL 2.0.9 pipeline (Li et al., 2003) with default settings was used to identify their relationships within and among species. Then, the protein sequences of one-to-one ortholog genes were aligned with muscle (Edgar, 2004) and were converted into nucleotide sequences using pal2nal (Suyama et al., 2006). Hypervariable regions of the alignments were removed with Gblocks 0.91b (Castresana, 2000) with default settings, and the remaining sequences were concatenated and fed into RAxML (Stamatakis, 2014) to reconstruct the relationships among these species. One hundred times of rapid bootstrap (Stamatakis et al., 2008) resampling were performed to access the robustness of the topology. Based on the topology and the alignment matrix, the divergence times among these species were estimated using MCMCTREE included in the PAML 4.9 package software (Yang, 2007) with the calibration time obtained by consulting the TimeTree database (Kumar et al., 2017). The calibration times were fetched from TimeTree (http://www.timetree.org/), including Callorhinchus milii and Danio rerio, 453–497 MYA, Lepisosteus oculatus and Danio rerio, 295–334 MYA, Tetraodon and Oryzias latipes, 165.2–149.85 MYA, Oryzias latipes and Takifugu rubripes, 104–145 MYA, Takifugu rubripes and Gasterosteus aculeatus, 99–127 MYA, and Takifugu rubripes and Tetraodon nigroviridis, 42–57 MYA. Based on the aforementioned results, the dynamics of the gene families reside in the genome including expansions and contractions were detected with café 4.2 (De Bie et al., 2006). Furthermore, candidate genes probably subject to positive selection were identified using CODEML by comparing the differences of likelihood values between model A with two different settings (fix_omega = 1 omega = 1 vs. fix_omega = 0 omega = 1.5) and the chi2 distribution.
The CDS sequences from the C. argus var. and C. argus genome were blasted against each other by BLASTN (Altschul, 2012). Genes from the two genomes with an alignment ratio larger than 80% were considered as shared genes. Genes with any hit were recognized as non-hit genes. To identify genome-specific genes, non-hit genes from the C. argus genome were aligned to the C. argus var. genome using exonerate (https://github.com/nathanweeks/exonerate) with parameters: –model est2genome –percent 80 –showtargetgff 1. If a gene had no hit on the C. argus var. genome, the gene was identified as C. argus specific genes. In the identical method, C. argus var. specific genes were also identified.
Based on the Illumina HiSeq X Ten platform, a total of 42.99 Gb cleaned data with ~67× coverage of estimated genome size was generated (Table 1). Using the high-quality whole-genome sequencing data, we applied the Kmer-based method for the genome size, heterozygosity, and repeat content estimation. The genome size was estimated using the Kmer-based method, and results are shown in Figure 2. Because 17mers with extremely low frequency most likely resulted from base errors in the PCR or sequencing, all 17mers with the frequency lower than 5 were excluded from the genome character estimation. As shown in Figure 2, we estimated that the genome size of C. argus var. was 668 Mb. We observed low heterozygosity in the Kmer plot, resulting in the whole-genome heterozygosity of 0.096%. The heterozygosity of C. argus var. was relatively smaller than for many teleosts (Xu et al., 2018a), even for other aquaculture species generated from gynogenesis (Wang et al., 2015), implying that the genetic diversity of the C. argus var. might be rather low in population.

Table 1 The statistics of the DNA and RNA sequencing data for the genome assembly and annotation.
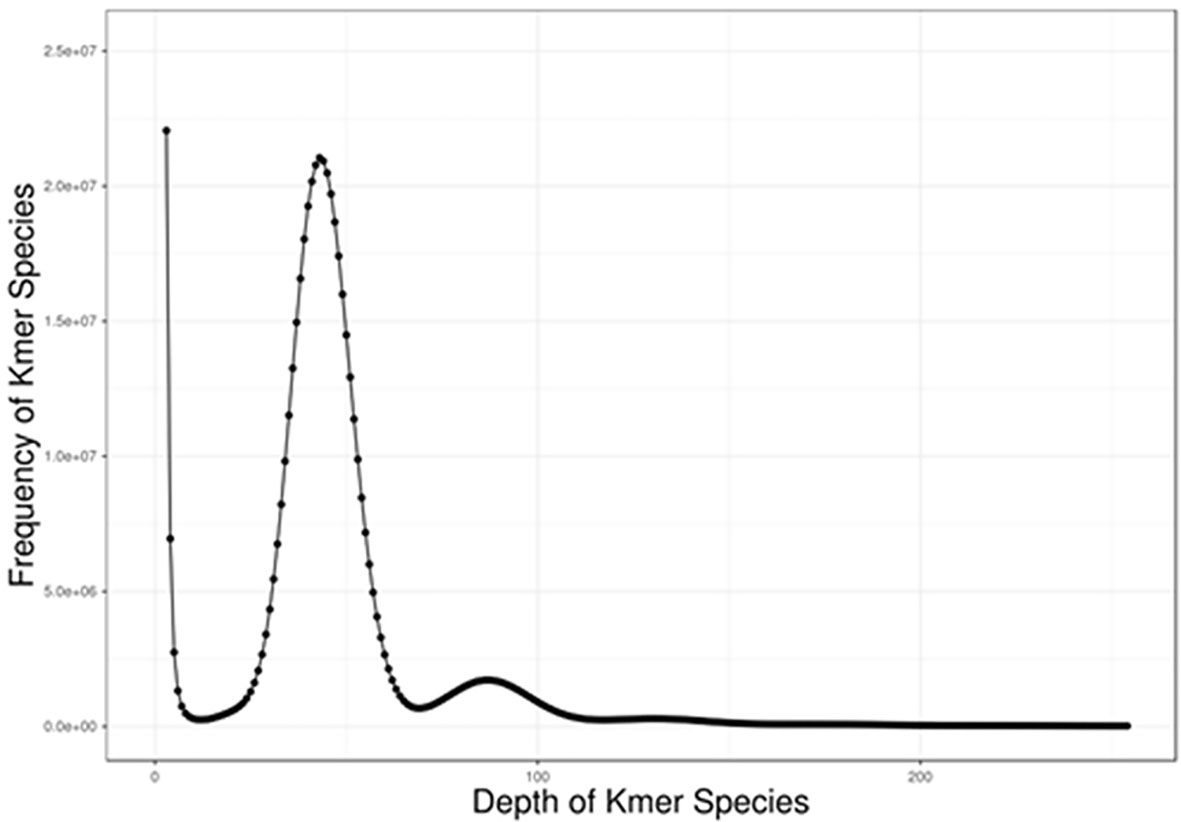
Figure 2 The frequency of 17mer distribution from the whole-genome Illumina sequencing data. The peak around 45 represented the frequency of homozygous Kmers and that around 90 referred to the frequency of repeated Kmers.
To de novo assemble the C. argus var. genome, we constructed and sequenced a 20-kb DNA library using the PacBio Sequel platform and obtained 4,650,237 subreads totaling 52.85 Gb and representing ~83× of estimated genome size (Table 1). The N50 length of the subreads was 18 kb with the maximal subread length of 94 kb (Supplementary Figure S1). After the preliminary assembly using Falcon 2.1.4, 640.6-Mb genomes with 749 contigs were obtained. The N50 and maximal length of contigs were 11.91 and 27.5 Mb, respectively (Supplementary Table S2). After rounds of polishing based on the long reads used for de novo assembly and short reads used for survey, the final genome was 644.1 Mb of 749 contigs with an N50 length of 11.98 Mb (Supplementary Table S2). The completeness of the assembled genome was validated by Benchmarking Universal Single-Copy Orthologs 3.0 (BUSCO 3.0) analysis using BUSCO v3.0 with the actinopterygii_odb9 database. As a result, 4,458 (97.2%) of the 4,584 BUSCO genes were completely identified in the genome with 4,334 (94.5%) single-copy and 124 (2.7%) multi-copy genes (Table 2), suggesting excellent completeness for the genome assembly.

Table 2 BUSCO analysis to validate the completeness of the genome assembly.
Although we obtained a genome with less than 1,000 contigs (Table 3), the genome sequences were still fragmented since the karyotype of C. argus was 2n = 48 according to the previous studies (Song et al., 2012; Zhong et al., 2016). The relative position and orientation along chromosomes of those contigs were crucial for the comparative genomic analysis, especially for the chromosome evolution studies. Many traditional scaffolding strategies were developed and reported to anchor contigs into chromosomes, such as genetic mapping, physical mapping, BAC sequencing, and large insert-size mate-pair sequencing; however, those methods were time-/labor-consuming and costly. In this work, we applied the Hi-C technique to assemble the first chromosome assembly of C. argus var.
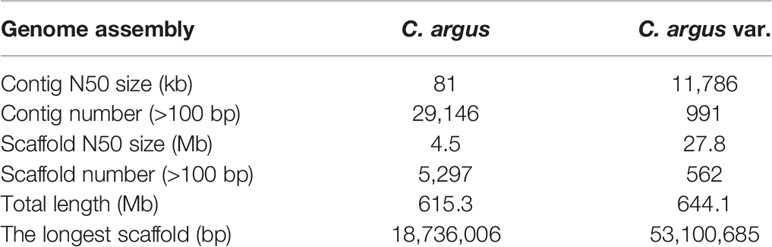
Table 3 Comparison of the two versions of genome assembly based on the short- and long-read sequencing technology, respectively.
From the Hi-C sequencing, we obtained more than 78 Gb of sequencing data, covering 122× of the C. argus var. genome (Table 1). As a result, a 644.1-Mb genome with 562 sequences and a contig/scaffold N50 length of 11.78/27.76 Mb were obtained (Table 3). It is worth noting that gaps among contigs were filled with 100-bp Ns in the genome; therefore, the gap lengths in the genome did not represent the real or estimated length. The total length of the top 24 longest sequences, representing 24 chromosomes of the C. argus var., was 617.6 Mb, covering more than 95.8% of contigs on the base level (Supplementary Figure S2). A total of 538 genome sequences were unplaced upon chromosome after the Hi-C analysis. The contig N50 length of unplaced sequences was 41.7 kb, which was significantly shorter than that of the genome level. We attributed the unplacement of those sequences to the insufficiency of interaction information with other contigs due to their short length.
More than 20% of the genomes were predicted as repetitive elements, and long interspersed nuclear elements (LINE) represented the most abundant repeat type in the genome (Table 4). For assisting the annotation of gene structures, about 10.8 Gb of transcriptome data (Table 1) representing almost all of the expressed genes from the 12 collected tissues was generated. Together with the other two annotation strategies including the de novo and homolog-based methods, we obtained 22,593 protein-coding genes in the C. argus var. genome at last (Table 5), of which 96.61% could be supported by transcripts with at least a 50% overlap. The annotation of gene structures was quality controlled by comparing to the closely related species. As shown in Figure 3, the gene structures of C. argus var. was comparable to those of A. testudineus, D. rerio, O. latipes, T. nigroviridis, and X. maculatus. Based on the chromosome assembly, the density of genes, repeat types, and GC contents were plotted along the chromosomes as in Figure S5. Generally, the distributions of repeats and genes are inversely correlated. Of all the predicted protein-coding genes, more than 91.9% were annotated to at least one public database (Table 6). We also compared the coding sequences of C. argus var. with those of C. argus and calculated the ks values for each gene. The median value of the ks values is 0.008, which is much larger than that of the distance of the COI gene but is still within the range of species (Zhou et al., 2015).
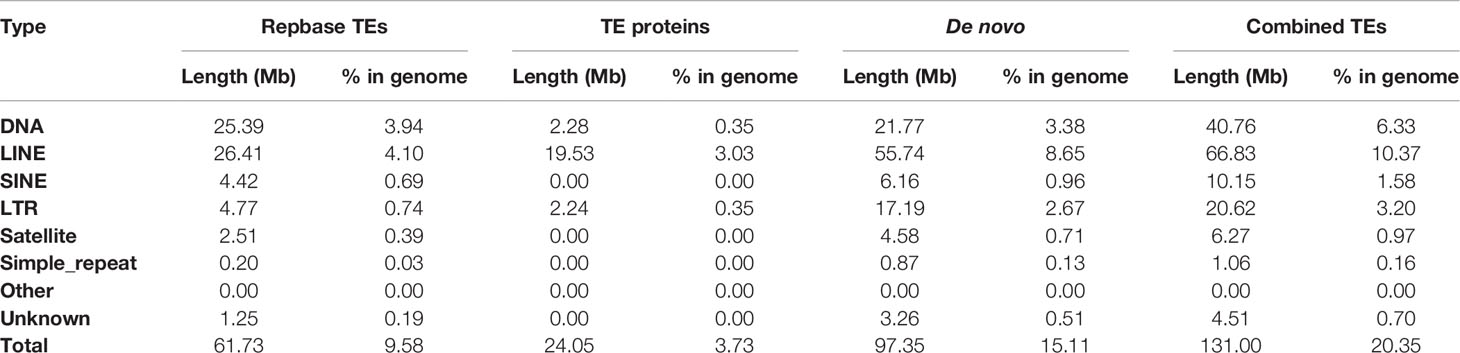
Table 4 The repetitive element predicted from the C. argus var. genome.
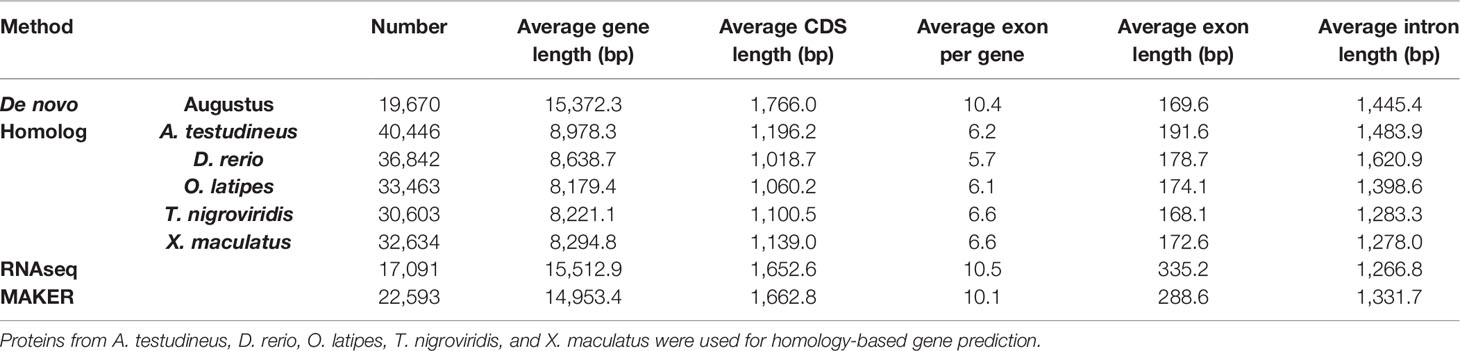
Table 5 The protein-coding gene annotation in the C. argus var. genome.
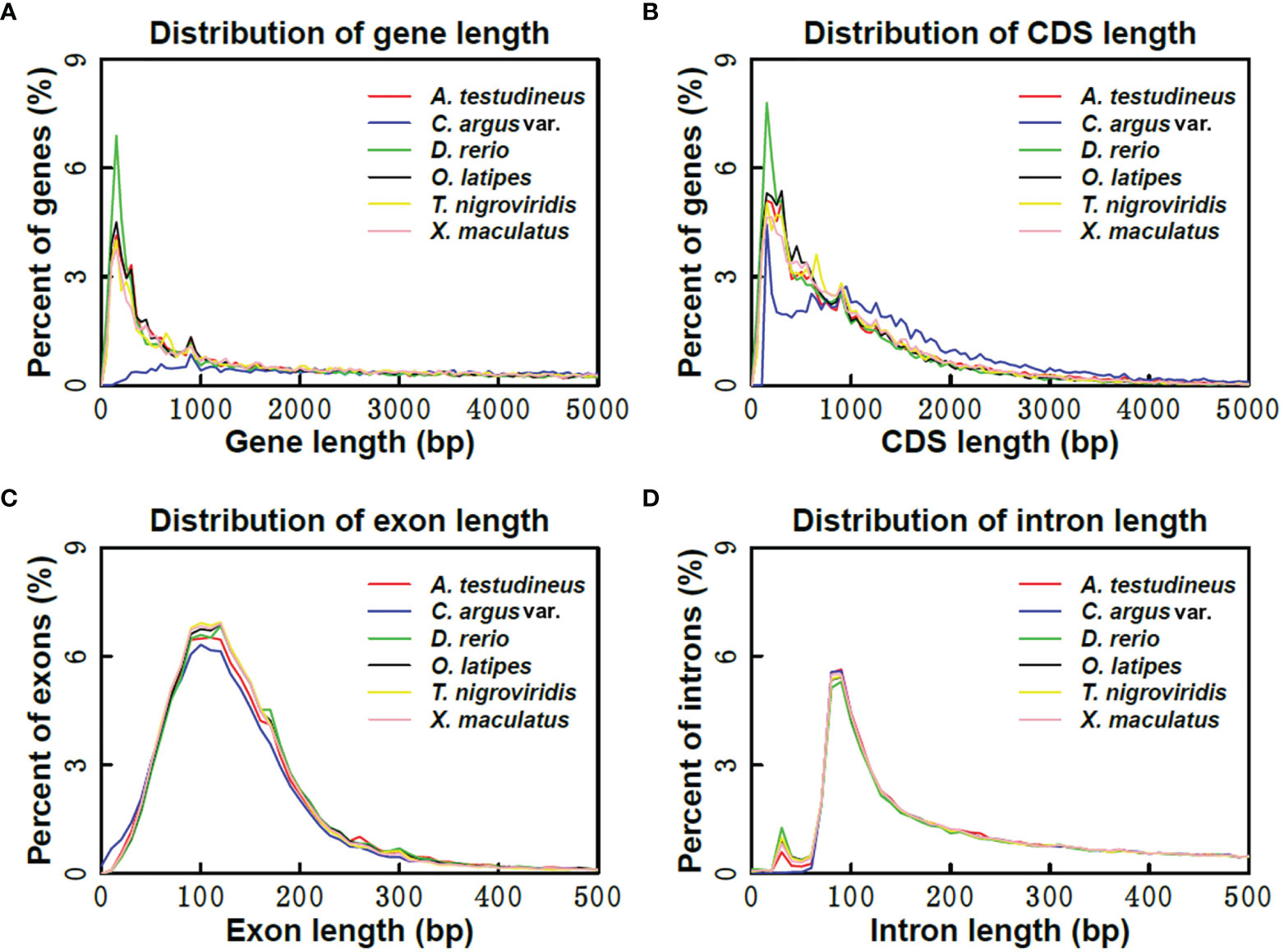
Figure 3 The gene structure comparison of C. argus var. to A. testudineus (Anabas testudineus), D. rerio, O. latipes, T. nigroviridis, and X. maculatus. (A) Gene length distribution comparison; (B) CDS length distribution comparison; (C) exon length distribution comparison; (D) intron length distribution comparison.
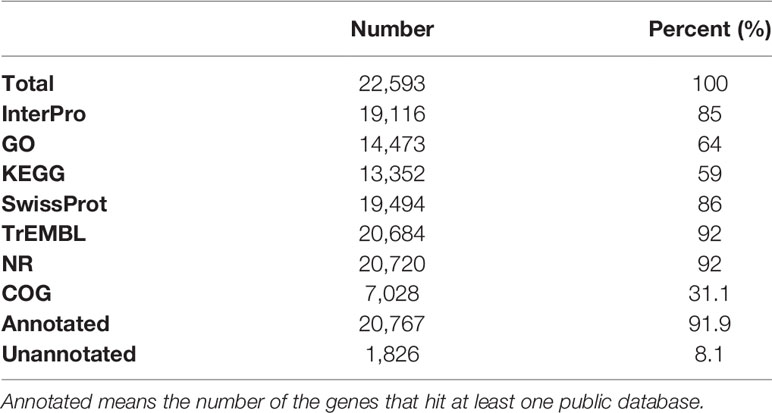
Table 6 The functional annotation statistics for the predicted protein-coding genes.
Although previous studies have reported one version of genome for C. argus, we found that our recent genome exhibited significant improvements on both continuity and completeness. Firstly, the published C. argus was assembled by short reads from next-generation sequencing technology (Xu et al., 2017), but our genome was assembled using long reads from the PacBio sequencing platform. The contig N50 length in our work (11.98 Mb) was more than 140 times higher than the previous version (81.4 kb), while the contig number was about 40 times smaller than that of the previous version (Table 3), indicating the remarkable updates on the reference genome continuity. As far as we know, the contig N50 length of 11.98 Mb for the new genome surpassed the majority teleost genomes, including model teleost species such as zebrafish and medaka. Secondly, the BUSCO comparison between our and previous genomes showed that 97.2% of BUSCO genes were identified in our genome, but only 82.9% were detected in the old genome, suggesting that our genome exhibited higher completeness. This might also explain that more protein-coding genes were predicated in this work. The continuity and completeness are crucial for a reference genome, since the fragmentation of the genome might break the continuity of the gene sequence in the genome and spoil the genome comparison among species, leading to incompleteness and inaccuracy alignments. Therefore, the merits of our genome make the new reference more suitable for gene and genome sequence analyses.
More importantly, our genome sequences were anchored into chromosomes by the Hi-C technique in this work, providing essential reference genomes for the genome evolution on chromosome levels. Similar to the contigs, the scaffold number of C. argus var. in this work was much lower whereas the contig N50 size is much bigger than that of C. argus (Table 3). With the development of functional genomics and the increasing research interests for the economic species, a large amount of genetic analyses for Channa argus, such as quantitative trait locus (QTL) and genome-wide association study (GWAS), will be performed. Those genetic analyses highly rely on the chromosome assembly.
Using the OrthoMCL 2.0.9 pipeline, a total of 3,166 single-copy ortholog groups were detected (Supplementary Figure S3). After alignment and removal of gapped regions, a concatenated alignment matrix with a length of 1,877,704 bps was created based on these genes. The recovered phylogeny showed that C. argus var. and another species belonging to Perciformes, A. testudineus, are sister species with high confidence (Figure 4). The phylogenetic tree of species in this work was consistent with previous study (Xu et al., 2017). Divergence time estimation showed that the two sister species diverged about 85.6 million years ago (Figure 4). The dynamics of members of gene families that reside in the genome may be the result of natural selection. A total of 514 and 2,664 gene families were probably subject to expansions and contractions within the genome (Supplementary Figure S4). The top enriched pathways of these expanded gene families are olfactory transduction, phagosome, and intestinal immune network for IgA production (Supplementary Table S3). On the contrary, the top enriched pathways of the contracted gene families are tight junction, cardiac muscle contraction, and NOD-like receptor signaling pathway (Supplementary Table S3). Another aspect of the impact of natural selection is the substitution of amino acids. Generally, the change of encoded amino acids is harmful to the species. However, some non-synonymous substitutions may increase the fitness of the species, especially when the living environment changes, and genes harboring these changes were termed positively selected genes (PSG). Using the branch-site model, a total of 214 genes were identified to be candidate PSGs in C. argus var., and they may be pivotal for the survival of the albino individuals. Functional analysis showed that genes participate in the pathways of non-homologous end-joining and protein export, and basal transcription factors were most significantly enriched (Supplementary Table S4). Especially, the genes participate in the pathways. Homologous recombination, mismatch repair, and DNA replication were also significantly enriched. These genes including rad50, rad51d, brcc36, msh6, and dna2 may protect the albino fish from ultraviolet radiation.
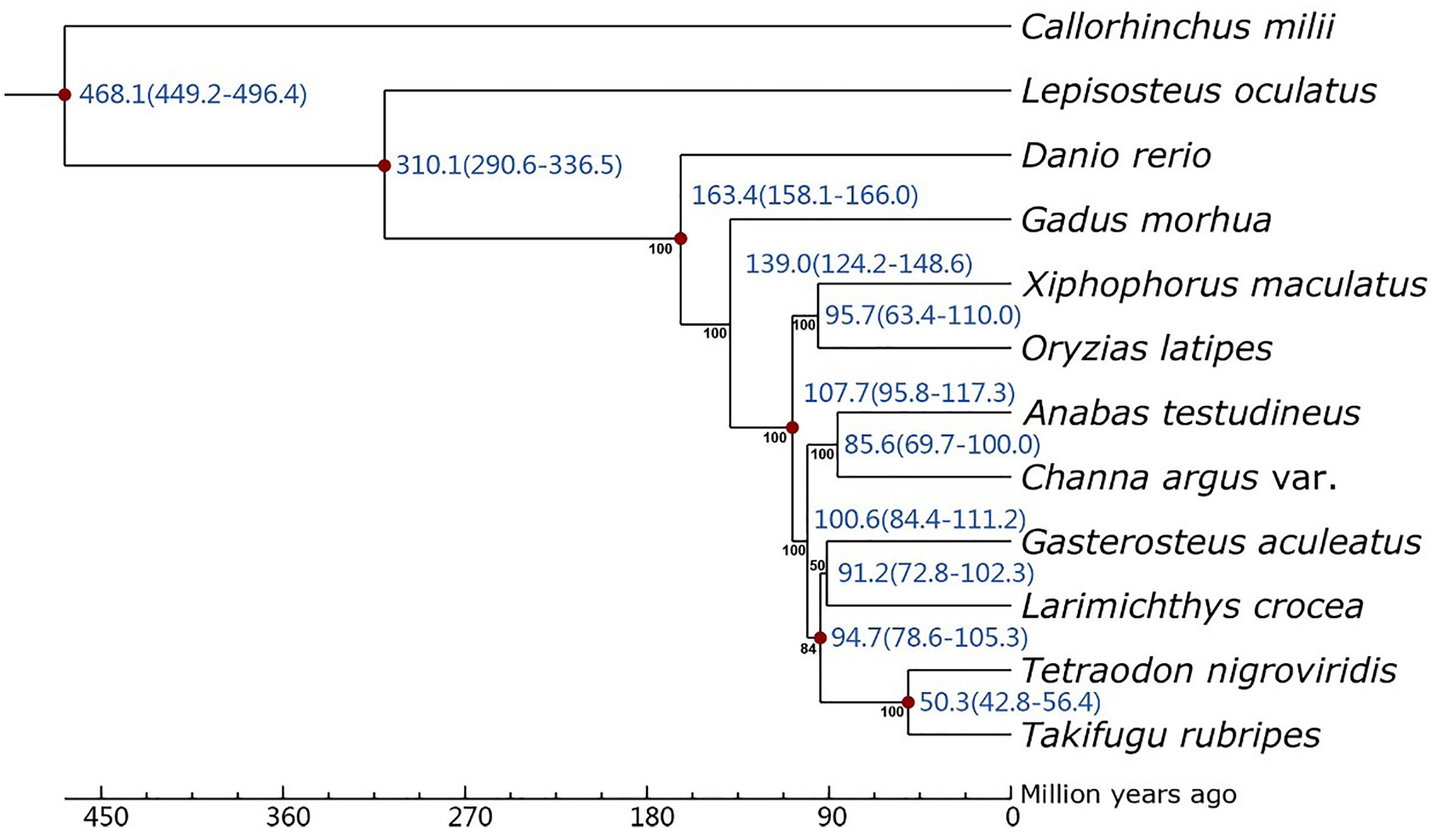
Figure 4 Phylogenetic relationships of C. argus var. and other fish species. Black numbers near nodes are bootstrap values estimated from 100 times of resampling. Blue number near nodes are the estimated divergence times with the 95% confidence interval. Nodes marked with red denotes the calibration time point.
Using sequence blast among genes from the C. argus var. and C. argus genomes, we found 425 and 136 specific genes for C. argus and C. argus var., respectively (Supplementary Tables S5, S6). As the top two groups of gene annotated in KEGG for C. argus, 26 immune-system and 22 signal transduction genes were identified (Figure 5A). To reveal the possible contribution of genome-specific genes for C. argus and C. argus var., functions of specific genes were enriched for each genome. We found that C. argus-specific genes were enriched on immunologic processes, such as antigen processing and presentation, suggesting that C. argus and C. argus var. might respond differently to pathogen infection (Figure 5B). Meanwhile, C. argus-specific genes were also significantly enriched on fatty acid metabolism, such as linoleic acid metabolism (Figure 5B). The result provided useful hints for the following C. argus and C. argus var. phenotype comparison. Interestingly, we found that adcy5 (adenylate cyclase 5) was specifically identified in the C. argus genome (Figure 5C). Previous studies have shown that adcy5 is required for melanophore and pigmentation patterns for fish (Kottler et al., 2015); therefore, the absence of gene of adcy5 might be related to the albinism of C. argus, which needs further validation in the following investigations.
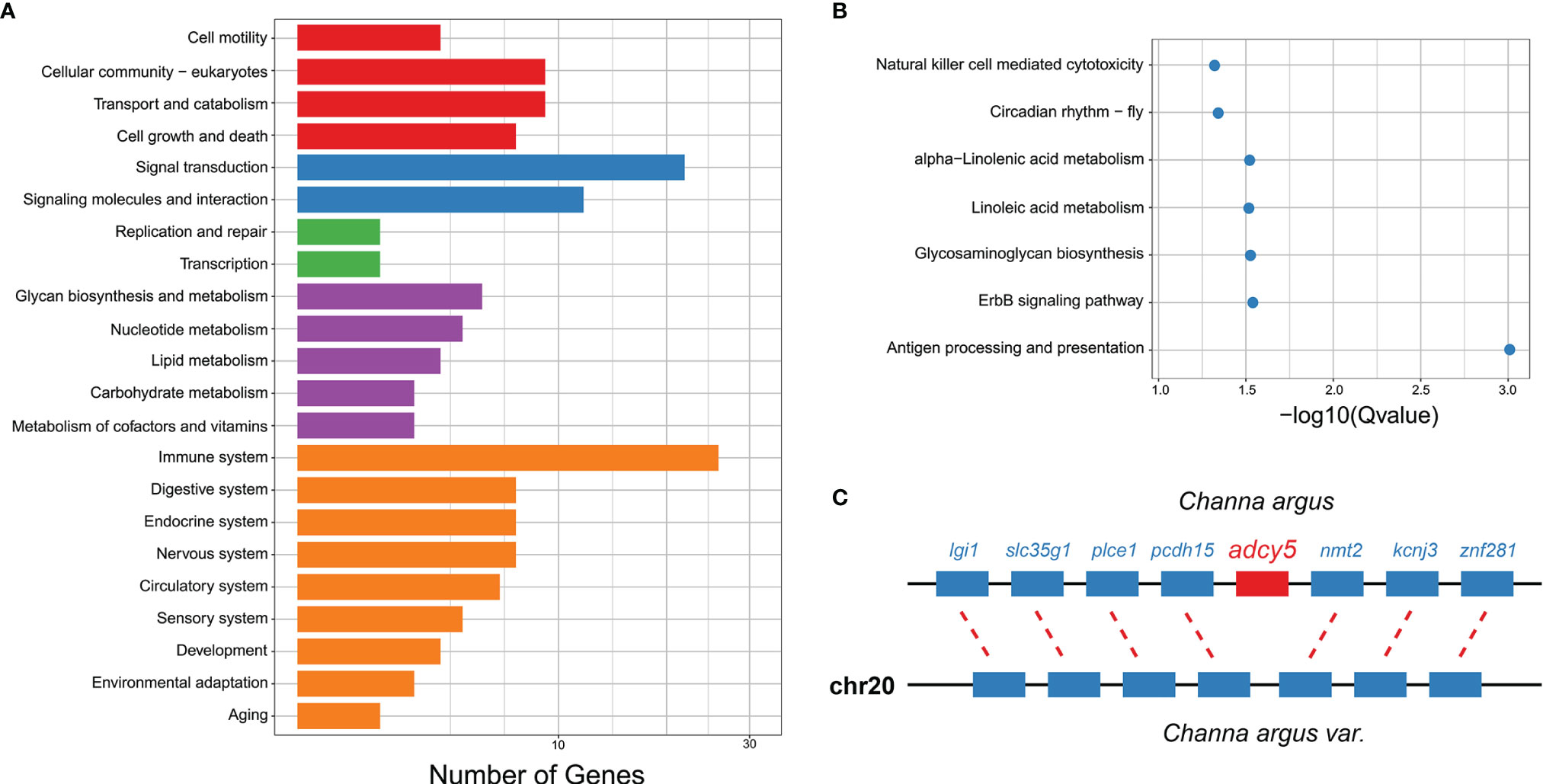
Figure 5 The C. argus var. and C. argus genome-specific gene identification and functional analysis. (A) The functional categories of KEGG annotations for candidate specific genes of C. argus. (B) The enrichment on KEGG annotation for candidate specific genes of C. argus. (C) The colinearity of neighboring genes to identify the absence of adcy5 in the C. argus var. genome.
On the basis of the long-read sequencing and Hi-C scaffolding technology, we de novo assembled a nearly chromosomal-level genome for C. argus var. The continuity and completeness of the newly assembled genome were significantly improved compared to the former assembly based on next-generation sequencing technology. We also annotated the genome and performed comparative analyses of the genome with other fish species. Phylogenetic analyses and divergence time estimations showed that C. argus var. and A. testudineus, the fish closest to have whole genome sequences publicly available at present, diverged about 85.6 million years ago. A number of expanded gene families and positively selected genes that reside in the C. argus var. genome were also detected, and these genes may be pivotal during the environmental adaptations of these albino individuals. Using comparative genomics, the putative genome-specific genes for C. argus var. and C. argus were detected and functionally analyzed. Based on our result, adcy5 (adenylate cyclase 5) was absent in the C. argus var. genome, which might be related to the albinism of C. argus var. Further investigations of these genes may provide insights into the molecular mechanisms of the albinism for fish, and even for other species including human. The high-quality genome of the albino fish C. argus var. provides a valuable resource for understanding the evolution events during fish evolution, especially for the understanding of fish albinism and their adaptation to the environment.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found as follows: NCBI [accession: PRJNA522012].
The animal study was reviewed and approved by the Animal Ethics Committee, Southwest University.
CZho, SX, and ML conceived and designed the study. XD, JW, JS, HY, and GL collected the samples. HL, YZo, GL, and GK performed the DNA sequencing and Hi-C experiments. HL and YZh performed the RNA sequencing. YZh, DY, and SX estimated the genome size, assembled the genome, and assessed the assembly quality. CZho, SX, and CZha performed the genome annotation and functional genomic analysis. CZho, SX, and ML wrote the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (No. 32072980), the Fundamental Research Funds for the Central Universities (No. XDJK2018C053), and the Financial Transfer Payment Project for Sichuan Province (2017NZYZF0089).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.839225/full#supplementary-material
Supplementary Figure 1 | The length distribution of subreads generated from the PacBio sequencing platform. The peak around 20 kb represented the insert length during the sequencing library construction.
Supplementary Figure 2 | The interaction frequency matrix among contigs generated from the Hi-C sequencing data. The interaction strength was colored by the logarithm of the contact density from red (high) to white (low).
Supplementary Figure 3 | Summaries of gene families classified using Orthomcl for each species.
Supplementary Figure 4 | The number of expanded and contracted gene families deduced using cafe for each branch.
Supplementary Figure 5 | The genome landscape for the C. argus var. From the outside to inside was GC content (pink), chromosome, gene density by number (blue line) and by length (blue heatmap), LINE density by number (red line) and by length (red heatmap), LTR density by number (green line) and by length (green heatmap), all repeat density (yellow) with tandem repeat density (purple). The inter-chromosome segment duplications were illuminated in the inner of the plot.
Abuín J. M., Pichel J. C., Pena T. F., Amigo J. (2015). BigBWA: Approaching the Burrows–Wheeler Aligner to Big Data Technologies. Bioinformatics 31, 4003–4005. doi: 10.1093bioinformatics/btv506
Aparicio S., Chapman J., Stupka E., Putnam N., Chia J., Dehal P., et al. (2002). Whole-Genome Shotgun Assembly and Analysis of the Genome of Fugu Rubripes. Science 297, 1301–1310. doi: 10.1126/science.1072104
Benson G. (1999). Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Birney E., Clamp M., Durbin R. (2004). GeneWise and Genomewise. Genome Res. 14, 988. doi: 10.1101/gr.1865504
Burton J. N., Adey A., Patwardhan R. P., Qiu R., Kitzman J. O., Shendure J. (2013). Chromosome-Scale Scaffolding of De Novo Genome Assemblies Based on Chromatin Interactions. Nat. Biotechnol. 31, 1119. doi: 10.1038/nbt.2727
Cantarel B. L., Korf I., Robb S. M., Parra G., Ross E., Moore B., et al. (2008). MAKER: An Easy-to-Use Annotation Pipeline Designed for Emerging Model Organism Genomes. Genome Res. 18, 188–196. doi: 10.1101/gr.6743907
Caro T. “The Colours of Extant Mammals,” in Seminars in Cell & Developmental Biology (Elsevier), 542–552.
Castresana J. (2000). Selection of Conserved Blocks From Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chaisson M. J., Tesler G. (2012). Mapping Single Molecule Sequencing Reads Using Basic Local Alignment With Successive Refinement (BLASR): Application and Theory. BMC Bioinf. 13, 238. doi: 10.1186/1471-2105-13-238
Chen F. D., Yang L. F. (2013). Channa Argus Nutritional Value and the Intensive Processing Development Prospect. Acad. Periodical Farm Products Process 4:51–3. doi: 10.3969/j.issn.1671-9646(x).2013.04.042.
Chin C.-S., Alexander D. H., Marks P., Klammer A. A., Drake J., Heiner C., et al. (2013). Nonhybrid, Finished Microbial Genome Assemblies From Long-Read SMRT Sequencing Data. Nat. Methods 10, 563. doi: 10.1038/nmeth.2474
Chin C.-S., Peluso P., Sedlazeck F. J., Nattestad M., Concepcion G. T., Clum A., et al. (2016). Phased Diploid Genome Assembly With Single-Molecule Real-Time Sequencing. Nat. Methods 13, 1050. doi: 10.1038/nmeth.4035
Conesa A., Götz S., García-Gómez J. M., Terol J., Talón M., Robles M. (2005). Blast2GO: A Universal Tool for Annotation, Visualization and Analysis in Functional Genomics Research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Courtenay W. R. J., Williams J. D. (2004). Snakeheads (Pisces, Channidae): A Biological Synopsis and Risk Assessment, US Department of the Interior. US Geolog. Surv. Circ. 43, 1251. doi: 10.3133/cir1251.
Dan C., Gong G., Wu J., Guo W., Li X., He Y., et al. (2018). Chromosomal-Level Assembly of Yellow Catfish Genome Using Third-Generation DNA Sequencing and Hi-C Analysis. GigaScience 7, 1–9. doi:10.1093/gigascience/giy120
De Bie T., Cristianini N., Demuth J. P., Hahn M. W. (2006). CAFE: A Computational Tool for the Study of Gene Family Evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
Edgar R. C. (2004). MUSCLE: Multiple Sequence Alignment With High Accuracy and High Throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Flicek P., Aken B. L., Beal K., Ballester B., Cáccamo M., Chen Y., et al. (2007). Ensembl 2008. Nucleic Acids Res. 36, D707–D714. doi: 10.1093/nar/gkm988
Ghosh S., Chan C. K. (2016). Analysis of RNA-Seq Data Using TopHat and Cufflinks. New York: Springer. doi: 10.1007/978-1-4939-3167-5_18
Glass M. L., Ishimatsu A., Johansen K. (1986). Responses of Aerial Ventilation to Hypoxia and Hypercapnia Inchanna Argus, an Air-Breathing Fish. J. Comp. Physiol. B 156 (3), 425–430. doi: 10.1007/BF01101105
Howe K., Clark M. D., Torroja C. F., Torrance J., Berthelot C., Muffato M., et al. (2013). The zebrafish reference genome sequence and its relationship to the human genome. Nature, 496(7446). doi: 10.1038/nature12111
Imakaev M., Fudenberg G., Mccord R. P., Naumova N., Goloborodko A., Lajoie B. R., et al. (2012). Iterative Correction of Hi-C Data Reveals Hallmarks of Chromosome Organization. Nat. Methods 9, 999. doi: 10.1038/nmeth.2148
Jurka J., Kapitonov V. V., Pavlicek A., Klonowski P., Kohany O., Walichiewicz J. (2005). Repbase Update, a Database of Eukaryotic Repetitive Elements. Cytogenetic Genome Res. 110, 462–467. doi: 10.1159/000084979
Kasahara M., Naruse K., Sasaki S., Nakatani Y., Qu W., Ahsan B., et al. (2007). The Medaka Draft Genome and Insights Into Vertebrate Genome Evolution. Nature 447, 714–719. doi: 10.1038/nature05846
Kottler V. A., Künstner A., Koch I., Fltenmeyer M., Langenecker T., Hoffmann M., et al. (2015). Adenylate Cyclase 5 is Required for Melanophore and Male Pattern Development in the Guppy (Poecilia Reticulata). Pigment Cell Melanoma Res. 28:545–558. doi: 10.1111/pcmr.12386
Kumar S., Stecher G., Suleski M., Hedges S. B. (2017). TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 34, 1812–1819. doi: 10.1093/molbev/msx116
Langmead B. (2010). Aligning Short Sequencing Reads With Bowtie. Curr. Protoc. Bioinf. 32, 11.17. doi: 10.1002/0471250953.bi1107s32
Li D. Y., Kang D. H., Yin Q. Q., Sun X. W., Liang L. Q. (2007). Microsatellite DNA Marker Analysis of Genetic Diversity in Wild Common Carp (CyprinuscarpioL.) Populations. J. Genes Genom. 34, 984–993. doi: 10.1016/S1673-8527(07)60111-8
Li L., Stoeckert C. J. Jr., Roos D. S. (2003). OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li Z., Xiang K., Zou Y.-C., Yue X.-J., Wang Y.-M., Qin C.-J., et al. (2016). Analysis on Karyotype and DNA Content of Opniocepnalus Argus. Freshwater Fisheries 46, 104–108.
Liu B., Shi Y., Yuan J., Hu X., Zhang H., Li N., et al (2013). Estimation of Genomic Characteristics by Analyzing K-Mer Frequency in De Novo Genome Projects. Quantitative Biol 35, 62–7.
Liu H. P., Xiao S. J., Wang D., Liu Y.-C., Zhou C.-W., Liu Q.-Y., et al. (2019). The sequence and de novo assembly of oxygymnocypris stewartii genome. Scientific Data 6, 190009. doi:10.1038/sdata.2019.9.
Marçais G., Kingsford C. (2011). A Fast, Lock-Free Approach for Efficient Parallel Counting of Occurrences of K-Mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Qin W. F., Jiang J. H. (2010). Experimental Study of the Influence of Wulifuyuantang on Wound Healing of Mice Operated. Chin. Med. Modern Distance Educ. China 8 (10), 189–190. doi:10.3969/j.issn.1672-2779.2010.10.166
Shi B. N., Deng Q. X. (1980). The history and formation of fish species in Jialing river. Joural of southwest China normal university (Natural Science) 2, 34–44. doi:10.1378/j.cnki.xsxb.1980.02.004
Song C., Liu S., Xiao J., He W., Zhou Y., Qin Q., et al. (2012). Advances in Polyploid Biology. Scientia Sin. Vitae 42, 173–184. doi: CNKI:SUN:JCXK.0.2012-03-002
Stamatakis A. (2014). RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stamatakis A., Hoover P., Rougemont J. (2008). A Rapid Bootstrap Algorithm for the RAxML Web Servers. Syst. Biol. 57, 758–771. doi: 10.1080/10635150802429642
Stanke M., Keller O., Gunduz I., Hayes A., Waack S., Morgenstern B. (2006). AUGUSTUS: Ab Initio Prediction of Alternative Transcripts. Nucleic Acids Res. 34, W435–W439.
Sun S., Wang Y., Zeng W. H., Du X., Li L., Hong X. N., et al. (2020). The Genome of Mekong Tiger Perch ( Datnioides Undecimradiatus ) Provides Insights Into the Phylogenetic Position of Lobotiformes and Biological Conservation. Sci. Rep. 10 (1), 8164. doi: 10.1038/s41598-020-64398-2
Suyama M., Torrents D., Bork P. (2006). PAL2NAL: Robust Conversion of Protein Sequence Alignments Into the Corresponding Codon Alignments. Nucleic Acids Res. 34, W609–W612. doi: 10.1093/nar/gkl315
Trapnell C., Pachter Lsalzberg S. L. (2009). TopHat: Discovering Splice Junctions With RNA-Seq. Bioinformatics. doi: 10.1093/bioinformatics/btp120
Walker B. J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., et al. (2014). Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS One 9, e112963. doi: 10.1371/journal.pone.0112963
Wang J. X., Zhao X. F., Zhou C. W., Liao Z. G.. (1992). Comparative Studies On the Ophiocephalus Argus Argus and O. A. Kimurai, With the Systematics of Ophiocephalus Argus. Transanctions Oceanol Limnol 2, 51–57.
Wang Y., Lu Y., Zhang Y., Ning Z., Li Y., Zhao Q., et al. (2015). The Draft Genome of the Grass Carp (Ctenopharyngodon Idellus) Provides Insights Into its Evolution and Vegetarian Adaptation. Nat. Genet. 47, 625–631. doi: 10.1038/ng.3280
Xiao M. X., Xia H., Bao F. (2015). Isolation and Characterization of 15 Microsatellite Loci for Ophiocephalusargus Cantor. Russ. J. Genet. 51, 1044–1047. doi: 10.1134/S1022795415090136
Xiao M. S., Bao F. Y., Zhao Y., Hu Q. S. (2019). Transcriptome Sequencing and De Novo Analysis of the Northern Snakehead, Ophiocephalus Argus. Genet. 98 (4). doi: 10.1007/s12041-019-1086-1
Xiao S. J., Mou Z., Fan D., Zhou H., Zou M., Zou Y., et al. (2020). Genome of Tetraploid Fish Schizothorax O'connori Provides Insights Into Early Re-Diploidization and High-Altitude Adaptation. iScience 23, 101–497. doi:10.1016/j.isci.2020.101497
Xu J., Bian C., Chen K., Liu G., Jiang Y., Luo Q., et al. (2017). Draft Genome of the Northern Snakehead, Channa Argus. Gigascience 6, 1–5. doi: 10.1093/gigascience/gix011
Xu S., Gao T., Liu J., Xiao S., Zhu S., Zeng X., et al. (2018a). A Draft Genome Assembly of the Chinese Sillago (Sillago Sinica), the First Reference Genome for Sillaginidae Fishes. GigaScience 7, 9. doi: 10.1093/gigascience/giy108
Xu Z., Wang H. (2007). LTR_FINDER: An Efficient Tool for the Prediction of Full-Length LTR Retrotransposons. Nucleic Acids Res. 35, W265–W268. doi: 10.1093/nar/gkm286
Xu S., Xiao S., Zhu S., Zeng X., Luo J., Liu J., et al. (2018b). A Draft Genome Assembly of the Chinese Sillago (Sillago Sinica), the First Reference Genome for Sillaginidae Fishes. GigaScience 7, giy108. doi: 10.1093/gigascience/giy108
Xu P., Zhang X., Wang X., Li J., Liu G., Kuang Y., et al. (2014). Genome Sequence and Genetic Diversity of the Common Carp, Cyprinus Carpio. Nat. Genet. 46, 1212–1219. doi: 10.1038/ng.3098
Xu S. Y., Lu Z. C., Cao B. B., Han D. D., Cai S. S., Han Z. Q., et al. (2021). Chromosome-Scale Genome Assembly of Brownspotted Flathead Platycephalus Sp.1 Provides Insights Into Demersal Adaptation in Flathead Fish[J]. Zoological Res. 42 (05), 660–665.
Yang Z. (2007). PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang X., Liu D., Liu F., Wu J., Zou J., Xiao X., et al. (2013). HTQC: A Fast Quality Control Toolkit for Illumina Sequencing Data. BMC Bioinf. 14, 33. doi: 10.1186/1471-2105-14-33
Yuan S. Y., Huo J. P., Chai Z. Y. (2005). Effects of Channa Argus and Channa Asiatica on Anti-Fatigue and Promoting the Growth of Muscle in Rats. Youjiang Med. J. 33, 109–111. doi: 10.3969/j.issn.1003-1383.2005.02.003
Zhang X., Yuan J., Sun Y., Li S., Gao Y., Yu Y., et al. (2019). Penaeid Shrimp Genome Provides Insights Into Benthic Adaptation and Frequent Molting. Nat. Commun. 10, 1–15. doi: 10.1038/s41467-018-08197-4
Zhong L., Kehan X., Yuanchao Z., Xingjian Y., Yongming W., Chuanjie Q., et al. (2016). Analysis on Karyotype and DNA Content of Opniocepnalus Argus. Freshwater Fisheries 46, 104–108. doi: 10.13721/j.cnki.dsyy.2016.03.018
Zhou C. W., Lei L., Deng X. X., Zheng Z. L., Zheng Y. H., Wu J., et al. (2018). Nutritional Composition Analysis and Evaluation of Ophicephalus Argus and Opniocepnalus Argus Var. Freshwater Fisheries 48, 83–89. doi: 10.13721/j.cnki.dsyy.2018.03.013
Zhou A., Wang C., Jiang W., Li Z., Chen Y., Xie S., et al. (2017). Genetic Comparison of Two Color Morphs of Northern Snakehead (Channa Argus) and Chnannidae Family. Mitochondrial DNA A DNA Mapp Seq Anal. 28, 971–973. doi: 10.1080/24701394.2016.1186668
Zhou Y., Zhuo X., Zou Q., Chen J., Zou J. (2015). Population Genetic Diversity of the Northern Snakehead (Channa Argus) in China Based on the Mitochondrial DNA Control Region and Adjacent Regions Sequences. Mitochondrial DNA 26, 341–349. doi: 10.3109/19401736.2014.908355
Zou Y. C., Wen Z. Y., Qin C. J., Li H. T., Wu J., Biwen B. W. (2017). Comparison of Nutritional Components in the Muscle of Opniocepnalus Argus of Different Bred Time. Acta Nutrimenta Sin. 39, 616–618. doi: 10.13325/j.cnki.acta.nutr.sin.2017.06.023
Keywords: the albino northern snakehead, genome, chromosome-scale assembly, PacBio, Hi-C
Citation: Zhou C, Li Y, Zhou Y, Zou Y, Yuan D, Deng X, Lei L, Su J, Zhu C, Ye H, Luo H, Lv G, Zhou X, Kuang G, Zhang C, Wu J, Zheng Z, Xiao S and Li M (2022) Chromosome-Scale Assembly and Characterization of the Albino Northern Snakehead, Channa argus var. (Teleostei: Channidae) Genome. Front. Mar. Sci. 9:839225. doi: 10.3389/fmars.2022.839225
Received: 19 December 2021; Accepted: 07 March 2022;
Published: 06 April 2022.
Edited by:
Xiaotong Wang, Ludong University, ChinaReviewed by:
Zhenkui Qin, Ocean University of China, ChinaCopyright © 2022 Zhou, Li, Zhou, Zou, Yuan, Deng, Lei, Su, Zhu, Ye, Luo, Lv, Zhou, Kuang, Zhang, Wu, Zheng, Xiao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shijun Xiao, c2hpanVuX3hpYW9AMTYzLmNvbQ==; Minghui Li, aW1oQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.