
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 14 March 2022
Sec. Ocean Observation
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.837515
This article is part of the Research TopicNatural and Artificial Radionuclides as Tracers of Ocean ProcessesView all 12 articles
 Karin Hain1*
Karin Hain1* Martin Martschini1Fadime Gülce1
Martin Martschini1Fadime Gülce1 Maki Honda1,2Johannes Lachner1,3
Maki Honda1,2Johannes Lachner1,3 Michael Kern1Johanna Pitters1Francesca Quinto4Aya Sakaguchi5Peter Steier1
Michael Kern1Johanna Pitters1Francesca Quinto4Aya Sakaguchi5Peter Steier1 Andreas Wiederin1
Andreas Wiederin1 Alexander Wieser1Akihiko Yokoyama6
Alexander Wieser1Akihiko Yokoyama6 Robin Golser1
Robin Golser1Recent major advances in Accelerator Mass Spectrometry (AMS) at the Vienna Environmental Research Accelerator (VERA) regarding detection efficiency and isobar suppression have opened possibilities for the analysis of additional long-lived radionuclides at ultra-low environmental concentrations. These radionuclides, including 233U, 135Cs, 99Tc, and 90Sr, will become important for oceanographic tracer application due to their generally conservative behavior in ocean water. In particular, the isotope ratios 233U/236U and 137Cs/135Cs have proven to be powerful fingerprints for emission source identification as they are not affected by elemental fractionation. Improved detection efficiencies allowed us to analyze all major long-lived actinides, i.e., 236U, 237Np, 239,240Pu, 241Am as well as the very rare 233U, in the same 10 L water samples of a depth profile from the northwest Pacific Ocean. For this purpose, a simplified and very flexible chemical purification procedure based on extraction chromatography (a single UTEVA® column) was implemented which can be extended by a DGA® column for Am purification. The procedure was validated with the reference materials IAEA-381/385. With the additional increase in ionization efficiency expected for the extraction of actinides as fluoride molecules from the AMS ion source, a further reduction of chemical processing may become possible. This method was successfully applied to an exemplary set of air filter samples. In order to determine the quantitative 237Np concentration reliably, a 236Np spike material is being developed in collaboration with the University of Tsukuba, Japan. Ion-Laser Interaction Mass Spectrometry (ILIAMS), a novel technique for the efficient suppression of stable isobaric background, has been developed at VERA and provides unprecedented detection sensitivity for the fission fragments 135Cs, 99Tc, and 90Sr. The corresponding setup is fully operational now and the isobar suppression factors of >105 achieved, in principle, allow for the detection of the mentioned radionuclides in the environment. Especially for 90Sr analysis, this new approach has already been validated for selected reference materials (e.g., IAEA-A-12) and is ready for application in oceanographic studies. We estimate that a sample volume of only (1–3) L ocean water is sufficient for 90Sr as well as for 135Cs analysis, respectively.
The application of long-lived anthropogenic radionuclides in oceanography requires ultra-sensitive detection methods to analyze trace concentrations at environmental levels while reducing the overall sample size. Smaller samples allow for the collection of a larger number of samples per cruise so that the resolution of the distribution of the radionuclides can be significantly improved. Such more detailed data is urgently needed to obtain a more accurate understanding of the complex system of ocean circulation. The interpretation of the measured data of a single nuclide tracer is often limited by mixing, dilution and sedimentation or scavenging processes. Analyzing as many nuclides as possible, which show different chemical migration behavior, from the same sample helps to disentangle the different environmental processes. For instance, it was shown that 236U, which is conservatively transported in the open oceans, is partly co-precipitated in coastal areas with high organic particle concentration by comparison with 237Np (López-Lora et al., 2020). In contrast, isotopic ratios, e.g., the plutonium (Pu) ratios 240,241Pu/239Pu [e.g., in Kelley et al. (1999), Zheng et al. (2012, 2013)], are very robust against chemical fractionation and hence, allow unambiguous identification of the emission source. The isotopic ratios of the two conservatively behaving elements uranium (U) and cesium (Cs), i.e., 233U/236U (Hain et al., 2020) and 137Cs/135Cs (Russell et al., 2015) have been introduced as two very promising oceanographic tracers because of their abilities for emission source assessment. The group of actinides is considered as especially interesting for oceanographic applications, as the individual elements show very different migration behavior depending on the ambient redox conditions. U and Np isotopes in general behave conservatively in ocean water and therefore are considered useful tracers for ocean dynamics (Dunk et al., 2002; Lindahl et al., 2005). On the other hand, Pu and Am are much more particle-reactive so that they are quickly removed from the water column by sedimentary mineral and organic particles (Lehto and Hou, 2011) and hence, they can serve as tracers for sedimentation processes (Tims et al., 2004). All the respective long-lived isotopes, i.e., 233,236U, 239,240Pu, 241Am, and 237Np, are available for routine analysis at the Vienna Environmental Research Accelerator (VERA) (Steier et al., 2019). However, quantification of the 237Np concentration still suffers from the lack of sufficiently pure spike material. Until such spike material is available, environmental 237Np concentrations are determined by using a non-isotopic internal 242Pu spike and an external 237Np/242Pu standard for normalization (Qiao et al., 2010; López-Lora et al., 2019), with the drawback of possible elemental fractionation during sample preparation and measurement. There is a multitude of chemical separation procedures for actinides available (e.g., López-Lora et al., 2019) but to our knowledge none of them allows the efficient extraction of the major long-lived nuclides including 241Am from the same small volume ocean sample (10 L).
As these long-lived actinides can be measured essentially background-free by Accelerator Mass Spectrometry (AMS), the minimum sample size is basically defined by the detection efficiency and the chemical yield of sample preparation. At the Vienna Environmental Research Accelerator (VERA) laboratory we have successfully increased the efficiency of the spectrometer by around one order of magnitude through several improvements of the setup and the measurement procedure (Steier et al., 2019). The AMS methodological development at VERA for actinide detection recently focused on the possibility to increase the negative ionization yield of U by adding the fluorine donator PbF2 to the sample matrix for the cesium sputter source. Fluoride compounds of U (e.g., UF5–) are then extracted from the ion source instead of the previously used oxide compounds (e.g., UO–) (Zhao et al., 2010; Cornett et al., 2015; Kazi et al., 2015). While, e.g., Hotchkis et al. (2019) report similarly high ionization efficiencies for UO– of several percent from their ion source, this is not observed by a number of other AMS laboratories including VERA. Our previous survey on U extraction confirmed an increase of the ionization yield using fluorides by at least a factor 5 for U standard materials (Kern et al., 2022). In addition, fluoride targets have been found to produce very strong UF5– currents within the first half hour of sputtering which then rapidly drop to zero output. Optimization of the ion source operation is still on-going to ensure more stable measurement conditions. Consequently, the applicability of this method to environmental samples regarding accuracy and precision had to be demonstrated.
Beyond that, the novel technique of Ion-Laser Interaction Mass Spectrometry (ILIAMS) (Martschini et al., 2019b) makes new radionuclides accessible at ultra-trace concentrations. First results presented here and in Martschini et al. (2021) show that this technique will allow for AMS-measurements of the fission products 99Tc, 135Cs, and 90Sr at environmental levels. Since these isotopes were previously inaccessible with other techniques in the general environment (135Cs), required huge instrumental effort in AMS (99Tc) or relied on slow and cumbersome sample preparation for radiometric detection (90Sr), ILIAMS is expected to facilitate much more widespread analysis of these tracers. The fission product technetium-99 (99Tc, T1/2 = 2.1 × 105 yr) is usually present as the highly soluble TcO4+ -ion in oxic waters, which is transported conservatively with the water masses in the open oceans similar to cesium (Cs). 99Tc was therefore proposed as oceanographic tracer for the North Atlantic Ocean early on (Aarkrog et al., 1987), where elevated concentrations due to emissions of the Sellafield reprocessing plant can be analyzed by less sensitive techniques, e.g., ICP-MS. Radiostrontium in the ocean is another viable conservative tracer that is generally even better stabilized than Cs as it is less adsorbed to particles (Povinec et al., 2012). However, its behavior in surface ocean water with possible removal by celeslite particle formation is still not fully understood and source terms in coastal areas are less straightforward to assess as fluvial input to the ocean from land deposition of strontium (Sr) plays a much stronger role than for Cs (Povinec et al., 2012). Being a chemical homolog of calcium, Sr is readily incorporated into the carbonate skeleton of corals which are considered a powerful archive for past climatic conditions. 90Sr archived in these time-resolved reservoirs can be used to estimate water transit times from the source of emission to the location of the collected coral core and as a proxy for past ocean chemistry (e.g., Toggweiler and Trumbore, 1985; Purdy et al., 1989). 135Cs, with a not well-known half-life between 1.3 and 3 Myrs (Singh et al., 2008; MacDonald et al., 2016), is a pure beta-emitter and therefore hardly detectable via radiometric methods. The low abundance of 135Cs with its high isobaric background of 135Ba makes it difficult to detect, also for mass spectrometric approaches. Due to this difficulty, most studies presenting 135Cs/137Cs ratios were performed on water with 135,137Cs/133Cs ratios of 10–8 or higher, i.e., close to sources of contamination (Zhu et al., 2020a). Two recent studies show first results on 135Cs concentration in general seawater from the west coast of Greenland. There, 40–50 L of ocean water were used for the successful detection with ICP-MS/MS (Zhu et al., 2021) and 200 mL with TIMS (Zhu et al., 2020b) characterized by a detection limit of 135Cs/133Cs ∼4 × 10–10. The residence time of Cs is comparable to that of U and amounts to 330 kyr for Cs (Broecker and Peng, 1982), so 135Cs can also be considered a conservative tracer which might have entered the marine environment due to the Fukushima accident (Zheng et al., 2014), representing a point source for radionuclide tracing.
This contribution at the one hand aims at giving an overview about the present status of method development at VERA toward quantitative analysis of the listed fission products in different compartments of the environment using ILIAMS. On the other hand, the new sample preparation and measurement procedure for routine analysis of actinides in ocean water samples will be discussed based on results for a water column from the Northwest Pacific Ocean. Furthermore, the reliability of the fluoride extraction method has been verified for 236U/238U results in air filters. Last but not least, we report on the current status of Np spike development at VERA within a joint project with the University of Tsukuba. This manuscript is intended to give potential users insight into the sample amount and preparation techniques required for the analysis of the respective mentioned isotopes with AMS at VERA. The presented procedures, especially those focusing on actinide detection, can be adopted by similar AMS facilities. This also holds for the suggested fission fragment tracers provided that suitable isobar suppression systems are implemented.
As 242Pu isotope standard, an aliquot of the certified spike solution IRMM-085 (Institute for Reference Materials and Measurements, Belgium) and as 243Am isotope standard, an aliquot of the Alpha Standard Solution (Eckert & Ziegler Nuclitec GmbH, Catalog No.: 7243, Source No.: 1637-61) were used.
A 95mTc spike, which serves as a chemical yield monitor for 99Tc extraction, was produced by proton irradiation of molybdenum (Mo) foils at the Maier-Leibnitz Laboratory in Munich and at the University of Cologne according to Wacker et al. (2004). The irradiated Mo foil was dissolved in aqueous H2O2 (30%, 35 mL) overnight, then purified using TEVA resin (Gülce, F. et al., in preparation) and the final 95mTc concentration was characterized by gamma-spectrometry.
The developed procedures for actinide extraction were verified using certified reference material available from the International Atomic Energy Agency (IAEA), i.e., Irish Sea water (IAEA-381) and sediment (IAEA-385). Literature values for 233,236U are not certified, neither for water nor for sediment, as well as for 237Np in sediment, but information values (Povinec et al., 2002; Pham et al., 2008; Hain et al., 2020) or individual studies (Lee et al., 2008; Goldstein et al., 2021) are available for comparison. The new procedures were applied to ocean water samples collected at a station in the northwest Pacific Ocean (BD04, 37°49 N, 143°53 E, maximum depth 600 m) collected during the cruise KH-12-4 (GP02) of the ship R/V Hakuho Maru as part of the GEOTRACES program on August 24/25, 2012. The original 20 L samples were split so that 10 L were used for actinide analysis while the second half was kept to determine the 99Tc concentration. Three samples from this water column had been analyzed before and additional details on the sampling procedure can be found in the corresponding publication (Hain et al., 2017). These samples had been also analyzed at VERA but applying a different actinide separation chemistry. Selected air filters collected in Austria (Meteorological Station Hohe Warte, Vienna, Austria) during the most active phase of atmospheric nuclear weapons testing, i.e., 1962–1965, were analyzed regarding their 233,236U concentrations. To assess the accuracy of chemical 90Sr extraction and subsequent AMS-measurement for various sample matrices, we applied our method to the certified IAEA reference material IAEA-A-12 (beef bone) with a recommended 90Sr content of 54.8 Bq/kg for a reference date of 15.12.1981 and to the IAEA Proficiency Test sample IAEA-TEL-2015-03_05 (Syrian soil) with a target value for the 90Sr content of (36.2 ± 2.7) Bq/kg on 01.01.2015. An overview of the analyzed sample materials and radionuclides is presented in Table 1.

Table 1. Summary of sample materials and radionuclides investigated within the scope of the present work.
The overall sample preparation procedure which is now routinely applied for the extraction of actinides from large volume ocean water samples (e.g., those from BD04) at VERA, is depicted in Figure 1. After adding the required isotopic spike solutions, i.e., 2 × 108 atoms each of 242Pu for 239,240Pu and 243Am for 241Am analysis, the volume is first reduced to half by evaporation. Actinides are then pre-concentrated from the remaining 5 L by a Fe(OH)3 co-precipitation which also removes the major salt load when the pH is carefully adjusted to 8.5. The column load solution for the UTEVA® resin is obtained by first taking up the sample in 3M HNO3-0.5M Al(NO3)3. The oxidation state of Pu and Np is then adjusted to +IV using a combination of sulfamic and ascorbic acid as well as NaNO2 according to Maxwell et al. (2010). Am, usually present in oxidation state +III in most chemical conditions, passes through the UTEVA (Eichrom Technologies) along with most other metal ions from the initial environmental matrix and the large surplus of iron used for the co-precipitation. Am is strongly adsorbed onto the DGA® resin (Eichrom Technologies) which is used to purify the Am fraction as especially Fe(III+) ions pass through the resin (Pourmand and Dauphas, 2010). After the separation of the UTEVA® and the DGA® cartridge, which are set up in tandem for loading of the sample solution and rinsing, Pu and Np are eluted from UTEVA® into the same fraction by complexation with oxalic acid, U is removed from UTEVA® by 0.02M HCl and Am is eluted from the DGA® cartridge with 0.25M HCl. The presented procedure is highly flexible and can be extended by an initial leaching step using aqua regia also for solid samples as applied to rubble or using 8M HNO3 for (sea and lake) sediment samples.
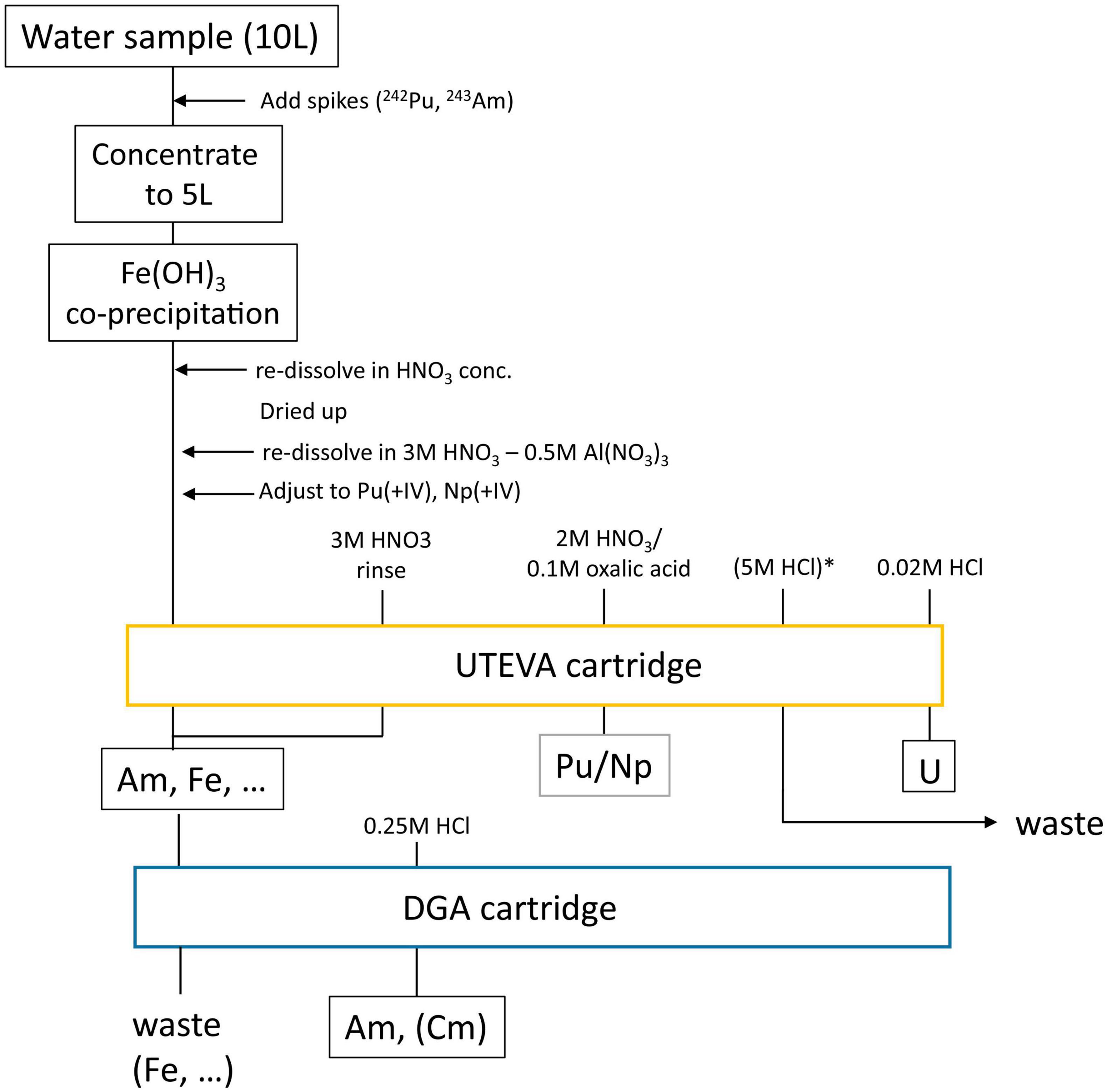
Figure 1. Flow chart of applied separation chemistry to extract and purify Pu/Np, U, and Am from large volume ocean water samples using extraction chromatography. * marks a rinsing step to remove Th in the final U fraction which is only required when 233U is to be analyzed.
The subsequent preparation of the AMS targets depends on whether actinides are to be analyzed in oxide form or the new approach of extracting actinides in fluoride form from the ion source is applied. In the routine procedure for oxide preparation, actinides are co-precipitated with 2 mg of Fe3+. Oxalates in the Pu/Np fraction need to be decomposed in a microwave oven beforehand, where the solution is slowly heated to 200°C and left at this temperature for 8 min. After centrifugation, the precipitate is washed three times and then dried at 80°C for at least 4 h. Finally, the dry Fe pellet is calcined at 800°C for 4 h in a quartz crucible and pressed into an Al sample holder. The target preparation procedure for fluoride extraction is under development, but has been already used for a comprehensive set of samples with added actinide spikes for the experiments described in section “Status of the Isotopic Spike for Quantitative 237Np Analysis.” Regarding environmental samples, the procedure has only been successfully applied to the U fraction so far, e.g., for the air filter samples [(Kern et al., 2022); this work]. In short, the fractions are eluted into Teflon tubes and about 200 μg of Fe is added. After evaporating the solution to dryness, one drop of acetone is added and the produced iron pellet is transferred to a quartz crucible using a pipette. The Fe is then calcined as described before and the small amount of Fe2O3 thus produced is carefully mixed with a large surplus of PbF2 (mass ratio ∼1:9) which is directly pressed into a Cu sample holder. Al sample holders cannot be used in this case, as PbF2 seems to react with Al under ion source conditions.
Several procedures for chemical Tc purification adjusted to Mass Spectrometry or even AMS have been reported (summarized in Shi et al. (2012)), most of which are based on TEVA® resin (Eichrom Technologies Inc.). However, the chemical Ru suppression obtained with a single TEVA column turned out to be insufficient to allow the analysis of 99Tc in certain environmental samples, e.g., peat bog samples. For this reason, a pre-treatment procedure based on LLX using 5% TIOA + Xylene for the pre-concentration of 99Tc from large volume ocean water samples with high salt load was developed which also achieved a very good Ru suppression (2 × 103) and very stable 99Tc recoveries (including purification with TEVA) of (89 ± 5%) (Gülce, F., in preparation). AMS targets are then prepared by adding 2 mg of Nb (in-house solution) to the Tc fraction which is then evaporated to dryness. We have performed experiments regarding the optimum temperature of the final calcination step, i.e., at 450 and 300°C, as there were indications for Tc losses at the higher temperature (99Tc Detection With Accelerator Mass Spectrometry). When using 95mTc as chemical yield monitor analyzed by gamma spectrometry to correct the final AMS result, losses of Tc during the preparation of the solid sputter matrix for AMS measurements are difficult to quantify as the geometry of the sample solution and the dry solid material required for AMS cannot be directly compared. Therefore, it is of importance that the recovery of these final preparation steps is stable and close to 100%. To ensure the same geometry for gamma spectrometry we compared the 95mTc count rates of samples after slow evaporation to dryness and after calcination, obtaining in this way the recovery of the calcination step.
Cs extraction and purification is based on the procedure described in Zok et al. (2021). A Cs extraction with Ammonium phosphomolybdate hydrate (AMP, Alfa Aesar) is followed by an anion exchange column [Dowex 1-X8 (100-200 mesh), Alfa Aesar] and a cation exchange column [AG 50W-X8 (100-200 mesh), Bio-Rad Laboratories, Inc.]. The overall process has a high yield of ca. 80%. The procedure might be simplified for ILIAMS assisted AMS measurements, which seem robust against content of other elements in the target including the presence of the Ba isobars. For the AMS target, the final purified Cs fraction in solution is mixed with a Cs carrier (Cs2SO4) and PbF2 needed for the extraction of CsF2– from the ion source. These components are mixed in Milli-Q water and dried at 70°C for several hours. Finally the dried powder is pressed into copper target holders for the AMS measurement. Until now, this procedure to produce AMS targets was only applied to organic samples.
The chemical sample preparation procedure for AMS of 90Sr has so far only been developed and tested for soil and bone samples. First, 1 mg of Sr carrier is added to the remaining ashed samples after a small part of the sample was taken to determine the amount of natural stable Sr by ICP-MS. The part with carrier added either undergoes microwave digestion or acid leaching with HNO3. For Sr-separation from the solution, we use Sr-resin cartridges (Eichrom Industries Inc.). Finally, SrF2 is precipitated from concentrated eluate by addition of 1 mL of HF. For soil samples, the typical sample size is 1–5 g and a recovery between 70 and 96% was achieved for different materials including soil or beef bone (M. Honda, in preparation). In principle, the same purification method can also be applied to marine samples like ocean water or corals after a pre-treatment step, i.e., either pre-concentration from liquid samples or acid decomposition of coral samples.
A detailed description of the routine analysis of actinides at VERA, in particular of the anthropogenic U isotopes, 236U and 233U, has been reported previously (Steier et al., 2019; Hain et al., 2020). While this is based on the extraction of the actinides as single negatively charged oxide molecules from the ion source, the procedure for fluoride extraction is identical in most parts. In short, the actinides extracted from the respective sputter matrix are injected into the accelerator after a first mass separation stage. The ions are stripped to positive charge states in a helium gas cell in the high-voltage terminal of the accelerator. Using a relatively high helium pressure in the terminal stripper to ensure the suppression of molecular background such as (235UH)3+, a stripping yield of around 21% is achieved for the selected charge state 3+ (U3+) (Steier et al., 2019). After passing additional filtering components including a second 90° magnet, a Wien filter, an electrostatic analyzer and a third 90° magnet, the remaining ions were identified in a Bragg type ionization chamber. The overall detection efficiency of the system for U atoms in the sputter target was 5 × 10–4 (Steier et al., 2019) assuming complete consumption of the sputter material. Usually, the majority of the measurement time is dedicated to the less abundant 233U and the whole material is not used up.
While the count rates of the rare U isotopes are normalized to the macroscopic 238U3+ ion current measurement to account for variations in detection efficiency and losses during chemical sample preparation, all the other actinides of interest are measured relative to the count rate of the respective internal isotopic spike. This presents a difficulty in the case of 237Np where the count rate has to be normalized to the non-isotopic 242Pu spike until now since no isotopic spike is currently available (Status of the Isotopic Spike for Quantitative 237Np Analysis). In that case, variations in the detection efficiency, in particular due to element specific changes of the ionization yield, have to be corrected by measuring an external 237Np/242Pu standard in addition. This sample is prepared from spike solutions with a defined 237Np/242Pu ratio by quantitative co-precipitation or just drying up the solution. In previous studies, it has been observed that in the Cs negative ion source of AMS the production of PuO– is usually enhanced compared to NpO– (Fifield, 2008; Quinto et al., 2015, 2017). Both the different ionization yield of Np and Pu and their possibly different chemical yield during chemical preparation of the sample may contribute to the higher relative output. Furthermore, a variability of such an enhancement factor has been suggested, in particular in a set of 5 groundwater samples (with an average enhancement factor of Pu/Np = 1.1 ± 0.1) (Quinto et al., 2017) and in a seawater sample as well as in a sample composed purely by reagents (enhancement factor of Pu/Np = 1.2 ± 0.5 and 1.2 ± 0.7, respectively) (Quinto et al., 2015). Such phenomenon requires further investigation in order to be fully understood.
Conventionally, AMS suppresses stable isobaric background (e.g., 99Ru in the case of 99Tc, 135Ba in the case of 135Cs) in the final detection setup by methods adopted from nuclear physics based on element specific energy loss in matter or the different deflection in a gas-filled magnet. Since the isobaric interference of the isotopes of interest usually stems from a neighboring element, this becomes increasingly difficult for heavier nuclides, where the relative difference in the nuclear charge decreases. While 10Be-10B separation nowadays is accomplished at compact 300 kV tandem accelerators, even the largest AMS facilities with terminal voltages >10 MV struggle to achieve the required isobar separation on the order of 106 or above for isobaric systems at ∼90 amu (Koll et al., 2019; Pavetich et al., 2019) and fail to do so at higher masses, e.g., in the actinide mass region. To overcome this limitation and make nuclides like the long-lived fission products accessible at medium-sized AMS-facilities with terminal voltages of 3 MV and below, an alternative approach using element-selective neutralization of anions via laser photodetachment was developed at VERA. The corresponding Ion-Laser Interaction Mass Spectrometry (ILIAMS) setup was successfully linked to the existing AMS setup at the VERA laboratory 5 years ago (Martschini et al., 2019b). In order to facilitate the necessary ion-laser interaction times, the ion beam is decelerated to an energy at eV levels inside a radiofrequency quadrupole ion guide filled with buffer gas and overlapped with a collinear laser therein. A recent review of this method and its achievements is given by Martschini et al. (2021). For “standard” AMS isobaric systems like 36Cl-36S and 26AlO-26MgO, the ILIAMS-technique provides unprecedented isobar suppression of 1011-1012 (Lachner et al., 2019, 2021).
Application of ILIAMS to the fission products 90Sr, 99Tc, and 135,137Cs has required a substantial development effort, which is certainly not yet finished. The prerequisite of this technique is the identification of a suitable molecular system in which the isotope of interest has a higher detachment energy than its isobaric interference. In this case, a laser characterized by a photon energy intermediate between the detachment energies of the molecular system of the isotope of interest and that of its isobar can be applied to neutralize or decompose the isobaric molecule while leaving the anionic molecule of the isotope of interest unaffected. Unfortunately, detachment energies of many molecules of elements in the fission product or actinide region are unknown and need to be determined experimentally. For the three isotopes discussed here, fluoride molecules have proven to be well suited while formerly used oxides did not match the criteria. Hence, an adaptation of sample chemistry and subsequent studies of the influence of ion source settings and composition of the matrix on the negative ion formation probability became necessary.
Apart from the additional filter step of ILIAMS, the measurement principle is very similar to the conventional actinide AMS. The trace isotope and a reference isotope (typically a stable isotope of the same element if available) are extracted as negative ions and sequentially injected through ILIAMS into the accelerator, where the ions are stripped to the 3+ or 4+ charge state using He gas. On the high energy side, abundant isotopes are measured in Faraday cups as ion currents and compared to the single particle detector count rates of the trace isotopes to deduce raw isotopic ratios, which are then normalized with the help of reference materials with known isotopic ratios. For new nuclides, suitable AMS reference materials are typically not available and have to be produced, e.g., by a dilution series of certified materials.
Accelerator Mass Spectrometry of 99Tc is complicated further by the lack of a stable Tc isotope for instrumental setup and quantification of results. Due to its similar mass, Nb has proven well-suited for beam tuning at high-energy AMS setups (Koll et al., 2019) extracted as NbO– as well as at VERA, using both NbO– and NbF5– beams. For oxide extraction, NbO– was also satisfactorily used for normalization of the 99Tc results which was associated with an uncertainty of around 30% (Koll et al., 2019). However, this bears similar problems as discussed above for the non-isotopic Np/Pu normalization and requires thorough assessment at VERA.
For the validation of the analytical method, the literature values for the concentrations of 239,240Pu, 237Np, and 241Am and the U isotope ratios of the analyzed reference materials are compared to our experimental values in Table 2. Samples were prepared by the procedure described in section “Chemical Extraction of Actinides (U, Pu, Np, and Am)” and actinides were measured in oxide form. No reference values for the 233U/238U and 233U/236U values are available until now. Apart from the 237Np concentration all isotope concentrations and ratios are well reproduced within at least 2 σ uncertainty corresponding to an average accuracy of 0.2 σ for 239Pu, 0.7 σ for 240Pu, and 1.1 σ for 241Am concentrations. The accuracy is given in terms of the literature value’s uncertainty. In comparison to the value published by Eigl et al. (2013) also using the VERA setup but a different sample preparation method, our present result shows an accuracy of 2.5 σ. However, both ratios obtained with VERA are significantly lower than the information value (5 σ accuracy). Possible reasons related to the high isotope ratio of the IAEA-381 material have already been discussed in Eigl et al. (2013). It demonstrates the need for an AMS ocean water reference material in the range of 236U/238U = 10–8. Accordingly, a very good accuracy of 0.15 σ was achieved for IAEA-385. While the IAEA-381 sample shows a deviation of 22% for the 237Np concentration which has been observed as typical precision for 237Np measurements when using a non-isotopic spike for normalization (Qiao et al., 2013; Quinto et al., 2015, 2017), the experimental value for IAEA-385 is 3 orders of magnitude lower than the published values. This is essentially equivalent to a complete loss of 237Np during sample preparation, which, however, is not reflected by the Pu spike, in this way leading to a wrong final result for the 237Np concentration in the initial sample. The inaccuracy of this result underlines the danger of using a Pu nuclide such as 242Pu as non-isotopic spike for 237Np determination in case the chemical yield of Np and Pu are unexpectedly different.
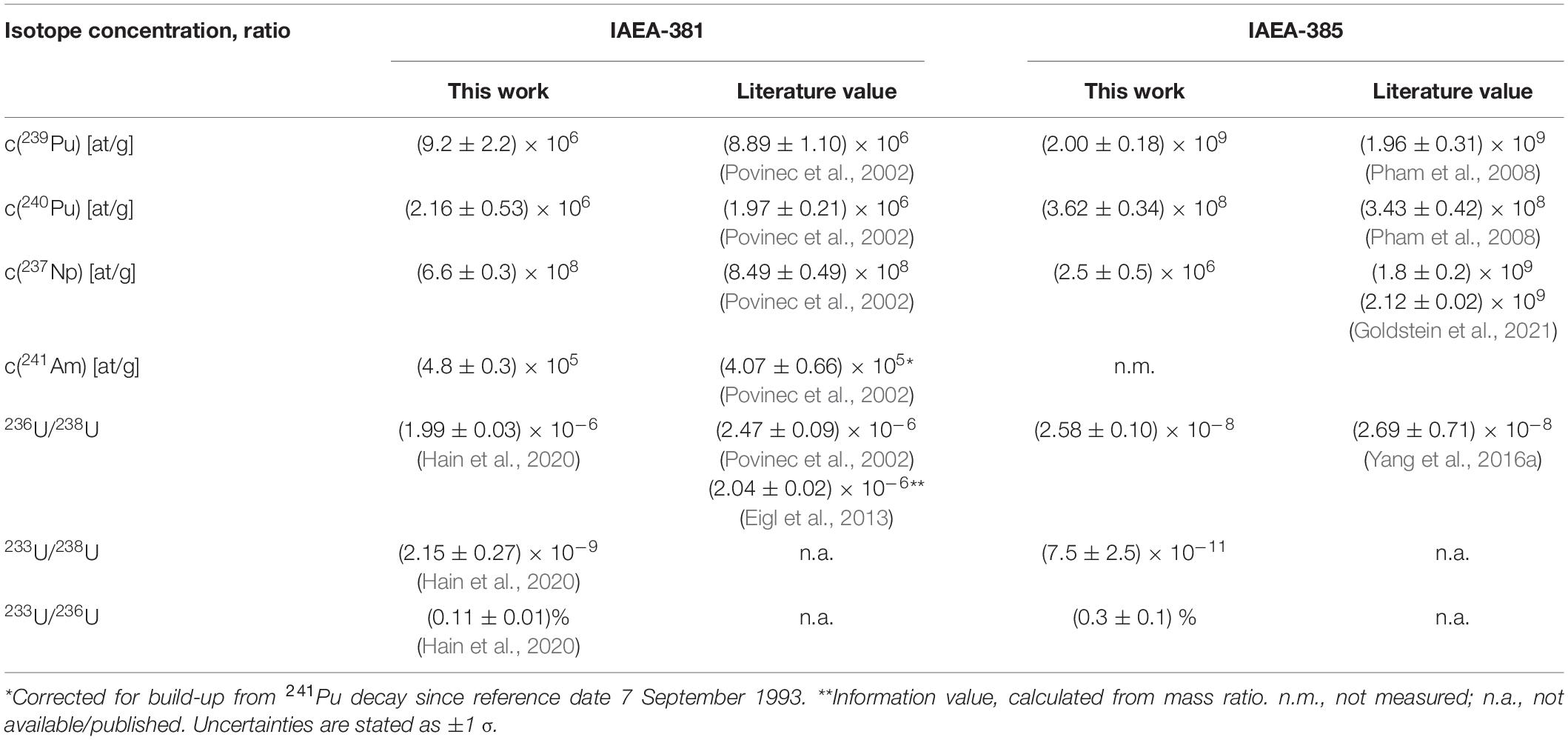
Table 2. Comparison of results obtained by the VERA routine analysis of long-lived actinides in reference materials with literature values.
The measurement results on the 239,240Pu and 241Am concentration (blank corrected) as well as the 236U/238U obtained for the water column at BD04 are plotted in Figures 2A–C. The calculated radionuclide ratios 240Pu/239Pu, 233U/236U, and 237Np/239Pu, which can serve as a fingerprint to distinguish different emission sources, are shown in Figures 2D–F. The values and further details on the measurement are listed in Supplementary Tables 1, 2. Regarding the shortcomings of normalizing 237Np count rates without isotopic spike, only the overall count rates including the process blank are compared without further interpretation of the absolute concentration, which therefore are not plotted in Figure 2. The new data for 239,240Pu obtained with the present procedure were combined with the data from the previous study (Hain et al., 2017) to show the complete depth profile. Selected AMS targets such as the U fraction of BD04-20m or the Pu fraction of BD04-200m were re-measured in a different measurement to verify the reproducibility of the investigated isotopic ratios and showed good agreement within 1 σ uncertainty. The concentrations of the two particle-reactive elements Pu and Am are increasing from the water surface to the maximum depth, while the increase by a factor of 24 from 5.4 × 103 atoms/kg to 1.3 × 105 atoms/kg is stronger for 241Am than for 239Pu (around factor 5). The 241Am count rate in the two surface water sample (0 and 20 m) are close to their corresponding blanks, i.e., BLKII and BLKI, respectively, (see Supplementary Table 2) leading to considerable uncertainties of the final 241Am concentrations in the two samples after blank correction. This means that the limiting factor for the multi-actinide measurements is not constituted by the AMS detection efficiency including chemical sample preparation, but by the background of the procedure blanks. It is important to note that previous studies usually used sample volumes of >50 L for multi-isotope analysis (Lee et al., 2003b; Povinec et al., 2003; Rozmaric et al., 2022). For the 236U/238U and 233U/238U ratios the respective blank levels are negligible compared to the uncertainty of the measurement result. A clear signal above the blank count rates was also observed for 237Np count rates, which shows the general feasibility of 237Np detection at VERA.
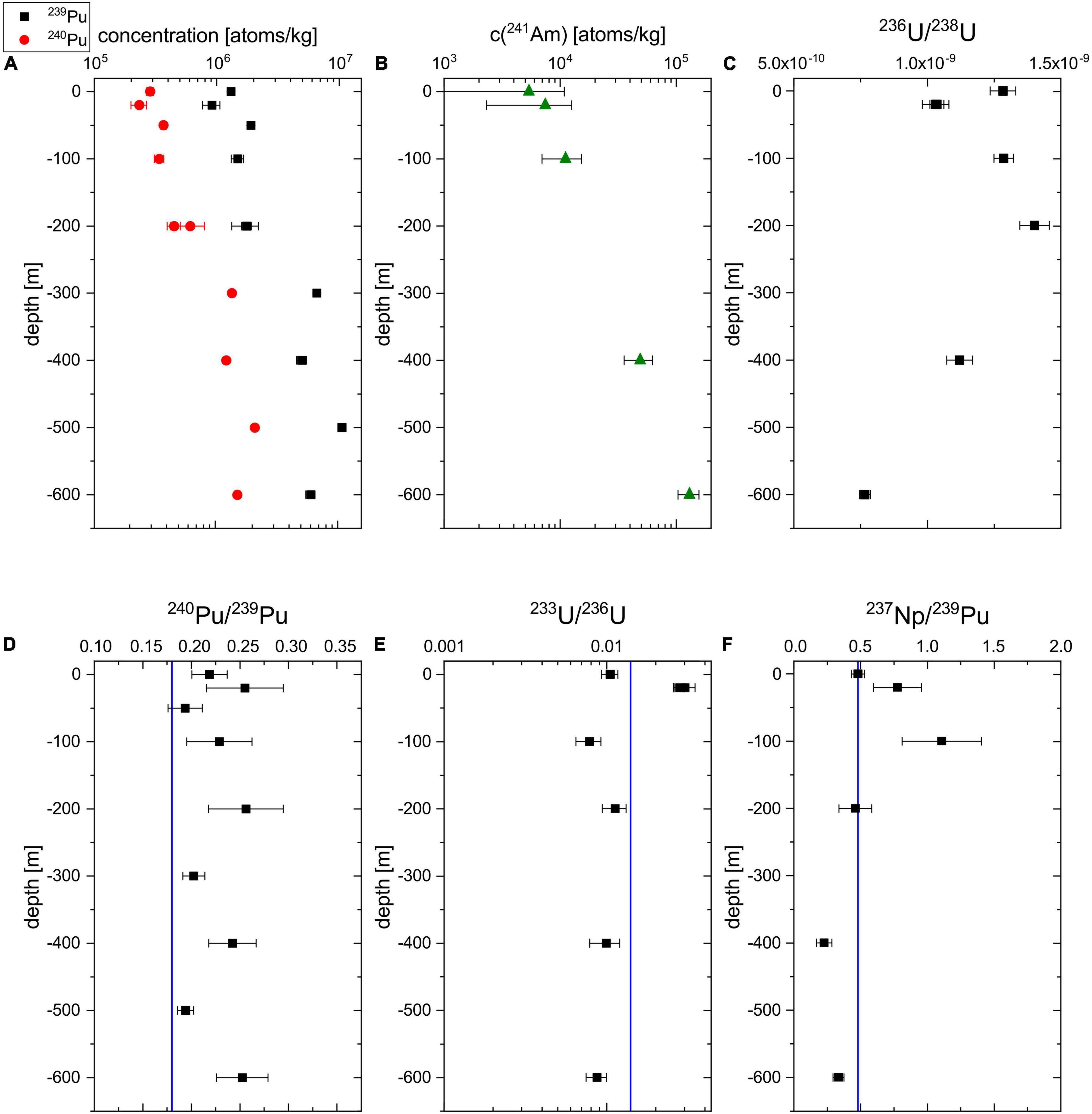
Figure 2. Depth profiles of the 239Pu (black) and 240Pu (red) concentration (A), the 241Am (green) concentration (B), the 236U/238U atom ratio (C), the 240Pu/239Pu atom ratio (D), the 233U/236U atom ratio (E), and the 237Np/239Pu count rate ratio (F) at the sampling station BD04. The vertical blue line indicates the literature value for global fallout of the respective radionuclide ratio (Kelley et al., 1999; Hain et al., 2020). For the Pu depth profiles, the new data has been combined with the data from a previous study (Hain et al., 2017).
The 236U/238U ratios around 1 × 10–9 in the topmost 600 m of the water column agree well with previously published data for the NW Pacific Ocean (Eigl et al., 2017). Also the presented surface 239+240Pu concentrations of 2 mBq/m3 fit into the rather large range of 239+240Pu concentrations reported before (Lee et al., 2003a; Hirose, 2009; Lindahl et al., 2010). The comparably low maximum concentrations of 15 mBq/m3 at 500 m depth (Hain et al., 2017) support the observation that the subsurface Pu maximum decreases in amplitude over the years due to sedimentation processes (Livingston et al., 2001). Our maximum 241Am concentration at 600 m corresponding to 6.6 mBq/m3 is also slightly below the concentration range of 7−22 mBq/m3 published for this depth in one of the very few studies on 241Am distributions in Pacific Ocean water (Povinec et al., 2003). In general, the observed depth profiles are compatible with the expected behavior of the different nuclides in the open ocean. The environmental behavior of actinides essentially depends on their oxidation state, i.e., the reduced species with oxidation state +3 and +4 are more particle-reactive than those with oxidation state +5 and +6 which quickly hydrolyze forming ionic bonds. The latter is the case for U6+ and Np5+, present in natural waters as the dissolved uranyl ion UO22+ and neptunyl ion NpO2+ (Lehto and Hou, 2011) respectively, so that especially Np is conservatively transported with the water masses. In the open sea, the mobility of U is further increased as it mainly exists as carbonate complex with the long residence time of 0.5 × 106 yrs (Dunk et al., 2002). Consequently, conservative radionuclides stay at the water surface if they entered by surface deposition as observed for 233,236U/238U in the presented depth profiles. Contrarily, Am present in oxidation state +3 and Pu, mainly existing in valence state +3 or +4 are considered as particle-reactive and are quickly removed from the water column by sedimentary mineral or organic particles (Lehto and Hou, 2011). Hence, the corresponding concentrations are lowest at the water surface and the stronger increase observed for 241Am proves the higher particle-reactivity compared to 239,240Pu at the given particle concentration in the water column. A detailed discussion of the distributions and the inventory does not appear prudent based on the limited data set from one sampling location only. As is typical for the Pacific Ocean, the 240Pu/239Pu atom ratios (Figure 2D) are elevated compared to the literature value for global fallout on the Northern Hemisphere of 0.180 ± 0.014 (Kelley et al., 1999) marked by the vertical blue line. This result suggests the presence of tropospheric close-in fallout from the Pacific Proving Grounds. The novel data on the 233U/236U depth profile in an ocean water column presented in Figure 2E presents a weighted mean of (1.04 ± 0.18) × 10–2 which is slightly lower (more than 1 σ) than the value for global nuclear weapons (1.40 ± 0.15) × 10–2 fallout published only recently in Hain et al. (2020) for the first time. As discussed in this initial publication on anthropogenic 233U and recently supported by data from Baltic Sea sediments (Lin et al., 2021), tropospheric fallout from smaller 233U fuelled fission devices might have led to a locally more varying 233U distribution. Being located west of the relevant Nevada test site, the Pacific Ocean might have received relatively small input from these smaller weapons but large amounts of 236U have been released at the Pacific Proving Grounds, which could be a possible explanation for the lower 233U/236U ratios in the presented water column. The uncorrected 237/239 count rate ratios are in reasonable agreement with the previously published value for nuclear weapons fallout of 0.48 ± 0.07 at northern latitudes (Kelley et al., 1999). Whereas the deviation from the literature value at 100 m is not significant due to the large uncertainties of the uncorrected experimental value, the measured ratios at greater depths, i.e., 400 and 600 m are clearly lower. This could be expected because of the strong chemical fractionation between the conservatively behaving Np, which has been found to stay at the surface (López-Lora et al., 2020), and the particle-reactive Pu with a subsurface maximum at around 1000 m depth (Livingston et al., 2001) in the water column. The indicated literature value is independent from chemical fractionation as the total fallout inventory of the two nuclides was compared in the corresponding study (Kelley et al., 1999).
Selected sample aliquots obtained by leaching of air filter ashes were used for the comparison of oxide and fluoride target preparation and extraction in the AMS ion source. The collection of the air filters, the chemical U purification and the results of the complete data set analyzed in oxide form will be published elsewhere (Wallner G. et al., submitted to J. Environ. Radioact.). It has to be noted, that nine times more solution was used for fluoride target preparation than for the oxides measurements, because the aim was to also analyze the much rarer 233U. This led to significantly better counting statistics of 236U, not attributable to fluoride extraction alone.
As shown in Figure 3, the 236U/238U ratios measured in oxide form can be very well reproduced by fluoride (UF5–) extraction for all samples as well as the in-house standard material Vienna-US-8 (236U/238U = 1.01 × 10–8) within 1σ uncertainty. The uncertainties plotted in this graph and listed in Supplementary Table 3 along with all the detailed information on both measurements also include systematic variations which usually dominate the overall uncertainty of AMS results. The final results for both methods show similar uncertainties therefore indicating that fluoride extraction did not induce a higher variability between the individual measurements. Also the 236U blank level for both methods is comparable, but the considerably higher 238U currents from the blanks need to be further investigated. A systematic input of 238U from the new reagents involved seems most probable. Recent measurements exclude the Cu sample holder as a significant source of 238U so that only PbF2 is left as a potential source. A direct comparison of the detection efficiency via the count rates and ion currents of the present experiment is not straightforward, as samples were split into two aliquots before chemical purification and might have suffered from different U losses during column separation. However, the consistently elevated 236U count rates and 238U ion currents (factor 1.7) taking into account that the initial Vienna-US-8 material was considerably diluted with PbF2 in a mass ratio 1:9 for fluoride extraction, point to an enhanced detection efficiency. A further increase in detection efficiency is in sight as first fluoride measurements of the mass-239/mass-238 ratio for the in-house standard material Vienna-KkU (Steier et al., 2008) lower presence of UH3+ molecules. In the experiment described in Steier et al. (2019) for oxide extraction, the stripper gas pressure was decreased from 1 Pa to 0.4 Pa for which the maximum transmission of 24% for charge state 3+ has been observed. Thereby, the m2393+/238U ratio increased from about 10–14 to 5 × 10–11. In the present experiments with fluoride extraction, we reached a 50 times lower m2393+/238U ratio at the lower stripper gas pressure of 0.4 Pa which indicates a lower formation of hydro-fluoride compounds.
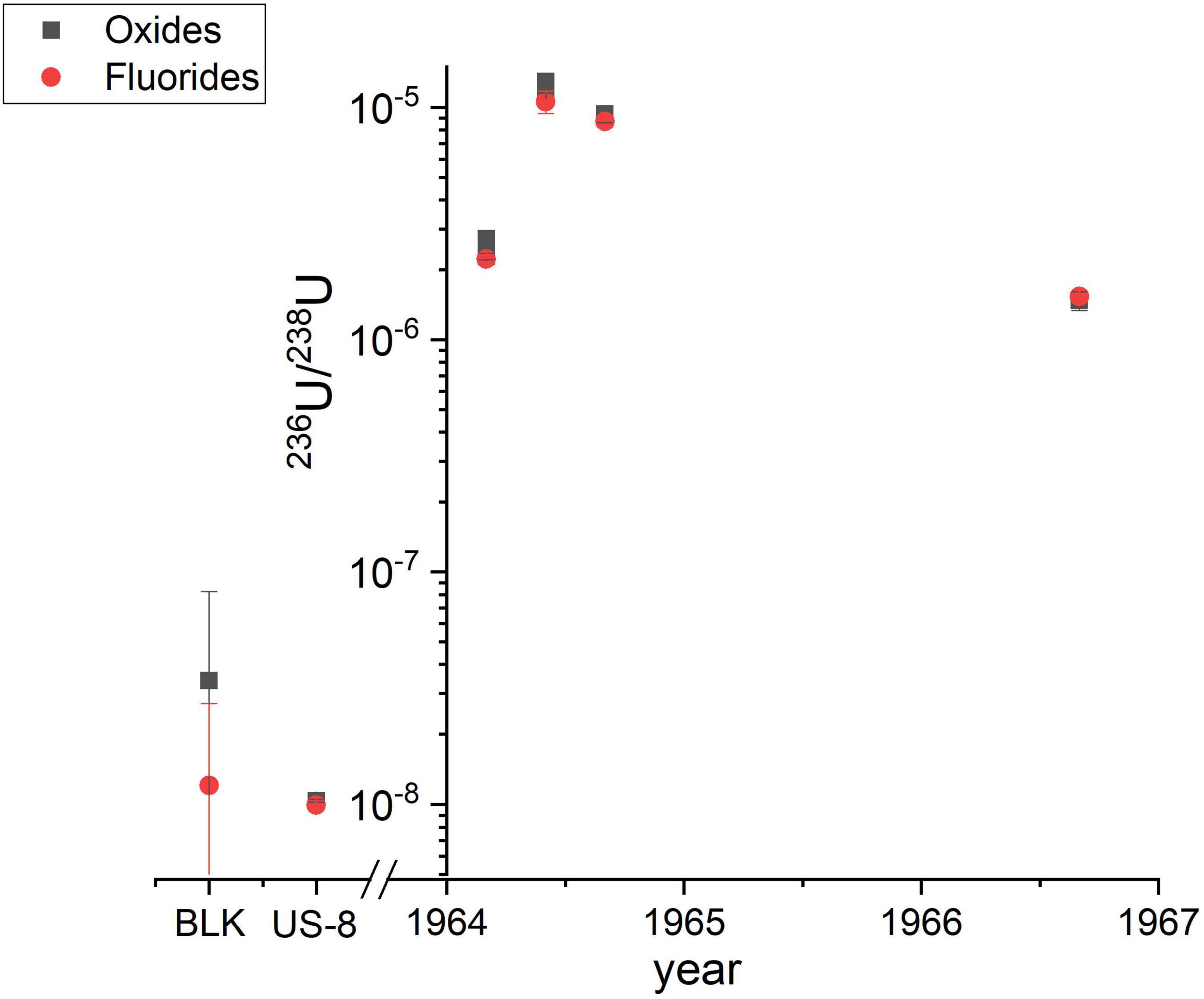
Figure 3. Comparison of the 236U/238U results for air filters, either obtained by UO– (black squares) or UF5– (red dots) extraction. “BLK” denotes the corresponding process blank and “US-8” is a VERA in-house standard for 236U (see main text).
Even though 237Np is well detectable above blank level at VERA as shown in section “Multi-Actinide Results From the Pacific Ocean,” and tailing from 238U does not interfere in the AMS measurement (m237/m238 < 1.8 × 10–13 measured on Vienna-KkU), a reliable quantitative analysis of 237Np concentrations requires a isotopic spike material which is sufficiently pure regarding 237Np (Jerome et al., 2014) or other by-products of the irradiation such as 236Pu (Efurd et al., 1991). The most promising spike candidate currently under development in a joint project with the University of Tsukuba (Japan) is 236Np produced via the bombardment of 232Th with 7Li in the 30–40 MeV range at the RIKEN Nishina Center. AMS measurements at VERA have confirmed the production of mass m236 above background and m237/m236 ratios down to (7.62 ± 0.08) × 10–2 for this material were measured. Whereas mass m237 is exclusively 237Np, on mass m236 there is potential isobaric interference. For the prospective spike material, the main isobar of concern is 236U co-produced either via neutron capture on 235U or the decay of the excited state 236mNp. Also 236Pu is built-up by the decay of 236mNp but usually can be easily suppressed by chemical purification. In order to be able to confirm the production of 236Np, a method to identify the isobaric composition of this irradiated material had to be developed.
As pointed out before, the admixture of PbF2– results in in situ fluoridization of the actinides (An) during the sputter process in the AMS ion source. We have systematically investigated the formation ratios of the AnF5–, AnF4O–, AnF3 O2–, AnF3O– AnF2O2–, and AnF4– molecular anions of the actinides U, Np, Pu, and Am. This effort has resulted in a novel isobar analysis technique based on the element specific AnF4–/AnF5– ratios that is capable of characterizing the elemental composition of the produced mass 236 in the prospective spike material (Zhao and Francisco, 2022; Wiederin, A. et al., in preparation). The observed NpF4–/NpF5– ratio in a sample with known isotopic composition was found to be a factor 10 different from the PuF4–/PuF5– and the UF4–/UF5–, respectively. First measurements based on this method of the irradiated material for Np spike production show a m236F4–/m236F5– ratio which is compatible with that of the reference NpF4–/NpF5– ratio within 1 σ.
The combination of SrF3– extraction from the ion source and ILIAMS-suppression of ZrF3– provides an isobaric suppression of 90Zr of >1010, which is sufficient to achieve a blank value of 90Sr/Sr ∼5 × 10–15 on all environmental soil and bone samples tested so far (Martschini et al., 2021). Based on the addition of 1 mg of stable Sr carrier for normalization and a total detection efficiency of 4 × 10–4, this is equivalent to a detection limit of <0.1 mBq, i.e., 105 atoms of 90Sr per sample. To our knowledge, this makes AMS with ILIAMS the most sensitive detection method for 90Sr so far as other techniques or setups achieve at most detection limits of 3 mBq (Bu et al., 2016).
Currently, we are in the process of assessing the accuracy of this method for different environmental materials. These tests are conducted with various IAEA reference materials such as IAEA-A-12 (beef bone) and IAEA-TEL-2015-03_05 (Syrian soil) and measured against an in-house AMS-reference materials prepared from a dilution series of an LSC-reference solution. Preliminary results from this study are shown in Figure 4. While the value of the measured 90Sr/Sr isotopic ration in the beef bone sample agrees perfectly well with its nominal value, the value measured in both soil samples is measured slightly too low compared to its nominal value. Whether the reason for this is due to some 90Sr loss in chemical sample preparation, some deviations in ICP-MS determination of intrinsic stable Sr in the soil sample required to calculate back to the 90Sr/Sr in the environmental sample or due to issues with our in-house-reference materials is currently under investigation.
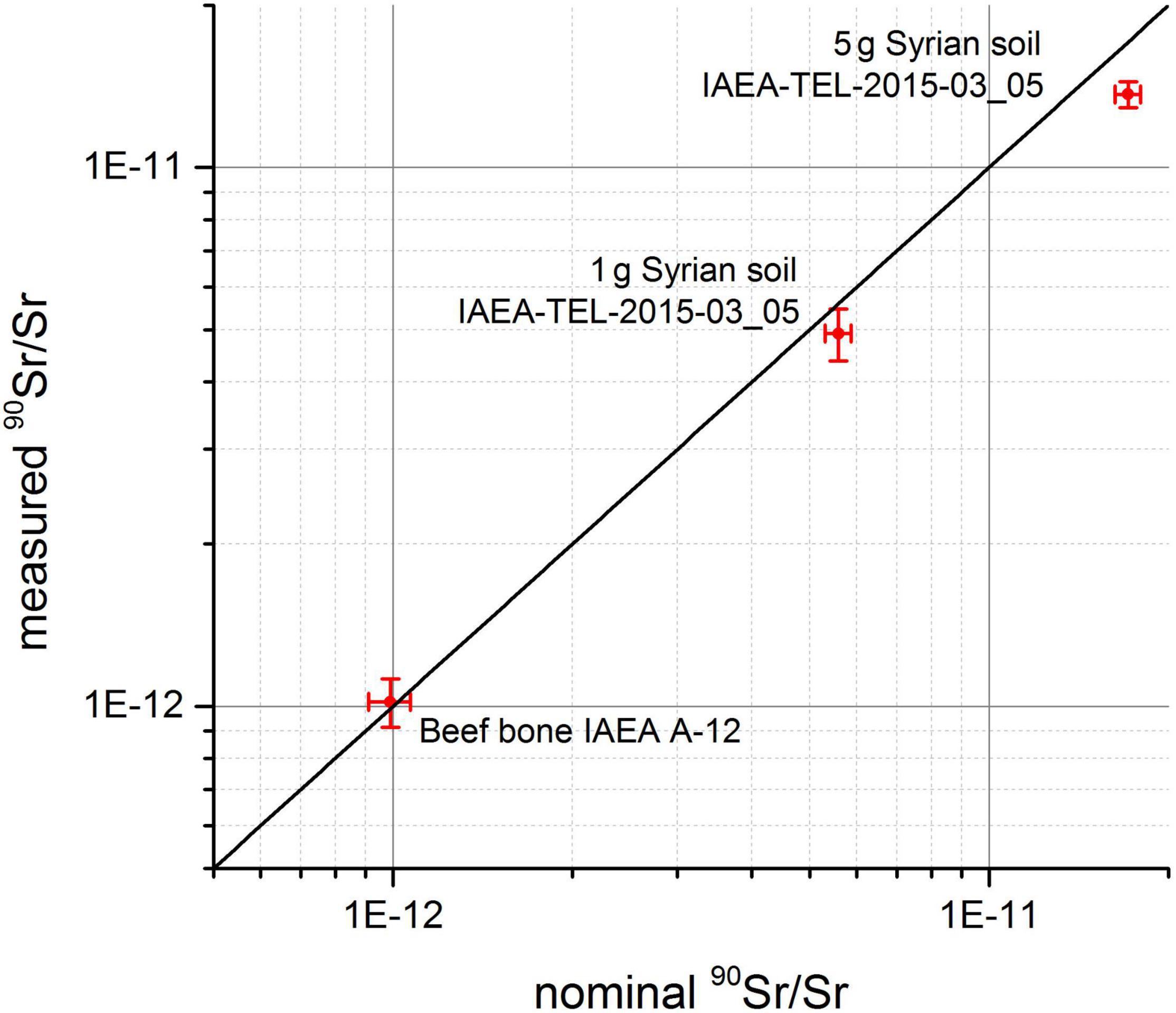
Figure 4. Comparison of nominal and measured 90Sr/Sr ratios for environmental IAEA-reference materials.
Based on the above detection limits, one can estimate the sample size of ocean water required for future analysis of 90Sr. Radiostrontium levels in surface water of the general ocean, e.g., the Pacific Ocean, are reported to be around 1 Bq/m3 at the beginning of the century with effective half residence times of 14–15 years (Povinec et al., 2012). Assuming reasonable 90Sr recovery in chemical sample preparation, this means that 1–3 L of ocean water should be sufficient to stay well above background for the next decades. As the Sr-content of 1 g of aragonitic coral skeleton is almost equivalent to the Sr-content of 1 L of ocean water at the time of coral growth (Purdy et al., 1989), a few g of coral are sufficient for similar analysis.
Using ILIAMS, the AMS system at VERA is very robust against the presence of the Ba isobars and other interferences, e.g., including the presence of Rb in the sample, which was reported to lead to molecular interferences for TIMS measurements (Zhu et al., 2020b). Additionally, because of the many energy and momentum filters, the AMS setup is not limited regarding the abundance sensitivity for the 135Cs/133Cs ratio by tailing effects of strong 133Cs signals. Therefore, we see a potential for the application of the ILIAMS assisted AMS measurements when oceanic data far from anthropogenic sources and close to natural 135Cs/133Cs levels are needed. Based on the performance parameters regarding 135Ba suppression and detection efficiency (Martschini et al., 2021; Wieser, A. et al. in preparation), we can now estimate the amount of ocean water sample required for the analysis of 135Cs with ILIAMS.
Similar to other fissiogenic radionuclides such as 129I (Fabryka-Martin, 1988), the main source of natural 135Cs is from spontaneous fission of 238U with a cumulative yield Ysf(135Cs) = 0.052. For the 135Cs estimation in the ocean we assume a continuous input of both U and Cs to the ocean via rivers. In addition, we assume that at their origin on the continent, 238U and 135Cs are in secular equilibrium, which is reached after approximately ten half-lives of 135Cs. Depending on the half-life of 135Cs used in this context, this will result in an atomic input ratio of (135Cs/238U)cont = (0.8–1.5) × 10–11. The radioactive lifetimes of 238U and 135Cs are much longer than their residence times in ocean water and both elements are removed from the water column before any significant build-up or decay can occur. Taking into account the difference in residence time τres for Cs and U we estimate a 135Cs concentration in the ocean water of,
Using an average 133Cs concentration of 1.3 × 1015 atoms/L (0.3 ppb) one can expect a natural 135Cs/133Cs background ratio in ocean water of (3–6) × 10–11. Due to the lower concentration of Cs relative to U in ocean water compared to the general crust, this value is higher than the expected 135Cs/133Cs ratio in the crust of ∼8 × 10–12 (Lachner et al., 2015). The expected 135Cs/133Cs background ratio in ocean water is still below the presently reported sensitivities for TIMS and ICP-MS/MS in ocean water samples, but can be reached using the high abundance sensitivity of 135Cs/133Cs < 10–11 and barium suppression of six orders of magnitude available with the ILIAMS assisted AMS facility VERA. With our present detection efficiency we expect that a sample of 1 L of ocean water is sufficient for an AMS measurement at VERA down to natural levels.
Figure 5 shows the results for the Tc recovery prepared with different amounts of Nb and calcined at the two different temperatures 450°C (orange) and 300°C (blue) for 1 h, respectively. The uncertainties are determined by the counting statistics of gamma spectrometry. While at 450°C variable losses of up to 25% were observed, the recovery is more stable at 300°C, independently of the amount of Nb used for the preparation of the target matrix. In the latter scenario, recoveries exceeded 90% for all samples. First test measurements with AMS did not indicate negative effects of the lower temperatures on the ion source output and the background for 99Tc.
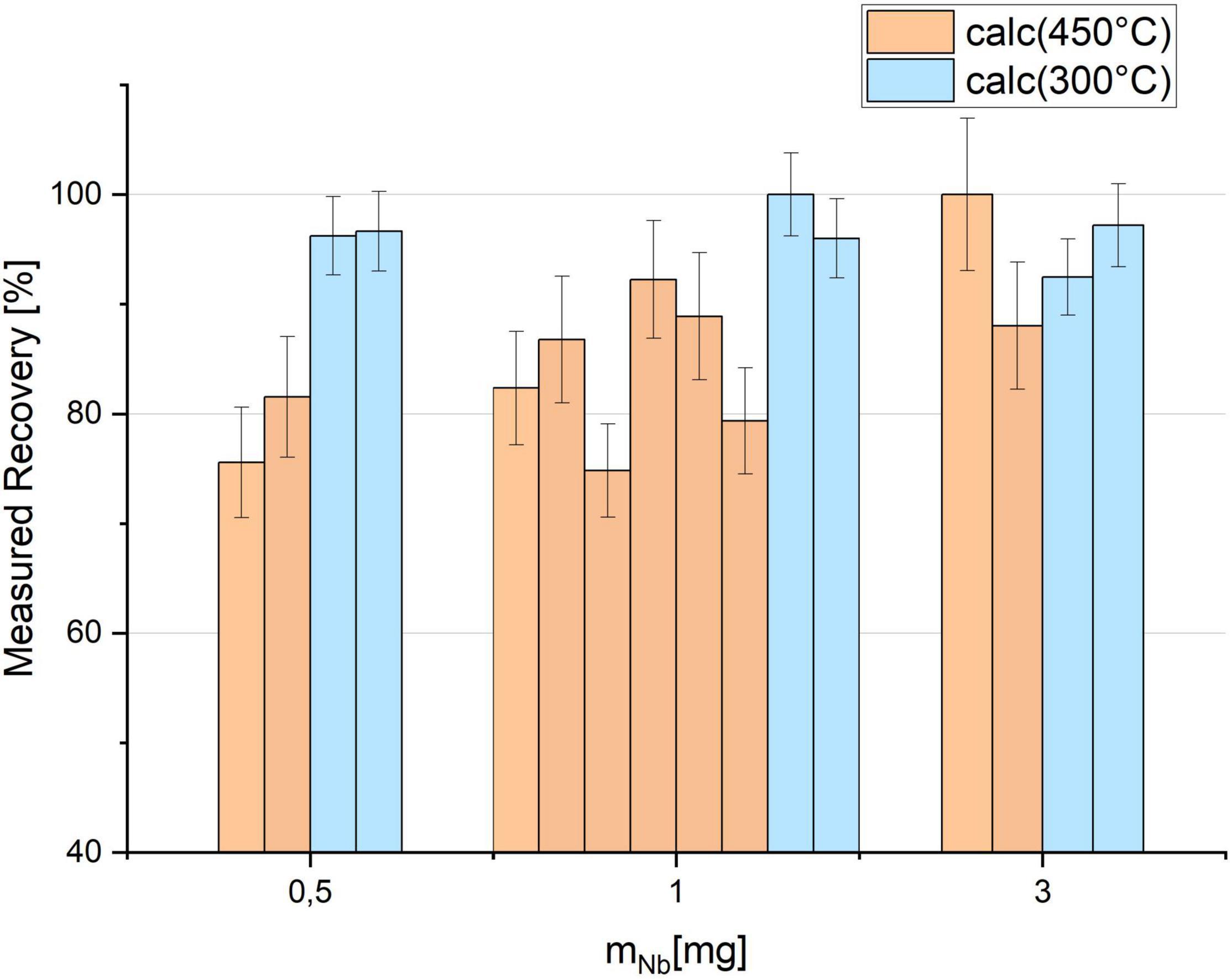
Figure 5. 95mTc recovery after calcination at 450 and 300°C in comparison measured by gamma counting. 95mTc was embedded in different amounts of Niobium (Nb) and prepared in individual crucibles.
The 99Tc concentrations in the water column at BD04 and a second sampling station around 1,500 km to the Northeast of BD04 were analyzed with the high-energy AMS setup at the Maier-Leibnitz Laboratory (MLL) in Munich which featured a 14 MV tandem accelerator and the Gas-filled analyzing magnet system “GAMS” (Koll et al., 2019; Quinto et al., 2019). The results obtained for BD04 of around 107 atoms/L, and below 5 × 106 atoms/L at greater distance to Japan (Hain, K., et al., in preparation) are in good agreement with previous studies with published values of 5 × 106 atoms/L for the transect Japan-Australia (Momoshima et al., 2005) and 7 × 107 atoms/L at the Fukuoka coast (Momoshima et al., 1995). For the GAMS setup an overall Ru suppression factor between 3 × 104 and 3 × 105 has been estimated in a previous study using samples with well-known Ru concentration (Quinto et al., 2019).
In comparison, first experiments with ILIAMS showed that TcF5– has to be used instead of TcO– for laser suppression of the isobar Ru (Martschini et al., 2021). Since Ru is among the stable elements with the lowest natural abundance with typical concentrations of 1 ppt in water and 10–100 ppt in sediment (Bekov et al., 1984), a 10 L ocean water sample or a 100 g sediment sample will both contain roughly 6 × 1013 atoms of Ru. In order to assess any difference in ion formation probabilities of TcF5– and RuF5– as well as laser suppression factors, we prepared two batches of samples from a clean Nb-solution, one spiked with a similar amount of Ru (7 × 1013 atoms), the other spiked with 1 × 1010 atoms of 99Tc. The count rates in the detector from procedure blanks prepared from the unspiked Nb-solution were below 0.01 cps for mass 99 amu, i.e., 99Tc3+ and 99Ru3+, respectively. Without laser suppression, the Ru-spiked samples showed 99Ru3+ count rates of 10–30 cps. Despite much lower atom concentrations, the Tc-spiked samples also yielded 8–10 cps of 99Tc3+ in the detector. Taking into account the natural abundance of 99Ru of 12.76% and assuming similar transmission through the system, this yields a Ru suppression vs. Tc of 250–900 by extraction of pentafluoride anions from the ion source. In combination with the Ru-suppression factor of ILIAMS of 105, the direct Ru-induced background signal of an environmental sample without separation chemistry would be equivalent to at most 3 × 105 atoms 99Tc in the sample. This is an excellent abundance sensitivity given that the sample preparation method gives us a substantial safety buffer by providing additional >103 isobar suppression [Extraction of Fission Products (Tc, Cs, and Sr)].
A highly flexible chemical separation procedure has been developed which allows for analysis of the complete nuclide vector of 233,236U, 239,240Pu, 241Am, and 237Np from a single ocean water sample at ultra-low levels (below 10–3 ppq for 241Am). In that way, the direct comparison of actinides with different migration behavior is possible to account for scavenging processes of radionuclides for the interpretation of the water mass transport. The new data on 233U/236U ratios in the Pacific Ocean support our previous finding that this ratio is around 10–2 in the general environment if no releases from nuclear power and reprocessing plants are present. Being located at a distance of 250 km north-east of the Fukushima Daiichi Nuclear Power Plant, where water masses are transported southward by the Oyashio Current, a contribution from the reactor accident at the sampling station seems rather unlikely. To be able to interpret such small deviations from the published 233U/236U ratio for nuclear weapons fallout correctly, a thorough study of the global 233U distribution and its inventory as well as the characterization of the weapons fallout source term is required. The present work shows that analysis of 233U/236U in ocean water remains challenging due to the very low abundance of 233U/238U in the lower 10–11 range in regions not directly affected by nuclear emissions. The 233U/238U ratio will decrease even further for larger depths (>600 m) as only little anthropogenic U is transported downward in the water column. Especially 233U analysis would benefit from an enhanced detection efficiency using UF5– extraction which is currently investigated at VERA. The results for environmental samples obtained from oxide extraction are well reproduced in fluoride measurements with a similar precision and an at least equally high efficiency. At the same time, the final steps for target preparation are considerably simplified as the co-precipitation and washing steps are omitted. For this reason, fluoride extraction can be considered advantageous for U analysis in large batches of environmental samples already at the present time, even for AMS facilities reaching already up to few percent ionization efficiencies for actinide-oxide extraction (Hotchkis et al., 2019). In addition, the reduced background on mass 236 and mass 239 caused by uranium-hydride molecules which have been found to survive the stripping process at lower stripper gas pressure (Lachner et al., 2013) can be perceived as clear advantage for any AMS facility. As the UF5– formation mainly depends on the presence of sufficient PbF2, it is also highly flexible regarding the target matrix and was successfully applied to environmental U fractions previously prepared as NdF3 precipitate for alpha spectrometry (unpublished). Next, experiments will compare aliquots obtained after chemical separation to assess the difference in detection efficiency more reliably and the procedure will be expanded to other actinides. With a further increase in detection efficiency, a full implementation of a multi-actinide approach where all the mentioned actinides are extracted from the same sputter target as already demonstrated for low-matrix and small volume samples (Quinto et al., 2015, 2017) is within reach.
Normalization of 237Np results to a non-isotopic spike using our rather simple chemical separation method did provide satisfactory results for water samples only. Provided that the ILIAMS method in combination with fluoride extraction for actinides facilitates suppression of isobars of 236Np as suggested by the observed fluoride formation patterns, the use of the 236Np isotopic spike will allow much more widespread and accurate 237Np analysis. Even AMS facilities without photodetachment capabilities will profit from this material as soon as potential isobaric contaminations and the 236Np/237Np ratio have been carefully quantified by VERA so that 237Np results for environmental samples can be corrected accordingly. Environmental samples are usually spiked with 1 × 108 atoms at VERA to ensure good counting statistics on the reference mass. This amount can be minimized to 1 × 107 if the chemical recovery of sample preparation is sufficiently high. However, with the best 236Np/237Np ratio achieved at present, this spike material would still add around 7.6 × 105 atoms of 237Np to the environmental sample which is in the order of magnitude of 237Np per kg water to be expected in the uppermost 1,000 m of water from the Pacific Ocean. Consequently, at least 10 L of water samples would be required to obtain reliable data on the 237Np concentrations with sufficiently low uncertainties to allow interpretation in the context of water mass transport. 237Np measurements are less challenging in the North Atlantic and Arctic Ocean where elevated 237Np concentrations have been detected due to releases from the Sellafield reprocessing plant (López-Lora et al., 2021).
Analysis of 90Sr in environmental samples using ILIAMS is ready for applications in bone and soil samples. It provides 30 times more abundance sensitivity compared to other techniques and omits the time-consuming sample preparation for radiometric techniques. The development of 135Cs and 137Cs measurements using ILIAMS are promising and the measurement of those isotopes in general ocean water is within reach. The robustness of AMS against interference from other elements and the strong suppression of the isobars may be an advantage over other MS techniques. In the near future, we plan to test our approach on coral samples and on ocean water reference materials such as IAEA-443, which will require adaptations to our sample chemistry procedure. It has to be noted that presently, there are no certified reference materials for the 135Cs/133Cs nor for the 135Cs/137Cs isotopic ratio available. However, the 135Cs/137Cs was already measured in various IAEA and NIST reference materials by various groups (Snow et al., 2015; Snow and Snyder, 2016; Bu et al., 2018, 2019; Zhu et al., 2020b) and their consensus values now are commonly used for method development and validation. An important validation step in the development of 135Cs measurement is to reproduce the given values for the 137Cs concentration. In this context, marine water materials certified for their 137Cs content, e.g., IAEA 381 and 443 (Irish Sea Water), would be of specific interest, but are presently not on stock at IAEA. Irish sea sediment (IAEA-385; reference date for decay correction: 2019-01-01) with a certified 137Cs activity of (19.8 ± 0.6) Bq/kg and a weighted mean of reported 135Cs/137Cs ratios of (1.15 ± 0.11) (Yang et al., 2016b; Zheng et al., 2016; Zhu et al., 2020a) thus presently seems to be the most suitable natural reference material for the advancement of Cs isotope measurements in the marine environment. In literature, we hardly find studies on 137Cs in coral cores, which reflects the inefficient incorporation of Cs in the coral skeleton. Previous data from coral material originating from a test site found detectable amounts of 137Cs in g-sized samples only during the peak activities of nuclear bomb testing (Noshkin et al., 1975). The detection of the 135Cs/137Cs ratio in coral samples not closely affected by anthropogenic sources is therefore expected to be a major challenge to be addressed with ILIAMS.
First experiments at VERA demonstrate that the abundance sensitivity achieved for 99Tc detection with the ILIAMS method is at least comparable to the former high-energy AMS setup GAMS at the MLL which already allowed the analysis of 99Tc concentrations in 10 L samples from the Pacific Ocean. Ongoing research focuses on finding a suitable normalization of the 99Tc count rate to obtain an absolute 99Tc concentration. Best precision will be achieved by using an isotopic spike analogous to the discussion of 237Np analysis as no elemental fractionation processes have to be taken into account. The potential candidate 97Tc (T1/2 = 1.6 × 106 yrs) has the disadvantage that it’s detection by mass spectrometry requires the suppression of the stable isobar 97Mo which is not only omnipresent in nature but especially in the ion source being a constituent of stainless steel. Consequently, an AMS measurement of 99Tc using a 97Tc spike has to ensure a highly efficient isobar suppression to higher and to lower atomic charge within the same measurement. Finding a combination of laser wavelength and molecular system with suitable electron affinities to fulfill this requirement is extremely challenging (Martschini et al., 2021). Also the alternative approach of using a spike of the mono-isotopic element Rh for normalization in combination with 95mTc as chemical yield monitor as proposed by Wacker et al. (2004) is currently being pursued. This normalization method has to be adjusted to fluoride extraction in order to allow its combination with ILIAMS and it has to be ensured that changes in the ion source output of RhF5– scale with TcF5–. Finally, also the normalization on the NbF5– current is being investigated in more detail. It has to be noted that the latter two normalization methods require the measurement of an external standard sample with well-known 99Tc/103Rh and 99Tc/93Nb ratio, respectively.
In addition, the complementary method of high-energy AMS is developed further in collaboration with our colleagues at the Heavy Ion Accelerator Facility (HIAF) of the Australian National University (ANU) to allow for environmental 99Tc detection in parallel to the ILIAMS approach. Since the closure of the MLL in Munich by the end of 2019, the HIAF facility houses the only remaining AMS setup with a 14 UD tandem accelerator reaching sufficiently high beam energies for the application of conventional isobar suppression and / or separation techniques. In contrast to the GAMS setup, AMS of 99Tc at HIAF uses an 8-anode ionization chamber for isobar identification (Martschini et al., 2019a). In principle, procedures for normalization used for measurements with the GAMS measurements can be adjusted to the HIAF setup.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://lehre4.rad.univie.ac.at/share/WWW_Exchange/public/H ain2021_frontiers_in_%20marine_sciences/.
KH: conceptualization, methodology, investigation and supervision for actinides, 99Tc, and Np spike, and writing – original draft. MM: conceptualization, methodology, investigation and supervision for 90Sr, 99Tc, and Np spike, and writing – original draft. FG: investigation on 99Tc sample preparation. MH: methodology, investigation and analysis for 90Sr samples, and writing – review. JL: conceptualization, methodology, investigation and supervision for 135Cs, and writing – original draft. MK: methodology, investigation, and fluoride extraction of actinides. JP: investigation and data evaluation for 99Tc. FQ: methodology, sample preparation actinides and 99Tc, and writing – review and editing. AS: conceptualization, methodology, and investigation chemical purification of 236Np. PS: AMS methodology for actinides, data interpretation, and writing – review and editing. AnW: conceptualization, methodology, investigation on Np spike characterization, and writing – original draft on Np spike and review. AlW: investigation and writing on 135Cs. AY: methodology and investigation 236Np production. RG: resources and ILIAMS methodology. All authors contributed to the article and approved the submitted version.
This project receives funding from the European Union’s Horizon 2020 Research and Innovation Program under grant agreement No. 824096 (Radiate). This work was partly funded by a bilateral project between the Austrian Science Fund (FWF): I 4803-N and Japan Society for the Promotion of Science (JSPS): JPJSBP120202001 as well as by the Austrian Science Fund (FWF): P 31614-N28 and Austrian Science Fund (FWF): P 22164-N20. The authors acknowledge financial support of the ILIAMS activities and the heavy isotope detection setup at VERA by the University of Vienna “Investitionsprojekte” program. This work was also partly supported by the JSPS Overseas Research Fellowships (No 201860538) and AS received funding from MEXT KAKENHI Grant Numbers 17H01874 and 21H03609 for parts of this work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
KH is very grateful to M. Yamada (Marine Ecology Research Institute, Japan) for providing the Pacific Ocean water samples. We thank T. Gamo from The University of Tokyo and the captain, officers, and crew of the R/V Hakuho Maru for their collaboration in the sampling during the KH-12-4 cruise. We acknowledge the support of M. Schiffer (University of Cologne) for the 95mTc production and S. Lehr (University of Vienna) for the preparation of the corresponding spike solution. We also thank G. Wallner (University of Vienna) for providing aliquots of the unique air filter samples for the fluoride-oxide comparison and a 90Sr-LSC-solution. Support by M. Kocadag and F. Mozina during the 90Sr sample preparation was highly appreciated. In addition, we thank Dorian Zok (Leibniz University Hannover, Germany) for providing comparison samples.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.837515/full#supplementary-material
Aarkrog, A., Boelskifte, S., Dahlgaard, H., Duniec, S., Hallstadius, L., Holm, E., et al. (1987). Technetium-99 and cesium-134 as long distance tracers in Arctic waters. Estuar. Coast. Shelf Sci. 24, 637–647. doi: 10.1016/0272-7714(87)90103-X
Bekov, G. I., Letokhov, V. S., Radaev, V. N., Baturin, G. N., Egorov, A. S., Kursky, A. N., et al. (1984). Ruthenium in the ocean. Nature 312, 748–750. doi: 10.1038/312748a0
Broecker, W. S., and Peng, T. H. (1982). Tracers In The Sea. New York, NY: Eldigio PressEldigio Press.
Bu, W., Ni, Y., Steinhauser, G., Zheng, W., Zheng, J., and Furuta, N. (2018). The role of mass spectrometry in radioactive contamination assessment after the Fukushima nuclear accident. J. Anal. At. Spectrom. 33, 519–546. doi: 10.1039/c7ja00401j
Bu, W., Tang, L., Liu, X., Wang, Z., Fukuda, M., Zheng, J., et al. (2019). Ultra-trace determination of the 135Cs/137Cs isotopic ratio by thermal ionization mass spectrometry with application to Fukushima marine sediment samples. J. Anal. At. Spectrom. 34, 301–309. doi: 10.1039/C8JA00380G
Bu, W., Zheng, J., Liu, X., Long, K., Hu, S., and Uchida, S. (2016). Mass spectrometry for the determination of fission products 135Cs, 137Cs and 90Sr: a review of methodology and applications. Spectrochim. Acta Part B 119, 65–75. doi: 10.1016/j.sab.2016.03.008
Cornett, R. J., Kazi, Z. H., Zhao, X.-L., Chartrand, M. G., Charles, R. J., and Kieser, W. E. (2015). Actinide measurements by AMS using fluoride matrices. Nucl. Instrum. Meth. in Phys. Res. B 361, 317–321. doi: 10.1016/j.nimb.2015.02.039
Dunk, R., Mills, R., and Jenkins, W. (2002). A reevaluation of the oceanic uranium budget for the Holocene. Chem. Geol. 190, 45–67. doi: 10.1016/S0009-2541(02)00110-9
Efurd, D. W., Roensch, F. R., Drake, J., and Perrin, R. E. (1991). Production, Separation, and Purification of 236Np and 236Pu. Radiochim. Acta 54, 159–162. doi: 10.1524/ract.1991.54.4.159
Eigl, R., Srncik, M., Steier, P., and Wallner, G. (2013). 236U/238U and 240Pu/239Pu isotopic ratios in small (2 L) sea and river water samples. J. Environ. Radioact. 116, 54–58. doi: 10.1016/j.jenvrad.2012.09.013
Eigl, R., Steier, P., Sakata, K., and Sakaguchi, A. (2017). Vertical distribution of 236U in the North Pacific Ocean. J. Environ. Radioact. 169-170, 70–78. doi: 10.1016/j.jenvrad.2016.12.010
Fabryka-Martin, J. T. (1988). Production Of Radionuclides In The Earth And Their Hydrogeologic Significance, With Emphasis On Chlorine-36 And Iodine-129. Ph.D. thesis. Tucson, AZ: University of Arizona.
Fifield, L. K. (2008). Accelerator mass spectrometry of the actinides. Quat. Geochronol. 3, 276–290. doi: 10.1016/j.quageo.2007.10.003
Goldstein, S. J., Price, A. A., Hinrichs, K. A., Lamont, S. P., Nunn, A. J., Amato, R. S., et al. (2021). High-precision measurement of U-Pu-Np-Am concentrations and isotope ratios in environmental reference materials by mass spectrometry. J. Environ. Radioact. 237:106689. doi: 10.1016/j.jenvrad.2021.106689
Hain, K., Faestermann, T., Fimiani, L., Golser, R., Gómez-Guzmán, J. M., Korschinek, G., et al. (2017). Plutonium isotopes (239−241Pu) dissolved in pacific ocean waters detected by accelerator mass spectrometry: no effects of the fukushima accident observed. Environ. Sci. Technol. 51, 2031–2037. doi: 10.1021/acs.est.6b05605
Hain, K., Steier, P., Froehlich, M. B., Golser, R., Hou, X., Lachner, J., et al. (2020). 233U/236U signature allows to distinguish environmental emissions of civil nuclear industry from weapons fallout. Nat. Commun. 11:1275. doi: 10.1038/s41467-020-15008-2
Hirose, K. (2009). Plutonium in the ocean environment: its distributions and behavior. J. Nucl. Radiochem. Sci. 10, 1_R7–1_R16. doi: 10.14494/jnrs.10.1_R7
Hotchkis, M., Child, D. P., Froehlich, M. B., Wallner, A., Wilcken, K., and Williams, M. (2019). Actinides AMS on the VEGA accelerator. Nucl. Instrum. Meth. in Phys. Res. B. 438, 70–76. doi: 10.1016/j.nimb.2018.07.029
Jerome, S. M., Ivanov, P., Larijani, C., Parker, D. J., and Regan, P. H. (2014). The production of neptunium-236g. J. Environ. Radioact. 138, 315–322. doi: 10.1016/j.jenvrad.2014.02.029
Kazi, Z. H., Cornett, R. J., Zhao, X., and Kieser, W. E. (2015). Comparison of the measurement of Pu and Am isotopes by AMS using fluoride and oxide anion beams. J. Anal. At. Spectrom. 30, 2235–2240. doi: 10.1039/C5JA00195A
Kelley, J. M., Bond, L. A., and Beasley, T. M. (1999). Global distribution of Pu isotopes and 237Np. Sci. Total Environ. 237-238, 483–500. doi: 10.1016/S0048-9697(99)00160-6
Kern, M., Hain, K. Prášek, T., Steier, P., Wiederin, A., and Golser, R. (2022). R-003 Increasing the negative ionization yield for the efficient detection of 233U and 236U by AMS (Version 2). Zenodo. doi: 10.5281/zenodo.6256795
Koll, D., Busser, C., Faestermann, T., Gómez-Guzmán, J. M., Hain, K., Kinast, A., et al. (2019). Recent developments for AMS at the Munich tandem accelerator. Nucl. Instrum. Meth. in Phys. Res. B 438, 180–183. doi: 10.1016/j.nimb.2018.05.002
Lachner, J., Christl, M., Vockenhuber, C., and Synal, H.-A. (2013). Detection of UH3+ and ThH3+ molecules and 236U background studies with low-energy AMS. Nucl. Instrum. Meth. in Phys. Res. B 294, 364–368. doi: 10.1016/j.nimb.2012.02.010
Lachner, J., Kasberger, M., Martschini, M., Priller, A., Steier, P., and Golser, R. (2015). Developments towards detection of 135Cs at VERA. Nucl. Instrum. Meth. in Phys. Res. B 361, 440–444. doi: 10.1016/j.nimb.2015.01.032
Lachner, J., Marek, C., Martschini, M., Priller, A., Steier, P., and Golser, R. (2019). 36Cl in a new light: AMS measurements assisted by ion-laser interaction. Nucl. Instrum. Meth. in Phys. Res. B 456, 163–168. doi: 10.1016/j.nimb.2019.05.061
Lachner, J., Martschini, M., Kalb, A., Kern, M., Marchhart, O., Plasser, F., et al. (2021). Highly sensitive 26Al measurements by Ion-Laser-InterAction Mass Spectrometry. Int. J. Mass Spectrom. 465:116576. doi: 10.1016/j.ijms.2021.116576
Lee, S. H., Povinec, P. P., Wyse, E., and Hotchkis, M. A. C. (2008). Ultra-low-level determination of 236U in IAEA marine reference materials by ICPMS and AMS. Appl. Radiat. Isot. 66, 823–828. doi: 10.1016/j.apradiso.2008.02.020
Lee, S.-H., La Rosa, J. J., Levy-Palomo, I., Oregioni, B., Pham, M. K., Povinec, P. P., et al. (2003b). Recent inputs and budgets of 90Sr, 137Cs, 239,240Pu and 241Am in the northwest Mediterranean Sea. Deep-Sea Res. II 50, 2817–2834. doi: 10.1016/S0967-0645(03)00144-9
Lee, S.-H., Gastaud, J., Povinec, P. P., Hong, G.-H., Kim, S.-H., Chung, C.-S., et al. (2003a). Distribution of plutonium and americium in the marginal seas of the northwest Pacific Ocean. Deep-Sea Res. II: Top. Stud. Oceanogr. 50, 2727–2750. doi: 10.1016/S0967-0645(03)00150-4
Lehto, J., and Hou, X. (2011). Chemistry And Analysis Of Radionuclides. Weinheim: Laboratory techniques and methodology Wiley-VCH-Verl.
Lin, M., Qiao, J., Hou, X., Dellwig, O., Steier, P., Hain, K., et al. (2021). 70-Year Anthropogenic Uranium Imprints of Nuclear Activities in Baltic Sea Sediments. Environ. Sci. Technol. 55, 8918–8927. doi: 10.1021/acs.est.1c02136
Lindahl, P., Lee, S.-H., Worsfold, P., and Keith-Roach, M. (2010). Plutonium isotopes as tracers for ocean processes: a review. Mar. Environ. Res. 69, 73–84. doi: 10.1016/j.marenvres.2009.08.002
Lindahl, P., Roos, P., Holm, E., and Dahlgaard, H. (2005). Studies of Np and Pu in the marine environment of Swedish-Danish waters and the North Atlantic Ocean. J. Environ. Radioact. 82, 285–301. doi: 10.1016/j.jenvrad.2005.01.011
Livingston, H. D., Povinec, P. P., Ito, T., and Togawa, O. (2001). “Plutonium in the environment,” in Proceedings of the Second International Symposium. (Amsterdam: Elsevier), 267–292.
López-Lora, M., Chamizo, E., Levy, I., Christl, M., Casacuberta, N., and Kenna, T. C. (2021). 236U, 237Np and 239,240Pu as complementary fingerprints of radioactiveeffluents in the western Mediterranean Sea and in the Canada Basin (Arctic Ocean). Sci. Total Environ. 765:142741. doi: 10.1016/j.scitotenv.2020.142741
López-Lora, M., Chamizo, E., Rožmarić, M., and Louw, D. C. (2020). Presence of 236U and 237Np in a marine ecosystem: the northern Benguela Upwelling System, a case study. Sci. Total Environ. 708:135222. doi: 10.1016/j.scitotenv.2019.135222
López-Lora, M., Levy, I., and Chamizo, E. (2019). Simple and fast method for the analysis of 236U, 237Np, 239Pu and 240Pu from seawater samples by accelerator mass spectrometry. Talanta 200, 22–30. doi: 10.1016/j.talanta.2019.03.036
MacDonald, C. M., Cornett, R. J., Charles, C. R. J., Zhao, X. L., and Kieser, W. E. (2016). Measurement of the 135Cs half-life with accelerator mass spectrometry and inductively coupled plasma mass spectrometry. Phys. Rev. C 93:014310. doi: 10.1103/PhysRevC.93.014310
Martschini, M., Hanstorp, D., Lachner, J., Marek, C., Priller, A., Steier, P., et al. (2019b). The ILIAMS project – An RFQ ion beam cooler for selective laser photodetachment at VERA. Nucl. Instrum. Meth. in Phys. Res. B 456, 213–217. doi: 10.1016/j.nimb.2019.04.039
Martschini, M., Fifield, L. K., Froehlich, M. B., Leckenby, G., Pavetich, S., Tims, S. G., et al. (2019a). New and upgraded ionization chambers for AMS at the Australian National University. Nucl. Instrum. Meth. in Phys. Res. B 438, 141–147. doi: 10.1016/j.nimb.2018.05.039
Martschini, M., Lachner, J., Hain, K., Kern, M., Marchhart, O., Pitters, J., et al. (2021). 5 years of ion-laser iinteraction mass spectrometry—status and prospects of isobar suppression in AMS by lasers. Radiocarbon 1–14. doi: 10.1017/RDC.2021.73
Maxwell, S. L., Culligan, B. K., Jones, V. D., Nichols, S. T., Bernard, M. A., and Noyes, G. W. (2010). Determination of 237Np and Pu isotopes in large soil samples by inductively coupled plasma mass spectrometry. Anal. Chim. Acta 682, 130–136. doi: 10.1016/j.aca.2010.10.001
Momoshima, N., Sayad, M., and Takashima, Y. (1995). Determination of 99Tc in coastal seawater collected in Fukuoka. Japan. J. Radioanal. Nucl. Chem. 197, 245–251. doi: 10.1007/bf02036003
Momoshima, N., Sayad, M., Yamada, M., Takamura, M., and Kawamura, H. (2005). Global fallout levels of 99Tc and activity ratio of 99Tc/137Cs in the Pacific Ocean. J. Radioanal. Nucl. Chem. 266, 455–460. doi: 10.1007/s10967-005-0931-2
Noshkin, V. E., Wong, K. M., Eagle, R. J., and Gatrousis, C. (1975). Transuranics and other radionuclides in Bikini Lagoon: concentration data retrieved from aged coral sections. Limnol. Oceanogr. 20, 729–742. doi: 10.4319/lo.1975.20.5.0729
Pavetich, S., Carey, A., Fifield, L. K., Froehlich, M. B., Halfon, S., Kinast, A., et al. (2019). 93Zr developments at the heavy ion accelerator Facility at ANU. Nucl. Instrum. Meth. in Phys. Res. B 438, 77–83. doi: 10.1016/j.nimb.2018.07.019
Pham, M. K., Sanchez-Cabeza, J. A., Povinec, P. P., Andor, K., Arnold, D., Benmansour, M., et al. (2008). A new certified reference material for radionuclides in irish sea sediment (IAEA-385). Appl. Radiat. Isot. 66, 1711–1717. doi: 10.1016/j.apradiso.2007.10.020
Pourmand, A., and Dauphas, N. (2010). Distribution coefficients of 60 elements on TODGA resin: application to Ca, Lu, Hf, U and Th isotope geochemistry. Talanta 81, 741–753. doi: 10.1016/j.talanta.2010.01.008
Povinec, P. P., Badie, C., and Baeza, A. (2002). Certified reference material for radionuclides in seawater IAEA-381 (Irish Sea Water). J. Radioanal. Nucl. Chem. 251, 369–374. doi: 10.1023/a:1014861620713
Povinec, P. P., Hirose, K., and Aoyama, M. (2012). Radiostrontium in the western North Pacific: characteristics, behavior, and the Fukushima impact. Environ. Sci. Technol. 46, 10356–10363. doi: 10.1021/es301997c
Povinec, P. P., Livingston, H. D., Shima, S., Aoyama, M., Gastaud, J., Goroncy, I., et al. (2003). IAEA’97 expedition to the NW Pacific Ocean—results of oceanographic and radionuclide investigations of the water column. Deep Sea Res. II 50, 2607–2637. doi: 10.1016/S0967-0645(03)00138-3
Purdy, C. B., Druffel, E. R., and Hugh, D. L. (1989). Anomalous levels of 90Sr and 239,240Pu in Florida corals: Evidence of coastal processes. Geochim. Cosmochim. Acta 53, 1401–1410. doi: 10.1016/0016-7037(89)90072-0
Qiao, J., Hou, X., Roos, P., and Miró, M. (2010). Rapid and simultaneous determination of neptunium and plutonium isotopes in environmental samples by extraction chromatography using sequential injection analysis and ICP-MS. J. Anal. At. Spectrom. 25:1769. doi: 10.1039/c003222k
Qiao, J., Hou, X., Roos, P., Lachner, J., Christl, M., and Xu, Y. (2013). Sequential injection approach for simultaneous determination of ultratrace plutonium and neptunium in urine with accelerator mass spectrometry. Anal. Chem. 85, 8826–8833. doi: 10.1021/ac4020319
Quinto, F., Blechschmidt, I., Garcia Perez, C., Geckeis, H., Geyer, F., Golser, R., et al. (2017). Multiactinide analysis with accelerator mass spectrometry for ultratrace determination in small samples: application to an in situ radionuclide tracer test within the colloid formation and migration experiment at the grimsel test site (Switzerland). Anal. Chem. 89, 7182–7189. doi: 10.1021/acs.analchem.7b01359
Quinto, F., Busser, C., Faestermann, T., Hain, K., Koll, D., Korschinek, G., et al. (2019). Ultratrace determination of 99Tc in small natural water samples by accelerator mass spectrometry with the gas-filled analyzing magnet system. Anal. Chem. 91, 4585–4591. doi: 10.1021/acs.analchem.8b05765
Quinto, F., Golser, R., Lagos, M., Plaschke, M., Schäfer, T., Steier, P., et al. (2015). Accelerator mass spectrometry of actinides in ground- and seawater: an innovative method allowing for the simultaneous analysis of U, NP, PU, AM, and CM isotopes below ppq levels. Anal. Chem. 87, 5766–5773. doi: 10.1021/acs.analchem.5b00980
Rozmaric, M., Chamizo, E., Louw, D. C., López-Lora, M., Blinova, O., Levy, I., et al. (2022). Fate of anthropogenic radionuclides (90Sr, 137Cs, 238Pu, 239Pu, 240Pu, 241Am) in seawater in the northern Benguela upwelling system off Namibia. Chemosphere 286, 131514. doi: 10.1016/j.chemosphere.2021.131514
Russell, B. C., Croudace, I. W., and Warwick, P. E. (2015). Determination of 135Cs and 137Cs in environmental samples: a review. Anal. Chim. Acta 890, 7–20. doi: 10.1016/j.aca.2015.06.037
Shi, K., Hou, X., Roos, P., and Wu, W. (2012). Determination of technetium-99 in environmental samples: a review. Anal. Chim. Acta 709, 1–20. doi: 10.1016/j.aca.2011.10.020
Singh, B., Rodionov, A. A., and Khazov, Y. L. (2008). Nuclear data sheets for A = 135. Nuclear Data Sheets 109, 517–698. doi: 10.1016/j.nds.2008.02.001
Snow, M. S., and Snyder, D. C. (2016). 135Cs/137Cs isotopic composition of environmental samples across Europe: environmental transport and source term emission applications. J. Environ. Radioact. 151, 258–263. doi: 10.1016/j.jenvrad.2015.10.025
Snow, M. S., Snyder, D. C., Mann, N. R., and White, B. M. (2015). Method for ultra-trace cesium isotope ratio measurements from environmental samples using thermal ionization mass spectrometry. Int. J. Mass Spectrom. 38, 17–24. doi: 10.1016/j.ijms.2015.03.006
Steier, P., Bichler, M., Keith Fifield, L., Golser, R., Kutschera, W., Priller, A., et al. (2008). Natural and anthropogenic 236U in environmental samples. Nucl. Instrum. Meth. in Phys. Res. B 266, 2246–2250. doi: 10.1016/j.nimb.2008.03.002
Steier, P., Hain, K., Klötzli, U., Lachner, J., Priller, A., Winkler, S., et al. (2019). The actinide beamline at VERA. Nucl. Instrum. Meth. in Phys. Res. B 458, 82–89. doi: 10.1016/j.nimb.2019.07.031
Tims, S., Hancock, G., Wacker, L., and Fifield, L. (2004). Measurements of Pu and Ra isotopes in soils and sediments by AMS. Nucl. Instrum. Meth. in Phys. Res. B 223-224, 796–801. doi: 10.1016/j.nimb.2004.04.147
Toggweiler, J. R., and Trumbore, S. (1985). Bomb-test 90Sr in Pacific and Indian Ocean surface water as recorded by banded corals. Earth Planet. Sci. Lett. 74, 306–314. doi: 10.1016/S0012-821X(85)80002-9
Wacker, L., Fifield, L., and Tims, S. (2004). Developments in AMS of 99Tc. Nucl. Instrum. Meth. in Phys. Res. B 223-224, 185–189. doi: 10.1016/j.nimb.2004.04.038
Yang, G., Tazoe, H., and Yamada, M. (2016b). Rapid determination of 135Cs and precise 135Cs/137Cs atomic ratio in environmental samples by single-column chromatography coupled to triple-quadrupole inductively coupled plasma-mass spectrometry. Anal. Chim. Acta 908, 177–184. doi: 10.1016/j.aca.2015.12.041
Yang, G., Tazoe, H., and Yamada, M. (2016a). Determination of 236U in environmental samples by single extraction chromatography coupled to triple-quadrupole inductively coupled plasma-mass spectrometry. Anal. Chim. Acta 944, 44–50. doi: 10.1016/j.aca.2016.09.033
Zhao, X.-L., and Francisco, B. (2022). Matrix-assisted production of actinide molecular anions for AMS. Nucl. Instrum. Meth. in Phys. Res. B 510, 10–19. doi: 10.1016/j.nimb.2021.10.013
Zhao, X.-L., Litherland, A. E., Eliades, J., Kieser, W. E., and Liu, Q. (2010). Studies of anions from sputtering I: survey of. Nucl. Instrum. Meth. in Phys. Res. B 268, 807–811. doi: 10.1016/j.nimb.2009.10.036
Zheng, J., Cao, L., Tagami, K., and Uchida, S. (2016). Triple-quadrupole inductively coupled plasma-mass spectrometry with a high-efficiency sample introduction system for ultratrace determination of 135Cs and 137Cs in environmental samples at femtogram levels. Anal. Chem. 88, 8772–8779. doi: 10.1021/acs.analchem.6b02150
Zheng, J., Tagami, K., and Uchida, S. (2013). Release of plutonium isotopes into the environment from the fukushima daiichi nuclear power plant accident: what is known and what needs to be known. Environ. Sci. Technol. 47, 9584–9595. doi: 10.1021/es402212v
Zheng, J., Tagami, K., Bu, W., Uchida, S., Watanabe, Y., Kubota, Y., et al. (2014). 135Cs/137Cs isotopic ratio as a new tracer of radiocesium released from the Fukushima nuclear accident. Environ. Sci. Technol. 48, 5433–5438. doi: 10.1021/es500403h
Zheng, J., Tagami, K., Watanabe, Y., Uchida, S., Aono, T., Ishii, N., et al. (2012). Isotopic evidence of plutonium release into the environment from the Fukushima DNPP accident. Sci. Rep. 2:304. doi: 10.1038/srep00304
Zhu, L., Hou, X., and Qiao, J. (2020a). Determination of ultralow Level 135Cs and 135Cs/137Cs ratio in environmental samples by chemical separation and triple quadrupole ICP-MS. Anal. Chem. 92, 7884–7892. doi: 10.1021/acs.analchem.0c01153
Zhu, L., Hou, X., and Qiao, J. (2021). Determination of low-level 135Cs and 135Cs/137Cs atomic ratios in large volume of seawater by chemical separation coupled with triple-quadrupole inductively coupled plasma mass spectrometry measurement for its oceanographic applications. Talanta 226:122121. doi: 10.1016/j.talanta.2021.122121
Zhu, L., Xu, C., Hou, X., Qiao, J., Zhao, Y., and Liu, G. (2020b). Determination of ultratrace level 135Cs and 135Cs/137Cs ratio in small volume seawater by chemical separation and thermal ionization mass spectrometry. Anal. Chem. 92, 6709–6718. doi: 10.1021/acs.analchem.0c00688
Keywords: Accelerator Mass Spectrometry, laser-photodetachment, isotopic signatures, Pacific Ocean, neptunium spike, Ion-Laser Interaction, actinides, fission products
Citation: Hain K, Martschini M, Gülce F, Honda M, Lachner J, Kern M, Pitters J, Quinto F, Sakaguchi A, Steier P, Wiederin A, Wieser A, Yokoyama A and Golser R (2022) Developing Accelerator Mass Spectrometry Capabilities for Anthropogenic Radionuclide Analysis to Extend the Set of Oceanographic Tracers. Front. Mar. Sci. 9:837515. doi: 10.3389/fmars.2022.837515
Received: 16 December 2021; Accepted: 10 February 2022;
Published: 14 March 2022.
Edited by:
Núria Casacuberta, ETH Zürich, SwitzerlandReviewed by:
Hans-Arno Synal, ETH Zürich, SwitzerlandCopyright © 2022 Hain, Martschini, Gülce, Honda, Lachner, Kern, Pitters, Quinto, Sakaguchi, Steier, Wiederin, Wieser, Yokoyama and Golser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karin Hain, a2FyaW4uaGFpbkB1bml2aWUuYWMuYXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.