 Yujia Yang
Yujia Yang Muyan Chen
Muyan Chen Yixin Wang
Yixin Wang Lina Sun
Lina Sun- 1The Key Laboratory of Mariculture, Ministry of Education, Ocean University of China, Qingdao, China
- 2Chinese Academy of Sciences (CAS), Key Laboratory of Marine Ecology and Environmental Sciences, Institute of Oceanology, Chinese Academy of Sciences (CAS), Qingdao, China
Introduction
Sea cucumber (Apostichopus japonicus) is a representative economic aquaculture species of echinoderms with a unique phylogeny and evolutionary classification. In China, the sea cucumber industry covered ~246,745 hectares, and the output was 171,000 tons (2020 China Fisheries Yearbook). Genetic resources include nucleotide and amino acid sequences, novel functional gene information, transcript isoforms, and molecular markers. Utilizing genetic resources provides materials for analyzing biological characteristics or performance traits. In A. japonicus, genome and transcriptome sequences provided genetic resources to investigate its morphological evolution, visceral regeneration, saponin biosynthesis, and aestivation regulation (Zhang et al., 2017; Li et al., 2018). The construction of genetic resources can also play a critical role in the breeding industry and germplasm preservation for sea cucumber. For instance, in molluscan aquaculture, genomic resources are helpful for selective breeding and produce new combinations of genotypes with enhanced performance (Guo, 2009). In addition, the genetic resources of wild fish species provide a large amount of initial genetic material for germplasm preservation and supplemental brood stock of new strains (Lind et al., 2012).
Transcriptome sequencing is one of the fundamental genetic resources to study gene expression and transcriptional regulatory mechanisms in A. japonicus. Since 2011, transcriptome sequencing of A. japonicus has been conducted by 454 sequencing (Sun et al., 2011) and Illumina HiSeq™ 2000 platform (Zhou et al., 2014). In A. japonicus, an extensive set of protein-coding genes and potential genetic markers were identified using transcriptome analysis (Zhou et al., 2014, 2016b). Furthermore, transcriptome analyses have revealed the global expression patterns during critical developmental processes or cellular responses to environmental factors. For instance, transcriptome analysis of sea cucumber during the anti-bacterial process provided a valuable database of the interactions of mRNA-miRNA and gene expression regulatory mechanisms (Zhang et al., 2013). Comparative transcriptome analysis of multiple sea cucumber tissues revealed the molecular mechanisms of larval development (Boyko et al., 2019), growth variation (Gao et al., 2017), color variation (Jo et al., 2016), tissue-specific expression (Zhou et al., 2016a), tissue development (Zhan et al., 2019) and regeneration (Sun et al., 2011), responses to environmental stress (Li et al., 2019), aestivation (Yang et al., 2021), evisceration behavior (Ding et al., 2019), tenderization (Dong et al., 2019), and immune responses (Guo et al., 2021).
The first generation of sequencing technology-Sanger sequencing, has been used to explore the gene sequences and structure in a few studies of sea cucumber. As second-generation sequencing has lower sequencing costs, higher sequencing throughput, and sequencing speed (Kchouk et al., 2017), many studies have been conducted with next-generation sequencing to investigate physiological and biological processes. However, the second-generation sequencing only provided short reads (Kchouk et al., 2017), which could not include the full-length transcripts and splicing isoforms. This problem has been solved by the third generation of sequencing, Pacbio Single Molecular Real-Time (SMRT) sequencing (Rhoads and Au, 2015). Pacbio SMRT Sequencing is a new high throughput sequencing strategy to explore longer reads (average 10–16 kb for PacBio RSII, average 10–14 kb for PacBio Sequel per SMRT cell) (Ardui et al., 2018) in either genomic or transcriptomic levels of organisms. Comparing with first and next generation sequencing technologies, Pacbio sequencing is real-time sequencing, which obtains the sequences of a target DNA molecule through the replication process (Rhoads and Au, 2015). During replication, 4 fluorescent-labeled dNTPs produce different light pulse that can identify the bases (Rhoads and Au, 2015). This technology has been widely used in sequencing genomes for many marine organisms. For example, Pacbio SMRT sequencing was used to obtain long reads and promote the sea cucumber reference genome assembly (Zhang et al., 2017; Li et al., 2018).
Without assembly via bioinformatic tools, the full-length transcriptome could provide complete transcripts (5′UTR, 3′UTR, polyA) without sequence fragmentation (Minoche et al., 2015). In addition, full-length transcriptome could identify alternative splicing isoforms (Arzalluz-Luque and Conesa, 2018) and novel functional genes (Wang et al., 2019). Full-length transcriptomes of several marine organisms were built in the last few years, including fishes (Ge et al., 2021) and shrimp (Katneni et al., 2020), etc. In these two species lacking a reference genome, without assembly from short reads, Pacbio sequencing helps construct an ideal full-length reference transcriptome to further analyze transcriptional regulation under various conditions or functional studies of desired economic traits (Katneni et al., 2020; Ge et al., 2021). However, a full-length transcriptome of an Echinoderm species, such as sea cucumber, is still lacking. Therefore, the full-length transcripts resources of A. japonicus could be a valuable database for diverse splicing isoforms, RNA processing status, the landscape of coding sequences, transcriptional factors, lncRNAs, and novel markers as SSRs in A. japonicus.
Data Description
Sample Collection, Library Preparation, and Pacbio SMRT Sequencing
Healthy adult A. japonicus were sampled from Weihai, Shandong Province, China. Five tissue samples (intestine, respiratory tree, muscle, gonads, calcareous ring) were, respectively, dissected from female and male sea cucumber and immediately frozen in liquid nitrogen and stored in −80°C freezer. The broodstock of A. japonicus were also collected from Weihai, and artificially spawned by flowing sea water at 16°C stimulation, then placed in the plastic buckets. The embryos were collected and cultured in filtered seawater at 18°C, and the developmental stages were examined using a light microscope. Fertilized embryos and different developmental stages (blastula, gastrula, Auricularia, doliolaria, pentactula) were collected, flash-frozen, and stored in −80°C freezer. An equal amount of total RNA from each sample was pooled together, and Poly-A selection was used to remove rRNA efficiently. The cDNA library was constructed by using the SMARTer PCR cDNA Synthesis Kit for Pacbio SMRT sequencing. SMRT libraries were constructed following the standard protocols. The quality passed libraries were sequenced using the Pacbio Sequel platform. Low-quality reads (Qphred <=20), adapter-related reads, and ambiguous reads (N ratio>10%) were filtered from raw reads. The classification of raw reads was listed in Supplementary Figure 1: clean reads (97.13%), reads containing N (0.20%), low-quality reads (0.31%), and adapter-related reads (2.36%).
Full-Length Transcriptome
The sequencing data was analyzed using PacBio SMRTlink V7.0. LoRDEC was used to accurately and efficiently correct long read errors (Salmela and Rivals, 2014). CD-HIT v4.6.8 was used to reduce sequence redundancy and improve the quality of full-length transcriptome (Fu et al., 2012). The similarity threshold was set at 95%, and the parameters were -c 0.95; -T 6; -G 0; -aL 0.00; -aS 0.99; -AS 30. The statistics of the full-length transcriptome are listed in Figure 1A. The full-length transcriptome of A. japonicus is ~85.75 Gbp, including 41,792 transcripts in total. The mean length of the transcripts is 2,052 bp. The minimum length of the transcripts is 95 bp, whereas the maximum length is 11,812 bp. The N50 of the full-length transcriptome is 2,524bp, and N90 is 1,116 bp. Approximately 39.16% of transcripts (16,366 transcripts) and 32.63% of genes (4,804 genes) are in the length interval of 1 k−2 kbp (Supplementary Figure 2).
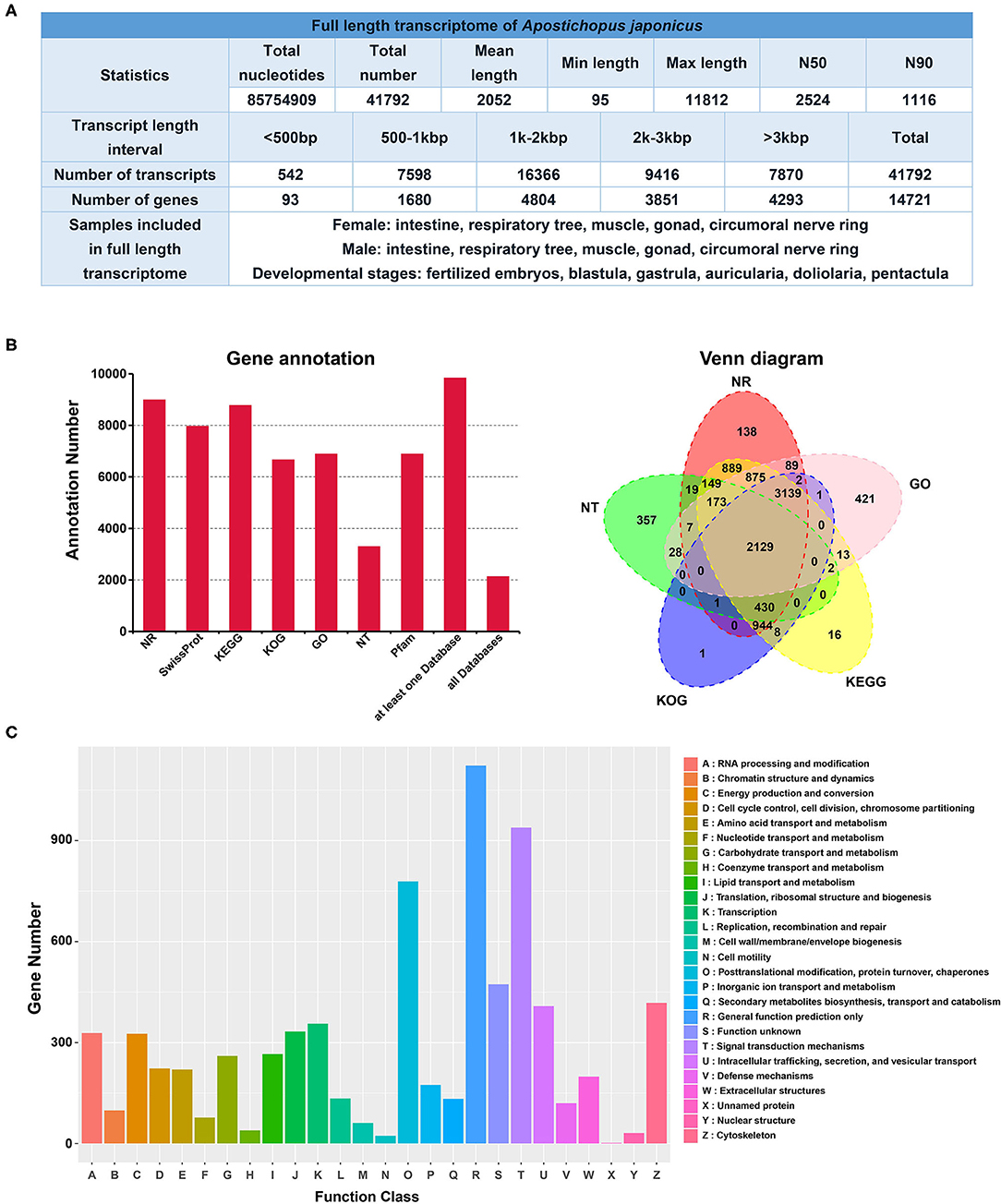
Figure 1. (A) Statistics of Apostichopus japonicus full-length transcriptome; (B) Gene annotation and Venn diagram of A. japonicus full-length transcriptome; (C) Classification of annotated genes using KOG databases.
Gene Annotation and Classification
The sequences after reducing sequence redundancy by CD-HIT were annotated against NCBI non-redundant protein sequence (NR), NCBI nucleotide sequence (NT), KEGG, Karyotic Ortholog Groups (KOG) (Tatusov et al., 2003), and GO (Ashburner et al., 2000) databases. We used diamond v0.8.36 for annotation against NR, KOG, and KEGG, and ncbi-blast-2.7.1+ against NT, and custom perl against GO. The full-length transcriptome was annotated with Nr (8,984 genes), Nt (3,295 genes), KOG (6,655 genes), KEGG (8,767 genes), GO (6,879 genes). We conducted BUSCO analyses to evaluate the non-redundant full-length transcriptome, and the assessment indicated that 59.2% of conserved genes were found using eukaryote lineage. A total of 9,831 genes are annotated based on five databases, which was ~1/3 of total annotated genes in the genome assembly (Zhang et al., 2017; Li et al., 2018). A total of 30,350 protein-coding genes were annotated in the genome assembly version (Zhang et al., 2017), whereas 29,451 protein-coding genes in another version of genome assembly (Li et al., 2018). A total of 2,129 genes are shared and found in five databases (Figure 1B). Using multiple databases (Nr, Nt, KEGG, KOG, and GO) for gene annotation, the full-length transcriptome of sea cucumber was well-annotated and ready for transcriptome analyses. After gene annotation with NR databases, genes in the full-length transcriptome of A. japonicus share the highest similarity with corresponding genes (over 4,000 genes) in the sea urchin Strongylocentrotus purpuratus (Supplementary Figure 3).
The other databases, including SwissProt, Pfam, Clusters of Orthologous Groups of proteins (COG), are used for gene classification. In addition, further analyses such as GO, KOG, and KEGG enrichment analysis of A. japonicus full-length transcriptome were conducted. Annotated with the KOG database, matched genes are categorized into function classes such as signal transduction mechanisms and posttranslational modification, protein turnover, chaperones (Figure 1C). GO term analyses of annotated genes from full-length transcripts are listed in Supplementary Figure 4. KEGG annotation of sea cucumber full-length transcriptome (Supplementary Figure 5) showed that 523 genes are categorized into transport and catabolism in cellular processes. Six hundred and eighty six genes are associated with signal transduction in the environmental information processing category. In the genetic information processing category, 384 genes are annotated in translation function. In terms of metabolism, 296 genes are associated with carbohydrate metabolism. In terms of organismal systems, 382 genes are endocrine system-related genes.
Isoforms and Gene Structure Analysis Utilizing Full-Length Transcriptome
The full-length transcriptome provides a data resource for gene isoforms in A. japonicus, which will be helpful for alternative splicing analyses under different conditions or desired economic traits. In other species, the full-length transcriptome was used to investigate novel transcripts and isoforms in unfertilized eggs (Mehjabin et al., 2019), early gametogenesis (Zhang et al., 2020), sex differentiation (Cui et al., 2021), economically phenotypes (Ali et al., 2021), environmental stress (Shi et al., 2020), and immune response (Zhang et al., 2019), etc. After removing redundant sequences, the isoform number per gene of 14,721 genes has been obtained from A. japonicus full-length transcriptome. Among the full-length non-redundant transcripts, a total of 8,299 genes have only one isoform (Figure 2). The other genes have more than one isoform, in detail, 2,481 genes (two isoforms), 1,281 genes (three isoforms), 699 genes (four isoforms), 452 genes (five isoforms), 289 genes (six isoforms), 218 genes (seven isoforms), 172 genes (eight isoforms), 135 genes (nine isoforms). The number of genes was decreased with the increase of isoform number per gene. Moreover, approximately 695 genes have more than ten isoforms (Figure 2).
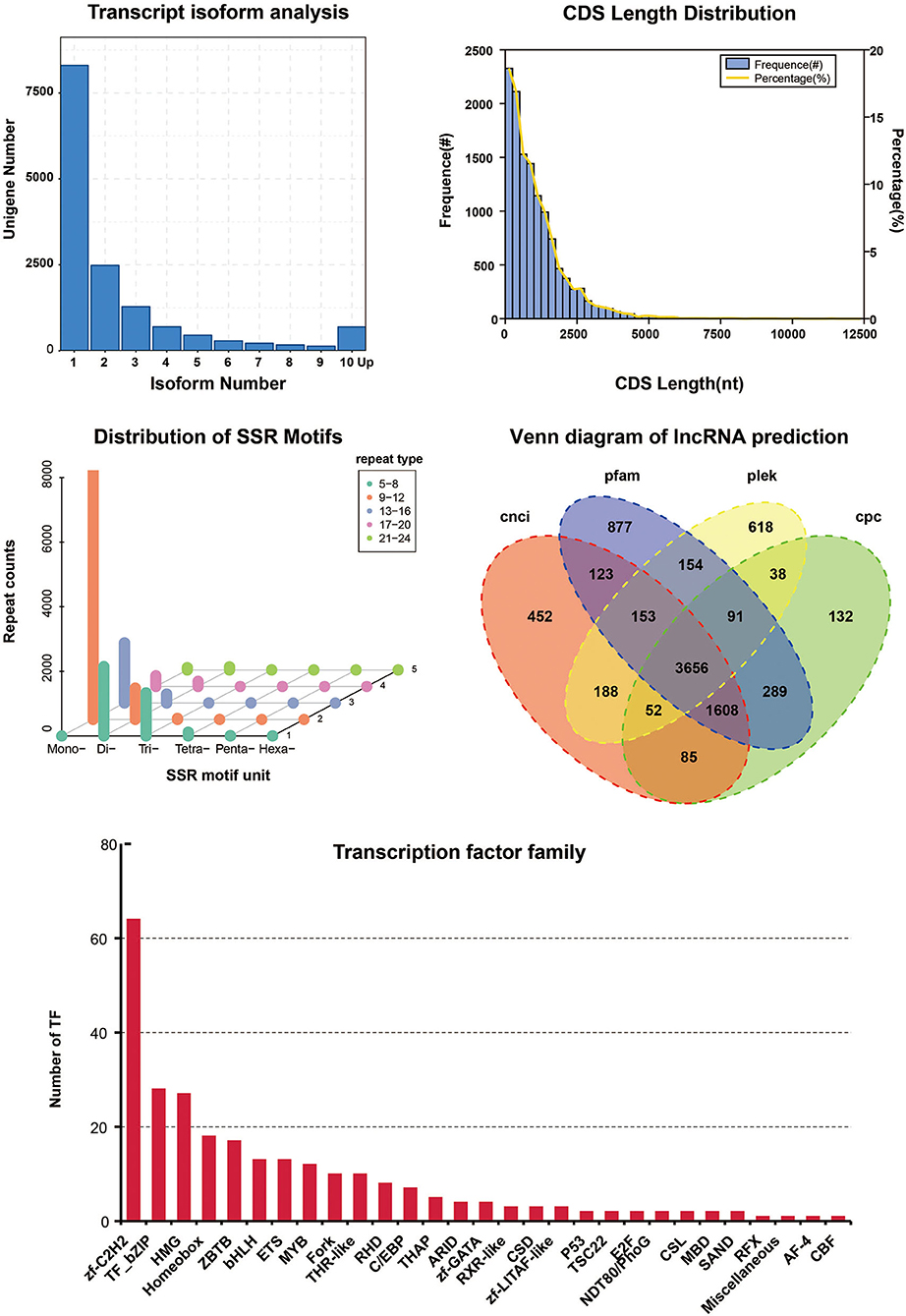
Figure 2. Utilization of A. japonicus full-length transcriptome. (1) Isoform number per gene in A. japonicus full-length transcriptome. (2) Coding region length distribution of A. japonicus full-length transcriptome. (3) Distribution of SSR motifs. (4) Venn diagram of lncRNA prediction; (5) Transcriptional factor prediction of A. japonicus full-length transcriptome.
The gene structure analysis was conducted based on CDS prediction, SSR analysis, lncRNA prediction, and transcriptional factor analysis. CDS prediction analysis was conducted using ANGEL software (Shimizu et al., 2006). In CDS prediction, the CDS length of over 90% is <2,500 bp (Figure 2). Additionally, full-length transcriptome has been helpful for marker discovery for simple sequence repeats. We used MISA (http://pgrc.ipk-gatersleben.de/misa/misa.html) to identify SSRs. The primary type of SSRs (>8,000 SSRs) was mono-nucleotide with 9–12 repeats, followed with di-nucleotide with 5–8 repeats (~2,000 SSRs) and mono-nucleotide with 13–16 repeats (~2,000 SSRs) (Figure 2). Furthermore, we used four methods to predict lncRNA in the full-length transcriptome. The lncRNAs were predicted by CNCI (Sun et al., 2013), CPC (Kong et al., 2007), Pfam (Finn et al., 2016), and PLEK (Li et al., 2014). A total number of 8,516 lncRNAs were found in the full-length transcriptome. A total of 6,317 lncRNAs, 5,951 lncRNAs, 6,951 lncRNAs, 4,950 lncRNAs were found using CNCI, CPC, Pfam, and PLEK, respectively. A total of 3,656 shared predicted lncRNAs using four methods (Figure 2). lncRNAs can interact with DNA, RNA, and proteins to regulate RNA splicing and translation of adjacent and distinct genes, and alter the chromatin structure and function (Statello et al., 2021). A resource of lncRNAs in sea cucumber will benefit the study of gene regulation by lncRNAs and their functions. For length distribution of lncRNA and mRNA, the peaks of lncRNA and mRNA are approximately 1,000 and 2,000 bp, respectively (Supplementary Figure 6). We also conducted transcriptome-wide identification of transcription factor families from A. japonicus full-length transcriptome using animalTFDB2.0 (Zhang et al., 2015). The top three transcription factor families are Zf-C2H2 (Zinc finger C2H2), TF_bZIP (basic leucine zipper), and HMG (High Mobility Group) (Figure 2). The analyses of transcription factor families in sea cucumber will gain a deeper understanding of their interactions with target genes and gene regulatory networks.
In the present study, the full length mRNA sequences were obtained from both adult female and male samples, including five tissues—intestine, respiratory tree, muscle, gonads, circumoral nerve ring, and several critical developmental stages (fertilized embryos, blastula, gastrula, Auricularia, doliolaria, pentactula), which will become one of the important genetic resources for both research and industry studies in sea cucumber Apostichopus japonicus.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI [accession: SRR17056084].
Author Contributions
LS conceived and designed the study and experiments and extracted total RNA for SMRT sequencing. YY, YW, and LS performed bioinformatics analysis. YY wrote manuscript. YW, MC, and LS revised manuscript. All authors have read and approved the final manuscript.
Funding
This research was supported by the National Natural Science Foundation of China (Grant Nos. 42076093 and 42106109), the Youth Innovation Promotion Association CAS (2019209).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.834255/full#supplementary-material
References
Ali, A., Thorgaard, G. H., and Salem, M. (2021). PacBio Iso-Seq improves the rainbow trout genome annotation and identifies alternative splicing associated with economically important phenotypes. Front. Genet. 12, 683408. doi: 10.3389/fgene.2021.683408
Ardui, S., Ameur, A., Vermeesch, J. R., and Hestand, M. S. (2018). Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic Acids Res. 46, 2159–2168. doi: 10.1093/nar/gky066
Arzalluz-Luque, Á., and Conesa, A. (2018). Single-cell RNAseq for the study of isoforms—how is that possible? Genome biology 19, 110. doi: 10.1186/s13059-018-1496-z
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Boyko, A. V., Girich, A. S., Eliseikina, M. G., Maslennikov, S. I., and Dolmatov, I. Y. (2019). Reference assembly and gene expression analysis of Apostichopus japonicus larval development. Sci. Rep. 9, 1131. doi: 10.1038/s41598-018-37755-5
Cui, W., Yang, Q., Zhang, Y., Farhadi, A., Fang, H., Zheng, H., et al. (2021). Integrative transcriptome sequencing reveals the molecular difference of maturation process of ovary and testis in mud crab Scylla paramamosain. Front. Marine Sci. 8, 288. doi: 10.3389/fmars.2021.658091
Ding, K., Zhang, L., Sun, L., Lin, C., Feng, Q., Zhang, S., et al. (2019). Transcriptome analysis provides insights into the molecular mechanisms responsible for evisceration behavior in the sea cucumber Apostichopus japonicus. Comparative Biochem. Physiol. Part D 30, 143–157. doi: 10.1016/j.cbd.2019.02.008
Dong, X., Qi, H., He, B., Jiang, D., and Zhu, B. (2019). RNA sequencing analysis to capture the transcriptome landscape during tenderization in Sea Cucumber Apostichopus japonicus. Molecules 24, 998. doi: 10.3390/molecules24050998
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2016). The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285. doi: 10.1093/nar/gkv1344
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gao, L., He, C., Bao, X., Tian, M., and Ma, Z. (2017). Transcriptome analysis of the sea cucumber (Apostichopus japonicus) with variation in individual growth. PLoS ONE 12, e0181471. doi: 10.1371/journal.pone.0181471
Ge, H., Zhang, H., Zhao, Q., Li, F., Gu, H., Liu, S., et al. (2021). Construction of a full-length transcriptome resource for the Chinese sucker (Myxocyprinus asiaticus), a rare protected fish, based on isoform sequencing (Iso-Seq). Front. Marine Sci. 8, 687. doi: 10.3389/fmars.2021.699504
Guo, L., Wang, Z., Shi, W., Wang, Y., and Li, Q. (2021). Transcriptome analysis reveals roles of polian vesicle in sea cucumber Apostichopus japonicus response to Vibrio splendidus infection. Comparative Biochem. Physiol. Part D 40, 100877. doi: 10.1016/j.cbd.2021.100877
Guo, X. (2009). Use and exchange of genetic resources in molluscan aquaculture. Rev. Aquaculture 1, 251–259. doi: 10.1111/j.1753-5131.2009.01014.x
Jo, J., Park, J., Lee, H.-G., Kern, E. M., Cheon, S., Jin, S., et al. (2016). Comparative transcriptome analysis of three color variants of the sea cucumber Apostichopus japonicus. Marine Genom. 28, 21–24. doi: 10.1016/j.margen.2016.03.009
Katneni, V. K., Shekhar, M. S., Jangam, A. K., Prabhudas, S. K., Krishnan, K., Kaikkolante, N., et al. (2020). Novel Isoform Sequencing based full-length transcriptome resource for Indian White Shrimp, Penaeus indicus. Front. Marine Sci. 7, 1053. doi: 10.3389/fmars.2020.605098
Kchouk, M., Gibrat, J.-F., and Elloumi, M. (2017). Generations of sequencing technologies: from first to next generation. Biol. Med. 9, 1000395. doi: 10.4172/0974-8369.1000395
Kong, L., Zhang, Y., Ye, Z.-Q., Liu, X.-Q., Zhao, S.-Q., Wei, L., et al. (2007). CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35, W345–W349. doi: 10.1093/nar/gkm391
Li, A., Zhang, J., and Zhou, Z. (2014). PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinformatics 15, 311. doi: 10.1201/b16589
Li, C., Fang, H., and Xu, D. (2019). Effect of seasonal high temperature on the immune response in Apostichopus japonicus by transcriptome analysis. Fish Shellfish Immunol. 92, 765–771. doi: 10.1016/j.fsi.2019.07.012
Li, Y., Wang, R., Xun, X., Wang, J., Bao, L., Thimmappa, R., et al. (2018). Sea cucumber genome provides insights into saponin biosynthesis and aestivation regulation. Cell Discovery 4, 1–17. doi: 10.1038/s41421-018-0030-5
Lind, C. E., Brummett, R. E., and Ponzoni, R. W. (2012). Exploitation and conservation of fish genetic resources in Africa: issues and priorities for aquaculture development and research. Rev. Aquaculture 4, 125–141. doi: 10.1111/j.1753-5131.2012.01068.x
Mehjabin, R., Xiong, L., Huang, R., Yang, C., Chen, G., He, L., et al. (2019). Full-length transcriptome sequencing and the discovery of new transcripts in the unfertilized eggs of Zebrafish (Danio rerio). G3 9, 1831–1838. doi: 10.1534/g3.119.200997
Minoche, A. E., Dohm, J. C., Schneider, J., Holtgräwe, D., Viehöver, P., Montfort, M., et al. (2015). Exploiting single-molecule transcript sequencing for eukaryotic gene prediction. Genome biology 16, 184. doi: 10.1186/s13059-015-0729-7
Rhoads, A., and Au, K. F. (2015). PacBio sequencing and its applications. Genomics Proteom. Bioinform. 13, 278–289. doi: 10.1016/j.gpb.2015.08.002
Salmela, L., and Rivals, E. (2014). LoRDEC: accurate and efficient long read error correction. Bioinformatics 30, 3506–3514. doi: 10.1093/bioinformatics/btu538
Shi, K., Li, J., Lv, J., Liu, P., Li, J., and Li, S. (2020). Full-length transcriptome sequences of ridgetail white prawn Exopalaemon carinicauda provide insight into gene expression dynamics during thermal stress. Sci. Total Environ. 747, 141238. doi: 10.1016/j.scitotenv.2020.141238
Shimizu, K., Adachi, J., and Muraoka, Y. (2006). ANGLE: a sequencing errors resistant program for predicting protein coding regions in unfinished cDNA. J. Bioinform. Comput. Biol. 4, 649–664. doi: 10.1142/S0219720006002260
Statello, L., Guo, C.-J., Chen, L.-L., and Huarte, M. (2021). Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 22, 96–118. doi: 10.1038/s41580-020-00315-9
Sun, L., Chen, M., Yang, H., Wang, T., Liu, B., Shu, C., et al. (2011). Large scale gene expression profiling during intestine and body wall regeneration in the sea cucumber Apostichopus japonicus. Compar. Biochem. Physiol. Part D 6, 195–205. doi: 10.1016/j.cbd.2011.03.002
Sun, L., Luo, H., Bu, D., Zhao, G., Yu, K., Zhang, C., et al. (2013). Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 41, e166. doi: 10.1093/nar/gkt646
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4, 1–14. doi: 10.1186/1471-2105-4-41
Wang, B., Kumar, V., Olson, A., and Ware, D. (2019). Reviving the transcriptome studies: an insight into the emergence of single-molecule transcriptome sequencing. Front. Genet. 10, 384. doi: 10.3389/fgene.2019.00384
Yang, Q., Zhang, X., Lu, Z., Huang, R., Tran, N. T., Wu, J., et al. (2021). Transcriptome and metabolome analyses of sea cucumbers Apostichopus japonicus in Southern China during the summer aestivation period. J. Ocean University China 20, 198–212. doi: 10.1007/s11802-021-4482-0
Zhan, Y., Lin, K., Ge, C., Che, J., Li, Y., Cui, D., et al. (2019). Comparative transcriptome analysis identifies genes associated with papilla development in the sea cucumber Apostichopus japonicus. Compar. Biochem. Physiol. Part D 29, 255–263. doi: 10.1016/j.cbd.2018.12.009
Zhang, H.-M., Liu, T., Liu, C.-J., Song, S., Zhang, X., Liu, W., et al. (2015). AnimalTFDB 2.0: a resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res. 43, D76–D81. doi: 10.1093/nar/gku887
Zhang, P., Li, C., Zhu, L., Su, X., Li, Y., Jin, C., et al. (2013). De novo assembly of the sea cucumber Apostichopus japonicus hemocytes transcriptome to identify miRNA targets associated with skin ulceration syndrome. PLoS ONE 8, e73506. doi: 10.1371/journal.pone.0073506
Zhang, X., Li, G., Jiang, H., Li, L., Ma, J., Li, H., et al. (2019). Full-length transcriptome analysis of Litopenaeus vannamei reveals transcript variants involved in the innate immune system. Fish Shellfish Immunol. 87, 346–359. doi: 10.1016/j.fsi.2019.01.023
Zhang, X., Sun, L., Yuan, J., Sun, Y., Gao, Y., Zhang, L., et al. (2017). The sea cucumber genome provides insights into morphological evolution and visceral regeneration. PLoS Biol. 15, e2003790. doi: 10.1371/journal.pbio.2003790
Zhang, X., Zhou, J., Li, L., Huang, W., Ahmad, H. I., Li, H., et al. (2020). Full-length transcriptome sequencing and comparative transcriptomic analysis to uncover genes involved in early gametogenesis in the gonads of Amur sturgeon (Acipenser schrenckii). Front. Zool. 17, 1–21. doi: 10.1186/s12983-020-00355-z
Zhou, X., Cui, J., Liu, S., Kong, D., Sun, H., Gu, C., et al. (2016a). Comparative transcriptome analysis of papilla and skin in the sea cucumber, Apostichopus japonicus. PeerJ 4, e1779. doi: 10.7717/peerj.1779
Zhou, X., Wang, H., Cui, J., Qiu, X., Chang, Y., and Wang, X. (2016b). Transcriptome analysis of tube foot and large scale marker discovery in sea cucumber, Apostichopus japonicus. Compar. Biochem. Physiol. Part D 20, 41–49. doi: 10.1016/j.cbd.2016.07.005
Keywords: genetic resource, full-length transcript, sea cucumber (Apostichopus japonicas), alternative splicing, gene structure analysis, gene annotation
Citation: Yang Y, Chen M, Wang Y and Sun L (2022) A Novel Full-Length Transcriptome Resource for Sea Cucumber Apostichopus japonicus Using Pacbio SMRT Sequencing. Front. Mar. Sci. 9:834255. doi: 10.3389/fmars.2022.834255
Received: 13 December 2021; Accepted: 02 February 2022;
Published: 24 February 2022.
Edited by:
Alexander Chong Shu-Chien, Universiti Sains Malaysia (USM), MalaysiaReviewed by:
Nyok Sean Lau, Universiti Sains Malaysia (USM), MalaysiaYaqing Chang, Dalian Ocean University, China
Copyright © 2022 Yang, Chen, Wang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lina Sun, c3VubGluYUBxZGlvLmFjLmNu