 Ran Xu
Ran Xu Yuhan Lü
Yuhan Lü Yan Tang1,2
Yan Tang1,2 Zhiwei Chen
Zhiwei Chen Chenxi Xu
Chenxi Xu Xiaodong Zheng
Xiaodong Zheng- 1Institute of Evolution and Marine Biodiversity, Ocean University of China, Qingdao, China
- 2Key Laboratory of Mariculture, Ministry of Education, Ocean University of China, Qingdao, China
Species delimitation by traditional morphological methods is challenging in cephalopods due to their flexible bodies, changeable pigment traits, and sometimes high morphological homoplasy. Molecular information provides important assistance for taxonomic decision. In this study, we applied three species delimitation methods on 132 cephalopods from 7 families along Chinese waters, and we found anunderestimated species diversity in these taxa. A maximum of 56 molecular operational taxonomic units was detected, and possible cryptic diversities were revealed in Loliolus beka, Uroteuthis edulis, Octopus minor, Amphioctopus fangsiao, and Hapalochlaena lunulate. This study provides molecular evidence for the hidden species diversity along Chinese waters, and it provides insight into further taxonomic research on these morphologically variable taxa.
Introduction
The Cephalopoda is the third largest class of Mollusca, comprising 845 known species (Hoving et al., 2014) and 134 known taxa records (69 genera, 35 families) in Chinese waters (Lu et al., 2012). Lacking hard structures, changing in colors, and with morphological homologies to closely related species are the major difficulties in determining cephalopod species. In addition, the arm length, the number of suckers on each arm, and the ratio of the mantle to the arm length are important morphological traits for accurate species classification in cephalopods. However, these traits can sometimes be misleading if different preservation methods were used. Some cephalopods have extremely different morphological characteristics between females and males, which further increase the difficulty for accurate morphological diagnosis. As a complement to traditional taxonomy diagnosis, DNA barcoding is currently applied in this class, but most studies have been devoted to identifying new species and phylogenetic analysis (Wiens, 2007). The growing concern over biodiversity threats and the growing availability of molecular data have led to a new focus on species delimitation to identify species accurately and quickly. The delimitation of multiple species using DNA barcoding relies on several different approaches, such as genetic distance and phylogenetic relationships. One of the most popular methods based on genetic distance is Automatic Barcode Gap Discovery (ABGD) (Puillandre et al., 2012), which applies an inferred barcode gap to partition the data and then recursively utilizes the limit and gap detected to decide the final partitions. The other two widely used tree-based methods are Bayesian Poisson Tree Processes Model (bPTP) and Species Tree and Classification Estimation Yarely (STACEY) (Jones, 2017). On a phylogenetic input tree, bPTP estimates hypothetical species boundaries using heuristics to maximize likelihood and Bayesian algorithm (Zhang et al., 2013), while STACEY uses a threshold and birth–death–collapse model to discriminate species in Bayesian framework under a multispecies coalescent model (Jones, 2017).
Recently, several studies reported cryptic diversities in the Cephalopoda, including Bobtail squids (Fernández-Álvarez et al., 2021), Graneledone taniwha (Ibáñez et al., 2020), Sepioteuthis lessoniana (Triantafillos and Adams, 2005), Loliolus beka (Dai et al., 2012), Uroteuthis duvauceli (Sales et al., 2013), Doryteuthis plei (Sales et al., 2017), and D. pealei (Sales et al., 2013). Two studies have examined cryptic diversity in Cephalopoda along Chinese waters (Dai et al., 2012; Zou and Li, 2016), which prompted us to conduct a larger study involving more specimens from a wider range of taxa. In this study, we collected 132 specimens covering 49 diagnostic morphospecies belonging to 7 families, and we used both distance-based method (ABGD) and tree-based methods (bPTP and STACEY) for molecular species delimitation. We hope that our results would provide useful implications for detecting marine biodiversity and enhancing species conservation.
Materials and Methods
Sample Collection and Morphospecies Identification
A total of 132 specimens were collected from 18 locations along Chinese waters during 2006–2021 (Figure 1 and Supplementary Table S1). The detailed information is shown in Supplementary Table S1. Specimens were identifed based on morphological characteristics (Jereb and Roper, 2010; Lu and Chung, 2017). All the specimens used in this study were preserved in 95% ethanol or stored at -30°C and deposited in Fisheries College, Ocean University of China. A portion of the mantle muscle tissue was removed and preserved in 100% alcohol for DNA extraction. Fixation and preservation were performed according to the guidelines (Sweeney and Roper, 1983). The undetermined specimens were named by letters according to their taxonomic status (Supplementary Table S1).
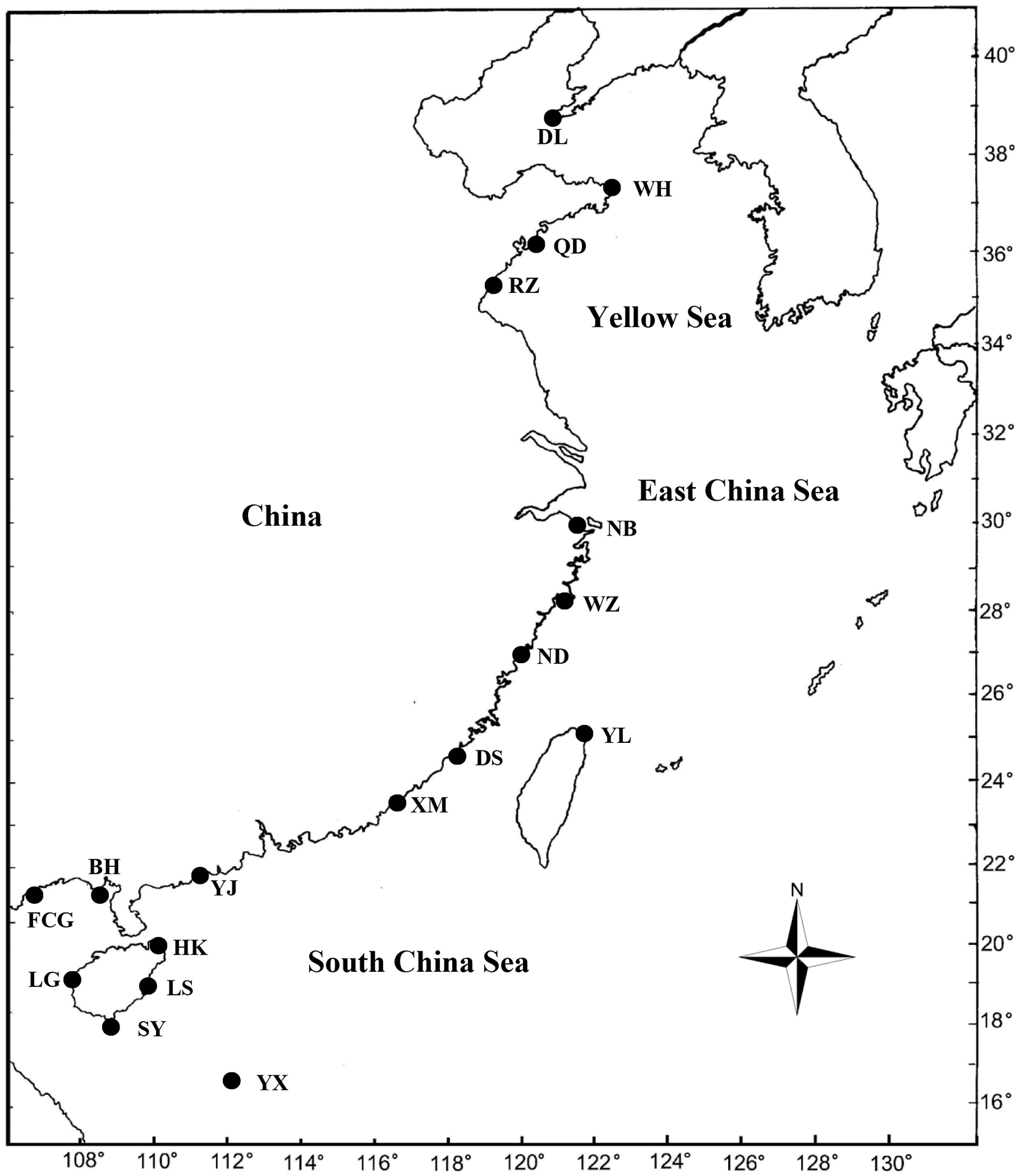
Figure 1 Map of collecting locations for 132 specimens in Chinese waters.A symbol may cover more than one collecting site. DL, Dalian; WH, Weihai; QD, Qingdao; RZ, Rizhao; NB, Ningbo; WZ, Wenzhou; ND, Ningde; YL, Yilan; DS, Dongshan; XM, Xiamen; YJ, Yangjiang; BH, Beihai; FCG, Fangchenggang; HK, Haikou; LG, Lingao; LS, Linshui.
DNA Extraction, PCR Amplification, and Sequencing
The total genomic DNA was extracted by mantle muscle tissue using improved CTAB method (Winnepenninckx, 1993). The cytochrome c oxidase subunit I (COI) fragments were amplified through universal primers LCO1490 and HCO2198 (Folmer et al., 1994). The polymerase chain reactions (PCRs) were carried out in a volume of 50 μl, which included 1 μl template DNA (∼100 ng/μl), 5 μl 10 mM dNTP, 5 μl 10× buffer (Mg2+ plus), 0.25 μl (1 U) rTaq DNA polymerase, 1 μl of each primer (10 μM), and sterile distilled H2O up to 36.75 μl. The processes of PCR amplification consisted of hot start of 94°C for 3 min; followed by 32 cycles of denaturation at 94°C for 45 s, annealing at 50°C for 1 min, extension at 72°C for 1 min; and followed by extension for 5 min at 72°C and holding at 4°C indefinitely. The PCR products were checked in 1.5% agarose gel. All PCR products were purified by EZ-10 Spin Column PCR Product Purification Kit (Sangon, Shanghai, China) following the manufacturer’s protocol. The purified products were sequenced using forward PCR primers on an ABI 3730xl at Sangon Biotech Company (Shanghai, China).
Sequence Analysis and Genetic Distance
A total of 132 sequences for 49 morphospecies were generated in the present study (Supplementary Table S1). The sequences were manually modified in SeqMan v.7.2 (DNASTAR software package) in DNAstar Lasergene (https://www.dnastar.com/software/). All modified sequences were imported into MEGA v.6.0 (Tamura et al., 2013) to be aligned by ClustalW based on amino acid sequences using default settings. The sequences were submitted to GenBank database (Supplementary Table S1). The uncorrected genetic distances (p-distances) between species (excluding the outgroup taxa) were calculated with MEGA6.0 (Tamura et al., 2013).
Barcode Tree Analysis and Species Delimitation
Nautilus pompilius was used as the outgroup for the phylogenetic analyses. Before the molecular phylogenetic analysis, nucleotide substitution models were selected according to the Bayesian information criterion in jModelTest (Posada, 2008). GTR+I+G+F were selected as the best suited models, and the phylogenetic tree was built in MrBayes v3.2 (Ronquist et al., 2012). In MrBayes-partitioned Bayesian analyses, after running 10 million generations for COI and sampling every 1,000 generations with the default prior and Markov chain Monte Carlo (MCMC) settings, the first 25% trees were discarded as burn-in. Additionally, a maximum likelihood tree was also constructed through IQTree with automatic-detect model (Trifinopoulos et al., 2016; available at http://iqtree.cibiv.univie.ac.at).
To estimate the number of molecular operational taxonomic units (MOTUs), we performed three methods: ABGD, STACEY, and bPTP. The ABGD analysis of MOTU detection was performed on the web interface (https://bioinfo.mnhn.fr/abi/public/abgd/) with the default settings by Jukes-Cantor (JC69), K2P, and simple distance (SD) models, the relative gap width was set to (X = 1.0) because the default value returned no results. The partition steps and number of bins were set to 100 and 50, respectively, and the default values were used for all the rest of the parameters. In the bPTP server (https://species.h-its.org/), the Bayesian tree from MrBayes and the maximum likelihood tree from IQTree were used as input trees, respectively. The MCMC generation was set to 500,000, which was enough to get a converged MCMC chain with the thinning of 100 and burn-in of 0.2. The STACEY template implemented in BEAST v2.6 (Bouckaert et al., 2019) was used, and a strict clock for each partition and Yule model species tree prior were selected. Four independent MCMC chains were run for 10 million generations and sampled every 5,000 generation. Because COI is a mitochondrial gene, the ploidy was set to 1. After running BEAST, the log files were then examined in Tracer v1.7.1 (Bouckaert et al., 2019), and a maximum clade credibility tree was generated after discarding 50% of all estimated trees in Tree Annotator v2.6 (Bouckaert et al., 2019). Finally, species delimitation based on the estimated trees were assessed by species DA (http://www.indriid.com/2014/speciesDA.jar) using a burn-in of 200 trees, a collapse height of 0.00001, and a cutoff of 1.0. All other parameters were set as default.
Results
Morphospecies Identification
The specimens used for this study were morphologically diagnosed by specialized taxonomists of Cephalopoda (see Acknowledgements). Aside from the certain species name, “sp.” (indeterminate species) and “cf.” (confer to the given species) were used for the uncertain species (Supplementary Table S1). Based on morphological diagnosis, the 132 specimens collected in this study were assigned to 49 morphospecies belonging to 7 families (Supplementary Table S1). Among them, Sepia sp.1, Octopus sp.1, Octopus sp.2, Octopus sp.3, Uroteuthis sp.1, Uroteuthis sp.2, Hapalochlaena sp., and Thaumoctopus sp. probably represent undescribed species, which require more species for formal description.
MOTU Estimation
Three species delimitation methods (ABGD, bPTP, and STACEY) were compared, and the number of MOTUs detected by each method is shown in Figure 2. The relationship between the undescribed morphospecies and the other species was indicated by the phylogenetic tree. Sepia sp. 1 and Sepia sp. 2 were sister species to Sepia esculenta, and intriguingly these two species were collected in the same location and were diagnosed as the same morphospecies. Uroteuthis sp. 1 and Uroteuthis sp. 2 showed a close relationship to Uroteuthis chinensis. Octopus sp. 1 formed a sister clade to the O. minor and O. sp. 2. Octopus sp. 3 formed a sister clade to Abdopus guangdongensis and O. cf. sasakii. Hapalochlaena sp. showed a close relationship to H. maculosa, and Thaumoctopus sp. showed a close relationship to T. mimicus. The MOTUs identified by the three different methods were highly overlapped. ABGD provides three distance measuring methods—Jukes–Cantor (JC69), Kimura (K80), and simple distance (SD)—for species delimitation, and the results could be varied with different prior intraspecific divergence (P). After partitioning, all three methods implemented in ABGD yielded the same MOTUs of 55 at the minimum P of 0.0129 (Figure 2). Five morphospecies, L. beka, U. edulis, A. fangsiao, H. lunulate, and O. minor showed two MOTUs inferred by ABGD. Consistently, STACEY assigned 1,665 trees (92.45%) from all the samples into the same 55 MOTUs (Figure 2). As shown in Figure 2 and Supplementary Figure 1, the phylogenetic trees based on Bayesian and maximum likelihood methods present an inconsistent topological structure, with the Bayesian tree splitting in Loliginidae. According to bPTP, 56 MOTUs were detected based on the tree generated by the Bayesian method (Supplementary Figure 1), while 55 MOTUs were detected based on the maximum likelihood tree (Figure 2). All three species delimitation methods shared the same 55 MOUTs, while one more MOTU in A. fangsiao I was revealed by bPTP based on the Bayesian tree (Figure 2).
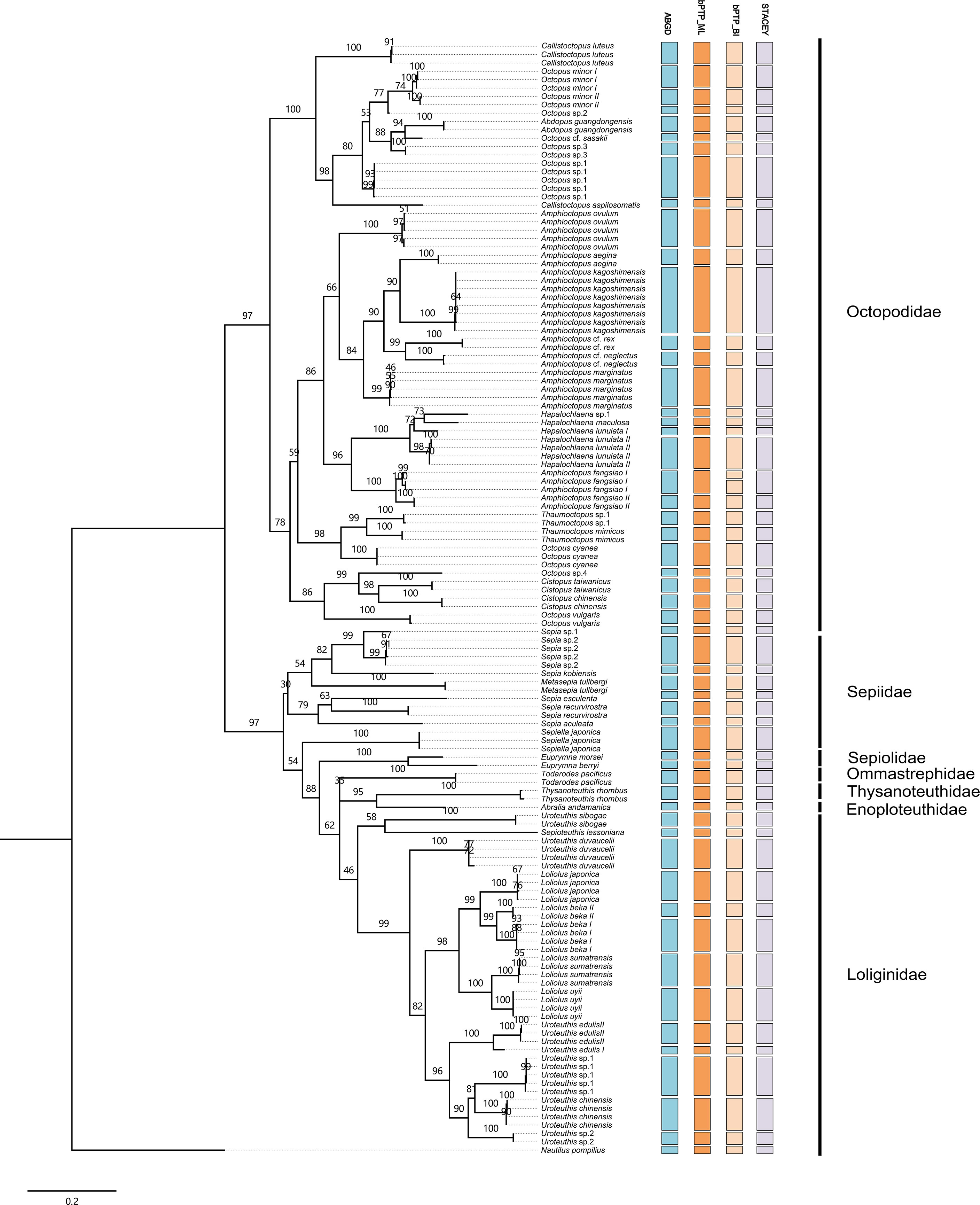
Figure 2 Summary of species delimitation results via Species Tree and Classification Estimation Yarely, automatic barcode gap discovery, and Bayesian Poisson tree processes model. The phylogenetic structure was induced by the Maximum Likelihood tree. Each column with a different color represents a species detected by molecular delimitation. The number in each node represents the bootstrap values.
Genetic Distance
The genetic distances between different species based on COI sequences are shown in Supplementary Table S2, and the p-distances between MOTUs ranged from 2.5 to 23.5%. As described above, two potential MOTUs were revealed in L. beka, U. edulis, O. minor, and H. lunulate by all three methods, and the average distances between two potentially cryptic species were 6.3, 6.2, 2.5, and 6.9%, respectively (Supplementary Table S2). For A. fangsiao, two MOTUs were suggested by ABGD and STACEY, with a genetic distance of 4.8%, while bPTP further suggested a MOTU in A. fangsiao I with a genetic distance of 1.6% between each other (Supplementary Table S2).
Discussion
In this study, we successfully used single-gene DNA barcoding for species delimitation in cephalopods along Chinese waters. Our results revealed an underestimated possible cryptic diversity in the Cephalopoda across a wide range of taxa. Moreover, we also found several undescribed morphospecies in genera Sepia, Uroteuthis, and Octopus.
In the family Loliginidae, four genera were collected, and all of them are important commercial species along the coast of China (Jereb and Roper, 2010). Loliginid monophyly was indicated by the maximum likelihood tree, in line with the results from other studies (Anderson, 2000; Jiang et al., 2018). Nevertheless, the Bayesian tree showed a subdivision within this family. A larger dataset with multiple genes would be helpful to solve such inconsistency. Despite such inconsistency in the phylogenetic tree, the species delimitation from these two methods showed similar results. These results were also in line with the results from the distance-based ABGD method, indicating that two different phylogenetic relationships inferred from the COI sequences might have a minimal influence on species delimitation. Although a single mitochondrial gene may not reflect the real taxa evolution, several studies in the Cephalopoda have successfully discriminated the cryptic species from a wide range of taxa using a single barcode (Eshragh and Leander, 2014; Van Nieuwenhove et al., 2019; Fernández-Álvarez et al., 2021).
Due to the considerable morphological variation in Loliginidae, cryptic diversity has also been reported in this family (Sales et al., 2013). When comparing all the obtained L. beka sequences, two well-supported branches were found (Figure 1). Cryptic species in L. beka have already been discovered in Zou and Li (2016) and Dai et al. (2012) using distance-based and character-based barcode analyses. Here we further confirmed the result with the tree-based approach. U. edulis is widely distributed in the Indo-Pacific, and it was reported that this species presents a considerably high morphologic and genetic variation (Yeatman and Benzie, 1993; Sin et al., 2009). Our results further indicated a possible cryptic diversity in U. edulis, and high genetic distances between two MOTUs (6.2%) were observed. U. edulis I was collected from Xiamen, while U. edulis II was collected from farther south Sanya. Considering that high morphological and genetic variation existed in this species, future studies involving more samples from different locations would help to further uncover the taxonomic uncertainties in this species. Another closely related species–U. chinensis—has been reported to be with high morphological similarities to U. edulis and high intraspecific diversity (Sin et al., 2009). In the present study, we found two undescribed species—U. sp. 1 and U. sp. 2—presented a close relationship to U. chinensis with the genetic divergence of 10.2 and 10.8%, respectively. However, we found no evidence of cryptic diversity in this species.
Hidden diversity was also discovered in genus Octopus, which belongs to the family Octopodidae that contains numerous undescribed highly contentious species (Norman and Lu, 2000; Söller et al., 2000). O. minor is characterized with a prominently nice, spoon-shaped terminal organ on the right arm 3 in mature males and has been morphologically classified into three subspecies: O. minor minor, O. minor typicus, and O. minor paradalis (Sasaki, 1929; Toll and Voss, 1998). In this study, the COI sequences of O. minor I showed a high similarity to the specimen classified as O. minor typicus in the research of Kaneko et al. (2011). However, according to our previous morphological study on 11 O. minor populations along Chinese waters (Gao et al., 2019), we found that O. minor I clustered together with the Northeast Chinese populations (O. minor II, known as O. minor) based on some morphological characteristics. Taken together, we speculated that O. minor I and O. minor II could most likely be the cryptic species. It was proposed that O. minor may comprise many morphological and genetic divergent species, namely, O. minor group. Therefore, a more comprehensive study on this species would be necessary and would help to uncover the taxonomy in this group. Although accurate classification still requires further morphological diagnosis, the molecular evidence provided straight and distinct clues for hidden diversity in this group.
Amphioctopus fangsiao is another taxonomically complicated species with different morphological types being reported, and two different varieties or subspecies have been recognized in a previous study: A. fangsiao var. typicus (with a brick-colored patch on the head between the eyes) and A. fangsiao var. echuanus (with a crescent-shaped patch on the head between the eyes) (Toll and Voss, 1998). In our result, A. fangsiao I showed a dumbbell-shaped patch, and A. fangsiao II showed an elliptical patch. At least two MOTUs were suggested by all three methods with a p-distance of 4.8% (Supplementary Table S1). However, due to some vague morphological descriptions, it is hard to determine whether our samples are the same as those of Toll and Voss’s (1998). According to recent studies, the genetic diversities and structures were observed in A. fangsiao between the northern and southern seas separated by the Yangtze River (Muhammad et al., 2019; Lashari et al., 2020). However, in this study, all the samples were from the same region (Xiapu, Fujian), eliminating the possibility that genetic differentiations were caused by geographical restrictions. Hence, we speculated that the species that we studied may represent a possible cryptic species in the A. fangsiao group, and the cryptic diversity in this species could be still underestimated.
Hapalochlaena genus, also known as blue-ringed octopus, includes only four confirmed species: H. lunulate, H. fasciata, H. maculosa, and H. nierstraszi (described only on one single specimen) (Norman and Lu, 2000; Norman et al., 2016; Lu and Chung, 2017; Morse et al., 2018). The taxonomic diversity in this genus is largely unknown. We found two MOTUs in H. lunulate, and high genetic distance was observed between two MOTUs (6.9%), indicating the possibility of cryptic species. In addition, we found overlooked biodiversity in this genus: an undocumented blue-ringed octopus Hapalochlaena sp. with distinct morphological characteristics compared to the four existing documented species, clearly two separated clades, and a relatively high p-distance; therefore, it could be a potentially undocumented species. Our results indicated that the species diversity in this genus are highly underestimated. Considering our poor understanding of the genus, a combination of molecular and morphological revision would be necessary in this genus. Moreover, our study covered several other unrecognized species belonging to the family Octopodidae that could be potentially new species in China seas. Four unrecognized species—Octopus sp. 1, Octopus sp. 2, Octopus sp. 3, and Thaumoctopus sp.—presented a relatively high p-distance in comparison to the other species in Octopodidae and highly supported clades, indicating the high possibility of undocumented species.
Large intraspecies morphological variations in cephalopods raise a great challenge to accurate species delimitation. In this study, we just provided the implications for species delimitations and clues for identifying potentially cryptic and new species. The barcoding evidence for potentially cryptic/new species was provided, and future works from more samples with both morphological diagnosis and multiple molecular markers would be necessary to generate more informative conclusions.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by the Experimental Animal Ethics Committee of the Ocean University of China.
Author Contributions
RX and XDZ conceived and designed this research. RX, YHL, YT, ZWC, CXX, and XYZ conducted the experiments. RX and YHL wrote the manuscript. XDZ supervised the work. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by research grants from the National Natural Science Foundation of China (32170536 and 31672257).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We gratefully thank Prof. Chung-Cheng Lu for morphological diagnosis. We also acknowledge Dr. Chun-wen Ho for providing partial sequences of Amphioctopus kagoshimensis from Taiwan island. The authors also thank Dian-hang Jiang for providing the sequence of Nautilus pompilius. The authors also acknowledge all the members in our team who did the sampling collection works.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.830381/full#supplementary-material
Supplementary Figure 1 | The Bayesian tree based on the cytochrome c oxidase subunit I sequences. The number in each node represents the posterior probability values.
References
Anderson F. E. (2000). Phylogeny and Historical Biogeography of the Loliginid Squids (Mollusca: Cephalopoda) Based on Mitochondrial DNA Sequence Data. Mol. Phylogenet. Evol. 15 (2), 191–214. doi: 10.1006/mpev.1999.0753
Bouckaert R., Vaughan T. G., Barido-Sottani J., Duchêne S., Fourment M., Gavryushkina A., et al. (2019). BEAST 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 15 (4), e1006650. doi: 10.1371/j.pcbi.1006650
Dai L., Zheng X., Kong L., Li Q. (2012). DNA Barcoding Analysis of Coleoidea (Mollusca: Cephalopoda) From Chinese Waters. Mol. Ecol. Res. 12 (3), 437–447. doi: 10.1111/j.1755-0998.2012.03118.x
Eshragh R., Leander B. S. (2015). Molecular Contributions to Species Boundaries in Dicyemid Parasites From Eastern Pacific Cephalopods. Mar. Biol. Res. 11 (4), 414–422. doi: 10.1080/17451000.2014.943241
Fernández-Álvarez F.Á., Sánchez P., Villanueva R. (2021). Morphological and Molecular Assessments of Bobtail Squids (Cephalopoda: Sepiolidae) Reveal a Hidden History of Biodiversity. Front. Mar. Sci. 7, 632261. doi: 10.3389/fmars.2020.632261
Folmer O., Black M., Hoeh W., Lutz R., Vrijenhoek R. (1994). DNA Primers for Amplification of Mitochondrial Cytochrome C Oxidase Subunit I From Diverse Metazoan Invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299.
Gao X. L., Xu R., Zhang Z. X., Zheng X. D. (2019). Morphological Variation Analysis of Octopus Minor Inthe Coastal Waters of China. J. Fish China 43, 1593–1602. doi: 10.11964/jfc.20181111521
Hoving H. J. T., Perez J. A. A., Bolstad K. S. R., Braid H. E., Evans A. B., Fuchs D., et al. (2014). The Study of Deep-Sea Cephalopods. Adv. Mar. Biol. 67, 235–359. doi: 10.1016/B978-0-12-800287-2.00003-2
Ibáñez C. M., Fenwick M., Ritchie P. A., Carrasco S. A., Pardo-Gandarillas M. C.. (2020). Systematics and Phylogenetic Relationships of New Zealand Benthic Octopuses (Cephalopoda: Octopodoidea). Front. Mar. Sci. 7, 182. doi: 10.3389/fmars.2020.00182
Jereb P., Roper C. F. (2010). “Cephalopods of the World. An Annotated and Illustrated Catalogue of Cephalopod Species Known to Date. Vol 2. Myopsid and Oegopsid Squids,” in FAO Species Catalogue for Fishery Purposes (Rome, Italy: FAO), 1–387.
Jiang L., Kang L., Wu C., Chen M., Lü Z. M. (2018). A Comprehensive Description and Evolutionary Analysis of 9 Loliginidae Mitochondrial Genomes. Hydrobiologia 808 (1), 115–124. doi: 10.1007/s10750-017-3377-y
Jones G. (2017). Algorithmic Improvements to Species Delimitation and Phylogeny Estimation Under the Multispecies Coalescent. J. Math. Biol. 74 (1-2), 447–467. doi: 10.1007/s00285-016-1034-0
Kaneko N., Kubodera T., Iguchis A. (2011). Taxonomic Study of Shallow-Water Octopuses (Cephalopoda: Octopodidae) in Japan and Adjacent Waters Using Mitochondrial Genes With Perspectives on Octopus DNA Barcoding. Malacologia 54 (1-2), 97–108. doi: 10.4002/040.054.0102
Lashari P., Wei C., Gong L., Liu L., Jiang L., Liu B., et al. (2020). A Mitogenomic Phylogeny and Genetic History of Amphioctopus Fangsiao (D’Orbigny 1839-1841) From China. J. Surv. Fish Sci. 6 (2), 1–16. doi: 10.18331/SFS2020.6.2.1
Lu C. C., Chung W. S. (2017). “Guide to the Cephalopods of Taiwan,” in National Museum of Natural Science (Taiwan: Huayu Nature Book Trade), 1–560.
Lu C., Zheng X., Lin X. (2012). “Diversity of Cephalopoda From the Waters of the Chinese Mainland and Taiwan,” in Proceedings of the 1st Mainland and Taiwan Symposium of Marine Biodiversity Studies. Eds. Lin M., Wang C. G. (Beijing: Ocean Press), 76–87.
Morse P., Kjeldsen S. R., Meekan M. G., Mccormick M. I., Finn J. K., Huffard C. L., et al. (2018). Genome-Wide Comparisons Reveal a Clinal Species Pattern Within a Holobenthic Octopod—The Australian Southern Blue-Ringed Octopus, Hapalochlaena Maculosa (Cephalopoda: Octopodidae). Ecol. Evol. 8 (4), 2253–2267. doi: 10.1002/ece3.3845
Muhammad F., Chen W., Liu L., Gong L., Du X., Shafi M., et al. (2019). Genetic Structure of Amphioctopus Fangsiao (Mollusca, Cephalopoda) in Chinese Waters Inferred From Variation in Three mtDNA Genes (ATPase 6, ND2, and ND5). Hydrobiologia 838 (1), 111–119. doi: 10.1007/s10750-019-03981-9
Norman M., Lu C. (2000). Preliminary Checklist of the Cephalopods of the South China Sea. Raffles Bull. Zool. 8, 539–567.
Norman M., Nabhitabhata J., Lu C. (2016). An Updated Checklist of the Cephalopods of the South China Sea. Raffles Bull. Zool. 34, 566–592.
Posada D. (2008). Jmodeltest: Phylogenetic Model Averaging. Mol. Biol. Evol. 25 (7), 1253–1256. doi: 10.1093/molbev/msn083
Puillandre N., Lambert A., Brouillet S., Achaz G. (2012). ABGD, Automatic Barcode Gap Discovery for Primary Species Delimitation. Mol. Ecol. 21 (8), 1864–1877. doi: 10.1111/j.1365-294X.2011.05239.x
Ronquist F., Teslenko M., van der Mark P., Ayres D. L., Darling A., Höhna S., et al. (2012). MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 61 (3), 539–542. doi: 10.1093/sysbio/sys029
Sales J. B. L., Rodrigues-Filho L. F. S., Ferreira Y. S., Carneiro J., Asp N. E., Shaw P. W., et al. (2017). Divergence of Cryptic Species of Doryteuthis Plei Blainvill(Loliginidae, Cephalopoda) in the Western Atlantic Ocean Is Associated With the Formation of the Caribbean Sea. Mol. Phylogenet. Evol. 106, 44–54. doi: 10.1016/j.ympev.2016.09.014
Sales J. B. L., Shaw P. W., Haimovici M., Markaida U., Cunha D. B., Ready J., et al. (2013). New Molecular Phylogeny of the Squids of the Family Loliginidae With Emphasis on the Genus Doryteuthis Nae: Mitochondrial and Nuclear Sequences Indicate the Presence of Cryptic Species in the Southern Atlantic Ocean. Mol. Phylogenet. Evol. 68 (2), 293–299. doi: 10.1016/j.ympev.2013.03.027
Sasaki M. (1929). A Monograph of Dibranchiate Cephalopods of the Japanese and Adjacent Waters. Jour. Coll. Agric. Hokkaido Univ. 201-357, 330.
Sin Y., Yau C., Chu K. (2009). Morphological and Genetic Differentiation of Two Loliginid Squids, Uroteuthis (Photololigo) Chinensis and Uroteuthis (Photololigo) Edulis (Cephalopoda: Loliginidae), in Asia. J. Exp. Mar. Biol. Ecol. 369 (1), 22–30. doi: 10.1016/j.jembe.2008.10.029
Söller R., Warnke K., Saint-Paul U., Blohm D. (2000). Sequence Divergence of Mitochondrial DNA Indicates Cryptic Biodiversity in Octopus Vulgaris and Supports the Taxonomic Distinctiveness of Octopus Mimus (Cephalopoda: Octopodidae). Mar. Biol. 136 (1), 29–35. doi: 10.1007/s002270050004
Sweeney M., Roper C. F. (1983). Techniques for Fixation, Preservation, and Curation of Cephalopods. Memoirs Natl. Museum Victoria 44, 29–48.
Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. (2013). MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 30 (12), 2725–2729. doi: 10.1093/molbev/mst197
Triantafillos L., Adams M. (2005). Genetic Evidence That the Northern Calamary, Sepioteuthis Lessoniana, Is a Species Complex in Australian Waters. ICES J. Mar. Sci. 62 (8), 1665–1670. doi: 10.1016/j.icesjms.2005.06.004
Trifinopoulos J., Nguyen L. T., von Haeseler A., Minh B. Q. (2016). W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 44 (W1), W232–W235. doi: 10.1093/nar/gkw256
Toll R. B., Voss G. L. (1998). The Systematic and Nomenclatural Status of the Octopodinae Described From the West Pacific Ocean. Smithson Contrib. Zool. 586, 457–474.
Van Nieuwenhove A. H. M., Ratsimbazafy H. A., Kochzius M. (2019). Cryptic Diversity and Limited Connectivity in Octopuses: Recommendations for Fisheries Management. PloS One 14 (5), e0214748. doi: 10.1371/journal.pone.0214748
Wiens J. J. (2007). Species Delimitation: New Approaches for Discovering Diversity. Syst. Biol. 56 (6), 875–878. doi: 10.1080/10635150701748506
Winnepenninckx B. (1993). Extraction of High Molecular Weight DNA From Molluscs. Trends Genet. 9, 407. doi: 10.1016/0168-9525(93)90102-N
Yeatman J., Benzie J. (1993). Cryptic Speciation in Loligo From Northern Australia. In: Okutani, T.e.a. (Ed.), Recent Advances in Cephalopod Fisheries Biology. Tokyo Univ. Press Tokyo, 641–652. doi: 10.1007/BF00699221
Zhang J., Kapli P., Pavlidis P., Stamatakis A. (2013). A General Species Delimitation Method With Applications to Phylogenetic Placements. Bioinformatics 29 (22), 2869–2876. doi: 10.1093/bioinformatics/btt499
Keywords: species delimitation, cryptic biodiversity, DNA barcoding, Cephalopoda, Chinese waters
Citation: Xu R, Lü Y, Tang Y, Chen Z, Xu C, Zhang X and Zheng X (2022) DNA Barcoding Reveals High Hidden Species Diversity of Chinese Waters in the Cephalopoda. Front. Mar. Sci. 9:830381. doi: 10.3389/fmars.2022.830381
Received: 07 December 2021; Accepted: 14 March 2022;
Published: 08 April 2022.
Edited by:
Anna Rita Rossi, Sapienza University of Rome, ItalyReviewed by:
Graziano Fiorito, Stazione Zoologica Anton Dohrn, ItalyWonchoel Lee, Hanyang University, South Korea
Copyright © 2022 Xu, Lü, Tang, Chen, Xu, Zhang and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaodong Zheng, eGR6aGVuZ0BvdWMuZWR1LmNu
†These authors have contributed equally to this work and share first authorship