 Jessica R. Zehnpfennig1
Jessica R. Zehnpfennig1 Colleen M. Hansel2
Colleen M. Hansel2 Scott D. Wankel2
Scott D. Wankel2 Cody S. Sheik3
Cody S. Sheik3 Dean J. Horton1
Dean J. Horton1 Carl H. Lamborg4
Carl H. Lamborg4 Deric R. Learman1*
Deric R. Learman1*
- 1Department of Biology, Institute for Great Lakes Research, Central Michigan University, Mount Pleasant, MI, United States
- 2Department of Marine Chemistry and Geochemistry, Woods Hole Oceanographic Institute Woods Hole, Woods Hole, MA, United States
- 3Large Lakes Observatory, Department of Biology, University of Minnesota-Duluth, Duluth, MN, United States
- 4Ocean Sciences Department, University of California, Santa Cruz, Santa Cruz, CA, United States
Microbial community dynamics are influenced not only by biological but also physical and chemical phenomena (e.g., temperature, sunlight, pH, wave energy) that vary on both short and long-time scales. In this study, samples of continental shelf waters of the northwest Atlantic Ocean were periodically collected from pre-sunrise to post-sunset and at multiple depths over summers of 2016 and 2017. Metatranscriptomic analyses revealed expression of photosynthetic genes in surface water samples corresponding to a diel relationship with sunlight. Photosynthetic genes originated from known phototrophs including Aureococcus, Ostreococcous, Synechocococus, and Prochlorococcus. Photosynthetic gene expression occurred pre-sunrise, suggesting the community initiates transcription before sunlight exposure, ostensibly to harvest energy more efficiently when the anticipated increase in light occurs. Transcripts from photoheterotrophic members of the SAR11 clade were also documented in surface samples, with rhodopsin expression being more abundant pre-sunrise and post-sunrise. Conversely, samples taken from the aphotic layer exhibited expression of transcripts related to nitrification that did not vary over the diel cycle. Nitrification gene transcripts, specifically amoA, nirK, hao, and norAB, were taxonomically related to well-known genera of ammonia oxidizers, such as Nitrospira, Candidatus Nitrosomarinus, Nitrosospira, and Nitrosopumilus. Overall, this study documents the role of light (varying with time and depth) in shaping the photosynthetic microbial community activity in the surface ocean, and further demonstrates that this diel regulation of photosynthesis is decoupled from the activity of the nitrifying microbial community in deeper and darker waters.
Introduction
Microbial communities change over short-term time scales to occupy ecological niches within marine ecosystems. Examining shifts in community gene expression over short-time periods can allow for insight into tightly coordinated ecosystem-level dynamics that orchestrate daily biogeochemical fluxes and into how they may respond in the face of rapid natural and anthropogenic environmental change. Changes in marine plankton community function are largely driven by variations in environmental conditions (e.g., physical, chemical) with implications for food web dynamics and global biogeochemical cycles (Barton et al., 2013). In the surface ocean, the relatively short time scales of diel changes in light intensity affect a multitude of different physical variables in the environment.
The function and activity of the photosynthetic community in the ocean surface has been shown to respond temporally with light exposure. As light intensity varies over the course of a day, the dynamics of photosynthesis, DOC production and utilization, nutrient uptake and regeneration and community metabolism should all be reflected in the corresponding dynamics of the metatranscriptome. For example, Aylward et al. (2015) compared daily metatranscriptome profiles of related microbial populations inhabiting the surface waters of a costal California upwelling region and oligotrophic North Pacific Subtropical Gyre (NPSG) and found that the dominant photoautotrophic microbes in each environment were central determinants of overall community transcriptome dynamics. The study also found that heterotrophic bacterial clades common to both ecosystems displayed conserved, genome-wide inter- and intra-taxon transcriptional patterns and diel cycles (Aylward et al., 2015). Changes in light also have been shown to impact amino acid metabolism, DNA processing, translation, transporters, and vitamin metabolism (Ottesen et al., 2013)—tightly regulated metabolic processes responding to intense resource competition. Viruses have also been shown to have diel oscillations in surface ocean microbial communities, resulting additional recourse competition within this ecosystem (Aylward et al., 2017). Overall, nutrient availability and the timing of growth and DNA replication are key factors playing into regulation of temporal dynamics in surface planktonic assemblages (Aylward et al., 2015). Similarly, Vislova et al. (2019) investigated depth-dependent changes in diel patterns for primary and secondary production, metabolic activity, and gene expression in marine microbial communities in the NPSG and found diel transcript oscillations in microbial communities decreased in abundance and regularity with depth and light levels.
Changes in light availability can also impact nitrifying microbial communities throughout the ocean water column. The first step of nitrification is the oxidation of ammonia to hydroxylamine catalyzed by amoA (Ahlgren et al., 2019) followed by the oxidation of hydroxylamine by hydroxylamine oxidoreductase (hao) to produce nitrite in ammonia oxidizing bacteria (Poret-Peterson et al., 2008). While bacterial nirK and norAB are involved in the conventional denitrification pathway, a number of nitrifying ammonia-oxidizing archaea (AOA) and ammonia-oxidizing bacteria (AOB) also possess these genes, which are believed to be involved in so-called nitrifier-denitrification (reduction of NO2– to NO and N2O) (Wijma et al., 2004; Kozlowski et al., 2014; Lehtovirta-Morley, 2018). As nitrogen availability often limits primary productivity in both marine and terrestrial ecosystems (Canfield et al., 2010), understanding of the nitrogen cycle is critical to understanding its role in regulating primary production in many regions. Diel periodicity of nitrogen cycling in the upper ocean has been shown in many previous studies (Wheeler and Kokkinakis, 1990; Cochlan et al., 1991; Bebout et al., 1994; Brandes et al., 2007; Ward, 2008; Devol, 2015; Wilson et al., 2017), however, much less is known about possible diel periodicity of nitrogen cycling in dark layers just beneath the euphotic ocean.
The coordinated activity of microbial communities operating over daily timescales results in temporal dynamics in the activity of specific microbial taxa and metabolic pathways on diel cycles. In terms of geographic range, metatranscriptomic studies have been conducted in different locations in both the Atlantic Ocean (Gilbert et al., 2010; Hewson et al., 2010), and Pacific Ocean (e.g., Poretsky et al., 2009; Shi et al., 2011; Ottesen et al., 2013, 2014). Further, many metatranscriptomic studies have focused on either one sample taken at one timepoint and depth (Frias-Lopez et al., 2008; Wu et al., 2013; Ji et al., 2018). To date, only one study has focused on spatial and temporal changes in microbial community gene expression (Vislova et al., 2019). While these studies used different sampling designs, many focused on the functionality of the microbial community and have examined essential metabolic functions that impact biogeochemical cycles, including carbon and nitrogen cycling (Frias-Lopez et al., 2008; Poretsky et al., 2009; Hewson et al., 2010; Shi et al., 2011; Ottesen et al., 2013, 2014; Wu et al., 2013; Aylward et al., 2015; Wilson et al., 2017; Ji et al., 2018; Vislova et al., 2019). Here, we expand upon these studies by examining how microbial community gene expression relating to photosynthesis and nitrification changes as a function of light as regulated over both space (i.e., depth in the water column) and time (i.e., diel cycle) at a single coastal site in the Continental shelf waters off New England (United States).
Materials and Methods
Sample Site and Collections
Water samples were collected from the North Atlantic Ocean, near the coast of New Jersey (United States) from the R/V Endeavor in August 2016 (cruise EN588, 39.417, –73.258) and August 2017 (cruise EN604, 39.188, –73.561). Samples were collected using a paint-sealed Sea-Bird rosette, outfitted with a Sea-Bird SBE 19 Plus V2 recording CTD (conductivity, temperature, depth) and 8-L X-Niskins (General Oceanics). The Niskins were acid-cleaned in the laboratory prior to use and “pre-soaked” for 12 h with local seawater collected at a shakedown station prior to deployment. This “clean” rosette was lowered on a non-metallic line (1/4′ O.D. vinyl-jacketed Vectran) and bottle closure at programmed depths controlled by a Sea-Bird Auto-firing Module. Sample depths were chosen based on the real-time water column profile data obtained from the ship’s CTD rosette system (Sea-Bird) that was deployed before the “clean rosette” and which also provided dissolved oxygen, salinity, photosynthetically active radiation (PAR), beam transmission, and fluorescence profiles. Samples were collected at three different depths for the 2016 cruise (5, 16, and 30 m) and 2017 cruise (3, 30, and 40 m) based on the PAR and fluorescence profiles. The 5 m samples of the 2016 cruise and the 3 m samples of the 2017 cruise were within surface sunlit layers (“photic”), the 16 m sample of the 2016 cruise was at the peak of the deep chlorophyll max (“DCM”) layer (as defined by the WETlabs ECO Chlorophyll Fluorometer), and the 30 m samples from both the 2016 and 2017 cruises and the 40 m samples from the 2017 cruise were collected at depths where the PAR was 1% or lower (hereafter “aphotic”). To examine changes at these depths as a function of relative sunlight intensity (sunlight terms are only relative as PAR was not collected at each time point), samples from the 2016 cruise were collected at 04:00 (pre-sunrise), 06:30 (post-sunrise), 14:00 (peak sunlight), 19:15 (pre-sunset), and 21:30 (post-sunset). For the 2017 cruise, samples were taken at 05:00 (pre-sunrise), 09:30 (post-sunrise), 14:00 (peak sunlight), 18:45 (pre-sunset) and 22:00 (post- sunset). For both cruises, samples were transferred from Niskin bottles to a 20 L carboy and immediately filtered as described below.
Samples for dissolved organic carbon (DOC) were passed through combusted 0.2 μm GF/F filters into acidified combusted vials and stored at 4°C until they were analyzed by a Shimadzu TOC-V analyzer (Kyoto, Japan). Samples (250 mL) for chlorophyll a quantification were collected on a 25 mm GF/F filters and stored at –20°C. Chlorophyll a was extracted from the filter in 90% ethanol in the dark at 4°C (9 h) measured with a 10AU fluorometer (Turner) with appropriate standards (Sigma) with the data being corrected for phaeopigments (Parsons and Strickland, 1972). Water samples for inorganic nitrogen speciation were filtered through a 0.2 μm filter, with aliquots stored frozen or analyzed immediately. Subsamples of filtered water were used for immediate shipboard measurement of ammonium (NH4+) by fluorescence using the OPA method (Holmes et al., 1999) and nitrite (NO2–) by spectrophotometry using the Griess reaction method (Pai et al., 2021), with detection limits of 0.02 and 0.1 μm, respectively. Frozen aliquots of nitrate (NO3) were measured in the lab by reduction to NO and quantification by chemiluminescence (Braman and Hendrix, 1989), with a detection limit of ∼0.7 μm.
Samples for RNA analysis were first filtered through a 2.7 μm filter (Whatman) and then a 0.2 μm Sterivex™ filter (EMD Millipore). The Sterivex filter was immediately treated with RNA later (Qiagen) and the samples were flash frozen in liquid nitrogen. Samples were then shipped on dry ice to Central Michigan University and stored at –80°C until processing.
RNA Extraction and Sequencing
RNA was extracted from each Sterivex filter using a MirVana miRNA isolation kits (Ambion) following the manufacturer’s instructions. The extracted RNA was treated with DNase (Qiagen, Valencia, CA, United States) and then cleaned with RNeasy MinElute kit (Qiagen, Valencia, CA, United States). Total RNA was quantified using a Qubit 2.0 fluorometer. For sequencing, RNA samples were sent to the Michigan State University’s Research Technology Support Facility Genomics Core (MSU RTSF). At the sequencing core, RNA was assessed with an Agilent 2100 Bioanalyzer RNA 6000 Pico Chip and RNA-Seq libraries were generated using the Ovation Complete Prokaryotic RNA-Seq System (NuGEN), which included a rRNA depletion step. Shotgun sequencing of the cDNA was performed on one lane of an Illuminia HiSeq 4000 with 2 × 150 bp chemistry. The resulting raw reads are deposited in the National Center for Biotechnology Information’s (NCBI) Short Read Archives (SRA), BioProject ID: PRJNA573842 (accession numbers SAMN12827068-SAMN12827096).
Metatranscriptomic de novo Assembly and Annotations
The resulting sequencing reads were quality checked with FastQC (Holmes et al., 1999) and trimmed using Trimmomatic v0.33, with the default parameters (Bolger et al., 2014). Paired-end reads were interleaved into a single file using Khmer v2.1.1 (Crusoe et al., 2015), and rRNA sequences were removed with SortMeRNA v. 2.0 (Kopylova et al., 2012). The resulting FASTA files were combined by depth and assembled using rnaSPAdes v. 3.13.0 (Bushmanova et al., 2019). QUAST v. 5.0 (Mikheenko et al., 2016) was used to assess the quality of the assembled files (Supplementary Table 1). The depth-specific transcriptome assemblies generated from rnaSPAdes v. 3.13.0 (Bushmanova et al., 2019) were then submitted to the Department of Energy’s Joint Genome Genomes Online Database (GOLD) (Mukherjee et al., 2019) for annotation (IMG Genome ID’s: 3300033603, 3300033604, 3300033605, 3300034405, and 3300034345). The three annotated datasets were then compared to each other using the JGI Institute Integrated Microbial Genomes (IMG) web interface. Functional groups (e.g., photosynthesis, carbon metabolism) from each layer were identified by the KEGG orthology (KO) database or the COG database (translation, ribosome structure, and biogenesis), both within IMG. Salmon v. 0.11.3 (Patro et al., 2017) was used to quantify the number of raw reads that matched to the annotated contigs in each of the layers and normalized the output as transcripts per million (TPM). The merged counts output file from Salmon for each assembly was then merged by locus tag to the genes identified and extracted in IMG using the statistical program R v. 3.5.1 (R Core Team, 2020). Taxonomic identifications (to their best match within the database) were performed by submitting the gene FASTA amino acid (.faa) files for each annotated gene to the GhostKOALA server (Kanehisa et al., 2016). Expression levels for each taxonomic group were found by aggregating the GhostKOALA output with the Salmon expression TPM counts to show total gene expression levels within each listed taxonomic group.
Results
Chemical and Physical Data
In both cruise years, surface water temperatures ranged between 27 and 25°C and decreased to 8°C at the deepest sampling depth (Figure 1). In 2016, light penetration (as PAR) declined sharply to undetectable levels in the first 12 m but measurable PAR was found down to ∼25 m in 2017 (Figure 1). Dissolved oxygen concentrations ranged from 228.23 μm at the surface to 196.33 μm at depth in 2016 and ranged from 227.98 μm at the surface to 206.54 μm at depth (Figure 1) in 2017. On average, chlorophyll-a concentration (extracted from water samples) and DOC concentrations were higher in 2016, when compared to 2017 (Supplementary Table 2). In addition, both ammonium and nitrite were low in the surface water samples but increased with depth in both years (Supplementary Table 2), while nitrate was undetectable across all depths.
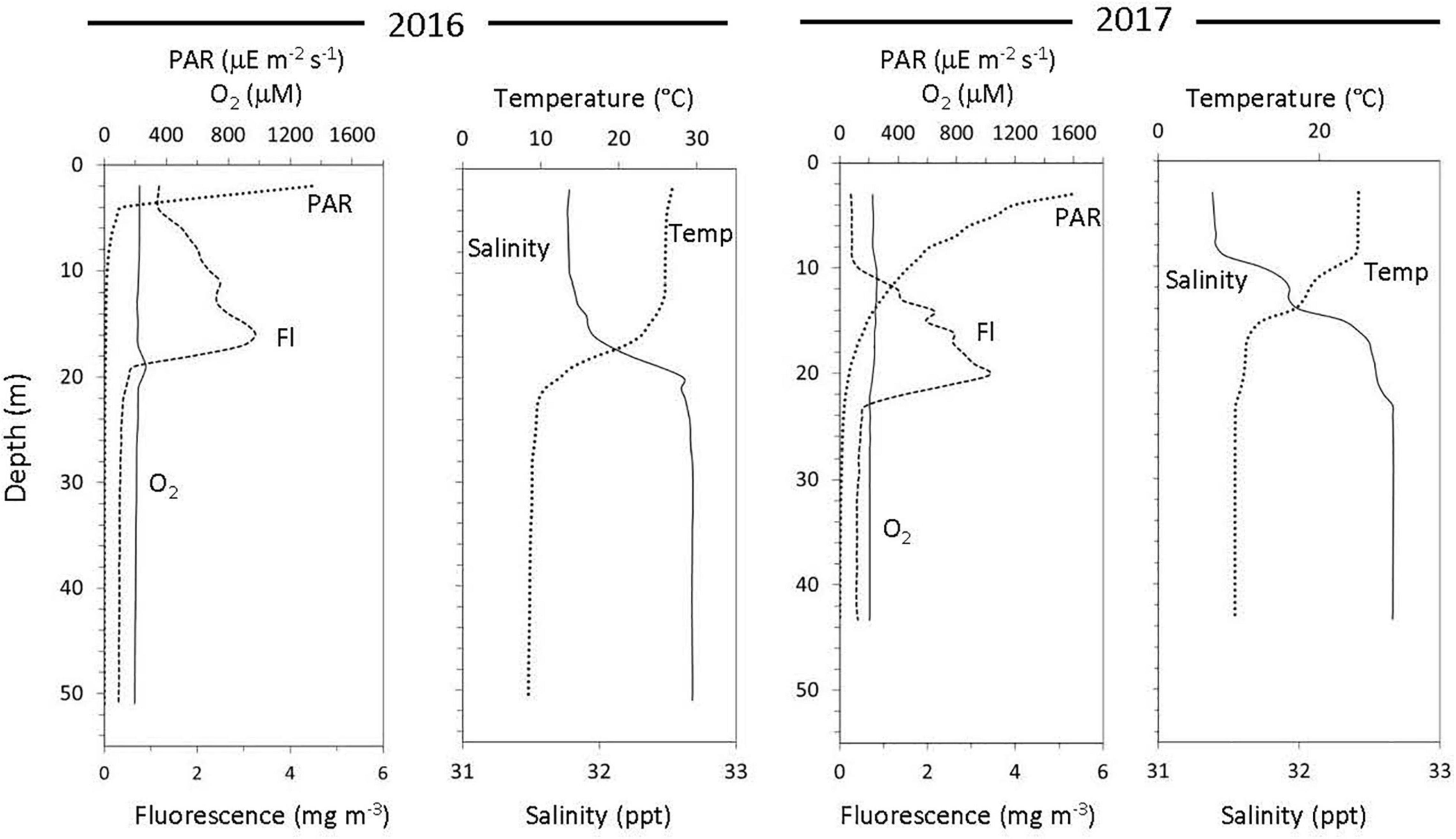
Figure 1. Water column physicochemical properties over 2 consecutive years at the same location off the coast of New Jersey.
Assembly
The 2016 cruise transcripts contained 1,761,728 annotated genes (Supplementary Table 3). By layer, the annotated gene counts were 492,496 (surface transcripts), 498,289 (DCM transcripts), and 770,943 (aphotic transcripts) (Supplementary Table 3). Of the protein coding genes, 36.86% had a function prediction associated with the annotation and 62.20% had no function prediction (Supplementary Table 4).
The 2017 cruise transcripts contained a total of 4,101,045 annotated genes (Supplementary Table 3). Gene annotations were 1,733,133 (surface transcripts), 2,367,912 (aphotic transcripts), and 1,629,343 (deep aphotic transcripts) (Supplementary Table 3). Of the protein coding genes, 29.91% had a function prediction associated with the annotation and 69.44% had no function prediction associated with the annotations (Supplementary Table 4).
Photosynthesis Gene Expression
During the 2016 cruise, gene transcripts annotated as Photosystem I and II in the surface layer (5 m) had measurable expression levels at each time point, including pre-sunrise and post-sunset. Within the surface waters, an increase in expression occurred post-sunrise and peaked at the time of peak sunlight (Figure 2). Similarly, expression of ATPases and ribosomal proteins had peak expression post sunrise with relatively high expression at peak sunlight (Supplementary Figure 1). The DCM layer (16 m) also had peak expression of both Photosystem I and II occurring post-sunrise (Figure 2).
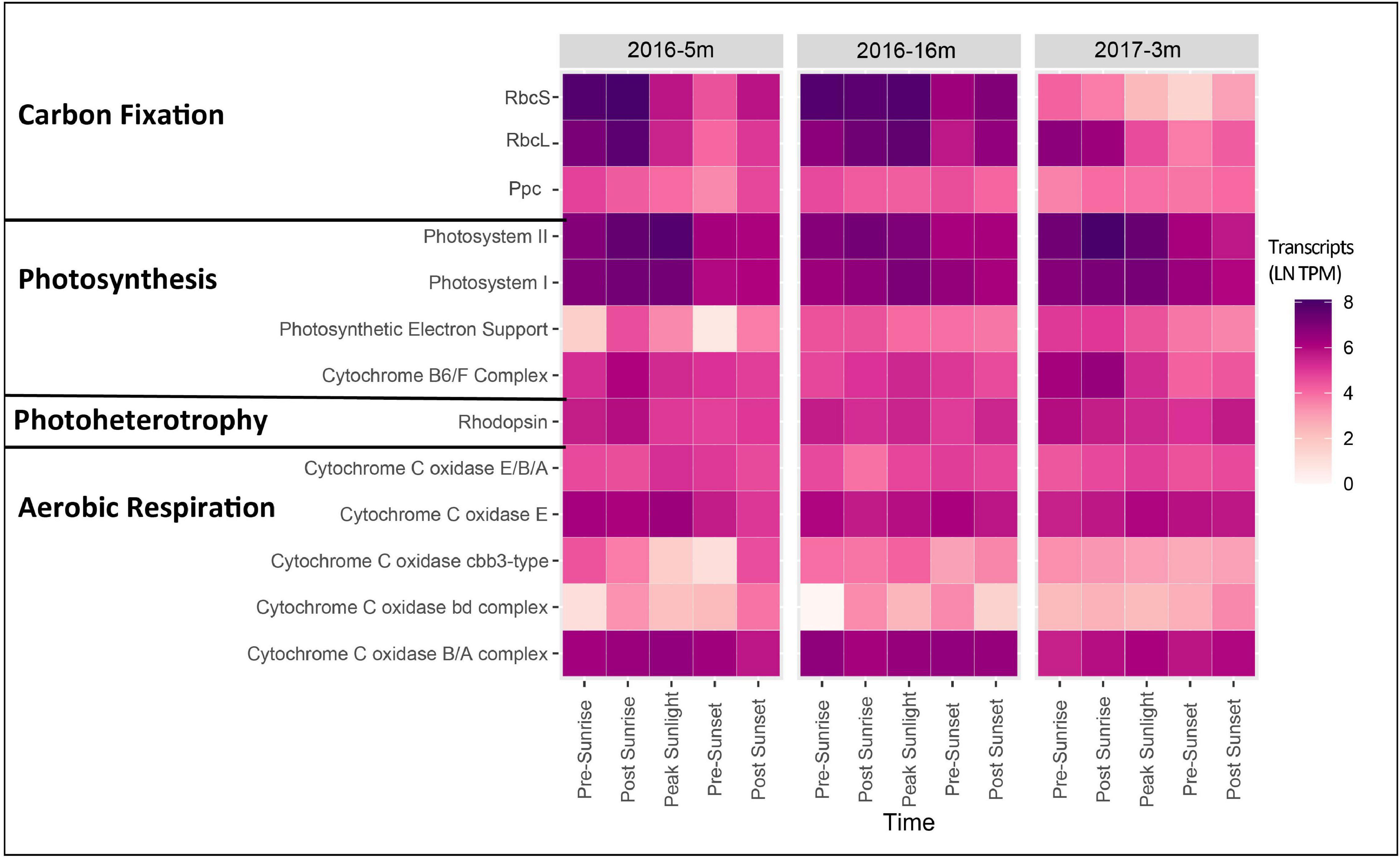
Figure 2. Heatmap showing gene expression [ln(TPM)] for genes associated with carbon fixation, photosynthesis, photoheterotrophy, and aerobic respiration for the photic layers of each cruise. Genes expression for all genes associated with each function was summed to give a final expression for each complex.
Transcripts of psbA, which encodes the D1 protein of photosystem II, in the surface water were allied with Stramenopile (Aureococcus), cyanobacteria (Synechocococus), and green algae (Ostreococcus). While expression of psbA associated with Synechococcus and Ostreococcus was highest during times of peak sunlight, Aureococcus psbA transcripts had highest levels before peak sunlight (Figure 3). Transcripts of psbA collected in the DCM were dominated by Aureococcus, with peak expression occurring post-sunrise (Figure 3). The taxa most highly expressing psaA, integral membrane protein of Photosystem I, in the surface layer was Aureococcus with the most expression occurring post-sunrise (Figure 3). Synechococccus had high psaA expression during times of sunlight, while Prochlorococcus had peak expression pre-sunset (Figure 3).

Figure 3. Dot plot showing gene expression in ln(TPM) for genes products annotated as photosystem I, photosystem II, RbcLS, cytochrome C oxidase B/A complex, and rhodopsins in the photic layers of each cruise for dominant organisms (Aureococus, Synehcococcus, Ostreococcus, Prochlorococcus, Alphaproteobacteria HIMB5, Alphaproteobacteria HIMB59, and Candidatus Pelagibacter). The colors in the figure are to help differentiate time points.
For the 2017 cruise, transcripts associated with Photosystem I and II surface layer were highest in the early morning (pre-sunrise through to peak sunlight) and then relatively decreased to post sunset (Figure 2). While Photosystem II transcript levels within surface water in 2016 were highest during peak sunlight, the highest transcript levels were observed post-sunrise in 2017 (Figure 2). Furthermore, in the 2017 surface samples the photosynthetic cytochrome B6/F complex showed peak expression pre- and post-sunrise with expression gradually declining throughout the day (Figure 2). ATPase and ribosomal protein expression also had relatively high expression pre- and post-sunrise (Supplementary Figure 1).
The surface water samples in the 2017 cruise showed the most dominant taxa expressing the psbA gene belonged to Aureococcus, Synechococcus, and Ostreococcus (Figure 3). Similarly, psaA surface transcripts were nearly consistent throughout the day for Aureococcus and Ostreococcus, but highest at peak sunlight for Synechococcus (Figure 3). Unlike the 2016 cruise photic samples, psbA and psaA were not expressed by Prochlorococcus in 2017 surface samples (Figure 3).
Photoheterotrophy Gene Expression
Photoheterotrophy was examined by assessing microbial rhodopsin expression. Specifically, rhodopsin was found in the photic layers in both years (Figure 2). Relative to the expression of genes annotated in PS I and II, rhodopsin expression in 2016 was lower, but peak expression was post sunrise in the surface layer and pre-sunrise in the DCM layer (Figure 2). The abundant rhodopsins were taxonomically identified as Alphaproteobacteria HIMB5, Alphaproteobacteria HIMB59, and Candidatus Peligibacter sp. (Figure 3). In the surface layer of the 2016 cruise, rhodopsin transcripts from Alphaproteobacteria HIMB5 showed expression throughout the day, with the highest peak expression occurring at post sunrise (Figure 3). There were no transcripts documented for other microbial rhodopsin (such as proteorhodopsin or halorhodopsin).
Rhodopsin expression patterns in 2017 were less obvious than 2016, and total rhodopsin transcripts within surface samples showed peak expression pre-sunrise (Figure 2). Similar to 2016, rhodopsins in the 2017 surface transcripts were taxonomically identified as Alphaproteobacteria HIMB5, Alphaproteobacteria HIMB59, and Candidatus Peligibacter (Figure 3) with peak expression either pre- or post-sunrise (Figure 3). Similar to 2016, there were no transcripts identified for other microbial rhodopsin (such as proteorhodopsin or halorhodopsin).
Ammonia Oxidation
Samples taken from depths deeper then 30 m from both years clearly documented the presence of genes and organisms associated with nitrification (Figure 4). In 2016, transcripts annotated as amoABC, norAB, and hao were highly expressed (Figure 4). Nitrosospira had the highest gene expression of amoA followed by Candidatus Nitrosomarinus (Figure 5). The most highly expressed transcripts of hao in the aphotic samples were taxonomically identified as Nitrosomonadaceae, Nitrosospira and Nitrosococcus (Figure 5). The major difference between samples collected in 2017 compared to 2016 was a lack of hao expression (Figure 5). Nitrosopumilus only expressed amoA at the 30 m depth and exhibited peak expression pre-sunset and post-sunset (Figure 5).
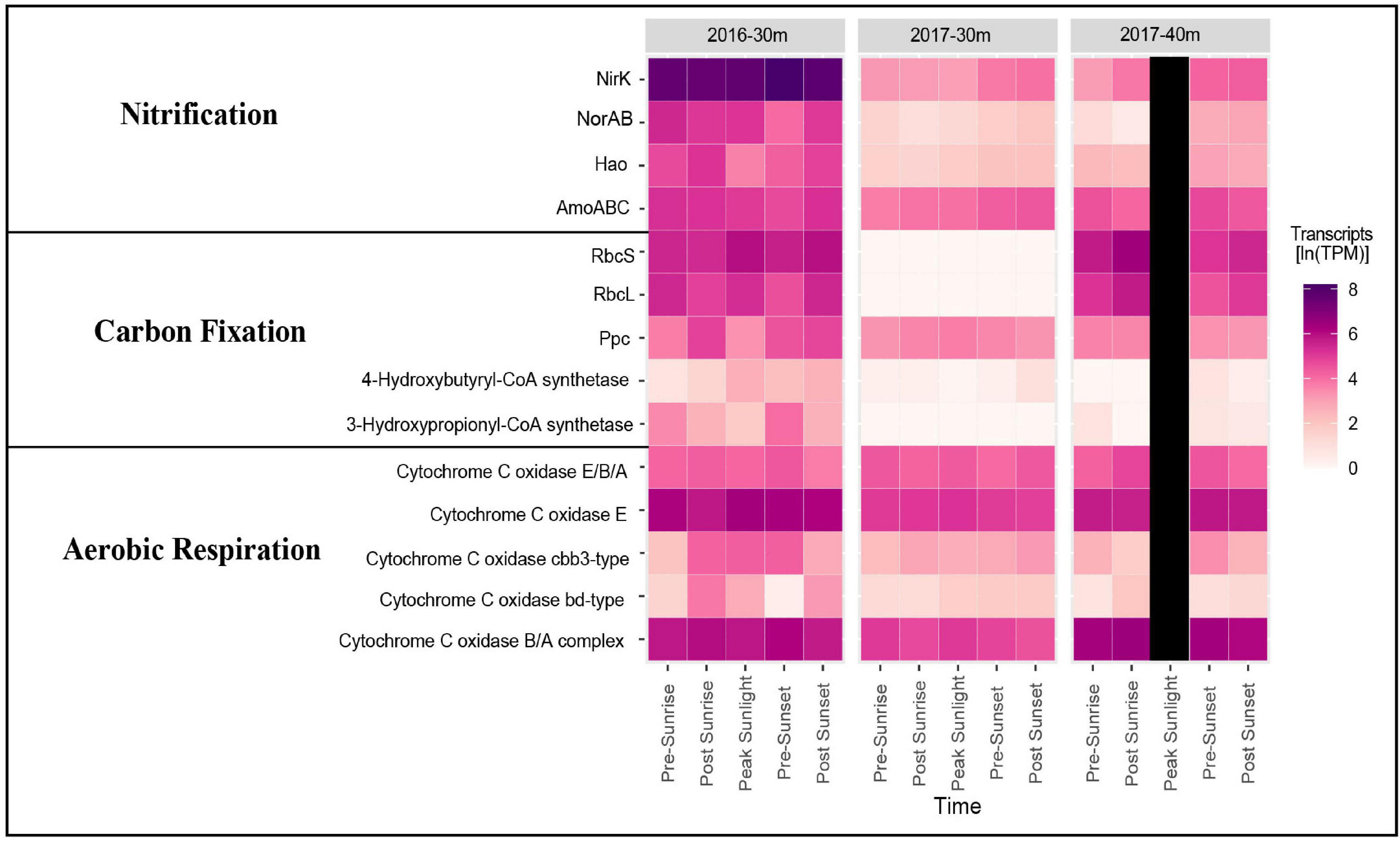
Figure 4. Heatmap showing gene expression in ln(TPM) for genes associated with nitrification, carbon fixation, and aerobic respiration in the euphotic layers of each cruise.
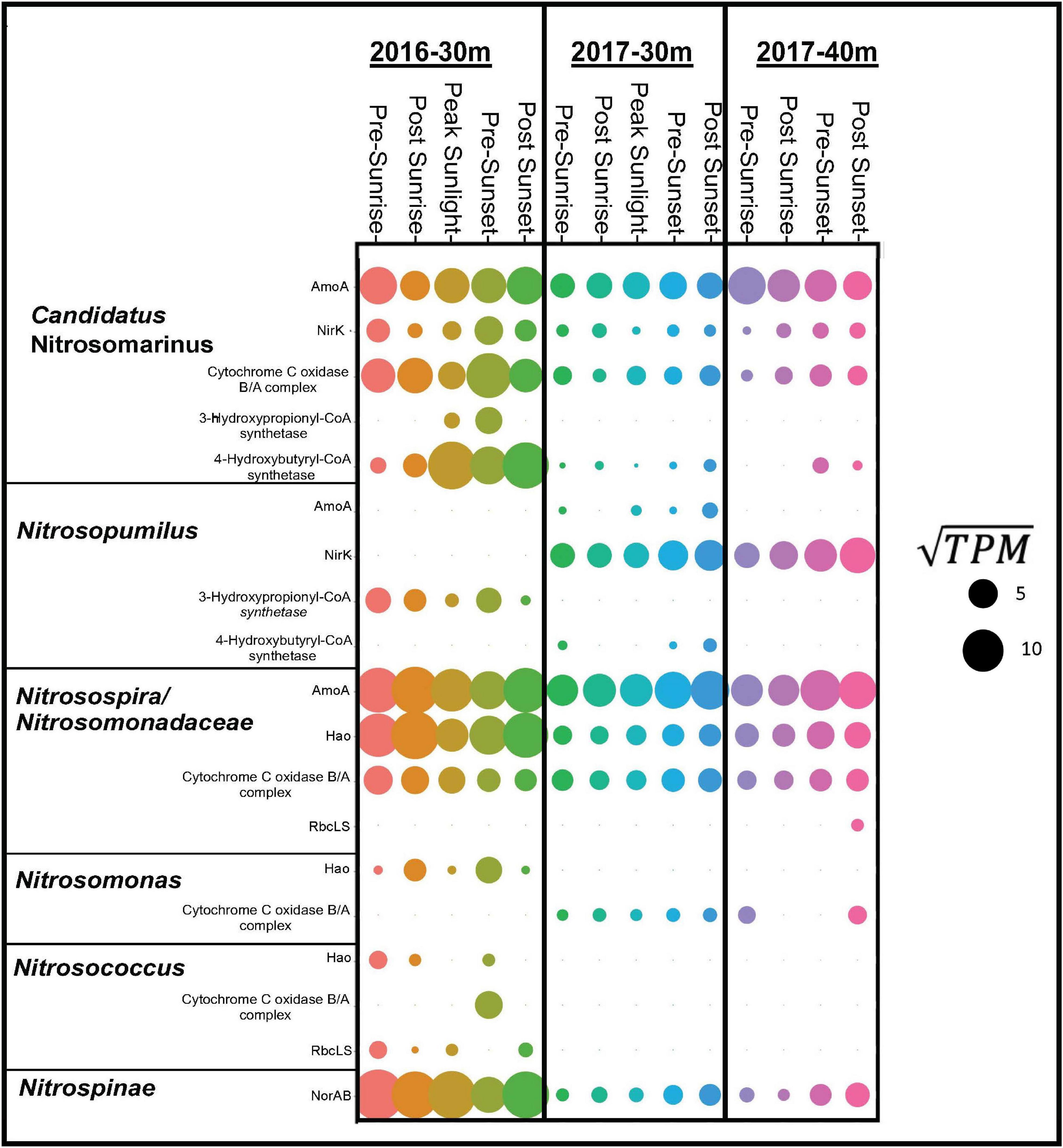
Figure 5. Dot plot showing gene expression in ln(TPM) for dominate taxa (Candidatus Nitrosomarinus, Nitrsopumilus, Nitrosospira, Nitrosomondaceae bacteria, Nitrosomonas, Nitrosococcus, and Nitrospinae) for gene products associated with nitrification, carbon fixation and aerobic respiration in euphotic layers of both cruises.
Discussion
It is well known that the availability of light regulates primary production by photosynthesis in the surface ocean (Poretsky et al., 2009; Hewson et al., 2010; Aylward et al., 2015). The energy and biomass generated via these processes varies temporally and spatially. As phototrophic organisms generate organic matter, exuded metabolites and detrital cellular material support a diverse heterotrophic community (Ottesen et al., 2014). Heterotrophic remineralization of organic matter produces reduced nitrogen compounds (especially NH4+, but also urea, cyanate) that stimulates and supports a chemolithoautotrophic community of nitrifying organisms, including AOA and bacteria (AOB), and nitrite-oxidizing bacteria (NOB) (Bebout et al., 1994; Zehr and Ward, 2002). The metabolic activity of each of these groups is reflected through expression of different metabolic pathways. Here, we present our examination of metatranscriptomic patterns gene expression of photosynthesis and nitrification over scales of space (depth) and time (over the course of a day and a yearly comparison).
Diel Cycling Along a Depth Gradient
Photosynthesis
In this coastal study, community transcripts related to photosynthesis (photosystem II and photosystem I) displayed peak gene expression occurring times of relatively higher sunlight (post-sunrise and peak-sunlight) and with lowest gene expression near sunset (Figure 2 and Supplementary Figure 1). Transcripts from photosynthetic organisms in the open ocean’s surface have been shown to vary with light intensity along a diel cycle (Frias-Lopez et al., 2008; Ottesen et al., 2013, 2014; Aylward et al., 2015), however, the current study did not always observe peak expression with peak sunlight. This difference could be due to variation in sample collection time, sample number, sequencing depth, or that the current study was conducted in a coastal marine ecosystem. While transcript peaks occurred near peak sunlight in this study, expression of photosynthetic genes (PS I and II) have also been documented to be expressed under conditions such as pre-sunrise (Hewson et al., 2010; Becker et al., 2019). Such dark expression likely reflects the physiological preparation of the community to efficiently harvest light as it becomes available after sunrise.
Expression of photosynthetic genes within surface waters was genetically similar to four known phototrophic microbes (Figure 3). The dominant (highest relative abundance) psbA (photosystem I) and psaA (photosystem II) transcripts in the surface and DCM layers belonged to the genera of Aureococcus, Ostreococcous, Synechocococus, and Prochlorococcus. The abundance of photosystem transcripts from certain taxa, such as Aureococcus in the DCM and Synechocococus in the surface, exhibited peak expression during peak sunlight. Similar to community transcript levels, Aureococcus and Ostreococcous within surface samples had relatively high expression of psbA and psaA pre-sunrise to peak sunlight. Both Ostreococcus (Ottesen et al., 2014; Aylward et al., 2015) and Prochlorococcus (Ottesen et al., 2013; Aylward et al., 2015; Vislova et al., 2019) have been shown to be dominant community members in surface waters of the Pacific Ocean with their expression following a diel pattern (synchronous with sunlight), but photosynthetic genes were still expressed pre-sunrise, as also observed here in the Atlantic Ocean.
Photoheterotrophy
Transcripts for production of rhodopsin, involved in harnessing light for energy (Spudich et al., 2000; Neutze et al., 2002; Pinhassi et al., 2016), maintained relatively stable expression throughout the day in photic zone samples of the 2016 cruise, with surface samples exhibiting highest levels of expression pre-sunset/post sunrise and the DCM samples showing high expression pre-sunrise and post sunset (Figure 2). Similar to photosynthic genes, the lack of peak expression during peak sunlight as seen in other studies could be due to differences in experimental design or ecosystem variability, however, two other studies have also documented peak transcript expression from phototrophs during darker periods (Ottesen et al., 2013; Becker et al., 2019). The abundant transcripts for the gene that produces rhodopsin observed in waters from the photic zone (surface and DCM) were taxonomically aligned with Alphaproteobacteria HIMB5, Alphaproteobacteria HIMB59, and Candidatus Peligibacter (Figure 3). All three of these taxa are members of the photoheterotrophic SAR11 clade (Grote et al., 2012; Zhao et al., 2017).
Ammonia Oxidation
While high levels of nitrification are generally not observed in the sunlit surface ocean, our data document transcripts suggesting an active nitrifying community at depths just below these sunlit layers (Figure 4). Taxonomic data from functional genes demonstrate that both AOA and AOB are active in the aphotic samples from the 2016 cruise (Figure 5). Both amoA and nirK allied to marine ammonia-oxidizing Thaumarchaeota Nitrosopumilus and Candidatus Nitrosomarinus (Herber et al., 2020). Transcripts of amoA and hao related to known AOB, Nitrosospira and Nitrosococcus (Hollibaugh et al., 2002; Klotz et al., 2006), and Nitrosomonadaceae (Prosser et al., 2014), were also identified (Figure 5). Vislova et al. (2019) also noted that activity of nitrifying taxa did not vary with diel periodicity.
Comparison of Year-to-Year Variability
While back-to-back cruises in the Fall of 2016 and 2017 provide an opportunity to compare yearly variability in diel activity at this coastal site, differences in physical (mixing) and chemical (oxygen) conditions were also evident and relevant (see Figure 1 and Supplementary Table 2). In particular, the waters in 2016 were warmer, more productive, and therefore had shallower light penetration than in 2017. As such, the transition from photic to aphotic depths (as defined here) began at 12 m in 2016 but was at 28 m in 2017. Further, the surface waters in 2017 were less mixed as observed in the defined layers in the temperature and salinity plots (Figure 1).
Similar expression trends in photosynthetic transcripts documented in 2016 were also observed in 2017. The 2017 cruise collected water from the surface and two deeper aphotic layers, and not the DCM as in 2016. Thus, comparisons between the years are solely focused on the photic and deeper, aphotic samples. Similar to the 2016 cruise, the dominant psbA (photosystem I) and psaA (photosystem II) transcripts in the surface layer of the 2017 cruise belonged to the genera of Aureococcus, Ostreococcous, and Synechocococus with expression being still relatively high pre-sunrise. Further, surface transcripts collected in 2017 documented the presence of photoheterotrophs (Alphaproteobacteria HIMB5, Alphaproteobacteria HIMB59, and Candidatus Peligibacter), with higher expression still occurring during darker times of day. The presence of both phototrophs and photoheterotrophs suggests a coupling of metabolic processes, which enable the community to both fix and metabolize carbon.
Over a 2-year period both AOA, Candidatus Nitrosomarinus, and AOB, Nitrosospira, were found in these coastal euphotic waters. AOB are thought to thrive in more nutrient rich ecosystems and AOA could thrive in more oligotrophic ecosystems (Santoro et al., 2008; Martens-Habbena et al., 2009) as AOA amoA genes have higher affinities for ammonia (Martens-Habbena et al., 2009; Straka et al., 2019). In the current study, primary controlling factors (e.g., temperature, pH, substrate concentrations) did not vary year to year. Thus, it is possible that the shelf waters examined here simply represent a midpoint along the continuum between the nutrient replete coastal waters (where AOB frequently outnumber AOA) and nutrient depleted waters further offshore, where AOA are typically the dominant nitrifiers, with physical conditions not allowing either group to establish dominance.
Periodic and daily transcriptional trends in microbial metabolism occur in marine communities as a function of light attenuation. As a result, microbial communities in the ocean’s surface follows a diel pattern of expression of gene transcripts related to metabolism. Interesting, photosynthetic genes described here also were expressed in pre-sunrise, ramping up for future times of more sunlight. However, below the photic zone, ocean microbial communities show more stable gene expression of nitrification. Given that both cruise (sampling separated by 1 year) transcripts showed similar expression trends, this suggests that in the photic zone microbial community function follows a consistent pattern through time. This work also focused on the coastal Atlantic Ocean, with other studies focusing on the Pacific Ocean (Ottesen et al., 2013, 2014; Aylward et al., 2015; Vislova et al., 2019), which suggests these expressional patterns occur globally.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA573842.
Author Contributions
JZ conducted the bioinformatics, analyzed the data, and drafted the figures. DH performed the laboratory work to extraction RNA from the samples. CH, SW, CL, and DL collected the samples. CS provided support and insight for bioinformatic analysis. CH and DL designed and planned the study. All authors contributed to writing the manuscript.
Funding
This work was supported by the National Science Foundation grant # 1355720 (CH and CL) and from Central Michigan University Faculty Research and Creative Endeavors Committee (DL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Carolyn Buchwald, Kevin Sutherland, Jennifer Karolewski, Gabriella Farfan, Kalina Grabb, and Kaitlin Bowman for shipboard sampling assistance, as well as the captain and crew of the R/V Endeavor who made sampling for this research possible. We also thank Julia Diaz and Sydney Plummer for collecting chlorophyll-a data and Carolyn Buchwald and Jennifer Karolewski for inorganic nitrogen measurements. We would also like to acknowledge the Michigan State University ICER team for the use of their HPCC. Additionally, we would like to thank John Johnston, Arkadiy Garber, Stephanie Gill, and Keith Palmer for their computational expertise. This is contribution #161 of the Central Michigan University Institute for Great Lakes Research.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.798877/full#supplementary-material
References
Ahlgren, N. A., Fuchsman, C. A., Rocap, G., and Fuhrman, J. A. (2019). Discovery of several novel, widespread, and ecologically distinct marine Thaumarchaeota viruses that encode amoC nitrification genes. ISME J. 13, 618–631. doi: 10.1038/s41396-018-0289-4
Aylward, F. O., Boeuf, D., Mende, D. R., Wood-Charlson, E. M., Vislova, A., Eppley, J. M., et al. (2017). Diel cycling and long-term persistence of viruses in the ocean’s euphotic zone. Proc. Natl. Acad. Sci. U.S.A. 114, 11446–11451. doi: 10.1073/pnas.1714821114
Aylward, F. O., Eppley, J. M., Smith, J. M., Chavez, F. P., Scholin, C. A., and DeLong, E. F. (2015). Microbial community transcriptional networks are conserved in three domains at ocean basin scales. Proc. Natl. Acad. Sci. U.S.A. 112, 5443–5448. doi: 10.1073/pnas.1502883112
Barton, A. D., Pershing, A. J., Litchman, E., Record, N. R., Edwards, K. F., Finkel, Z. V., et al. (2013). The biogeography of marine plankton traits. Ecol. Lett. 16, 522–534. doi: 10.1111/ele.12063
Bebout, B. M., Paerll, H. W., Baue, J. E., and Canfield, D. E. (1994). “Nitrogen cycling in microbial mat communities: the quantitative importance of N-fixation and other sources for N for primary productivity,” in Microbial Mats: Structure, Development and Environmental Significance: Series G: Ecological Sciences, eds L. Stal and P. Caumette (Berlin: Springer-Verlag), 265–271.
Becker, J. W., Hogle, S. L., Rosendo, K., and Chisholm, S. W. (2019). Co-culture and biogeography of Prochlorococcus and SAR11. ISME J. 13, 1506–1519. doi: 10.1038/s41396-019-0365-4
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Braman, R. S., and Hendrix, S. A. (1989). Nanogram nitrite and nitrate determination in environmental and biological materials by vanadium(III) reduction with chemiluminescence detection. Anal. Chem. 61, 2715–2718. doi: 10.1021/ac00199a007
Brandes, J. A., Devol, A. H., and Deutsch, C. (2007). New developments in the marine nitrogen cycle. Chem. Rev. 107, 577–589. doi: 10.1021/cr050377t
Bushmanova, E., Antipov, D., Lapidus, A., and Prjibelski, A. D. (2019). rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. Gigascience 8:giz100. doi: 10.1093/gigascience/giz100
Canfield, D. E., Glazer, A. N., and Falkowski, P. G. (2010). The evolution and future of earth’s nitrogen cycle. Science 330, 192–196. doi: 10.1126/science.1186120
Cochlan, W. P., Harrison, P. J., and Denman, K. L. (1991). Diel periodicity of nitrogen uptake by marine phytoplankton in nitrate-rich environments. Limnol. Oceanogr. 36, 1689–1700. doi: 10.4319/lo.1991.36.8.1689
Crusoe, M. R., Alameldin, H. F., Awad, S., Boucher, E., Caldwell, A., Cartwright, R., et al. (2015). The khmer software package: enabling efficient nucleotide sequence analysis. F1000Res 4:900. doi: 10.12688/f1000research.6924.1
Devol, A. H. (2015). Denitrification, anammox, and N2 production in marine sediments. Annu. Rev. Mar. Sci. 7, 403–423. doi: 10.1146/annurev-marine-010213-135040
Frias-Lopez, J., Shi, Y., Tyson, G. W., Coleman, M. L., Schuster, S. C., Chisholm, S. W., et al. (2008). Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. U.S.A. 105, 3805–3810. doi: 10.1073/pnas.0708897105
Gilbert, J. A., Meyer, F., Schriml, L., Joint, I. R., Mühling, M., and Field, D. (2010). Metagenomes and metatranscriptomes from the L4 long-term coastal monitoring station in the Western English Channel. Stand. Genomic Sci. 3, 183–193. doi: 10.4056/sigs.1202536
Grote, J., Thrash, J. C., Huggett, M. J., Landry, Z. C., Carini, P., Giovannoni, S. J., et al. (2012). Streamlining and core genome conservation among highly divergent members of the SAR11 clade. mBio 3:e00252-12. doi: 10.1128/mBio.00252-12
Herber, J., Klotz, F., Frommeyer, B., Weis, S., Straile, D., Kolar, A., et al. (2020). A single Thaumarchaeon drives nitrification in deep oligotrophic Lake Constance. Environ. Microbiol. 22, 212–228. doi: 10.1111/1462-2920.14840
Hewson, I., Poretsky, R. S., Tripp, H. J., Montoya, J. P., and Zehr, J. P. (2010). Spatial patterns and light-driven variation of microbial population gene expression in surface waters of the oligotrophic open ocean. Environ. Microbiol. 12, 1940–1956. doi: 10.1111/j.1462-2920.2010.02198.x
Hollibaugh, J. T., Bano, N., and Ducklow, H. W. (2002). Widespread distribution in Polar oceans of a 16S rRNA gene sequence with affinity to nitrosospira-like ammonia-oxidizing bacteria. Appl. Environ. Microbiol. 68:7. doi: 10.1128/AEM.68.3.1478-1484.2002
Holmes, R. M., Aminot, A., Kérouel, R., Hooker, B. A., and Peterson, B. J. (1999). A simple and precise method for measuring ammonium in marine and freshwater ecosystems. Can. J. Fish. Aquat. Sci. 56, 1801–1808. doi: 10.1139/f99-128
Ji, N., Lin, L., Li, L., Yu, L., Zhang, Y., Luo, H., et al. (2018). Metatranscriptome analysis reveals environmental and diel regulation of a Heterosigma akashiwo (Raphidophyceae) bloom: metatranscriptome profiling of Heterosigma akashiwo. Environ. Microbiol. 20, 1078–1094. doi: 10.1111/1462-2920.14045
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Klotz, M. G., Arp, D. J., Chain, P. S. G., El-Sheikh, A. F., Hauser, L. J., Hommes, N. G., et al. (2006). Complete genome sequence of the marine, chemolithoautotrophic, ammonia-oxidizing bacterium Nitrosococcus oceani ATCC 19707. Appl. Environ. Microbiol. 72, 6299–6315. doi: 10.1128/AEM.00463-06
Kopylova, E., Noé, L., and Touzet, H. (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217. doi: 10.1093/bioinformatics/bts611
Kozlowski, J., Price, J., and Stein, L. (2014). Revision of N2O-producing pathways in the ammonia-oxidizing bacterium Nitrosomonas europaea ATCC 19718. Appl. Environ. Microbiol. 80:6. doi: 10.1128/AEM.01061-14
Lehtovirta-Morley, L. E. (2018). Ammonia oxidation: ecology, physiology, biochemistry and why they must all come together. FEMS Microbiol. Lett. 365:fny058. doi: 10.1093/femsle/fny058
Martens-Habbena, W., Berube, P. M., Urakawa, H., de la Torre, J. R., and Stahl, D. A. (2009). Ammonia oxidation kinetics determine niche separation of nitrifying Archaea and bacteria. Nature 461, 976–979. doi: 10.1038/nature08465
Mikheenko, A., Saveliev, V., and Gurevich, A. (2016). MetaQUAST: evaluation of metagenome assemblies. Bioinformatics 32, 1088–1090. doi: 10.1093/bioinformatics/btv697
Mukherjee, S., Stamatis, D., Bertsch, J., Ovchinnikova, G., Katta, H. Y., Mojica, A., et al. (2019). Genomes OnLine database (GOLD) v.7: updates and new features. Nucleic Acids Res. 47, D649–D659. doi: 10.1093/nar/gky977
Neutze, R., Pebay-Peyroula, E., Edman, K., Royant, A., Navarro, J., and Landau, E. M. (2002). Bacteriorhodopsin: a high-resolution structural view of vectorial proton transport. Biochim. Biophys. Acta 1565, 144–167. doi: 10.1016/S0005-2736(02)00566-7
Ottesen, E. A., Young, C. R., Eppley, J. M., Ryan, J. P., Chavez, F. P., Scholin, C. A., et al. (2013). Pattern and synchrony of gene expression among sympatric marine microbial populations. Proc. Natl. Acad. Sci. U.S.A. 110, E488–E497. doi: 10.1073/pnas.1222099110
Ottesen, E. A., Young, C. R., Gifford, S. M., Eppley, J. M., Marin, R., Schuster, S. C., et al. (2014). Multispecies diel transcriptional oscillations in open ocean heterotrophic bacterial assemblages. Science 345, 207–212. doi: 10.1126/science.1252476
Pai, S.-C., Su, Y.-T., Lu, M.-C., Chou, Y., and Ho, T.-Y. (2021). Determination of nitrate in natural waters by vanadium reduction and the griess assay: reassessment and optimization. ACS EST Water 1, 1524–1532. doi: 10.1021/acsestwater.1c00065
Parsons, J. D. H., and Strickland, T. R. (1972). A Practical Handbook of Seawater Analysis. Ottawa, ON: Fisheries Research Board of Canada.
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon: fast and bias-aware quantification of transcript expression using dual-phase inference. Nat Methods 14, 417–419. doi: 10.1038/nmeth.4197
Pinhassi, J., DeLong, E. F., Béjà, O., González, J. M., and Pedrós-Alió, C. (2016). Marine bacterial and archaeal ion-pumping rhodopsins: genetic diversity, physiology, and ecology. Microbiol. Mol. Biol. Rev. 80:26. doi: 10.1128/MMBR.00003-16
Poret-Peterson, A. T., Graham, J. E., Gulledge, J., and Klotz, M. G. (2008). Transcription of nitrification genes by the methane-oxidizing bacterium, Methylococcus capsulatus strain Bath. ISME J. 2, 1213–1220. doi: 10.1038/ismej.2008.71
Poretsky, R. S., Hewson, I., Sun, S., Allen, A. E., Zehr, J. P., and Moran, M. A. (2009). Comparative day/night metatranscriptomic analysis of microbial communities in the North Pacific subtropical gyre. Environ. Microbiol. 11, 1358–1375. doi: 10.1111/j.1462-2920.2008.01863.x
Prosser, J. I., Head, I. M., and Stein, L. Y. (2014). “The family Nitrosomonadaceae,” in The Prokaryotes: Alphaproteobacteria and Betaproteobacteria, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin: Springer), 901–918. doi: 10.1007/978-3-642-30197-1_372
R Core Team. (2020). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Santoro, A. E., Francis, C. A., Sieyes, N. R. D., and Boehm, A. B. (2008). Shifts in the relative abundance of ammonia-oxidizing bacteria and archaea across physicochemical gradients in a subterranean estuary. Environ. Microbiol. 10, 1068–1079. doi: 10.1111/j.1462-2920.2007.01547.x
Shi, Y., Tyson, G. W., Eppley, J. M., and DeLong, E. F. (2011). Integrated metatranscriptomic and metagenomic analyses of stratified microbial assemblages in the open ocean. ISME J. 5, 999–1013. doi: 10.1038/ismej.2010.189
Spudich, J. L., Yang, C.-S., Jung, K.-H., and Spudich, E. N. (2000). Retinylidene proteins: structures and functions from archaea to humans. Annu. Rev. Cell Dev. Biol. 16, 365–392. doi: 10.1146/annurev.cellbio.16.1.365
Straka, L. L., Meinhardt, K. A., Bollmann, A., Stahl, D. A., and Winkler, M.-K. H. (2019). Affinity informs environmental cooperation between ammonia-oxidizing archaea (AOA) and anaerobic ammonia-oxidizing (Anammox) bacteria. ISME J. 13, 1997–2004. doi: 10.1038/s41396-019-0408-x
Vislova, A., Sosa, O. A., Eppley, J. M., Romano, A. E., and DeLong, E. F. (2019). Diel oscillation of microbial gene transcripts declines with depth in oligotrophic ocean waters. Front. Microbiol. 10:2191. doi: 10.3389/fmicb.2019.02191
Ward, B. B. (2008). “Nitrification in marine systems,” in Nitrogen in the Marine Environment, eds D. G. Capone, D. A. Bronk, M. R. Mulholland, and E. J. Carpenter (New York, NY: Elsevier), 199–261. doi: 10.1016/B978-0-12-372522-6.00005-0
Wheeler, P. A., and Kokkinakis, S. A. (1990). Ammonium recycling limits nitrate use in the oceanic subarctic Pacific. Limnol. Oceanogr. 35, 1267–1278. doi: 10.4319/lo.1990.35.6.1267
Wijma, H. J., Canters, G. W., de Vries, S., and Verbeet, M. P. (2004). Bidirectional catalysis by copper-containing nitrite reductase. Biochemistry 43, 10467–10474. doi: 10.1021/bi0496687
Wilson, S. T., Aylward, F. O., Ribalet, F., Barone, B., Casey, J. R., Connell, P. E., et al. (2017). Coordinated regulation of growth, activity and transcription in natural populations of the unicellular nitrogen-fixing cyanobacterium Crocosphaera. Nat Microbiol 2:17118. doi: 10.1038/nmicrobiol.2017.118
Wu, J., Gao, W., Johnson, R. H., Zhang, W., and Meldrum, D. R. (2013). Integrated metagenomic and metatranscriptomic analyses of microbial communities in the Meso- and bathypelagic realm of North Pacific Ocean. Mar. Drugs 11, 3777–3801. doi: 10.3390/md11103777
Zehr, J. P., and Ward, B. B. (2002). Nitrogen cycling in the ocean: new perspectives on processes and paradigms. AEM 68, 1015–1024. doi: 10.1128/AEM.68.3.1015-1024.2002
Keywords: transcriptomics, marine microbiology, diel cycles, nitrification, photosyhthesis
Citation: Zehnpfennig JR, Hansel CM, Wankel SD, Sheik CS, Horton DJ, Lamborg CH and Learman DR (2022) Diel Patterns in Marine Microbial Metatranscriptomes Reflect Differences in Community Metabolic Activity Over Depth on the Continental Shelf of the North Atlantic. Front. Mar. Sci. 9:798877. doi: 10.3389/fmars.2022.798877
Received: 20 October 2021; Accepted: 12 January 2020;
Published: 03 February 2022.
Edited by:
Jose M. Gonzalez, University of La Laguna, SpainReviewed by:
Taichi Yokokawa, Japan Agency for Marine-Earth Science and Technology (JAMSTEC), JapanDirk de Beer, Max Planck Society, Germany
Frank O’Neill Aylward, Virginia Tech, United States
Copyright © 2022 Zehnpfennig, Hansel, Wankel, Sheik, Horton, Lamborg and Learman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deric R. Learman, ZGVyaWMubGVhcm1hbkBjbWljaC5lZHU=