 Zhen Wu
Zhen Wu Wai Hin Lee2,3
Wai Hin Lee2,3 Senjie Lin
Senjie Lin- 1Shenzhen Branch, Guangdong Laboratory of Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen, China
- 2State Key Laboratory of Marine Pollution, City University of Hong Kong, Kowloon, Hong Kong SAR, China
- 3Department of Biomedical Sciences, City University of Hong Kong, Kowloon, Hong Kong SAR, China
- 4State Key Laboratory of Marine Environmental Science, Xiamen University, Xiamen, China
- 5Department of Marine Sciences, University of Connecticut, Groton, CT, United States
- 6Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai), Zhuhai, China
- 7Research Centre for the Oceans and Human Health, City University of Hong Kong Shenzhen Research Institute, Shenzhen, Hong Kong SAR, China
- 8Office of the President, The Open University of Hong Kong, Shenzhen, Hong Kong SAR, China
Dinoflagellates, including harmful algal bloom species, are known to co-exist with and rely upon bacteria but how the microbiome changes with the physiologies of the cognate dinoflagellates is poorly understood. Here, we used 16S rRNA gene meta-barcoding to characterize the bacterial community in the cultures of Gambierdiscus balechii, a ciguatoxin-producing benthic dinoflagellate, under different nitrogen (N)-nutrient conditions and at different ciguatoxin-producing growth. The high-throughput sequencing of a total of 12 libraries generated 926,438 reads which were classified into 16 phyla. We observed a shift of the G. balechii-associated microbiome from N-replete to low-N conditions and from the early (low toxin) to the late exponential (high toxin) growth stage. Common across these conditions were species from families Rhodobacteraceae and Flavobacteriaceae. Species abundant in the low-N condition mainly included Planctomyces, Ekhidna, and Lactobacillus. Dominant or highly abundant microbial taxa in the high toxin-producing stage (N-replete, late exponential stage) were Oceanococcus and Marinoscillum. Under this condition, one Rhizobiales bacterium, Oricola, also increased in relative abundance. Our study documents the high diversity and dynamics of the G. balechii-associated microbiome, and identifies condition-specific sub-communities: the core (constitutive) microbiome that stably co-exists with G. balechii, the bacterial lineages that are responsive to N-nutrient variations, and species whose abundances are correlated with toxin content of the dinoflagellate. These findings demonstrate that particular bacterial groups are responsive to N-nutrient or toxicity changes of G. balechii and thus will be useful for further investigations on the associated microbiome’s interactions with benthic dinoflagellates and functions in the course of benthic harmful algae blooms.
Introduction
Harmful algal blooms caused by dinoflagellates and many lineages of algae, toxic or non-toxic, create negative impacts to the coastal economy, marine ecosystems and public health (Watson et al., 2015), and have increased in frequency and severity due to global climate and environmental changes (Vidyarathna and Granéli, 2013). Benthic dinoflagellates in the genus Gambierdiscus are able to produce ciguatoxins (CTXs) and hence responsible for ciguatera fish poisoning (CFP) in humans who consume CTX-contaminated fishes (Ansdell, 2014). CFP is one of the most severe tropical diseases worldwide, causing a series of syndromes in humans including short-term gastrointestinal disorders, cardiovascular disorders, long-lasting neurological disorders (Authority, 2010), and even paralysis, coma and death (Friedman et al., 2017). As many as 50,000 people worldwide are adversely affected by CFP each year (Ansdell, 2014).
Dinoflagellates usually co-exist and functionally interact with many microorganisms, and they together produce a dynamic microbial ecosystem (Patin et al., 2020). In the association, dinoflagellates provide the microbiome with organic carbon matter, while the associated bacteria metabolize the organic matter and recycle nutrients for the host dinoflagellates (Landa et al., 2017). In some cases, dinoflagellates can obtain vitamins from the associated bacteria (Tang et al., 2010). In other cases, bacteria are food sources for dinoflagellates capable of phagotrophy (Li et al., 2014). In addition, secretion of signaling molecules from bacteria can control cellular or communication processes in phytoplankton (Delucca and McCracken, 1977; Nakanishi et al., 1996; Amin et al., 2015). The presence of microbes in a dinoflagellate culture can even modulate the gene expression profile of dinoflagellates (Moustafa et al., 2010). Such interactions can regulate the growth of dinoflagellates and that of the associated microbiome (Tarazona-Janampa et al., 2020). The tight association between dinoflagellates and bacteria could explain why dinoflagellate cultures usually grow better in xenic than in axenic conditions. The associated microbiome has been suspected of either producing or modifying some dinoflagellate-derived toxins (Tosteson et al., 1989; Gallacher and Smith, 1999; Kodama et al., 2006; Tarazona-Janampa et al., 2020).
Some bacteria were observed to promote growth or toxicity of Gambierdiscus spp., with examples of Alteromonas sp. (Sakami et al., 1999), Marinobacter hydrocarbonoclasticus (Wang et al., 2018), and Bacillus anthracis (Wang et al., 2018). However, the general microbiome assembly and dominant lineages responding to varying Gambierdiscus physiologies (e.g., nutrient stress, growth stage, toxicity level) are less understood. In the present study, we conducted experiments with different N-nutrient conditions on xenic cultures of a toxic G. balechii strain and high-throughput sequencing of 16S rRNA gene (16S rDNA) for samples in two different growth stages. The primary goals were to identify the core (constitutive) G. balechii-associated microbiome, the N-nutrient responsive microbiome, and G. balechii growth stage (and toxin level)-specific microbiome.
Materials and Methods
Algal Culture and N-Nutrient Treatment
Gambierdiscus balechii was isolated from Marakei Island, Republic of Kiribati and cultured in State Key Laboratory of Marine Pollution, City University of Hong Kong (Dai et al., 2017). Specifically, seaweed samples were collected in Marakei Island, the Republic of Kiribati in November 2012 and then were shaken vigorously for about 1 min to detach the epiphytic cells. Samples containing detritus were sieved through 120-μm filters and then through 20-μm filters. The residue on 20-μm filters was rinsed with filtered seawater and transferred into a T25 cell culture flask (Thermo Fisher Scientific) containing filtered seawater. Single-cell isolation was then carried out using the micropipetting technique. Isolated cells were dispensed into wells of microwell plates (Thermo Fisher Scientific, IN, United States) filled with the L1 medium (30 psu; without NaSiO3; Guillard and Hargraves, 1993); prepared with filtered (0.22 μm; Millipore Corp.) seawater from the sampling site. After 30 days of incubation at a temperature of 25 ± 1°C under a 14: 10 h light: dark cycle with a photon flux of 100 μm photons m–2 s–1, surviving clones were transferred to the freshly prepared L1 medium in a T25 cell culture flask (Thermo Fisher Scientific, IN, United States) and cultured with a salinity of 30 ± 1 ‰ under the same temperature and illumination conditions as the initial incubation.
Prior to the commencement of the experiment, G. balechii was cultured for 50 days to consume as much N as possible. To investigate potential changes of the associated microbiome, the N-nutrient condition of G. balechii was varied. As described above, the N nutrient-replete (N-replete) group was grown in the L1 medium (with 882 μM NaNO3, without NaSiO3 addition). For the cultures of low-N content, the L1 medium was modified by reducing nitrate concentration to 1/200 that in L1 (4.41 μM). Both the N-replete and the low-N treatments were performed in triplicate, each at the volume of 800 mL in 1,000-mL flasks.
Measurements of Growth Rate and Chlorophyll (Chl) a Content
From day 1, a 1-mL sample was collected every two days from each of the cultures, fixed in Lugol’s solution, and examined microscopically with a Sedgewick-R after counting chamber for cell counts (Lin et al., 2012). The average daily population growth rate (μ) was calculated as μ = ln (N2/N1)/(t2−t1), where N2 and N1 are cell concentrations on day t2 and day t1, respectively. In addition, a 5 to 10 mL sample was filtered onto a Whatman GF/F membrane (25 mm diameter; Merck KGaA, Darmstadt, Germany). Chl a was extracted from the cell-containing membrane in 90% acetone for 24 h in the dark at 4°C, and determined through a microplate reader (BMG Polarstar Optima) (Ritchie, 2006).
Determination of P-CTX-1 Equivalents Using Mouse Neuroblastoma Assay
Cells were harvested on day 43 and day 61, when cultures were in the early and late exponential stages, respectively. A 200-mL sample was taken from each culture and centrifuged at 3,901× g for 5 min. Toxins were extracted from the harvested G. balechii cells according to procedures described by Dai et al. (2017). Toxin content was determined using mouse neuroblastoma assay with Neuro-2a cells (ATCC ® CCL-131™), as previously reported (Chan et al., 2011; Xu et al., 2014). Purified P-CTX-1 was used as a standard for calibration with concentrations ranging from 0.00975 to 0.31200 ppb. The detection limit in this assay was 1.37 × 10–2 fg P-CTX-1 eq cell–1. The assay was conducted twice for each sample, and toxin contents from triplicates of Gambierdiscus cultures were averaged and presented as P-CTX-1 eq cell–1.
High-Throughput Sequencing of Microbiome 16S rDNA Sequencing
Ten-mL samples were collected from the triplicate G. balechii cultures from groups N-replete and low-N in the early (μ = 0.13 and 0.04 d–1, respectively) and late exponential stages (μ = 0.04 and 0.01 d–1, respectively), and were filtered onto 0.22-μm nitrocellulose membranes (Merck Millipore, Darmstadt, Germany). The membranes were then transferred into a 2-mL micro-centrifuge tube and immersed in 1 ml of DNA lysis buffer (containing 0.1 M EDTA, 1% sodium dodecyl sulfate, 8 μg of lysozyme). Samples incubation (at 55°C) and DNA extraction followed a CTAB protocol combined with the Zymo DNA Clean & Concentrator kit (Zymo Research Corp., Orange CA, United States) as described previously (Zhang et al., 2005).
From each DNA extract, 16S rDNA in the V4-V5 region was amplified from each culture using primers of 515F (forward primer, 5′-GTGCCAGCMGCCGCGG-3′) and 907R (reverse primer 5′-CCGTCAATTCMTTTRAGTTT-3′). PCR was carried out as the following: 95°C for 2 min, followed by 30 cycles of incubation at 95°C (30 s), 55°C (30 s), and 72°C (30 s), with a final extension cycle of 5 min at 72°C. All PCR reactions used Phusion ® High Fidelity PCR Master Mix (New England Biolabs). The amplicons were checked on an agarose gel, where the major band positioned between 300-400 bp was recovered. The retrieved DNA was purified, uniquely barcoded for each sample, and pooled for sequencing on the IonS5™XL sequencing platform (Novogene Bioinformatics Technology Co., Ltd., Hong Kong).
Data Analysis
The high-throughput sequencing reads were sorted into samples based on their unique barcodes, and were trimmed of barcodes, and primer sequences. Sequences of all 12 samples are available in the Sequence Read Archive (SRA), National Center for Biotechnology Information (NCBI1 under the accession number PRJNA578018. Quality filtering on the trimmed reads was performed following the Cutadapt quality control process (Martin, 2011). Chimera sequences were detected using the UCHIME algorithm (Edgar et al., 2011) by comparing reads with the Gold database (Version microbiomeutil-r201105192) and then was removed. The resulting high quality sequences were grouped into Operational Taxonomic Units (OTUs) at 97% sequence similarity via the UPARSE software (Version 7.0.1001; Edgar, 2013). Silva Database (Version 132; Quast et al., 2012) was used for taxonomic annotation based on the Mothur software (Schloss et al., 2009), and OTUs were denominated at the domain, phylum, class, order, family, and genus levels. Their relative abundances were estimated based on their read counts normalized to the total number of good quality reads. In order to study the phylogenetic relationship of different OTUs and the difference of the dominant species in different sample groups, multiple sequence alignment was carried out via the MUSCLE software (Version 3.8.31; Edgar, 2004). Shannon, Simpson Chao 1, ACE, and Good’s coverage diversity indices were calculated using QIIME (Version 1.7.0; Caporaso et al., 2010) and displayed with the R software (Version 2.15.3; R Core Team, 2013). To evaluate inter-sample differences in species complexity, beta diversity based on weighted UniFrac was calculated with the QIIME software (Version1.7.0; Caporaso et al., 2010). An unweighted pair-group method with arithmetic (UPGMA) tree was constructed using the weighted UniFrac distance matrix which was also conducted with QIIME (Version 1.7.0). A distance matrix of weighted UniFrac among samples obtained was then transformed to a new set of orthogonal axes, by which the maximum variation factor is demonstrated by the first principal coordinate and the second maximum one by the second principal coordinate. Weighted UniFrac principal coordinate analysis (PCoA) was displayed with the weighted correlation network analysis (WGCNA) package, stats packages, and ggplot2 package in the R software (Version 2.15.3; R Core Team, 2013). Venn diagram was displayed using R (Version 2.15.3; R Core Team, 2013) and hierarchical clustering was displayed by QIIME (Version 1.7.0; Caporaso et al., 2010). The abundance distribution heatmaps of the 35 most dominant orders or genera that have the most sequences were generated by R (Version 2.15.3; R Core Team, 2013). The relative abundance of species is indicated by a Z value in each row of the heatmap after standardization. No further data filtration was conducted in the current study so singletons and low-abundance samples might remain.
Correlation tests using bivariate non-parametric Spearman rank’s correlation coefficient and Mann-Whitney U test were conducted with the SPSS Statistics 17.0 software package. A correlation between two items was considered statistically robust if the Spearman correlation coefficient (|r|) was ≥ 0.5 and p < 0.05 (Niven and Deutsch, 2012). All data were presented as means with standard deviation calculated from the triplicate cultures under each of the N conditions and growth stages.
Results
Culture Growth, Chl a, and Toxin Content
Gambierdiscus balechii cell concentrations were the same initially in both the N-replete and N-groups but became higher in the N-replete than the low-N group after 34 days. The N-replete group displayed a maximum cell concentration of 3,015 cells mL–1 (n = 3) (Table 1). In contrast, the maximum cell concentration for the low-N culture was significantly lower (557 cells mL–1; n = 3; Mann-Whitney U test, p < 0.05) (Table 1).
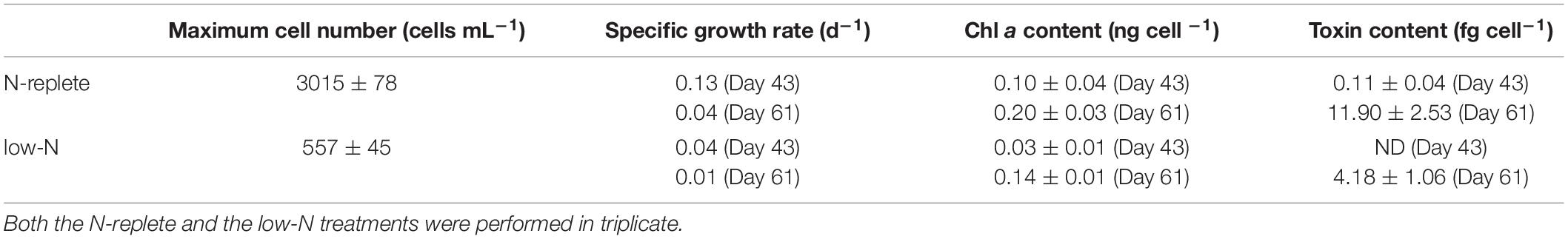
Table 1. Physiological parameters of G. balechii under N-replete and low-N conditions.
Toxin and Chl a contents of G. balechii cells were higher in the late exponential growth stage (day 61) than the early exponential stage (day 43) in both N conditions and higher in the N-replete than in the low-N groups (Table 1). In the late exponential stage of G. balechii, toxin content in N-replete group was 11.90 fg P-CTX-1 eqcell–1 (n = 3) and Chl a content was 0.14 ng cell –1 (μ = 0.04 d–1; n = 3) while 4.18 fg P-CTX-1 eq cell–1 (n = 3; Mann-Whitney U test, p < 0.05) of toxin content and 0.03 ng cell –1 (n = 3; Mann-Whitney U test, p < 0.05) of Chl a content was detected on day 43 when μ was 0.01 d–1 (n = 3) under the low-N condition. In the early exponential growth stage, toxin content was not detectable in the low-N cultures (μ = 0.04 d–1; n = 3) and 0.11 fg P-CTX-1 eq cell–1 (n = 3) in the N-replete cultures (μ = 0.13 d–1; n = 3) (Table 1). For the Chl a contents, 0.20 ng cell –1 (n = 3) was detected in the N-replete cultures while 0.10 ng cell –1 (n = 3) was detected in the low-N cultures.
Microbiome Profiles
Sequencing of the total of 12 libraries generated 926,438 reads which were classified into 16 phyla. Good’s coverage values, the sample completeness indicator (Chao and Jost, 2012), were approximately 1 in all treatment groups, indicating that the sequencing depth was sufficient to capture most of the taxa in the bacterial communities (Table 2). A total of 926,438 clean reads were obtained from the 12 samples collected from the early and late exponential growth stages (EE, LE) in the two N treatment groups, with sequences per sample ranging from 55,761 to 90,251 (Table 2). The taxonomy annotation analysis resulted in a total of 54 species from 16 phyla of bacteria. These sequences were clustered into 3,879 OTUs based on 97% identity criterion, with an average of 323 OTUs per sample (Table 2). No significant difference (Mann-Whitney U test, P > 0.05) of bacterial diversity represented by OTU numbers, Shannon index, Simpson index, Chao1 index, or ACE index was detected between different N treatment groups or growth stages, suggesting that the bacterial diversity was not affected by N nutrient limitation or growth stages.
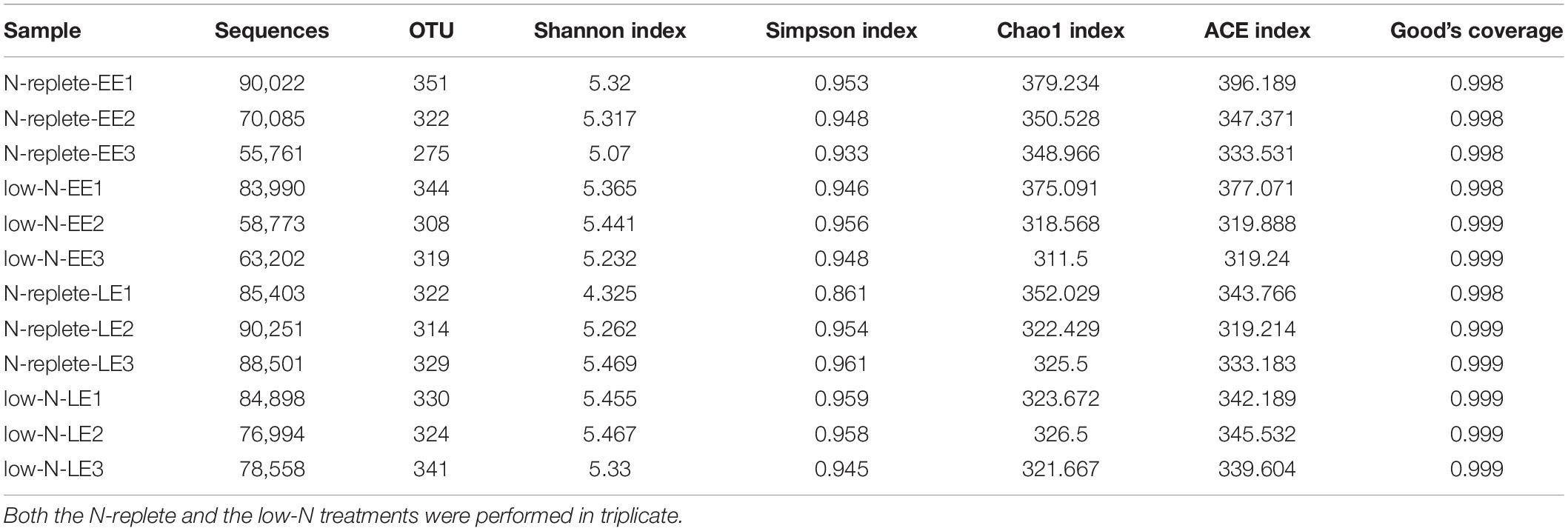
Table 2. OTU numbers and alpha diversity indices of the bacterial communities from G. balechii cultures under N-replete and low-N conditions during the early and late exponential growth stages.
PCoA analysis (Figure 1) showed that the —low-N-LE samples clustered together and were separated from other samples, indicating that the overall bacterial community varied with N treatments and growth stages. However, in the early exponential stage, the N-replete microbiome was close to the low-N microbiome, consistent with the corresponding similarity of G. balechii cell concentrations, indicating that the physiological status of the alga did not yet diverge much in the early exponential phase between the two N-nutrient treatments. In addition, one of the triplicates for the N-replete-LE group was markedly deviant from the other two replicates, for unknown reasons (Figure 1).
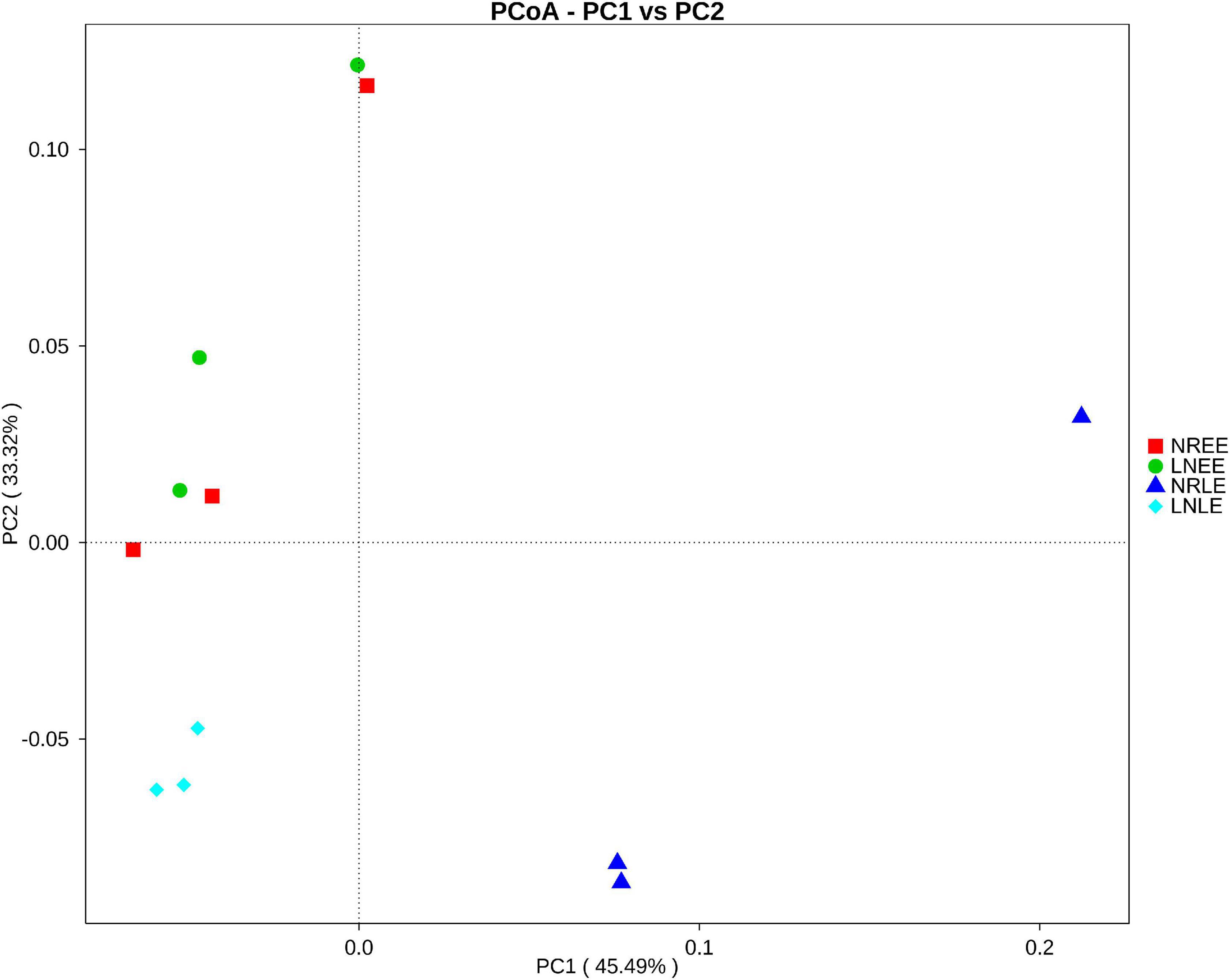
Figure 1. Weighted UniFrac PCoA plot of the bacterial community from G. balechii cultures under N-replete and low-N conditions during the early and late exponential growth stages. Both the N-replete and the low-N treatments were performed in triplicate. NREE: N-replete samples in the early exponential growth stage; LNEE: low-N samples in the early exponential growth stage; NRLE: N-replete samples in the late exponential growth stage; LNLE: low-N samples in the late exponential growth stage.
The relative abundance of Proteobacteria, Bacteroidetes, and Planctomycetes were the three most dominant phyla, representing around 90% in each of the N treatment groups (Figure 2). The most abundant phylum, Proteobacteria, contributed more than 70% in each N treatment of G. balechii. The contribution of Bacteroidetes increased markedly, from an average percentage of 4.31 to 9.39%, in the N-replete treatment from the early to the late exponential stage, but stayed relatively stable in the low-N treatments with an average percentage of 6.76 and 6.70% in the two growth stages, respectively (Figure 2). The abundance of Planctomycetes increased from 5.64 to 18.32% on average in the low-N groups from the early to the late exponential stage but ranged from 8.05 to 9.40% in the time course in the N-replete groups (Figure 2).
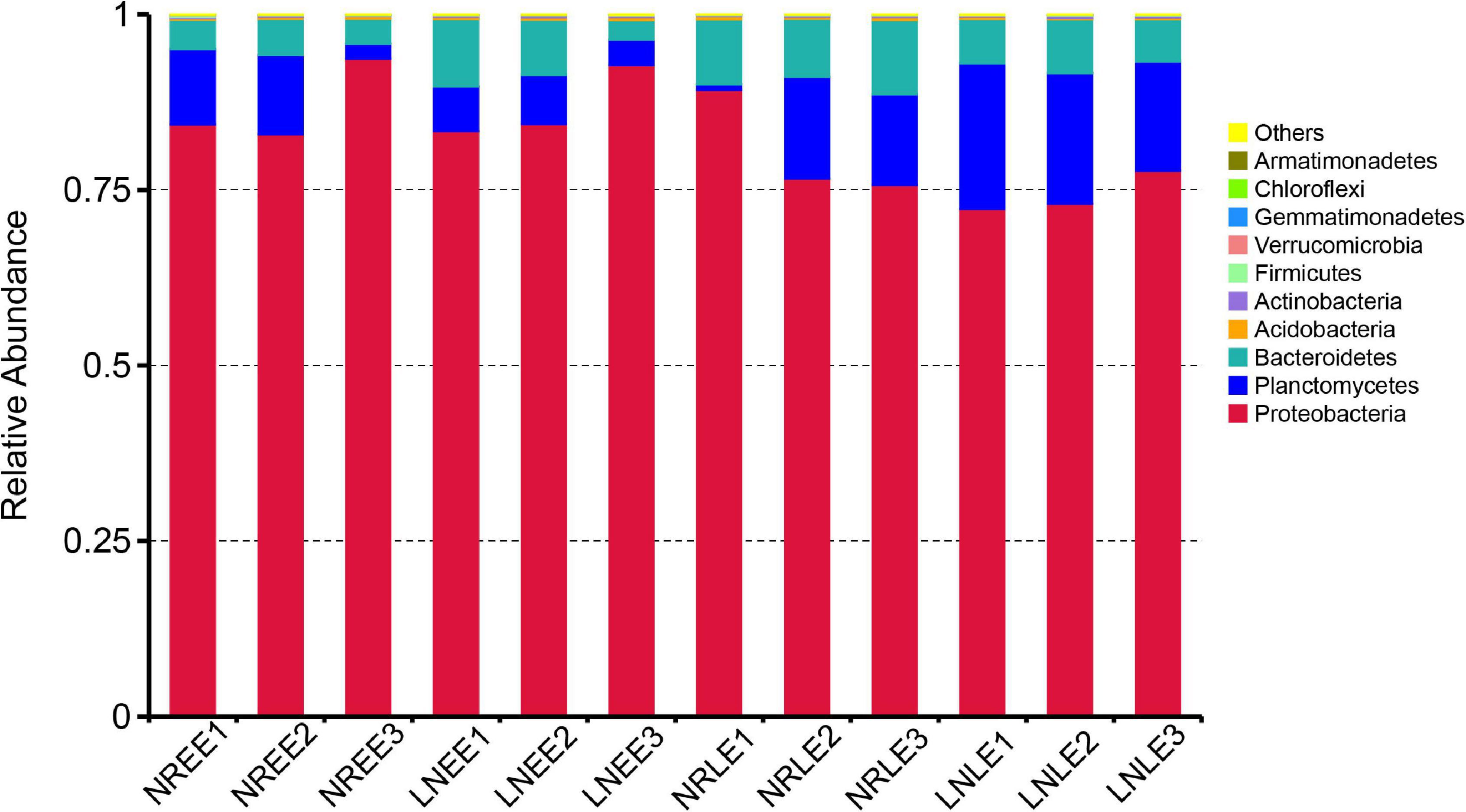
Figure 2. Relative abundance of different bacterial phyla in the G. balechii cultures under N-replete and low-N conditions during the early and late exponential stages. Both the N-replete and the low-N treatments were performed in triplicate. NREE, N-replete samples in the early exponential growth stage; LNEE, low-N samples in the early exponential growth stage; NRLE, N-replete samples in the late exponential growth stage; LNLE, low-N samples in the late exponential growth stage.
Common and Condition-Responsive Components of the Microbiome
The bacterial compositions were highly similar in general across all four conditions, sharing 285 OTUs (Figure 3). At the same time, 47, 28, 36, and 49 OTUs (totally 160 OTUs) were unique to groups N-replete-EE, N-replete-LE, low-N-EE, and low-N-LE, respectively (Figure 3). From these 285 common OTUs, 140 genera were identified, including 35 abundant genera, of which 11 were similar in abundance across all four conditions (Mann-Whitney U test, no significant difference, P > 0.05; Supplementary Table 1). These included Labrenzia and Muricauda, among the top 10 most abundant genera (Supplementary Table 1). These 11 genera potentially are the core constituents of the G. balechii-associated microbiome.
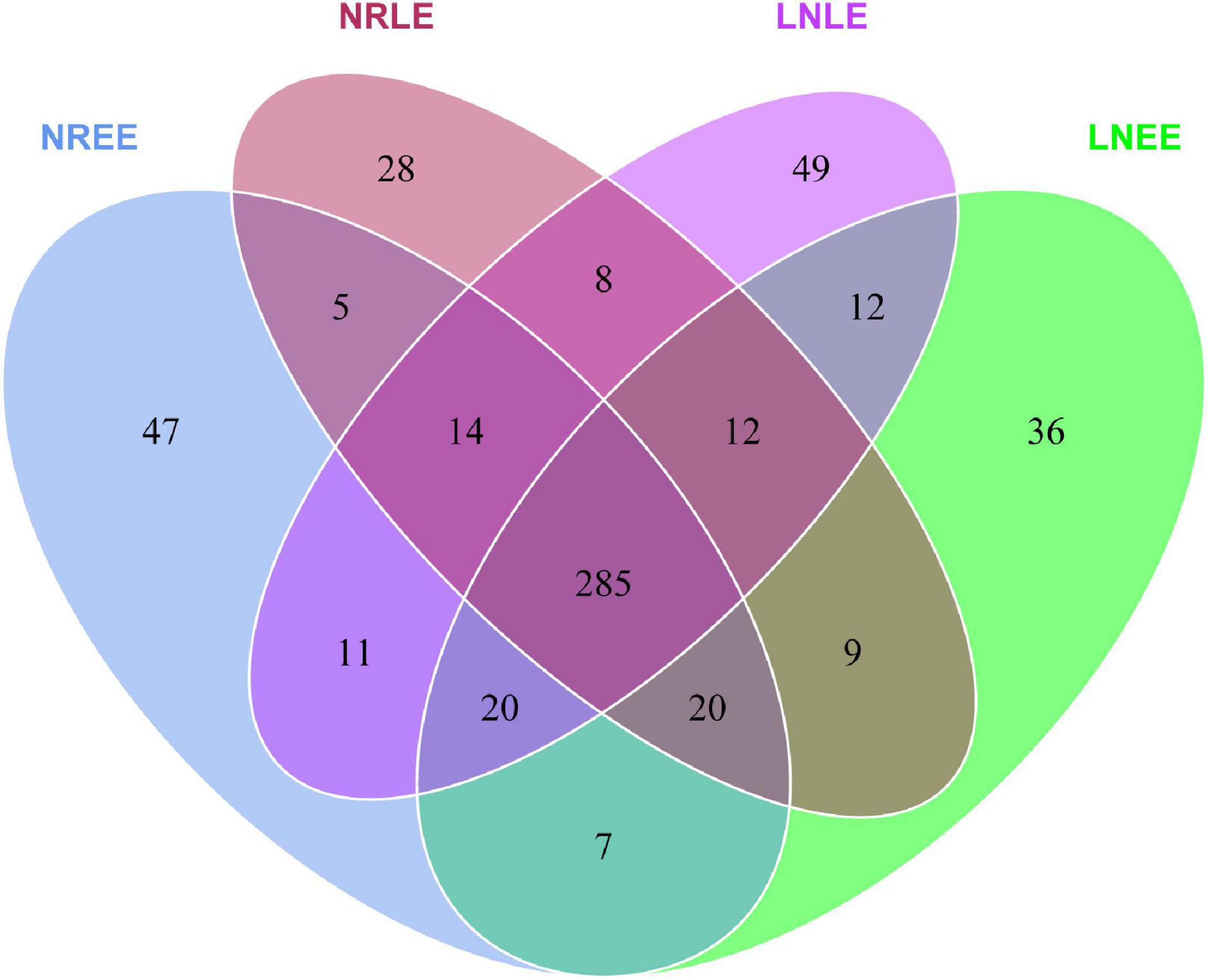
Figure 3. Venn diagrams showing the shared and specific OTUs in groups N-replete-EE, N-replete-LE, low-N-EE, and low-N-LE. Both the N-replete and the low-N treatments were performed in triplicate. NREE, N-replete samples in the early exponential growth stage; LNEE, low-N samples in the early exponential growth stage; NRLE, N-replete samples in the late exponential growth stage; LNLE, low-N samples in the late exponential growth stage.
In addition to the 28-49 OTUs being exclusive to one of the four treatment groups (Figure 3), some of the common taxa changed in abundance with varying conditions. The abundance distribution of the dominant 35 orders with more sequences among all samples was displayed in the species abundance heatmap (Figure 4A). These taxa were grouped into three major clusters (A–C) across the four treatment groups (n = 3) (Figure 4A). These bacterial orders showed specific abundance characteristics in different N treatments. Specifically, three orders in Cluster A were dominant in group low-N-LE while the other six, such as Alteromonadales, were dominant in group N-replete-EE (n = 3; Figure 4A). Ten orders were most abundant in group N-replete-LE (n = 3), which fell into Cluster B (Figure 4A). For instance, the abundance of Rhizobiales was the highest in group N-replete-LE, but it was only 10% in groups N-replete-EE, low-N-EE, and low-N-LE (n = 3; Figure 4A). Cluster C included 16 orders whose abundances were relatively low in groups N-replete-EE and N-replete-LE, but were higher under N deficiency (n = 3; Figure 4A). Flavobacteriales and Planctomycetales, in particular, were less abundant in groups N-replete-EE and N-replete-LE but were dominant in group low-N, especially in low-N-LE (n = 3; Figure 4A).
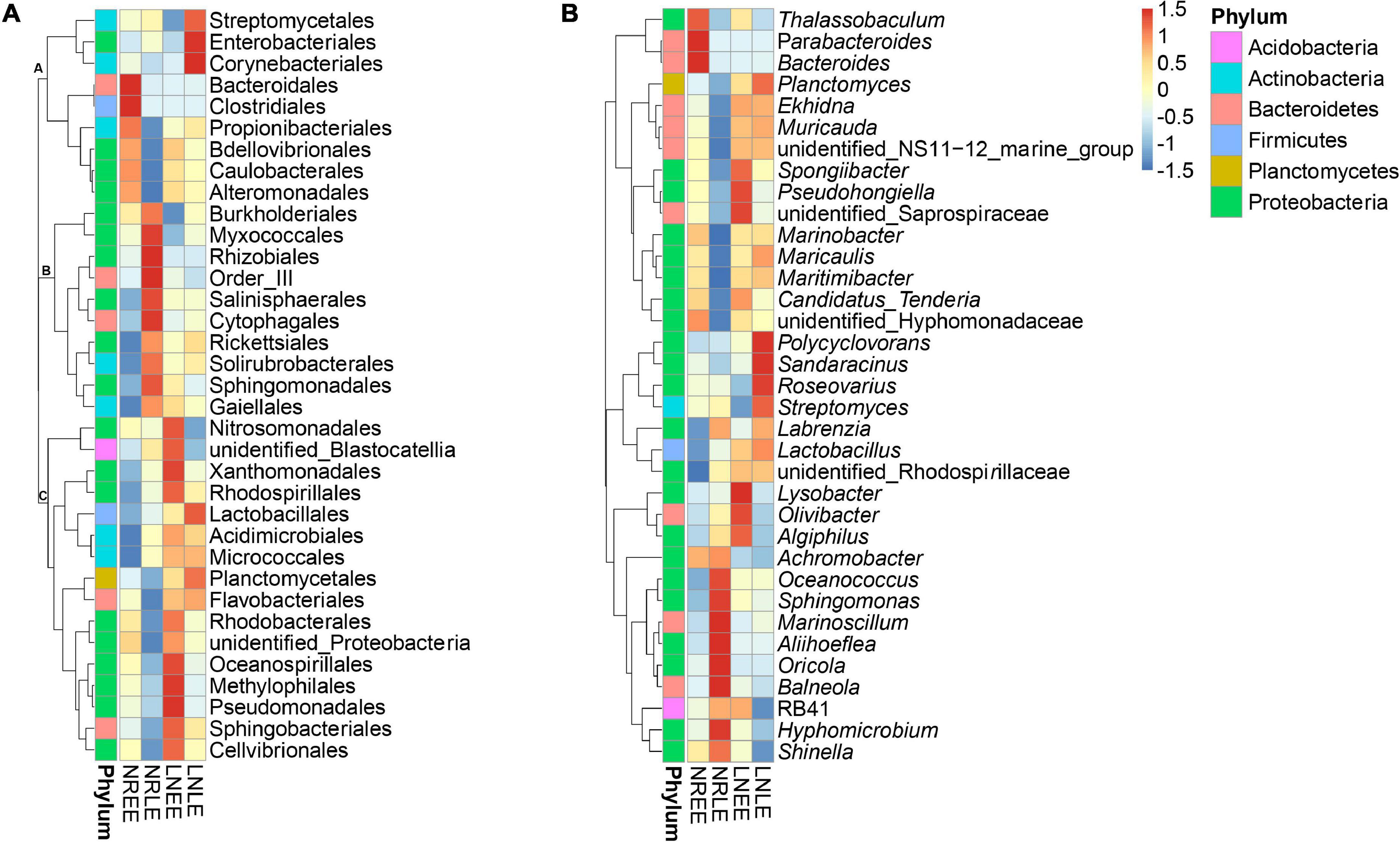
Figure 4. The distribution heatmap of bacterial abundance clusturing in the N-replete and low-N conditions during the early and late exponential stages. Thirty-five orders (A) and genera (B) were classified as differentially abundant and grouped by similarity of patterns. Red color indicates increased abundance while blue indicates reduced abundance. Letters (A-C) indicate clusters of similar patterns. Both the N-replete and the low-N treatments were performed in triplicate. NREE, N-replete samples in the early exponential growth stage; LNEE, low-N samples in the early exponential growth stage; NRLE, N-replete samples in the late exponential growth stage; LNLE, low-N samples in the late exponential growth stage.
At the genus level, the high-abundant genera in the N-replete culture groups included Achromobacter and Shinella (Figure 4B), and that in the low-N groups were mainly Planctomyces, Ekhidna, and Lactobacillus (Figure 4B). These, along with the condition-exclusive taxa, are potentially N nutrient-responsive components of the G. balechii-associated microbiome.
Dominant Bacterial Groups Correlated With Toxin Content in Gambierdiscus balechii
Taxonomic analysis showed a shift of bacterial community at the genus level from the early exponential stage (low toxin content) to the late exponential stage (high toxin content) (Figure 4B). The Spearman correlation analysis between the abundance of the 35 dominant genera and the toxin content indicated that both positive and negative correlations were found (Table 3). Of these, Oceanococcus (r = 0.606, p < 0.05; Table 3) and Marinoscillum (r = 0.761, p < 0.05; Table 3) showed positive correlations with toxin content of G. balechii (Table 3). These genera were dominant in group N-replete-LE (Figure 4B), the condition under which toxin content was the highest of all the treatments (11.90 fg P-CTX-1 eq cell–1, Table 1). One Rhizobiales genus, Oricola, also showed a significant positive correlation with the toxin content of G. balechii (r = 0.634, p < 0.05; Table 3). On the contrary, negative correlations were seen between some bacterial genera and the toxin content of G. balechii. For instance, Spongiibacter, Candidatus Tenderia, Pseudohongiella, and Thalassobaculum responded negatively to the toxin content as they showed negative correlations (−0.9 < rs < −0.5, p < 0.05; Table 3).
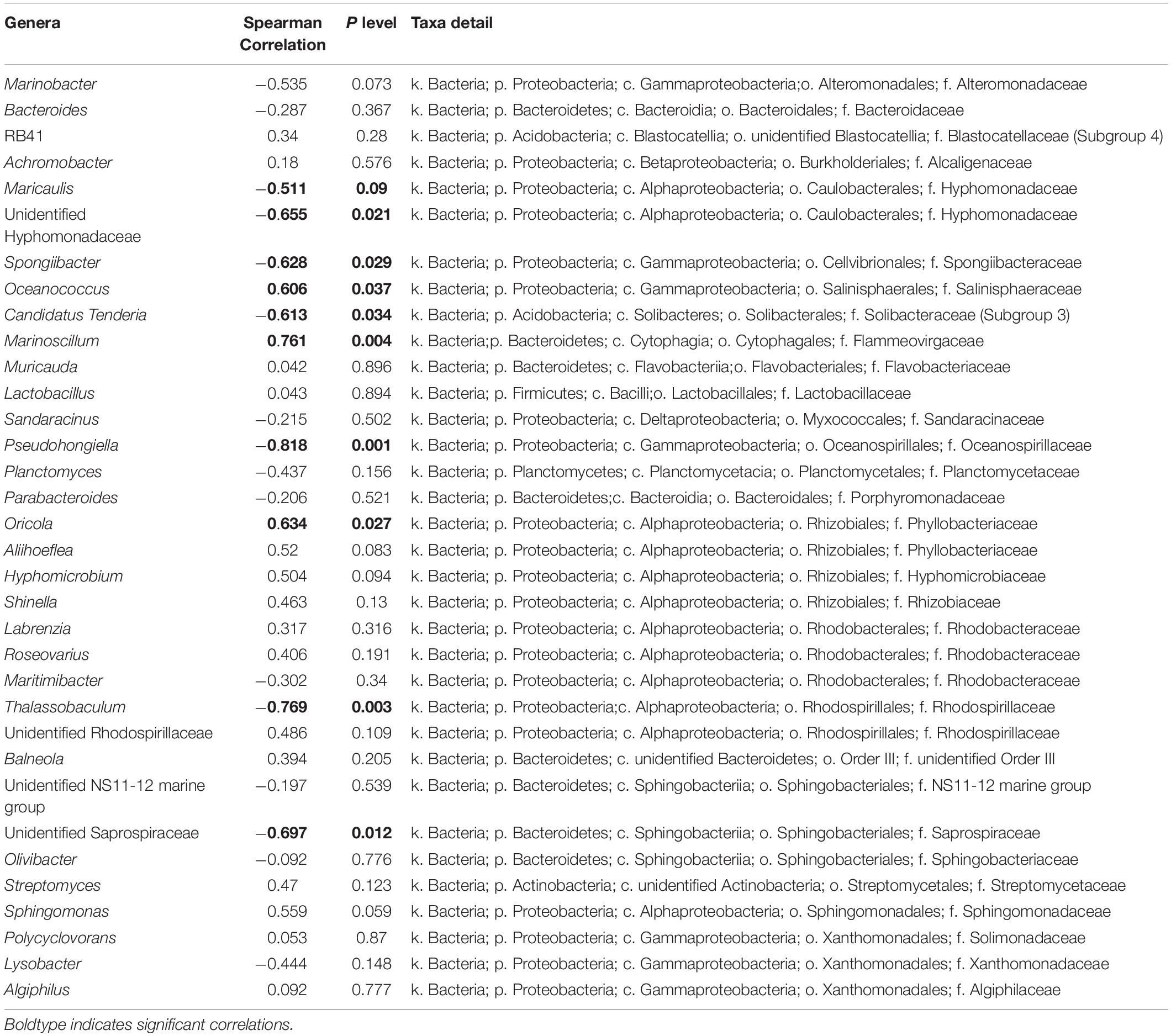
Table 3. Spearman correlation analysis between toxin content and abundance of the 35 dominant genera.
Discussion
This study profiled the G. balechii-associated microbiome compositions under N-replete and low-N conditions and their succession from the early to the late exponential growth stages. With the data, the relationship between the microbiome structure and G. balechii physiological condition as well as toxin content was explored. It is important to note that before the experiments in this study, G. balechii was cultured for 50 days to consume as much N as possible; therefore, prior selection bias on the microbiome might have occured.
Another caveat of this study is that N contents G. balechii cells and the medium were not measured. However, cultures under N-replete and low-N conditions exhibited significantly different levels of measured physiological parameters, including cell growth rate, Chl a content, and toxin content, indicating that the N nutrition contrasted sharply between the two treatments. Of these measured parameters, Chl a is an N-rich compound and its content is tightly coupled with N concentration in algae (Li et al., 2008). Under such an experimental setting, for the microbiome, our study showed that some members were shaped by the growth phases of G. balechii while some others appeared to be responsive to N condition or in correlation with toxin content.
Identification of Core (Constitutive) Component of Gambierdiscus balechii-Associated Microbiome
Arguably, bacterial taxa that are common and consistently abundant across different culturing and physiological conditions of G. balechii can be regarded as the core or stable constituents of the associated microbiome of the dinoflagellate. This notion is similar to previous work on Prorocentrum (Wang et al., 2017; Kim et al., 2021), Alexandrium spp. (Sörenson et al., 2019), and Symbiodiniacean species (Lawson et al., 2018; Nitschke et al., 2020). In the present study, we found that Labrenzia and Muricauda were the most abundant and common bacteria shared across all cultures. Labrenzia was frequently reported to be present with microalgae and corals and has also been identified as the core component of the Symbiodiniaceae species (Lawson et al., 2018). This genus is notable for the production of dimethylsulfoniopropionate (Curson et al., 2017), the high concentration of which likely plays a role in stress tolerance of microalgae (Deschaseaux et al., 2014). Muricauda and other Flavobacteriaceae are known to associate with dinoflagellates, and some of them can degrade both high and low molecular weight compounds exuded from microalgae and secrete surface proteins that might facilitate their attachment (Buchan et al., 2014).
The presence of Labrenzia and Muricauda in all treatment groups from both growth stages in the present study suggests that they are likely the core microbiome associated with the G. balechii strain across our experimental conditions. Whether these bacteria are common in natural assemblages will need further investigation.
N-Responsive Microbiome in Gambierdiscus balechii Culture
Our high throughput sequencing results indicate that some of the G. balechii-associated microbial taxa are potentially responsive to N nutrient conditions. First, 160 OTUs were found exclusively in one of the two N conditions. In addition, among the taxa in the 35 most abundant and commonly present orders, three orders in cluster A and the majority in cluster C were promoted markedly in low-N treatment relative to the N-replete groups, indicating that they are better adapted to or more competitive under N deficiency directly or indirectly through the organic compounds produced and released by G. balechii. Most of these bacteria came from Proteobacteria whereas other phylotypes included Acidobacteria, Firmicutes, Actinobacteria, and Bacteroidetes. One of the bacterial orders from cluster C, Flavobacteriales, appears to be able to metabolize organic phosphorus (P) for phytoplankton and provide phosphate under the low-inorganic P condition (Wang et al., 2017). Additionally, Maritimibacter of Rhodobacterales, showing high abundances in low-N groups, has been reported to be abundant in the Fe-limited culture of the dinoflagellate Akashiwo sanguinea (Li et al., 2019). Considering that Flavobacteriales and Rhodobacterales species are usually abundant in coastal waters and can be thriving in algal blooms (Buchan et al., 2014), it is possible that these types of bacteria provide nutrients for microalgae under low-nutrient environments by quickly recycling ambient organic nitrogen. Besides, G. balechii may ingest cluster A and cluster C bacteria to survive N deficiency as its mixotrophic potential exists (Price et al., 2016).
Nevertheless, the abundance of cluster B bacteria was notably high in group N-replete-LE while remaining relatively low under N deficiency. One of the cluster B phylotypes, Rhizobiales, contains symbiont species of plants and some algae that are able to fix N2 for their hosts under N limitation (Mitsustin and Sil’nikova, 1968; Dittami et al., 2014; Kim et al., 2014); however, they did not respond to N deficiency in the present study, suggesting they may not be able to fix N2 for G. balechii. Albeit less known, Rhizobiales bacteria have been reported in other dinoflagellates as well, including Symbiodinium minutum (Shoguchi et al., 2013), Prorocentrum lima (Biebl et al., 2006), Alexandrium (Biebl et al., 2006; Palacios et al., 2006), and Akashiwo sanguinea (Chen et al., 2021). Besides, a Rhizobium species has been isolated from G. balechii cultures, and shown not to contain the conserved N2-fixing enzyme gene, nifH (Wu et al., 2021).
Bacteria in Correlation With Toxin Production of Gambierdiscus balechii
Many dinoflagellate species are able to produce potent neurotoxins and accumulating evidence has shown that some bacteria may play a role in this process of dinoflagellates, such as the okadaic acid -producing Prorocentrum spp. (Perez et al., 2008), the dinophysistoxin-producing Dinophysis (Perez et al., 2008), toxin-producing Ostreopsis lenticularis (Tosteson et al., 1989), and CTX-producing Gambierdiscus spp. (Wang et al., 2018). In this study, we observed that some bacteria showed a positive correlation of relative abundance (from the sequencing results) with toxin content in G. balechii cultures, including Marinoscillum and Oceanococcus, both of which are aquatic bacteria that have been identified from dinoflagellate-associated microbiomes (Lu et al., 2000; Wang et al., 2017; Guidi et al., 2018; Lawson et al., 2018). Besides, one Rhizobiales bacterium, Oricola, was also positively correlated with the toxin contents of G. balechii. Likewise, a high proportion of Rhizobiales species was detected in the toxic P. lima cultures, yet the evidence is lacking if it could promote toxin production in P. lima (Prokic et al., 1998). These bacteria might be involved in the toxin production directly or indirectly through metabolizing algal toxins to yield known congeners or chemically modified derivatives from the same toxin classes as previously described (Stewart et al., 1998; Hold et al., 2001; Smith et al., 2001; Kodama et al., 2006). It is also possible that algal toxins serve as nutrients for these bacteria (Sison-Mangus et al., 2016).
By contrast, some bacteria were intensely inhibited by G. balechii toxins. These mainly included Spongiibacter, Candidatus Tenderia, Pseudohongiella, and Thalassobaculum. Antagonism may exist between G. balechii and these bacteria as toxins could act as a selective agent to alter the associated bacterial community. Additionally, some bacteria can be antagonistic to algae, and their presence can provoke an increase in phycotoxin production as a specific response to the bacteria’s presence (Kaczmarska et al., 2005; Su et al., 2011).
Among the bacterial genera detected, Marinobacter is a common dinoflagellate-associated bacterium found in both cultures and field samples (Seibold et al., 2001; Green et al., 2006; Bolch et al., 2011; Tarazona-Janampa et al., 2020). Bacterial species of this genus have been found to alter toxin profiles of dinoflagellates (Wang et al., 2018) or possess genes related to paralytic shellfish toxin biosynthesis (Yang et al., 2019). In this study, no clear relationship was found between the abundance of this bacterial genus and the toxin content of G. balechii. The difference between studies might reflect the different roles of different Marinobacter species associated with different species of dinoflagellates.
N or other nutrient concentrations can affect metabolisms of algae and thus influence algal growth and toxin synthesis, which can ultimately influence the composition and diversity of the algae-associated bacterial community. Our study shows that N-nutrient conditions are associated with particular bacterial groups and related to the toxin synthesis level in G. balechii. However, the role of N-nutrient status in influencing toxin synthesis in dinoflagellates remains to be further elucidated. Nevertheless, it is also possible that the bacteria correlated with toxin content in G. balechii can influence toxin synthesis in this dinoflagellate directly or indirectly. These possibilities remain to be disentangled in future research using interdisciplinary methods such as the hologenomic approach to obtain the genomes of both the symbiotic bacteria and dinoflagellates.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA578018.
Author Contributions
ZW and SL: conceptualization and methodology. ZW and WL: investigation. ZW and ZL: data curation, software, and visualization. ZW: writing—original draft. ZL, SL, and PL: writing—review and editing. SL and PL: supervision. ZW, SL, and PL: project administration. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Meng Yan and Hua Zhang for their kind assistance in revising this manuscript. The study was supported by the Collaborative Research Fund from the Research Grant Council (C1012-15G) of Hong Kong, National Key Research and Development Program of China grant (2017YFC1404302), and National Natural Science Foundation of China (42106093).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.760553/full#supplementary-material
Footnotes
References
Amin, S., Hmelo, L., Van Tol, H., Durham, B., Carlson, L., Heal, K., et al. (2015). Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature 522, 98–101. doi: 10.1038/nature14488
Authority, E. F. S. (2010). Scientific Opinion on marine biotoxins in shellfish – Cyclic imines (spirolides, gymnodimines, pinnatoxins and pteriatoxins). EFSA Panel on Contaminants in the Food Chain. EFSA J. 8, 1–39.
Biebl, H., Tindall, B. J., Pukall, R., Lünsdorf, H., Allgaier, M., and Wagner-Döbler, I. (2006). Hoeflea phototrophica sp. nov., a novel marine aerobic alphaproteobacterium that forms bacteriochlorophyll a. Int. j. syst. Evol. Microbiol. 56, 821–826. doi: 10.1099/ijs.0.63958-0
Bolch, C. J., Subramanian, T. A., and Green, D. H. (2011). The toxic dinoflagellate Gymnodinium catenatum (Dinophyceae) requires marine bacteria for growth 1. J. Phycol. 47, 1009–1022. doi: 10.1111/j.1529-8817.2011.01043.x
Buchan, A., LeCleir, G. R., Gulvik, C. A., and González, J. M. (2014). Master recyclers: features and functions of bacteria associated with phytoplankton blooms. Nat. Rev. Microbiol. 12, 686–98. doi: 10.1038/nrmicro3326
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. methods 7, 335–6. doi: 10.1038/nmeth.f.303
Chan, W. H., Mak, Y. L., Wu, J. J., Jin, L., Sit, W. H., Lam, J. C., et al. (2011). Spatial distribution of ciguateric fish in the Republic of Kiribati. Chemosphere 84, 117–123. doi: 10.1016/j.chemosphere.2011.02.036
Chao, A., and Jost, L. (2012). Coverage-based rarefaction and extrapolation: standardizing samples by completeness rather than size. Ecology 93, 2533–2547. doi: 10.1890/11-1952.1
Chen, H., JIang, J., Jiang, F., Li, S., and Hu, Z. (2021). Temporal variability of free-living microbial culturability and community composition after an Akashiwo sanguinea bloom in Shenzhen. China. Ecotoxicol. 30, 975–985. doi: 10.1007/s10646-021-02407-4
Curson, A., Liu, J., Martínez, A. B., Green, R. T., Chan, Y., Carrión, O., et al. (2017). Dimethylsulfoniopropionate biosynthesis in marine bacteria and identification of the key gene in this process. Nat. Microbiol. 2:17009. doi: 10.1038/nmicrobiol.2017.9
Dai, X., Mak, Y. L., Lu, C.-K., Mei, H.-H., Wu, J. J., Lee, W. H., et al. (2017). Taxonomic assignment of the benthic toxigenic dinoflagellate Gambierdiscus sp. type 6 as Gambierdiscus balechii (Dinophyceae), including its distribution and ciguatoxicity. Harmful Algae 67, 107–118. doi: 10.1016/j.hal.2017.07.002
Delucca, R., and McCracken, M. D. (1977). Observations on interactions between naturally-collected bacteria and several species of algae. Hydrobiologia 55, 71–75.
Deschaseaux, E. S., Jones, G. B., Deseo, M. A., Shepherd, K. M., Kiene, R., Swan, H., et al. (2014). Effects of environmental factors on dimethylated sulfur compounds and their potential role in the antioxidant system of the coral holobiont. Limnol. Oceanogr. 59, 758–768.
Dittami, S. M., Barbeyron, T., Boyen, C., Cambefort, J., Collet, G., Delage, L., et al. (2014). Genome and metabolic network of “Candidatus Phaeomarinobacter ectocarpi” Ec32, a new candidate genus of Alphaproteobacteria frequently associated with brown algae. Front. Genet. 5:241. doi: 10.3389/fgene.2014.00241
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. methods 10, 996–8. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Friedman, M. A., Fernandez, M., Backer, L. C., Dickey, R. W., Bernstein, J., Schrank, K., et al. (2017). An updated review of ciguatera fish poisoning: clinical, epidemiological, environmental, and public health management. Mar. Drugs 15:72. doi: 10.3390/md15030072
Gallacher, S., and Smith, E. A. (1999). Bacteria and paralytic shellfish toxins. Protist 150, 245–255. doi: 10.1016/s1434-4610(99)70027-1
Green, D. H., Bowman, J. P., Smith, E. A., Gutierrez, T., and Bolch, C. J. (2006). Marinobacter algicola sp. nov., isolated from laboratory cultures of paralytic shellfish toxin-producing dinoflagellates. Int. J. Syst. Evol. Microbiol. 56, 523–527. doi: 10.1099/ijs.0.63447-0
Guillard, R., and Hargraves, P. (1993). Stichochrysis immobilis is a diatom, not a chrysophyte. Phycologia 32, 234–236.
Hold, G. L., Smith, E. A., Rappë, M. S., Maas, E. W., Moore, E. R., Stroempl, C., et al. (2001). Characterisation of bacterial communities associated with toxic and non-toxic dinoflagellates: Alexandrium spp. and Scrippsiella trochoidea. FEMS Microbiol. Ecol. 37, 161–173.
Kaczmarska, I., Ehrman, J. M., Bates, S. S., Green, D. H., Léger, C., and Harris, J. (2005). Diversity and distribution of epibiotic bacteria on Pseudo-nitzschia multiseries (Bacillariophyceae) in culture, and comparison with those on diatoms in native seawater. Harmful Algae 4, 725–741. doi: 10.1016/j.hal.2004.10.001
Kim, B.-H., Ramanan, R., Cho, D.-H., Oh, H.-M., and Kim, H.-S. (2014). Role of Rhizobium, a plant growth promoting bacterium, in enhancing algal biomass through mutualistic interaction. Biomass Bioenergy 69, 95–105. doi: 10.1016/j.biombioe.2014.07.015
Kim, D. D., Wan, L., Cao, X., Klisarova, D., Gerdzhikov, D., Zhou, Y., et al. (2021). Metagenomic insights into co-proliferation of Vibrio spp. and dinoflagellates Prorocentrum during a spring algal bloom in the coastal East China Sea. Wat. Res. 204:117625. doi: 10.1016/j.watres.2021.117625
Kodama, M., Doucette, G. J., and Green, D. H. (2006). “Relationships between bacteria and harmful algae,” in Ecology of Harmful Algae, Ecological Studies, Vol. Vol. 189, eds E. Granéli and J. T. Turner (Berlin: Springer-Verlag), 243–255. doi: 10.1007/978-3-540-32210-8_19
Landa, M., Burns, A. S., Roth, S. J., and Moran, M. A. (2017). Bacterial transcriptome remodeling during sequential co-culture with a marine dinoflagellate and diatom. ISME j. 11, 2677–2690. doi: 10.1038/ismej.2017.117
Lawson, C. A., Raina, J. B., Kahlke, T., Seymour, J. R., and Suggett, D. J. (2018). Defining the core microbiome of the symbiotic dinoflagellate, Symbiodinium. Environ. Microbiol. Rep. 10, 7–11. doi: 10.1111/1758-2229.12599
Li, D., Zhang, H., Fu, L., An, X., Zhang, B., Li, Y., et al. (2014). A novel algicide: evidence of the effect of a fatty acid compound from the marine bacterium, Vibrio sp. BS02 on the harmful dinoflagellate, Alexandrium tamarense. PLoS One 9:e91201. doi: 10.1371/journal.pone.0091201
Li, S., Chen, M., Chen, Y., Tong, J., Wang, L., Xu, Y., et al. (2019). Epibiotic bacterial community composition in red-tide dinoflagellate Akashiwo sanguinea culture under various growth conditions. FEMS Microbiol. Ecol. 95, fiz057. doi: 10.1093/femsec/fiz057
Li, Y., Horsman, M., Wang, B., Wu, N., and Lan, C. Q. (2008). Effects of nitrogen sources on cell growth and lipid accumulation of green alga Neochloris oleoabundans. App. Microbiol. Biotechnol. 81, 629–636. doi: 10.1007/s00253-008-1681-1
Lin, X., Zhang, H., Huang, B., and Lin, S. (2012). Alkaline phosphatase gene sequence characteristics and transcriptional regulation by phosphate limitation in Karenia brevis (Dinophyceae). Harmful Algae 17, 14–24. doi: 10.1016/j.hal.2012.02.005
Lu, Y. H., Chai, T. J., and Hwang, D. F. (2000). Isolation of bacteria from dinoflagellate Alexandrium minutum and their effects on algae toxicity. J. Nat. Toxins. 9, 409–417.
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 1138-1143. doi: 10.1089/cmb.2017.0096
Mitsustin, E., and Sil’nikova, V. (1968). Biological fixation of atmospheric nitrogen. Moscow: Izdatel’stvo ’Nauka.
Moustafa, A., Evans, A. N., Kulis, D. M., Hackett, J. D., Erdner, D. L., Anderson, D. M., et al. (2010). Transcriptome profiling of a toxic dinoflagellate reveals a gene-rich protist and a potential impact on gene expression due to bacterial presence. PloS one 5:e9688. doi: 10.1371/journal.pone.0009688
Nakanishi, K., Nishijima, M., Nishimura, M., Kuwano, K., and Saga, N. (1996). Bacteria that induce morphogenesis in Ulva pertusa (Chlorophyta) grown under axenic conditions. J. Phycol. 32, 479–482.
Nitschke, M. R., Fidalgo, C., Simões, J., Brandão, C., Alves, A., Serôdio, J., et al. (2020). Symbiolite formation: a powerful in vitro model to untangle the role of bacterial communities in the photosynthesis-induced formation of microbialites. ISME J. 14, 1533–1546. doi: 10.1038/s41396-020-0629-z
Niven, E. B., and Deutsch, C. V. (2012). Calculating a robust correlation coefficient and quantifying its uncertainty. Comput. Geosci. 40, 1–9.
Palacios, L., Arahal, D. R., Reguera, B., and Marín, I. (2006). Hoeflea alexandrii sp. nov., isolated from the toxic dinoflagellate Alexandrium minutum AL1V. Int. J. Syst. Evol. Microbiol. 56, 1991–1995. doi: 10.1099/ijs.0.64238-0
Patin, N. V., Brown, E., Chebli, G., Garfield, C., Kubanek, J., and Stewart, F. J. (2020). Microbial and chemical dynamics of a toxic dinoflagellate bloom. PeerJ 8:e9493. doi: 10.7717/peerj.9493
Perez, R., Liu, L., Lopez, J., An, T., and Rein, K. S. (2008). Diverse bacterial PKS sequences derived from okadaic acid-producing dinoflagellates. Marine drugs 6, 164–179. doi: 10.3390/md20080009
Price, D. C., Farinholt, N., Gates, C., Shumaker, A., Wagner, N. E., Bienfang, P., et al. (2016). Analysis of Gambierdiscus transcriptome data supports ancient origins of mixotrophic pathways in dinoflagellates. Environ. Microbiol. 18, 4501–4510. doi: 10.1111/1462-2920.13478
Prokic, I., Brümmer, F., Brigge, T., Görtz, H. D., Gerdts, G., Schütt, C., et al. (1998). Bacteria of the genus Roseobacter associated with the toxic dinoflagellate Prorocentrum lima. Protist 149, 347–357. doi: 10.1016/S1434-4610(98)70041-0
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic acids res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Available online at https://www.R-project.org/
Ritchie, R. J. (2006). Consistent sets of spectrophotometric chlorophyll equations for acetone, methanol and ethanol solvents. Photosynth. Res. 89, 27–41. doi: 10.1007/s11120-006-9065-9
Sakami, T., Nakahara, H., Chinain, M., and Ishida, Y. (1999). Effects of epiphytic bacteria on the growth of the toxic dinoflagellate Gambierdiscus toxicus (Dinophyceae). J. exp. marine biol. ecol. 233, 231–246. doi: 10.1016/s0022-0981(98)00130-0
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Seibold, A., Wichels, A., and Schütt, C. (2001). Diversity of endocytic bacteria in the dinoflagellate Noctiluca scintillans. Aquat. microbial ecol. 25, 229–235. doi: 10.3354/ame025229
Shoguchi, E., Shinzato, C., Kawashima, T., Gyoja, F., Mungpakdee, S., Koyanagi, R., et al. (2013). Draft assembly of the Symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr. Biol. 23, 1399–1408. doi: 10.1016/j.cub.2013.05.062
Sison-Mangus, M. P., Jiang, S., Kudela, R. M., and Mehic, S. (2016). Phytoplankton-associated bacterial community composition and succession during toxic diatom bloom and non-bloom events. Front. Microbiol. 7:1433. doi: 10.3389/fmicb.2016.01433
Smith, E. A., Grant, F., Ferguson, C. M., and Gallacher, S. (2001). Biotransformations of paralytic shellfish toxins by bacteria isolated from bivalve molluscs. Appl. Environ. Microbiol. 67, 2345–2353. doi: 10.1128/AEM.67.5.2345-2353.2001
Sörenson, E., Bertos-Fortis, M., Farnelid, H., Kremp, A., Krüger, K., Lindehoff, E., et al. (2019). Consistency in microbiomes in cultures of Alexandrium species isolated from brackish and marine waters. Environ. Microbiol. Rep. 11, 425–433. doi: 10.1111/1758-2229.12736
Stewart, J. E., Marks, L., Gilgan, M., Pfeiffer, E., and Zwicker, B. (1998). Microbial utilization of the neurotoxin domoic acid: blue mussels (Mytilus edulis) and soft shell clams (Mya arenaria) as sources of the microorganisms. Can. J. microbiol. 44, 456–464.
Su, J., Yang, X., Zhou, Y., and Zheng, T. (2011). Marine bacteria antagonistic to the harmful algal bloom species Alexandrium tamarense (Dinophyceae). Biol. Cont. 56, 132–138.
Tang, Y. Z., Koch, F., and Gobler, C. J. J. P. O. T. N. A. O. S. (2010). Most harmful algal bloom species are vitamin B1 and B12 auxotrophs. Honolulu: University of Hawaii.
Tarazona-Janampa, U. I., Cembella, A. D., Pelayo-Zárate, M. C., Pajares, S., Márquez-Valdelamar, L. M., Okolodkov, Y. B., et al. (2020). Associated bacteria and their effects on growth and toxigenicity of the dinoflagellate Prorocentrum lima species complex from epibenthic substrates along mexican coasts. Front. Marine Sci. 7:569. doi: 10.3389/fmars.2020.00569
Tosteson, T., Ballantine, D., Tosteson, C., Hensley, V., and Bardales, A. (1989). Associated bacterial flora, growth, and toxicity of cultured benthic dinoflagellates Ostreopsis lenticularis and Gambierdiscus toxicus. Appl. Environ. Microbiol. 55, 137–141. doi: 10.1128/aem.55.1.137-141.1989
Vidyarathna, N. K., and Granéli, E. (2013). Physiological responses of Ostreopsis ovata to changes in N and P availability and temperature increase. Harmful algae 21, 54–63.
Wang, B., Yao, M., Zhou, J., Tan, S., Jin, H., Zhang, F., et al. (2018). Growth and toxin production of Gambierdiscus spp. can be regulated by quorum-sensing bacteria. Toxins 10, 257. doi: 10.3390/toxins10070257
Wang, C., Lin, X., Li, L., Lin, L., and Lin, S. (2017). Glyphosate shapes a dinoflagellate-associated bacterial community while supporting algal growth as sole phosphorus source. Front. Microbiol. 8:2530. doi: 10.3389/fmicb.2017.02530
Watson, S. B., Whitton, B. A., Higgins, S. N., Paerl, H. W., Brooks, B. W., and Wehr, J. D. (2015). Harmful algal blooms, Freshwater Algae of North America. Amsterdam: Elsevier, 873–920.
Wu, Z., Yang, X., Lin, S., Lee, W. H., and Lam, P. K. (2021). A Rhizobium bacterium and its population dynamics under different culture conditions of its associated toxic dinoflagellate Gambierdiscus balechii. Marine Life Sci. Technol. Volume 3, 542–551.
Xu, Y., Richlen, M. L., Morton, S. L., Mak, Y. L., Chan, L. L., Tekiau, A., et al. (2014). Distribution, abundance and diversity of Gambierdiscus spp. from a ciguatera-endemic area in Marakei. Repub. Kiribati. Harmful Algae 34, 56–68.
Yang, Q., Jiang, Z., Zhou, X., and Zhang, X. (2019). Draft genome sequence of Marinobacter sp. strain LZ-6, isolated from the toxic dinoflagellate Alexandrium catenella. Microbiol. Resour. Announ. 8, e795–e719. doi: 10.1128/MRA.00795-19
Keywords: Gambierdiscus balechii, ciguatoxin, 16S rRNA, bacterial community, nitrogen
Citation: Wu Z, Lee WH, Liu Z, Lin S and Lam PKS (2022) Microbiome Associated With Gambierdiscus balechii Cultures Under Different Toxicity Conditions. Front. Mar. Sci. 9:760553. doi: 10.3389/fmars.2022.760553
Received: 18 August 2021; Accepted: 17 January 2022;
Published: 09 February 2022.
Edited by:
Punyasloke Bhadury, Indian Institute of Science Education and Research Kolkata, IndiaReviewed by:
Allan Douglas Cembella, Alfred Wegener Institute Helmholtz Centre for Polar and Marine Research (AWI), GermanyXinguo Shi, Fuzhou University, China
Copyright © 2022 Wu, Lee, Liu, Lin and Lam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Senjie Lin, c2VuamllLmxpbkB1Y29ubi5lZHU=; Paul K. S. Lam, cGtzLmxhbUBjaXR5dS5lZHUuaGs=